

Summary
Environmental and host‐associated microbial communities are complex ecosystems, of which many members are still unknown. Hence, it is challenging to study community dynamics and important to create model systems of reduced complexity that mimic major community functions. Therefore, we developed MiMiC, a computational approach for data‐driven design of simplified communities from shotgun metagenomes. We first built a comprehensive database of species‐level bacterial and archaeal genomes (n = 22 627) consisting of binary (presence/absence) vectors of protein families (Pfam = 17 929). MiMiC predicts the composition of minimal consortia using an iterative scoring system based on maximal match‐to‐mismatch ratios between this database and the Pfam binary vector of any input metagenome. Pfam vectorization retained enough resolution to distinguish metagenomic profiles between six environmental and host‐derived microbial communities (n = 937). The calculated number of species per minimal community ranged between 5 and 11, with MiMiC selected communities better recapitulating the functional repertoire of the original samples than randomly selected species. The inferred minimal communities retained habitat‐specific features and were substantially different from communities consisting of most abundant members. The use of a mixture of known microbes revealed the ability to select 23 of 25 target species from the entire genome database. MiMiC is open source and available at https://github.com/ClavelLab/MiMiC.
MiMiC is a computational approach for data‐driven design of simplified communities from shotgun metagenomes. It is open‐source and available at https://github.com/ClavelLab/MiMiC.

Introduction
Microbial communities are ubiquitous and influence many fundamental processes ranging from carbon and nitrogen cycles in water and soil to health and disease regulation in host‐associated habitats (Thompson et al., 2017; Vujkovic‐Cvijin et al., 2020). A major bottleneck for the study of these communities is the vast number of microbes that are still unknown (Hug et al., 2016). This prevents accurate assessment of community dynamics and interactions with the environment. Moreover, the tremendous complexity of these communities, due to hundreds of members and the possible interactions between them, renders the task of understanding how they establish and function very difficult. Hence, being able to design simplified communities of microbes as proxy for the native community of interest is important, albeit not an easy task. Such simplified (or synthetic) communities can be used in modelling or experimental approaches (e.g. continuous culture or gnotobiology) to highlight fundamental concepts underlying relationships between community members, evolutionary processes, or mechanisms of interactions with environmental factors or host species (Payne et al., 2012; Brugiroux et al., 2016; Bauer et al., 2017; Noronha et al., 2019; Tanoue et al., 2019; Streidl et al., 2021).
A variety of examples of such synthetic communities have been published for several habitats, including plant roots (Armanhi et al., 2017; Niu et al., 2017; Vorholt et al., 2017; Herrera Paredes et al., 2018; Zhang et al., 2019), soils (Kleyer et al., 2017; Puentes‐Tellez and Falcao Salles, 2018; Zhalnina et al., 2018) and gastrointestinal tracts (Schaedler et al., 1965; Becker et al., 2011; Petrof et al., 2013; Brugiroux et al., 2016; Calatayud Arroyo et al., 2018). Experimental approaches to build minimal communities include incremental, function‐driven selection of taxa in vitro or in vivo (functional enrichments) or tailored assemblage of previously isolated axenic strains (community assembly) (Clavel et al., 2017). Functional enrichment has the advantage of directly providing communities that carry out the desired function, as published already in the context of plant fitness or the induction of specific immune responses (Atarashi et al., 2013; Herrera Paredes et al., 2018; Stein et al., 2018), but they are experimentally demanding because of the necessity to test the given function after each round of enrichment. In contrast, the drawback of community assembly, the approach that is most commonly followed, is that the selection of strains is knowledge‐driven, i.e. based on ease of cultivation, availability of genomic information, an educated opinion on phylogenetic diversity, known functions and occurrence of taxa in the ecosystem of interest. Developing methods towards data‐driven design of synthetic communities of microbes would open new avenues by providing tailored synthetic community compositions fitted to the specific need of individual studies. Considering functional redundancy within complex microbial ecosystems, i.e. several community members carry out the same given function (Tian et al., 2020), favouring minimal communities that best represent the array of functions expressed rather than the original taxonomic profile of complex microbial communities is a sound objective (Johns et al., 2016; Eng and Borenstein, 2019; McCarty and Ledesma‐Amaro, 2019).
In this context, the present study aimed at creating and benchmarking a bioinformatic pipeline, called MiMiC, for automated prediction of synthetic community compositions mimicking the functional repertoire of the input microbial ecosystems.
Results and discussion
Overall concept of MiMiC
The rationale behind MiMiC is to infer the composition of a synthetic community of prokaryotes based on individual metagenomic profiles. Therefore, the pipeline processes (meta)genomic data into vectors of protein families (Pfams) used as the foundation for an iterative scoring process to determine a short list of best matching genomes from a comprehensive database. A schematic overview of the pipeline and tools used can be seen in Fig. 1A, experimental details are provided in the methods section, and all scripts and data are accessible via the project‐specific repository: https://github.com/ClavelLab/MiMiC. The current genome database consists of 22 627 species‐level genomes from bacteria and archaea spanning a total of 53 phyla and representing an average of 2523 ± 452 Pfams each (Fig. 1B). Ecosystem‐specific genome databases can also be used, as currently included for the human (n = 803) (Zou et al., 2019), mouse (n = 104) (www.dsmz.de/miBC) (Lagkouvardos et al., 2016) and pig intestine (n = 111) (www.dsmz.de/pibac) (Wylensek et al., 2020). After selecting a minimal number of genomes (either pre‐set by the user or determined in silico as detailed in the methods) from the database by maximizing the ratio of matches‐to‐mismatches to the input metagenome, MiMiC returns their NCBI RefSeq genome accession numbers along with various genome‐derived statistics. Running MiMiC with 50 iterations against the entire reference genome database using a metagenomic Pfam binary vector as input returned results within an average of 11 min by a computer system with 32GB RAM and 12 cores operating on linux (x86_64‐pc‐linux‐gnu) and using R version 3.6.3 (2020‐02‐29).
Fig. 1.
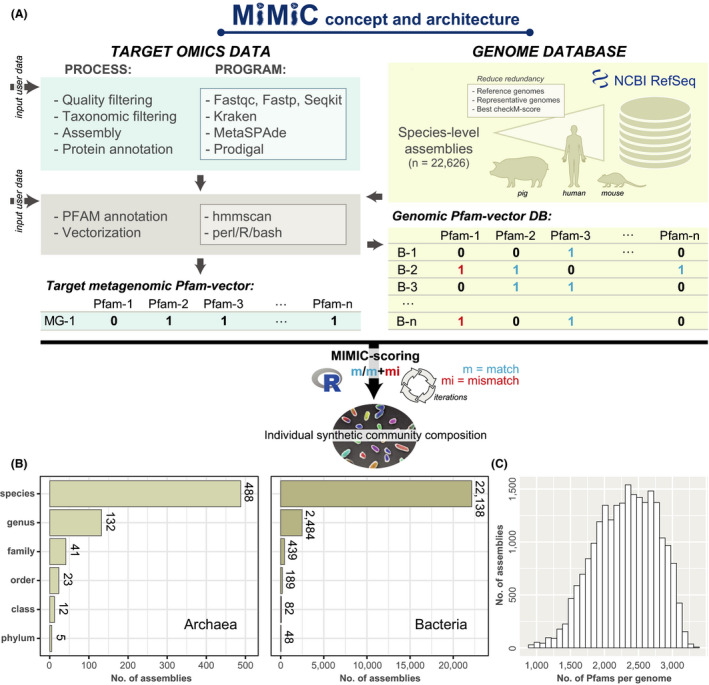
Schematic overview of the architecture and content of MiMiC. See methods section for all details.
A. (Meta)genomic reads are quality‐checked and processed into binary vectors of protein families (Pfams). The resulting metagenomic profile (MG‐1) is used for iterative selection of a minimal number of genomes (calculated by a knee point approach) from the database (B‐1 to B‐n) that best cover the functional potential of the input data (highest number of matches; least number of mismatches).
B. The genome database currently consists of 22 627 species‐level genomes of archaea and bacteria spanning a total of 53 phyla, with an average of approximately 2500 Pfams per genome.
Functional metagenomic profiles to infer synthetic community composition
In order to test MiMiC, we processed a set of 937 shotgun metagenomes from six different microbial habitats: marine water, soil, human tongue and the intestine of humans, mice and pigs (see the methods for details). The rationale was to select shotgun profiles representing very distinct (e.g. environmental vs. host‐associated) but also more closely related ecosystems (host‐specific microbiomes) obtained from published studies.
Expectedly, multidimensional analysis of functional profiles from these metagenomes showed a clear distinction between environmental and host‐associated microbiomes (Fig. 2A). The generated binary Pfam‐based profiles also allowed to differentiate gut microbiomes from the three host species analysed (Fig. 2B). A prerequisite for inferring the composition of simplified consortia from complex native communities is the ability to cover a sufficiently high fraction of metagenomic functions using reference genomes. For all ecosystems, the median metagenomic Pfam coverage by the entire database was close to 100% (Fig. 2C). While this covered fraction decreased when using the host‐specific database for gut microbiomes, all median values remained > 90% and all single values > 75%.
Fig. 2.
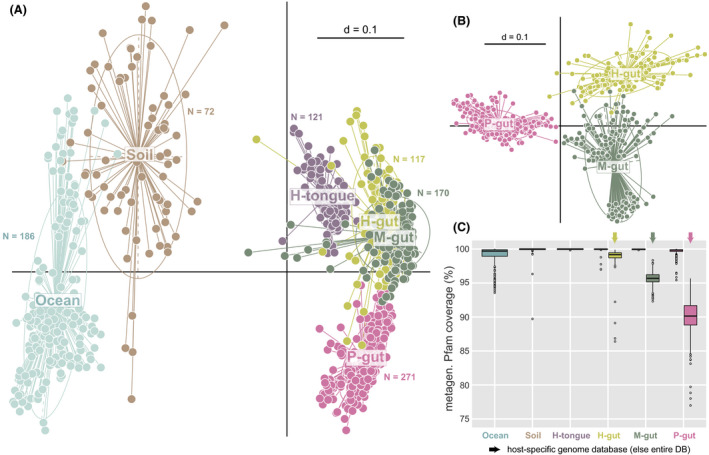
Input metagenomes from six different types of complex microbial communities.
A. Multi‐dimensional plot based on the presence/absence of Pfams (Jaccard index) in each individual metagenome (dots) from two environmental (marine water and soil) and four host‐associated metagenomes: the tongue and gut of humans (H) and the gut of mice (M) and pigs (P). The number of computed metagenomes per environment is indicated next to the corresponding cluster of samples. P < 0.01 as tested by permutational multivariate analysis of variance using distance matrices with the function adonis in R.
B. Same as in A showing further host species delineations between gut microbiomes.
C. Coverage of all metagenomes per habitat category (i.e. percentage of metagenomic Pfams also present in the reference MiMiC‐processed genomes) by the entire database (DB) or host‐specific collections of genomes for the human, mouse and pig intestine.
Altogether, these data show that binary Pfam‐based annotation of shotgun metagenomes retained enough resolution to distinguish even closely related ecosystems and the established genome databases can be used as a robust foundation for synthetic community design.
Features of the inferred minimal consortia
MiMiC predictions were performed on all 937 individual metagenomes aforementioned. Knee point calculation (see methods) to determine the optimal number of species within the inferred synthetic communities after 50 iterations revealed lowest (n = 7) and highest (n = 10) median diversity for soil and pig gut microbiomes respectively (Fig. 3A). The cumulative metagenomic coverage by the inferred synthetic communities was > 80% for all habitats, with a sharp increase in function coverage (up to 75% of metagenomic Pfams) observed already for the first four species selected (Fig. 3B). Each individually generated synthetic community was then compared with 100 sets of randomly selected species for each metagenome, resulting in a total of 7200–27 100 random sets for each ecosystem. The cumulative functional coverage was always significantly higher (+10–15%) for MiMiC predictions vs. the random sets (Fig. 3C). In contrast, the fraction of mismatches was significantly lower for MiMiC predictions in two cases (human tongue and gut), while equal for soil and mouse gut and higher for marine water and pig gut.
Fig. 3.
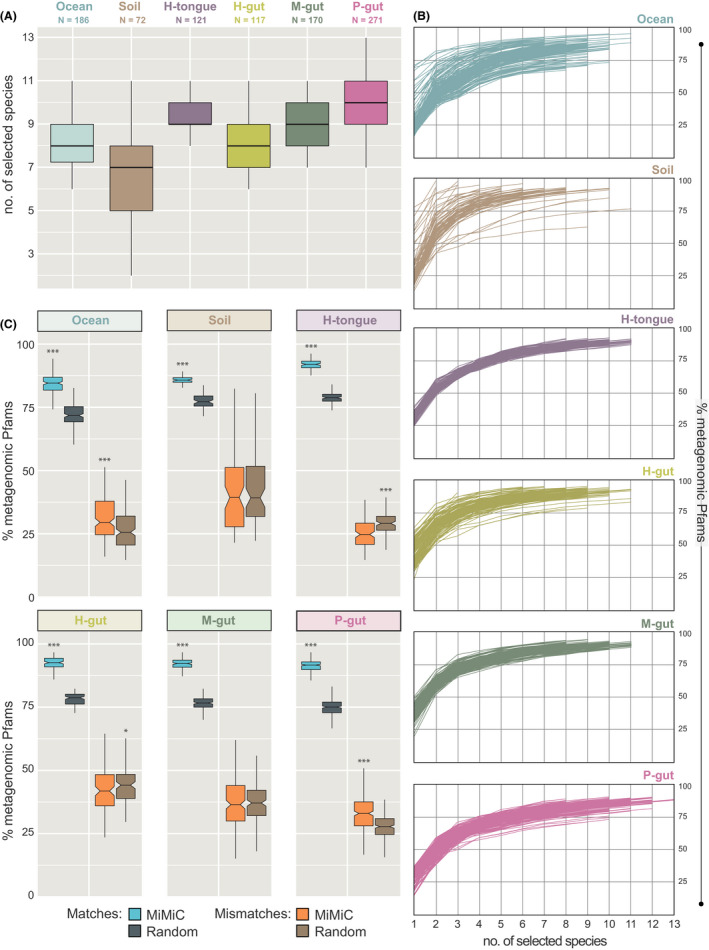
Output of MiMiC analysis on complex communities.
A. Distribution of the number of species per minimal consortium according to knee point‐based calculation after 50 iterations. The number of samples considered is indicated below the name of each community category.
B. Coverage of metagenomic profiles (y‐axis) according to incremental selection of genomes by MiMiC until sample‐specific knee points (x‐axis = rank of genome selection).
C. Performance (% of matches and mismatches) of MiMiC outputs compared with 100 sets of an equal number of species randomly selected from the entire database for each individual metagenome. P‐values were calculated using the Wilcoxon rank‐sum test; ***P < 0.001.
The synthetic communities created were then assessed in terms of composition respective to their ecosystem of origin. Multidimensional binary plots of the presence/absence of selected species per individual community showed that the main difference between environmental (marine water and soil) and host‐associated ecosystems previously observed with the native metagenomic profiles was conserved after MiMiC predictions (Fig. 4A). The distinction among host‐associated communities was less clear, with marked inter‐individual differences especially in the case of human tongue samples. Nonetheless, simplified communities from the pig intestine were most distinct from the others (Fig. 4B). Ecosystem‐specific species were identified by reporting the 10 most prevalent species per habitat, e.g. those species that were most often selected across all input metagenomes for the given habitat (Fig. 4C). This analysis showed no overlap between environmental and host‐associated ecosystems. A few common species were observed between habitats within each of these two main ecosystem types, while each type of communities was characterized by four to seven uniquely selected species. Strikingly, soil samples were represented by species with an overly low prevalence, indicating marked inter‐sample differences in composition, as illustrated in the native metagenomic profiles (Fig. 2A) and the wide range of species diversity within the synthetic communities (Fig. 3A).
Fig. 4.
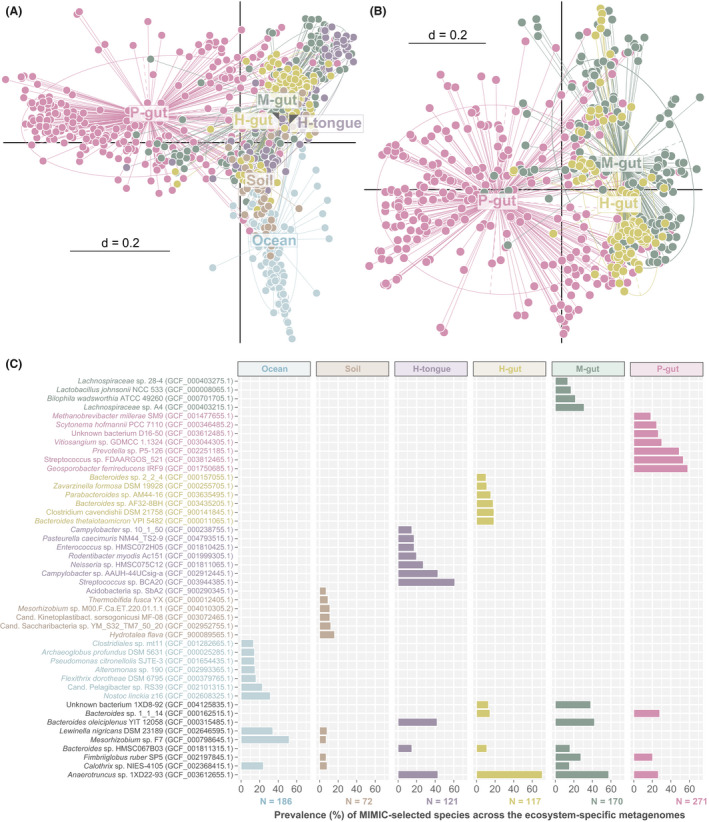
Ecosystem‐specific synthetic communities.
A. Multi‐dimensional plot based on the presence/absence of species (Jaccard index) selected by MiMiC within individual minimal consortia (dots), each corresponding to one input metagenome from six ecosystem types. The centroid of each type is indicated directly by the label or else by grey arrowheads within the label in case of overlaps. P < 0.01 as tested by permutational multivariate analysis of variance using distance matrices with the function adonis in R.
B. Same as in A showing further host species delineations between gut microbiomes.
C. Species identity (with sequence accession in brackets) of the 10 most prevalent genomes selected by MiMiC for each habitat type, prevalence being defined as the percentage of minimal microbial consortia containing the given species. The total number of metagenomes/minimal consortia considered per habitat category is shown below the x‐axis. Species identities are written in habitat‐specific colours if unique and in black if shared between habitats.
Comparison with alternative strategies for genome selection
We further evaluated the relevance of the MiMiC selection strategy using the metagenomic data from the pig intestine (N = 271), as we have recently established a comprehensive collection of gut bacterial isolates and corresponding genomes from pigs (Wylensek et al., 2020), allowing host‐specific analysis and taxonomic annotation of the data. The standard MiMiC procedure described above was compared with synthetic communities selected based on (i) most abundant members (Most abund.), at a number of species equal to that selected by MiMiC (knee point method); (ii) genomes with the greatest number of additional Pfam matches with the input metagenome, i.e. mismatches were not considered (m‐only); and (iii) excluding the first selected genome (skip 1st). The metagenomic Pfam fraction covered by the respective synthetic communities was highest with the ‘m‐only’ approach, albeit at the expense of a substantial proportion of functions not present in the original metagenomes (> 20%), justifying the consideration of mismatches in the selection process (Fig. 5A). The fraction of mismatches was also significantly higher for the ‘Most abund.’ and ‘Skip 1st’ strategies, although the magnitude of differences to MiMiC was much lower than for ‘m‐only’ and the metagenomic coverage was slightly higher for the ‘Most abund.’ strategy. These subtle changes in terms of percentages of Pfams translated into more evident differences when looking at the diversity of top‐10 most prevalent species selected across all 271 metagenomes by each method (Fig. 5B). MiMiC selected only 6 of the 10 most abundant taxa, highlighting the functional input of lower abundant taxa. Species selected on the basis of matches only were drastically different from the other approaches (in particular, no species in common with MiMiC), likely due to the preferred selection of functionally pluripotent species. Skipping the species most often ranked no. 1 (i.e. for which the genome was most often selected first), namely Streptococcus alactolyticus, favoured the selection of the species Roseburia porci within the top‐10 species. The little differences observed between these two methods (MiMiC Vs. Skip 1st) in terms of coverage values (Fig. 5A) highlight that overall Pfam profiles can be compensated between several species with a synthetic community. This is further discussed below in the section ‘Recommendations and outlooks’.
Fig. 5.
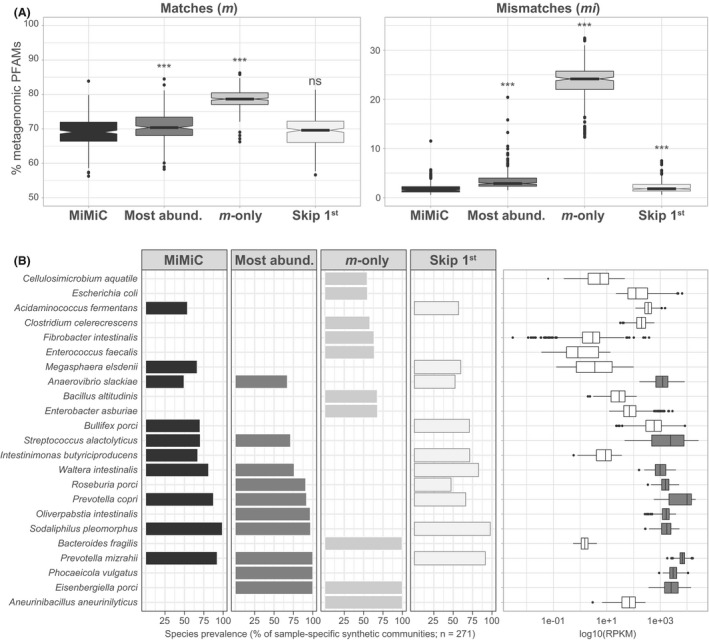
Comparison with alternative strategies for genome selection. Data were generated using the 271 pig metagenomes (Xiao et al., 2016) and genomes from the pig intestinal bacterial collection (Wylensek et al., 2020).
A. Fraction of matches (m) and mismatches (mi) within the synthetic communities as a percentage of metagenomic Pfams for each of the following four methods: (i) MiMiC (see detailed description in the methods; (ii) synthetic communities based on most abundant members (Most abund.), at a number of species equal to that selected by MiMiC (knee point method); (ii) calculated using only Pfam matches between any genome and the input metagenome, i.e. mismatches were not considered (m‐only); (iii) excluding the first selected genome (skip 1st).
B. List of species selected by the different methods. The 10 most prevalent species, i.e. most often selected by the given method across all input metagenomes, are shown. The bars indicate their respective prevalence (% of 271). The relative abundance of each species is shown as logarithmic values of RPKM (reads per kilo base per million mapped reads).
Benchmarking against a reference community
To assess the performance of MiMiC further, we used metagenomic data from the mock community MBARC (Singer et al., 2016), which consists of 23 bacterial and 3 archaeal species. Iterations were run against the entire genome database (n = 22 627; including the target genomes) until full functional coverage was reached, which happened at a number of 68 species (Fig. 6A). Knee point determination returned a number of 25 target species, which indirectly confirms the relevance of this approach, as close to the number of reference taxa within this community. Of note, one of the 26 target species, namely Nocardiopsis dassonvillei, was absent from the input metagenomic reads and can thus not be considered as a community member within the dataset tested. Comparison of cumulative functional coverage and numbers of mismatches at the theoretical number of species (n = 26) between the MiMiC prediction and 100 sets of randomly selected species confirmed the superiority of targeted community design (Fig. 6B). Interestingly, from the MiMiC species‐rank (i.e. the order at which genomes are selected) 19 onwards, we observed an increased amount of both matches and mismatches in the MiMiC prediction (blue dots) compared with the random sets (black box plots). Four of the seven species selected during the late iterations corresponded to species characterized by a low relative abundance within the MBARC metagenome analysed (Fig. 6C), suggesting that such taxa contribute to higher mismatch values due to their incomplete occurrence within metagenomic reads. Altogether, of the 25 target species present within the dataset, 23 were selected by MiMiC (Fig. 6C).
Fig. 6.
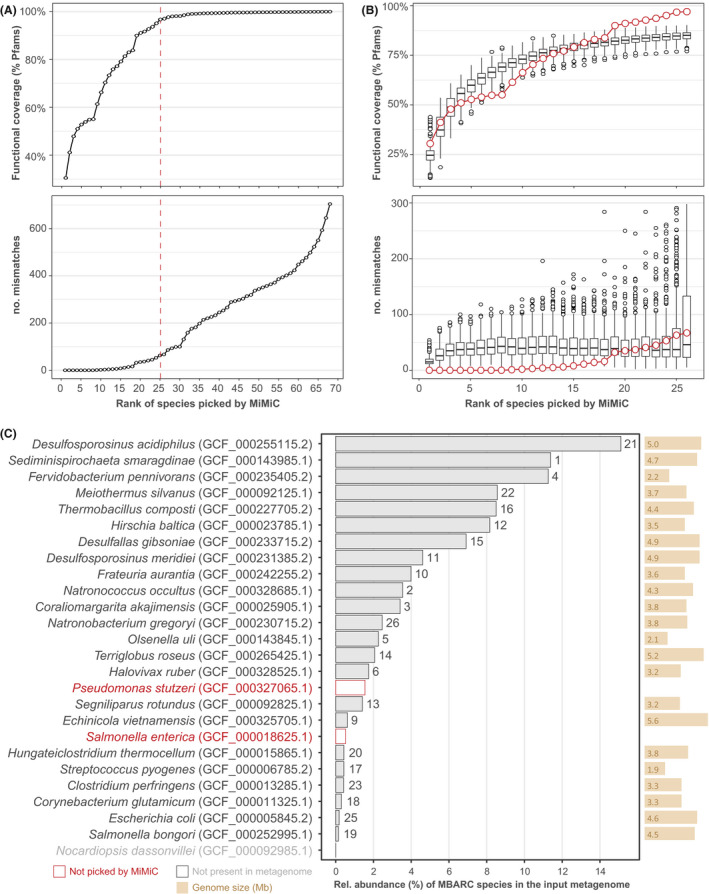
Mock community analyses.
A. Functional coverage and number of mismatches across the total iterations required to cover all Pfams from the input reference metagenome. The dashed red line represents the knee point determined on functional coverage.
B. MiMiC output (red dots) against 100 sets of randomly picked genomes until the number of 26 taxa.
C. Strain identification (with sequence accession in brackets) of the target species within the mock community sorted in decreasing relative abundance within the shotgun sequencing data (x‐axis, in %) as reported in the original paper (Singer et al., 2016). The rank (i.e. iteration) at which each species was selected by MiMiC is indicated on top of the bars.
Recommendations and outlooks
Despite its usefulness by enabling individualized synthetic community design based on functional profiles, the proposed approach has some limitations that we transparently present here and will be addressed in further versions of the tool.
MiMiC is reference‐based; i.e. predictions are dependent on the quality of the genome database. However, the pool and diversity of genomes publicly available are growing exponentially and the reference dataset will be regularly updated, including microbes other than prokaryotes such as fungi (Richard and Sokol, 2019). Moreover, using ecosystem‐specific genome databases can help restricting predictions to those taxa most relevant for that ecosystem. Users may also modify the provided databases by applying their own taxonomic filters to restrict the search to relatively high taxonomic ranks contained in an input metagenome, e.g. families. This would narrow the pool of reference genomes to ecosystem‐relevant taxa while retaining enough diversity to not antagonize the concept of this tool: providing consortia mimicking ecosystem functions and not taxonomic profiles. This will, however, come at the cost of additional computation time required for the taxonomic annotation of metagenomic reads.
The current genome selection approach is intrinsically limited by the generally low standard annotation power of genomes (without manual curation) and the functional resolution of Pfams, i.e. core functions outnumber specific functional features, inflating the possible functional redundancy between selected genomes. A future approach that considers the rareness of functions among microbes may better recapitulate the individuality of ecosystems.
Determination of the number of species to be contained in the output synthetic community is not an easy task. For instance, the output of knee point calculation is influenced by the number of scoring iterations, which we recommend keeping below 50, as the steep increase in metagenomic coverage occurs across the first 10 selected taxa. Additional parameters beyond metagenomic coverage will also be considered in the future to determine the number of species within synthetic communities. As an alternative to sample‐specific determination of the number of species to be included in a synthetic community, users can also run the MiMiC script by setting a defined number of iterations corresponding to the wished number of species without considering knee point calculation.
Over the last few years, two algorithms for designing minimal microbial communities have been published. The first aimed for desired metabolic capacities based on integer linear programming (Eng and Borenstein, 2016). While a step‐forward, this approach is limited due to complete metabolic pathways being targeted, which may be prevented by genome completeness and functional annotation quality (Parks et al., 2015; Karp et al., 2018). Moreover, communities of surprisingly low complexity were inferred using random selection of metabolite pairs (substrate product), and no information was provided on the complexity of usage and computation time. The second method aimed to generate communities that would be stable within a chemostat based on a user‐provided list of strains with known quorum sensing and bacteriocin production and sensitivity values (Karkaria et al., 2021). This method was designed for application to engineered strains, for which such data would be known due to having been designed into the strains genome, and not for environmental isolates. Neither of the methods aim to generate ecosystem representative minimal communities, making MiMiC a unique addition to the existing suite of bioinformatic tools currently available. Future incorporation of systems biology approaches into MiMiC will provide in silico validation of community structure and species interactions prior in experimental testing (Bauer et al., 2017; Venturelli et al., 2018).
Conclusion
Synthetic communities are very important experimental models to study microbiomes but very few approaches have been developed for their design. We share a bioinformatic tool to create synthetic community compositions that mimic the functional prokaryotic potential within input metagenomes, which we hope will facilitate experimental studies in many fields of microbiology and biotechnology.
Experimental procedures
Datasets
Genomes
All bacterial and archaeal genome assemblies (.faa) available in the NCBI RefSeq database (Haft et al., 2018) as of February 2019 (n = 156 800) and host‐specific genomes from published studies (n = 1018) (Lagkouvardos et al., 2016; Zou et al., 2019; Wylensek et al., 2020) were considered to build the MiMiC genome database. As a few species, such as pathogens, have been repeatedly sequenced, redundancy within the NCBI‐derived genome database was reduced in a stepwise manner: (i) reference genomes were kept whenever existing, defined as manually selected high‐quality genome assemblies that NCBI and the community identified as being important, and for which experimental data and extensive proteome support from Uniprot exist (UniProt, 2021); (ii) for species without a reference genome, representative genomes were kept whenever available, defined as computationally or manually selected genomes among the best genomes available for a species or clade; (iii) for those species without reference or representative genomes but for which several assemblies were available, checkm v.1.0.18 (Parks et al., 2015) was used to select the genome with highest completeness; (iv) for species with only one assembly available, this unique genome was kept in the database. This resulted in a total of 22 627 species‐level assemblies in the final database.
Mock community
The defined mixture of prokaryotes referred to as Mock Bacteria Archaea Community (MBARC‐26) (Singer et al., 2016) was used for validation. Shotgun metagenomic Illumina reads were downloaded using the SRA accession number SRX1836716.
Metagenomes
In total, 937 metagenomes from six different microbial habitats were processed and analysed in the present study. Human‐derived metagenomes were retrieved from the Integrative Human Microbiome Project (iHMP‐Consortium, 2019). Communities from the tongue (habitat 1; n = 121) and faeces (habitat 2; n = 117) of healthy individuals were selected due to their distinct microbial compositions. Mouse gut metagenomes (habitat 3; n = 170) including multiple genetic backgrounds and housing conditions (Xiao et al., 2015) were downloaded from the European Nucleotide Archive (ENA), project PRJEB7759. Pig faecal metagenomes (habitat 4; n = 271) from the original metagenomic gene catalogue for this host species (Xiao et al., 2016), including animals from China, Denmark and France, were downloaded from ENA, project PRJEB11755. Assemblies from marine water samples (habitat 5; n = 186) collected from 68 different sites worldwide from the surface and at the deep chlorophyll maximum were downloaded from ENA, project PRJEB4352 (Carradec et al., 2018). For soil samples (habitat 6; n = 72), assemblies from different studies were downloaded from the NCBI with the following accessions: PRJEB10725, PRJNA13699, PRJNA202911, PRJNA269960, PRJNA271842, PRJNA295927, PRJNA342745, PRJNA358809, PRJNA376086, PRJNA422409, PRJNA489261 (Tringe et al., 2005; Johnston et al., 2016; Meier et al., 2016).
Raw data processing
Genomes
Whenever PGAP‐annotated assemblies (Tatusova et al., 2016) were available from NCBI RefSeq, files with the suffix ‘translated_cds.faa.gz’ were downloaded. Else, raw genomic fastq reads were assembled using spades v.3.13.0 with default settings (Bankevich et al., 2012) and assemblies were annotated using prodigal v.2.6.3 (Hyatt et al., 2010) with the following settings: ‐c (closed‐end genes only), ‐m (do not predict genes including ‘N’ nucleotides). Quality was assessed using checkm v.1.0.18 (Parks et al., 2015), retaining only genomes with > 90% completeness and < 5% contamination. For all genomes, annotated proteins were assigned a protein family (Pfam) (El‐Gebali et al., 2019) using hmmscan v.3.2.1 based on database version 17.0 (containing 17 929 Pfams) with the gathering threshold score cut‐offs applied (‐cut_ga) (Finn et al., 2011).
Metagenomes
Whenever appropriate (i.e. no protein sequence files available), the quality of fastq data was ensured using fastp v.0.14.1 (Chen et al., 2018), including adaptor sequence removal (default option), phred score filtering (min. 40% bases >q15), 3′‐end trimming (average q20 at a window size of 16), and read length filtering (exclude <35 nt). For those metagenomes from host‐associated ecosystems, host‐specific reads were removed (Bushnell, 2014). Reads were then assembled using spades v.3.13.0 with (‐meta) default options (Bankevich et al., 2012). Contigs were filtered at a minimum length of 500 nt using seqkit v.0.10.0 (Shen et al., 2016). Proteins were annotated using prodigal v.2.6.3 with gene modelling parameters (‐c, ‐m, ‐q, ‐f, sco, ‐d) (Hyatt et al., 2010) and then assigned to Pfams as mentioned above for genomes.
Vectorization and scoring
For each individual genome and metagenome, Pfam accessions from the hmmscan output were parsed into a text file using custom perl and R scripts, resulting in qualitative functional profiles consisting of binary vectors of 1 (Pfam present) and 0 (Pfam absent) across all Pfams contained within the reference set (n = 17 929).
The rationale for selecting genomes from the database as members of a synthetic community representing the input metagenome was to maximize the number of matches (m) and minimize the number of mismatches (mi) between a given genomic and metagenomic Pfam profile. Therefore, a genome‐specific score defined as ‘m/m+mi’ was calculated, where ‘m’ is the number of Pfams present in both the genome and the metagenome, and ‘mi’ is the number of Pfams present in the genome only (not in the metagenome). This process was iterative: (i) the genome with the highest score was selected first; (ii) the Pfams covered by this first genome were zeroed in both the target metagenome and throughout the genome database to ensure further selection was based on the addition of as‐yet‐missing functions; (iii) the next genome with highest score was selected and the process continued.
The total number of taxa to be selected can be either set by the user or calculated in silico. In the latter case, the script was let to run until the cumulative metagenomic coverage (i.e. the fraction of metagenomic Pfams covered by those from selected genomes) reaches a stable plateau, and the number of taxa corresponding to the elbow point was calculated using a knee point approach using the method uik (unit invariant knee) (Christopoulos, 2016). To assess the representation of MiMiC predictions, the species selected as described above for each microbial habitat were compared with artificial communities consisting of the same number of randomly selected species.
Outputs and statistics
Each MiMiC prediction reports the genome IDs of selected strains (i.e. RefSeq assembly accessions or study‐specific labels/accessions in the case of individual databases) along with genome‐derived parameters, e.g. size, Pfam number, number/percentage of matches/mismatches (per iteration and cumulative). Functional profiles of the different microbial ecosystems were examined by generating distance matrices from the Pfam‐based vectors using the Jaccard index (Jaccard, 1901) followed by multidimensional scaling (MDS) plotting using cmdscale (Mead, 1992). Statistical differences were assessed by permutational multivariate analysis of variance using distance matrices with the function adonis in R. Statistical significances between MiMiC and random outputs were tested using Wilcoxon rank‐sum tests.
Authors' contributions
NK, IL and TC designed the study. NK and TCAH performed computation work. NK and TCAH analysed data. NK, TCAH and TC interpreted results. DH and IL provided guidance and access to essential infrastructure. TCAH, DH and TC secured funding. TC supervised the project. NK generated the original figures and drafted the methods. TC wrote the manuscript. NK and TCAH corrected and improved the original draft. All authors reviewed the final version and agreed with its content.
Conflicts of interest
TC has ongoing scientific collaborations with Cytena GmbH and HiPP GmbH and is member of the scientific advisory board of Savanna Ingredients GmbH.
Acknowledgements
Genomic and metagenomic data processing was supported by resources at the RWTH Compute Cluster under project rwth2608.
Microb. Biotechnol. (2021) 14(4), 1757–1770
Funding information
TC received funding from the German Research Foundation (DFG, Deutsche Forschungsgemeinschaft)—project no. 403224013, SFB 1382. DH was also funded by the DFG—project no. 395357507, SFB 1371. TCAH and NK were partially funded by a START grant from the University Hospital RWTH Aachen.
Data availability statement
All codes and data generated in the present work are available https://github.com/ClavelLab/MiMiC.
References
- Armanhi, J.S.L. , de Souza, R.S.C. , Damasceno, N.B. , de Araujo, L.M. , Imperial, J. , and Arruda, P. (2017) A community‐based culture collection for targeting novel plant growth‐promoting bacteria from the sugarcane microbiome. Front Plant Sci 8: 2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi, K. , Tanoue, T. , Oshima, K. , Suda, W. , Nagano, Y. , Nishikawa, H. , et al. (2013) Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500: 232–236. [DOI] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A.A. , Dvorkin, M. , Kulikov, A.S. , et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. J Comput Biol 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, E. , Zimmermann, J. , Baldini, F. , Thiele, I. , and Kaleta, C. (2017) BacArena: Individual‐based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput Biol 13: e1005544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, N. , Kunath, J. , Loh, G. , and Blaut, M. (2011) Human intestinal microbiota: characterization of a simplified and stable gnotobiotic rat model. Gut Microbes 2: 25–33. [DOI] [PubMed] [Google Scholar]
- Brugiroux, S. , Beutler, M. , Pfann, C. , Garzetti, D. , Ruscheweyh, H.‐J. , Ring, D. , et al. (2016) Genome‐guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat Microbiol 2: 16215. [DOI] [PubMed] [Google Scholar]
- Bushnell, B. (2014) BBMap: A Fast, Accurate, Splice‐Aware Aligner. [WWW document]. URL https://www.osti.gov/servlets/purl/1241166. [Google Scholar]
- Calatayud Arroyo, M. , Van de Wiele, T. , and Hernandez‐Sanabria, E. (2018) Assessing the viability of a synthetic bacterial consortium on the in vitro gut host‐microbe interface. J Vis Exp 137: 57699. 10.3791/57699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carradec, Q. , Pelletier, E. , Da Silva, C. , Alberti, A. , Seeleuthner, Y. , Blanc‐Mathieu, R. , et al. (2018) A global ocean atlas of eukaryotic genes. Nat Commun 9: 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Zhou, Y. , Chen, Y. , and Gu, J. (2018) fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics 34: i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos, D.T. (2016) Introducing Unit Invariant Knee (UIK) as an objective choice for elbow point in multivariate data analysis techniques. SSRN eLibrary. 10.2139/ssrn.3043076 [DOI] [Google Scholar]
- Clavel, T. , Lagkouvardos, I. , and Stecher, B. (2017) From complex gut communities to minimal microbiomes via cultivation. Curr Opin Microbiol 38: 148–155. [DOI] [PubMed] [Google Scholar]
- El‐Gebali, S. , Mistry, J. , Bateman, A. , Eddy, S.R. , Luciani, A. , Potter, S.C. , et al. (2019) The Pfam protein families database in 2019. Nucleic Acids Res 47: D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng, A. , and Borenstein, E. (2016) An algorithm for designing minimal microbial communities with desired metabolic capacities. Bioinformatics 32: 2008–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng, A. , and Borenstein, E. (2019) Microbial community design: methods, applications, and opportunities. Curr Opin Biotechnol 58: 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Clements, J. , and Eddy, S.R. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft, D.H. , DiCuccio, M. , Badretdin, A. , Brover, V. , Chetvernin, V. , O'Neill, K. , et al. (2018) RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res 46: D851–D860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera Paredes, S. , Gao, T. , Law, T.F. , Finkel, O.M. , Mucyn, T. , Teixeira, P. , et al. (2018) Design of synthetic bacterial communities for predictable plant phenotypes. PLoS Biol 16: e2003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug, L.A. , Baker, B.J. , Anantharaman, K. , Brown, C.T. , Probst, A.J. , Castelle, C.J. , et al. (2016) A new view of the tree of life. Nat Microbiol 1: 16048. [DOI] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G.L. , Locascio, P.F. , Land, M.L. , Larimer, F.W. , and Hauser, L.J. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iHMP‐Consortium (2019) The integrative human microbiome project. Nature 569: 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaccard, P. (1901) Étude Comparative de La Distribution Florale Dans Une Portion Des Alpes et Des Jura. Bull La Soc Vaudoise Sci Nat 7: 547–579. [Google Scholar]
- Johns, N.I. , Blazejewski, T. , Gomes, A.L. , and Wang, H.H. (2016) Principles for designing synthetic microbial communities. Curr Opin Microbiol 31: 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, E.R. , Rodriguez‐R, L.M. , Luo, C. , Yuan, M.M. , Wu, L. , He, Z. , et al. (2016) Metagenomics reveals pervasive bacterial populations and reduced community diversity across the Alaska Tundra ecosystem. Front Microbiol 7: 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkaria, B.D. , Fedorec, A.J.H. , and Barnes, C.P. (2021) Automated design of synthetic microbial communities. Nat Commun 12: 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp, P.D. , Weaver, D. , and Latendresse, M. (2018) How accurate is automated gap filling of metabolic models? BMC Syst Biol 12: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleyer, H. , Tecon, R. , and Or, D. (2017) Resolving species level changes in a representative soil bacterial community using microfluidic quantitative PCR. Front Microbiol 8: 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagkouvardos, I. , Pukall, R. , Abt, B. , Foesel, B.U. , Meier‐Kolthoff, J.P. , Kumar, N. , et al. (2016) The Mouse Intestinal Bacterial Collection (miBC) provides host‐specific insight into cultured diversity and functional potential of the gut microbiota. Nat Microbiol 1: 16131. [DOI] [PubMed] [Google Scholar]
- McCarty, N.S. , and Ledesma‐Amaro, R. (2019) Synthetic biology tools to engineer microbial communities for biotechnology. Trends Biotechnol 37: 181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead, A.L. (1992) Review of the development of multidimensional scaling methods. J R Stat Soc D 41: 27–39. [Google Scholar]
- Meier, M.J. , Paterson, E.S. , and Lambert, I.B. (2016) Use of substrate‐induced gene expression in metagenomic analysis of an aromatic hydrocarbon‐contaminated soil. Appl Environ Microbiol 82: 897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, B. , Paulson, J.N. , Zheng, X. , and Kolter, R. (2017) Simplified and representative bacterial community of maize roots. Proc Natl Acad Sci USA 114: E2450–E2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noronha, A. , Modamio, J. , Jarosz, Y. , Guerard, E. , Sompairac, N. , Preciat, G. , et al. (2019) The Virtual Metabolic Human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res 47: D614–D624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, D.H. , Imelfort, M. , Skennerton, C.T. , Hugenholtz, P. , and Tyson, G.W. (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne, A.N. , Zihler, A. , Chassard, C. , and Lacroix, C. (2012) Advances and perspectives in in vitro human gut fermentation modeling. Trends Biotechnol 30: 17–25. [DOI] [PubMed] [Google Scholar]
- Petrof, E.O. , Gloor, G.B. , Vanner, S.J. , Weese, S.J. , Carter, D. , Daigneault, M.C. , et al. (2013) Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome 1: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puentes‐Tellez, P.E. , and Falcao Salles, J. (2018) Construction of effective minimal active microbial consortia for lignocellulose degradation. Microb Ecol 76: 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard, M.L. , and Sokol, H. (2019) The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol 16: 331–345. [DOI] [PubMed] [Google Scholar]
- Schaedler, R.W. , Dubs, R. , and Costello, R. (1965) Association of Germfree mice with bacteria isolated from normal mice. J Exp Med 122: 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, W. , Le, S. , Li, Y. , and Hu, F. (2016) SeqKit: a cross‐platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One 11: e0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, E. , Andreopoulos, B. , Bowers, R.M. , Lee, J. , Deshpande, S. , Chiniquy, J. , et al. (2016) Next generation sequencing data of a defined microbial mock community. Sci Data 3: 160081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, R.R. , Tanoue, T. , Szabady, R.L. , Bhattarai, S.K. , Olle, B. , Norman, J.M. , et al. (2018) Computer‐guided design of optimal microbial consortia for immune system modulation. eLife 7: 30916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streidl, T. , Karkossa, I. , Segura Munoz, R.R. , Eberl, C. , Zaufel, A. , Plagge, J. , et al. (2021) The gut bacterium Extibacter muris produces secondary bile acids and influences liver physiology in gnotobiotic mice. Gut Microbes 13: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue, T. , Morita, S. , Plichta, D.R. , Skelly, A.N. , Suda, W. , Sugiura, Y. , et al. (2019) A defined commensal consortium elicits CD8 T cells and anti‐cancer immunity. Nature 565: 600–605. [DOI] [PubMed] [Google Scholar]
- Tatusova, T. , DiCuccio, M. , Badretdin, A. , Chetvernin, V. , Nawrocki, E.P. , Zaslavsky, L. , et al. (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44: 6614–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, L.R. , Sanders, J.G. , McDonald, D. , Amir, A. , Ladau, J. , Locey, K.J. , et al. (2017) A communal catalogue reveals Earth's multiscale microbial diversity. Nature 551: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, L. , Wang, X.‐W. , Wu, A.‐K. , Fan, Y. , Friedman, J. , Dahlin, A. , et al. (2020) Deciphering functional redundancy in the human microbiome. Nat Commun 11: 6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe, S.G. , von Mering, C. , Kobayashi, A. , Salamov, A.A. , Chen, K. , Chang, H.W. , et al. (2005) Comparative metagenomics of microbial communities. Science 308: 554–557. [DOI] [PubMed] [Google Scholar]
- UniProt, C. (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49: D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturelli, O.S. , Carr, A.V. , Fisher, G. , Hsu, R.H. , Lau, R. , Bowen, B.P. , et al. (2018) Deciphering microbial interactions in synthetic human gut microbiome communities. Mol Syst Biol 14: e8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorholt, J.A. , Vogel, C. , Carlstrom, C.I. , and Muller, D.B. (2017) Establishing causality: opportunities of synthetic communities for plant microbiome research. Cell Host Microbe 22: 142–155. [DOI] [PubMed] [Google Scholar]
- Vujkovic‐Cvijin, I. , Sklar, J. , Jiang, L. , Natarajan, L. , Knight, R. , and Belkaid, Y. (2020) Host variables confound gut microbiota studies of human disease. Nature 587: 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylensek, D. , Hitch, T.C.A. , Riedel, T. , Afrizal, A. , Kumar, N. , Wortmann, E. , et al. (2020) A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat Commun 11: 6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, L. , Estellé, J. , Kiilerich, P. , Ramayo‐Caldas, Y. , Xia, Z. , Feng, Q. , et al. (2016) A reference gene catalogue of the pig gut microbiome. Nat Microbiol 1: 161. [DOI] [PubMed] [Google Scholar]
- Xiao, L. , Feng, Q. , Liang, S. , Sonne, S.B. , Xia, Z. , Qiu, X. , et al. (2015) A catalog of the mouse gut metagenome. Nat Biotechnol 33: 1103–1108. [DOI] [PubMed] [Google Scholar]
- Zhalnina, K. , Zengler, K. , Newman, D. , and Northen, T.R. (2018) Need for laboratory ecosystems to unravel the structures and functions of soil microbial communities mediated by chemistry. MBio 9: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Liu, Y.‐X. , Zhang, N.a. , Hu, B. , Jin, T. , Xu, H. , et al. (2019) NRT1.1B is associated with root microbiota composition and nitrogen use in field‐grown rice. Nat Biotechnol 37: 676–684. [DOI] [PubMed] [Google Scholar]
- Zou, Y. , Xue, W. , Luo, G. , Deng, Z. , Qin, P. , Guo, R. , et al. (2019) 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat Biotechnol 37: 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All codes and data generated in the present work are available https://github.com/ClavelLab/MiMiC.