

Abstract
Rationale:
Patients with and without cardiovascular diseases have been shown to be at risk of influenza-mediated cardiac complications. Recent clinical reports support the notion of a direct link between laboratory-confirmed influenza virus infections and adverse cardiac events.
Objective:
Define the molecular mechanisms underlying influenza virus-induced cardiac pathogenesis after resolution of pulmonary infection and the role of necroptosis in this process.
Methods and Results:
Hearts from wild-type and necroptosis deficient (MLKL-KO) mice were dissected twelve days after initial Influenza A virus (IAV) infection when viral titers were undetectable in the lungs. Immunofluorescence microscopy and plaque assays showed presence of viable IAV particles in the myocardium without generation of interferon responses. Global proteome and phosphoproteome analyses using high resolution accurate mass based LC-MS/MS and label-free quantitation showed that the global proteome as well as the phosphoproteome profiles were significantly altered in IAV-infected mouse hearts in a strain independent manner. Necroptosis deficient mice had increased survival and reduced weight loss post-IAV infection, as well as increased antioxidant and mitochondrial function, indicating partial protection to IAV infection. These findings were confirmed in vitro by pre-treatment of human and rat myocytes with antioxidants or necroptosis inhibitors, which blunted oxidative stress and mitochondrial damage after IAV infection.
Conclusions:
This study provides the first evidence that the cardiac proteome and phosphoproteome are significantly altered post pulmonary influenza infection. Moreover, viral particles can persist in the heart after lung clearance, altering mitochondrial function and promoting cell death without active replication and interferon responses. Finally, our findings show inhibition of necroptosis or prevention of mitochondrial damage as possible therapeutic interventions to reduce cardiac damage during influenza infections.
Keywords: Necroptosis, influenza A, mitochondria, oxidative stress, proteomics, cell death, phosphoproteomics, cardiac metabolism, Animal Models of Human Disease, Cardiotoxicity, Cell Signaling/Signal Transduction, Oxidant Stress
Graphical Abstract
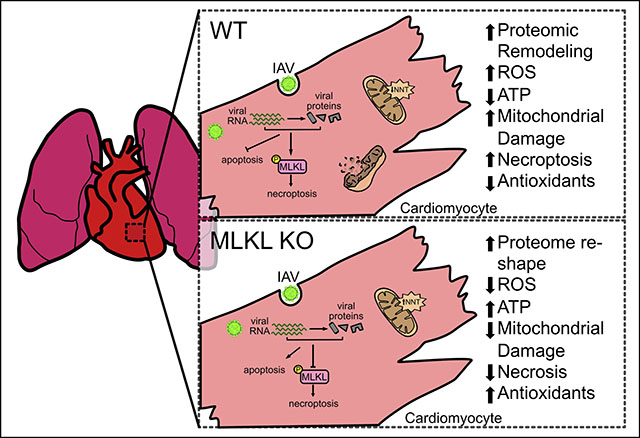
INTRODUCTION
The top three global causes of death worldwide are heart disease, cancer and respiratory tract (RT) infections1, 2. RT infections are commonly caused by influenza virus, a class of enveloped RNA viruses that trigger an acute respiratory illness occurring in local outbreaks, seasonal epidemics and/or pandemics3, 4. In the United States alone, influenza infections lead to over 500,000 hospitalizations, and over 36,000 deaths each year5, 6. A key factor contributing to the high mortality rate are major adverse cardiac events (MACE) that occur during hospitalization or convalescence, which are more likely to arise in patients with pre-existing cardiac conditions7. Clinical studies have shown that elderly patients admitted to the hospital for severe viral and/or bacterial pneumonia experience 10–25% mortality rates; of these, up to 40% perish within one year8, and up to 20% experience MACE that can last for up to 10 years after discharge9. Accumulating evidence has tightened the association between MACE and infectious diseases including influenza, pneumococcal pneumonia, or bacteremia9–12. The majority of the clinical and basic research regarding MACE and pneumonia have studied community acquired pneumonia caused by the Gram-positive bacterium Streptococcus pneumoniae13, but not respiratory infections from seasonal, epidemic, or pandemic influenza viruses10. Therefore, it is essential to untangle the potential molecular links between viral pulmonary infections and alterations to cardiac biology, as illustrated by the two most recent pandemics caused by H1N1 influenza (2009) and SARS-CoV-2 (2019), when many patients experienced adverse cardiac events such as cardiac infarct, myocarditis, ischemic injury and stroke10, 14.
While the molecular and epidemiological effects of influenza infection in pulmonary tissue are well-documented, the molecular effects of influenza in other organs remains unclear15, 16. Influenza virus has been shown to be cardiotropic in multiple models of disease17–25, with virus disseminating from the lungs to cardiac tissue, especially during severe infections, to cause myocarditis, ischemia, and cardiac dysfunction18–21, 24. Fatalities from typical seasonal influenza have shown myocarditis in up to 48% of all autopsies22–25, and studies from the 1918 H1N1 influenza pandemic showed severe cardiac damage in 90% of fatalities17. The 1918 influenza strain was later used to infect macaques, and viral particles disseminated from the lungs to the heart after acute respiratory distress and persisted in cardiac tissue up to 8 days post infection19, the last time point collected. This suggests that pandemic influenza strains may lead to more exacerbated, prolonged cardiac pathology17, 19, 24, 25. In rodent models, both lab-adapted influenza H1N1 strains like A/Puerto Rico/8/1934 (PR8) and pandemic strains A/California/7/2009 and A/England/195/2009 induce clinical signs (weight loss) as well as histological and molecular markers of pulmonary disease (inflammation and cell death)26–28. In this study, we utilize the mouse adapted A/California/7/2009 strain as a prototype pandemic strain and PR8 as a point of comparison. While cardiac tropism by influenza viruses has been observed for over a century, the underlying molecular mechanisms that promote cardiac pathologies remain undefined.
Cell death is a key step in host response to viral and bacterial pathogens29. The ability of cells to undergo programmed cell death has extensive influence on recovery. One major programmed cell death pathway with direct implications in host responses to viral pathogens is programmed necrosis, i.e. necroptosis30. Necroptosis is a highly inflammatory form of cell death regulated by the receptor-interacting serine/threonine-protein kinases (RIPK) 1 and 3, and the effector molecule mixed lineage kinase domain-like protein (MLKL). Importantly, necroptosis (especially the activity of RIPK3) evolutionarily promotes host survival during viral infections, in contrast to its detrimental role during severe bacterial infection31, 32. During influenza A virus (IAV) infections, the virus utilizes self-encoded proteins to block canonical apoptosis. Host cells then attempt to halt viral replication by expressing RIPK3 to initiate necroptosis and/or re-initiate apoptosis15. This suggests that inhibition of the necroptosis effector molecule MLKL may promote apoptotic activity, which may in turn enhance host antiviral responses. To date, necroptosis has not been implicated in cardiac responses to influenza infection.
Here, to test if influenza virus infection has a prolonged effect on cardiac health during convalescence, we employed a mouse model of IAV infection. We combined proteomic, phosphoproteomic, and molecular techniques to describe the molecular changes in the heart after clearance of pulmonary influenza infection. Finally, we used mice deficient in necroptosis to investigate the role of this pathway in cardiac tissue alterations after pulmonary resolution of influenza infection.
METHODS
Data Availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request. All data from the proteomic analyses have been made publicly available at ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD017936, PXD016412, and PXD015992.
Supplemental Methods and data.
Supplemental figures and detailed experimental procedures are available online.
RESULTS
Influenza infection induces cardiomyocyte necroptosis, oxidative stress and cellular metabolic breakdown without active viral replication.
To determine if myocytes are susceptible to influenza virus infection, we challenged rat H9c2 myocytes and human AC16 cardiomyocytes with pandemic influenza A H1N1 A/California/7/2009 (IAV) or A/Puerto Rico/8/1934 (PR8) and assessed cellular cytotoxicity 48h after infection. We observed increased cellular cytotoxicity (Fig. 1A, Online Figure I. A, B) without active viral replication (Online Figure I. C), suggesting that myocytes could die upon IAV infection while promoting abortive replication33, 34. Necroptosis has been shown to have a crucial role in suppressing viral replication in vitro and in vivo15, 35. 48h after the IAV challenge of rat and human cardiomyocytes, we observed increased immunofluorescent staining of phosphorylated MLKL, the necroptosis effector molecule (Fig. 1B–C, Online Figure I. D), indicating enhanced necroptosis activity in IAV-infected myocytes. Pre-treatment of myocytes with necroptosis inhibitors ponatinib, pazopanib36 or necrosulfonamide37 led to a significant decrease in cell cytotoxicity (Fig. 1D–F). Taken together, necroptosis inhibition can partially rescue IAV-induced myocyte cell death.
Figure 1: Influenza A virus infection leads to programmed necrosis, oxidative stress and metabolic breakdown in cardiomyocytes.
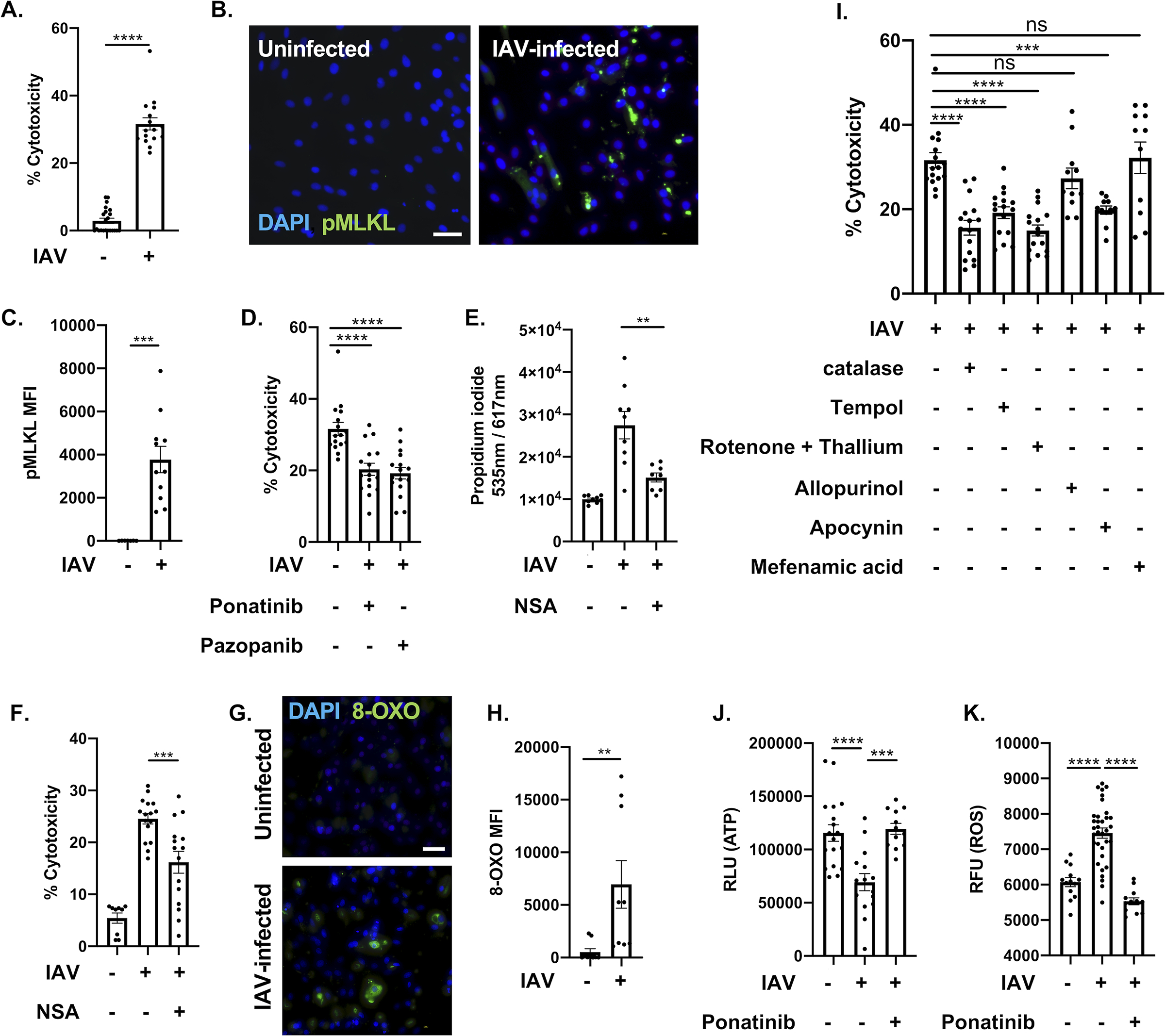
(A) LDH release of H9c2 rat myocytes were infected with an MOI of 0.1 of IAV strain Influenza A virus/California/7/2009 (IAV) for 48-h. (B) Immunofluorescent staining for pMLKL (green) in H9c2 rat myocytes infected with IAV. Cell nucleus was stained in blue. White bar = 50 μm. (C) Mean fluorescence intensity (MFI) of pMLKL. (D) LDH cytotoxicity assay of H9c2 infected with IAV at an MOI of 0.1 for 48-h. Cells were treated with two necroptosis inhibitors: ponatinib and pazopanib (10 μM each) for 1h before infection. (E) Propidium iodide and (F) LDH cytotoxicity assay staining of human AC16 myocytes infected with IAV at an MOI of 0.1 for 48-h. Cells were treated with MLKL inhibitor necrosulfonamide (10 μM each) for 1h before infection. (G) 8-oxoguanine (8-OXO, green) was stained in H9c2 rat myocytes infected with IAV. Cell nucleus was stained in blue. White bar = 50 μm. (H) Mean fluorescence intensity (MFI) of 8-OXO staining. (I) LDH cytotoxicity assay of H9c2 infected with IAV at an MOI of 0.1 for 48-h. Cells were treated with Catalase (10 μM), Tempol (10 μM), Rotenone + Thallium trifluoroacetate (Rot tha, 10 nM/mL/10 nM/mL), Allopurinol (Allu, 10 nM/mL), Apocynin (Apo, 1 μM/mL), Mefenamic acid (Mefe, 20 nM/mL). (J) ATP levels and (K) ROS/superoxide levels of IAV-infected H9c2 myocytes treated with ponatinib (10 μM), compared to mock treated and uninfected H9c2. RLU: relative light unit of luminescence. Mann-Whitney U tests were applied for two-group comparisons. Kruskal-Wallis test with Dunn’s multiple-comparison post-test. Asterisks denote the level of significance observed: * = p ≤ 0.05; ** = p ≤ 0.01; *** = p ≤ 0.001, **** = p ≤ 0.0001.
During pulmonary infection, IAV promotes oxidative stress that leads to tissue damage, enhanced inflammatory responses, and cell death38. To determine if IAV infection in cardiac tissue leads to oxidative stress induced damage, we stained H9c2 myocytes 48h post-IAV infection against 8-Oxoguanine (8-OXO), a marker of DNA damage resulting from reactive oxygen species (ROS)39. We saw a significant increase of 8-OXO staining, indicating elevated ROS-mediated damage (Fig. 1G–H). We then attempted to rescue this by pre-treating myocytes with general antioxidants (catalase or tempol), mitochondria-dependent [rotenone + thallium trifluoroacetate (mitochondria) or apocynin (NADPH)], or mitochondria-independent [allopurinol (xanthine oxidase-dependent) or mefenamic acid (cyclooxygenase-dependent)] antioxidants prior to IAV infection. Pre-treatment with general antioxidants or mitochondria-dependent ROS inhibitors protected myocytes from IAV-induced cell death, but pre-treatment with mitochondria-independent ROS inhibitors had no effect (Fig. 1I), indicating that toxic cellular ROS originated from the mitochondria. IAV-infected H9c2 and AC16 myocytes showed decreased ATP production (Fig. 1J, Online Figure I. E) that could be rescued by necroptosis inhibition (Fig. 1J). We also observed a stark decrease in ROS production in IAV-infected myocytes after necroptosis inhibition (Fig. 1K). Taken together, our data suggest that influenza infection induces necroptosis and promotes mitochondria-dependent oxidative stress.
MLKL deficiency improves antioxidant and mitochondrial activity.
It has been shown that RIPK3 KO mice or MLKL/FADD double-KO mice are more susceptible to IAV infection than wild-type, whereas MLKL KO mice were not susceptible to lethal disease15. Recently, our group showed that inhibition of MLKL can protect against pulmonary injury induced during bacterial infection secondary to IAV38. Together, these reports suggest that RIPK3 is required for an efficient response against influenza and that inhibition of MLKL can be an efficient approach to reduce IAV-induced necrotic cell damage and virus dissemination15, 30, 40. To test if necroptosis inhibition could potentially protect against IAV-induced cardiac damage we analyzed baseline proteomes of uninfected WT (C57BL/6) and MLKL deficient mice (MLKL KO) using High-Resolution Accurate-Mass (HR/AM) LC-MS/MS platform and a label-free quantitation (LFQ)41, 42. 278 proteins were significantly changed between the two groups (Online Figure II. A). Among them, up-regulated proteins in MLKL KO hearts are largely associated with oxidation-reduction, fatty acid/glutathione metabolism, responses to oxidative stress, and general metabolic processes after Gene Ontology (GO) analysis (Online Figure II. B). Global inhibition of MLKL leads to a baseline increase in mitochondrial and antioxidant activity, which suggests that mice lacking MLKL may be protected against the cellular changes induced by IAV infection (Fig. 1).
It has been shown that during S. pneumoniae infection, bacteria can translocate to cardiac tissue and modulate local inflammatory responses13. However, it is not known whether IAV can translocate to heart, or if virus persists in in cardiac tissue after its clearance from the lungs. To test the potential persistence of the virus in the heart, we designed a study in which 6-to-8-week-old WT and MLKL KO mice43 were infected with 250 plaque forming units (PFU) of IAV strain A/California/7/2009 and followed for twelve days (Fig. 2A). Ten days after initial infection, IAV was undetectable in the pulmonary tissue (Online Figure III.), and MLKL KO mice showed increased survival (Fig. 2B) and reduced weight loss (Fig. 2C) during the acute phase of the infection. After 10 days, 76% of WT mice (n = 13) and 92% of MLKL KO mice (n = 12) had recovered from the initial infection (Fig. 2B–C). At day twelve, hearts were collected. Hematoxylin and eosin staining of frozen sections showed some structural changes in cardiac tissue of uninfected vs IAV-infected mice (Online Figure IV. A). Immunofluorescence staining (Online Figure IV. B) and plaque assays (Fig. 2D) showed presence of IAV viral particles in both WT and MLKL KO cardiomyocytes twelve days after initial infection. To test if the presence of IAV drives inflammation in the cardiac tissue during convalescence, we measured the level of inflammation-associated cytokines via enzyme-linked immunosorbent assay (ELISA). No significant changes in TNF-α, IFN-ƛ IFN-Ɣ, IFN-α, IL-1β, IL-10 (Fig. 2F) or IFN-β (below level of detection <5 pg/mL, Online Figure V.) were observed. TNF and IFN responses have been shown to promote necroptosis during viral infection44. We tested whether cytokine production during acute infection may exacerbate IAV-induced tissue damage by pre-treating human AC16 cardiomyocytes with either TNF-α or IFN-β before challenge with IAV. Inhibition of MLKL with necrosulfonamide protected cardiomyocytes after challenge with either cytokine in combination with IAV (Online Figure VI. A–B). Inhibition of TNF receptor 1 or blocking of TNF-α by pre-treatment of cells with R7050 or SPD304, respectively, reduced influenza-induced cell death in vitro (Online Figure VI. C). During acute infection, cardiac damage is exacerbated by TNF production, but cytokine levels return to baseline during convalescence despite persistence of IAV in cardiac tissue.
Figure 2: Influenza virus persists in mice hearts during convalescence without exacerbated inflammatory response.
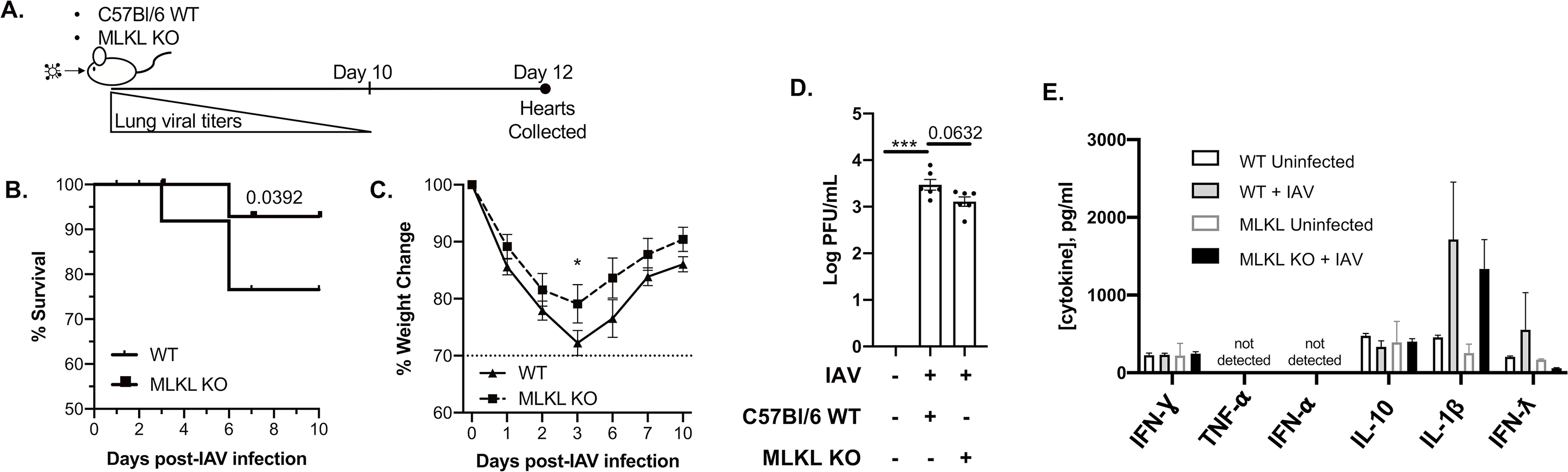
(A) Timeline for mice infection and heart collection. Male and female wild-type and MLKL KO 6-to-8-week-old C57Bl/6 mice were intranasally infected with A/California/7/2009 (IAV) at day 0. Mice were euthanized and heart tissue was collected at day 12 (n = 10–12, 5–6 mice of each sex). (B) survival and (C) percent weight changes up to 10 days post IAV infection. (D) Viral titers were measured for WT C57Bl/6 and MLKL KO mice 12 days post initial IAV infection (n = 6, 3 of each sex) by the plaque assay and plotted in Log PFU/mL. (E) Levels of IFNƔ, TNF⍺, IFN⍺, IL-10, IFN-ƛ and IL-1-β measured by ELISA (pg/mL, n = 3–6, representative data for each sex of mice infected with IAV or challenged with vehicle (PBS) control). Log-rank (Mantel-Cox) test for survival. Kruskal-Wallis test with Dunn’s multiple-comparison post-test. Asterisks denote the level of significance observed: * = p ≤ 0.05; ** = p ≤ 0.01; *** = p ≤ 0.001.
Recently, it has been shown that both apoptosis and necroptosis are present in pulmonary tissue after influenza infection15. Pre-treatment of H9c2 myocytes with pan-caspase inhibitor Z-vad-fmk before IAV infection also protected against cell death (Online Figure VII. A), indicating a requirement for caspase activity in cardiomyocyte death. A stark increase in active caspase-1 and Gasdermin-D was observed in both WT and MLKL KO mice (Online Figure VII. B), suggesting an additional role for caspase activity45 in IAV-induced cardiomyocyte death. Caspase-3 activation was notably increased in MLKL KO hearts after influenza infection (Online Figure VII. C) and in IAV-infected AC16 human cardiomyocytes pretreated with necrosulfonamide (Online Figure VII. D–F), suggesting that blocking necroptosis redirects cells to apoptosis.
Influenza virus infection leads to global proteome remodeling during convalescence.
To gain a comprehensive understanding of the effects of influenza infection on cardiac tissue and how necroptosis influences this process, we investigated the proteome-level changes in cardiac tissue after IAV clearance from the lungs. Hearts from C57BL/6 WT and MLKL KO mice (uninfected or 12 days post IAV infection) were analyzed by label-free quantitative proteomics41, 42. In the global proteome (no filters applied, 1,592 total quantified proteins; Online Table I, Online Figure VIII. and IX), biological replicates within groups correlated tightly (Pearson r = 0.969 ± 0.013, n = 12), suggesting excellent experimental reproducibility (Online Figure VIII. A). Unsupervised hierarchical clustering (Online Figure VIII. B) and principal component analysis (PCA) (Online Figure VIII. C) of the unfiltered proteome showed segregation between experimental groups. Further filtering of the global proteomes yielded 295 proteins that were significantly different in at least one of the groups by ANOVA (permutation-based FDR < 0.01) (Fig. 3A). These proteins clustered into seven main groups based on their abundance profiles among the four conditions (Fig. 3A). GO enrichment analysis of proteins in each cluster revealed oxidation-reduction processes enriched in clusters 2, 4, 5, and 7 and collectively contained 50 mitochondrial proteins (Online Table II), consistent with our observation of altered mitochondrial function in vitro.
Figure 3: Influenza infection induces cardiac proteome remodeling.
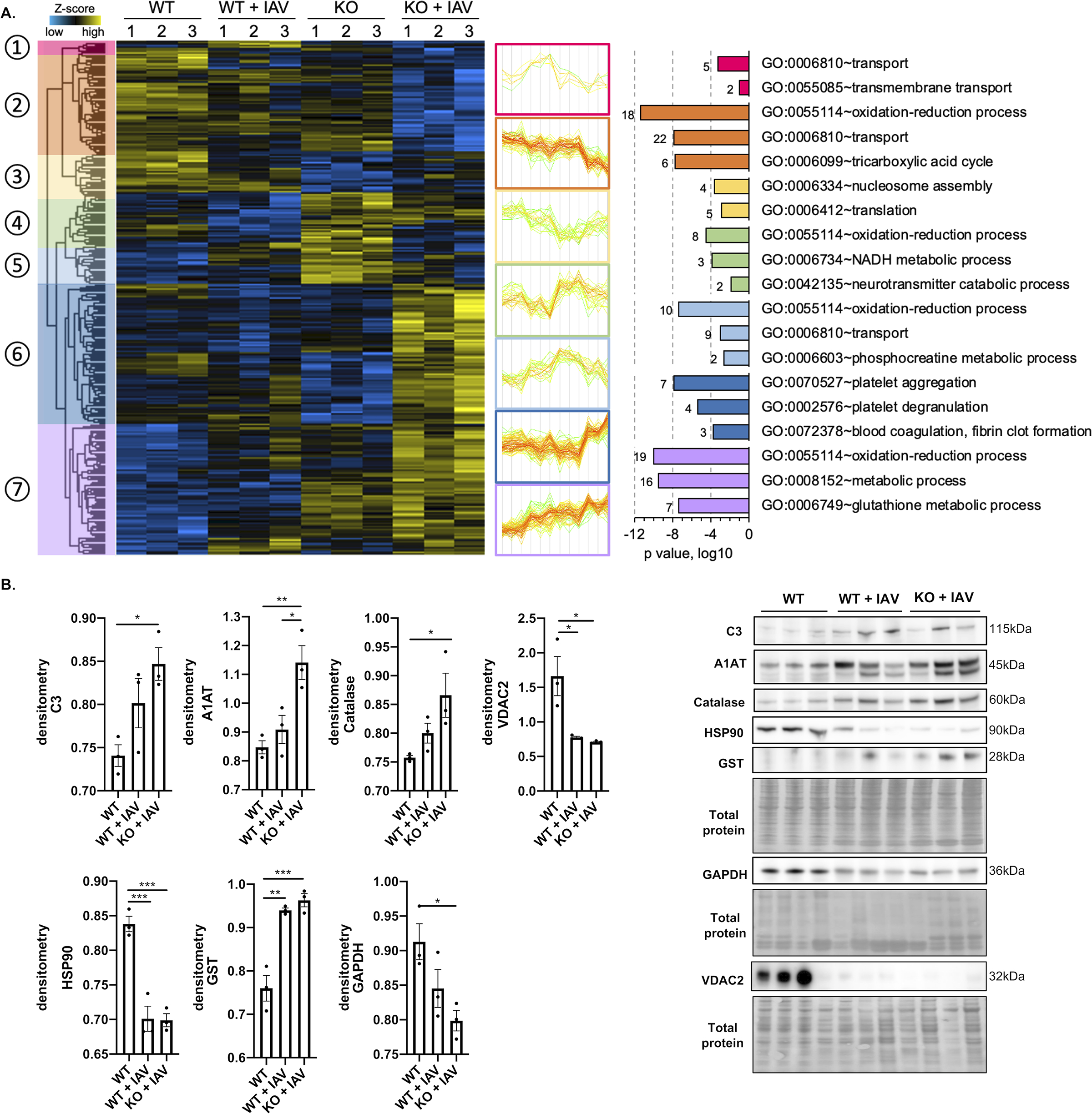
Male and female 6-to-8-week-old wild-type and MLKL KO C57Bl/6 mice were intranasally infected with A/California/7/2009 (IAV) or mock challenged with vehicle (PBS) and hearts excised at day 12 post-infection. (A) Proteomic changes of mice hearts after IAV infection or mock challenge (n = 3 per condition). Hierarchical clustering of LFQ intensities of 295 significantly changed proteins (ANOVA, FDR < 0.01) revealed seven distinct clusters. Their abundance profiles among the groups were plotted in the middle panel. Enriched GO biological process terms are indicated for each marked cluster on the right panel. (B) Immunoblots for complement C3 (C3), AIAT, Catalase, HSP90, GST, GAPDH and VDAC and histograms of protein level quantification (n = 3 per group). Proteomic data of mice is representative from 2 separate experiments done with 3 mice of each sex; no sex based differences were observed. Kruskal-Wallis test with Dunn’s multiple-comparison post-test. Asterisks denote the level of significance observed: * = p ≤ 0.05; ** = p ≤ 0.01; *** = p ≤ 0.001.
The three main clusters with the most total proteins are 2, 6, and 7, which have 59, 80, and 75 proteins, respectively. Proteins in cluster 2 showed a decreased abundance in IAV-infected hearts. Several proteins in this cluster are associated with cardiac muscle contraction, including Troponin I (Tnn3), Tropomyosin-1, and Cysteine-rich protein 3 (Csrp3) (Online Table II–V). Subunits from ATP synthase and members of the mitochondrial electron transport chain were also found in this cluster, indicating broadly decreased energy production in cardiac tissues during IAV infection, consistent with our in vitro observation (Fig. 1I). Moreover, several enzymes involved in the TCA cycle were seen, including subunits or components from Pyruvate dehydrogenase (PdhA), Malate dehydrogenase (Mdh), and Isocitrate dehydrogenase (Idh). Another cluster enriched with metabolic enzymes is cluster 4, which contains a different subunit of Idh, Glyceraldehyde-3-phosphate dehydrogenase (Gapdh), and 2-oxoglutarate dehydrogenase (Ogdh), all of which have similar regulation trends. Cluster 4 also contains Heat shock protein 90 (Hsp90), an important molecular chaperone that promotes cell maturation, cytoskeletal maintenance, and proper regulation of cell cycle control and signal transduction46. Down regulation of Hsp90 in both IAV-infected WT and MLKL KO cardiac tissue suggests altered heart protein homeostasis due to virus infection (Fig. 3A).
Cluster 7 showed an inverse trend of protein abundance change compared to cluster 2, where the IAV-infected hearts have proteins up-regulated, and those in MLKL KO mice are higher than in WT mice (Fig. 3A). This cluster includes proteins that regulate glutathione metabolism, including several glutathione transferases, and proteins with antioxidant activities, such as superoxide dismutase (Sod) and Catalase (Cat). Changes in these proteins may indicate an attempt of cardiac tissue to mitigate oxidative stress caused by influenza infection47. Another interesting group of proteins up-regulated in this cluster were inhibitors of endopeptidase activities, including Serpina1b, Serpina1d, and Serpina3k. Protease activity is required for influenza virus activation48, and an up-regulation of protease inhibitors may be an alternative strategy cardiac tissues undertake to combat influenza invasion. Cluster 6 contains proteins drastically increased only in IAV-infected MLKL KO hearts. This cluster is enriched in proteins that function in platelet aggregation, such as fibrinogens, and innate immune proteins, such as complement C3 and protein S100-A10. Several significantly changed proteins were validated by immunoblot (Fig. 3B). Importantly, we observed that hearts of mice infected with IAV strain PR8 showed similar proteomic (Online Figure X) and GO (Online Figure XI) changes to those infected with strain A/California/7/2009, with both infected tissues differing from uninfected hearts.
The cardiac proteome of IAV-infected MLKL KO mice is reshaped during convalescence.
To investigate the response of cardiac tissue to IAV infection when necroptosis is blocked, we compared the proteomes of IAV-infected WT (WT+IAV) and IAV-infected MLKL KO (KO+IAV) hearts (Fig. 4A). Among the 1,029 proteins quantified in the two groups, 220 of them were significantly different by at least 1.5-fold, with 158 and 62 proteins up- and down-regulated in the KO+IAV hearts, respectively. The protein with highest increase was NAD(P) transhydrogenase (Nnt), an inner mitochondrial membrane protein that maintains the mitochondrial NADPH level and can mediate the oxidative stress defense. Decreased cardiac Nnt has been closely associated with heart failure49. In addition to antioxidants, proteins associated with glutathione metabolism (Glutathione transferase (GST) and Glutathione peroxidase), inflammatory or immune response (Alpha-1-acid glycoprotein 1, Complement C3), and regulation of protease functions (Serpina3k, Serpina1d) were also found to be highly up-regulated in IAV-infected MLKL KO hearts. Interestingly, components in the regulation of proteasomal protein catabolic process and regulation of phagocytosis were also enriched (Fig. 4B), suggesting that blocking necroptosis may re-activate alternative pathways in the heart to fight against IAV.
Figure 4: MLKL deficiency leads to proteome reshape in mice hearts during convalescence.
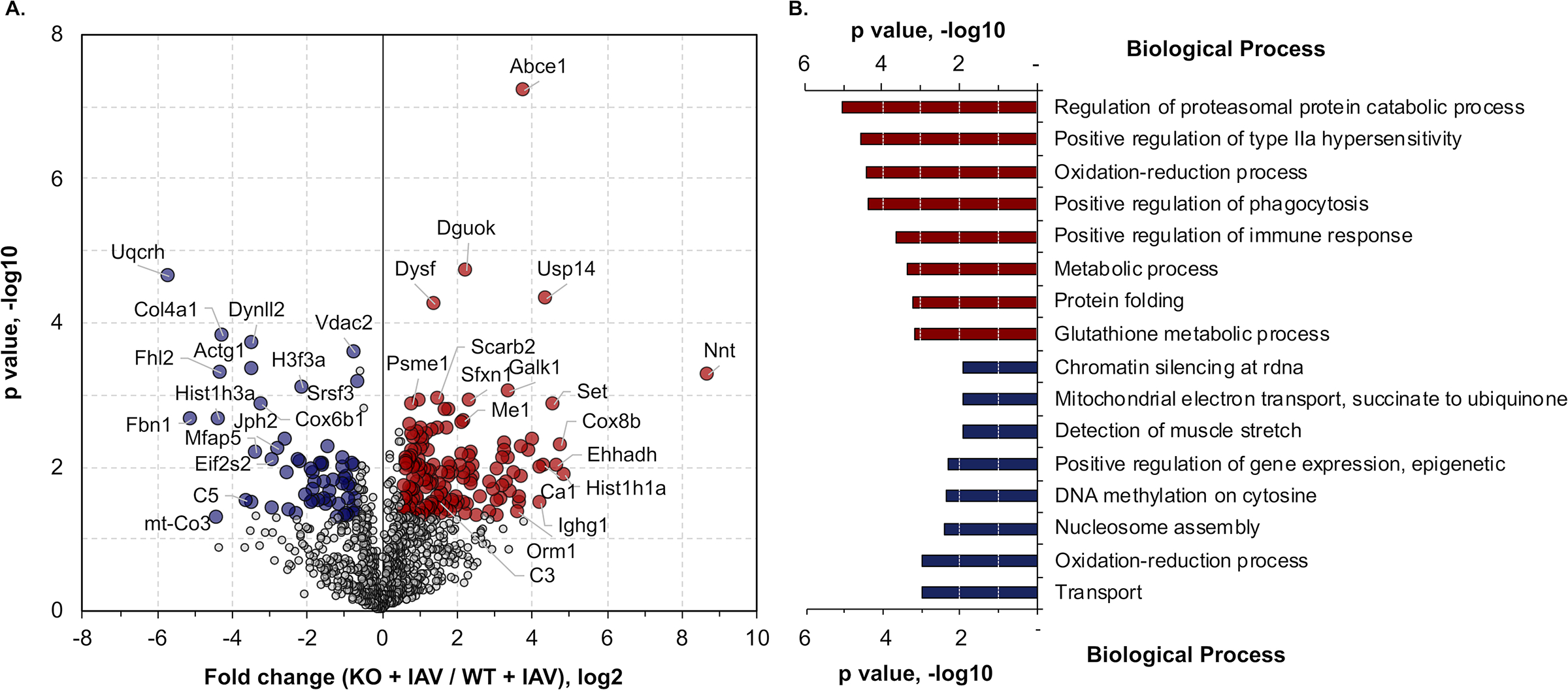
(A) Quantitative comparison of IAV infected WT and MLKL KO mice (n = 3 per group). Fold change of the proteins (x-axis) and their significance (p value, y-axis) were plotted. Up- and down-regulated proteins (1.5-fold cutoff) were highlighted in red and blue, respectively. (B) GO enrichment analysis of the significantly changed proteins as shown in panel A. Top 8 enriched biological terms of each group were displayed. Proteomic data of control and infected mice is representative from 2 separate experiments done with 3 mice of each sex; no sex based differences were observed.
Influenza virus infection affects phosphorylation in cardiac tissue proteins.
Using immunoblot analysis, we found the phosphorylation of AKT, CaMKII and AMPK⍺, kinases known to be important in regulating cell growth, apoptosis, and cardiac function and development50 (Fig. 5A–B), decreased in both infected WT and MLKL KO cardiac tissue compared to uninfected hearts (Fig. 5A–B). To further evaluate the alteration of cardiac protein phosphorylation during influenza infection in an unbiased manner, we used TiO2-based phosphopeptide enrichment for global phosphoproteome analyses51. In total, we identified 2,340 unique phosphopeptides (probability score > 0.75) corresponding to 898 phosphoproteins from the four groups of heart samples. Known phosphosites, as well as novel ones, were identified from our study (Online Tables V and VI). Interestingly, nearly 2/3 of them were only identified in the phosphoproteome but not the global proteome (Online Figure XII. A), possibly due to the lower abundance of phosphopeptides that can only be detected after enrichment. The majority of the 591 unique phosphoproteins were found to be associated with mRNA processing and splicing, including splicing factors and RNA binding motif proteins (e.g. Rbm10, Rbm17, Rbm25, Rbm37, Srsf6, Srsf9). The 307 proteins that were detected in both global proteomic and phosphoproteomic methods were strongly associated with cardiac muscle contraction and myofibril assembly process, including myosin light chain kinase 3 (Mylk3), tropomyosin (Tpm1) and myopalladin (Mypn) (Online Figure XII. A). The phosphorylation states of these proteins have also been implicated in heart diseases such as cardiomyopathy52.
Figure 5: Influenza infection leads to downregulation of functional proteins in the heart.
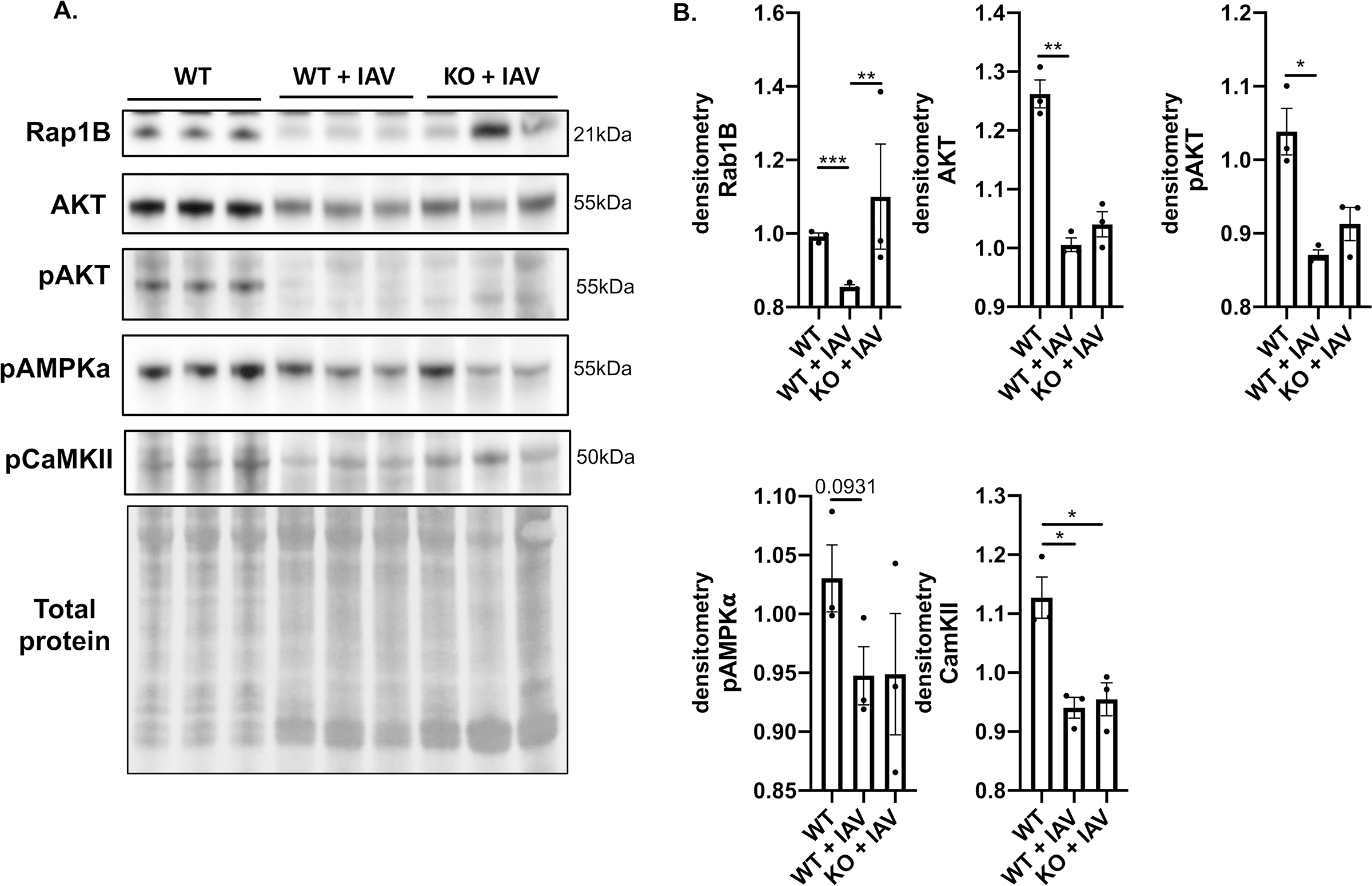
Male and female 6-to-8-week-old wild-type and MLKL KO mice were intranasally infected with A/California/7/2009 (IAV) or mock challenged with vehicle (PBS) and hearts excised at day 12 post-infection. (n = 3 per group, representative of 2 experiments for a total n =6, with 3 mice of each sex). Heart tissue was collected and immunoblots were performed against Rap1B, AKT, pAKT, pAMPKa, and pCaMKII, presented with quantification of relative protein level. Kruskal-Wallis test with Dunn’s multiple-comparison post-test. Asterisks denote the level of significance observed: * = p ≤ 0.05; ** = p ≤ 0.01.
Quantitative assessment of the phosphoproteomic data (Permutation-based FDR < 0.05) resulted in 1,691 phosphopeptides that were significantly different among the four groups (Online Figure XII. B). The majority of phosphopeptides (~97%) were decreased in IAV-infected hearts, regardless of genotype, suggesting a general protein dephosphorylation during IAV infection that may result in probable impact on cardiac function. Fig. 6A illustrates selected proteins with significantly different phosphorylation levels between the four groups. Proteins with decreased phosphorylation after infection include kinases such as Akt1 (S124), AMPK (e.g., Prkab1, S108; Prkab2, S183), PKA (e.g., Prkaca, T198), protein kinase C β (Prkcb, T500) and δ (Prkcd, S662), and apoptosis-associated proteins such as Pea-1553, Gja1 (Connexin-43)54, and Bcl-2 family proteins (Bclaf1 and Bag3)55, suggesting possible dysregulation in intercellular communication and cell proliferation. Several mitochondrial proteins showed varied phosphorylation trends during infection: upregulated phosphoproteins include ADP/ATP translocase 1 (Ant1) and pyruvate dehydrogenase E1 component subunit α (Pdha1), and down-regulated ones included Vdac1, Bckdha, and Cytochrome b-c1 complex subunit 1 (Uqcrc1) (Fig. 6). Finally, cardiomyocytes may not be the only cells affected by IAV infection, as changes in Rap1B (Fig. 5, 6) indicate altered vascular function. Rap1b is involved in modulating basal vascular tone and blood pressure56. Of interest, staining endothelial cells (CD144, VE Cadherin)57 or cardiac fibroblasts (gp38)58 and IAV in frozen cardiac sections suggests IAV may be able to infect both cell types during in vivo infection (Online Figure XIII. A). When NRK-49F fibroblasts and human umbilical vein endothelial cells (HUVECs) were infected with influenza virus, only endothelial cells showed significantly increased cytotoxicity upon infection (Online Figure XIII. B–C).
Figure 6: Influenza virus infection leads to changes in phosphorylation of cardiac proteins.
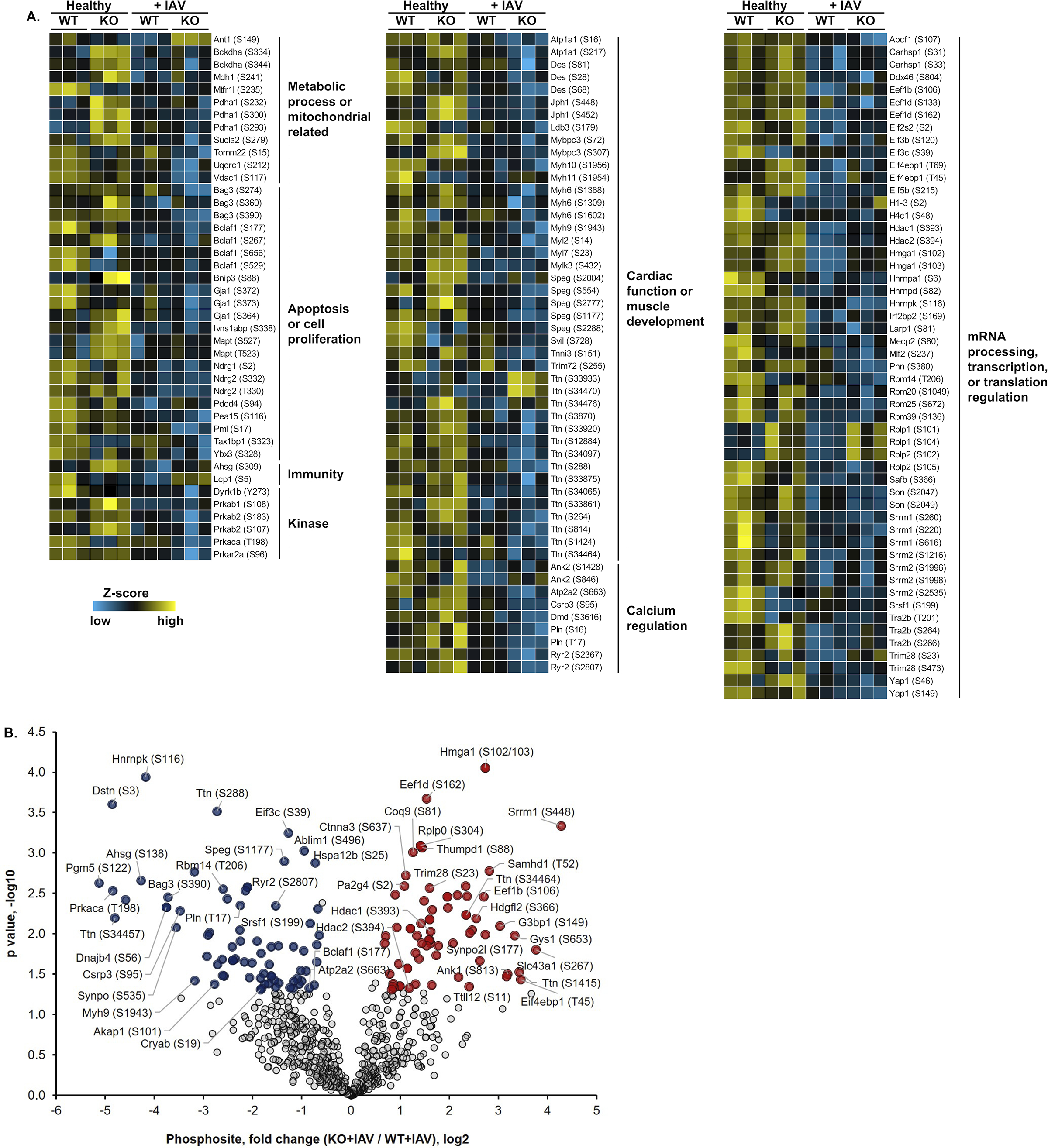
(A) Quantitative phosphoproteomic profiling of mouse heart. Plotted here are selected proteins and their identified phosphosites (indicated in the parentheses) grouped by protein functions or related pathways. (B) Quantitative comparison of IAV infected or mock infected (PBS challenge) WT and MLKL KO mice (n = 3 per group). Fold change of the phosphosites (x-axis) and their significance (p value, y-axis) were plotted. Up- and down-regulated proteins (1.5-fold cutoff) are highlighted in red and blue, respectively. Phosphoproteomic data is representative from 2 separate experiments done with 3 mice of each sex, no sex based differences were observed.
To better understand how necroptosis affects phosphorylation in cardiac tissue after IAV infection, pair-wise comparison of the KO+IAV group to the WT+IAV group identified 72 and 76 phosphopeptides that were up- and down-regulated, respectively, by at least 1.5-fold (Fig. 6B). The up-regulated phosphoproteins include histone deacetylases (HDAC) 1 and 2. Similar HDAC phosphorylation patterns have been implicated in regulating cardiac morphogenesis and growth59. Among proteins with down-regulated phosphorylation in the KO+IAV group, we found several apoptosis-associated proteins, including Bcl-family proteins (Bclaf1 and Bag3), protein PML, and alpha-crystallin (Cryab)60. Interestingly, membrane proteins that mediate Ca2+ homeostasis in cardiomyocytes, including the complex formed by sarcoplasmic reticulum Ca2+ ATPase (SERCA), phospholamban (Pln)61, and ryanodine receptor 2 (Ryr2), were all found to be down-regulated in phosphorylation. The phosphorylation intensity of identified peptides can be found in Online Tables V–VI. These results provide functional perspective of the cardiac proteins and pathways that are altered within necroptosis-deficient mice in response to influenza infection.
Inhibition of the mitochondrial permeability transition pore (MPTP) and replenishing of nicotinamide adenine dinucleotide (NAD) protects cardiomyocytes from influenza-mediated injury. Several mitochondrial proteins related to stress response or oxidation-reduction processes were identified in our proteome and phosphoproteome analyses. MPTP has been shown to maintain Ca2+ homeostasis and mediate both necrotic cell death (e.g. necroptosis62 and pyroptosis63) and apoptosis62, 64, 65. MPTP has also been implicated in major cardiac pathologies such as ischemia and reperfusion injuries66. We also observed significant changes in the phosphorylation states of proteins regulating MPTP activity, Voltage-dependent anion channel 1 (VDAC1) and ADP/ATP translocase 1 (Ant1) (Fig. 5, 6). VDAC proteins localize at the outer membrane of mitochondria and regulate metabolite, ion, and ATP/ADP translocation across the membrane65. Additionally, one initiator of mitochondrial damage and necroptosis is depletion of nicotinamide adenine dinucleotide (NAD+)64, 65. Therefore, we hypothesized that therapeutically increasing NAD+ levels, antioxidant treatment, or inhibition of MPTP may rescue IAV-induced cardiomyocyte death. Pre-treatment of rat H9c2 myocytes with either nicotinamide to directly increase NAD+, TRO 1962267 to inhibit VDAC and indirectly decrease leak of NAD+ from mitochondria, or the general antioxidant N-acetylcysteine (NAC), significantly lowered cytotoxicity and cellular ROS, and increased cellular ATP after IAV infection (Fig. 7A–C). Necroptosis and oxidative stress inhibition also showed protection against mitochondrial membrane permeabilization induced by IAV infection in AC16 myocytes (Fig. 7D). Together, these data demonstrate that IAV infection leads to necroptosis-associated mitochondrial damage, ROS production, and NAD+ depletion in cardiomyocytes.
Figure 7: Inhibition of the MPTP and replenishment of NAD+ leads to cardiomyocyte protection after IAV infection.
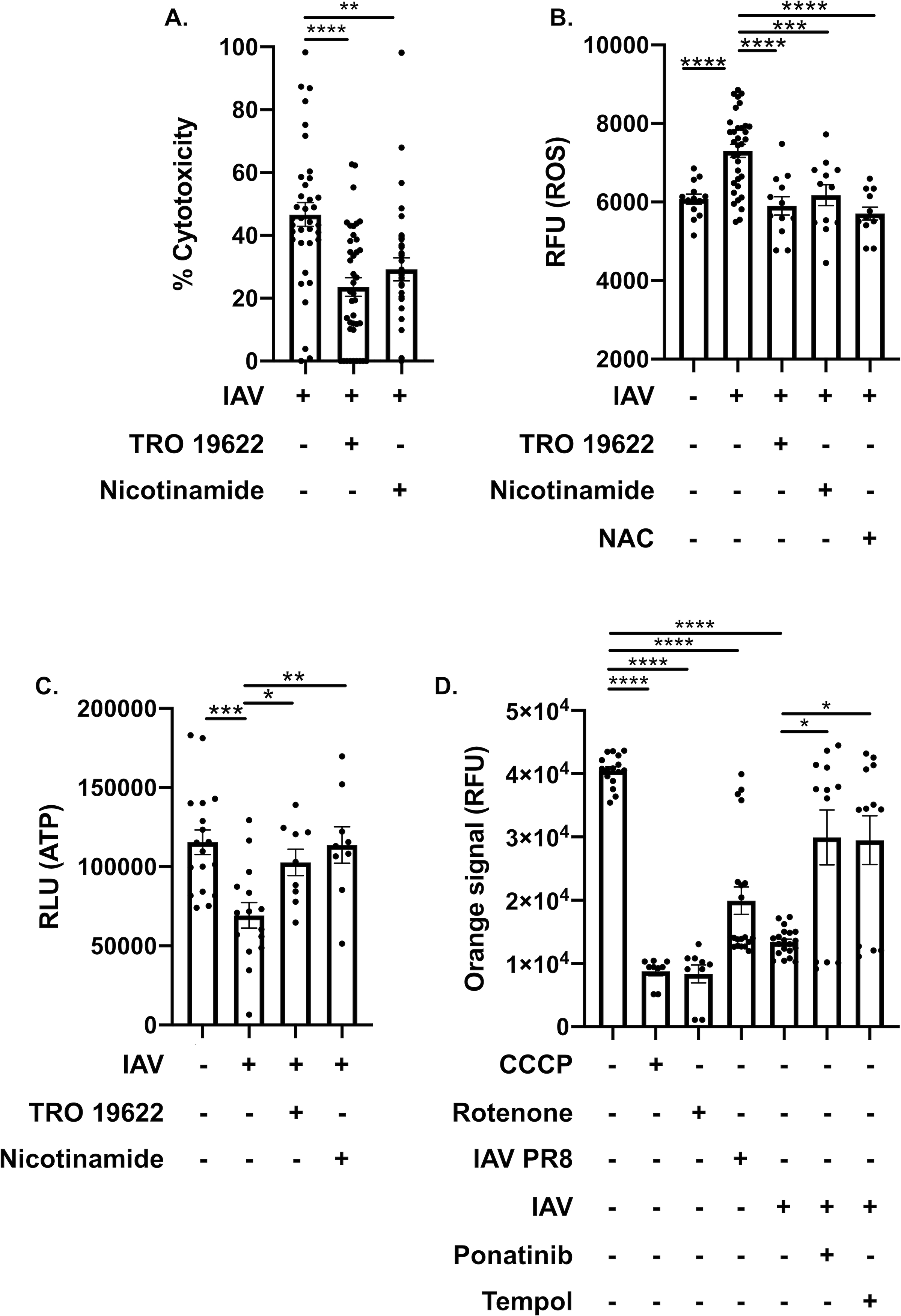
(A) Cytotoxicity of H9c2 myocytes treated with TRO 19622 (10 μM) or nicotinamide (1 mM) infected with A/California/7/2009 (IAV). (B) ROS/superoxide levels and (C) ATP levels of IAV-infected H9c2 myocytes treated with TRO 19622, nicotinamide with same concentration in (A) or n-acetylcysteine (10 μM), compared to uninfected or mock treated H9c2. RLU: relative light unit of luminescence. (D) Mitochondrial membrane permeabilization assay as measured via changes in RFU: relative fluorescent units of human AC16 cardiomyocytes infected with A/California/7/2009 (IAV) or PR8 and treated with rotenone, tempol or ponatinib. Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) positive control for mitochondrial membrane permeabilization. Kruskal-Wallis test with Dunn’s multiple-comparison post-test. Asterisks denote the level of significance observed: * = p ≤ 0.05; ** = p ≤ 0.01; *** = p ≤ 0.001, **** = p ≤ 0.0001.
DISCUSSION
Increasingly strong association of MACE with influenza infection68 demands a better understanding of the underlying molecular mechanisms driving this phenotype. Here, we provide the first evidence that viral particles can be detected in the myocardium during convalescence, indicating influenza virus persistence in other organs after its clearance from the lungs. Multiple studies have shown that cellular blocks can lead to abortive influenza virus replication in several different cell types33, 34. This is supported by our study that shows IAV abortive replication in cardiomyocytes. How the virus translocates to cardiac tissue remains to be determined, but the pronounced effect it has on the heart merits further investigation.
While there are limitations in the presented study, mainly due to the use of experimental animal models and murine or human cells in vitro, the presented data offers compelling experimental evidence of the effects of influenza infection in cardiac tissue. Differences in infection outcomes between murine models and human hosts occur due to the complexity and differences of the human immune system and its murine counterpart. In humans, severe respiratory infection from viruses can lead to tachycardia, hypotension, vascular inflammation, myocarditis, cardiac arrhythmias and increase risk of death. The processes identified in this report are common to both mice and human allowing for the translation of important observations upon validation69–72. Here we present a comprehensive report of the underlying molecular effects of influenza infection in cardiac tissue during convalescence using a multi-proteomic approach (Online Figure XIV). The global proteomic comparison between uninfected and IAV-infected WT hearts showed upregulation of pathways involved in metabolism and oxidative stress, indicating a high-stress, low-energy state of the heart during convalescence. Several endopeptidase inhibitor proteins were also up-regulated in IAV-infected hearts, providing possible novel targets73 to treat IAV-driven cardiac dysfunction. This study serves as a valuable resource for future studies to pinpoint molecular intervention in cardiac pathology after IAV infection.
Phosphoproteomic analysis provided further insight into the functional state of cardiac proteins. The down-regulation of overall phosphorylation in IAV-infected hearts was consistent with a lower energy state, as seen in the global proteome analysis. Furthermore, many kinases and their substrate proteins that are important for cell proliferation, apoptosis, and intercellular communication had reduced phosphorylation. These data support the notion of decreased mitochondrial function and potential dysregulation of cardiomyocyte cellular and metabolic homeostasis65, 74. Using cultured cardiomyocytes, we corroborated our proteomic data, showing that IAV can directly infect cardiomyocytes. IAV infection leads to cell death, oxidative stress, and mitochondrial damage, which is sensitive to treatment with either ROS inhibitors directed against mitochondrial function or repressing NAD+ depletion. Future studies should test the role of influenza infection in cardiac function and fibrosis by using left ventricular pressure-volume analyses, 2-D echocardiography and pathology assessments.
Finally, we observed that necroptosis modulates cardiac protein expression and functional profiles upon IAV infection. Necroptosis deficiency did not completely abolish the effects of IAV infection in the heart, suggesting that there are other molecular pathways essential for these changes. However, MLKL KO mice had a significantly altered cardiac proteome upon IAV infection compared with IAV-infected WT mice. Further studies using WT mice from intercrossed heterozygous animals may be required to fully define the role of necroptosis in modulation of influenza driven cardiac changes. Necroptosis inhibition led to an increase in NAD(P) transhydrogenase, protease inhibitors, and elevated detoxifying proteins and innate immunity, potential strategies that may protect cardiac tissue. Indeed, changes in mitochondrial integrity and oxidative stress proteins were partially rescued in IAV-infected MLKL KO mice when compared to IAV-infected WT mice. In vitro cardiac infection showed that necroptosis inhibition was beneficial, as cell toxicity and ROS generation were reduced, and production of ATP was promoted. Inhibition of MLKL leads to the activation of apoptosis via RIPK375 and its beneficial during influenza infection in the lungs15, however, inhibition of RIPK3 as a therapeutic target in this case may detrimental due to its role in activating apoptosis38. Blocking of necroptosis was shown to increase apoptotic activity, suggesting a possible target to reduce necrotic cardiac injury. Our data also shows that pandemic influenza A H1N1 A/California/7/2009 and A/Puerto Rico/8/1934 alter similar pathways in mice hearts, with the pandemic strain showing a more profound effect, supporting our initial hypothesis that pandemic strains may be more cardiotropic. Thus, future studies could further elucidate similarities and differences between epidemic and pandemic influenza strains.
In conclusion, this report provides comprehensive evidence that influenza infection can modify the molecular homeostasis of the heart. Our results provide new insights into the prolonged cardiac effects after viral epidemics and pandemics. These results could also translate to other pathogenic viruses that frequently cause pulmonary infections. For example, clinical evidence from cases of SARS-CoV-2, causative of coronavirus disease (COVID)-19 have shown systemic manifestations by the virus. Recent reports have indicated the possibility of a direct effect of COVID-19 in causing myocarditis, fatal arrhythmias and other adverse cardiac events. This may be due in part to the presence of SARS-CoV-2 viral particles in cardiac tissue, a phenotype observed in patients that succumbed to infection, and could be further exacerbated indirectly via systemic inflammation76, 77. Our results provide possible underlying mechanisms that may be shared by these respiratory viruses. Future studies are required to define similarities and differences between these pandemic viruses.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The incidence of cardiovascular events increases after respiratory infections.
The cell death pathway of necroptosis promotes inflammation and tissue injury.
What New Information Does This Article Contribute?
Influenza virus can be found in mouse hearts after resolution of lung infection.
Cardiac influenza virus presence during convalescence leads to proteome and phosphoproteome remodeling.
Necroptosis inhibition promotes mitochondrial function, proteome and phosphoproteome reshaping and reduces cardiomyocyte damage after influenza infection.
Respiratory infections have been linked to adverse cardiac events. However, the role of influenza viral infection in the context of cardiac health during convalescence has not been explored. Influenza infection of cardiomyocytes showed changes to mitochondrial function, increase oxidative stress and cellular toxicity. The cellular changes in myocytes promoted the cell death mechanism of necroptosis. We identify that the proteomic and phosphoproteomic profiles of mouse hearts taken during convalescence showed significant remodeling. Deletion of the necroptosis effector molecule MLKL led to increase mitochondrial function and decrease cardiomyocyte death. Furthermore, necroptosis inhibition led to a re-shaped proteome and phosphoproteome. Our data collectively raise the possibility that therapies targeting oxidative stress and necroptosis may be effective in reducing the long-term effects of influenza infection in the heart.
ACKNOWLEDGEMENT
We thank Drs. Carlos J. Orihuela, and Kevin Harrod for their assistance and for providing materials used in the presented report.
SOURCES OF FUNDING
N.G-J. and Y.Y. were supported by J. Craig Venter Institute Start-up funds. N.G-J. was also supported by NIH grant 1R21AI148722–01A1. R.P.G. is supported by NIH grant 5T32DE014318–16.
Nonstandard Abbreviations and Acronyms:
- MACE
Major adverse cardiac events
- MPTP
Mitochondrial permeability transition pore
- IAV
Influenza A virus
- MLKL
Mixed Lineage Kinase Domain Like Pseudokinase
- CAP
community acquired pneumonia
- RIPK
receptor-interacting serine/threonine-protein kinases
- 8-OXO
8-Oxoguanine
- ROS
reactive oxygen species
- HR/AM LC-MS/MS
High-Resolution Accurate-Mass
- LFQ
Label-free quantitation
- GO
Gene Ontology
- PFU
Plaque forming units
Footnotes
DISCLOSURES
The authors declare no competing financial interests.
SUPPLEMENTAL MATERIALS
Expanded Materials & Methods
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Mortality GBD, Causes of Death C. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the global burden of disease study 2015. Lancet. 2016;388:1459–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heron M Deaths: Leading causes for 2017. National vital statistics reports. 2019;68 [PubMed] [Google Scholar]
- 3.Paules C, Subbarao K. Influenza. Lancet. 2017;390:697–708 [DOI] [PubMed] [Google Scholar]
- 4.Noda T [orthomyxoviruses]. Uirusu. 2012;62:219–228 [DOI] [PubMed] [Google Scholar]
- 5.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the united states. JAMA. 2003;289:179–186 [DOI] [PubMed] [Google Scholar]
- 6.Rolfes MA, Foppa IM, Garg S, Flannery B, Brammer L, Singleton JA, Burns E, Jernigan D, Olsen SJ, Bresee J, Reed C. Annual estimates of the burden of seasonal influenza in the united states: A tool for strengthening influenza surveillance and preparedness. Influenza Other Respir Viruses. 2018;12:132–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain S, Self WH, Wunderink RG, Fakhran S, Balk R, Bramley AM, Reed C, Grijalva CG, Anderson EJ, Courtney DM, Chappell JD, Qi C, Hart EM, Carroll F, Trabue C, Donnelly HK, Williams DJ, Zhu Y, Arnold SR, Ampofo K, Waterer GW, Levine M, Lindstrom S, Winchell JM, Katz JM, Erdman D, Schneider E, Hicks LA, McCullers JA, Pavia AT, Edwards KM, Finelli L, Team CES. Community-acquired pneumonia requiring hospitalization among u.S. Adults. N Engl J Med. 2015;373:415–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corrales-Medina VF, Taljaard M, Fine MJ, Dwivedi G, Perry JJ, Musher DM, Chirinos JA. Risk stratification for cardiac complications in patients hospitalized for community-acquired pneumonia. Mayo Clin Proc. 2014;89:60–68 [DOI] [PubMed] [Google Scholar]
- 9.Musher DM, Rueda AM, Kaka AS, Mapara SM. The association between pneumococcal pneumonia and acute cardiac events. Clin Infect Dis. 2007;45:158–165 [DOI] [PubMed] [Google Scholar]
- 10.Musher DM, Abers MS, Corrales-Medina VF. Acute infection and myocardial infarction. N Engl J Med. 2019;380:171–176 [DOI] [PubMed] [Google Scholar]
- 11.Kwong JC, Schwartz KL, Campitelli MA, Chung H, Crowcroft NS, Karnauchow T, Katz K, Ko DT, McGeer AJ, McNally D, Richardson DC, Rosella LC, Simor A, Smieja M, Zahariadis G, Gubbay JB. Acute myocardial infarction after laboratory-confirmed influenza infection. N Engl J Med. 2018;378:345–353 [DOI] [PubMed] [Google Scholar]
- 12.Warren-Gash C, Blackburn R, Whitaker H, McMenamin J, Hayward AC. Laboratory-confirmed respiratory infections as triggers for acute myocardial infarction and stroke: A self-controlled case series analysis of national linked datasets from scotland. Eur Respir J. 2018;51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown AO, Mann B, Gao G, Hankins JS, Humann J, Giardina J, Faverio P, Restrepo MI, Halade GV, Mortensen EM, Lindsey ML, Hanes M, Happel KI, Nelson S, Bagby GJ, Lorent JA, Cardinal P, Granados R, Esteban A, LeSaux CJ, Tuomanen EI, Orihuela CJ. Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog. 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long B, Brady WJ, Koyfman A, Gottlieb M. Cardiovascular complications in covid-19. Am J Emerg Med. 2020;38:1504–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm, Verbist K, Gough PJ, Bertin J, Hartmann BM, Sealfon SC, Kaiser WJ, Mocarski ES, Lopez CB, Thomas PG, Oberst A, Green DR, Balachandran S. Ripk3 activates parallel pathways of mlkl-driven necroptosis and fadd-mediated apoptosis to protect against influenza a virus. Cell Host Microbe. 2016;20:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, Hernandez M, Quinones-Falconi F, Bautista E, Ramirez-Venegas A, Rojas-Serrano J, Ormsby CE, Corrales A, Higuera A, Mondragon E, Cordova-Villalobos JA, Influenza IWGo. Pneumonia and respiratory failure from swine-origin influenza a (h1n1) in mexico. N Engl J Med. 2009;361:680–689 [DOI] [PubMed] [Google Scholar]
- 17.BALDWIN LUCKE MDTW MD; EDWIN KIME MD Pathologic anatomy and bacteriology of influenza. Arch Intern Med (Chic). 1919;24:154–237 [Google Scholar]
- 18.Fislova T, Gocnik M, Sladkova T, Durmanova V, Rajcani J, Vareckova E, Mucha V, Kostolansky F. Multiorgan distribution of human influenza a virus strains observed in a mouse model. Arch Virol. 2009;154:409–419 [DOI] [PubMed] [Google Scholar]
- 19.Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, Feldmann F, Alimonti JB, Fernando L, Li Y, Katze MG, Feldmann H, Kawaoka Y. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323 [DOI] [PubMed] [Google Scholar]
- 20.Kodama M Influenza myocarditis. Circ J. 2010;74:2060–2061 [DOI] [PubMed] [Google Scholar]
- 21.Kotaka M, Kitaura Y, Deguchi H, Kawamura K. Experimental influenza a virus myocarditis in mice. Light and electron microscopic, virologic, and hemodynamic study. Am J Pathol. 1990;136:409–419 [PMC free article] [PubMed] [Google Scholar]
- 22.Oseasohn R, Adelson L, Kaji M. Clinicopathologic study of thirty-three fatal cases of asian influenza. N Engl J Med. 1959;260:509–518 [DOI] [PubMed] [Google Scholar]
- 23.Paddock CD, Liu L, Denison AM, Bartlett JH, Holman RC, Deleon-Carnes M, Emery SL, Drew CP, Shieh WJ, Uyeki TM, Zaki SR. Myocardial injury and bacterial pneumonia contribute to the pathogenesis of fatal influenza b virus infection. J Infect Dis. 2012;205:895–905 [DOI] [PubMed] [Google Scholar]
- 24.Ukimura A, Ooi Y, Kanzaki Y, Inomata T, Izumi T. A national survey on myocarditis associated with influenza h1n1pdm2009 in the pandemic and postpandemic season in japan. J Infect Chemother. 2013;19:426–431 [DOI] [PubMed] [Google Scholar]
- 25.Ukimura A, Satomi H, Ooi Y, Kanzaki Y. Myocarditis associated with influenza a h1n1pdm2009. Influenza Res Treat. 2012;2012:351979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Groves HT, McDonald JU, Langat P, Kinnear E, Kellam P, McCauley J, Ellis J, Thompson C, Elderfield R, Parker L, Barclay W, Tregoning JS. Mouse models of influenza infection with circulating strains to test seasonal vaccine efficacy. Front Immunol. 2018;9:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartmann BM, Li W, Jia J, Patil S, Marjanovic N, Martinez-Romero C, Albrecht RA, Hayot F, Garcia-Sastre A, Wetmur JG, Moran TM, Sealfon SC. Mouse dendritic cell (dc) influenza virus infectivity is much lower than that for human dcs and is hemagglutinin subtype dependent. J Virol. 2013;87:1916–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Numata M, Mitchell JR, Tipper JL, Brand JD, Trombley JE, Nagashima Y, Kandasamy P, Chu HW, Harrod KS, Voelker DR. Pulmonary surfactant lipids inhibit infections with the pandemic h1n1 influenza virus in several animal models. J Biol Chem. 2020;295:1704–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujikura D, Miyazaki T. Programmed cell death in the pathogenesis of influenza. Int J Mol Sci. 2018;19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nailwal H, Chan FK. Necroptosis in anti-viral inflammation. Cell Death Differ. 2019;26:4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, Restrepo MI, Dube PH, Bergman MA, Orihuela CJ. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 2017;24:917–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, Orihuela CJ. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PLoS Pathog. 2015;11:e1005337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ioannidis LJ, Verity EE, Crawford S, Rockman SP, Brown LE. Abortive replication of influenza virus in mouse dendritic cells. J Virol. 2012;86:5922–5925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marvin SA, Russier M, Huerta CT, Russell CJ, Schultz-Cherry S. Influenza virus overcomes cellular blocks to productively replicate, impacting macrophage function. J Virol. 2017;91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Downey J, Pernet E, Coulombe F, Allard B, Meunier I, Jaworska J, Qureshi S, Vinh DC, Martin JG, Joubert P, Divangahi M. Ripk3 interacts with mavs to regulate type i ifn-mediated immunity to influenza a virus infection. PLoS Pathog. 2017;13:e1006326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fauster A, Rebsamen M, Huber KV, Bigenzahn JW, Stukalov A, Lardeau CH, Scorzoni S, Bruckner M, Gridling M, Parapatics K, Colinge J, Bennett KL, Kubicek S, Krautwald S, Linkermann A, Superti-Furga G. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 2015;6:e1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of rip3 kinase. Cell. 2012;148:213–227 [DOI] [PubMed] [Google Scholar]
- 38.Gonzalez-Juarbe N, Riegler AN, Jureka AS, Gilley RP, Brand JD, Trombley JE, Scott NR, Platt MP, Dube PH, Petit CM, Harrod KS, Orihuela CJ. Influenza-induced oxidative stress sensitizes lung cells to bacterial-toxin-mediated necroptosis. Cell Rep. 2020;32:108062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fortini P, Pascucci B, Parlanti E, D’Errico M, Simonelli V, Dogliotti E. 8-oxoguanine DNA damage: At the crossroad of alternative repair pathways. Mutat Res. 2003;531:127–139 [DOI] [PubMed] [Google Scholar]
- 40.Balachandran S, Rall GF. Benefits and perils of necroptosis in influenza virus infection. J Virol. 2020;94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu Y, Sikorski P, Bowman-Gholston C, Cacciabeve N, Nelson KE, Pieper R. Diagnosing inflammation and infection in the urinary system via proteomics. J Transl Med. 2015;13:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y, Sikorski P, Smith M, Bowman-Gholston C, Cacciabeve N, Nelson KE, Pieper R. Comprehensive metaproteomic analyses of urine in the presence and absence of neutrophil-associated inflammation in the urinary tract. Theranostics. 2017;7:238–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase mlkl mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453 [DOI] [PubMed] [Google Scholar]
- 44.Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, Andrake M, Vogel P, Sigal LJ, tenOever BR, Thomas PG, Upton JW, Balachandran S. Dai senses influenza a virus genomic rna and activates ripk3-dependent cell death. Cell Host Microbe. 2016;20:674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee S, Hirohama M, Noguchi M, Nagata K, Kawaguchi A. Influenza a virus infection triggers pyroptosis and apoptosis of respiratory epithelial cells through the type i interferon signaling pathway in a mutually exclusive manner. J Virol. 2018;92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taiyab A, Rao Ch M. Hsp90 modulates actin dynamics: Inhibition of hsp90 leads to decreased cell motility and impairs invasion. Biochim Biophys Acta. 2011;1813:213–221 [DOI] [PubMed] [Google Scholar]
- 47.Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, Epstein PN. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes. 2004;53:1336–1343 [DOI] [PubMed] [Google Scholar]
- 48.Klenk HD, Rott R, Orlich M, Blodorn J. Activation of influenza a viruses by trypsin treatment. Virology. 1975;68:426–439 [DOI] [PubMed] [Google Scholar]
- 49.Sheeran FL, Rydstrom J, Shakhparonov MI, Pestov NB, Pepe S. Diminished nadph transhydrogenase activity and mitochondrial redox regulation in human failing myocardium. Biochim Biophys Acta. 2010;1797:1138–1148 [DOI] [PubMed] [Google Scholar]
- 50.Garcia D, Shaw RJ. Ampk: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017;66:789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu X, Tian L, Li J, Zhang Y, Han V, Li Y, Xu X, Li H, Chen X, Chen J, Jin W, Xie Y, Han J, Zhong CQ. Investigation of receptor interacting protein (rip3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol Cell Proteomics. 2012;11:1640–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Filomena MC, Yamamoto DL, Caremani M, Kadarla VK, Mastrototaro G, Serio S, Vydyanath A, Mutarelli M, Garofalo A, Pertici I, Knoll R, Nigro V, Luther PK, Lieber RL, Beck MR, Linari M, Bang ML. Myopalladin promotes muscle growth through modulation of the serum response factor pathway. J Cachexia Sarcopenia Muscle. 2020;11:169–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Renganathan H, Vaidyanathan H, Knapinska A, Ramos JW. Phosphorylation of pea-15 switches its binding specificity from erk/mapk to fadd. Biochem J. 2005;390:729–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang RY, Laing JG, Kanter EM, Berthoud VM, Bao M, Rohrs HW, Townsend RR, Yamada KA. Identification of camkii phosphorylation sites in connexin43 by high-resolution mass spectrometry. J Proteome Res. 2011;10:1098–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lakshmikanthan S, Zieba BJ, Ge ZD, Momotani K, Zheng X, Lund H, Artamonov MV, Maas JE, Szabo A, Zhang DX, Auchampach JA, Mattson DL, Somlyo AV, Chrzanowska-Wodnicka M. Rap1b in smooth muscle and endothelium is required for maintenance of vascular tone and normal blood pressure. Arterioscler Thromb Vasc Biol. 2014;34:1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dejana E Endothelial cell-cell junctions: Happy together. Nat Rev Mol Cell Biol. 2004;5:261–270 [DOI] [PubMed] [Google Scholar]
- 58.Stellato M, Czepiel M, Distler O, Blyszczuk P, Kania G. Identification and isolation of cardiac fibroblasts from the adult mouse heart using two-color flow cytometry. Front Cardiovasc Med. 2019;6:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kee HJ, Kook H. Roles and targets of class i and iia histone deacetylases in cardiac hypertrophy. J Biomed Biotechnol. 2011;2011:928326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitra A, Basak T, Datta K, Naskar S, Sengupta S, Sarkar S. Role of alpha-crystallin b as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 2013;4:e582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gustavsson M, Verardi R, Mullen DG, Mote KR, Traaseth NJ, Gopinath T, Veglia G. Allosteric regulation of serca by phosphorylation-mediated conformational shift of phospholamban. Proc Natl Acad Sci U S A. 2013;110:17338–17343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM, Molkentin JD. Necroptosis interfaces with momp and the mptp in mediating cell death. PLoS One. 2015;10:e0130520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, Son J, Yu JW. Mptp-driven nlrp3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019;26:213–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Graham BH, Craigen WJ. Genetic approaches to analyzing mitochondrial outer membrane permeability. Curr Top Dev Biol. 2004;59:87–118 [DOI] [PubMed] [Google Scholar]
- 65.Kerner J, Lee K, Tandler B, Hoppel CL. Vdac proteomics: Post-translation modifications. Biochim Biophys Acta. 2012;1818:1520–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miao J, Huang Z, Liu S, Li X, Jia P, Guo Y, Wu N, Jia D. Hydroxytyrosol protects against myocardial ischemia reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Exp Ther Med. 2019;17:671–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzalez S, Berthelot J, Jiner J, Perrin-Tricaud C, Fernando R, Chrast R, Lenaers G, Tricaud N. Blocking mitochondrial calcium release in schwann cells prevents demyelinating neuropathies. J Clin Invest. 2017;127:1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Filgueiras-Rama D, Vasilijevic J, Jalife J, Noujaim SF, Alfonso JM, Nicolas-Avila JA, Gutierrez C, Zamarreno N, Hidalgo A, Bernabe A, Cop CP, Ponce-Balbuena D, Guerrero-Serna G, Calle D, Desco M, Ruiz-Cabello J, Nieto A, Falcon A. Human influenza a virus causes myocardial and cardiac-specific conduction system infection associated with early inflammation and premature death. Cardiovasc Res. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Madjid M, Safavi-Naeini P, Solomon SD, Vardeny O. Potential effects of coronaviruses on the cardiovascular system: A review. JAMA Cardiol. 2020;5:831–840 [DOI] [PubMed] [Google Scholar]
- 70.Mai F, Del Pinto R, Ferri C. Covid-19 and cardiovascular diseases. J Cardiol. 2020;76:453–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nguyen JL, Yang W, Ito K, Matte TD, Shaman J, Kinney PL. Seasonal influenza infections and cardiovascular disease mortality. JAMA Cardiol. 2016;1:274–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gopal R, Marinelli MA, Alcorn JF. Immune mechanisms in cardiovascular diseases associated with viral infection. Front Immunol. 2020;11:570681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laporte M, Naesens L. Airway proteases: An emerging drug target for influenza and other respiratory virus infections. Curr Opin Virol. 2017;24:16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siasos G, Tsigkou V, Kosmopoulos M, Theodosiadis D, Simantiris S, Tagkou NM, Tsimpiktsioglou A, Stampouloglou PK, Oikonomou E, Mourouzis K, Philippou A, Vavuranakis M, Stefanadis C, Tousoulis D, Papavassiliou AG. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann Transl Med. 2018;6:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase ripk3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360 [DOI] [PubMed] [Google Scholar]
- 76.Yancy CW, Fonarow GC. Coronavirus disease 2019 (covid-19) and the heart-is heart failure the next chapter? JAMA Cardiol. 2020 [DOI] [PubMed] [Google Scholar]
- 77.Lindner D, Fitzek A, Brauninger H, Aleshcheva G, Edler C, Meissner K, Scherschel K, Kirchhof P, Escher F, Schultheiss HP, Blankenberg S, Puschel K, Westermann D. Association of cardiac infection with sars-cov-2 in confirmed covid-19 autopsy cases. JAMA Cardiol. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, Carano RA, Cao TC, van Bruggen N, Bernstein L, Lee WP, Wu X, DeVoss J, Zhang J, Jeet S, Peng I, McKenzie BS, Roose-Girma M, Caplazi P, Diehl L, Webster JD, Vucic D. Ripk3 deficiency or catalytically inactive ripk1 provides greater benefit than mlkl deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Webster JD, Kwon YC, Park S, Zhang H, Corr N, Ljumanovic N, Adedeji AO, Varfolomeev E, Goncharov T, Preston J, Santagostino SF, Patel S, Xu M, Maher J, McKenzie BS, Vucic D. Rip1 kinase activity is critical for skin inflammation but not for viral propagation. J Leukoc Biol. 2020;107:941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin Y-H, Eguez RV, Torralba MG, Singh H, Golusinski P, Golusinski W, Masternak M, Nelson KE, Freire M, Yu Y. Self-assembled strap for global proteomics and salivary biomarker discovery. J. Proteome Res. 2019;18:1907–1915 [DOI] [PubMed] [Google Scholar]
- 81.Yu Y, Pieper R. Using proteomics to identify inflammation during urinary tract infection. Methods Mol Biol. 2019;2021:259–272 [DOI] [PubMed] [Google Scholar]
- 82.Thingholm TE, Jørgensen TJD, Jensen ON, Larsen MR. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nature Protocols. 2006;1:1929–1935 [DOI] [PubMed] [Google Scholar]
- 83.Yu Y, Pieper R. Urinary pellet sample preparation for shotgun proteomic analysis of microbial infection and host-pathogen interactions. Methods Mol. Biol 2015;1295:65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tyanova S, Temu T, Cox J. The maxquant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protocols. 2016;11:2301–2319 [DOI] [PubMed] [Google Scholar]
- 85.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed maxlfq. Mol. Cell. Proteomics 2014;13:2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat Protoc. 2009;4:44–57 [DOI] [PubMed] [Google Scholar]
- 87.Dormitzer PR, Suphaphiphat P, Gibson DG, Wentworth DE, Stockwell TB, Algire MA, Alperovich N, Barro M, Brown DM, Craig S, Dattilo BM, Denisova EA, De Souza I, Eickmann M, Dugan VG, Ferrari A, Gomila RC, Han L, Judge C, Mane S, Matrosovich M, Merryman C, Palladino G, Palmer GA, Spencer T, Strecker T, Trusheim H, Uhlendorff J, Wen Y, Yee AC, Zaveri J, Zhou B, Becker S, Donabedian A, Mason PW, Glass JI, Rappuoli R, Venter JC. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci Transl Med. 2013;5:185ra168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. All data from the proteomic analyses have been made publicly available at ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD017936, PXD016412, and PXD015992.