

Abstract
Root depth, as an important component of root architecture, plays a significant role in growth, grain yield determination and abiotic stress tolerance in crop plants, but its genetic basis remains poorly elucidated. In this study, a panel composed of 323 wheat (Triticum aestivum L.) accessions was assessed for variation in root depth and genotyped with the Wheat 660K SNP Array. GWAS (genome‐wide association study) detected significant association between a 125 bp miniature inverted‐repeat transposable element (MITE) in the promoter of the TaVSR1‐B gene with root depth at the booting stage. We showed that the MITE repressed TaVSR1‐B expression by DNA methylation and H3K27 tri‐methylation. The roles of TaVSR1‐B in root growth were verified by altered expression of the gene in transgenic wheat, rice and a tavsr1 TILLING mutant. Increased TaVSR1‐B expression made the root elongation zone shorter and the differentiation zone longer, leading to deeper root. This work provides novel insight into the genetic basis of variation in root depth and a promising target for genetic improvement of root architecture in wheat.
Keywords: wheat, DNA methylation, H3K27me3, MITE, root depth, VSR
Introduction
Wheat (Triticum aestivum L.) is the most widely cultivated crop in the world, providing about 19% of the global calories consumed by humans (Godfray et al., 2010; Ray et al., 2013). With the increasing global demand for food and aggravated effects of abiotic stress, including drought and nutrient deficiency, there is an urgent need for breeding wheat cultivars with high and stable yields (Tester and Langridge, 2010; Wang et al., 2020). Roots play a vital role in plant development, not only for absorbing water and nutrients, but also for perceiving stress signals from the soil (Aida et al., 2004; Promchuea et al., 2017). Therefore, understanding root morphology and function is vitally important for wheat improvement.
Recently, several QTLs and genes for root‐related traits in wheat were identified using forward genetic approaches (Liu et al., 2013; Maccaferri et al., 2016; Soriano and Alvaro, 2019). For example, VERNALIZATION1 and TaMOR modulate root system architecture in wheat (Li et al., 2016; Voss‐Fels et al., 2018). On the other hand, GWAS has made significant contributions in wheat genetic research following the development of the Wheat 660K SNP Array, which consists of 630 517 SNPs with advantages of high efficiency and genome specificity (Cavanagh et al., 2013; Li et al., 2020; Zhou et al., 2018). However, the problem of observing root phenotype and the complexity of the wheat genome have hampered efficient identification of regulatory genes for root‐related traits in this crop species (Ayalew et al., 2018; Cane et al., 2014; Pang et al., 2020).
The booting stage is a key stage of yield determination, and root depth is an important aspect of root architecture (Li et al., 2019b; Wasson et al., 2012). In this study, a previously genotyped panel of 323 wheat accessions was used in a GWAS based on the Wheat 660K SNP Array. We identified TaVSR1‐B as a key regulatory gene for root depth at the booting stage. Enhanced expression of TaVSR1‐B conferred a deeper root in both transgenic wheat and rice, whereas a tavsr1 mutant has shallower root. The presence of a 125 bp MITE insertion in the TaVSR1‐B promoter was associated with shallower root, lower TaVSR1‐B expression, higher DNA methylation and H3K27 tri‐methylation. This work uncovered a function of TaVSR1‐B in root development, and the results could facilitate the optimization of root architecture in wheat breeding.
Results
TaVSR1‐B is associated with root depth at the booting stage
We previously used a panel of 323 wheat accessions to identify the genomic regions controlling root traits at late growth stages in wheat (Li et al., 2019a). In this study, we used the same data to study the genetic basis regulating root depth at the booting stage. The mean value for root depth was 57.4 cm, and the coefficient of variation and broad‐sense heritability were 12.4% and 78.7%, respectively, suggesting that the root depth at the booting stage was suitable for genetic analysis. In consideration of the population structure (Q) and kinship (K), marker–trait relationships were analysed using mixed linear model (MLM). A haplotype block with peak SNP (89 435 662‐89 562 498 bp) on chromosome 2B (Figure S1) was significantly associated with root depth at the booting stage (Figure 1). Six candidate genes were predicted in this block (Table S1). Two significant SNPs were located within the promoter region, and one significant SNP was within the coding region of TraesCS2B02G122400 (Table S2), which encodes a vacuolar sorting receptor protein sharing high protein sequence homology with Arabidopsis VSR1. The wheat gene is hereafter named TaVSR1‐B (Figure S2).
Figure 1.
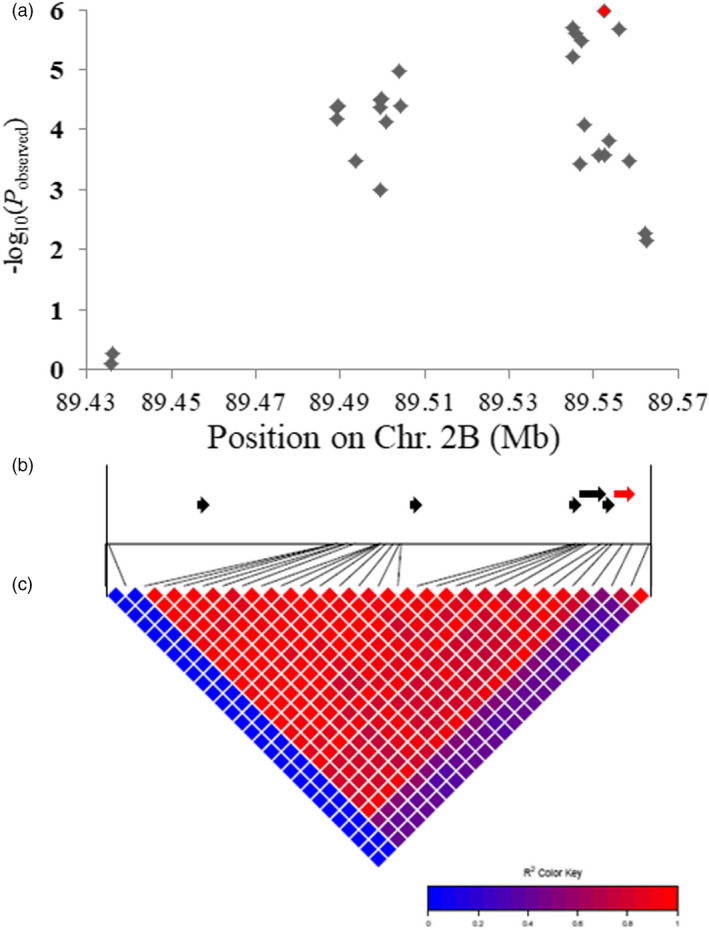
A locus on chromosome 2B was significantly associated with root depth at the booting stage. (a) Regional Manhattan block surrounding the TaVSR1‐B genomic region (89 435 662–89 562 498 bp). The SNP are shown by diamonds. The InDel at −1845 bp in the TaVSR1‐B promoter is shown with a red diamond. (b) Filtered gene models (IWGSC_v1.1) within the block region are indicated as arrows. The position of TaVSR1‐B (TraesCS2B02G122400) is indicated by a red arrow. (c) Linkage disequilibrium heat map of the chromosome region.
TaVSR1‐B is involved in regulation of root development in wheat
Previous studies showed that atvsr1 mutants had defective seedling establishment (Zouhar et al., 2010) and that the atvsr1 plants were smaller than the wild‐type (WT) plants when grown under normal conditions (Wang et al., 2015). We speculated that TaVSR1‐B might affect root growth in wheat. We obtained three TILLING mutants of durum wheat cv. Kronos (Krasileva et al., 2017) (detailed information in Table S3; Figure S2) for phenotyping. The mutants were backcrossed twice to the WT Kronos in order to eliminate background noise. When grown in polyvinyl chloride tubes containing soil, the mutant roots were significantly shallower than those of the WT at the booting stage (Figure S3). These results supported the prediction that TaVSR1‐B might be responsible for the observed differences in root phenotype.
We analysed the spatiotemporal expression pattern of the TaVSR1‐B gene in wheat cv. Hanxuan 10 (Hap4). TaVSR1‐B showed higher expression in roots at the seedling stage, and in spikes and roots at the booting stage (Figure 2a). This expression pattern suggested that TaVSR1‐B may also play a role in spike development, in addition to root development.
Figure 2.
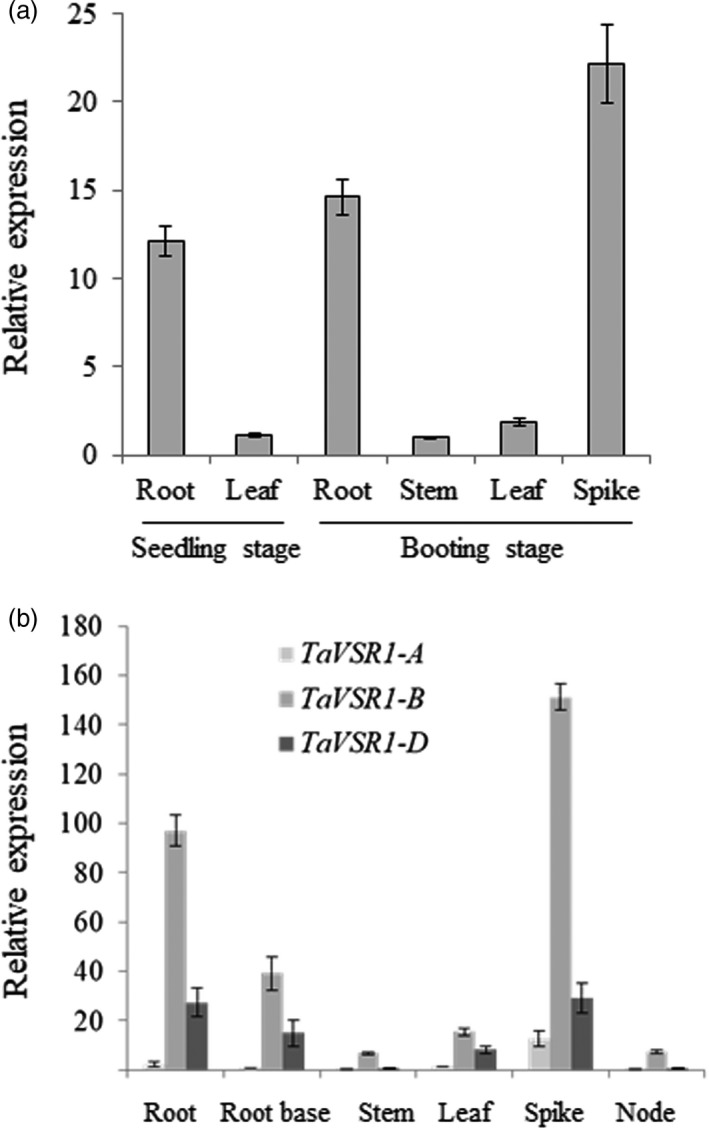
Expression pattern of TaVSR1s in wheat. (a) Expression pattern of TaVSR1‐B in tissues of cv. Hanxuan 10 at the seedling and booting stages. (b) Expression patterns of TaVSR1 genes in cv. Hanxuan 10 at the booting stage. Error bars, ± SE.
It is notable that our GWAS identified a strong correlation between TaVSR1‐B (located on chromosome 2B) and root depth in hexaploid wheat, but not the homoeologs on chromosomes 2A and 2D (TaVSR1‐A and ‐D), suggesting that these homoeologs might have diverged functions. To verify this hypothesis, we investigated the expression levels of TaVSR1‐B in roots of the three homoeologs. Expression pattern analysis showed that TaVSR1‐B had the highest expression level, TaVSR1‐D next and TaVSR1‐A, the lowest expression in all examined tissues of wheat cv. Hanxuan 10 at the booting stage (Figure 2b). These data suggested that TaVSR1‐B might play a predominant role among the three TaVSR1 homoeologs in root depth determination due to its higher expression level.
Sequence polymorphism of TaVSR1‐B
To identify the causal variation in root depth, we sequenced TaVSR1‐B in a diverse set of 32 accessions identified by simple sequence repeat (SSR) markers from a germplasm panel (Zhang et al., 2017). A 6.3 kb genomic region, including promoter, coding region and 5'‐ and 3'‐UTR sequences of TaVSR1‐B, was analysed. Forty polymorphic sites were identified, including a 125 bp InDel (insertion/deletion) located 1845 bp upstream of the start codon of TaVSR1‐B, 11 synonymous SNPs, 2 non‐synonymous SNPs in the signal peptide region and 26 SNPs in the promoter, UTR and intron regions (Table S4). These variations in the germplasm were identified as four TaVSR1‐B haplotypes named Hap1 to Hap4. The natural population of 323 wheat accessions was also classified into four haplotypes based on 3 polymorphic sites: the InDel at −1845 bp, the SNP at −1569 bp and the SNP at 1840 bp (Table S5; Figure 3a). Notably, Hap1 and Hap2 genotypes with InDel‐1845 had significantly shallower roots than those of Hap3 and Hap4 without the InDel (Figure 3b, c). Hap1 and Hap3 genotypes encode the same protein named TaVSR1‐B‐1/3, whereas Hap2 and Hap4 genotypes encode a different protein, named TaVSR1‐B‐2/4 (Figure S2).
Figure 3.
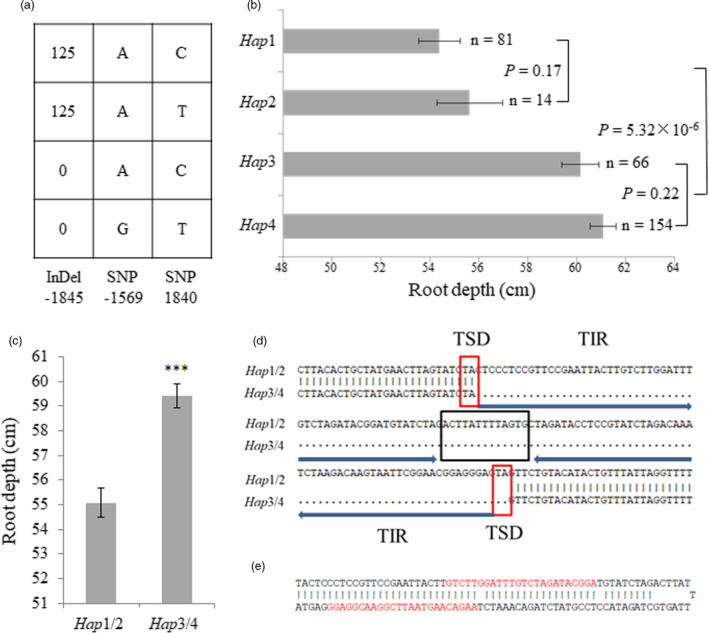
Haplotypes of TaVSR1‐B and root depth phenotypes at the booting stage for the entire test panel. (a) Four TaVSR1‐B haplotypes were identified. When a series of consecutive variants were in complete LD, only one is shown. (b) Root depth phenotypes of different haplotypes. n denotes the number of genotypes per haplotype. Statistical significance was determined by a two‐sided t‐test. (c) Root depth difference between Hap1/2 and Hap3/4. Error bars, ± SE. Statistical significance was determined by a two‐sided t‐test, *** P < 0.001. (d) DNA sequence difference between Hap1/2 and Hap3/4 caused by a 125 bp MITE inserted in the TaVSR1‐B promoter region. Target site duplications (TSDs) are indicated by red boxes. The loop is indicated by a black box. Two terminal inverted repeats (TIRs) are indicated by arrows. (e) Predicted hairpin structure of the MITE consisting of two 57 bp stems and a 13 bp loop. Two 24‐nt small RNA sequences found in the IWGSC Small RNA Database matching the MITE are highlighted in red.
In order to clarify whether variations in the TaVSR1‐B coding region affect gene function, we compared the protein functions of TaVSR1‐B‐1/3 and TaVSR1‐B‐2/4. Transmembrane domain prediction showed that transmembrane probability of TaVSR1‐B‐1/3 in the signal peptide region was much higher than that of TaVSR1‐B‐2/4 (Figures S4 and S5a, b). However, both TaVSR1‐B‐1/3 and TaVSR1‐B‐2/4 co‐localized with AtVSR1 in PVC/MVB (pre‐vacuolar compartment or multi‐vesicular body) of wheat protoplasts (Figure S5c, d). It appeared that these variations in signal peptides had no effect on sub‐cellular localization of TaVSR1‐B. We therefore assumed that InDel‐1845 plays a key role in affecting root depth at the booting stage despite the similarity of the protein sequences encoded by the four haplotypes.
Sequence and structure analysis of InDel‐1845 revealed that it comprised a long‐terminal inverted repeat (TIR, each 55 bp), a 13 bp loop and a target site duplication (TSD) of two nucleotides (TA) at the end. There was another TSD before the insertion (Figure 3d). This 125 bp DNA sequence can form a stem‐loop structure (Figure 3e). Two 24‐nt small RNA sequences found in the Small RNA Database of the IWGSC (International Wheat Genome Sequencing Consortium) aligned to the MITE (Figure S6). All these features indicated that it belonged to the Tc1/Mariner superfamily of MITE that consists of TIRs and TSDs, with preferential insertion at TA (Jiang et al., 2004; Lu et al., 2012). The MITE was present in the promoter of TaVSR1‐B allele for shallow root, in cultivars such as Drysdale (Hap1), but was absent in the allele for deep root in cultivars represented by Hanxuan 10 (Hap4).
Effect of the MITE insertion on canopy temperature in wheat germplasm panel
Previous research demonstrated that root depth was significantly correlated with CT (canopy temperature) at both booting and grain filling (Li et al., 2019b, 2019a). CT can therefore be used as an indicator of root development because root system depth affects the uptake of soil water, thereby affecting transpiration and finally CT differences. An association analysis of the MITE insertion and CT on the full panel of 323 accessions showed that the MITE insertion was also significantly associated with CT (Table S6). MITE+ accessions had significantly higher CT than MITE− accessions in 12 of 16 comparisons, that is combinations of 10 environmental situations and the booting or grain filling stage (Figure S7a). Correspondingly, the root systems of MITE+ accessions were significantly shorter than those of MITE− accessions at both growth stages (Figure S7b). These results supported the notion that the MITE insertion in the TaVSR1‐B promoter directly affects root depth and indirectly affects transpiration and CT.
The MITE insertion correlates with lower TaVSR1‐B expression
Since the MITE insertion is located in the promoter of TaVSR1‐B, we hypothesized that it might affect TaVSR1‐B expression. Real‐time RT‐PCR analysis of root samples from ten MITE+ and ten MITE− genotypes randomly selected from the full germplasm panel (Table S7) and grown in polyvinyl chloride tubes showed that the MITE+ genotypes had significantly lower TaVSR1‐B expression than the MITE− genotypes in roots at the booting stage (Figure 4a), suggesting that the MITE insertion represses expression of TaVSR1‐B.
Figure 4.
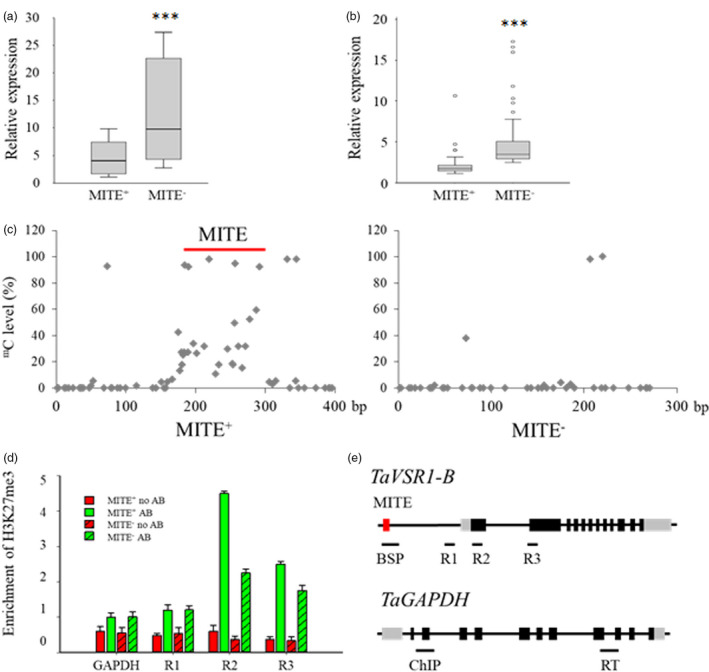
Comparisons of TaVSR1‐B expression, DNA methylation and H3K27me3 status between MITE+ and MITE− haplotypes of wheat. (a) TaVSR1‐B expression in roots of MITE+ and MITE− haplotypes at the booting stage. Ten accessions of each of MITE+ and MITE− haplotypes were assayed. Error bars, ± SE. Statistical significance was determined by a two‐sided t‐test, ***P < 0.001. (b) TaVSR1‐B expression in MITE+ and MITE− haplotypes in roots of seedlings of all 323 accessions in the test panel. In box plots, centre values are medians, lines indicate variability outside the upper and lower quartiles, and circles denote outliers. Error bars, ± SE. Statistical significance was determined by a two‐sided t‐test, ***P < 0.001. (c) Average DNA methylation status determined by bisulphite sequencing. The 20 accessions used for determining the expression of TaVSR1‐B haplotypes were used. The MITE region is indicated by a red line. (d) H3K27me3 methylation status detected by ChIP–qPCR assays. The same 20 accessions were used. The H3K27me3 status of the TaGAPDH was detected in parallel as a negative control. Anti‐H3 was used as an internal reference in the ChIP–qPCR assay. AB indicates with antibody; no AB indicates without antibody. Error bars, ± SE. (e) Position for detecting epigenetic status. The 125 bp MITE insertion is illustrated by the red box, 5'‐ and 3'‐UTR regions by grey boxes and exons by black boxes. The positions of bisulphite sequencing (BSP), ChIP‐qPCR (R1‐R3) in the genomic region of TaVSR1‐B and ChIP‐qPCR (ChIP), real‐time (RT) in the TaGAPDH are indicated by black lines selected based on H3K27me3 of Chinese Spring (Figure S9).
To substantiate this notion, we compared the expression levels of TaVSR1‐B in seedling root samples of all 323 accessions in the test panel. Real‐time RT‐PCR results revealed that the MITE+ genotypes had significantly lower TaVSR1‐B expression than the MITE− genotypes (Figure 4b and Table S8) and that presence of the MITE was correlated with the shallower root phenotype (Figure 3c).
The MITE insertion correlates with DNA and histone methylation differences
Previous studies showed that MITEs have regulatory roles in nearby gene expression by changing DNA and/or histone methylation status of genes (Castelletti et al., 2014; Gardiner et al., 2015; Mao et al., 2015). To better understand why the MITE insertion is correlated with lower TaVSR1‐B expression, we checked the DNA and histone methylation status of TaVSR1‐B in MITE+ and MITE− genotypes. We found that the positions of bisulphite sequencing (BSP) in the vicinity of the MITE insertion in the TaVSR1‐B promoter were hyper‐methylated in the MITE+ genotypes compared with the MITE− genotypes (Figure 4c). H3K9me2 and H3K27me3, as repressive histone markers, play an important role in regulating gene expression (Li et al., 2019c; Ramirez‐Gonzalez et al., 2018). As shown in Figure S8, there was low level of H3K9me2 distributed around the region. Based on H3K27me3 enrichment in the wheat cv. Chinese Spring (Figure S9), three regions (R1‐R3) spanning the TaVSR1‐B gene and promoter were analysed. Chromatin immunoprecipitation (ChIP) using H3K27me3 antibody followed by real‐time PCR analysis demonstrated that R2, nearest to the translation start site, was hyper‐methylated in the MITE+ genotypes compared with the MITE− genotypes (Figure 4d, e). These results further supported the premise that variations in the TaVSR1‐B promoter affect root development through modifying DNA and histone methylation.
The MITE insertion in wheat introgression lines
To further validate the effect of the MITE insertion on root development in wheat, we constructed a BC3F4 introgression population derived from a shallow rooted cv. Drysdale (with MITE insertion, MITE+) and a deep rooted cv. Hanxuan 10 (without MITE insertion, MITE−). Among 160 introgression lines (ILs), there were 6 MITE+ genotypes, 153 MITE− genotypes and 1 heterozygote (Figure 5a). To determine the phenotypic contributions of the alleles to root development in a more quantitative way, the primary root lengths of seedlings were determined. Apparently, genotypes with MITE+ had significantly shorter primary roots than MITE− genotypes (Figure 5b, c). Moreover, qRT‐PCR expression analysis of the 160 hexaploid introgression lines showed that TaVSR1‐B expression in seedling roots was higher in the MITE− genotypes, including Hanxuan 10, than in the MITE+ genotypes, including Drysdale (Figure 5d). Additionally, we also checked H3K27me3 and DNA methylation status of TaVSR1‐B in the ILs and observed a similar trend as that in 323 accessions population (Figure 5e, f).
Figure 5.
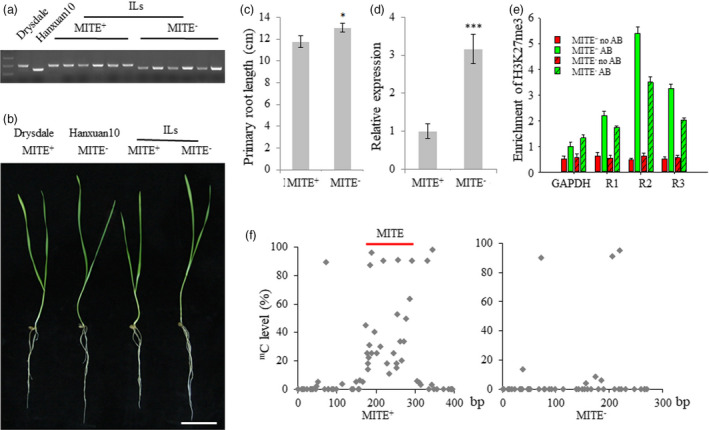
Effect of the MITE insertion in TaVSR1‐B on roots in an IL population. (a) Representative comparison of genotyping based on the 125 bp MITE insertion in the parents and IL population. The DNA band from MITE+ genotypes was 517 bp; and the band from MITE− genotypes was 392 bp. (b) Phenotypes of two‐week‐old seedlings of two TaVSR1‐B haplotypes in the ILs. Bar, 5 cm. (c) Average primary root lengths of two TaVSR1‐B haplotypes in the ILs. Error bars, ± SE. Statistical significance was determined by a two‐sided t‐test, *P < 0.05. (d) Average relative expression in roots of two TaVSR1‐B haplotypes. Error bars, ± SE. Statistical significance was determined by a two‐sided t‐test, ***P < 0.001. (e) Average H3K27me3 methylation status detected by anti‐H3K27me3 ChIP–qPCR assays. The six homozygous MITE+ accessions and randomly selected six homozygous of MITE− accessions were used. Error bars, ± SE. (f) Average DNA methylation status determined by bisulphite sequencing. The same 12 accessions were used in this experiment.
Increased expression of TaVSR1‐B confers deeper roots
Given that the MITE insertion was negatively correlated with TaVSR1‐B expression and root depth, we generated transgenic cv. Fielder expressing the coding sequence of TaVSR1‐BHap 2/4 driven by MITE+ (Hap2) or MITE− (Hap4) promoters. Transgenic lines were assayed using pCAMBIA1391 vector primers (Figure S10a). Lines with Hap4 showed significantly increased expression of TaVSR1‐B, and lines with Hap2 showed similar expression of TaVSR1‐B compared with that of the WT (Figure S10b). This result verified the notion that MITE+ has a significant effect on TaVSR1‐B expression in the transgenic wheat. Phenotypic analysis showed that the Hap4‐4/9 transgenic lines (MITE−) had deeper roots and higher dry weights of root and shoot than the Hap2−‐5/7 transgenic lines (MITE+) and the WT (Figure 6 and Figure S10c). There were no significant differences for other agronomic traits, including crown root number, plant height, spike number per plant, grain number per spike and 1000‐kernel weight, between WT and transgenic lines irrespective of being grown in polyvinyl chloride tubes or in the field (Figures S10d‐h and S11).
Figure 6.
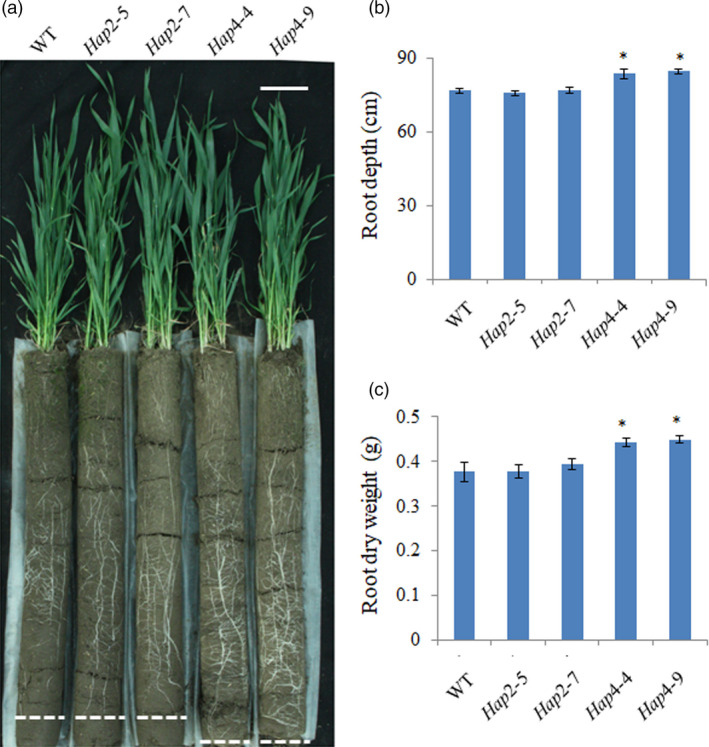
Seedling phenotypes of two transgenic TaVSR1‐B haplotype wheat lines. Seedlings (a), root depth (b), and root dry weight (c) of transgenic Hap2 (MITE+) and Hap4 (MITE−) lines. Data were based on three biological replications, nine seedlings per line per replicate. Bar, 10 cm. Error bars, ± SE. Statistical significance was determined by a two‐sided t‐test, * P < 0.05.
We also constructed vectors representing the four haplotypes based on pCAMBIA1391 and transformed them into rice. As expected, transgenic rice lines with vector 3 or 4 (MITE−) showed higher TaVSR1‐B expression levels than lines with vector 1 or 2 (MITE+) (Figure S12). These results indicated that the MITE insertion had a significant repressive effect on TaVSR1‐B expression in rice as well. Consistently, transgenic rice lines with Hap3 or Hap4 (MITE−) had deeper roots than comparable lines with Hap1, Hap2 (both MITE+) or WT (Figure S13). Taken together, heterologous expression of TaVSR1‐B without the MITE insertion conferred deeper roots while with the MITE insertion did not.
TaVSR1‐B regulates root depth by influencing the ratio of elongation zone to differentiated zone
We next examined the structure of root tip to investigate the cellular basis of TaVSR1‐B in regulating root depth. We found that there was no significant difference between transgenic line Hap4‐9 and wild type in the meristematic zone, regardless of zone length, average cell length or cell number. However, the elongation zone of TaVSR1‐B transgenic lines was shorter and contained smaller cells than the wild type, whereas the differentiation zone was longer and contained more cells than the wild type (Figure 7). These results suggested that TaVSR1‐B might accelerate transition of cells in the elongation zone to cells in the differentiation zone.
Figure 7.
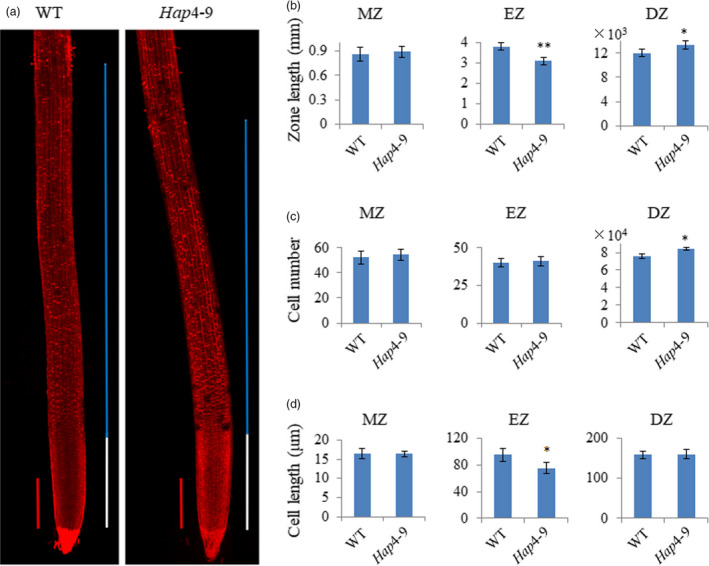
Root morphology of transgenic TaVSR1‐B Hap4 wheat. (a) PI staining of roots of 2‐week‐old WT and Hap4‐9 transgenic seedlings. The meristematic zone (MZ) is marked by a white line, and the elongation zone (EZ) by a blue line. Red bars, 500 μm. Each image was made by fusing four photographs of the same root. (b) Zone length of MZ, EZ and differentiation zone (DZ); (c) Cell number of MZ, EZ and DZ; (d) Average cell length of MZ, EZ and DZ. At least 10 roots were measured in each of three repeated experiments. Error bars, ± SE. Statistical significance was determined by a two‐sided t‐test, * P < 0.05, ** P < 0.01.
Previous studies demonstrated that AtVSR1 was likely involved in biogenesis of PVCs by transporting cargo from the PVC (or MVB) to the vacuole (Shimada et al., 2003). We examined whether TaVSR1‐B took part in formation of central vacuoles in the root cells, but detected no significant differences between wild type and line Hap4‐9 at the MZ or EZ (Figure S14). These results hinted that TaVSR1‐B might affect other processes rather than formation of vacuoles.
Discussion
Root depth, an important aspect of root architecture, has a strong connection with plant growth, grain yield and abiotic stress tolerance. Consequently, optimization of root architecture could be a significant objective for wheat breeders. Booting is the key growth period for establishing grain yield potential in wheat, as the root depth at this stage is significantly correlated with yield per plant and canopy temperature (Li et al., 2019a). As large‐scale phenotyping of root depth in field conditions in technically challenging and prohibitively labour intensive, we investigated root depth at the booting stage using a polyvinyl chloride tube method in this study. Our GWAS identified TaVSR1‐B, which encodes a vacuolar sorting receptor protein, as an important regulator of root depth in wheat at the booting stage.
Previous studies reported that VSRs (vacuolar sorting receptors) play a universal role in sorting vacuolar‐soluble proteins (Niemes et al., 2010; Pelham and Rothman, 2000; Reyes et al., 2011). In Arabidopsis, there are seven family members (Lousa et al., 2012; Robinson, 2014) not only involved in sorting storage proteins targeted to the protein storage vacuole (PSV) in seeds but also involved in sorting soluble lytic vacuolar (LV) and PSV proteins in vegetative cells (Lee et al., 2013). VSR1, first known as Arabidopsis thaliana epidermal growth factor receptor‐like protein (AtELP1), showed highest expression in roots (Ahmed et al., 1997), although its function was unknown. Recent studies showed that VSR1 was essential for germination vigour, seedling establishment (Zouhar et al., 2010), pollen tube growth (Wang et al., 2010) and osmotic stress‐induced ABA biosynthesis (Wang et al., 2015). Thus, VSR may have multiple functions in plant growth. Here, we proved that TaVSR1‐B is involved in determination of root architecture in wheat. However, we did not find evidence for TaVSR1‐B in regulating vacuole formation. Instead, we found that TaVSR1‐B accelerates transition of cells in the elongation zone to cells in the differentiation zone, hence producing deeper roots (Figure 7). Further studies are required to elucidate the precise molecular mechanisms of TaVSR1‐B in regulating root growth in wheat.
More than 85% of the wheat genome is comprised of transposable elements (TEs) (International Wheat Genome Sequencing Consortium, 2018) that are present in about half of all genes (Zhao et al., 2017). Numerous studies have documented important roles of TEs in reshaping the genomes, evolution and ecological adaptation of various plants (Cui and Cao, 2014; Slotkin and Martienssen, 2007). For example, a TE (Hopscotch) inserted in the regulatory region of teosinte branched1 (tb1) increases apical dominance in maize (Studer et al., 2011). In maize, within the Vgt1 region, a major QTL regulating flowering time, the contrasting QTL alleles showed 29 SNPs and InDels and one 143‐bp insertion into the Gaspé Flint allele of a MITE transposon belonging to the Tourist family (Salvi et al., 2007). A CACTA‐like TE in the ZmCCT promoter attenuates photoperiod sensitivity (Yang et al., 2013). A terminal‐repeat retrotransposon in miniature (TRIM) element in the promoter of Ms2 causes male sterility in wheat (Xia et al., 2017). Previous studies also reported that insertion of a MITE in the wheat Vrn‐A1 promoter led to an altered vernalization requirement made through the miRNA pathway (Gorafi et al., 2016; Yu et al., 2014), while insertion of TaMITE81 in the 5'‐UTR of TaCHS7BL (chalcone synthase (CHS) caused its low expression, leading to reduced anthocyanin biosynthesis and a white grain phenotype (Xi et al., 2016). In this study, we found that a 125 bp MITE insertion in the promoter region TaVSR1‐B was responsible for shorter root depth at the booting stage of growth. We further showed that the MITE insertion in the promoter of TaVSR1‐B represses its expression by modulating DNA and histone methylation. Our findings highlight an important role of TEs in regulating wheat root development.
Breeding wheat for diverse target ecological areas requires different root architecture. Here, we found that the presence/absence of a MITE in the promoter of TaVSR1‐B‐affected root depth at the booting stage. Thus, appropriate TaVSR1‐B alleles (MITE+ or MITE−) could be used to breed wheat cultivars fit different target ecological areas. For example, as phosphorus is mainly found in the shallow soil, nitrogen is distributed in the deep soil, and moisture content in different soil layers varies according to natural precipitation and irrigation conditions, the target TaVSR1‐B allele can be chosen to gain the optimal root architecture on the basis of soil water, fertilizer and other conditions in the target area.
Experimental procedures
GWAS of root depth
The germplasm panel used in this study was a panel of 323 wheat accessions including 12 landraces, 36 advanced lines and 275 modern varieties previously genotyped with the Wheat 660K SNP Array. A total of 395 675 SNPs with missing rates ≤0.2 and minor allele frequency ≥0.05 were used for GWAS (Li et al., 2019a). Root depth and statistical analyses were performed as described by Li et al. (2019a). Briefly, polyvinyl chloride tubes (11 cm in diameter, and 100 cm length for booting plants, 180 cm length for grain filling plants) were pre‐buried into a soil pit. Matching polyethylene (PE) bags loaded with soil were inserted into the tubes. The soil moisture was maintained at 80 ± 5% of field capacity. Eight seeds of each accession were sown in each soil column with three replications in October, and seedlings were thinned to three plants. The PE bags were withdrawn from the tubes at the booting or grain filling stages, and root depths were recorded for deepest root tip. The relationships between markers and traits were analysed using TASSEL 5.0 software based on mixed linear model (MLM), in which the population structure (Q), kinship (K) and PCA were estimated as previously described (Li et al., 2019a). Based on the Bonferroni correction, the suggestive P value threshold was 2.53 × 10–6 (= 1/395 675), which was considered as the threshold for association.
Canopy temperature and its association with genotype
The entire test panel was planted in 10 environments (year × location × treatment) at Changping (40°13'N, 116°13'E) and Shunyi (40°23'N, 116°56'E), Beijing, over two growing seasons (2015–2017) for measurement of canopy temperature (CT) under two water regimes, drought‐stressed (DS) and well‐watered (WW) with or without heat‐stress (HS) treatment at the booting (B) and grain filling (G) stages. The experimental unit was a 2‐m long, four‐row plot with 30 cm between rows and 40 seeds planted per row. DS plots were rain‐fed (the amount of rainfall was shown in Figure S15) whereas WW plots were thrice irrigated with 750 m3/ha at the pre‐overwintering, booting and flowering stage when the amounts of rainfall were insufficient. Heat stress (HS) was simulated at Shunyi by covering the plots with polythene sheets at the flowering stage to increase temperature. Association analysis between phenotype and genotype was performed as described by Li et al. (2019a).
Phylogenetic tree construction for VSR1
Using the full‐length amino acid sequence of TaVSR1‐B as query, VSR1 family members were identified from different plant species by BLAST search of proteins in the NCBI database. Homologous VSR1 proteins were aligned using DNAMAN. The full‐length sequences were used to construct a phylogenetic tree by the maximum likelihood method using Molecular Evolutionary Genetics Analysis (MEGA) software version 5.2 (http://www.megasoftware.net).
Phenotypic analysis of Kronos TILLING mutants
Three TILLING mutants of durum wheat cv. Kronos were obtained from Professor Daolin Fu (Shandong Agricultural University, China), who has received approval from Dr. Jorge Dubcovsky at UC Davis, the inventor of the Kronos mutant lines. These mutants were twice backcrossed with Kronos to eliminate noise from the genetic background prior to phenotyping. Seeds of Kronos (WT) and its mutants were planted in soil columns in polyvinyl chloride PVC tubes in March. Root depth at the booting stage and statistical analyses were performed as described above.
Sequence polymorphism of TaVSR1‐B
The genomic region of TaVSR1‐B (6.3 kb), including promoter (2.3 kb), coding regions (including introns) and 5' and 3'‐UTR sequences were amplified from 32 widely variable wheat accessions identified by dispersed simple sequence repeat (SSR) markers. Purified PCR products were cloned into pEASY‐Blunt vectors and transformed into E. coli. Twelve positive clones were sequenced with an ABI 3730 DNA Analyzer. Sequence polymorphism was analysed using SeqMan software (DNASTAR, USA).
Analysis of TaVSR1‐B protein structure
Signal peptides in the TaVSR1‐B protein were predicted through Signalp Server v.5.0 (http://www.cbs.dtu.dk/services/SignalP), and transmembrane domains were predicted by TMHMM Server v.2.0 (http://www.cbs.dtu.dk/services/TMHMM/).
Sub‐cellular localization
The full‐length cDNA of TaVSR1‐Bs was fused at restriction enzyme sites of Spe I and Kpn I upstream of GFP in the pCAMBIA1300 vector under control of the CaMV 35S promoter. The constructs and AtVSR1‐mCherry were co‐transformed into wheat protoplasts from 2‐week‐old seedlings based on the PEG‐mediated method (Yoo et al., 2007), then incubated at 25 °C in darkness for 16 h. Imaging was performed with a Zeiss LSM880 confocal microscope.
TaVSR1s gene expression analysis
Root and leaf tissues of wheat cv. Hanxuan 10 from 2‐week‐old seedlings and different tissues at the booting stage (root, root base, stem, node, leaf and spike) were harvested to analyse the spatiotemporal expression pattern of the TaVSR1 genes. To detect effects of allelic variants on expression level of TaVSR1‐B, two sets of ten randomly selected accessions with and without MITE (Table S7) were grown in the polyvinyl chloride tubes and roots were sampled at the booting stage. Root samples from all 323 accessions in the test panel were harvested from 2‐week‐old seedlings. Total RNAs were extracted, and real‐time PCR analysis was performed. The TaGAPDH gene was used as an internal control to normalize the data.
Bisulphite sequencing
Genomic DNA isolated from the above 20 accessions was used for bisulphite sequencing (Table S7). One microgram of genomic DNA from each sample was treated by bisulphite with the EZ DNA Methylation‐Gold Kit (Zymo Research, Orange, CA, D5005), following the manufacturer’s protocol. After bisulphite conversion, the DNA was used as template for PCR. Amplified PCR fragments were then cloned into the pEASY‐T1 cloning vector (TRANS, CT101) and transformed into E. coli for sequencing. At least ten clones of each accession were sequenced. DNA methylation status was the mean value for accessions of each MITE+ or MITE− haplotype.
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed following a published protocol (Saleh et al., 2008) using whole‐plant tissues of 2‐week‐old seedlings from the above 20 accessions (Table S7). Briefly, 5 μl of anti‐H3 (Abcam, ab1791, http://www.abcam.com/) or anti‐H3K27me3 (Abcam, ab6002) antibodies were used for each ChIP assay. ChIP products were dissolved in TE, and the amount of immunoprecipitated chromatin was determined by real‐time PCR. TaGAPDH was used as the internal control. The relative abundance was normalized to the amount of DNA immunoprecipitated by H3 antibody. H3K27me3 methylation status was the mean value of accessions with each MITE+ or MITE− haplotype.
Allelic effect of TaVSR1‐B in an introgression population
A BC3F4 population was used as the plant material to validate the effects of TaVSR1‐B alleles. The population was constructed by using a deep root genotype Hanxuan 10 as the recurrent parent, and a shallow root genotype Drysdale as the donor parent, through three randomly backcrossing generations followed by three selfing generations. One hundred and sixty introgression lines (ILs) were used in the validation of TaVSR1‐B allele effects. The genotypes of the ILs were identified by separating the PCR products of the MITE insertion at the TaVSR1‐B locus on 2% agarose gel. Primary root samples were collected from 2‐week‐old seedlings grown hydroponically to measure root length and to detect gene expression differences of two haplotypes. Six homozygous MITE+ accessions and six randomly selected homozygous MITE− accessions were used to measure DNA methylation and H3K27me3 status as mentioned above.
Production of transgenic wheat
Four kinds of vectors based on pCAMBIA1391 were constructed for transgenesis. Four fragments were amplified: TaVSR1‐BHap 1/3 cDNA from cDNA of wheat cv. Drysdale (Hap1), TaVSR1‐BHap 2/4 cDNA from cDNA of wheat cv. Hanxuan 10 (Hap4), TaVSR1‐B promoter with MITE insertion (MITE+) from DNA of Drysdale and TaVSR1‐B promoter without MITE insertion (MITE−) from DNA of Hanxuan 10. Then, combinations of different cDNAs and promoters were amplified together based on the Splice Overlap Extension (SOE) PCR method (Ward et al., 1993) and inserted into pCAMBIA1391 through seamless DNA cloning using In‐Fusion® HD Cloning Plus (638910, TaKaRa, http://www.takara‐bio.com/). In detail, Vector 1 representing Hap1 consisted of TaVSR1‐BHap 1/3 cDNA driven by the TaVSR1‐B MITE+ promoter; Vector 2 representing Hap2 consisted of TaVSR1‐BHap 2/4 cDNA driven by the TaVSR1‐B MITE+ promoter; Vector 3 representing Hap3 consisted of TaVSR1‐BHap 1/3 cDNA driven by the TaVSR1‐B MITE− promoter; and Vector 4 representing Hap4, consisted of TaVSR1‐BHap 2/4 cDNA driven by the TaVSR1‐B MITE− promoter. All primers used in this study are listed in Table S9. Transgenic wheat cv. Fielder lines were produced by infection with Agrobacterium EHA105 with Vector 2 and Vector 4. Transgenic T0, T1 and T2 wheat seeds were selected by 50 μg/mL hygromycin B. Transgenic‐positive and sibling transgenic‐negative (WT) plants were confirmed in each generation by PCR analysis. The roots of 2‐week‐old homozygous T3 wheat lines were used to detect expression of TaVSR1‐B through real‐time PCR. At least 20 independent lines were obtained for each vector. Transgenic lines Hap2‐5 and Hap2‐7 (Vector 2) and Hap4‐4 and Hap4‐9 (Vector 4) were selected for further analyses.
Production of transgenic rice
Transgenic rice cv. Kitaake lines were produced by infection with Agrobacterium EHA105 with above four kinds of vectors. Transgenic T0, T1 and T2 rice seeds were selected by 50 μg/mL hygromycin B. Transgenic‐positive and sibling transgenic‐negative (WT) plants were confirmed in each generation by PCR. The roots of 2‐week‐old homozygous T3 rice lines were used to measure expression of TaVSR1‐Bs through real‐time PCR. At least 20 independent lines were obtained for each vector. Four lines, Hap1‐3, Hap2‐12, Hap3‐8 and Hap4‐10 were selected for further analyses.
Phenotypic analysis of transgenic lines
Homozygous T3 transgenic wheat lines and WT Fielder were grown in soil column for phenotyping root traits as described above. The traits measured at booting included root depth, crown root number, root dry weight and shoot dry weight. The same materials were also grown in the field as 1 m single row plots spaced 30 cm apart, and with 10 plants per row. Crown root numbers at booting, plant height, spike number per plant and grain number per spike were measured at the adult stage; thousand kernel weight was recorded post‐harvest.
Homozygous T3 transgenic rice lines and WT Kitaake were used for phenotyping. The rice plants were grown in peat soil in plastic containers (90 × 35 × 30 cm). Each container held 3 rows spaced 10 cm apart, with 8 plants in each row with normal field management. There were three replications with 10 plants per line investigated in each replicate. Similar traits were investigated as for wheat.
Wheat root morphology
The transgenic wheat lines and WT were germinated and grown in water for observing root phenotypes. Roots of 2‐week‐old seedlings were stained with 10 μg/mL propidium iodide (PI) for 5 min, photographed using a Zeiss LSM880 confocal microscope, analysed and jointed by ImageJ (National Institutes of Health, http://rsb.info.nih.gov/ij/). Zone lengths, cell numbers and cell lengths were measured for the meristematic zone (MZ, between the root cap and first elongating cell), elongation zone (EZ, from the first elongating cell to the first root hair) and differentiation zone (DZ, from the first root hair to the merging of root and hypocotyl). At least 10 roots were analysed per line in each of three repeated experiments.
Roots were stained by 0.03% Neutral Red for 5 min and washed three times before MZ and EZ images were collected using a Zeiss LSM880 confocal microscope with a 543 nm laser for excitation. At least 10 roots were observed per line in each of three repeated experiments.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
J.W. and R.J. designed the experiments, analysed the results and wrote the manuscript; J.W. performed most of the experiments. L.L. participated in the data analysis and phenotyping of root morphology and CT; X.M. provided advice on the experiments; C.L., X.Y., Y.X. and Z.Z. helped with propagation of the plant materials and expression analysis of TaVSR1s. The project was conceived, planned and supervised by R.J. All authors discussed the results and provided feedback on the manuscript.
Supporting information
Figure S1 Genome‐wide association study of wheat root depth at the booting stage.
Figure S2 Sequence alignment and phylogenetic tree of VSR1 proteins.
Figure S3 Roots of vsr1 mutants.
Figure S4 Signal peptide analysis of different haplotypes using SignalP‐5.0 Server.
Figure S5 Transmembrane domain prediction and sub‐cellular localization of TaVSR1‐B‐1/3 and TaVSR1‐B‐2/4.
Figure S6 sRNAs aligned to the 125 bp MITE in the promoter of TaVSR1‐B.
Figure S7 Phenotypic comparisons of accessions with or without the MITE insertion in multiple environments in the test panel.
Figure S8 H3K9me2 status in wheat Chinese Spring around TaVSR1‐B (TraesCS2B02G122400) region.
Figure S9 H3K27me3 status in wheat cv. Chinese Spring.
Figure S10 Phenotypes of TaVSR1‐B Hap2 (MITE+) and Hap4 (MITE−) transgenic wheat lines grown in polyvinyl chloride tubes.
Figure S11 Phenotype of TaVSR1‐B Hap2 (MITE+) and Hap4 (MITE−) transgenic wheat lines grown in the field.
Figure S12 Phenotypes of four TaVSR1‐B haplotypes in transgenic rice.
Figure S13 Seedling phenotypes of four transgenic TaVSR1‐B rice haplotypes.
Figure S14 Vacuoles in wheat roots stained with Neutral Red.
Figure S15 The amount of rainfall in the whole growing seasons.
Table S1 Annotation of candidate genes in the chromosome 2B block.
Table S2 Candidate SNP in the TaVSR1‐B block on chromosome 2B detected by GWAS.
Table S3 Details of the cv. Kronos (tetraploid variety) TILLING mutants.
Table S6 Association of the MITE insertion with canopy temperature.
Table S7 Sets of 10 accessions with MITE+ or MITE− TaVSR1‐B haplotypes randomly selected from the entire test panel and used for measuring TaVSR1‐B expression at the booting stage, and DNA methylation and H3K27me3 status.
Table S4 Forty polymorphic sites in TaVSR1‐B identified from a diverse selection of 32 wheat accessions representing four haplotypes.
Table S5 Haplotypes of all 323 wheat accessions in the test panel.
Table S8 Relative expression of TaVSR1‐B in 2‐week‐old seedling roots from the test panel.
Table S9 Primers used in the study.
Acknowledgements
We appreciate Dr. Yulong Ren (Institute of Crop Sciences, Chinese Academy of Agricultural Sciences (ICS CAAS), China) for providing the AtVSR1‐mCherry vector to co‐locate TaVSR1 protein; Professor Daolin Fu (Shandong Agricultural University, China) for providing Kronos TILLING mutant seeds; Drs Genying Li (Shandong Academy of Agricultural Sciences, China) and Xingguo Ye (ICS CAAS) for helping us to get transgenic wheat plants; Dr. Chuanyin Wu (ICS CAAS) for helping us to get transgenic rice plants. We thank Professor Robert A. McIntosh (Plant Breeding Institute, University of Sydney, Australia) for revising the manuscript. We also appreciate Professor Haiyang Wang (Biotechnology Research Institute, CAAS) for his valuable discussions and revisions to the paper. This research was supported by grants from the National Natural Science Foundation of China (31801347) and the National Key R&D Program of China (2017YFD0300202).
Wang, J. , Li, L. , Li, C. , Yang, X. , Xue, Y. , Zhu, Z. , Mao, X. and Jing, R. (2021) A transposon in the vacuolar sorting receptor gene TaVSR1‐B promoter region is associated with wheat root depth at booting stage. Plant Biotechnol. J., 10.1111/pbi.13564
Contributor Information
Xinguo Mao, Email: maoxinguo@caas.cn.
Ruilian Jing, Email: jingruilian@caas.cn.
References
- Ahmed, S.U. , BarPeled, M. and Raikhel, N.V. (1997) Cloning and subcellular location of an Arabidopsis receptor‐like protein that shares common features with protein‐sorting receptors of eukaryotic cells. Plant Physiol. 115, 311–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aida, M. , Beis, D. , Heidstra, R. , Willemsen, V. , Blilou, I. , Galinha, C. , Nussaume, L. et al. (2004) The PLETHORA genes mediate patterning of the Arabidopsis root stem cell niche. Cell, 119, 109–120. [DOI] [PubMed] [Google Scholar]
- Ayalew, H. , Liu, H. , Borner, A. , Kobiljski, B. , Liu, C. and Yan, G. (2018) Genome‐wide association mapping of major root length QTLs under PEG induced water stress in wheat. Front. Plant Sci. 9, 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane, M.A. , Maccaferri, M. , Nazemi, G. , Salvi, S. , Francia, R. , Colalongo, C. and Tuberosa, R. (2014) Association mapping for root architectural traits in durum wheat seedlings as related to agronomic performance. Mol. Breed. 34, 1629–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelletti, S. , Tuberosa, R. , Pindo, M. and Salvi, S. (2014) A MITE transposon insertion is associated with differential methylation at the maize flowering time QTL Vgt1. G3 Genes Genome. Genet. 4, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh, C.R. , Chao, S. , Wang, S. , Huang, B.E. , Stephen, S. , Kiani, S. , Forrest, K. et al. (2013) Genome‐wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc. Natl Acad. Sci. USA, 110, 8057–8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, X. and Cao, X. (2014) Epigenetic regulation and functional exaptation of transposable elements in higher plants. Curr. Opin. Plant Biol. 21, 83–88. [DOI] [PubMed] [Google Scholar]
- Gardiner, L.J. , Quinton‐Tulloch, M. , Olohan, L. , Price, J. , Hall, N. and Hall, A. (2015) A genome‐wide survey of DNA methylation in hexaploid wheat. Genome Biol. 16, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfray, H.C. , Beddington, J.R. , Crute, I.R. , Haddad, L. , Lawrence, D. , Muir, J.F. , Pretty, J. et al. (2010) Food security: the challenge of feeding 9 billion people. Science, 327, 812–818. [DOI] [PubMed] [Google Scholar]
- Gorafi, Y.S. , Eltayeb, A.E. and Tsujimoto, H. (2016) Alteration of wheat vernalization requirement by alien chromosome‐mediated transposition of MITE. Breed Sci. 66, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Wheat Genome Sequencing Consortium . (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science, 361, 661–673. [DOI] [PubMed] [Google Scholar]
- Jiang, N. , Feschotte, C. , Zhang, X.Y. and Wessler, S.R. (2004) Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs). Curr. Opin. Plant Biol. 7, 115–119. [DOI] [PubMed] [Google Scholar]
- Krasileva, K.V. , Vasquez‐Gross, H.A. , Howell, T. , Bailey, P. , Paraiso, F. , Clissold, L. , Simmonds, J. et al. (2017) Uncovering hidden variation in polyploid wheat. Proc. Natl Acad. Sci. USA, 114, E913–E921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. , Jang, M. , Song, K. , Kang, H. , Lee, M.H. , Lee, D.W. , Zouhar, J. et al. (2013) Functional identification of sorting receptors involved in trafficking of soluble lytic vacuolar proteins in vegetative cells of Arabidopsis . Plant Physiol. 161, 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. , Liu, D. , Li, Q. , Mao, X. , Li, A. , Wang, J. , Chang, X. et al. (2016) Overexpression of wheat gene TaMOR improves root system architecture and grain yield in Oryza sativa . J. Exp. Bot. 67, 4155–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Peng, Z. , Mao, X. , Wang, J. , Chang, X. , Reynolds, M. and Jing, R. (2019a) Genome‐wide association study reveals genomic regions controlling root and shoot traits at late growth stages in wheat. Ann. Bot. 124, 993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X.X. , Ingvordsen, C.H. , Weiss, M. , Rebetzke, G.J. , Condon, A.G. , James, R.A. and Richards, R.A. (2019b) Deeper roots associated with cooler canopies, higher normalized difference vegetation index, and greater yield in three wheat populations grown on stored soil water. J. Exp. Bot. 70, 4963–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Xu, X. , Liu, W. , Li, X. , Yang, X. , Ru, Z. and Li, L. (2020) Dissection of superior alleles for yield‐related traits and their distribution in important cultivars of wheat by association mapping. Front. Plant Sci. 11, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Wang, M. , Lin, K. , Xie, Y. , Guo, J. , Ye, L. , Zhuang, Y. et al. (2019c) The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements. Genome Biol. 20, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X.L. , Li, R.Z. , Chang, X.P. and Jing, R.L. (2013) Mapping QTLs for seedling root traits in a doubled haploid wheat population under different water regimes. Euphytica, 189, 51–66. [Google Scholar]
- Lousa, C.D. , Gershlick, D.C. and Denecke, J. (2012) Mechanisms and concepts paving the way towards a complete transport cycle of plant vacuolar sorting receptors. Plant Cell, 24, 1714–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, C. , Chen, J.J. , Zhang, Y. , Hu, Q. , Su, W.Q. and Kuang, H.H. (2012) Miniature inverted‐repeat transposable elements (MITEs) have been accumulated through amplification bursts and play important roles in gene expression and species diversity in Oryza sativa . Mol. Biol. Evol. 29, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri, M. , El‐Feki, W. , Nazemi, G. , Salvi, S. , Cane, M.A. , Colalongo, M.C. , Stefanelli, S. and et al. (2016) Prioritizing quantitative trait loci for root system architecture in tetraploid wheat. J. Exp. Bot. 67, 1161–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, H.D. , Wang, H.W. , Liu, S.X. , Li, Z. , Yang, X.H. , Yan, J.B. , Li, J.S. et al. (2015) A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat. Commun. 6, 8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemes, S. , Labs, M. , Scheuring, D. , Krueger, F. , Langhans, M. , Jesenofsky, B. , Robinson, D.G. et al. (2010) Sorting of plant vacuolar proteins is initiated in the ER. Plant J. 62, 601–614. [DOI] [PubMed] [Google Scholar]
- Pang, Y.L. , Liu, C.X. , Wang, D.F. , St Amand, P. , Bernardo, A. , Li, W.H. , He, F. et al. (2020) High‐resolution genome‐wide association study identifies genomic regions and candidate genes for important agronomic traits in wheat. Mol. Plant, 13, 1311–1327. [DOI] [PubMed] [Google Scholar]
- Pelham, H.R.B. and Rothman, J.E. (2000) The debate about transport in the Golgi ‐ Two sides of the same coin? Cell, 102, 713–719. [DOI] [PubMed] [Google Scholar]
- Promchuea, S. , Zhu, Y. , Chen, Z. , Zhang, J. and Gong, Z. (2017) ARF2 coordinates with PLETHORAs and PINs to orchestrate ABA‐mediated root meristem activity in Arabidopsis . J. Integr. Plant Biol. 59, 30–43. [DOI] [PubMed] [Google Scholar]
- Ramirez‐Gonzalez, R.H. , Borrill, P. , Lang, D. , Harrington, S.A. , Brinton, J. , Venturini, L. et al. (2018) The transcriptional landscape of polyploid wheat. Science, 361, eaar6089. [DOI] [PubMed] [Google Scholar]
- Ray, D.K. , Mueller, N.D. , West, P.C. and Foley, J.A. (2013) Yield trends are insufficient to double global crop production by 2050. PLoS One, 8, e66428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes, F.C. , Buono, R. and Otegui, M.S. (2011) Plant endosomal trafficking pathways. Curr. Opin. Plant Biol. 14, 666–673. [DOI] [PubMed] [Google Scholar]
- Robinson, D.G. (2014) Trafficking of vacuolar sorting receptors: new data and new problems. Plant Physiol. 165, 1417–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh, A. , Alvarez‐Venegas, R. and Avramova, Z. (2008) An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat. Protoc. 3, 1018–1025. [DOI] [PubMed] [Google Scholar]
- Salvi, S. , Sponza, G. , Morgante, M. , Tomes, D. , Niu, X. , Fengler, K.A. , Meeley, R. et al. (2007) Conserved noncoding genomic sequences associated with a flowering‐time quantitative trait locus m maize. Proc. Natl Acad. Sci. USA, 104, 11376–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada, T. , Fuji, K. , Tamura, K. , Kondo, M. , Nishimura, M. and Hara‐Nishimura, I. (2003) Vacuolar sorting receptor for seed storage proteins in Arabidopsis thaliana . Proc. Natl Acad. Sci. USA, 100, 16095–16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin, R.K. and Martienssen, R. (2007) Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285. [DOI] [PubMed] [Google Scholar]
- Soriano, J.M. and Alvaro, F. (2019) Discovering consensus genomic regions in wheat for root‐related traits by QTL meta‐analysis. Sci. Rep. 9, 10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer, A. , Zhao, Q. , Ross‐Ibarra, J. and Doebley, J. (2011) Identification of a functional transposon insertion in the maize domestication gene tb1 . Nat. Genet. 43, 1160–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester, M. and Langridge, P. (2010) Breeding technologies to increase crop production in a changing world. Science, 327, 818–822. [DOI] [PubMed] [Google Scholar]
- Voss‐Fels, K.P. , Robinson, H. , Mudge, S.R. , Richard, C. , Newman, S. , Wittkop, B. , Stahl, A. et al. (2018) VERNALIZATION1 modulates root system architecture in wheat and barley. Mol. Plant, 11, 226–229. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Sun, S. , Ge, W. , Zhao, L. , Hou, B. , Wang, K. , Lyu, Z. et al. (2020) Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science, 368, eaba5435. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Tse, Y.C. , Law, A.H.Y. , Sun, S.S.M. , Sun, Y.B. , Xu, Z.F. , Hillmer, S. et al. (2010) Vacuolar sorting receptors (VSRs) and secretory carrier membrane proteins (SCAMPs) are essential for pollen tube growth. Plant J. 61, 826–838. [DOI] [PubMed] [Google Scholar]
- Wang, Z.Y. , Gehring, C. , Zhu, J.H. , Li, F.M. , Zhu, J.K. and Xiong, L.M. (2015) The Arabidopsis vacuolar sorting receptor1 is required for osmotic stress‐induced abscisic acid biosynthesis. Plant Physiol. 167, 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, R.L. , Hawkins, N.J. , Wakefield, D. , Atkinson, K. and Biggs, J.C. (1993) Production of a functional single‐chain Fv fragment from the pan‐leukocyte antibody WM65 using splicing by asymmetric PCR. Exp. Hematol. 21, 660–664. [PubMed] [Google Scholar]
- Wasson, A.P. , Richards, R.A. , Chatrath, R. , Misra, S.C. , Prasad, S.V. , Rebetzke, G.J. , Kirkegaard, J.A. et al. (2012) Traits and selection strategies to improve root systems and water uptake in water‐limited wheat crops. J. Exp. Bot. 63, 3485–3498. [DOI] [PubMed] [Google Scholar]
- Xi, X. , Li, N. , Li, S. , Chen, W. , Zhang, B. , Liu, B. and Zhang, H. (2016) The characteristics and functions of a miniature inverted‐repeat transposable element TaMITE81 in the 5' UTR of TaCHS7BL from Triticum aestivum . Mol. Genet. Genomics, 291, 1991–1998. [DOI] [PubMed] [Google Scholar]
- Xia, C. , Zhang, L. , Zou, C. , Gu, Y. , Duan, J. , Zhao, G. , Wu, J. et al. (2017) A TRIM insertion in the promoter of Ms2 causes male sterility in wheat. Nat. Commun. 8, 15407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q. , Li, Z. , Li, W.Q. , Ku, L.X. , Wang, C. , Ye, J.R. , Li, K. et al. (2013) CACTA‐like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the postdomestication spread of maize. Proc. Natl Acad. Sci. USA, 110, 16969–16974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo, S.D. , Cho, Y.H. and Sheen, J. (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2, 1565–1572. [DOI] [PubMed] [Google Scholar]
- Yu, M. , Carver, B.F. and Yan, L. (2014) TamiR1123 originated from a family of miniature inverted‐repeat transposable elements (MITE) including one inserted in the Vrn‐A1a promoter in wheat. Plant Sci. 215–216, 117–123. [DOI] [PubMed] [Google Scholar]
- Zhang, B. , Xu, W. , Liu, X. , Mao, X. , Li, A. , Wang, J. , Chang, X. et al. (2017) Functional conservation and divergence among homoeologs of TaSPL20 and TaSPL21, two SBP‐box genes governing yield‐related traits in hexaploid wheat. Plant Physiol. 174, 1177–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, G. , Zou, C. , Li, K. , Wang, K. , Li, T. , Gao, L. , Zhang, X. et al. (2017) The Aegilops tauschii genome reveals multiple impacts of transposons. Nat. Plants, 3, 946–955. [DOI] [PubMed] [Google Scholar]
- Zhou, S. , Zhang, J. , Che, Y. , Liu, W. , Lu, Y. , Yang, X. , Li, X. et al. (2018) Construction of Agropyron Gaertn. genetic linkage maps using a wheat 660K SNP array reveals a homoeologous relationship with the wheat genome. Plant Biotechnol. J. 16, 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouhar, J. , Munoz, A. and Rojo, E. (2010) Functional specialization within the vacuolar sorting receptor family: VSR1, VSR3 and VSR4 sort vacuolar storage cargo in seeds and vegetative tissues. Plant J. 64, 577–588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Genome‐wide association study of wheat root depth at the booting stage.
Figure S2 Sequence alignment and phylogenetic tree of VSR1 proteins.
Figure S3 Roots of vsr1 mutants.
Figure S4 Signal peptide analysis of different haplotypes using SignalP‐5.0 Server.
Figure S5 Transmembrane domain prediction and sub‐cellular localization of TaVSR1‐B‐1/3 and TaVSR1‐B‐2/4.
Figure S6 sRNAs aligned to the 125 bp MITE in the promoter of TaVSR1‐B.
Figure S7 Phenotypic comparisons of accessions with or without the MITE insertion in multiple environments in the test panel.
Figure S8 H3K9me2 status in wheat Chinese Spring around TaVSR1‐B (TraesCS2B02G122400) region.
Figure S9 H3K27me3 status in wheat cv. Chinese Spring.
Figure S10 Phenotypes of TaVSR1‐B Hap2 (MITE+) and Hap4 (MITE−) transgenic wheat lines grown in polyvinyl chloride tubes.
Figure S11 Phenotype of TaVSR1‐B Hap2 (MITE+) and Hap4 (MITE−) transgenic wheat lines grown in the field.
Figure S12 Phenotypes of four TaVSR1‐B haplotypes in transgenic rice.
Figure S13 Seedling phenotypes of four transgenic TaVSR1‐B rice haplotypes.
Figure S14 Vacuoles in wheat roots stained with Neutral Red.
Figure S15 The amount of rainfall in the whole growing seasons.
Table S1 Annotation of candidate genes in the chromosome 2B block.
Table S2 Candidate SNP in the TaVSR1‐B block on chromosome 2B detected by GWAS.
Table S3 Details of the cv. Kronos (tetraploid variety) TILLING mutants.
Table S6 Association of the MITE insertion with canopy temperature.
Table S7 Sets of 10 accessions with MITE+ or MITE− TaVSR1‐B haplotypes randomly selected from the entire test panel and used for measuring TaVSR1‐B expression at the booting stage, and DNA methylation and H3K27me3 status.
Table S4 Forty polymorphic sites in TaVSR1‐B identified from a diverse selection of 32 wheat accessions representing four haplotypes.
Table S5 Haplotypes of all 323 wheat accessions in the test panel.
Table S8 Relative expression of TaVSR1‐B in 2‐week‐old seedling roots from the test panel.
Table S9 Primers used in the study.