

Abstract
Infant high-grade gliomas appear clinically distinct from their counterparts in older children, indicating that histopathologic grading may not accurately reflect the biology of these tumors. We have collected 241 cases under 4 years of age, and carried out histologic review, methylation profiling, and custom panel, genome, or exome sequencing. After excluding tumors representing other established entities or subgroups, we identified 130 cases to be part of an “intrinsic” spectrum of disease specific to the infant population. These included those with targetable MAPK alterations, and a large proportion of remaining cases harboring gene fusions targeting ALK (n = 31), NTRK1/2/3 (n = 21), ROS1 (n = 9), and MET (n = 4) as their driving alterations, with evidence of efficacy of targeted agents in the clinic. These data strongly support the concept that infant gliomas require a change in diagnostic practice and management.
SIGNIFICANCE:
Infant high-grade gliomas in the cerebral hemispheres comprise novel subgroups, with a prevalence of ALK, NTRK1/2/3, ROS1, or MET gene fusions. Kinase fusion–positive tumors have better outcome and respond to targeted therapy clinically. Other subgroups have poor outcome, with fusion-negative cases possibly representing an epigenetically driven pluripotent stem cell phenotype.
INTRODUCTION
The prognosis of pediatric high-grade gliomas (HGG) remains dismal, with a 5-year survival rate of only approximately 20% for children ages 0 to 14 years (1). They are strongly associated with unique location-dependent mutations in histone 3 variants H3.3 (H3F3A) and H3.1 (HIST1H3B/C) including two recurrent amino acid substitutions (K27M and G34R/V; refs. 2, 3), which together account for nearly half of all pediatric HGG and identify robust biological subgroups (4, 5). Histone wild-type cases are comprised of a highly diverse set of tumors, ranging from those with some of the highest somatic mutational burdens in human cancer (patients with biallelic mismatch repair deficiency syndrome; refs. 6, 7) to others seemingly driven by single genetic events, often gene fusions (8). The latter are particularly found in cases originally diagnosed as high-grade glioma at an infant age (9).
The definition of an infant used in pediatric neuro-oncology varies, but typically refers to children younger than 3 to 5 years (10); congenital cases are generally defined as being present at birth (11). The most frequent types of infant brain tumor are medulloblastomas, ependymomas, and low-grade gliomas (LGG; ref. 12). The latter include the relatively common pilocytic astrocytomas, but also other rarer entities such as desmoplastic infantile ganglioglioma/astrocytoma (DIGG/DIA; ref. 13). Tumors reported as HGG appear to be associated with significant differences in clinical outcome, with infant HGG (even with incomplete resection and without irradiation) showing a significantly improved survival compared with those in older children (8, 14–17), which may indicate the presence of a distinct, overlapping group of tumors where histopathologic grading may not be representative of clinical behavior.
Treatment outcomes also reflect these differences; the Baby POG I study found four children younger than 3 years of age who were diagnosed with a malignant glioma, underwent 24 months of chemotherapy without radiation treatment, and did not develop recurrent disease (10). A 5-year overall survival rate of 59% was reported in infants with HGG after prolonged chemotherapy treatment alone, and in another study 16 patients diagnosed with HGG and treated with focal radiotherapy showed a 5-year overall survival rate of 66% (11). Five reported cases of patients with congenital glioblastomas who survived surgery (with only one patient receiving a gross total resection) all showed a better outcome than expected (18), and two infant cases who both underwent subtotal resection of their tumors and did not receive any adjuvant therapy postoperatively saw regression of the residual tumors (19). The improved outcome both with chemotherapy and with surgery alone is particularly significant in this age group when considering the risk of declining cognition (13) and the development of leukoencephalopathy post–radiation treatment (11).
Previous studies have hinted at different histologic features within infant high-grade gliomas. High densities of “minigemistocytic-shaped” cells with abundant mitoses and absent necrosis were described previously (20), with others showing moderately hypercellular, mitotic, and necrotic tumors with cellular monotony and a lack of significant pleomorphism, and some showing a more spindled appearance (18, 19).
Current molecular data is limited, but EGF receptor (EGFR) and platelet-derived growth factor receptor A (PDGFRA) expression is reported as uniformly low in congenital glioblastomas (GBM), with a low level or absence of copy-number alterations in these genes (18, 21). TP53 and PTEN mutations, CDKN2A/B deletions, and other copy-number alterations often seen in older children are also not typically found in infant HGG (22). Occasional BRAFV600E mutations are found, particularly in DIGG/DIA (23), whereas histone and IDH1 mutations are rare. Methylation profiling indicates that the infant group may display a more LGG-like methylation pattern, with a 2-year survival of 74% (8). The most common somatic alterations seen in infants appear to be gene fusion events, particularly involving NTRK1/2/3. Although not specific to brain tumors (24), these were found to span both LGG and HGG in large-scale studies in children, with novel QKI–NTRK2 and NACC2–NTRK2 fusions found in pilocytic astrocytomas (25, 26), and AGBL4–NTRK2, TPM3–NTRK1, and ETV6–NTRK3 fusions found in patients with HGG who were less than 3 years of age (9). More recently, several case reports have identified additional receptor tyrosine kinase (RTK) gene fusions in infant gliomas of differing histologies (17, 27–35).
In this study, we collected the largest series of infant gliomas (exclusive of pilocytic astrocytomas) assembled to date and present a classification system based on integrated methylation profiling, fusion gene analysis, mutation detection, and histologic review, with preclinical and clinical evidence of effective targeting of the driving alterations in these unique entities.
RESULTS
Refinement of an Intrinsic Set of Infant Hemispheric Gliomas
We collected a unique series of 241 gliomas from patients under the age of four years at diagnosis from multiple centers around the world, with a view to exclude a priori pilocytic astrocytomas and other well-characterized, low-grade lesions with clear molecular markers (Fig. 1A). To ensure this, we searched for pathognomonic structural variants using a variety of sequencing platforms including whole-genome, exome, and RNA sequencing and a novel custom capture panel (Fig. 1B). We identified 28 cases to be excluded, mostly due to presence of KIAA1549–BRAF fusions (n = 22), the vast majority of which were collected as otherwise unspecified cerebellar astrocytomas (Supplementary Table S1). We also identified three cases of FGFR1 tandem duplication (including glioneuronal tumors), two MYB/MYBL1 fusions, and a case with MN1–BEND2 (representing the novel entity of HGNET–MN1; ref. 36). Of the remaining 213 cases, a further 13 were excluded on the basis of clear Heidelberg classifier matches to other nonglioma central nervous system (CNS) tumors from methylation array profiling data (Fig. 1C). These included two ependymomas, two HGNET–BCORs, an ETMR, and others (Supplementary Fig. S1). A further 9 cases failed array quality control and were excluded from further analysis. Finally, our series of 191 cases were projected onto a reference set of gliomas comprising multiple entities. Sixty-one of these infant samples most readily clustered with a known high- or low-grade subtype, leaving 130 infant gliomas for further analysis that we define as our “intrinsic set” (Fig. 1D), as they comprise a novel grouping of tumors with key clinical and molecular features in common, as we describe below.
Figure 1.

Defining an intrinsic set of infant gliomas. A, Flow diagram providing an overview of the inclusion and exclusion criteria for the assembled cohort of 241 samples from patients younger than 4 years. B, Fusion gene analysis by a variety of means allowed for the identification of 28 fusions marking clearly defined entities that were subsequently excluded from further analysis. C, Methylation array profiling and analysis by the Heidelberg classifier excluded a further 12 cases closely resembling non-glioma entities or failing quality control (n = 9). D, t-statistic based stochastic neighbor embedding (t-SNE) projection of the remaining cases highlighted 61 samples which clustered with previously reported high or low grade glioma subtypes, leaving an intrinsic set of 130 infant gliomas for further characterization by more histopathological assessment and in-depth sequencing. E, Anatomic location of infant gliomas after exclusion of pathognomonic fusions and non-glioma entities by methylation profiling (n = 130). Left, sagittal section showing internal structures; right, external view highlighting cerebral lobes. Each circle represents a single case and is colored by the glioma subgroup it most closely clusters with, defined by the key below. F, Kaplan–Meier plot of overall survival of cases separated by methylation subgroups DIGG (desmoplastic infantile ganglioglioma/astrocytoma), IHG (infantile hemispheric glioma), LGG (other low-grade glioma subgroups), and HGG (other high-grade glioma subgroups; n = 102). P value is calculated by the log-rank test (P = 0.0566 for HGG vs. rest). G, t-statistic based t-SNE projection of a combined methylation dataset comprising the intrinsic set of the current study (n = 130, circled) plus a reference set of glioma subtypes (n = 1,652). The first two projections are plotted on the x and y axes, with samples represented by dots colored by subtype according to the key provided.
The infant glioma cases excluded on the basis of methylation profiling (n = 61) were found to have arisen in anatomic areas of the CNS appropriate for the relative subgroup assignment, such as diffuse midline glioma K27M-mutant cases in the pons, pilocytic astrocytoma-like cases in the cerebellum, and PXA-like cases in the cerebral hemispheres (Fig. 1E), and were often accompanied by the expected genetic alteration. Interestingly, the remaining intrinsic set included the vast majority of those patients diagnosed younger than the age of 1 year (49/63, 78%; overall median of intrinsic set = 7.2 months). These cases scored most highly as two named subgroups in the current version (v11b4) of the methylation classifier—DIGG/DIA and the poorly defined infantile hemispheric glioma (IHG; Supplementary Table S2). The vast majority of these cases were found in the cortex, DIGG/DIAs particularly in the frontal lobe. These cases were found to have a significantly improved outcome compared with cases classified as HGG, with a median overall survival similar to those considered LGG (Fig. 1F), with the important caveat that detailed treatment information was not available across the cohort. The HGG subtype exclusions were predominantly >1 year old and showed a tendency toward a worse outcome than the other infant tumors (P = 0.0567, log-rank test). This remaining intrinsic group of tumors formed a continuum that clustered clearly apart from other glioma subgroups in a t-statistic based stochastic neighbor embedding (t-SNE) projection based upon methylation array data from the glioma reference set (n = 1,652; Fig. 1G). Many of these cases did not unequivocally classify as either IHG or DIGG/DIA despite their tight clustering, suggesting that the reference classes for these tumors likely need expanding and updating.
IHGs Are Defined by Presence or Absence of RTK Fusions
Additional gene sequencing (panel, exome, or genome) was available for 65 cases, including 41 of the intrinsic set, all of whom had fusion analysis by panel or RNA sequencing. Samples excluded as representing other glioma subtypes were found to harbor mutations consistent with such tumors, including IDH1R132H, H3F3AK27M, and HIST1H3BK27M, as well as common cosegregating variants in TP53, NF1, PTEN, PIK3CA, and ACVR1, deletions of CDKN2A/B, and amplification of PDGFRA (Fig. 2A). These were almost entirely absent from the intrinsic set. Instead, 25 of 41 (61%) cases harbored fusions in either ALK (n = 10), NTRK1/2/3 (n = 2, 2, and 8, respectively) ROS1 (n = 2), or MET (n = 1), usually in the absence of other alterations (Fig. 2B). The fusion-positive cases were mostly classified as IHGs (n = 21) or low-scoring DIGG/DIAs (n = 4). Although ALK fusions were restricted to the intrinsic set, we observed NTRK fusions in other glioma subtypes (especially NTRK2, n = 3). We additionally observed an FGFR1–TACC1 fusion in the IDH1/TP53 case (Fig. 2A). High-scoring DIGG/DIAs and “DIGG/DIA-like” tumors were found with BRAFV600E (n = 3) or PIK3R1 mutations and isolated mutations in bromodomain-containing genes (BRD8, BRD4, BRD2) and others (Fig. 2B). A single case harbored amplifications in both MYC and MYCN, in addition to TP53 and PIK3CA mutations. Although a proportion (<25%) of tumors were found with whole-arm DNA copy-number changes, the majority of intrinsic cases harbored few if any large-scale copy-number alterations (Supplementary Fig. S2A; Supplementary Table S3).
Figure 2.
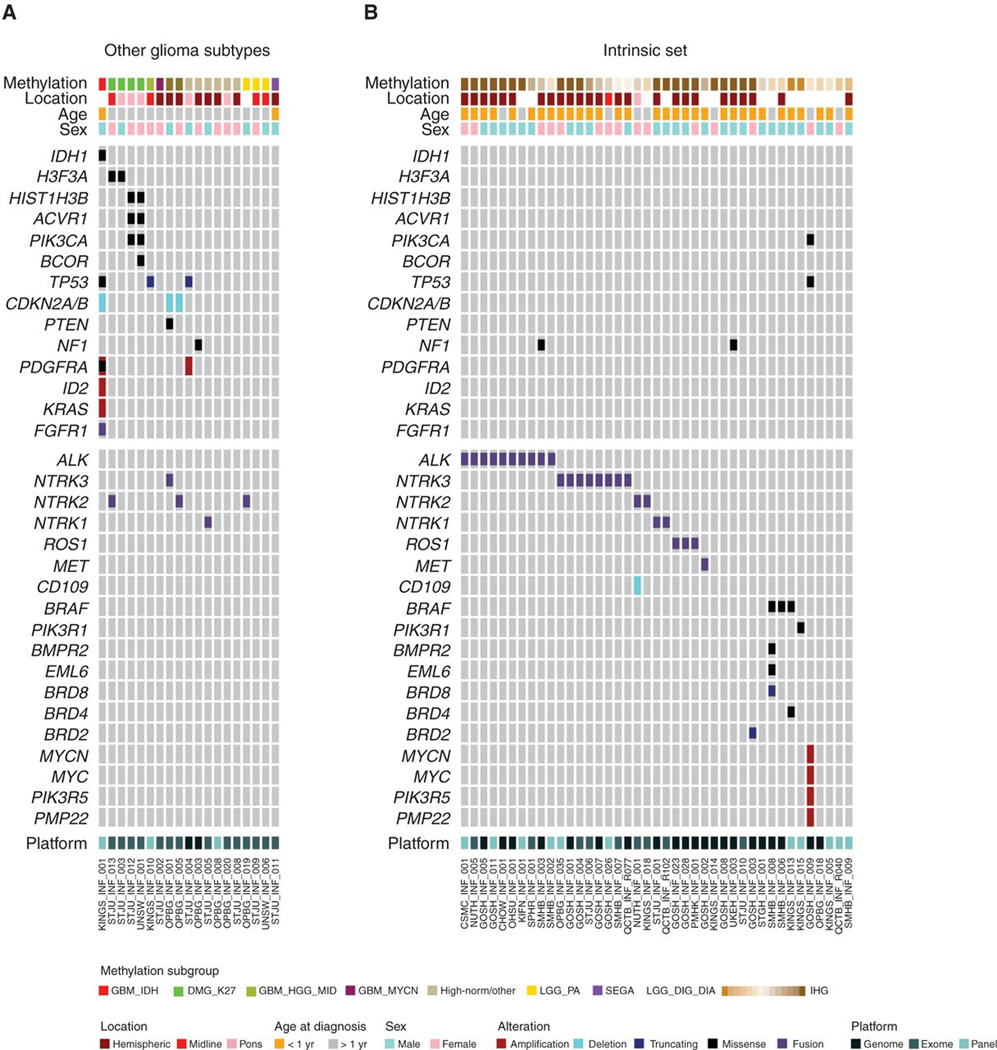
Mutations in infant gliomas. A, OncoPrint representation of an integrated annotation of single-nucleotide variants, DNA copy-number changes, and structural variants for infant gliomas excluded as other subgroups (n = 24). B, OncoPrint representation of an integrated annotation of single-nucleotide variants, DNA copy-number changes, and structural variants for infant gliomas in the intrinsic set (n = 41). Samples are arranged in columns with genes labeled along rows. Clinicopathologic and molecular annotations are provided as bars according to the included key.
There were no differences in the number of copy-number changes between fusion-positive and fusion-negative cases (P = 0.567, t test; Supplementary Fig. S2B). Notably, the only significant focal differences were those marking common gene fusions at the ALK and NTRK3 loci (Supplementary Fig. S2C). A novel and refined copy-number analysis from the methylation array data identified such breakpoints in either intrachromosomal (short gains or losses) or interchromosomal (imbalances) RTK fusion events in 53/71 (75%) cases across the whole cohort (Fig. 3A–C). Across the intrinsic set as a whole, 65 of 130 (50%) cases were found to harbor structural variants targeting ALK, NTRK1/2/3, ROS1, or MET (46/80, 57.5% IHGs), compared with 18 of the other 111 cases in the original series (16%; P < 0.0001, Fisher exact test; Supplementary Fig. S3A–S3C; Supplementary Table S4). Where possible, these were validated through a combination of genome, RNA, and/or Sanger sequencing, and were frequently accompanied by detectable focal DNA copy-number breakpoints within the fusion partners, as exemplified for ETV6–NTRK3 (Fig. 3D) and the novel ZC3H7A–ALK fusions (Fig. 3E). The most commonly targeted genes in the intrinsic set included NTRK1/2/3, predominantly ETV6–NTRK3 but also recurrent EML4–NTRK3 and TPM3–NTRK1 fusions (Fig. 3F). NTRK2 was found with numerous novel partners (e.g., KCTD16–NTRK2 and AGBL4–NTRK2) but was largely seen in other glioma subtypes occurring in the appropriate anatomic locations (e.g., H3K27M in midline regions; Supplementary Table S4), suggesting an important difference in NTRK2 compared with NTRK1/3 fusion–positive cases. ALK fusions were the most common (n = 39), were largely restricted to the intrinsic set, and included both intra- and interchromosomal rearrangements (Fig. 3G), including both previously reported (PPP1CB–ALK, EML4–ALK, HIP1–ALK, PRKAR2A–ALK, SPTBN1–ALK) and novel fusions (MAD1L1–ALK, MAP2–ALK, MSI2–ALK, SPECC1L1–ALK, SYNDIG1L–ALK, ZC3H7A–ALK, CLIP2A–ALK; Supplementary Table S4). Within the intrinsic set, there was a trend toward the presence of any fusion conferring a longer overall survival compared with those without (P = 0.0687, log-rank test; Fig. 3H).
Figure 3.

Copy number–associated fusion genes in infant gliomas. A, Segmented DNA copy-number heat map for ALK breakpoint cases, plotted according to chromosomal location. Pink, gain; blue, loss. B, Segmented DNA copy-number heat map for ROS1 breakpoint cases, plotted according to chromosomal location. Pink, gain; blue, loss. C, Segmented DNA copy-number heat map for MET breakpoint cases, plotted according to chromosomal location. Pink, gain; blue, loss. D, ETV6–NTRK3. Cartoon representation of the fusion structure, with reads on either side of the breakpoint colored by gene partner and taken from an Integrated Genome Viewer snapshot. Below this is a Sanger sequencing trace spanning the breakpoint. Underneath are copy-number plots (log2 ratio, y-axis) for chromosomal regions spanning the breakpoints (x-axis). Points are colored red for copy-number gain, blue for loss, and grey for no change. The smoothed values are overlaid by the purple line. E, ZC3H7A–ALK. Cartoon representation of the fusion structure, with reads on either side of the breakpoint colored by gene partner and taken from an Integrated Genome Viewer snapshot. Below this is a Sanger sequencing trace spanning the breakpoint. Underneath are copy-number plots (log2 ratio, y-axis) for chromosomal regions spanning the breakpoints (x-axis). Points are colored red for copy-number gain, blue for loss, and gray for no change. The smoothed values are overlaid by the purple line. F, Circos plot of gene fusions targeting NTRK1 (light orange), NTRK2 (orange), and NTRK3 (dark orange). Lines link fusion gene partners according to chromosomal location, represented by ideograms arranged around the circle. G, Circos plot of gene fusions targeting ALK (dark blue). Lines link fusion gene partners according to chromosomal location, represented by ideograms arranged around the circle. H, Kaplan–Meier plot of overall survival of cases separated by fusion event (n = 63). P value is calculated by the log-rank test (P = 0.085 for any fusion vs. none).
With whole-genome sequencing of fusion-negative cases failing to identify consistent genetic drivers of this subtype of the disease (Supplementary Fig. S4), we turned to the methylation data to further explore the heterogeneity within infant HGG. Hierarchical clustering on the basis of differential probes associated with the most common genetic alterations found, resulting in the separation of distinct sets of IHG subgroups in addition to clear DIGG/DIA and “DIGG/DIA-like” tumors (Supplementary Fig. S5A). Despite the presence of recurrent NTRK fusions, these infant gliomas clustered apart from mesenchymal tumors harboring ETV6–NTRK3, including infantile fibrosarcoma and congenital mesoblastic nephroma (ref. 37; Supplementary Fig. S5B). Running methylation-based gene ontology analysis on the differentially methylated regions (Supplementary Table S5) highlighted little overlap between ALK fusion, NTRK fusion, and fusion-negative cases (total 9.5%; Supplementary Fig. S5C). ALK fusion cases were significantly associated with dysregulation of genes associated with glutamate receptors, synapses, signal transduction, and morphogenic stages of development (Fig. 4A), whereas NTRK fusion cases were linked with genes controlling neuronal differentiation and the earliest stages of embryogenesis, as well as signaling via the JNK cascade (Fig. 4B). In contrast, fusion-negative cases were predominantly associated with the response to multiple endogenous stimuli, particularly the TGFβ pathway, and the regulation of stem cell pluripotency and cell fate (Fig. 4C). Although only exploratory due to the small sample sizes, and needing independent validation in an independent cohort, as exemplars of the differential epigenetic regulation of key genes controlling these processes in the distinct subgroups, we observed consistent reduction in methylation at CpG sites governing expression of WNT5A in ALK fusion cases (Fig. 4D), STAT1 in NTRK fusion cases (Fig. 4E), and TP63 in fusion-negative samples (Fig. 4F; Supplementary Table S5). This resulted in differential protein expression as assessed by multilabeled immunofluorescence with antibodies directed against these targets, with representative examples shown for WNT5A and STAT1 in ALK-fusion (Fig. 4G) and NTRK-fusion (Fig. 4H) cases, respectively. Using a NanoString assay for the 30 most differentially methylated genes between subgroups, we were able to distinguish ALK/NTRK fusion–positive and –negative subgroups in a series of 21 infant HGG for which we had sufficient material (Fig. 4I). Notably, we did not observe TP63 protein expression in any of our samples, although differential overexpression of the transcript was observed for fusion-negative cases.
Figure 4.

Epigenetic alterations in fusion-positive and fusion-negative infant gliomas. A, Differential methylation-based gene ontology analysis for ALK-fusion cases, represented in bar plots of −log10 P value for labeled highest-scoring categories (top) and aggregated ontology networks (bottom). B, Differential methylation-based gene ontology analysis for NTRK-fusion cases, represented in bar plots of −log10 P value for labeled highest-scoring categories (top) and aggregated ontology networks (bottom). C, Differential methylation-based gene ontology analysis for fusion-negative cases, represented in bar plots of −log10 P value for labeled highest scoring categories (top) and aggregated ontology networks (bottom). Node size is proportional to the number of genes, shading represents −log10 P value (darker is higher). Thickness of connecting lines reflects the percentage of overlapping genes. D, Genome browser view of the WNT5A locus, with lower methylation, provided as bar plots, in selected ALK-fusion (blue) cases compared to NTRK-fusion (orange) and fusion-negative (gray) cases. E, Genome browser view of the STAT1 locus, with lower methylation, provided as bar plots, in selected NTRK-fusion (orange) cases compared with ALK-fusion (blue) and fusion-negative (gray) cases. F, Genome browser view of the TP63 locus, with lower methylation provided as bar plots, in selected fusion-negative (gray) cases compared with ALK-fusion (blue) and NTRK-fusion (orange) cases. Chromosomal ideograms are provided, with the red bar indicating the cytoband in which the locus is found. Differentially methylated probes are highlighted by the red box. G, Immunofluorescence staining of an antibody directed against WNT5A (white) in an EML4–ALK fusion infant glioma case, UOLP_INF_001. DAPI is used as a counterstain. Scale bar, 50 μm. H, Immunofluorescence staining of an antibody directed against STAT1 (green) in an ETV6–NTRK3 fusion infant glioma case, GOSH_INF_007. DAPI is used as a counterstain. Scale bar, 50 μm. I, Heat map representing gene expression values from a NanoString assay of 30 most differentially methylated genes between ALK-fusion (blue), NTRK-fusion (orange), and fusion-negative (gray) cases. Expression values are colored according to the scale provided.
Histologic examination of those tumors classified as IHG revealed highly cellular astrocytic tumors with cells arranged in uniform sheets throughout the section (Supplementary Fig. S6A–S6C). Cytologically, spindled nuclei (Supplementary Fig. S6D), an occasional ganglion cell component (Supplementary Fig. S6E), or gemistocytic-like cells (Supplementary Fig. S6F) could be seen either focally or throughout the tumor. Tumors frequently showed a superficial hemispheric location, often involving the meninges, and had a well-defined border with adjacent normal brain. Palisading necrosis (Supplementary Fig. S6G), microvascular proliferation, and mild to moderate nuclear pleomorphism were almost universally seen. In some cases, a more nodular architecture was observed (Supplementary Fig. S6H and S6I). Rarely, some showed less cellularity (Supplementary Fig. S6J), and mineralization, calcification, or xanthomatous change could be observed (Supplementary Fig. S6K). Consistent with these features, 67 of 80 (84%) IHG cases were originally diagnosed as a high-grade glioma, although a variety of other diagnoses were included in the original pathology reports (Supplementary Fig. S6L). A summary of the histologic findings is given in Supplementary Table S6, with no statistically significant difference in features assessed between fusion-positive and fusion-negative subgroups. The number of mitoses observed was highly variable, and proliferation as assessed by Ki-67 staining highlighted cases presenting with both frequently (Supplementary Fig. S6M) and sparsely positive nuclei (Supplementary Fig. S6N). There was a significantly elevated Ki-67 index in NTRK fusion–positive compared with fusion-negative IHG cases (P = 0.0479, t test), although not for ALK (P = 0.3622, t test; Supplementary Fig. S6O). Notably, the NTRK (median = 22.5) and ALK (median = 15.6) fusion–positive indices are at the upper end of values reported (38) for older patients with grade IV (median = 15.8) and grade III (median = 11.8) glioblastomas and anaplastic astrocytomas, with fusion-negative cases (median = 5.6) closer to grade II astrocytomas (median = 3.0).
Generation and Preclinical Testing of an ALK Fusion–Driven In vivo Model
To assess the tumorigenic potential of the most commonly detected ALK gene fusion variant (PPP1CB–ALK) in a model system, we attempted to generate an in vivo model using two complementary somatic gene transfer-based methods (RCAS/Ntv-a viral gene transfer) and in utero electroporation (IUE; Fig. 5A). When using the RCAS approach with injection of cells producing PPP1CB–ALK-containing virus at p0 on a Cdkn2a-null background, tumor formation was rare (2/19 mice), and occurred only after 300 days. In contrast, in utero electroporation at E14.5 with PPP1CB–ALK alone was able to generate consistent tumor formation with 100% penetrance, albeit with a relatively long latency of more than 250 days. Although not commonly found in the human disease, when combined with CRISPR/Cas9-mediated knockout (KO) of either Trp53 or Cdkn2a for practical purposes, we observed highly efficient tumor formation with a median survival of 32 and 52 days, respectively (Fig. 5B). PPP1CB–ALK mice ± Cdkn2a-KO gave rise to tumors which reflected the human setting, including the typical foci of palisading necrosis, mitotic activity, glial cytology, and/or clear astrocytic differentiation (Supplementary Fig. S7A). All tumors would be classified as high-grade astrocytomas or glioblastomas. Staining for the HA epitope tag included at the C-terminus of the ALK fusion protein in the IUE/Cdkn2a-KO setting indicated widespread expression of the fusion protein, with invasion of individual tumor cells into the brain parenchyma (Supplementary Fig. S7B).
Figure 5.

Preclinical modelling of ALK-fused glioma. A, Schematic representation of the in vivo modeling workflow. IUE, in utero electroporation; KD, kinase domain. B, Kaplan–Meier curve of injected animals using IUE and p0-RCAS method: PPP1CB–ALK only IUE, PPP1CB–ALK + Trp53-KO IUE, PPP1CB–ALK + Cdkn2a-KO IUE and PPP1CB–ALK p0-RCAS only. *, P < 0.05; **, P < 0.01. C and D, Effect of targeted ALK inhibition on growth of allografted PPP1CB–ALK + Cdkn2a-KO mouse tumor cells in vivo. p.i., post injection. E, Targeted inhibition significantly prolonged the survival of PPP1CB– ALK + Cdkn2a-KO allografted mice compared with temozolomide or vehicle controls. Two mice in the lorlatinib group were sacrificed due to technical complications with drug delivery, with no tumor being evident upon dissection of the brain. ***, P < 0.001. F, Clinical history of DKFZ_INF_307, with confirmed MAD1L1–ALK fusion. Timeline of clinical interventions is provided below, with treatment shaded in gray. Axial T2 MRI scans from diagnosis and successive surgeries and chemotherapeutic regimens are provided, in addition to treatment with the ALK inhibitor ceritinib, with tumor circled in red.
To test the potential efficacy of targeted ALK inhibition in the context of this tumor model, we first dissociated tissue from a murine tumor into a single-cell suspension for growth in neurosphere (serum-free, nonadherent) conditions. Four different ALK inhibitors (crizotinib, ceritinib, alectinib, lorlatinib) were then tested for in vitro growth-inhibitory effects, representing different generations of inhibitor either approved for clinical use or currently in trials. Although all inhibitors showed a significant growth-inhibitory effect at nanomolar concentrations (ref. 17; Supplementary Fig. S7C), there were differences in potency between the different compounds (Supplementary Table S7).
Because of its clear in vitro efficacy and reportedly good blood–brain barrier penetration (an important consideration for clinical translation for brain tumors), lorlatinib was chosen as the primary candidate for in vivo testing in our preclinical ALK fusion model. For this purpose, adult CD1 mice were allografted with short-term in vitro–cultured PPP1CB–ALK;Cdkn2a−/− cells and monitored for tumor growth using bioluminescence imaging (BLI). At the start of treatment (14 days after injection), mice were stratified into temozolomide (standard chemotherapy), vehicle control, or lorlatinib arms, based on consecutive ranking (highest BLI signal to lorlatinib, 2nd highest to control, 3rd highest to temozolomide, and so on). Although temozolomide was found to slow tumor growth in comparison with vehicle control, all tumors in these two treatment arms continued to grow. In contrast, all but one lorlatinib-treated animal displayed a significant reduction in BLI signal compared with the pretreatment baseline (Fig. 5C and D). This imaging response corresponded with a significant increase in survival in the lorlatinib-treated group compared with the two control arms (P < 0.0001; although all tumors regrew after stopping treatment after 28 days, with all mice ultimately needing to be sacrificed due to onset of tumor symptoms; Fig. 5E). No significant difference in body weight was observed between mice on the different treatment arms (data not shown), and the compounds were generally well tolerated. A similar experiment was performed using lorlatinib versus temozolomide in mice transplanted with cells from an ALK fusion–only mouse tumor. This also revealed a significant tumor regression (Supplementary Fig. S7D) and survival increase with lorlatinib (P = 0.004, log-rank test), with one animal showing prolonged survival at last follow-up, 8 months postinjection (∼6 months after end of treatment; Supplementary Fig. S7E). Overall, these findings provide a strong preclinical rationale for the potential use of targeted ALK inhibition in a clinical setting. For one of the cases in our study, DKFZ_INF_307, we have been able to demonstrate this directly. Here, a 1-month-old boy underwent a left craniotomy with gross total resection, and was diagnosed with glioblastoma (WHO grade IV). He underwent successive rounds of HIT SKK/ACNS and temozolomide chemotherapy, eventually showing progressive disease after both. He was found to have a MAD1L1–ALK fusion and was started on ceritinib, resulting in stable residual disease for nearly two years to date (Fig. 5F).
Patient-Derived Models and Clinical Experience with NTRK Inhibitors
Finally, we explored the utility of treating RTK fusion–positive infant gliomas with targeted inhibitors. We established two primary patient-derived cell cultures from infant glioma specimens with either TPM3–NTRK1 or ETV6–NTRK3 fusions (Fig. 6A) and compared their in vitro sensitivities to three small-molecule inhibitors of TRKA/B/C with two fusion-negative pediatric glioma cultures (Fig. 6B). NTRK fusion–positive cells were more sensitive to entrectinib, crizotinib, and milciclib, with differential sensitivities ranging from 2- to 9-fold over fusion-negative cells (P = 0.0253, crizotinib; P = 0.0786, entrectinib; P = 0.0141, milciclib; Supplementary Table S7), and reduction in downstream signaling via phospho-AKT and phospho-ERK (Supplementary Fig. S7F). The infant glioma models were not tumorigenic after multiple orthotopic implantation experiments in immunodeficient mice, precluding in vivo assessment (data not shown).
Figure 6.

Preclinical and clinical experience with TRK inhibitors in fusion-positive infant glioma. A, Light microscopy image of two patient-derived infant glioma cell cultures, harboring either TPM3–NTRK1 (QCTB-R102; light orange) or ETV6–NTRK3 (QCTB-R077; dark red) fusions. B, Concentration–response curves for three TRK inhibitors tested against two NTRK fusion–positive infant glioma cell cultures (QCTB-R102, TPM3–NTRK1, light orange; QCTB-R077, ETV6–NTRK3, dark red) and two fusion-negative glioma cultures (QCTB-R006, light gray; QCTB-R059, dark gray). Concentration of compound is plotted on a log scale (x-axis) against cell viability (y-axis). Mean plus SE are plotted from at least n = 3 experiments. C, Clinical history of OPBG_INF_035, with confirmed ETV6-NTRK3 fusion. Timeline of clinical interventions is provided below, with TRK inhibitor treatment shaded in gray. Diagnosis, postbiopsy, pre/postsurgery, post-crizotinib, and post-larotrectinib axial T2 MRI scans are provided, with tumor circled in red. D, Clinical history of MSKC_INF_006, with confirmed ETV6–NTRK3 fusion. Timeline of clinical interventions is provided below, with TRK inhibitor treatment shaded in gray. Diagnosis and post-larotrectinib post-contrast axial T1 MRI scans are provided, with tumor circled in red.
Clinical treatment with TRK inhibitors was given to two patients in our cohort with ETV6–NTRK3 fusions. The first case, OPBG_INF_035, was a girl diagnosed with a large frontal mass at 36 weeks gestation (Fig. 6C). It was a large, heterogenous mass with solid, cystic, and hemorrhagic components. A biopsy was performed after birth and it was diagnosed histologically as a glioblastoma (WHO grade IV). The child subsequently received chemotherapy (methotrexate, vincristine, etoposide, cyclophosphamide, thiotepa) before undergoing a subtotal resection 3 months later. An ETV6–NTRK3 fusion was identified in the DNA from both the biopsy and resection specimens, and 4 months postsurgery the child was commenced on crizotinib. An MRI scan performed after 9 months of treatment with crizotinib showed a 56% reduction in the size of the remaining solid component of the tumor compared with the postsurgery MRI scan (RANO criteria size reduction of >50% and stable). After an additional 3 months of treatment with larotrectinib, the remaining solid component showed a further reduction in size, now reaching 73% (Supplementary Fig. S8A). Clinically, the child remains well. The second patient, MSKC_INF_006, presented with a generalized seizure at age 11 months (Fig. 6D). An MRI scan revealed a pontine mass with central hemorrhage. The child underwent surgery and a gross total resection was achieved. Histologically the tumor was diagnosed as a low-grade neuroepithelial neoplasm. The child developed a recurrence, at which point vincristine and carboplatin were commenced and a complete response was achieved. However, the tumor progressed two years after the original resection; a further gross total resection was achieved and the child was treated with larotrectinib after an ETV6–NTRK3 fusion was identified, with the aim of preventing further recurrence. To date, the child remains well with no evidence of recurrence after 12 months of treatment.
Notably, the patients from whose tumors our primary cell lines were derived have both received only surgery to date, and remain well. QCTB_INF_R077 was diagnosed with a tumor in the left frontoparietal lobe in utero and underwent biopsy and subsequent resection shortly after birth. Histologically, the tumor was reported as a primary neuroepithelial tumor. The child was not treated with any adjuvant therapy. At 5.5 years old, there has been no progression or relapse and the child has stable disease (Supplementary Fig. S8B). The second patient, QCTB_INF_R102, presented with a tumor in the left temporal lobe at age 8 months. He subsequently received a gross total resection, with the tumor diagnosed as a ganglioglioma (WHO grade I). He also did not receive any adjuvant therapy post resection and is currently 4 years old and remains stable under regular surveillance (Supplementary Fig. S8C).
In summary, diffuse infant gliomas represent distinct disease entities marked by characteristic clinicopathologic profiles and in most cases clinically actionable gene fusions (Fig. 7).
Figure 7.

Summary of infant HGG subgroups.
DISCUSSION
Malignant glioma presenting in infancy represents a specific clinical challenge, involving diagnostic uncertainty and a hesitancy to aggressively treat given the reported superior outcomes compared with older children, coupled with the high risk of neurocognitive deficits (39). This is compounded by a lack of biological understanding due to the rarity of these tumors. The present international collaborative study brings together the largest collection of tumors originally reported as high-grade or diffuse gliomas in this age group, in contrast with another recent multi-institutional study that was predominantly comprised of low-grade tumors (17). Our study uniquely includes methylation and gene-expression data, and allows for refinement of subgroups within the malignant spectrum of disease with important clinical management implications; we also present experience of clinical responses with targeted agents even after progression on standard chemotherapies.
A first key finding relates to the difficulty of differential diagnoses in these very young children, with approximately 10% of cases unequivocally classifying as other tumor entities on the basis of methylation profiling (40) or the presence of pathognomonic gene fusions (36), even after discounting misdiagnosed or misassigned pilocytic astrocytomas. Often this uncertainty is reflected in the original pathology report, with atypical features highlighted. However, the highly heterogeneous nature of high-grade glial tumors provides for a broadly inclusive category in the current WHO classification, which in many cases may result in what is considered to be a relatively uncontroversial histologic diagnosis despite widely varying morphologies. Similarly, combined genetic and epigenetic analyses reveal a third of remaining cases to be biologically identical to known high- or low-grade glioma subtypes, with substantially different prognoses reflective of the known clinical course of the relevant tumor categories. Together, these data make the important points that histopathologic evaluation alone is insufficient to predict outcome, and that high-grade gliomas predominantly occurring in older childhood may also present in the infant population with little survival benefit from standard treatment protocols.
After these exclusions, there remains what we define as an intrinsic set of infant gliomas, which are largely restricted to the cerebral hemispheres and occur in the youngest patients, usually younger than 12 months old. These patients, despite more than three quarters unequivocally reported as WHO grade III or IV astrocytoma, have an overall survival more akin to lower-grade tumors, yet lack the key molecular features of both HGG and LGG. They appear to form a biological continuum of disease between the recognized MAPK-driven desmoplastic lesions (DIGG/DIA), which may respond clinically to targeted BRAFV600E inhibitors even after previous chemotherapy (41), and a novel assignation of diffuse infant hemispheric glioma. This latter end of the spectrum is strikingly defined by nearly two thirds of tumors harboring fusions in genes encoding the RTKs ALK, NRTK1/2/3, ROS1, and MET. Although structural variants involving these genes within the age group have been described in case reports (27–35) and a recent larger study (n = 29; ref. 17), the current report represents a uniquely powerful study of these rare tumors by accumulating a series of 82 infant cases with RTK fusions with full methylation profiles.
Molecularly, these events included interstitial microdeletions such as those at chromosome 2p23 resulting in the fusion of CCDC88A or PPP1CB and ALK (17, 34) and at 6q21 fusing ROS1 and GOPC (previously known as FIG, and originally described in an adult GBM cell line; ref. 42); additional focal DNA copy-number losses targeted MET at 7q31 (43). There were multiple instances of interchromosomal copy-number gains fusing ALK to a series of novel partners, including MAD1L1 (7p22), ZC3H7A (16p13), MSI2 (17q22), SYNDIG1 (20p11), and SPECC1L (22q11), as well as the intrachromosomal EML4–ALK fusion that is well characterized in non–small cell lung cancer and others (44). The NTRK genes had a variety of interchromosomal partners, with around half of cases marked by a DNA copy imbalance at either locus. Notably, NTRK2 fusions (also described in LGG; refs. 25, 26, 45) were largely found in tumors classifying as other glioma subtypes, as were the previously described FGFR–TACC fusions (46).
Histopathologically, within the context of HGGs, certain common features of the intrinsic infant hemispheric gliomas could be recognized. Cases tended to have a relatively uniform architecture, with marked pleomorphism. There was an enrichment of gemistocytic-like cells, as has been reported for a case with ZCCHC8–ROS1 fusion (29) and a predominance of spindle cell differentiation, reminiscent of mesenchymal tumors with NTRK fusions (47) and also described in an ETV6–NTRK3 infant glioma (35). Our NTRK fusion cases in this study clustered distinctly from ETV6–NTRK3-positive infantile fibrosarcoma and congenital mesoblastic nephroma, however, suggesting a distinct origin. Several cases also had ependymal differentiation, consistent with two cases with ALK fusions (KTN1–ALK and CCDC88A–ALK) reported as not easily fitting the established WHO brain tumor entities (34). Notably, CCDC88A–ALK cases have been reported clinically as both low- and high-grade glioma; however, the same study found tumors generated by overexpressing the fusion in xenografted immortalized human astrocytes to have a high proliferative index, glial marker expression, and pseudopalisading necrosis (17), suggestive of high-grade lesions in common with our in utero electroporation modeling approach. A further case report described a KIF5B–ALK fusion in an infant with microglial proliferation, spindle cells with scattered mitotic figures, and a mixed inflammatory infiltrate of scattered lymphocytes, plasma cells, and eosinophils, indicating potential microglioma or gliofibroma (31). The recognition of tumors in this series that biologically resemble DIGG/DIA (WHO grade I) is compatible with their histology, in that some cases have been described as presenting with a poorly differentiated component (39). The case with ZCCHC8–ROS1 fusion was also described to display a cellular element within a fusocellular desmoplastic component (29), and we noted focal ganglion cells in our series. However, despite these differences, it is still not possible at the present time to define clear histology-only criteria which can reliably distinguish between these molecularly defined intrinsic infant tumors and other glioma subtypes in the same age group.
The presence of recurrent ALK/NTRK/ROS1/MET fusions represents clearly targetable alterations, in common with subgroups of adult epithelial tumors (48, 49), and their identification through screening approaches and routine diagnostic sequencing panels (50–53) makes them amenable to selection for clinical trials despite their rarity. The distinct morphologic variants, the restricted spatial and temporal patterns of presentation, and the specificity of oncogenic events largely in the absence of other mutations or large-scale chromosomal rearrangements suggests an exquisite developmental susceptibility for transformation that would account for this rare subgroup of tumors.
Multiple ALK partners are associated with synapse formation and activity (CCDC88A, HIP1, SYNDIG1), neuronal cytoskeletal reorganization (CCDC88A, SPECC1L), and microtubule assembly (MAP2, PRKAR2A, EML4), as well as PI3K–MAPK signaling (PPP1CB, CCDC88A, SPECC1L) and cell-cycle progression (MAD1L1; refs. 54–62). Thus, in addition to the activated kinase activity of the ALK receptor itself, these fusions likely disrupt key regulatory processes in neurodevelopment, as exemplified by the differential methylation of genes controlling these processes that we observed. The most common ALK fusion, PPP1CB–ALK, was found to be tumorigenic when introduced in prenatal, although largely not postnatal, mice, further demonstrating the importance of developmental context associated with the oncogenicity of these alterations.
ALK fusion–positive tumors were found to be sensitive to targeted ALK inhibition in vitro and in vivo, resulting in tumor shrinkage and extension of survival in the latter in contrast to the standard chemotherapeutic agent temozolomide. Excitingly, this experience was mirrored in the clinic, whereby a child diagnosed at 1 month old experienced stable disease for nearly two years on targeted therapy after progressing on two successive chemotherapy protocols, including temozolomide. Critically, NTRK fusion cases were also found to respond to targeted inhibitors in patient-derived models in vitro as well as in children treated clinically, in common with isolated reported cases (35), whereby for example a 3-year-old girl who had failed multiple therapies including chemotherapy and radiotherapy showed near-total resolution of primary and metastatic lesions after treatment with larotrectinib. If validated in larger trials, such agents may represent attractive options to spare the long-term sequelae of chemotherapy and radiotherapy, while maintaining the generally good prognosis of these patients (27, 30, 33).
Despite the frequency of alterations identified, not all of the intrinsic infant gliomas were found to harbor RTK fusions. These fusion-negative cases (at least on the basis of the platforms used in this study) had a lower proliferation index compared with NTRK fusion–positive cases, but a worse prognosis under standard treatment. Although we could identify no apparent recurrent genetic driver of this subgroup, even with whole-genome sequencing of a subset of cases, there were clear epigenetic differences compared with fusion-positive cases, with dysregulated gene networks associated with the regulation of stem cell pluripotency, plausibly suggesting an immature progenitor cell phenotype for these genetically bland lesions. In contrast, NTRK-fusion cases were associated with an embryonic, neuronal developmental program, and ALK-fusion cases with later AMPA-receptor synaptic plasticity signatures.
Further work is needed to explore all intrinsic infant glioma subgroups, in particular the fusion-negative cases. However, it is clear that these tumors harbor unique biology with associated clinicopathologic differences and should no longer be diagnosed or treated in the same way as their older counterparts. Maximal safe surgical resection remains the aim of treatment, regardless of subtype (17). However, our study has shown that RTK fusions can be found across all subgroups (although they are more frequently seen in the IHG group) and so screening (initially via copy-number profiling with subsequent validation) will help to identify patients who may be eligible for targeted therapy or clinical trials.
METHODS
Cases
All patient samples included were classified as gliomas (WHO grade II, III or IV) age <4 years old (including congenital cases) from all CNS locations (including spinal tumors). Cases were excluded if they had been diagnosed as a pilocytic astrocytoma with a known BRAF fusion or mutation. Ependymal, embryonal, mesenchymal, and germ cell tumors were also excluded. Samples were received from national collaborators (Great Ormond Street Hospital, London, n = 33; King’s College Hospital, London, n = 21; University Hospitals Bristol, n = 9; Newcastle Royal Infirmary, n = 6; St George’s Hospital, London, n = 4) and international collaborators (German Cancer Research Center [DKFZ], n = 86; Ospedale Pediatrico Bambino Gesù, n = 37; St. Jude Children’s Research Hospital, Memphis, n = 17; Memorial Sloan Kettering Cancer Center, New York, n = 6; Queensland Children’s Tumor Bank, Brisbane, n = 5; Universitätsklinikum Hamburg-Eppendorf, n = 5; Children’s Cancer Institute, Sydney, n = 2; Children’s Hospital of Wisconsin, Milwaukee, n = 2; Emory University Hospital, Atlanta, n = 1; St. Petersburg Hospital No. 6, n = 1; Wake Forest School of Medicine, Winston-Salem, n = 1; The Chinese University of Hong Kong, n = 1; Children’s National Medical Center, Washington, DC, n = 1; Chaim Sheba Medical Center, Tel Aviv, n = 1; Oregon Health & Science University, Portland, n = 1; University of Ljubljana, n = 1). Where possible, a hematoxylin and eosin (H&E) slide, 10 unstained sections, formalin-fixed paraffin-embedded (FFPE) tissue rolls, or frozen tissue was provided for each case. In some cases, data alone were provided. A total of 241 cases were entered into the study. Eight cases from King’s College and St George’s Hospital London (8) and ten cases from St. Jude Children’s Hospital in Memphis (9) have been previously published. All patient samples were collected under full Research Ethics Committee approval at each participating center.
Nucleic Acid Extraction
DNA was extracted from frozen tissue by homogenization prior to following the DNeasy Blood & Tissue Kit protocol (QIAGEN). DNA was extracted from FFPE pathology blocks after manual macrodis-section using the QIAamp DNA FFPE Tissue Kit protocol (QIAGEN). Concentrations were measured using a Qubit fluorometer (Life Technologies). RNA was extracted by following the RNeasy Mini Kit protocol (QIAGEN), and quantified using a Nanodrop 2000 Spectrophotomer (Thermo Fisher Scientific).
Methylation Profiling
The quantity and quality of DNA varied between cases with FFPE samples yielding less (range for FFPE: 11.0–2960.0 ng, range for fresh frozen: 211.0–5358.0 ng). Methylation analysis was performed when > 150 ng of DNA was extracted, using either Illumina 450K or EPIC BeadArrays at DKFZ (Heidelberg, Germany), University College London (UCL) Great Ormond Street Institute of Child Health or St. Jude Children’s Research Hospital. Data from Illumina 450K or EPIC arrays was preprocessed using the minfi package in R (v11b4). DNA copy number was recovered from combined intensities using the conumee package. The Heidelberg brain tumor classifier (molecularneuropa thology.org; ref. 40) was used to assign a calibrated score to each case, associating it with one of the 91 tumor entities which feature within the current classifier (v4). Clustering of beta values from methylation arrays was performed based upon correlation distance using a ward algorithm. DNA copy number was derived from combined log2 intensity data based upon an internal median processed using the R packages minfi and conumee to call copy number in 15,431 bins across the genome. Gene ontology analysis of differentially methylated regions was carried out using methylGSA (rdrr.io/bioc/methylGSA/), adjusting the number of CpGs for each gene by weighted resampling and Wallenius noncentral hypergeometric approximation in methylgometh (63). Ontology networks were constructed using ShinyGO (bioinformatics.sdstate.edu/go/).
Fusion Panel
A custom fusion panel consisting of 22 genes associated with fusions in pediatric brain tumors (ALK, BCOR, BRAF, c11orf95, C19MC, CIC, ETV6, FGFR1/2/3, FOXR2, KIAA1549, MET, MN1, MYB, MYBL1, NTRK1/2/3, RAF, RELA, TPM3, and YAP1) was designed with a library of probes to ensure adequate coverage of the specified regions (Roche Sequencing Solutions; ref. 64). Where available, 100 to 200 ng of DNA was used for library preparation using KAPA Hyper and HyperPlus Kit (Kapa Biosystems) and SeqCap EZ adaptors (Roche). Following fragmentation, DNA was end-repaired, A-tailed, and indexed adaptors ligated. DNA was amplified, multiplexed, and hybridized using 1 μg of the total precapture library DNA. After hybridization, capture libraries were amplified and sequencing was performed on a MiSeq and NextSeq (Illumina). Quality control, variant annotation, deduplication, and metrics were generated for each sample. The raw list of candidates provided by Manta (https://github.com/Illumina/manta) was filtered for more than 2 reads covering both genes, common false-positive base pairs (bp) positions/fusions outside of the capture set at both ends, common breakpoint/false positives within 10 bp, common false-positive gene pairs, fusions within the same gene and homologous sequences greater than 10 bp. Breakdancer was used to confirm all the breakpoints in all samples. Sequences either side of the break points were annotated to look for repetitive elements. A BLAT score was obtained to remove loci which were not uniquely mapped. Integrative Genomics Viewer was used to view the fusions.
DNA and RNA Sequencing
DNA was sequenced either as whole genome or captured using Agilent SureSelect whole exome v6 or a custom panel of 329 genes known to present in an unselected series of pediatric high-grade glioma (8). Library preparation was performed using 50 to 200 ng of genomic DNA. Following fragmentation, DNA was end-repaired and A-tailed, and indexed adapters ligated. DNA was amplified, multiplexed, and hybridized using 1 μg of total precapture library. After hybridization, capture libraries were amplified and sequencing was performed on a NextSeq500 (Illumina) with 2 × 150 bp paired-end reads following the manufacturer’s instructions. Ribosomal RNA was depleted from 500 to 2,000 ng of total RNA from FF and FFPE using NEBNext rRNA Depletion Kit. Following first-strand synthesis and directional second-strand synthesis, resulting cDNAs were used for library preparation using NEBNext Ultra II Directional RNA Library Prep Kit from Illumina performed as per the manufacturer’s recommendations. Exome capture reads were aligned to the hg19 build of the human genome using bwa v0.7.12 (bio-bwa.sourceforge.net), and PCR duplicates removed with PicardTools 1.94 (pcard.sourceforge. net). Single-nucleotide variants were called using the Genome Analysis Tool Kit v3.4–46 based upon current best practices using local realignment around indels, downsampling, and base recalibration with variants called by the Unified Genotyper (broadinstitute.org/gatk/). Variants were annotated using the Ensembl Variant Effect Predictor v74 (ensembl.org/info/docs/variation/vep) incorporating SIFT (sift.jcvi.org) and PolyPhen (genetics.bwh.harvard.edu/pph2) predictions, COSMIC v64 (sanger.ac.uk/genetics/CGP/cosmic/), dbSNP build 137 (ncbi.nlm.nih.gov/sites/SNP), ExAc, and ANNOVAR annotations. RNA sequences were aligned to hg19 and organized into de novo spliced alignments using bowtie2 and TopHat version 2.1.0 (ccb.jhu.edu/software/tophat). Fusion transcripts were detected using chimerascan version 0.4.5a filtered to remove common false positives.
PCR/Sanger Sequencing Validation
PCR to validate fusion breakpoints was carried out using primers obtained from Integrated DNA Technologies. PCR products were cleaned using the ExoProStar S 20 (Sigma-Aldrich) and were sent for Sanger sequencing (DNA Sequencing and Services, University of Dundee, United Kingdom). Sequences were analyzed manually with 4Peaks (Nucleobytes).
NanoString Gene Expression Analysis
The top 30 genes with the most differentially methylated regions between ALK-fusion, NTRK-fusion, and fusion-negative cases were selected for an mRNA expression analysis using a custom nCounter platform and nDesign (NanoString). Specimen RNA was mixed in hybridization buffer with CodeSets and hybridized overnight at 65°C. Samples wash reagents and imaging cartridge were processed on the nCounter Prep Station and imaged on the nCounter Digital Analyzer according to the manufacturer’s instructions. Data were normalized with NanoStringNorm v1.2.1 using variance stabilizing normalization. Heat maps were made by clustering the median centered expression values or a correlation matrix based on Euclidean distance using a Ward D2 algorithm.
Immunofluorescence
Paraffin-embedded tissue sections were deparaffinized in three changes of xylene and ethanol. Heat-mediated antigen retrieval was performed (Dako S1699, pH 6.0) and tissue slides were permeabilized with 0.5% Triton X-100 solution for 10 minutes at room temperature and then blocked with appropriate serum according to the species of secondary antibody for 1 hour at room temperature. For STAT1 staining (AHO0832, Invitrogen, 1:800), Alexa Fluor 488 Tyramide Super Boost Kit was used (B40941, Invitrogen) and antibody was incubated at 37°C for 30 minutes. For WNT5A (MA5–15502, Invitrogen, 1:800) and TP63 (39692, Cell Signaling Technology, 1:900) staining, samples were incubated at 37°C for 30 minutes. Sample slides were then washed in PBS three times and incubated with DyLight 649 (DI-2649, Vector, 1:100) and Alexa Fluor 555 (A31572, Invitrogen, 1:300) conjugated secondary antibodies for an hour at room temperature. Nuclei were counterstained with DAPI and samples mounted with Vectashield (H1000, Vector Laboratories) and examined using Zeiss Axio Scan.Z1 automated fluorescence slide scanner.
Histology and IHC
Histologic review was undertaken according to the WHO Classification of Tumors of the Central Nervous System (2016; ref. 65). Each case was reviewed blinded to the molecular features with a predetermined set of criteria to assess for the presence of histologic features characteristic of gliomas such as necrosis, mitotic figures, and stromal and astrocytic morphology. Any unusual features not previously associated with these tumors, including unusual nuclear morphology, was noted. These features were then rereviewed in the context of any molecular results identified. IHC for Ki-67 (M7240, DAKO, 1:100) was carried out using pressure-mediated antigen retrieval and the Envision detection system (DAKO K5007). Slides were mounted using Leica CV Ultra mounting medium, imaged using the high-throughput scanning microscope AxioScan Z1, and quantified using Definiens software.
Novel ALK Fusion Mouse Model
A PPP1CB–ALK fusion construct was cloned into either an RCAS or a pT2K vector using RNA from a human glioma sample as a template. After cDNA synthesis and PCR amplification, the ends of the product were cut with EcoRI and XhoI (for cloning into pT2K) or NotI and ClaI (for RCAS) and ligated into the target vector using the Takara Ligation mixture (Clontech). Bacterial amplification and QIAprep Spin Miniprep Kit (QIAGEN) were performed according to the manufacturer’s instructions to isolate the cloned plasmid. The DNA was sequenced using Sanger Sequencing at GATC Biotech and protein expression was confirmed on Western blot after transfection of DF-1 cells with the vector.
In Utero Electroporation.
After confirming that the expression vector contained the right inserts, embryos of CD1 mice were injected with plasmid into the fourth ventricle and electroporated in utero at E14.5. The PPP1CB–ALK fusion plasmid was used alone or in combination with CRISPR guide RNAs against Cdkn2a. Because of the incorporated IRES-Luciferase reporter on the pT2K vector, mice with successful integration of the transgene could be assessed at postnatal day 3 using bioluminescence imaging on an IVIS imager (PerkinElmer). Mice were sacrificed upon first signs of tumor-related symptoms according to humane endpoint criteria. H&E and IHC staining was performed according to standard protocols on 3-μm sections.
RCAS.
Four days before the calculated birth date, early-passage DF-1 fibroblasts for virus production were plated at 2–3 × 105 cells/T25 flask in 5 mL DMEM with 10% FCS + 1% penicillin/streptomycin + 1% Glutamax at 5% CO2 at 39°C. One day after, the cells were transfected with the RCAS construct as follows: 4 μg of the RCAS plasmid was incubated in 200 μL of room temperature OptiMEM and 10 μL FuGene transfection reagent. After a 15-minute incubation time, this mixture was slowly added to the settled DF-1 cells, mixed well by gently moving the flask and placed back in the incubator. An RCAS-GFP plasmid was always run in parallel in a separate flask to check for transfection success. On the day of birth, the transfected DF-1 cells were harvested using 10× Trypsin-EDTA and counted using the automated cell counter TC20. Cells (4 × 105 in 1 μL) were used for injection into newborn Ntv-a;Cdkn2a−/−;Ptenfl/fl pups at p0. The required amount of cells, depending on the size of the litter, was eluted in DMEM culture medium. The pups were taken out of the cage in a sterile hood and injected into the striatum with 1 μL of the DF-1 cell solution using a 10 μL Hamilton syringe. Mice were sacrificed upon first signs of tumor-related symptoms according to humane endpoint criteria. All animal protocols were approved by the relevant authority (Regierungspräsidium Karlsruhe) under registration numbers G-212/16 and G-168/17.
In vitro Culture and Compound Testing of Murine Tumor Cells
Murine ALK fusion–positive tumors were dissected immediately postmortem, mechanically dissociated, and then filtered through a 40-μm cell strainer. Cells were then plated in vitro in 10-cm dishes and grown as spheres in a 1:1 mix of Neurobasal-A and DMEM/F-12 media containing 1% 1 mol/L HEPES buffer solution, 1% 100 mmol/L sodium pyruvate MEM, 1% 10 mmol/L MEM nonessential amino acids solution, 1% GlutaMAX and 1% antibiotic–antimycotic supplemented with 2% B27, 2 μg/mL heparin solution, 10 ng/mL H-PDGF-AA, 20 ng/mL recombinant human bFGF, and 20 ng/mL recombinant human EGF. For splitting, cells were dissociated with Accumax at 37°C for 5 minutes.
For in vitro drug testing, primary sphere culture cells were plated at 1 × 104 cells/well in 80 μL growth factor–containing medium/well in 96-well plates. Triplicates per drug concentration (20 μL total volume for each) were added 24 hours after seeding the cells. The drug concentrations ranged between 1 nmol/L and 30 μmol/L. Corresponding DMSO concentrations were plated as controls, to which the treated wells were normalized. The ALK inhibitors crizotinib, alectinib, ceritinib, and lorlatinib were used. All compounds were purchased from Selleck Chemicals and initially diluted in DMSO to either a 10 mmol/L or 1 mmol/L stock, which were stored at −80°C. A CellTiter-Glo assay (Promega) was used as a readout of compound efficacy. This assay was conducted 72 hours after drugs were added to the cells. For this purpose, 50 μL of CellTiter-Glo substrate was added to each well using a multichannel pipette, and plates were incubated for 15 minutes while shaking in the dark. After that time, the luminescence signal per well was measured using a Mithras LB940 microplate reader. The respective DMSO control value was subtracted from the drug’s value to normalize the readout. The GI50 curves show the mean ± SD of the triplicates per condition measured. Representative results from duplicate experiments are shown.
Western Blot Analysis
Cells were incubated in complete media with vehicle or increasing concentrations of entrectinib (0.1, 1, 10 μmol/L), and protein was collected 4 hours post-treatment. Samples were lysed using lysis buffer (Cell Signaling Technology) containing phosphatase inhibitor cocktail (Sigma) and protease inhibitor cocktail (Roche Diagnostics). Following quantification using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), cell extracts were loaded for Western blot analysis. Membranes were incubated with primary antibody (1:1,000) overnight at 4°C, and horseradish peroxidase secondary antibody (Amersham Biosciences) for 1 hour at room temperature. Signal was detected with ECL Prime Western blotting detection agent (Amersham Biosciences), visualized using Hyperfilm ECL (Amersham Biosciences) and analyzed using an X-ray film processor in accordance with standard protocols. Primary antibodies used were phospho-AKT (Ser473; Cell Signaling Technology; #4060), phosphop44/42 (Thr202/Tyr204; Cell Signaling Technology; #4370), AKT (Cell Signaling Technology, #9272), p44/42 (Cell Signaling Technology; #9102), GAPDH (Cell Signaling Technology; #2118).
In vivo Compound Testing
To test the effectiveness of ALK inhibition in vivo, 6-week-old CD1 mice were intracranially allografted with 5 × 105 mouse PPP1CB–ALK tumor cells (see above) to give a more standardized latency of tumor formation and to avoid having to administer treatment to very young animals. The chosen inhibitor was lorlatinib based on the in vitro results, as well as HCl and temozolomide as vehicle control and standard of care, respectively. Dosing and treatment schedules were described previously (66). Tumor growth was monitored using BLI on an IVIS imager (PerkinElmer). The tumors were allowed to develop for two weeks before animals were stratified into three treatment groups based on their luciferase signal (rank 1, 4, 7, etc., being assigned to lorlatinib, rank 2, 5, 8, etc., to temozolomide, and rank 3, 6, 9, etc., to vehicle control). Animals were monitored daily for symptoms or abnormal behavior and weighed three times a week, and were sacrificed upon first signs of tumor-related symptoms according to humane endpoint criteria.
Novel Patient-Derived NTRK Fusion Models
Each cell culture was initiated using the following method: Tissue was first minced using a sterile scalpel followed by enzymatic dissociation with LiberaseTL for 10 minutes at 37°C. Cells were grown under stem cell conditions, as two-dimensional adherent cultures on laminin and laminin/fibronectin. Cells were cultured in a serum-free medium, tumor stem media consisting of 1:1 Neurobasal(-A), and DMEM:F12 supplemented with HEPES, NEAA, Glutamaxx, sodium pyruvate, and B27(-A), human bFGF (20 ng/mL), human EGF (20 ng/mL), human PDGF-AA (10 ng/mL) and PDGF-BB (10 ng/mL), and heparin (2 ng/mL). Control lines QCTB-R006 (9.5 years, male, frontal lobe GBM, wild-type) and QCTB-R059 (10.4 years, female, thalamic, H3F3AK27M mutant) were also grown as adherent cultures (laminin and laminin-fibronectin). Cells were dissociated enzymatically with accutase and counted using a Beckman-Coulter ViCell cell viability analyzer. For intracranial implantation, all experiments were performed in accordance with the local ethical review panel, the UK Home Office Animals (Scientific Procedures) Act 1986, the United Kingdom National Cancer Research Institute guidelines for the welfare of animals in cancer research and the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines (67, 68). Single-cell suspensions were obtained immediately prior to implantation in NOD.Cg-Prkdcscid Il2rgtm1WjI/SzJ (NSG) mice (Charles River). Animals were anesthetized with intraperitoneal ketamine (100 mg/kg)/xylazine (16 mg/kg) and maintained under 1% isoflurane (0.5 L/minute). Animals were depilated at the incision site and Emla cream 5% (lidocaine/prilocaine) was applied on the skin. A subcutaneous injection of buprenorphine (0.03 mg/kg) was given for general analgesia. The cranium was exposed via midline incision under aseptic conditions, and a 31-gauge burr hole drilled above the injection site. Mice were then placed on a stereotactic apparatus for orthotopic implantation. The coordinates used for the cortex were x = −2.0, z = +1.0, y = −2.5 mm from bregma. Three hundred thousand cells in 5 μL were stereotactically implanted using a 25-gauge SGE standard fixed needle syringe (SGE 005000) at a rate of 2 μL/minute using a digital pump (HA1100, Pico Plus Elite, Harvard Apparatus). At the completion of infusion, the syringe needle was allowed to remain in place for at least 3 minutes, and then manually withdrawn slowly to minimize backflow of the injected cell suspension. An intraperitoneal injection of the reversing agent atipamezole (1 mg/kg) diluted in Hartmann solution for rehydration was administered. Mice were monitored until fully recovered from surgery and given Carprofen (analgesia) in a gel diet for 48 hours postsurgery. Mice were weighed twice a week and imaged by 1H MRI on a horizontal bore Bruker Biospec 70/20 system equipped with physiologic monitoring equipment (SA Instruments) using a 2 cm × 2 cm mouse brain array coil. Anesthesia was induced using 3% isoflurane delivered in oxygen (1 L/minute) and maintained at 1% to 2%. Core body temperature was maintained using a thermoregulated water-heated blanket.
In vitro Compound Testing of Patient-Derived Cells
Cells were seeded (3,000–5,000 cells per well) into laminin or laminin–fibronectin-coated 96-well plates and treated with different TRK inhibitors at concentration ranging from 0 to 20 μmol/L for 8 days. The drugs used were entrectinib (RXDX-101, Selleckchem), crizotinib (PF-02341066, Selleckchem), and milciclib (PHA-848125, Selleckchem). Each assay was performed in three independent biological replicates of three technical replicates each. Cell viability was assessed with CellTiter-Glo using a FLUOstar Omega plate reader (BMG, LABTECH). Data was analyzed and IC50 values were calculated using GraphPad Prism software.
Statistical Analysis
Statistical analysis was carried out using R 3.5.0 (www.r-project.org) and GraphPad Prism 7. Categorical comparisons of counts were carried out using Fisher exact test; comparisons between groups of continuous variables employed Student t test or ANOVA. Univariate differences in survival were analyzed by the Kaplan–Meier method and significance was determined by the log-rank test. All tests were two-sided and a P value of less than 0.05 was considered significant.
Data Availability
All newly generated data have been deposited in the European Genome–phenome Archive (www.ebi.ac.uk/ega) with accession number EGAS00001003532 (sequencing) or ArrayExpress (www.ebi.ac.uk/arrayexpress/) with accession numbers E-MTAB-7802 and E-MTAB-7804 (methylation arrays). Curated gene-level copy number, mutation data, and RNA-sequencing data are provided as part of the pediatric-specific implementation of the cBioPortal genomic data visualization portal (pedcbioportal.org).
Supplementary Material
Acknowledgments
This work was supported by the CRIS Cancer Foundation and the INSTINCT network funded by The Brain Tumour Charity, Great Ormond Street Children’s Charity, and Children with Cancer UK, Cancer Research UK. The authors acknowledge NHS funding to the National Institute for Health Research Biomedical Research Centre at The Royal Marsden and the ICR, the NIHR Great Ormond Street Hospital Biomedical Research Centre, research nurse funding by the Experimental Cancer Medicines Centre (ECMC) Paediatric Network, as well as CRUK support to the Cancer Imaging Centre at the ICR and Royal Marsden in association with the MRC and Department of Health (England; C1060/A16464). Further funding support was provided by the German Children’s Cancer Foundation (DKKS, project “MNP2.0 – Improving the Diagnostic Accuracy of Pediatric Brain Tumors,” and support for the German Brain Tumor Reference Center of the DGNN, grant 2014.17) and the PedBrain Tumour Project contributing to the International Cancer Genome Consortium, funded by German Cancer Aid (109252) and by the German Federal Ministry of Education and Research (BMBF, grant #01KU1201A), and the DKFZ-MOST Cooperation Program. We would like to thank Laura von Soosten (DKFZ) for technical assistance and Richard Buus (ICR) and the Breast Cancer Now NanoString facility for conducting the NanoString gene expression profiling. The authors thank Brain UK for provision of cases and clinical information. The authors thank the Cure Brain Cancer Foundation, Australian Lions Childhood Cancer Research Foundation, and Lions Club International Foundation (LCIF). Some of the results are in part based upon data generated by Lions Kids Cancer Genome Project (LKCGP) Partners. The authors thank the German Cancer Research Center (DKFZ) Genomics and Proteomics Core Facility and the Hartwell Center at St. Jude Children’s Research Hospital for technical support. The authors acknowledge funding from the American, Lebanese and Syrian-Associated Charities (ALSAC). The Queensland Children’s Tumour Bank is funded by the Children’s Hospital Foundation (Queensland). This work was funded in part by the Marie-Josée and Henry R. Kravis Center for Molecular Oncology and the National Cancer Institute Cancer Center Core Grant No. P30-CA008748. We gratefully acknowledge the members of the Memorial Sloan Kettering Molecular Diagnostics Service in the Department of Pathology. M. Snuderl acknowledges funding from the Friedberg Charitable Foundation, the Making Headway Foundation, and the Sohn Conference Foundation. A. Korshunov is supported by the Helmholtz Association Research Grant (Germany). M. Vinci is a CwCUK Fellow (grant number 16–234). S.J. Baker acknowledges funding support from the NIH (CA096832).
M. Hubank is a consultant/advisory board member for Merck, Boehringer Ingelheim, Guardant, Bristol-Myers Squibb, and Astra-Zeneca and has received speakers bureau honoraria from Roche Diagnostics. J. Knipstein is medical director at PRA Health Sciences and a consultant at Atheneum Partners. U. Kordes is a principal investigator for Novartis. I.J. Dunkel is a pediatric oncology steering committee member at AstraZeneca and is a consultant/advisory board member for Celgene, Roche, Apexigen, and Bayer. M. Preusser has received honoraria from Bayer, Bristol-Myers Squibb, Novartis, Gerson Lehrman Group (GLG), CMC Contrast, GlaxoSmithKline, Mundipharma, Roche, BMJ Journals, MedMedia, AstraZeneca, AbbVie, Lilly, Medahead, Daiichi Sankyo, Sanofi, Merck Sharp & Dohme, and Tocagen, and reports receiving commercial research grants from Boehringer Ingelheim, Bristol-Myers Squibb, Roche, Daiichi Sankyo, Merck Sharp & Dohme, Novocure, GlaxoSmithKline, and AbbVie. C. Haberler is a consultant/advisory board member at Bayer and Roche. J. Schittenhelm has ownership interest in Qiagen shares. P.H. Driever is a consultant/advisory board member at Novartis. O. Witt is a consultant/advisory board member at Novartis USA, Roche, Janssen Research & Development, SK Life Science, and Bristol-Myers Squibb. D.S. Ziegler has received speakers bureau honoraria from Bayer. M.A. Karajannis is a consultant/advisory board member at CereXis, Recursion Pharmaceuticals, and Bayer. D.R. Hargrave is a consultant for entrectinib and is a consultant/advisory board member for larotrectinib. L.V. Marshall has received speakers’ bureau honoraria from Bayer. A. von Deimling has received royalties for antibodies for clone H09 from Dianova (IDH1R132H) and for clone VE1 from Roche/Ventana (BRAFV600E). C.M. Kramm reports receiving a commercial research grant from Novartis and is a consultant/advisory board member for Novartis and Boehringer Ingelheim. F. Sahm has received speakers bureau honoraria from Illumina, Agilent, and Medac. D. Capper has ownership interest in a patent pending on DNA methylation–based tumor classification.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
M. Clarke, A. Mackay, and B. Ismer contributed equally to this article.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed by the other authors.
REFERENCES
- 1.Jones C, Perryman L, Hargrave D . Paediatric and adult malignant glioma: close relatives or distant cousins? Nat Rev Clin Oncol 2012;9: 400–13. [DOI] [PubMed] [Google Scholar]
- 2.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012;482:226–31. [DOI] [PubMed] [Google Scholar]
- 3.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012;44:251–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones C, Baker SJ. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer 2014;14:651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012;22:425–37. [DOI] [PubMed] [Google Scholar]
- 6.Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 2016;34:2206–11. [DOI] [PubMed] [Google Scholar]
- 7.Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D, et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet 2015;47:257–62. [DOI] [PubMed] [Google Scholar]
- 8.Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 2017;32: 520–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 2014;46:444–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duffner PK, Horowitz ME, Krischer JP, Burger PC, Cohen ME, Sanford RA, et al. The treatment of malignant brain tumors in infants and very young children: an update of the Pediatric Oncology Group experience. Neuro Oncol 1999;1:152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lafay-Cousin L, Strother D. Current treatment approaches for infants with malignant central nervous system tumors. Oncologist 2009;14:433–44. [DOI] [PubMed] [Google Scholar]
- 12.Isaacs H Jr. Perinatal (fetal and neonatal) astrocytoma: a review. Childs Nerv Syst 2016;32:2085–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gelabert-Gonzalez M, Serramito-Garcia R, Arcos-Algaba A. Desmoplastic infantile and non-infantile ganglioglioma. Review of the literature. Neurosurg Rev 2010;34:151–8. [DOI] [PubMed] [Google Scholar]
- 14.Dufour C, Grill J, Lellouch-Tubiana A, Puget S, Chastagner P, Frappaz D, et al. High-grade glioma in children under 5 years of age: a chemotherapy only approach with the BBSFOP protocol. Eur J Cancer 2006; 42:2939–45. [DOI] [PubMed] [Google Scholar]
- 15.Geyer JR, Finlay JL, Boyett JM, Wisoff J, Yates A, Mao L, et al. Survival of infants with malignant astrocytomas. A Report from the Childrens Cancer Group. Cancer 1995;75:1045–50. [DOI] [PubMed] [Google Scholar]
- 16.Grundy RG, Wilne SH, Robinson KJ, Ironside JW, Cox T, Chong WK, et al. Primary postoperative chemotherapy without radiotherapy for treatment of brain tumours other than ependymoma in children under 3 years: results of the first UKCCSG/SIOP CNS 9204 trial. Eur J Cancer 2010;46:120–33. [DOI] [PubMed] [Google Scholar]
- 17.Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 2019;10:4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macy ME, Birks DK, Barton VN, Chan MH, Donson AM, Kleinschmidt-Demasters BK, et al. Clinical and molecular characteristics of congenital glioblastoma. Neuro Oncol 2012;14:931–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeshima H, Kawahara Y, Hirano H, Obara S, Niiro M, Kuratsu J. Postoperative regression of desmoplastic infantile gangliogliomas: report of two cases. Neurosurgery 2003;53:979–83. [DOI] [PubMed] [Google Scholar]
- 20.Haberler C, Slavc I, Czech T, Prayer D, Pirker C, Budka H, et al. Malignant predominantly minigemistocytic glioma in two infants: a distinctive glioma variant? Neuropathol Appl Neurobiol 2007;33: 169–78. [DOI] [PubMed] [Google Scholar]
- 21.Gielen GH, Gessi M, Buttarelli FR, Baldi C, Hammes J, zur Muehlen A, et al. Genetic analysis of diffuse high-grade astrocytomas in infancy defines a novel molecular entity. Brain Pathol 2015;25:409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anestis DM, Tsitsopoulos PP, Ble CA, Tsitouras V, Tsonidis CA. Congenital glioblastoma multiforme: an unusual and challenging tumor. Neuropediatrics 2017;48:403–12. [DOI] [PubMed] [Google Scholar]
- 23.Wang AC, Jones DTW, Abecassis IJ, Cole BL, Leary SES, Lockwood CM, et al. Desmoplastic infantile ganglioglioma/astrocytoma (DIG/DIA) are distinct entities with frequent BRAFV600 mutations. Mol Cancer Res 2018;16:1491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016;1:e000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 2013;45:927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013;45:602–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aghajan Y, Levy ML, Malicki DM, Crawford JR. Novel PPP1CB-ALK fusion protein in a high-grade glioma of infancy. BMJ Case Rep 2016;2016:pii:bcr2016217189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chmielecki J, Bailey M, He J, Elvin J, Vergilio JA, Ramkissoon S, et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res 2017;77:509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cocce MC, Mardin BR, Bens S, Stutz AM, Lubieniecki F, Vater I, et al. Identification of ZCCHC8 as fusion partner of ROS1 in a case of congenital glioblastoma multiforme with a t(6;12)(q21;q24.3). Genes Chromosomes Cancer 2016;55:677–87. [DOI] [PubMed] [Google Scholar]
- 30.Kiehna EN, Arnush MR, Tamrazi B, Cotter JA, Hawes D, Robison NJ, et al. Novel GOPC(FIG)-ROS1 fusion in a pediatric high-grade glioma survivor. J Neurosurg Pediatr 2017;20:51–5. [DOI] [PubMed] [Google Scholar]
- 31.Maruggi M, Malicki DM, Levy ML, Crawford JR. A novel KIF5B-ALK fusion in a child with an atypical central nervous system inflammatory myofibroblastic tumour. BMJ Case Rep 2018;2018:pii: bcr226431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakano Y, Tomiyama A, Kohno T, Yoshida A, Yamasaki K, Ozawa T, et al. Identification of a novel KLC1-ROS1 fusion in a case of pediatric low-grade localized glioma. Brain Tumor Pathol 2019;36:14–9. [DOI] [PubMed] [Google Scholar]
- 33.Ng A, Levy ML, Malicki DM, Crawford JR. Unusual high-grade and low-grade glioma in an infant with PPP1CB-ALK gene fusion. BMJ Case Rep 2019;12:pii:e228248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olsen TK, Panagopoulos I, Meling TR, Micci F, Gorunova L, Thorsen J, et al. Fusion genes with ALK as recurrent partner in ependymoma-like gliomas: a new brain tumor entity? Neuro Oncol 2015;17: 1365–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziegler DS, Wong M, Mayoh C, Kumar A, Tsoli M, Mould E, et al. Brief report: potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br J Cancer 2018;119:693–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DT, Capper D, et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 2016;164:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koelsche C, Mynarek M, Schrimpf D, Bertero L, Serrano J, Sahm F, et al. Primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features share a highly distinct methylation profile and DICER1 mutations. Acta Neuropathol 2018;136:327–37. [DOI] [PubMed] [Google Scholar]
- 38.Johannessen AL, Torp SH. The clinical value of Ki-67/MIB-1 labeling index in human astrocytomas. Pathol Oncol Res 2006;12:143–7. [DOI] [PubMed] [Google Scholar]
- 39.El-Ayadi M, Ansari M, Sturm D, Gielen GH, Warmuth-Metz M, Kramm CM, et al. High-grade glioma in very young children: a rare and particular patient population. Oncotarget 2017;8:64564–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature 2018;555:469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Tilburg CM, Selt F, Sahm F, Bachli H, Pfister SM, Witt O, et al. Response in a child with a BRAF V600E mutated desmoplastic infantile astrocytoma upon retreatment with vemurafenib. Pediatr Blood Cancer 2018;65:e26893. [DOI] [PubMed] [Google Scholar]
- 42.Charest A, Lane K, McMahon K, Park J, Preisinger E, Conroy H, et al. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosomes Cancer 2003; 37:58–71. [DOI] [PubMed] [Google Scholar]
- 43.International Cancer Genome Consortium PedBrain Tumor Project. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat Med 2016;22:1314–20. [DOI] [PubMed] [Google Scholar]
- 44.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6. [DOI] [PubMed] [Google Scholar]
- 45.Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 2016;131:833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012;337:1231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis JL, Lockwood CM, Stohr B, Boecking C, Al-Ibraheemi A, DuBois SG, et al. Expanding the spectrum of pediatric NTRK-rearranged mesenchymal tumors. Am J Surg Pathol 2019;43:435–45. [DOI] [PubMed] [Google Scholar]
- 48.Farago AF, Azzoli CG. Beyond ALK and ROS1: RET, NTRK, EGFR and BRAF gene rearrangements in non-small cell lung cancer. Transl Lung Cancer Res 2017;6:550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Tejpar S, Sartore-Bianchi A, et al. ALK, ROS1, and NTRK rearrangements in metastatic colorectal cancer. J Natl Cancer Inst 2017;109(12). [DOI] [PubMed] [Google Scholar]
- 50.Davare MA, Henderson JJ, Agarwal A, Wagner JP, Iyer SR, Shah N, et al. Rare but recurrent ROS1 fusions resulting from chromosome 6q22 microdeletions are targetable oncogenes in glioma. Clin Cancer Res 2018;24:6471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gatalica Z, Xiu J, Swensen J, Vranic S. Molecular characterization of cancers with NTRK gene fusions. Mod Pathol 2019;32:147–53. [DOI] [PubMed] [Google Scholar]
- 52.Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist 2017;22:1478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R. Analysis of NTRK Alterations in Pan-Cancer Adult and Pediatric Malignancies: Implications for NTRK-Targeted Therapeutics. JCO Precis Oncol 2018;2018: 10.1200/PO.18.00183. doi: 10.1200/PO.18.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McArthur GA, Laherty CD, Queva C, Hurlin PJ, Loo L, James L, et al. The Mad protein family links transcriptional repression to cell differentiation. Cold Spring Harb Symp Quant Biol 1998;63:423–33. [DOI] [PubMed] [Google Scholar]
- 55.Gripp KW, Aldinger KA, Bennett JT, Baker L, Tusi J, Powell-Hamilton N, et al. A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair. Am J Med Genet A 2016;170:2237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dehmelt L, Halpain S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol 2005;6:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saadi I, Alkuraya FS, Gisselbrecht SS, Goessling W, Cavallesco R, Turbe-Doan A, et al. Deficiency of the cytoskeletal protein SPECC1L leads to oblique facial clefting. Am J Hum Genet 2011;89:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilson NR, Olm-Shipman AJ, Acevedo DS, Palaniyandi K, Hall EG, Kosa E, et al. SPECC1L deficiency results in increased adherens junction stability and reduced cranial neural crest cell delamination. Sci Rep 2016;6:17735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kalashnikova E, Lorca RA, Kaur I, Barisone GA, Li B, Ishimaru T, et al. SynDIG1: an activity-regulated, AMPA-receptor-interacting transmembrane protein that regulates excitatory synapse development. Neuron 2010;65:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Metzler M, Li B, Gan L, Georgiou J, Gutekunst CA, Wang Y, et al. Disruption of the endocytic protein HIP1 results in neurological deficits and decreased AMPA receptor trafficking. EMBO J 2003;22:3254–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakai T, Nagai T, Tanaka M, Itoh N, Asai N, Enomoto A, et al. Girdin phosphorylation is crucial for synaptic plasticity and memory: a potential role in the interaction of BDNF/TrkB/Akt signaling with NMDA receptor. J Neurosci 2014;34:14995–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, et al. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell 2005;9:389–402. [DOI] [PubMed] [Google Scholar]
- 63.Ren X, Kuan PF. methylGSA: a Bioconductor package and Shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics 2019;35:1958–9. [DOI] [PubMed] [Google Scholar]
- 64.Mackay A, Burford A, Molinari V, Jones DTW, Izquierdo E, Brouwer-Visser J, et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade glioma from the HERBY phase II randomized trial. Cancer Cell 2018;33:829–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 66.Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer Discov 2016;6:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 2010;8:e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer 2010;102:1555–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All newly generated data have been deposited in the European Genome–phenome Archive (www.ebi.ac.uk/ega) with accession number EGAS00001003532 (sequencing) or ArrayExpress (www.ebi.ac.uk/arrayexpress/) with accession numbers E-MTAB-7802 and E-MTAB-7804 (methylation arrays). Curated gene-level copy number, mutation data, and RNA-sequencing data are provided as part of the pediatric-specific implementation of the cBioPortal genomic data visualization portal (pedcbioportal.org).