

Summary
Inhibition of histone deacetylases causes rapid and robust acetylation of histones. In this case, histone acetylation is likely increased on nearly every nucleosome, and the per-cell DNA/chromatin yield in chromatin immunoprecipitation (ChIP) experiments is significantly increased. Spike-in controls are essential for normalizing ChIP sequencing (ChIP-seq) data to capture this massive effect. Here, we report a detailed protocol of H3K27-ac ChIP-seq in human cells with chromatin from an ancestral species as a spike-in control.
For complete details on the use and execution of this protocol, please refer to Wu et al. (2021).
Subject areas: Bioinformatics, Sequence analysis, Cell Biology, Cell-based Assays, Genomics, ChIP-seq
Graphical abstract
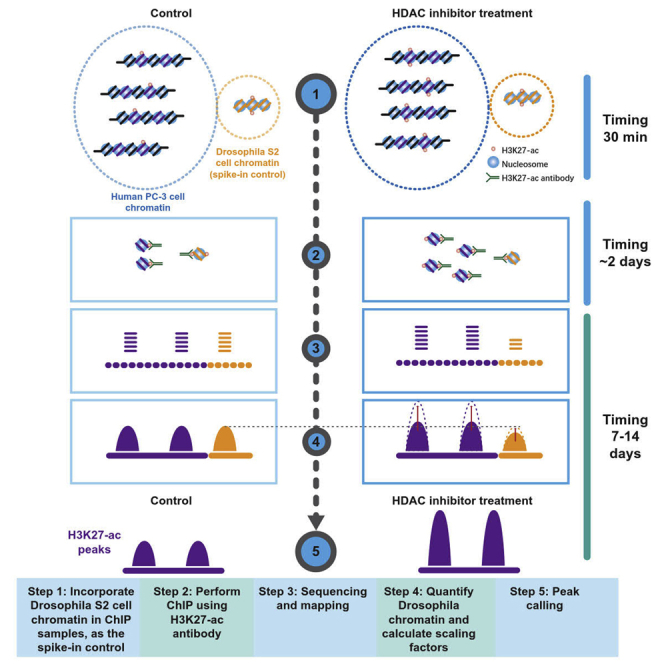
Highlights
-
•
Spike-in control is essential for ChIP-seq to capture massive histone acetylation
-
•
The protocol contains detailed steps for a spike-in controlled H3K27ac ChIP-seq
-
•
The protocol can be applied to capture massive changes in other histone modifications
-
•
An online tool “SPIKER” is created to help analyze spike-in ChIP-seq data
Inhibition of histone deacetylases causes rapid and robust acetylation of histones. In this case, histone acetylation is likely increased on nearly every nucleosome, and the per-cell DNA/chromatin yield in chromatin immunoprecipitation (ChIP) experiments is significantly increased. Spike-in controls are essential for normalizing ChIP sequencing (ChIP-seq) data to capture this massive effect. Here, we report a detailed protocol of H3K27-ac ChIP-seq in human cells with chromatin from an ancestral species as a spike-in control.
Before you begin
The protocol below describes steps of a spike-in controlled H3K27-ac ChIP-seq experiment in human prostate cancer cell line PC-3. We have also applied the same procedures to profile H3K27-ac modification in C4-2 cells. In principle, this protocol can also be applied to capture massive changes in other histone modifications in different cell types.
Used aberrations
HDAC: histone deacetylase; DMSO: dimethyl sulfoxide; SAHA: suberoylanilide hydroxamic acid; PBS: phosphate buffer saline; IP: immunoprecipitation; ChIP: chromatin immunoprecipitation
Preparation 1: Determine the global change of histone acetylation and the necessity to perform spike-in ChIP
Timing: about 2 days
Because the main experiment is time consuming and not intended for normal ChIP-seq, we suggest only to apply spike-in ChIP-seq to capture significant global changes of histone modifications. Therefore, it is necessary to profile global changes of histone acetylation (or other types of histone modification) quantitatively.
-
1.(Prepare DMSO and SAHA solution) Cell culture and treatment of HDAC inhibitor.
-
a.PC-3 cells are grown in two 3.5-cm culture dishes until the cell confluence is approximately 70%.
-
b.Dish 1 is added with DMSO; Dish 2 is treated with 1 μM SAHA.
-
c.The treatments last for 12 h.
-
a.
-
2.(Prepare 0.5% Triton X 100 and 0.2 N HCl, cool down the centrifuge to 4°C) Acid extraction of histones.
-
a.Cells are collected and washed with ice cold 1× PBS before they are lysed with 0.5% Triton X 100 (v/v) for 10 min on ice.
-
b.Centrifuge at 1,000 × g for 10 min at 4°C, discard the supernatant.
-
c.The cell nuclei pellet is re-suspended in 0.2 N HCl for 16 h at 4°C. The acid extracted sample is centrifuged and supernatant is reserved for protein quantification.
-
a.
Pause point: Protein samples for western blotting can be stored at −20°C for 1 week.
-
3.(Prepare 15% SDS polyacrylamide gel and anti-H3K27-ac antibody) Western blotting detecting histone acetylation.
-
a.Load 20 μg of each acid extracted histone sample into a sodium dodecyl sulfate polyacrylamide gel (a 1.5-mm thick, 15% polyacrylamide gel).
-
b.Separate each sample by gel electrophoresis (80V voltage for 30 min to concentrate samples and 100V for 60 min to separate samples) and transfer proteins onto nitrocellulose membranes (15V for 30 min using common semi-dry transferring system).
-
c.The membranes are incubated with primary histone acetylation antibody, for example, anti-H3K27-ac, for 16 h at 4°C and subsequently probed with horseradish peroxidase-conjugated secondary antibody. The immunoreactive blots are visualized using a chemiluminescence reagent (Thermo Fisher Scientific, catalog number 32106).
-
a.
-
4.
Determine the global change of histone acetylation.
For the typical result (Figure 1), the sample “SAHA treatment” yields much stronger blotting intensity than “DMSO control”. Result like this indicates that HDAC inhibitor SAHA induces a robust increase in H3K27-ac modification. To capture this effect in ChIP-seq, it is essential to perform spike-in controlled ChIP-seq as described in the following protocol.
Figure 1.

PC-3 cells are treated with DMSO or 1 μM SAHA for 12 h before acid extraction of histones and western blotting
Preparation 2: Prepare target cell chromatin and Drosophila cell chromatin (spike-in control) for ChIP and verification of antibodies
If the necessity of spike-in ChIP-seq has been determined, you should prepare Drosophila cell chromatin (as spike-in control) and ChIP grade antibody.
-
5.Preparation of Drosophila S2 cells.
-
a.6×107 Drosophila S2 cells (purchased from ATCC, catalog number CRL-1963) are cultured in Schneider’s Drosophila media (Life Technologies, catalog number 21720-024) supplemented with 10% FBS in 6×10-mm culture dishes (1×107 cells in one dish) and cultured at 21°C without additional CO2.
-
b.Acid extract of S2 cell histones as described in Preparation Step 2 (Usually we extracted histones from 1×107 cells in one dish); freeze the samples in −20°C for further antibody verification.
-
a.
-
6.(Prepare 11% Formaldehyde Solution, cool down the centrifuge to 4°C) Growing human prostate cancer PC-3 cells in 10-cm culture dishes until the cell confluence is approximately 70%. Set two groups for these cells. One group is treated with DMSO and the other treated with 1 μM SAHA. The treatments last for 12 h 5×107 PC-3 cells are needed for each group (combined as one ChIP-seq sample). For both S2 cells and PC-3 cells adherently grown in culture dishes, following steps are performed:
-
a.Add 1/10 volume of fresh 11% Formaldehyde Solution to plates.
-
b.Swirl plates briefly and let them sit at 21°C for 10 min.
-
c.Add 1/20 volume of 2.5 M glycine to plates to quench formaldehyde.
-
d.Rinse cells twice with 5 mL 1× PBS. Harvest cells using silicon scraper.
-
e.Pool cells in 50 mL conical tubes and spin at 1,000 × g for 5 min at 4°C. Discard supernatant and resuspend pellet in 5 mL 1× PBS per 5×107 cells.
-
f.Transfer 5×107 cells to 15 mL conical tube and spin at 1,000 × g for 5 min at 4°C. Discard supernatant.
-
g.Flash freeze cells in liquid nitrogen and store pellet at −80°C. (Pause point here)
-
a.
-
7.(Prepare LB1, LB2 and LB3 solutions, turn on the sonicator) Cell nucleus sonication: For both S2 cells and PC-3 cell pellets, following steps are performed:
-
a.Resuspend each pellet of 5×107 cells in 2.5 mL of LB1. Rock at 4°C for 10 min. Spin at 1,000 × g for 5 min at 4°C.
-
b.Resuspend each pellet in 2.5 mL of LB2. Rock gently at 21°C for 10 min. Pellet nuclei by spinning at 1,000 × g for 5 min at 4°C.
-
c.Resuspend each pellet in each tube in 1.5 mL LB3.
-
d.Sonicate suspension with a microtip attached to Misonix 3000 sonicator. Samples should be kept in an ice water bath during sonication. To decrease foaming, initially set output power to 4 and increase manually to final power 7 during first burst; sonicate 7 cycles of 30 s ON and 60 s OFF.Note: You will probably need to optimize sonication conditions. These are suggested starting parameters. Shearing varies greatly depending on cell type, growth conditions, quantity, volume, crosslinking, and equipment. Depending on the precise experiment, we use power settings as high as 9, anywhere from 3–12 cycles and variable ratios of time ON and time OFF. In general, we look for the lowest settings that result in sheared DNA that ranges from 100–600 bp in size.
-
e.Add 150 μL of 10% Triton X-100 to sonicated lysate. Split into two 1.5 mL centrifuge tubes. Spin at 11,000 × g for 10 min at 4°C to pellet debris.
-
f.Combine supernatants from the two 1.5 mL centrifuge tubes in a new tube for immunoprecipitation. Samples here could be stored at 4°C for 24 h.
-
g.Save 50 μL of cell nucleus lysate from each sample to measure the DNA concentrations via the Qubit dsDNA quantification system.
-
a.
-
8.Verify the specificity and efficiency of the antibody (here for example, anti-histone H3K27-ac).
-
a.Perform immunoprecipitation (IP) using the anti-histone H3K27-ac antibody in the nucleus lysate of S2 and PC-3 cells respectively. We use Roche Immunoprecipitation kit (cat#11719394001) to carry out the IP. The dilution rate of the antibody in this step is the same as in the ChIP procedures.
-
b.Carry out western blotting, with the same anti-histone H3K27-ac antibody, of both the acid extracted histones (Preparation Step 2 and Preparation Step 5b) and the IP products (Preparation Step 8a).
-
c.The anti-histone H3K27-ac antibody should recognize and precipitate H3K27-ac marker in both Drosophila S2 and PC-3 cells.
-
a.
CRITICAL: The efficiency of one antibody should vary among different species. However, to perform a successful spike-in controlled ChIP, the antibody used in ChIP must demonstrate similar efficiency in both target cells (here PC-3 cells) and control cells (here Drosophila S2 cells).
Key resources table
| REAGENT or RESOURCE |
SOURCE |
IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Polyclonal Anti-Histone H3 (1 in 1000) | Cell Signaling Technology | Cat# 9715; RRID:AB_331563 |
| Rabbit polyclonal Anti-Histone H3K27-ac | Abcam | Cat# ab4729; RRID:AB_2118291 |
| Chemicals, peptides, and recombinant proteins | ||
| Vorinostat (SAHA) | Cayman | Cat# 10009929 |
| Deposited data | ||
| ChIP-seq data | GEO database | GSE137209: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137209. |
| Experimental models: Cell lines | ||
| Human: PC-3 | ATCC | CRL-1435 |
| Drosophila: S2 | ATCC | CRL-1963 |
| Software and algorithms | ||
| SPIKER | https://spiker.readthedocs.io/en/latest/index.html | N/A |
| MACS2 | https://pypi.org/project/MACS2/ | N/A |
| Bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | N/A |
| Samtools | http://www.htslib.org/ | N/A |
| UCSC genome browser | http://genome.ucsc.edu/ | N/A |
| Other | ||
| Polyacrylamide gel electrophoresis and transferring system | Bio-Rad | N/A |
| Sonicator | Misonix 3000 | N/A |
| A rotator working at 4°C | N/A | N/A |
| A centrifuge that spins to 11,000 × g at 4°C | N/A | N/A |
| Thermoshaker | N/A | N/A |
| dsDNA quantification system | Qubit 4 | N/A |
Materials and equipment
Other solutions
| Formaldehyde solution | Final concentration | Amount |
|---|---|---|
| 1M Hepes-KOH, pH 7.5 | 50 mM | 2.5 mL |
| 5M NaCl | 100 mM | 1 mL |
| 0.5M EDTA | 1 mM | 50.0 μL |
| 0.5M EGTA | 0.5 mM | 100.0 μL |
| 37% Formaldehyde | 11% | 100.0 μL |
| ddH2O | n/a | 31.5 mL |
| Total | n/a | 50 mL |
| Block solution | Final concentration | Amount |
|---|---|---|
| 10×PBS | 1×PBS | 10 mL |
| BSA | 0.5% BSA (w/v) | 500.0 mg |
| ddH2O | n/a | 90 mL |
| Total | n/a | 100 mL |
| LB1 | Final concentration | Amount |
|---|---|---|
| 1M Hepes-KOH, pH 7.5 | 50 mM | 5 mL |
| 5M NaCl | 140 mM | 2.8 mL |
| 0.5M EDTA | 1 mM | 0.2 mL |
| 50% glycerol | 10% | 20.0 mL |
| 10% NP-40 | 0.5% | 5.0 mL |
| 10% Triton X-100 | 0.25% | 2.5 mL |
| ddH2O | n/a | 64.5 mL |
| Total | n/a | 100 mL |
| LB2 | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 8.0 | 10 mM | 1 mL |
| 5M NaCl | 200 mM | 4 mL |
| 0.5M EDTA | 1 mM | 0.2 mL |
| 0.5M EGTA | 0.5 mM | 0.1 mL |
| ddH2O | n/a | 94.7 mL |
| Total | n/a | 100 mL |
| LB3 | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 8.0 | 10 mM | 1 mL |
| 5M NaCl | 100 mM | 2 mL |
| 0.5M EDTA | 1 mM | 0.2 mL |
| 0.5M EGTA | 0.5 mM | 0.1 mL |
| 10% Na-Deoxycholate | 0.1% | 1 mL |
| 20% N-lauroylsarcosine | 0.5% | 2.5 mL |
| ddH2O | n/a | 93.2 mL |
| Total | n/a | 100 mL |
| Wash Buffer | Final concentration | Amount |
|---|---|---|
| 1M Hepes-KOH, pH 7.6 | 50 mM | 12.5 mL |
| 5M LiCl | 500 mM | 25 mL |
| 0.5M EDTA | 1 mM | 0.5 mL |
| 10% NP-40 | 1% | 25 mL |
| 10% Na-Deoxycholate | 0.7% | 17.5 mL |
| ddH2O | n/a | 169.5 mL |
| Total | n/a | 250 mL |
| Elution Buffer | Final concentration | Amount |
|---|---|---|
| 1M Tris-HCl, pH 8.0 | 50 mM | 5 mL |
| 0.5M EDTA | 10 mM | 2 mL |
| 10% SDS | 1% | 10 mL |
| ddH2O | n/a | 83 mL |
| Total | n/a | 100 mL |
Note: Store these solutions at 22°C–26°C; the maximum time for storage is 90 days.
Step-by-step method details
Incorporate Drosophila S2 cell chromatin in ChIP samples of human prostate cancer PC-3 cells
The information of pan-chromatin changes would be lost in the PCR-based amplification of DNA library. To retrieve the lost information, it is necessary to use the constant amount of Drosophila chromatin to precipitate alongside with the PC-3 cell chromatin.
-
1.(Prepare the block solution) Pre-block and binding of antibody to magnetic beads.
-
a.Add 100 μL Dynal magnetic beads to microfuge tube. Add 1 mL block solution. Set up 1 tube per IP.
-
b.Collect the beads using magnetic stand. Remove supernatant.
-
c.Wash beads in 1.5 mL block solution two more times.
-
d.Resuspend beads in 250 μL block solution and add 10 μg of antibody.
-
e.Incubate for 16 h on a rotating platform at 4°C.
-
f.Next day, wash beads as described above (3 times in 1 mL block solution).
-
g.Resuspend in 100 μL block solution.
-
a.
-
2.Combine Drosophila S2 cell and human PC-3 cell chromatin from Preparation 2.
-
a.For each spike-in H3K27-ac ChIP experiment (in either DMSO or SAHA treated cells), a 20:1 ratio of PC-3 versus Drosophila S2 cell chromatin was used (according to the DNA quantification results). Typically, we got ∼300 μg DNA in sonicated chromatin from 5×107 PC3 cells (in a volume of ∼1.5 mL). Therefore, the S2 cell chromatin containing 15 μg Drosophila DNA would be added.Note: Once Drosophila S2 and human cells were combined, the sample was treated as a single ChIP-seq sample throughout the experiment until completion of DNA sequencing.
-
b.Add 100 μL antibody/magnetic bead mix to 1.5 mL chromatin mixture produced above.
-
c.Gently mix for 16 h on rotator or rocker at 4°C.
-
a.
-
3.(Prepare Wash Buffer, TE Buffer and Elution Buffer, set thermoshaker to 65°C) Wash, elution, and cross-link reversal after ChIP.
-
a.Pre-chill one 1.5-mL microfuge tube for each ChIP.
-
b.Transfer half the volume of the ChIP material (steps 2b and 2c) to a pre-chilled tube.
-
c.Let tubes sit in magnetic stand to collect the beads. Remove supernatant and add remaining IP. Let tubes sit again in magnetic stand to collect the beads.
-
d.Add 1 mL Wash Buffer to each tube. Remove tubes from magnetic stand and shake or agitate tube gently to resuspend beads. Replace tubes in magnetic stand to collect beads. Remove supernatant. Wash 7 more times.Note: Exact number of washes depends on quality of antibody and may need to be optimized for each antibody. For most antibody we used, washing 6 times or more is applicable.
-
e.Wash once with 1 mL TE buffer that contains 50 mM NaCl.
-
f.Spin at 1,000 × g for 3 min at 4°C and remove any residual TE buffer.
-
g.Add 210 μL of elution buffer.
-
h.Elute at 65°C for 15 min. Resuspend beads every 2 min with brief vortex.
-
i.Spin down beads at 11,000 × g for 1 min at 21°C.
-
j.Remove 200 μL of supernatant and transfer to new tube. Reverse crosslink of this IP DNA by incubating at 65°C for 16 h.
-
a.
-
4.(set thermoshaker to 55°C) Digestion of cellular protein/RNA and extraction of ChIP-DNA.
-
a.Add 200 μL of TE buffer to each tube of IP to dilute SDS in elution buffer.
-
b.Add 8 μL of 10 mg/mL RNaseA (0.2 μg/mL final concentration).
-
c.Mix and incubate at 37°C for 2 h.
-
d.Add 4 μL of 20 mg/mL proteinase K (0.2 μg/mL final concentration).
-
e.Mix and incubate at 55°C for 2 h.
-
f.Add 400 μL phenol: chloroform: isoamyl alcohol (P:C: IA) and transfer the mixture to a new 2-mL tube. Then centrifuge the sample at 12,000 × g for 10 min to separate phases.
-
g.Transfer 400 μL aqueous layer to new centrifuge tube containing 16 μL of 5M NaCl (200 mM final concentration) and 1.5 μL of 20 μg/μL glycogen (30 μg total).
-
h.Add 800 μL EtOH. Incubate for 30 min at −80°C.
-
i.Spin at 11,000 × g for 10 min at 4°C to pellet DNA. Wash pellets with 500 μL of 80% EtOH.
-
j.Dry pellets and resuspend each in 70 μL of 10mM Tris-HCl, pH 8.0.
-
k.Save 15 μL of ChIP product for future checkpoints or verification.
-
l.Measure DNA concentration of ChIP products with Qubit dsDNA quantification system. (Here is a Safe Stopping Point, DNA samples can be stored at −80°C for one month.)
-
a.
Analyzing PC-3 cell ChIP-DNA with spike-in Drosophila DNA
The ChIP-sequencing generally takes 4–55 h, but the exact time can vary greatly depending on the workload of the sequencing core, the model of the sequencers (NextSeq, NovaSeq) used, the flow cells used, and the availability of the computational resources. The analysis time also varies significantly commensurate to personal experiences and different type of tertiary analyses (Figure 2). A walkthrough example to analyze spike-in ChIP-seq data are provided in our online documentation (https://spiker.readthedocs.io/en/latest/index.html)
-
5.
Sequencing. We use The NEB Next Ultra II DNA Library Preparation Kit to convert our ChIP DNA (containing spike-in Drosophila DNA) into libraries for next-generation sequencing on the Illumina platform. To be general, the dsDNA from step 4 is end repaired, 5′ phosphorylated and added with a dA tail at the 3′ ends. Then the processed DNA is ligated with NEBNext Adaptors (adaptor is 1:10 diluted) and U excision is performed. We do clean up of the products without size selection, following the kit instruction. We amplify these DNA to a library yield of ∼1000 ng using PCR (We use unique dual index E6440S that has both forward and reverse primers, PCR cycles is 15). Libraries were sequenced at 30 million fragment reads per sample following Illumina’s standard protocol using the Illumina cBot and HiSeq 3000/4000 PE Cluster Kit. Base-calling is performed using Illumina’s RTA version 2.7.7. Raw reads in FASTQ format are usually delivered from the sequencing core. If pre-aligned BAM files are delivered, we convert them back into FASTQ files using Samtools (e.g., samtools fastq -1 read_1.fq -2 read_2.fq -0 /dev/null -s /dev/null -n input.bam).
-
6.Preprocessing and mapping. After QC, reads are mapped to the composite reference genome comprised of human and Drosophila chromosomes (human/hg38 + Drosophila/dm6).
-
a.Download the human reference genome sequences (the current version is GRCh38 or hg38) and Drosophila reference genome sequences (the current version is “BDGP Release 6 + ISO1 MT” or dm6) reference genome sequences and saved as “hg38.fa.gz” and “dm6.fa.gz”, respectively.
-
b.Uncompress “hg38.fa.gz” and “dm6.fa.gz”.
-
c.Because both human and Drosophila genomes have the chromosome X (named “chrX”) and chromosome Y (named “chrY”), we need to modify the Drosophila chromosome IDs to make them different from humans. In this case, we add “dm6_” to each chromosome ID of the Drosophila. For example, we change Drosophila’s “chrX” into “dm6_chrX”, “chr2L” into “dm6_chr2L”, and so forth. Although changing the human chromosome IDs is technically equivalent at this step (for example, changing human “chr1” into “hg38_chr1”), doing this brings inconvenience even problems present in downstream analyses.
-
d.Concatenate the human reference genome sequences (hg38) and Drosophila reference genome sequences (dm6) into a single FASTA file – the composite reference genome named “hg38_dm6.fa”.
-
e.Index the “hg38_dm6.fa” using the “bowtie2-build” command: bowtie2-build hg38_dm6.fa hg38_dm6
-
f.Map reads to the composite reference genome using Bowtie2. Using Samtools, convert the output alignments into BAM format, sort the BAM files by position, and then index them. Using other short reads aligners such as BWA is fine, as long as the composite reference genome is indexed accordingly.
-
g.Repeat this step for all biological replicates of ChIP and control samples.
-
a.
Figure 2.

Workflow of spike-in ChIP-DNA sequencing and analysis
A step-by-step implementation is given below. Assuming ‘wget’, ‘gunzip’, ‘sed’, ‘bowtie2’, ‘bowtie2-build’ and ‘samtools’ commands are already available in a Linux/Unix environment.
# Step (a): Download reference genome sequences.
$ wget http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz
$ wget http://hgdownload.soe.ucsc.edu/goldenPath/dm6/bigZips/dm6.fa.gz
# Step (b): Uncompress the reference genome files.
$ gunzip hg38.fa.gz
$ gunzip dm6.fa.gz
# Step (c): Change Drosophila chromosome IDs.
$ sed 's/chr/dm6_chr/g' dm6.fa > dm6_renameID.fa
# Step (d): Concatenate the human and Drosophila genome files.
$ cat hg38.fa dm6_renameID.fa > hg38_dm6.fa
# Step (e): Run "bowtie2-build" command to build index files.
# "hg38_dm6" is the prefix of index files, which is needed in the next step.
$ bowtie2-build hg38_dm6.fa hg38_dm6
# Step (f): Use "bowtie2" to map reads to the composite reference genome (i.e., hg38_dm6.fa); save the alignments in BAM format; sort and index the BAM file using samtools.
# For paired-end reads
$ bowtie2 --threads 4 -x GRCh38_dm6 -1 read_1.fastq.gz -2 read_2. fastq.gz | samtools view -Sbh - > output.bam
# For single-end reads
$ bowtie2 --threads 4 -x GRCh38_dm6 -U read.fastq.gz | samtools view -Sbh - > output.bam
# sort the BAM file using samtools
$ samtools sort -@ 4 output.bam > output.sorted.bam
# index the BAM file using samtools
$ samtools index -@ 4 output.sorted.bam
# Step (g) : Repeat steps (a-f) for other samples.
-
7.Quantify Drosophila chromatin. The amount of Drosophila chromatin in the sequencing library can be measured by the number of reads that are aligned to the Drosophila genome. Reads in each BAM file can be classified into four categories including:
-
a.“Human reads”: reads can only map to the human reference genome. These reads are used for peak calling.
-
b.“Drosophila reads”: reads can only map to the Drosophila genome. These reads are used to calculate the scaling factors.
-
c.“Both”: reads can be mapped to both human and the Drosophila genomes. These reads are tossed out.
-
d.“Neither (or unmapped)”: reads cannot reliably map to the composite reference genome. Reads with mapping quality (MAPQ) < 30 are also included in this category.
-
a.
Run “split_bam.py” in the SPIKER package (https://spiker.readthedocs.io/en/latest/index.html) to classify reads and split the “composite” BAM files into Human and Drosophila BAM files.
-
8.
Calculate scaling factors. For both peak calling (i.e., comparing ChIP to control samples) and quantitative analysis (i.e., comparing different ChIP samples), the number of “Drosophila reads” calculated from different samples need to normalize to the same level. For example, if we want to normalize the number of “Drosophila reads” to 1 million, the scaling factor for a particular sample with 800,000 Drosophila reads is 1000000/800000 = 1.25.
-
9.
Peak calling. The peak calling procedure is described in https://spiker.readthedocs.io/en/latest/usage.html. Depending on the histone marker, ENCODE narrowPeak or gappedPeak are generated. bedGraph and bigwig files are also generated for visualization on UCSC genome browser. A demo dataset and step-by-step walkthrough is also available: https://spiker.readthedocs.io/en/latest/walkthrough.html
Expected outcomes
In regular ChIP-seq procedures, both peak calling and quantitative analysis assume that the same number of cells yield the same amount of DNAs (or chromatin) under different conditions. However, this assumption does not hold sometimes (Chen et al., 2015). For example, when the histone methyltransferases, such as EZH2 for H3K27 (Cao et al., 2002), SETD2 for H3K36 (Pfister et al., 2014), DOT1L for H3K79 (Orlando et al., 2014), are mutated or dysregulated. The per-cell DNA/chromatin yield in ChIP experiments is significantly reduced. In these scenarios, conventional normalization and analytic methods fail or compromise the power to identify peaks or detect epigenetic changes. We have also noticed this issue when studying the site-specific histone acetylation changes after HDAC inhibitor treatment (Wu et al., 2021).
Typical spike-in ChIP-seq results from our study are shown in Figure 3. Figure 3A shows the number of sequencing reads, from each ChIP-seq experiment performed in PC-3-GDC-R cells, which are mapped to human or Drosophila genome. Based on this information, we calculated the normalization factors for each ChIP-seq tracks. Figure 3B shows the ChIP-seq tracks without spike-in normalization. Apparently, these tracks hardly reflect the overall increase of H3K27-ac after HDAC inhibitor SAHA treatment. Figure 3C shows the ChIP-seq tracks with spike-in normalization (multiplied by the factors in Figure 3A). After normalization, ChIP-seq tracks become reflecting the increase of H3K27-ac after SAHA treatment.
Figure 3.

Typical outcomes of spike-in ChIP-seq
(A) The number of ChIP-DNA sequencing reads from a spike-in ChIP experiment. This experiment was performed in PC-3 GDC0068 resistant cell lines (PC-3 GDC-R) treated with or without HDAC inhibitor SAHA.
(B) H3K27-ac profiles in a specific region of PC-3 cell genome. These ChIP-seq tracks are not normalized with spike-in control.
(C) H3K27-ac profiles in the same region of PC-3 cell genome as in (B). However, these ChIP-seq tracks are normalized with spike-in control.
Limitations
The precision level of Spike-in controlled ChIP-seq depends on the efficiency of antibody to recognize histone modification marker in both target and control cells. If the antibody has much weaker affinity with the histone modification in the control cells, the result is vulnerable to the ratio of target cell: control cell. Although we used a 20:1 ratio in this protocol, a lower ratio is also recommended.
Troubleshooting
Problem 1
Cell growth might be significantly inhibited after SAHA treatment, which makes the cell number of “SAHA treatment” group much less than control group. (Related to Preparation step 1)
Potential solution
HDAC inhibitors including SAHA slow down growth of normal cells and induce cell death in cancer cells. However, with spike-in control, differences of cell number would have much less impact on the final results. To achieve better results, we recommend to shorten SAHA treatment time to no more than 12 h.
Problem 2
The global change of some histone modifications is not significant by western blotting detection. (Related to Preparation step 4)
Potential solution
We have to clearly determine the significancy of global change of target histone modification, hence to make decision whether or not to perform Spike-in ChIP. Acid extract of histones is suitable for histone acetylation detection but may not for other modification types. SAHA induced increase in histone acetylation is detectable by western blotting. However, for other modification like site-specific histone methylation, mass spectrum-based detection is necessary.
Problem 3
The histone modification to be detected is rare or not conserved in Drosophila. (Related to Preparation step 5)
Potential solution
Drosophila chromatin is preferred but not the only choice as spike-in control. If you decide to use the chromatin of alternative species as spike-in, make sure the antibody you use could recognize the target modification in that species.
Problem 4
Drosophila specific DNA reads are too few in the total ChIP-DNA reads, which makes the normalization inaccurate. (Related to step 2)
Potential solution
Increase Drosophila S2 cell input ratio in step 2a. If necessary, use more cells per ChIP or combine multiple paralleled ChIP-DNA into one to increase the DNA for sequencing (To avoid Drosophila spike-in DNA lost in the library amplification).
Problem 5
The ChIP-DNA yield is low (lower than 5 ng/sample for this case, related to step 4).
Potential solution
If the total ChIP-DNA yield is low, normalization based on spike-in control is inaccurate. To achieve greater DNA yield, increase the input cell number to 2×108 may help. You might also have to increase the antibody used per sample.
Problem 6
How to do normalization using the Drosophila reads? (Related to step 8)
Potential solution
The normalization strategies depend on the data and study design.
Normalize to the minimum (downsampling). If there is a sample with the minimum “Drosophila reads” as well as “human reads”, we could use this sample as the reference (the sample-D in the table below), and “human reads” from all the other samples can be “down-sampled” to the same level as this sample. Advantages: downsampling can be performed at BAM files. After downsampling, the analyses can be handled the same as regular ChIP-seq data. Disadvantages: discard many reads and decrease statistical power of peak calling. This approach is applicable to a dataset where the numbers of reads are similar.
| Sample | Before normalization |
Scaling factors | After normalization |
||
|---|---|---|---|---|---|
| Human_reads (million) | Drosophila_reads (million) | Human_reads (million) | Drosophila_reads (million) | ||
| A | 55 | 2.5 | 2/2.5 = 0.8 | 55∗0.8 = 44 | 2 |
| B | 53 | 2.1 | 2/2.1 = 0.9524 | 53∗0.9524 = 50.48 | 2 |
| C | 52 | 2.4 | 2/2.4 = 0.8333 | 52∗0.8333 = 43.33 | 2 |
| D | 51 | 2 | 1 | 51 | 2 |
Normalize to the median. All samples can be normalized to the median value of Drosophila reads. In the example above, median (2.5, 2.1, 2.4, 2) = 2.25. Advantages: Half of the samples are scaled up and half of the samples are scaled-down, signals after normalization may not off the scale too much. Disadvantages: not suitable for integration analysis, as different datasets would have different median values.
| Sample | Before normalization |
Scaling factors | After normalization |
||
|---|---|---|---|---|---|
| Human_reads (million) | Drosophila_reads (million) | Human_reads (million) | Drosophila_reads (million) | ||
| A | 55 | 2.5 | 2.25/2.5 = 0.9 | 55∗0.9 = 49.5 | 2.25 |
| B | 53 | 2.1 | 2.25/2.1 = 1.0714 | 53∗1.0714 = 56.78 | 2.25 |
| C | 52 | 2.4 | 2.25/2.4 = 0.9375 | 52∗0.9375 = 48.75 | 2.25 |
| D | 51 | 2 | 2.25/2 = 1.125 | 51∗1.125 = 57.375 | 2.25 |
Normalize to a common value (such as 1 million). Advantage: good for integration and visualization of multiple, heterogeneous datasets. Disadvantages: some datasets might be disproportionately scaled up or down.
| Sample | Before normalization |
Scaling factors | After normalization |
||
|---|---|---|---|---|---|
| Human_reads (million) | Drosophila_reads (million) | Human_reads (million) | Drosophila_reads (million) | ||
| A | 55 | 2.5 | 1/2.5 = 0.4 | 55∗0.4= 22 | 1 |
| B | 53 | 2.1 | 1/2.1 = 0.4762 | 53∗0.4762 = 25.24 | 1 |
| C | 52 | 2.4 | 1/2.4 = 0.4166 | 52∗0.4166 = 21.66 | 1 |
| D | 51 | 2 | 1/2 = 0.5 | 51∗0.5 = 25.5 | 1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Haojie Huang (huang.haojie@mayo.edu).
Materials availability
This study did not generate any new unique reagents.
Data and code availability
This study did not generate datasets/codes.
Acknowledgments
We would like to thank Dr. Jeong-Heon Lee at the Epigenetic Development Laboratory at the Center for Individualized Medicine at Mayo Clinic for technical advice for spike-in ChIP. This work was supported in part by grants from the National Institutes of Health (CA130908 and CA203849 to H.H.) and the Mayo Clinic Foundation (to H.H.).
Author contributions
D.W. and L.W. wrote the experimental steps and data analysis sections of the protocol, respectively. H.H., D.W., and L.W. edited and finalized the protocol.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Di Wu, Email: wudi@fudanpci.org.
Haojie Huang, Email: huang.haojie@mayo.edu.
References
- Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Chen K., Hu Z., Xia Z., Zhao D., Li W., Tyler J.K. The overlooked fact: Fundamental need for spike-in control for virtually all genome-wide analyses. Mol. Cell. Biol. 2015;36:662–667. doi: 10.1128/MCB.00970-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando D.A., Chen M.W., Brown V.E., Solanki S., Choi Y.J., Olson E.R., Fritz C.C., Bradner J.E., Guenther M.G. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 2014;9:1163–1170. doi: 10.1016/j.celrep.2014.10.018. [DOI] [PubMed] [Google Scholar]
- Pfister S.X., Ahrabi S., Zalmas L.P., Sarkar S., Aymard F., Bachrati C.Z., Helleday T., Legube G., La Thangue N.B., Porter A.C. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7:2006–2018. doi: 10.1016/j.celrep.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Yan Y., Wei T., Ye Z., Xiao Y., Pan Y., Orme J.J., Wang D., Wang L., Ren S. An acetyl-histone vulnerability in PI3K/AKT inhibition-resistant cancers is targetable by both BET and HDAC inhibitors. Cell Rep. 2021;34:108744. doi: 10.1016/j.celrep.2021.108744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate datasets/codes.