

Summary
The lambda phage Red proteins Redα/Redβ/Redγ and Rac prophage RecE/RecT proteins are powerful tools for precise and efficient genetic manipulation but have been limited to only a few prokaryotes. Here, we report the development and application of a new recombineering system for Burkholderia glumae and Burkholderia plantarii based on three Rac bacteriophage RecET‐like operons, RecETheBDU8, RecEThTJI49 and RecETh1h2eYI23, which were obtained from three different Burkholderia species. Recombineering experiments indicated that RecEThTJI49 and RecETh1h2eYI23 showed higher recombination efficiency compared to RecETheBDU8 in Burkholderia glumae PG1. Furthermore, all of the proteins currently categorized as hypothetical proteins in RecETh1h2eYI23, RecEThTJI49 and RecETheBDU8 may have a positive effect on recombination in B. glumae PG1 except for the h2 protein in RecETh1h2eYI23. Additionally, RecETYI23 combined with exonuclease inhibitors Pluγ or Redγ exhibited equivalent recombination efficiency compared to Redγβα in Escherichia coli, providing potential opportunity of recombineering in other Gram‐negative bacteria for its loose host specificity. Using recombinase‐assisted in situ insertion of promoters, we successfully activated three cryptic non‐ribosomal peptide synthetase biosynthetic gene clusters in Burkholderia strains, resulting in the generation of a series of lipopeptides that were further purified and characterized. Compound 7 exhibited significant potential anti‐inflammatory activity by inhibiting lipopolysaccharide‐stimulated nitric oxide production in RAW 264.7 macrophages. This recombineering system may greatly enhance functional genome research and the mining of novel natural products in the other species of the genus Burkholderia after optimization of a protocol.
Here, we report the development and application of a new recombineering system for Burkholderia glumae and Burkholderia plantarii based on three Rac bacteriophage RecET‐like operons, RecETheBDU8, RecEThTJI49, and RecETh1h2eYI23, which were obtained from three different Burkholderia species. Using recombinase‐assisted in situ insertion of promoters, we successfully activated three cryptic nonribosomal peptide synthetase biosynthetic gene clusters in Burkholderia strains, resulting in the generation of a series of lipopeptides that were further purified and characterized. This recombineering system may greatly enhance functional genome research and the mining of novel natural products in the other species of the genus Burkholderia after optimization of a protocol.

Introduction
Phage‐encoded homologous recombination systems, either Redα/Redβ/Redγ from the lambda phage Red operon or RecE/RecT from Rac prophage, have been employed for the genetic manipulation of Escherichia coli (Zhang et al., 1998; Fu et al., 2012; Wang et al., 2014; Abbasi et al., 2020) and some closely related bacteria, such as Salmonella enterica (Bunny et al., 2002), Shigella flexneri (Beloin et al., 2003), Klebsiella pneumoniae (Wei et al., 2012), Agrobacterium tumefaciens (Hu et al., 2014) and Escherichia albertii (Egan et al., 2016). However, in more distant species, their application has been limited due to apparent host specificities. Nevertheless, this recombinant DNA technology, termed recombinogenic engineering or recombineering, can be a powerful genetic tool for deletion, insertion, replacement, point mutation, multi‐fragment assembly and direct cloning of large DNA fragments (Fu et al., 2012; Wang et al., 2014, 2016, 2018a). Compared with homologous recombination‐based genetic engineering of recA‐dependent genetic engineering of E. coli, recombineering has a significant advantage in that it utilizes shorter homology arms (~ 50 bp), which can be included in synthetic oligonucleotides (Zhang et al., 1998).
Redα and RecE are 5′–3′ exonucleases that generate 3′‐ended, single‐stranded DNA (ssDNA) overhangs, and Redβ and RecT are ssDNA annealing proteins (SSAP) that bind to the ssDNA forming a recombinogenic proteonucleic filament, which is used in recombination (Carter and Radding, 1971; Kolodner et al., 1994; Karakousis et al., 1998). Redα/Redβ are reported to favour homologous recombination between a linear and a circular DNA molecule in E. coli, whereas RecE/RecT are more efficient at linear–linear homologous recombination (Fu et al., 2012). Redγ, identified only in lambda phage and not in Rac prophage, forms a dimer to mimic DNA to significantly enhance the recombination efficiency of Redα/Redβ by inhibiting the exonuclease and helicase activities of the RecBCD complex, which rapidly degrades linear double‐stranded DNA (dsDNA; Taylor and Smith, 1980). Although the RecET operon does not contain a Redγ equivalent, the inclusion of Redγ with RecE/RecT has been shown to promote recombination efficiency (Fu et al., 2012). The Redγ‐like protein Pluγ from Photorhabdus luminescens can also inhibit the RecBCD complex in both Photorhabdus and E. coli (Yin et al., 2015).
Microorganisms serve as an excellent source of structurally diverse natural products, many of which possess important biological activities, such as antibacterial, antifungal, antiviral and anticancer properties (Newman and Cragg, 2012). Burkholderia, a Gram‐negative genus belonging to the β‐proteobacteria, encompasses more than 100 species that colonize a wide range of environments and that have highly diverse symbiotic associations (Coenye and Vandamme, 2003; Compant et al., 2008; Estrada‐de los Santos et al., 2018; Kunakom and Eustáquio, 2019). Burkholderia species are well‐known as pathogens of humans, animals, and plants, as well as for bioremediation, biocontrol, plant growth promotion, and biopesticidal properties (Depoorter et al., 2016). Burkholderia sensu lato (s.l.) is a large and complex group, and has been divided into six genera Burkholderia sensu stricto (s.s.), Paraburkholderia, Caballeronia, Robbsia, Mycetohabitans and Trinickia (Estrada‐de los Santos et al., 2018; Table S1). In this study, Burkholderia sensu stricto (s.s.) is represented by Burkholderia.
Burkholderia strains synthesize numerous bioactive compounds (Kunakom and Eustáquio, 2019), such as the anticancer drug FK228 (Liu et al., 2012); the structural analogs burkholdacs, thailandepsins and spiruchostatins (Crabb et al., 2008; Biggins et al., 2011); the antibacterial compounds thailandamides (Nguyen et al., 2008) and capistruin (Knappe et al., 2009); and the antifungal agents pyrrolnitrin (Hammer et al., 1999), cepapafungins and cepacidines (Lim et al., 1994). Furthermore, genome analyses of Burkholderia species and development of a variety of bioinformatic tools such as antibiotics & Secondary Metabolite Analysis SHell (antiSMASH) have demonstrated that Burkholderia is an untapped reservoir of bioactive natural products, especially polyketides (PKs), non‐ribosomal peptides (NRPs) and hybrid NRP/PKs (Esmaeel et al., 2016; Ren et al., 2017; Esmaeel et al., 2018). Several groups have reported the application of the lambda Red recombination system in Burkholderia strains (Jia et al., 2010; Kang et al., 2011; Moebius et al., 2012). However, the long homology arms (> 500 nt) required, and the low recombination efficiency limit its extended application in Burkholderia strains. Thus, highly efficient and simple genetic tools that would enable the activation of cryptic biosynthetic gene clusters (BGCs) in Burkholderia species are needed to accelerate the discovery of novel bioactive natural products.
Recent study from our group described the recombinase Redβα7029 from strain DSM7029 for use in recombination in Burkholderia strains (Wang et al., 2018b). However, DSM7029 has been subsequently reclassified as Schlegelella brevitalea DSM7029, a new species of the genus Schlegelella, which belongs to the family Comamonadaceae, instead of the genus Burkholderia, which belongs to the family Burkholderiaceae (Tang et al., 2019). The recombination efficiency of a RecET‐like recombinase (RecEThBDU8) in Burkholderia sp. BDU8 has also been reported as very low, with colony numbers per millilitre (cnpm) of less than 10 in Paraburkholderia rhizoxinica HKI 454 (Wang et al., 2018b), which has been subsequently reclassified into the genus Mycetohabitans (Estrada‐de los Santos et al., 2018).
In this work, three new recombination systems, comprising Rac bacteriophage RecET‐like operons, were discovered in the genera Burkholderia and Caballeronia: RecETheBDU8 from Burkholderia sp. BDU8 which is clustered within the Burkholderia pseudomallei group, RecEThTJI49 from Burkholderia sp. TJI49 which is clustered within the unclassified Burkholderia group, and RecETh1h2eYI23 from Burkholderia cordobensis YI23 which is clustered within the genus Caballeronia (Draghi et al., 2014). RecEThTJI49 and RecETh1h2eYI23 showed higher recombineering efficiency than RecETheBDU8 did in B. glumae PG1. Hence, recombineering with RecEThTJI49 and RecETh1h2eYI23 was systematically optimized, and the efficiency of various recombinase combinations was further tested in E. coli and B. glumae PG1. These recombineering systems were introduced into different strains of Burkholderia for genome engineering by inserting functional promoters to activate cryptic NRPS BGCs of which three were successfully activated. Three new (compounds 1–3) and four known (compounds 4–7) lipopeptides were identified from the recombinant B. plantarii DSM9509 mutant DSM9509::PApra‐BGC4. Compounds 3 and 7 exhibited potential anti‐inflammatory activity by inhibiting lipopolysaccharide (LPS)‐stimulated nitric oxide (NO) production in RAW 264.7 macrophages.
Results and discussion
Endogenous phage recombinase pairs in Burkholderia
Burkholderia and Burkholderia phage genomes were searched for candidate DNA recombination proteins with Position‐Specific Iterative BLAST (PSI‐BLAST) using the coding sequences of Redβ, RecT and Pluβ as queries in the non‐redundant protein sequence database (Altschul et al., 1997). Three RecET‐like operons, RecETheBDU8, RecEThTJI49 and RecETh1h2eYI23, were identified (Fig. 1 and Table S4). RecETheBDU8 from Burkholderia sp. BDU8 harbours a four‐gene operon predicted to encode: the YqaJ viral recombinase family protein RecEBDU8 (protein ID: KVE53656.1; locus tag: WS71_06320), which is equivalent to RecE; the recombinase RecTBDU8 (protein ID: KVE53655.1; locus tag: WS71_06315), equivalent to RecT; and the two hypothetical proteins hBDU8 (protein ID: KVE53654.1; locus tag: WS71_06310) and eBDU8 (protein ID: KVE53653.1; locus tag: WS71_06305). The second operon, RecEThTJI49 from Burkholderia sp. TJI49, is predicted to encode the following three proteins: the hypothetical protein RecETJI49 (protein ID: EGD06616.1; locus tag: B1M_00520), which was 24% identical to RecE; the phage‐related DNA recombination protein RecTTJI49 (protein ID: EGD06615.1; locus tag: B1M_00515); and the hypothetical protein hTJI49 (protein ID: EGD06614.1; locus tag: B1M_00510). The third operon, RecETh1h2eYI23 from Burkholderia cordobensis YI23, contains genes encoding the putative 5′‐3′ specific dsDNA exonuclease RecEYI23 (protein ID: AET91062.1; locus tag: BYI23_B004550); the putative recombinase protein RecTYI23 (protein ID: AET91060.1; locus tag: BYI23_B004530), which showed significant similarity to RecT (sequence identity of 46% in a 108‐amino acid region); three hypothetical proteins h1YI23 (protein ID: AET91061.1; locus tag: BYI23_B004540), h2YI23 (protein ID: AET91063.1; locus tag: BYI23_B004560) and eYI23 (protein ID: AET91059.1; locus tag: BYI23_B004520). The three pairs of recombinases (RecE/RecT homologues) from these operons were chosen for the development of the recombineering systems in B. glumae PG1.
Fig. 1.
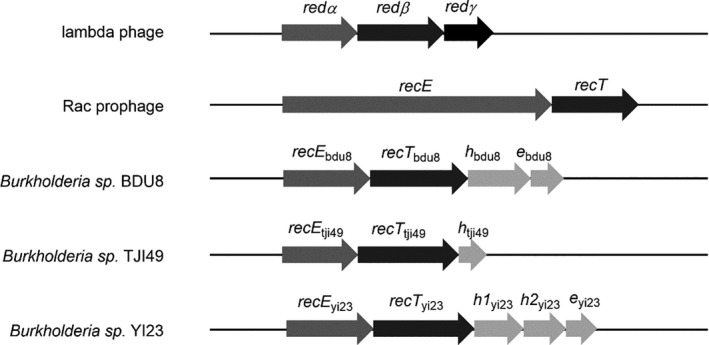
Red/ET recombinase pairs from lambda phage and Rac prophage and their homologues in Burkholderia. Arrows with the same shade represent genes with similar functions or classification. All genes are drawn to scale.
Optimization of transformation efficiency in B. glumae PG1
In E. coli, transformation efficiency is important when manipulating DNA in vivo (Sharan et al., 2009), and the recombinant proteins are usually induced when the cells enter log phase growth (Fu et al., 2010; Yin et al., 2019, 2015). In order to optimize transformation efficiency for Burkholderia, a B. glumae PG1 growth curve was first plotted at 30°C (Fig. 2A). Overnight cultures were diluted to OD600 = 0.1 to start the growth‐monitored cultures. After approximately 2 h, the B. glumae PG1 culture entered log phase, and the plasmid pBBR1‐Rha‐Firefly‐kan was transformed into electrocompetent cells prepared at different time points. The cells prepared at 2 h and room temperature yielded the most transformants (Fig. 2B). Transformation efficiency in B. glumae PG1 using different electroporation solutions and DNA amounts was also tested (Fig. 2C and D), with the highest transformation efficiency obtained using S solution at room temperature. The cells yielded the most transformants using 1500 ng DNA with B. glumae PG1 in S solution, although 500 ng DNA was sufficient for transformation. Thus, efficient transformation conditions for B. glumae PG1 were established.
Fig. 2.
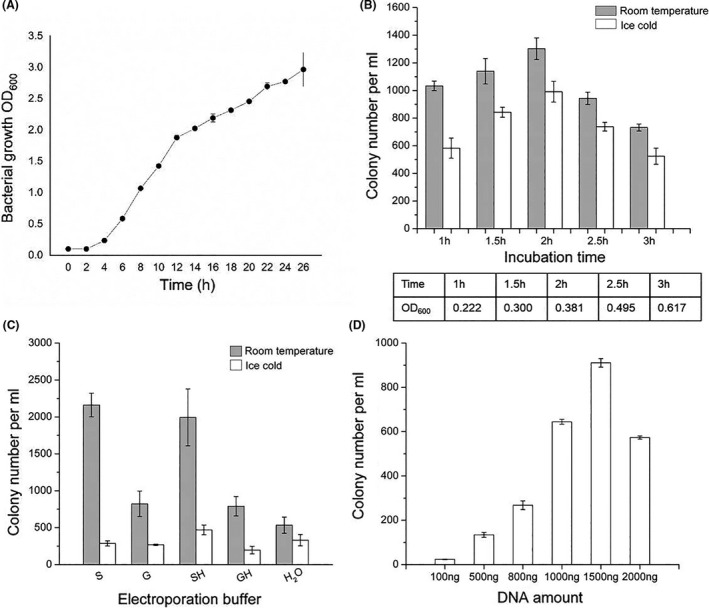
Optimization of transformation efficiency in B. glumae PG1. (A) Growth curve of B. glumae PG1. The optical density at 600 nm (OD600) was measured every 2 h from a starting OD600 of 0.1. (B–D) Transformation efficiency comparison in B. glumae PG1 using different (B) incubation times and temperatures, (C) electroporation solutions and (D) DNA amounts. For (C), the competent cells were treated with different electroporation solutions: H2O, double‐distilled water; S, 10% sucrose (w/v); G, 10% glycerol (v/v); SH, 10% sucrose (w/v) + 2 μM HEPES; GH, 10% glycerol (v/v) + 2 μm HEPES. Error bars, SD; n = 3.
Efficiency of recombineering systems
The recombinase expression plasmids (Fig. 3 and Table S2) used to evaluate homologous recombination efficiencies were based on a broad host range origin of replication (pBBR1; Antoine and Locht, 1992), and the rhaR‐rhaS PRha inducible promoter (Egan and Schleif, 1993, 1994) was inserted into these plasmids to control the expression of the recombination operons. We first compared the recombination efficiency of three operons, RecETheBDU8, RecEThTJI49 and RecETh1h2eYI23, using E. coli Redγβα, S. brevitalea DSM7029 Redβα7029, and Pseudomonas RecTEPsy, BAS, and GBAS as reference. The assays were plasmid modification in E. coli and the genome modification in B. glumae PG1 (Fig. 3). For transformation of E. coli, we used conditions described in our previous study (Fu et al., 2012), and for transformation of B. glumae, PG1, we used the optimized conditions described above.
Fig. 3.
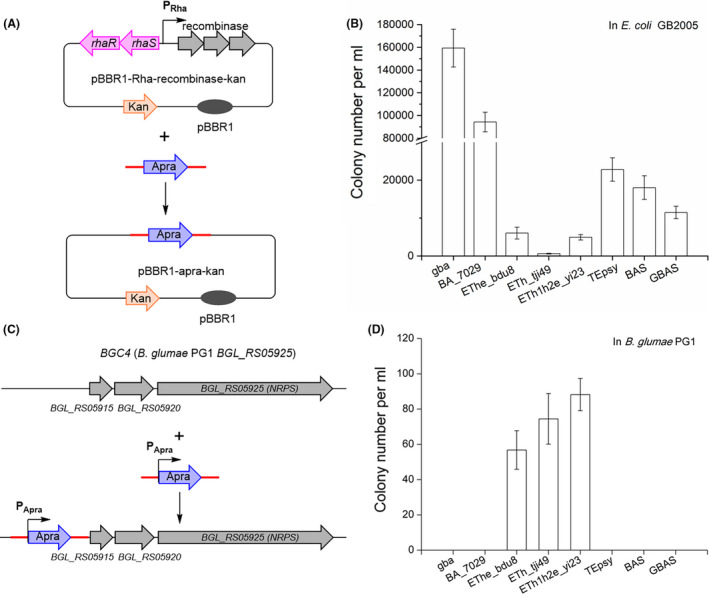
Recombineering with different protein combinations in E. coli and B. glumae PG1.
A. Diagram of plasmid modification (linear plus circular homologous recombination, LCHR) in E. coli. A PCR product carrying an apramycin resistance gene (apra) flanked by 80 bp homology arms (represented by red lines) was integrated into the expression plasmid, replacing the recombinase genes, the Rha promoter, and the rhaS and rhaR genes. gba, Redγβα of E. coli; BA_7029, Redβα7029; EThe_bdu8, RecET homologs and hypothetical proteins of Burkholderia sp. BDU8; ETh_tji49, RecET homologs and a hypothetical protein of Burkholderia sp. TJI49; ETh1h2e_yi23, RecET homologs and hypothetical proteins of Burkholderia cordobensis YI23; TEpsy, RecTE homologs from P. syringae pv. tomato DC3000; BAS, Redβα and SSB protein from P. aeruginosa phage vB_PaeP_Tr60_Ab31; GBAS, Redγ combined with Redβα and SSB protein from P. aeruginosa phage vB_PaeP_Tr60_Ab31.
B. Functional characterization of recombinases in E. coli using an LCHR assay.
C. Diagram of genome modification of B. glumae PG1. An apra gene flanked by 80 bp homology arms (red lines) was inserted upstream of the BGL_RS05915 gene in B. glumae PG1.
D. Functional characterization of recombinase combinations in B. glumae PG1 using a genome modification assay. Error bars, SD; n = 3.
In the plasmid modification assay with E. coli, a PCR product carrying an apramycin resistance gene (apra) flanked by 80 bp homology arms (Fig. 3A, red lines) was integrated into an expression plasmid, replacing the recombinase genes, the Rha promoter, and the rhaS and rhaR genes and resulting in the plasmid product pBBR1‐apra‐kan (Fig. 3A). Results are based on counting apramycin‐resistant colonies followed by verification of pBBR1‐apra‐kan by restriction analysis. In the genome modification assay with B. glumae PG1, an apramycin resistance gene (apra) flanked by 80 bp homology arms (red lines) was inserted before the gene BGL_RS05915 in B. glumae PG1 (Fig. 3C). Results are based on counting apramycin‐resistant colonies followed by PCR verification (Fig. S2).
All eight recombinase operons exhibited recombination activity in E. coli (Fig. 3B), with Redγβα showing the highest efficiency followed by Redβα7029. Although Redβα7029 exhibited high efficiency in S. brevitalea DSM7029 (Wang et al., 2018b), it showed very low recombineering efficiency when compared to that of RecEThTJI49 and RecETh1h2eYI23 in Burkholderia glumae PG1 (Fig. 3D). Recombination operons from Burkholderia, especially RecEThTJI49, showed very low efficiency in E. coli. However, in B. glumae PG1, only RecETh1h2eYI23, RecEThTJI49 and RecETheBDU8 were functional (Fig. 3D). As RecEThTJI49 and RecETh1h2eYI23 had the highest recombineering efficiency in B. glumae PG1, these two operons were selected for further optimization of recombineering.
Recombination efficiency of different recombinase combinations
In the search for recombination proteins in Burkholderia and Burkholderia phage genomes, the hypothetical proteins in the RecETheBDU8, RecEThTJI49 and RecETh1h2eYI23 operons were considered to be functionally associated with their neighbouring recombinase. Therefore, to test the function of the hypothetical proteins, we generated a series of constructs to evaluate recombinase activity with or without the different hypothetical proteins (Table S2). RecETBDU8, RecETTJI49 and RecETYI23 were also combined with either Redγ or Pluγ to determine whether these two proteins could function in synergy with the Burkholderia RecET proteins (Table S2). The recombination efficiency of these recombinase expression plasmids was compared in E. coli and B. glumae PG1 (Fig. 4).
Fig. 4.
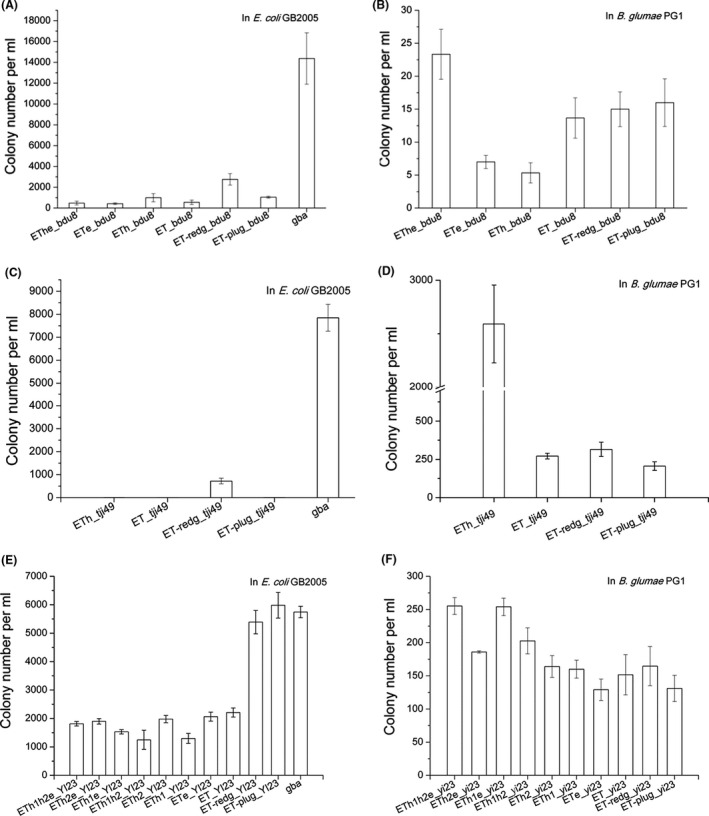
Recombination efficiencies of various recombinase combinations in (A, C, E) E. coli and (B, D, F) B. glumae PG1. Efficiencies were compared using complete or partial Burkholderia recombinase operons or combinations of the Burkholderia RecE/T proteins with proteins from other recombinase systems. GBA was included for comparison in the E. coli assays. (A, B) ETheBDU8 combinations. (C, D) EThTJI49 combinations. (E, F) ETh1h2eYI23 combinations. Error bars, SD; n = 3.
In E. coli, Redγ significantly increased the recombination efficiency of RecETBDU8, RecETTJI49 and RecETYI23, whereas the hypothetical proteins did not. Pluγ also markedly enhanced the efficiency of RecETBDU8 and RecETYI23, although it failed to function synergistically with RecETTJI49. Notably, the efficiency of RecETYI23 was as equivalent as that of Redγβα in E. coli when a host nuclease inhibitor was provided (Fig. 4E).
In B. glumae PG1, removal of any of the hypothetical proteins decreased the efficiency of the three operons, with the exception of h2 from the RecETh1h2eYI23 operon, which did not show any effect on recombination (Fig. 4F). The efficiency of RecETBDU8, RecETTJI49 and RecETYI23 combined with Redγ or Pluγ was lower than that of RecETheBDU8, RecEThTJI49 and RecETh1h2eYI23, indicating that the complete native operons were more efficient in B. glumae PG1.
Optimization of recombineering in B. glumae PG1
Next, we optimized the electroporation procedure for RecEThTJI49 and RecETh1h2eYI23 for recombineering in B. glumae PG1 (Figs 5 and S3). For both RecEThTJI49 and RecETh1h2eYI23, optimal recombination efficiency was achieved using an incubation time of 2 h, an induction time of 40 min and an induction temperature of 35°C (Figs 5A–C and S3A–C). RecEThTJI49 exhibited optimal recombination efficiency with ice‐cold 10% (v/v) glycerol as the electroporation solution whereas ice‐cold water was more effective for RecETh1h2eYI23 (Figs 5D and S3D). Titration of DNA amounts from 100 ng to 2000 ng indicated that increases from 800–2000 ng had no significant effect on the recombination efficiency of either RecEThTJI49 or RecETh1h2eYI23 (Figs 5E and S3E), and therefore, the saturation amount of DNA was 800 ng for B. glumae PG1. Various lengths of the homology arms were also tested, and although RecEThTJI49 and RecETh1h2eYI23 exhibited highest efficiency with lengths of 125 and 150, 100 bp was sufficient for both operons in B. glumae PG1 (Figs 5F and S3F). PCR verification of recombination with RecEThTJI49 using homology arms of different lengths confirmed that all recombinants were correct. From the above results, optimal recombineering conditions of RecEThTJI49 and RecETh1h2eYI23 in B. glumae PG1 were obtained.
Fig. 5.
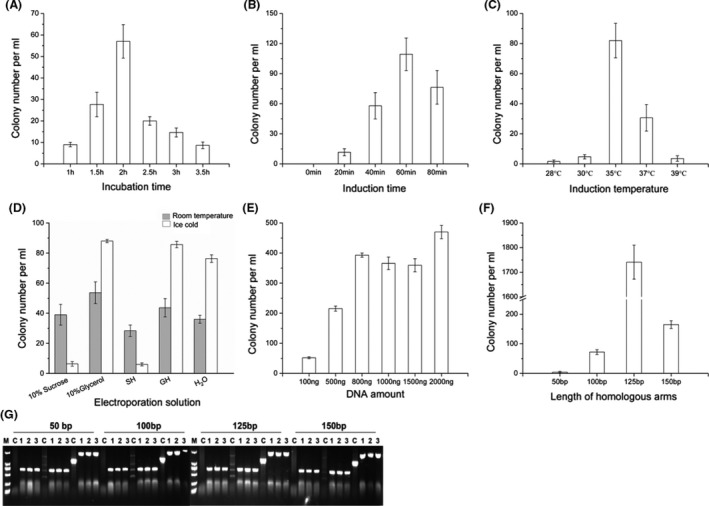
Optimization of recombination efficiency of RecEThTJI49 in B. glumae PG1 for genome modification. (A–F) Recombination efficiency comparisons of RecEThTJI49 using different (A) incubation times, (B) induction times, (C) induction temperatures, (D) electroporation solutions, (E) DNA concentrations and (F) lengths of the homology arms. (G) PCR verification of recombinants obtained using different lengths of homology arms. M, Takara DL2000 DNA ladder. H2O, double‐distilled water; S, 10% (w/v) sucrose; G, 10% (v/v) glycerol; SH, 10% (w/v) sucrose + 2 μM HEPES; GH, 10% (v/v) glycerol + 2 μm HEPES. Error bars, SD; n = 3.
In situ activation of cryptic NRPS BGCs in Burkholderia strains
Insertion of a functional promoter can precisely and successfully activate cryptic BGCs (Myronovskyi and Luzhetskyy, 2016). In this study, cryptic NRPS/PKS BGCs were examined by comparative metabolite analysis using promoter insertion activated mutants, inactivated mutants and wild‐type strains (Table S6). The activated mutants were constructed by inserting an antibiotic selection marker upstream of the first NRPS‐PKS gene. The DNA sequence of the modules of the core NRPS‐PKS genes in the target BGC was interrupted by the same antibiotic marker to generate a completely inactivated mutant (Fig. 6). Using this approach, we generated inactivated and activated mutants of three NRPS/PKS BGCs of B. plantarii DSM9509 and three NRPS/PKS BGCs of B. glumae DSM9512, respectively. The metabolite profiles of these activated mutants, inactivated mutants and wild‐type strains were compared using HPLC‐MS (Figs 7, S5 and S6), and the products of three cryptic NRPS BGCs, BGC4 and BGC11 of B. plantarii DSM9509 and BGC9 of B. glumae DSM9512, were detected in the activated mutants, indicating successful activation by promoter insertion. Thus, in this study, the success rate of this strategy of using recombinase‐assisted in situ insertion of promoters to activate cryptic BGCs was about 50%.
Fig. 6.
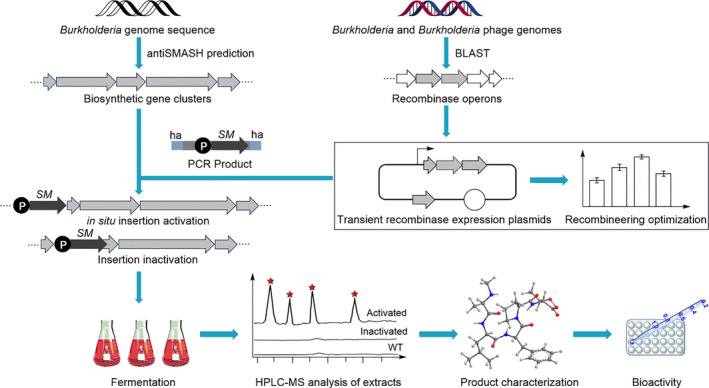
Workflow of the development of an efficient recombineering system and its application to activate cryptic biosynthetic gene clusters (BGCs). ‘SM’, selectable marker; ‘P’, promoter; ‘ha’, homology arms.
Fig. 7.
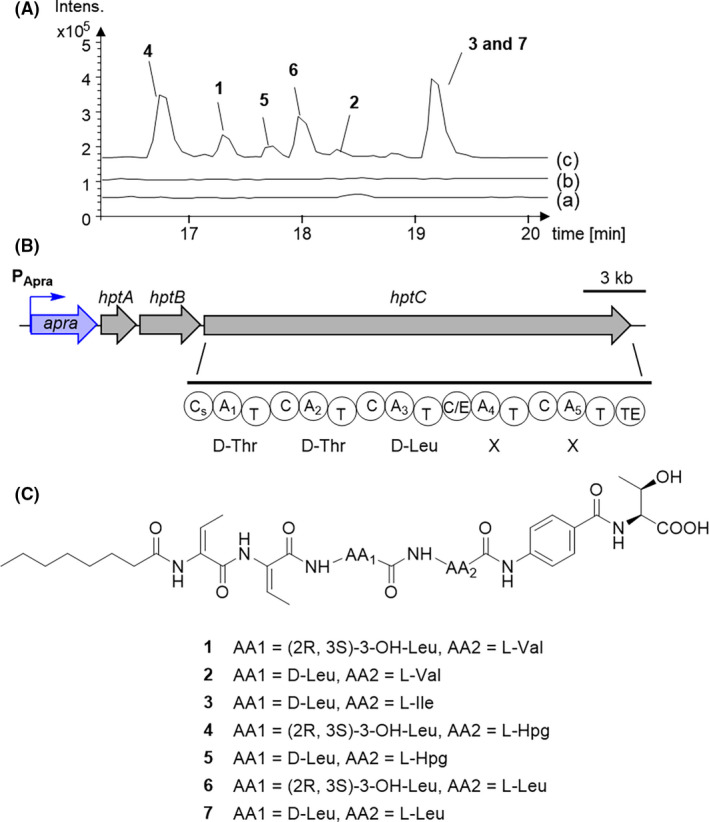
Mining of a cryptic BGC in B. plantarii DSM9509.
A. HPLC‐MS analysis (BPC) of extracts from (a) DSM 9509 wild type, (b) BGC4 inactivated mutant of DSM9509‐DSM9509::PApra‐BGC4Δmodule2 and (c) BGC4 activated mutant of DSM 9509‐DSM9509::PApra‐BGC4.
B. Structure of BGC4 and NRPS module architecture of hptC. The structure shows the promoter (P‐Apra) inserted upstream of the first gene in the BGC. The predicted NRPS modules and A domain are shown for hpt. A, adenylation domain; C, condensation domain; C/E, dual condensation/epimerization domain; CS, starter condensation domain; T, peptidyl carrier domain; TE, thioesterase domain.
C. Complete structures of compounds 1–7 produced by DSM9509::PApra‐BGC4.
Identification of new lipopeptides from DSM9509::PApra‐BGC4
The seven lipopeptides 1–7 were obtained from the recombinant B. plantarii DSM9509 mutant DSM9509::PApra‐BGC4 (Fig. 7). BGC4 (hpt) was similar to those of hgd BGC from B. gladioli pv. agaricicola and hgm BGC from B. glumae (Fig. 7 and Table S6). The hptC gene encodes an NRPS predicted to form a five‐amino acid lipopeptide. Furthermore, the first condensation (C) domain was deduced to be a starter type condensation (Cs) domain that can incorporate an acyl group as the first unit (Rausch et al., 2007). Thus, the lipopeptide was predicted to be Acyl‐Thr‐Thr‐Leu‐X‐X, which differs from haereogladin and haereoglumin.
The structures of the new lipopeptides (1–3) were elucidated by comprehensive analysis using nuclear magnetic resonance (NMR), high‐resolution electrospray ionization mass spectrometry (HRESIMS), MS/MS data and Marfey’s analysis. Compound 1 was isolated as a white, amorphous powder, and its molecular formula, C38H58N6O10, was established from a pseudomolecular ion peak at m/z 759.4278 [M + H]+ in its HRESIMS profile (Fig. S16), which suggested 17 indices of hydrogen deficiency. The infrared spectroscopy (IR) spectrum of 1 exhibited absorption bands due to OH and NH (3315 cm−1), C=O (1637 cm−1) and C=C (1538 cm−1) groups (Fig. S18). 1H NMR, 13C NMR, distortionless enhancement by polarization transfer (DEPT)‐135, DEPT‐90 and heteronuclear single quantum coherence (HSQC) spectra showed 38 carbon signals, including 11 nonprotonated carbons (seven carbonyl and four olefinic), 13 methines (two olefinic and four aromatic), six methylenes and eight methyls (Figs S8–S12). Analysis of the 1H,1H−COSY (correlated spectroscopy) correlations revealed a spin system of octanoate (Oct), two units of dehydrobutyrine (Dhb), β‐OH‐leucine (β‐OH‐Leu), valine, p‐amino benzoate (PABA) and threonine (Thr; Fig. S13). The peptide sequence was determined to be Oct/Dhb/Dhb/β‐OHLeu/Val/PABA/Thr by further analysis of the heteronuclear multiple bond correlation spectroscopy (HMBC) correlations from amide protons and adjacent carbonyl groups (Fig. S14). This conclusion is in agreement with the MS/MS fragmentation pattern (Fig. S17). The configuration of the Dhb units was determined as having nuclear Overhauser effect (NOE) correlations of H3‐4 in Dhb1 with H‐N in the adjacent Dhb2, H3‐4 in Dhb2 with H‐N in the adjacent β‐OH Leu (Figs S7 and S15). The absolute configurations of the amino acids were established by Marfey’s method as L‐Val, (2R,3S)‐3‐OH‐Leu and L‐Thr, which is consistent with bioinformatics analysis of the C domains (Table S13). The structure of compound 1 was found to be similar to that of haereoplantin D, which was published during the preparation of this manuscript, except that a l‐Leu was replaced by l‐Val in 1. Thus, compound 1 was named haereoplantin F.
Compound 2 exhibited an HRESIMS [M + H]+ ion peak at m/z 743.4334 (calc. 743.4338), indicating one less O‐atom than 2 (Fig. S30). An examination of the NMR spectroscopic data (Figs S21–S29) showed the structure of 2 to be similar to that of 1, except for the presence of the methylene protons at δ H 1.720 and 1.530 ppm and δ C 39.58 ppm, respectively. Further analysis of the 1H,1H−COSY and HMBC correlations and MS/MS fragmentation pattern determined the presence of a Leu in 2 instead of the 3‐OH‐Leu in 1. The complete structure of 2 was further determined by Marfey’s analysis (Table S13). Compound 2 was named haereoplantin G, and its structure was elucidated as shown. Compound 3, named haereoplantin H, showed NMR data similar to that of 2, except for the presence of an Ile instead of the Val. This structure was also confirmed by NMR, HRESIMS and MS/MS fragmentation studies (Figs S35–S44).
The structures of the known compounds 4 (haereoplantin A), 5 (haereoplantin C), 6 (haereoplantin D) and 7 (haereoglumin B) were elucidated based on comparison of their NMR and MS data with those reported in the literature (Thongkongkaew et al., 2018; Yoshimura et al., 2020).
Compounds 8 (985 [M + H]+) and 9 (971 [M + H]+) were both detected in the recombinant B. glumae DSM9512 mutant DSM9512::PApra‐BGC9 and B. plantarii DSM9509 mutant DSM9509::PApra‐BGC11 (Table S6, Figs S5 and S6). Compound 8 was determined to be burrioglumin B by comparing the NMR data and MS/MS fragmentation patterns (Thongkongkaew et al., 2018). Only 0.4 mg of compound 9 was obtained, which was not enough for NMR analysis. However, 9 was deduced to be burrioglumin A by MS/MS fragmentation studies and bioinformatic analysis (Thongkongkaew et al., 2018).
Bioactivity analysis
Compounds 1–7 were evaluated for their ability to inhibit NO production in LPS‐stimulated RAW264.7 cells using the Griess assay (Sun et al., 2015). Notably, none of the compounds displayed cytotoxicity with murine macrophage RAW264.7 cells using the MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐2H‐tetrazolium bromide, Sigma] assay (IC50 > 48 μM; Table S14; Alley et al., 1988), which suggested that any inhibition of NO production would not be due to cytotoxicity. As shown in Fig. 8, compound 3, which has a d‐Leu and an l‐Ile, and compound 7, which has a d‐Leu and an l‐Leu, showed strong inhibition of NO generation activity. Compound 7 exhibited the highest potency, and the inhibition increased in a concentration‐dependent manner, with inhibition rates of 28.29%, 44.56% and 66.43% at 5, 10 and 20 μM, respectively (Fig. 8).
Fig. 8.
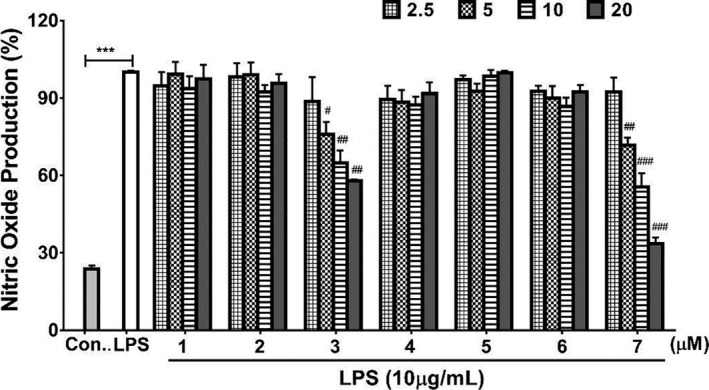
Effects of compounds 1–7 on LPS‐stimulated NO production in RAW264.7 cells. NO production in the absence of LPS was used as negative control. LPS‐stimulated NO production in the absence of added compounds (‘LPS’ bar) was used as the positive control and was considered 100% production. Patterned bars indicate different amounts of the compounds in micrograms. ***P < 0.001 compared with the control group; # P < 0.05, ## P < 0.01 and ### P < 0.001 compared with the LPS group. Error bars, SD; n = 3.
As LPS‐induced oxidative stress plays a pivotal role in inflammation, we further investigated the effects of compounds 1–7 on LPS‐induced production of reactive oxygen species (ROS) production. The results showed that pretreatment with 20 μM of compound 7 significantly decreased LPS‐induced ROS production in RAW264.7 cells (Fig. S47). Our findings suggest that compounds with a d‐Leu and either an l‐Leu or an l‐Ile have potentially important anti‐inflammatory activities in LPS‐stimulated macrophages and that they may be promising lead compounds for developing anti‐inflammatory therapies.
Concluding remarks
Recombineering, developed in E. coli in 1998 (Abbasi et al., 2020), has been efficiently used to engineer the genome of E. coli and several genetically close species. However, apparent host‐specific factors limited the wider application of recombineering in other species. In recent years, we and others have explored recombination systems for some genetically distant bacteria, such as the RecETPsy system from Pseudomonas syringae pv. DC3000 (Swingle et al., 2010), BAS from Pseudomonas aeruginosa phage Ab31, and TEPsy from Pseudomonas syringae pv. syringae B728a (Yin et al., 2019), Pluγβα from Photorhabdus luminescens (Yin et al., 2015) and Redβα7029 from S. brevitalea DSM7029 (Wang et al., 2018b).
The Burkholderia genus includes both human and plant pathogens and environmentally important species. Burkholderia strains have been reported to be new sources of natural products with a myriad of cryptic BGCs (Lim et al., 1994; Hammer et al., 1999; Crabb et al., 2008; Nguyen et al., 2008; Knappe et al., 2009; Biggins et al., 2011), emphasizing the need for an efficient Burkholderia recombineering system that enables the discovery of natural products by genome engineering. In this study, we have described a recombineering system based on three RecET‐like operons, RecETheBDU8 from Burkholderia sp. BDU8, RecEThTJI49 from Burkholderia sp. TJI49 and RecETh1h2eYI23 from Burkholderia cordobensis YI23. All three operons encode RecE and RecT homologues and hypothetical proteins, which may function similarly to Redγ or Pluγ (Fig. 1 and Table S4). Our experiments to optimize the efficiency of recombineering led to the following conclusions.
All three operons exhibited recombination function in B. glumae PG1, although RecETh1h2eYI23 and RecEThTJI49 showed higher efficiency compared to RecETheBDU8. Additionally, five other non‐Burkholderia recombineering operons, that is Redγβα, Redβα7029, RecTEPsy, BAS and GBAS, were inferior to the Burkholderia operons in B. glumae PG1. Therefore, these three recombinase operons provide a more suitable foundation for efficient genome engineering systems in Burkholderia.
In our previous work, BAS and TEPsy combined with Redγ and Pluγ, respectively, remarkably enhanced recombination efficiency in Pseudomonas aeruginosa (Yin et al., 2019). The addition of Redγ into Redβα7029 also significantly increased recombination efficiency in both E. coli and S. brevitalea DSM 7029 (Wang et al., 2018b). In this current study, the addition of Redγ into the RecETBDU8, RecETTJI49, or RecETYI23 operons, and Pluγ into RecETBDU8 or RecETYI23 significantly increased recombination efficiency in E. coli. However, in B. glumae PG1, neither Redγ nor Pluγ increased the efficiency of the three Burkholderia operons. These findings suggest that Redγ and Pluγ may alter the function of the RecBCD complex by temporarily blocking its exonuclease activity in E. coli but not in B. glumae PG1.
With the exception of the hypothetical protein h2YI23, removal of any of the hypothetical proteins of these three recombinase operons decreased recombination efficiency in B. glumae PG1, suggesting a role for these proteins in recombination. This finding is consistent with a previous study showing that RecEThBDU8, which is missing the hypothetical protein ‘e’, had very low recombination efficiency in Paraburkholderia rhizoxinica HKI 454 (Wang et al., 2018b). Nevertheless, as it had no effect on recombination efficiency, h2YI23 could be omitted to yield the optimal combination RecETh1eYI23. Notably, the hypothetical proteins did not enhance recombination efficiency in E. coli. Thus, we suggest that the cooperation of these hypothetical proteins with the Burkholderia RecE and RecT homologues might proceed via a new mechanism, although further experiments are needed to clarify their role.
Finally, the combination of Redγ or Pluγ with RecETYI23 were as efficient as Redγβα in E. coli for recombineering, revealing RecETYI23 may have relaxed host specificity which worth further investigation of its utility in other Gram‐negative bacteria.
Overall, through the evaluation of different configurations and the optimization of the RecEThTJI49, RecETh1eYI23 and RecETh1h2eYI23 operons in B. glumae PG1, our study indicates that high recombineering efficiency can be achieved in Burkholderia and that these operons may be applied to engineer the genome of Burkholderia strains. Characterization of cryptic BGCs identified by genome sequencing remains a significant challenge because most of these clusters are not expressed in laboratory cultures. Mining of cryptic BGCs from native producers, which likely possess all the metabolic and biosynthetic requirements, is comparatively easier than from heterologous hosts. Recently, promoter reengineering of cryptic BGCs has resulted in the successful activation of BGCs and production of new natural products (Montiel et al., 2015; Zhang et al., 2017). In our study, cryptic BGCs were activated by using our recombineering system to insert a promoter upstream of the target BGCs. Using this recombineering system, pathway‐specific regulatory genes can be manipulated to activate cryptic BGCs or improve production of weakly expressed BGCs.
In conclusion, we developed a recombineering system for Burkholderia glumae and the related B. plantarii based on three Rac bacteriophage RecET‐like operons, RecETheBDU8, RecEThTJI49 and RecETh1h2eYI23. We then demonstrated the successful application of this recombineering system by inserting a promoter upstream of the main biosynthetic genes in cryptic BGCs, leading to activation of these BGCs and the identification of three new lipopeptides. Compound 7 exhibited significant potential anti‐inflammatory activity by inhibiting LPS‐stimulated NO production in RAW 264.7 macrophages. This recombineering system may work for the other species of the genus Burkholderia after optimization of a protocol and enhance functional genome research and mining of novel natural products.
Experimental procedures
Strains, plasmids and reagents
The wild‐type bacterial strains, the mutants and plasmids used in this work are listed in Table S2. All the expression plasmids used here to evaluate homologous recombination efficiencies are based on the pBBR1 origin (Antoine and Locht, 1992) and the rhamnose inducible promoter PRhaSR (Wang et al., 2016). The plasmids were constructed by recombineering either in E. coli GB08‐red for linear plus circular homologous recombination (LCHR) or in E. coli GB05‐dir linear plus linear homologous recombination (LLHR; Fu et al., 2012). When T4 DNA ligase was used, the DNA ligation products were dialyzed and then electroporated in E. coli GB2005. Genes encoding different recombinases were amplified using polymerase chain reaction (PCR) products from corresponding genomic DNA or synthesized according to the original sequences in Genbank by Sangon Biotech (Shanghai), in China. Oligonucleotides were synthesized by Sangon Biotech (Shanghai), in China (Table S3). Restriction enzymes, DNA polymerases and DNA markers were supplied by New England Biolabs (Lpswich, MA, USA). The antibiotics were purchased from Invitrogen (Carlsbad, MA, USA). E. coli was cultured in Luria–Bertani (LB) broth or on LB agar plates (1.2% (m/v) agar). Burkholderia species were cultured in CYMG (8 g l−1 Casein peptone, 4 g l−1 Yeast extract, 4.06 g l−1 MgCl2·2H2O, 10 ml l−1 glycerin) broth or agar plates. The concentration of the required antibiotics is listed in Table S5.
General experimental procedures
Optical rotations were acquired by an Anton Paar MCP 200 polarimeter at 20°C. UV data were recorded using a UV‐2550 spectrophotometer (Shimadzu, Japan). IR spectra were measured on a Nicolet iN 10 Micro FTIR spectrometer. 1H and 13C NMR, DEPT, and 2D NMR spectra were recorded on a Bruker AvanceIII 600 MHz with TCI cryoprobe using TMS as an internal standard. HRESIMS spectra were measured on a Bruker Impact HD microTOF Q III mass spectrometer (BrukerDaltonics, Bremen, Germany) using the standard ESI source. HPLC‐MS was operated using a Thermo Scientific Dionex Ultimate 3000 system coupled with the Bruker amazon SL Ion Trap mass spectrometry (Bruker Corporation, Bremen, Germany), controlled by Hystar v3.2 and Chromeleon Xpress software. A Thermo Scientific™ Acclaim™ C18 column (2.1 × 100 mm, 2.2 μm) was used. The mobile phase consisted of H2O containing 0.1% (v/v) FA and ACN. Semipreparative HPLC was performed using an ODS column [Bruker ZORBAX SB‐C18, 250 × 10 mm, 5 μm, 2 ml min−1]. TLC and column chromatography (CC) were performed on plates precoated with silica gel GF254 (10–40 μm) and over silica gel (200–300 mesh, Qingdao Marine Chemical Factory, Qingdao, Shandong, China) and Sephadex LH‐20 (GE Healthcare, Pittsburgh, PA, USA) respectively.
Bioinformatic analysis
The coding sequences of Redβ (WP_001350280.1), RecT (WP_000166319.1) and Pluβ (WP_011147155.1) homologs were examined in the NCBI non‐redundant protein sequence database using PSI‐BLAST (Altschul et al., 1997). RecTYI23 was taken as an example to illustrate the method of finding its genomic position. (i) The protein ID of RecTYI23, AET91060.1, was obtained after PSI‐BLAST search; (ii) the locus tag of recTYI23 , BYI23_B004530, was found by searching the protein ID in GenBank database; (iii) the genomic position of recTYI23 was determined by searching the locus tag BYI23_B004530 in the genome sequence of Burkholderia cordobensis YI23. Only adjacent Redαβ‐like, RecET‐like or Pluαβ‐like recombinases present in either a Burkholderia or Burkholderia phage genome were selected in Table S4. Additionally, three RecET‐like recombinase operons (RecETheBDU8, RecEThTJI49 and RecETh1h2eYI23) containing hypothetical proteins were selected in this study.
Construction of recombinase expression plasmids
All the recombinase expression plasmids (Table S2) used to evaluate homologous recombination efficiencies are based on the pBBR1 origin and under the control of rhaR‐rhaS PRha inducible promoter (Antoine and Locht, 1992; Egan and Schleif, 1993, 1994). The original plasmid pBBR1‐Rha‐Redgba‐kan was digested with HindIII and NdeI to give linear fragment. The operon EThe_bdu8 with an AseI digestion site at each end was synthesized into a pUC57‐amp vector. The synthesized plasmid pUC57‐amp‐EThe_bdu8 was digested by AseI to expose the terminal homology arms to the digested (HindIII and NdeI) fragment of pBBR1‐Rha‐Redgba‐kan. Then, these two fragments were co‐transformed into GB05‐dir to construct pBBR1‐Rha‐EThe_bdu8‐kan. Constructions of the other two recombinase expression plasmids pBBR1‐Rha‐ETh_tji49‐kan and pBBR1‐Rha‐ETh1h2e_yi23‐kan were similar to that of pBBR1‐Rha‐EThe_bdu8‐kan respectively. The complete nucleotide sequences for pBBR1‐Rha‐ETh_tji49‐kan and pBBR1‐Rha‐ETh1h2e_yi23‐kan have been deposited in Addgene under accession numbers 166669 and 166670 respectively.
Different restriction sites have been added to each end of the h or e gene during synthesis of these three operons. Thus, the recombinase expression plasmids of different combinations were constructed by digestion and ligation. For example, pBBR1‐Rha‐EThe_bdu8‐kan was first digested by BamHI to remove the hypothetic protein coding gene ‘h’. Then, the above liner fragment was circularized by T4 DNA ligase to yield the plasmid pBBR1‐Rha‐ETe_bdu8‐kan.
RecETBDU8, RecETTJI49 and RecETYI23 combined with Redγ or Pluγ, respectively, were constructed by recombineering using ccdB for counterselection (Wang et al., 2014). For example, pBBR1‐Rha‐EThe_bdu8‐kan was first digested by XbaI to remove the hypothetic protein coding genes ‘h’ and ‘e’ to give a linear fragment. This linear fragment and cm‐ccdB with homology arms were co‐transformed into GBdir‐gyrA462 to construct the plasmid pBBR1‐Rha‐EThe_bdu8‐cm‐ccdB‐kan. Then, this plasmid and Redγ with homology arms were co‐transformed into GB08‐red to construct pBBR1‐Rha‐ET‐redγ_bdu8‐kan.
All the recombinants were selected on LB plates containing suitable antibiotics incubate at 37°C. Correct clones were verified by restriction analysis of the plasmids and sequencing of the regions comprising ‘redγ with homology arms’ or ‘pluγ with homology arms’.
Transformation in B. glumae PG1
For B. glumae PG1, overnight cultures were diluted to OD600 = 0.1 and 1 ml of the fresh cultures were grown at 30°C, 950 rpm for different time (1, 1.5, 2, 2.5 and 3 h). Cells were then centrifuged at 10 000 rpm for 1 min at room temperature or ice cold. The pellet was resuspended in room temperature or ice‐cold solution s (S solution, G solution, SH solution, GH solution and ddH2O) and centrifuged again (10 000 rpm, at room temperature or ice cold, 1 min). This was repeated twice more. The cell pellet was suspended in 30 μl ice‐cold S solution, and 200 ng of the plasmid pBBR1‐Rha‐Firefly‐kan was added. Electroporation was performed using ice‐cold cuvettes (1 mm) and an Eppendorf 2510 electroporator (1300 V). Then, 1 ml LB medium was added after electroporation. The cells were incubated at 30°C for 2 h with shaking at 950 rpm and then spread on LB plates containing 5 μg ml−1 kanamycin.
Electrocompetent cells preparation and recombineering
Various recombinase expression plasmids were electroporated into E. coli and B. glumae PG1 respectively. The E. coli electrocompetent cells were prepared according to our established protocol (Fu et al., 2012). For B. glumae PG1, overnight cultures containing the expression plasmids were diluted into 20 ml LB medium with kanamycin (5 μg ml−1). The starting OD600 value was 0.1. 1 ml of the fresh culture was grown at 30°C, 200 rpm for 2 h. After addition of the inducer l‐(+)‐rhamnose to a final concentration of 1.0 mg ml−1, the cells were grown at 35°C, 950 rpm for 60 min. After the OD600 was normalized, cells were then centrifuged for 1 min at 10 000 rpm at 2°C. The supernatants were discarded, and the cell pellets were resuspended in 1 ml of ice‐cold 10% (v/v) glycerol solution for cells containing pBBR1‐Rha‐ETh_tji49‐kan and 1 ml of ice‐cold ddH2O for cells containing pBBR1‐Rha‐ETh1h2e_yi23‐kan and centrifuged. The washing procedure was repeated one more time. Then, cells were resuspended in 30 µl of washing solution (ice cold), and PCR product (500 ng) was added. Electroporation was performed using ice‐cold cuvettes (1 mm) and an Eppendorf 2510 electroporator (1300 V). Then, LB medium (1 ml) was added after electroporation. The cells were incubated at 30°C for 2 h with shaking (950 rpm) and then spread on LB plates containing appropriate antibiotics.
Fermentation and extraction of compounds from Burkholderia strains
Liquid seed cultures of wild‐type and engineered strains were inoculated from a plate in 1.3 ml CYMG tubes and then incubated at 30°C for 18 h with shaking (950 rpm). Seed cultures were diluted at the ratio of 1:100 into 50 ml of CYMG broth in 250 ml baffled flasks, and the flash cultures were incubated at 30°C, 200 rpm. Incubation was continued for 48 h, and then, 2% (v/v) of absorber resin Amberlite XAD‐16 was added and incubated for 24 h continually. The biomass and XAD‐16 were harvested at maximum speed in an Eppendorf 5240R centrifuge for 10 min centrifugation, and the crude extracts were extracted with 50 ml methanol. Finally, the extract was concentrated in vacuo and redissolved in 1 ml MeOH for further HPLC‐MS analysis.
HPLC‐MS analysis of extracts from Burkholderia strains
The HPLC system was performed using a Thermo Scientific™ Acclaim™ C18 column (2.1 × 100 mm, 2.2 μm, 0.2 ml min−1) with gradient elution. UV spectra were recorded on a DAD detector with wavelength ranging from 190 to 400 nm. The MS was measured on a Bruker amazon SL Ion Trap mass spectrometry (Bruker Corporation) using the standard ESI source. Mass spectra were acquired in centroid mode ranging from 100 to 1500 m/z with positive‐mode electrospray ionization and auto MS2 fragmentation. HPLC parameters were as follows: solvent A, H2O with 0.2% (v/v) TFA; solvent B, 0.1% (v/v) TFA in acetonitrile (ACN); gradient at a constant flow rate of 0.2 ml min−1, 0–5 min, 5% (v/v) B; 5–45 min, 5%–95% (v/v) B; 45–50 min, 95% (v/v) B; or 0–5 min, 5% (v/v) B; 5–25 min, 5%–95% (v/v) B; 25–30 min, 95% (v/v) B detection by UV spectroscopy at 190–400 nm.
Isolation and purification of compounds from DSM9509::PApra‐BGC4
DSM9509::PApra‐BGC4 was cultured in 50 ml CYMG in 250 ml flasks (Total 20 l medium) at 30°C, 200 rpm for two days. The resin XAD‐16 was then added into the fermentation broth. After three days, the biomass and XAD‐16 were harvested and extracted with methanol. The MeOH extract (32.4 g) was separated by silica gel column chromatography (MeOH−CH2Cl2, 1:20 to 1:1) to give two fractions (Frs 1–2). Fr. 1 (9.5 g) was separated over RP C18 lobar column (MeOH/H2O, 5:5 to 9:1) to obtain six fractions; Frs. 1.1–1.6. Fr. 1.2 (510 mg) was purified by semi‐preparative HPLC (ODS; Bruker ZORBAX SB‐C18, 5 μm, 250 × 10 mm, ACN−H2O, 36:64, 2.5 ml min−1) to yield 1 (5.7 mg, t R = 13.2 min) and 4 (21.0 mg, t R = 12.6 min). Fr. 1.3 (250 mg) was separated by semi‐preparative HPLC (ACN−H2O, 38:62, 2.5 ml min−1) to afford 5 (7.8 mg, t R = 13.1 min) and 6 (5.8 mg, t R = 13.8 min). Fr. 1.4 (340 mg) was separated by semi‐preparative HPLC (ACN−H2O, 45:55, 2.5 ml min−1) to yield 2 (12.7 mg, t R = 13.9 min), 3 (5.9 mg, t R = 14.4 min) and 7 (6.3 mg, t R = 14.7 min).
Marfey’s analysis of the amino acid constituents of new compounds
A 300–400 µg sample of compound was hydrolysed in 6N HCl at 60°C for 24 h. The acid hydrolysates of 1–7 were redissolved in H2O (50 μl), and then, 0.25 μM L‐FDAA in 100 μl of acetone was added, followed by 1 N NaHCO3 (25 μl). The mixtures were heated for 1 h at 40°C. After cooling to room temperature, the reaction was quenched by the addition 2 N HCl (25 μl). Finally, the resulting solution was filtered through a small 2.5 μm filter and analysed by LC‐MS using Acclaim™ RSLC 120 C18 column (2.1 × 100 mm 2.2 μm) with a linear gradient of ACN and 0.1% (v/v) aqueous formic acid with different elution conditions (5%–95% (v/v) ACN in 15 min (3‐OH‐Leu), 5%–55% (v/v) ACN at a flow rate of 0.3 ml min−1 and UV detection at 330 nm. Amino acid standards were derivatized with L‐FDAA in a similar manner. Each chromatographic peak was identified by comparing its retention times and molecular weight for the l‐FDAA derivatives of the l‐ and d‐amino acid standards (Fujii et al., 1997a, 1997b,1997a, 1997b).
Bioactivity assay
Cell culture
Murine macrophage RAW264.7 cells were purchased from the Shanghai Institutes for Biological Science (SIBS, Shanghai, China). The cells were cultured in DMEM (Hyclone, Waltham, MA, USA) with 100 units ml−1 streptomycin, 100 units ml−1 penicillin (Gibco, Waltham, MA, USA) and 10% (v/v) foetal bovine serum (Livning, Beijing, China) at 37°C in a humidified environment with 5% (v/v) CO2 in cell incubator.
MTT assay
In vitro cytotoxicity was determined by the MTT assay. Murine macrophage RAW264.7 cells were seeded in 96‐well plates at a density of 5 × 103 cells well−1. After 24 h of culture, compounds 1–7 were added at various concentrations (0.375, 0.75, 1.5, 3, 6 or 12 μmol l−1). Then, cells were treated with 10 μl MTT (5 g l−1; Sigma, Darmstadt, German) for 4 h, and the medium was replaced by 150 μl DMSO (Sigma, Darmstadt, German). The absorbance was determined at 570 nm using a VERSA max microplate reader (Molecular Devices, San Jose, CA, USA). Cells viability was calculated from the percentage relative to the absorbance of control group.
Measurement of NO production
RAW 264.7 cells were seeded in 96‐well plates for 24 h. Then, cells were treated with compounds (0–20 μM) for 30 min, followed by co‐treatment with LPS (10 μg ml−1) for another 24 h. The Nitric Oxide assay kit with Griess reagents (Beyotime, Lot: S0021, Shanghai, China) was used to examine cellular supernatant nitrite accumulation, which represents cellular NO levels. The OD value of 540 nm absorbance was detected with a microplate reader at 540 nm.
Measurement of intracellular ROS level
The ROS level was detected using the DCFH‐DA assay, strictly followed the guidance (Beyotime, Lot: S0033, Shanghai, China). Briefly, cells were cultured in 6‐well plates. After the appropriate treatments, 10 μM DCFH‐DA was added and the cells were incubated for 20 min at 37°C in the dark. After 20 min, cells were washed three times to remove unloaded probe. A FACS Calibur flow cytometer (Becton, Dickinson and Company, New York, NJ, USA) was used to immediately detect fluorescence via the FL1‐H channel. Results were calculated assuming that control absorbance was 1.0.
Statistical analysis
All experiments were performed at least thrice. Statistical analysis was performed with ANOVA (spss 17.0, Chicago, IL, USA) followed by Tukey’s t‐test. A P‐values < 0.05 were considered to be statistically significant.
Conflict of interest
The authors declare that there are no conflict of interests.
Supporting information
Table S1. Taxonomy of Burkholderia sensu lato (s.l.) and representative strains.
Table S2. Strains, plasmids and mutants in this work.
Table S3. Oligonucleotides.
Table S4. Recombinase‐exonuclease pairs in Burkholderia species.
Table S5. Antibiotic concentrations used in different strains.
Table S6. Putative structures of lipopeptides deduced from biosynthesis gene clusters among Burkholderia species.
Table S7. Physical data of 1.
Table S8. The 1H (600 MHz) and DEPTQ (150 MHz) data of 1 in DMSO‐d6.
Table S9. Physical data of 2.
Table S10. The 1H (600 MHz) and 13C NMR (150 MHz) data of 2 in DMSO‐d6.
Table S11. Physical data of 3.
Table S12. The 1H (600 MHz) and DEPTQ (150 MHz) data of 3 in DMSO‐d6.
Table S13. Retention times of amino acids derivatized with Marfey’s reagent (L‐FDAA).
Table S14. Cytotoxicity effect of compounds 1–7.
Fig. S1. Construction of recombinase expression plasmids.
Fig. S2. PCR verification of insertion of apramycin resistance gene (apra) before the gene BGL_RS05915 in B. glumae PG1.
Fig. S3. Optimization of recombination efficiency of ETh1h2eYI23 in B. glumae PG1 for genome modification.
Fig. S4. Diagram for verification and metabolic analysis of BGC4 activation and inactivation in B. plantarii DSM9509.
Fig. S5. Diagram for construction, verification and metabolic analysis of BGC9 activation and inactivation in DSM9512.
Fig. S6. Diagram for construction, verification and metabolic analysis of BGC11 activation and inactivation in DSM9509.
Fig. S7. Key 1H‐1H COSY, HMBC and NOESY correlations of 1.
Fig. S8. 1H NMR spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S9. DEPTQ spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S10. DEPT135 spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S11. DEPT90 spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S12. HSQC spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S13. 1H‐1H COSY spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S14. HMBC spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S15. NOESY spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S16. HRESIMS spectrum of 1.
Fig. S17. MS/MS fragmentation analysis and spectrum of 1.
Fig. S18. IR spectrum of 1.
Fig. S19. UV spectra of 1.
Fig. S20. Key 1H‐1H COSY, HMBC and NOESY correlations of 2.
Fig. S21. 1H NMR spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S22. 13C NMR spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S23. DEPTQ spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S24. DEPT135 spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S25. DEPT90 spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S26. HSQC spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S27. 1H‐1H COSY spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S28. HMBC spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S29. NOESY spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S30. HRESIMS spectrum of 2.
Fig. S31. MS/MS fragmentation analysis and spectrum of 2.
Fig. S32. IR spectrum of 2.
Fig. S33. UV spectra of 2.
Fig. S34. Key 1H‐1H COSY, HMBC and NOESY correlations of 3.
Fig. S35. 1H NMR spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S36. DEPTQ spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S37. DEPT135 spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S38. DEPT90 spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S39. HSQC spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S40. 1H‐1H COSY spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S41. HMBC spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S42. NOESY spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S43. HRESIMS spectrum of 3.
Fig. S44. MS/MS fragmentation analysis and spectrum of 3.
Fig. S45. IR spectrum of 3.
Fig. S46. UV spectra of 3.
Fig. S47. Effect of compounds 1–7 on LPS‐induced ROS production.
Acknowledgements
We are grateful for the support from the National Key R&D Programme of China (2018YFA0900400, 2019YFA0904000, 2019YFA0905700); the National Natural Science Foundation of China (31170050, 31670097, 31970119); the 111 Project (B16030); the Shandong Provincial Natural Science Foundation of China (ZR2020MC015, ZR2018ZC2261, ZR2017MC031); the Taishan Scholar Programme of Shandong Province; the Fundamental Research Funds of Shandong University (2018GN021); and the Open Project Programme of the State Key Laboratory of Bio‐based Material and Green Papermaking (KF201825). We thank Zhifeng Li, Jing Zhu, and Qu Jingyao from the Analysis & Testing Center of SKLMT (State Key laboratory of Microbial Technology, Shandong University) for assistance with HRESIMS.
Microb. Biotechnol. (2021) 14(4), 1809–1826
Funding information
We are grateful for the support from the National Key R&D Programme of China (2018YFA0900400, 2019YFA0904000, 2019YFA0905700); the National Natural Science Foundation of China (31170050, 31670097, 31970119); the 111 Project (B16030); the Shandong Provincial Natural Science Foundation of China (ZR2020MC015, ZR2018ZC2261, ZR2017MC031); the Taishan Scholar Programme of Shandong Province; the Fundamental Research Funds of Shandong University (2018GN021); and the Open Project Programme of the State Key Laboratory of Bio‐based Material and Green Papermaking (KF201825).
Contributor Information
Youming Zhang, Email: zhangyouming@sdu.edu.cn.
Aiying Li, Email: aiyl@sdu.edu.cn.
Jun Fu, Email: fujun@sdu.edu.cn.
References
- Abbasi, M.N. , Fu, J. , Bian, X. , Wang, H. , Zhang, Y. , Li, A. , et al. (2020) Recombineering for genetic engineering of natural product biosynthetic pathways. Trends Biotechnol 38: 715–728. [DOI] [PubMed] [Google Scholar]
- Alley, M.C. , Scudiero, D.A. , Monks, A. , Hursey, M.L. , Czerwinski, M.J. , Fine, D.L. , et al. (1988) Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 48: 589–601. [PubMed] [Google Scholar]
- Altschul, S.F. , Madden, T.L. , Schaffer, A.A. , Zhang, J. , Zhang, Z. , Miller, W. , et al. (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine, R. , and Locht, C. (1992) Isolation and molecular characterization of a novel broad‐host‐range plasmid from Bordetella bronchiseptica with sequence similarities to plasmids from gram‐positive organisms. Mol Microbiol 6: 1785–1799. [DOI] [PubMed] [Google Scholar]
- Beloin, C. , Deighan, P. , Doyle, M. , and Dorman, C.J. (2003) Shigella flexneri 2a strain 2457T expresses three members of the H‐NS‐like protein family: characterization of the Sfh protein. Mol Genet Genomics 270: 66–77. [DOI] [PubMed] [Google Scholar]
- Biggins, J.B. , Gleber, C.D. , and Brady, S.F. (2011) Acyldepsipeptide HDAC inhibitor production induced in Burkholderia thailandensis . Org Lett 13: 1536–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunny, K. , Liu, J. , and Roth, J. (2002) Phenotypes of lexA mutations in Salmonella enterica: evidence for a lethal lexA null phenotype due to the Fels‐2 prophage. J Bacteriol 184: 6235–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, D.M. , and Radding, C.M. (1971) The role of exonuclease and beta protein of phage lambda in genetic recombination. II. Substrate specificity and the mode of action of lambda exonuclease. J Biol Chem 246: 2502–2512. [PubMed] [Google Scholar]
- Coenye, T. , and Vandamme, P. (2003) Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ Microbiol 5: 719–729. [DOI] [PubMed] [Google Scholar]
- Compant, S. , Nowak, J. , Coenye, T. , Clément, C. , and Barka, E.A. (2008) Diversity and occurrence of Burkholderia spp. in the natural environment. FEMS Microbiol Rev 32: 607–626. [DOI] [PubMed] [Google Scholar]
- Crabb, S.J. , Howell, M. , Rogers, H. , Ishfaq, M. , Yurek‐George, A. , Carey, K. , et al. (2008) Characterisation of the in vitro activity of the depsipeptide histone deacetylase inhibitor spiruchostatin A. Biochem Pharmacol 76: 463–475. [DOI] [PubMed] [Google Scholar]
- Depoorter, E. , Bull, M.J. , Peeters, C. , Coenye, T. , Vandamme, P. , and Mahenthiralingam, E. (2016) Burkholderia: an update on taxonomy and biotechnological potential as antibiotic producers. Appl Microbiol Biotechnol 100: 5215–5229. [DOI] [PubMed] [Google Scholar]
- Draghi, W.O. , Peeters, C. , Cnockaert, M. , Snauwaert, C. , Wall, L.G. , Zorreguieta, A. , et al. (2014) Burkholderia cordobensis sp. nov., from agricultural soils. Int J Syst Evol Microbiol 64: 2003–2008. [DOI] [PubMed] [Google Scholar]
- Egan, M. , Ramirez, J. , Xander, C. , Upreti, C. , and Bhatt, S. (2016) Lambda red‐mediated recombineering in the attaching and effacing pathogen Escherichia albertii . Biol Proced Online 18: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan, S.M. , and Schleif, R.F. (1993) A regulatory cascade in the induction of rhaBAD. J Mol Biol 234: 87–98. [DOI] [PubMed] [Google Scholar]
- Egan, S.M. , and Schleif, R.F. (1994) DNA‐dependent renaturation of an insoluble DNA binding protein. Identification of the RhaS binding site at rhaBAD. J Mol Biol 243: 821–829. [DOI] [PubMed] [Google Scholar]
- Esmaeel, Q. , Pupin, M. , Kieu, N.P. , Chataigne, G. , Bechet, M. , Deravel, J. , et al. (2016) Burkholderia genome mining for nonribosomal peptide synthetases reveals a great potential for novel siderophores and lipopeptides synthesis. MicrobiologyOpen 5: 512–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmaeel, Q. , Pupin, M. , Jacques, P. , and Leclere, V. (2018) Nonribosomal peptides and polyketides of Burkholderia: new compounds potentially implicated in biocontrol and pharmaceuticals. Environ Sci Pollut Res Int 25: 29794–29807. [DOI] [PubMed] [Google Scholar]
- Estrada‐de los Santos, P. , Palmer, M. , Chávez‐Ramírez, B. , Beukes, C. , Steenkamp, E.T. , Briscoe, L. , et al. (2018) Whole genome analyses suggests that Burkholderia sensu lato contains two additional novel genera (Mycetohabitans gen. nov., and Trinickia gen. nov.): implications for the evolution of diazotrophy and nodulation in the Burkholderiaceae . Genes 9: 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, J. , Bian, X. , Hu, S. , Wang, H. , Huang, F. , Seibert, P.M. , et al. (2012) Full‐length RecE enhances linear‐linear homologous recombination and facilitates direct cloning for bioprospecting. Nat Biotechnol 30: 440–446. [DOI] [PubMed] [Google Scholar]
- Fu, J. , Teucher, M. , Anastassiadis, K. , Skarnes, W. , and Stewart, A.F. (2010) A recombineering pipeline to make conditional targeting constructs. Methods Enzymol 477: 125–144. [DOI] [PubMed] [Google Scholar]
- Fujii, K. , Ikai, Y. , Oka, H. , Suzuki, M. , and Harada, K. (1997a) A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: combination of Marfey's method with mass spectrometry and its practical application. Contraception 84: 549–557. [Google Scholar]
- Fujii, K. , Ikai, Y. , Mayumi, T. , Oka, H. , Suzuki, M. , and Harada, K. (1997b) A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Elucidation of limitations of Marfey's method and of its separation mechanism. Anal Chem 69: 3346–3352. [Google Scholar]
- Hammer, P.E. , Burd, W. , Hill, D.S. , Ligon, J.M. , and van Pee, K. (1999) Conservation of the pyrrolnitrin biosynthetic gene cluster among six pyrrolnitrin‐producing strains. FEMS Microbiol Lett 180: 39–44. [DOI] [PubMed] [Google Scholar]
- Hu, S. , Fu, J. , Huang, F. , Ding, X. , Stewart, A.F. , Xia, L. , and Zhang, Y. (2014) Genome engineering of Agrobacterium tumefaciens using the lambda Red recombination system. Appl Microbiol Biotechnol 98: 2165–2172. [DOI] [PubMed] [Google Scholar]
- Jia, B. , Yang, J.K. , Liu, W.S. , Li, X. , and Yan, Y.J. (2010) Homologous overexpression of a lipase from Burkholderia cepacia using the lambda Red recombinase system. Biotechnol Lett 32: 521–526. [DOI] [PubMed] [Google Scholar]
- Kang, Y. , Norris, M.H. , Wilcox, B.A. , Tuanyok, A. , Keim, P.S. , and Hoang, T.T. (2011) Knockout and pullout recombineering for naturally transformable Burkholderia thailandensis and Burkholderia pseudomallei . Nat Protoc 6: 1085–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakousis, G. , Ye, N. , Li, Z. , Chiu, S.K. , Reddy, G. , and Radding, C.M. (1998) The beta protein of phage lambda binds preferentially to an intermediate in DNA renaturation. J Mol Biol 276: 721–731. [DOI] [PubMed] [Google Scholar]
- Knappe, T.A. , Linne, U. , Robbel, L. , and Marahiel, M.A. (2009) Insights into the biosynthesis and stability of the lasso peptide capistruin. Chem Biol 16: 1290–1298. [DOI] [PubMed] [Google Scholar]
- Kolodner, R. , Hall, S.D. , and Luisi‐DeLuca, C. (1994) Homologous pairing proteins encoded by the Escherichia coli recE and recT genes. Mol Microbiol 11: 23–30. [DOI] [PubMed] [Google Scholar]
- Kunakom, S. , and Eustáquio, A.S. (2019) Burkholderia as a source of natural products. J Nat Prod 82: 2018–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, Y. , Suh, J.W. , Kim, S. , Hyun, B. , Kim, C. , and Lee, C.H. (1994) Cepacidine A, a novel antifungal antibiotic produced by Pseudomonas cepacia. II. Physico‐chemical properties and structure elucidation. J Antibiot (Tokyo) 47: 1406–1416. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Wang, C. , and Cheng, Y. (2012) FK228 from Burkholderia thailandensis MSMB43. Acta Crystallogr Sect E Struct Rep Online 68: o2757–o2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moebius, N. , Ross, C. , Scherlach, K. , Rohm, B. , Roth, M. , and Hertweck, C. (2012) Biosynthesis of the respiratory toxin bongkrekic acid in the pathogenic bacterium Burkholderia gladioli . Chem Biol 19: 1164–1174. [DOI] [PubMed] [Google Scholar]
- Montiel, D. , Kang, H.S. , Chang, F.Y. , Charlop‐Powers, Z. , and Brady, S.F. (2015) Yeast homologous recombination‐based promoter engineering for the activation of silent natural product biosynthetic gene clusters. Proc Natl Acad Sci USA 112: 8953–8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myronovskyi, M. , and Luzhetskyy, A. (2016) Native and engineered promoters in natural product discovery. Nat Prod Rep 33: 1006–1019. [DOI] [PubMed] [Google Scholar]
- Newman, D.J. , and Cragg, G.M. (2012) Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75: 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, T.A. , Ishida, K. , Jenke‐Kodama, H. , Dittmann, E. , Gurgui, C. , Hochmuth, T. , et al. (2008) Exploiting the mosaic structure of trans‐acyltransferase polyketide synthases for natural product discovery and pathway dissection. Nat Biotechnol 26: 225–233. [DOI] [PubMed] [Google Scholar]
- Rausch, C. , Hoof, I. , Weber, T. , Wohlleben, W. , and Huson, D.H. (2007) Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol Biol 7: 78–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, H. , Wang, B. , and Zhao, H. (2017) Breaking the silence: new strategies for discovering novel natural products. Curr Opin Biotechnol 48: 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan, S.K. , Thomason, L.C. , Kuznetsov, S.G. , and Court, D.L. (2009) Recombineering: a homologous recombination‐based method of genetic engineering. Nat Protoc 4: 206–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X. , Jiang, C. , Ma, L. , Zhao, X. , Chang, J. , Zheng, B. , et al. (2015) 3β‐Angeloyloxy‐8β,10β‐dihydroxyeremophila‐7(11)‐en‐12,8α‐lactone inhibits lipopolysaccharide‐induced nitric oxide production in RAW264.7 Cells. Biol Pharma Bull 38: 836–843. [DOI] [PubMed] [Google Scholar]
- Swingle, B. , Bao, Z. , Markel, E. , Chambers, A. , and Cartinhour, S. (2010) Recombineering using RecTE from Pseudomonas syringae . Appl Environ Microbiol 76: 4960–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, B. , Yu, Y. , Liang, J. , Zhang, Y. , Bian, X. , Zhi, X. , and Ding, X. (2019) Reclassification of ‘Polyangium brachysporum’ DSM 7029 as Schlegelella brevitalea sp. nov. Int J Syst Evol Microbiol 69: 2877–2883. [DOI] [PubMed] [Google Scholar]
- Taylor, A. , and Smith, G.R. (1980) Unwinding and rewinding of DNA by the RecBC enzyme. Cell 22: 447–457. [DOI] [PubMed] [Google Scholar]
- Thongkongkaew, T. , Ding, W. , Bratovanov, E. , Oueis, E. , Garcia‐Altares, M. , Zaburannyi, N. , et al. (2018) Two types of threonine‐tagged lipopeptides synergize in host colonization by pathogenic Burkholderia species. ACS Chem Biol 13: 1370–1379. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Bian, X. , Xia, L. , Ding, X. , Müller, R. , Zhang, Y. , et al. (2014) Improved seamless mutagenesis by recombineering using ccdB for counterselection. Nucleic Acids Res 42: e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Li, Z. , Jia, R. , Hou, Y. , Yin, J. , Bian, X. , et al. (2016) RecET direct cloning and Redalphabeta recombineering of biosynthetic gene clusters, large operons or single genes for heterologous expression. Nat Protoc 11: 1175–1190. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Li, Z. , Jia, R. , Yin, J. , Li, A. , Xia, L. , et al. (2018a) ExoCET: exonuclease in vitro assembly combined with RecET recombination for highly efficient direct DNA cloning from complex genomes. Nucleic Acids Res 46: 2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Zhou, H. , Chen, H. , Jing, X. , Zheng, W. , Li, R. , et al. (2018b) Discovery of recombinases enables genome mining of cryptic biosynthetic gene clusters in Burkholderiales species. Proc Natl Acad Sci USA 115: E4255–e4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, D. , Wang, M. , Shi, J. , and Hao, J. (2012) Red recombinase assisted gene replacement in Klebsiella pneumoniae . J Ind Microbiol Biotechnol 39: 1219–1226. [DOI] [PubMed] [Google Scholar]
- Yin, J. , Zheng, W. , Gao, Y. , Jiang, C. , Shi, H. , Diao, X. , et al. (2019) Single‐stranded DNA‐binding protein and exogenous RecBCD inhibitors enhance phage‐derived homologous recombination in Pseudomonas. iScience 14: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, J. , Zhu, H. , Xia, L. , Ding, X. , Hoffmann, T. , Hoffmann, M. , et al. (2015) A new recombineering system for Photorhabdus and Xenorhabdus . Nucleic Acids Res 43: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura, A. , Covington, B.C. , Gallant, E. , Zhang, C. , Li, A. , and Seyedsayamdost, M.R. (2020) Unlocking cryptic metabolites with mass spectrometry‐guided transposon mutant selection. ACS Chem Biol 15: 2766–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M.M. , Wong, F.T. , Wang, Y. , Luo, S. , Lim, Y.H. , Heng, E. , et al. (2017) CRISPR‐Cas9 strategy for activation of silent Streptomyces biosynthetic gene clusters. Nat Chem Biol 13: 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Buchholz, F. , Muyrers, J.P. , and Stewart, A.F. (1998) A new logic for DNA engineering using recombination in Escherichia coli . Nat Genet 20: 123–128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Taxonomy of Burkholderia sensu lato (s.l.) and representative strains.
Table S2. Strains, plasmids and mutants in this work.
Table S3. Oligonucleotides.
Table S4. Recombinase‐exonuclease pairs in Burkholderia species.
Table S5. Antibiotic concentrations used in different strains.
Table S6. Putative structures of lipopeptides deduced from biosynthesis gene clusters among Burkholderia species.
Table S7. Physical data of 1.
Table S8. The 1H (600 MHz) and DEPTQ (150 MHz) data of 1 in DMSO‐d6.
Table S9. Physical data of 2.
Table S10. The 1H (600 MHz) and 13C NMR (150 MHz) data of 2 in DMSO‐d6.
Table S11. Physical data of 3.
Table S12. The 1H (600 MHz) and DEPTQ (150 MHz) data of 3 in DMSO‐d6.
Table S13. Retention times of amino acids derivatized with Marfey’s reagent (L‐FDAA).
Table S14. Cytotoxicity effect of compounds 1–7.
Fig. S1. Construction of recombinase expression plasmids.
Fig. S2. PCR verification of insertion of apramycin resistance gene (apra) before the gene BGL_RS05915 in B. glumae PG1.
Fig. S3. Optimization of recombination efficiency of ETh1h2eYI23 in B. glumae PG1 for genome modification.
Fig. S4. Diagram for verification and metabolic analysis of BGC4 activation and inactivation in B. plantarii DSM9509.
Fig. S5. Diagram for construction, verification and metabolic analysis of BGC9 activation and inactivation in DSM9512.
Fig. S6. Diagram for construction, verification and metabolic analysis of BGC11 activation and inactivation in DSM9509.
Fig. S7. Key 1H‐1H COSY, HMBC and NOESY correlations of 1.
Fig. S8. 1H NMR spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S9. DEPTQ spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S10. DEPT135 spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S11. DEPT90 spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S12. HSQC spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S13. 1H‐1H COSY spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S14. HMBC spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S15. NOESY spectrum (600 MHz) of 1 in DMSO‐d6.
Fig. S16. HRESIMS spectrum of 1.
Fig. S17. MS/MS fragmentation analysis and spectrum of 1.
Fig. S18. IR spectrum of 1.
Fig. S19. UV spectra of 1.
Fig. S20. Key 1H‐1H COSY, HMBC and NOESY correlations of 2.
Fig. S21. 1H NMR spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S22. 13C NMR spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S23. DEPTQ spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S24. DEPT135 spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S25. DEPT90 spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S26. HSQC spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S27. 1H‐1H COSY spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S28. HMBC spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S29. NOESY spectrum (600 MHz) of 2 in DMSO‐d6.
Fig. S30. HRESIMS spectrum of 2.
Fig. S31. MS/MS fragmentation analysis and spectrum of 2.
Fig. S32. IR spectrum of 2.
Fig. S33. UV spectra of 2.
Fig. S34. Key 1H‐1H COSY, HMBC and NOESY correlations of 3.
Fig. S35. 1H NMR spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S36. DEPTQ spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S37. DEPT135 spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S38. DEPT90 spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S39. HSQC spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S40. 1H‐1H COSY spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S41. HMBC spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S42. NOESY spectrum (600 MHz) of 3 in DMSO‐d6.
Fig. S43. HRESIMS spectrum of 3.
Fig. S44. MS/MS fragmentation analysis and spectrum of 3.
Fig. S45. IR spectrum of 3.
Fig. S46. UV spectra of 3.
Fig. S47. Effect of compounds 1–7 on LPS‐induced ROS production.