

Abstract
Nonmelanoma skin cancer (NMSC) such as cutaneous squamous cell carcinoma (cSCC) is caused by solar ultraviolet (SUV) exposure and is the most common cancer in the United States. T-LAK cell-originated protein kinase (TOPK), a serine-threonine kinase is activated by SUV irradiation and involved in skin carcinogenesis. Strategies with research focusing on the TOPK signaling pathway and targeted therapy in skin carcinogenesis may helpful for the discovery of additional treatments against skin cancer. In this study, we found that TOPK can directly bind to and phosphorylate c-Jun (as one of the core member of AP-1) at Ser63 and Ser73 after SSL exposure in a JNKs-independent manner. TOPK knocking down, or HI-TOPK-032 (TOPK specific inhibitor) attenuated colony formation and cell proliferation of skin cancer cells. Phosphorylated levels of c-Jun were overexpressed in human AK and cSCC compared with normal skin tissues, and HI-TOPK-032 inhibited the phosphorylation of c-Jun in SCC cell line in a dose-dependent manner. Furthermore, HI-TOPK-032 decreased SSL-induced AP-1 transactivation activity. Moreover, acute SSL-induced inflammation was attenuated by the topical application of HI-TOPK-032 in SKH1 hairless mice. Importantly, HI-TOPK-032 suppressed chronic SSL-induced skin carcinogenesis and c-Jun phosphorylation levels in SKH1 hairless mice. Our results demonstrate that TOPK can phosphorylate and activate c-Jun at Ser63 and Ser73 in the process of skin carcinogenesis and HI-TOPK-032 could be used as a potential chemopreventive drug against cSCC development.
Introduction
Skin cancer is the most common malignancy in the United States. Each year there are more new cases of skin cancer than the combined incidence of cancers of the breast, prostate, lung, and colon [1]. Nonmelanoma skin cancers (NMSC), which include basal cell carcinomas and squamous cell carcinomas (SCC) [2], are the most common skin cancers and about 90% of NMSC are associated with exposure to solar ultraviolet (SUV) radiation from the sun [3]. Therefore, understanding the mechanisms of SUV-induced skin carcinogenesis and finding effective strategies for preventing skin cancer is important.
Signaling cascades that lead to the activation of mitogen-activated protein kinases (MAPK) play an important role in UV-induced cutaneous SCC (cSCC) [4]. Serine-threonine kinase T-LAK cell-originated protein kinase (TOPK/PBK) is a member of the MAPK kinase family, which is involved in tumor development, cancer growth, apoptosis, and inflammation [5]. Only a few papers have been published about molecular mechanisms of TOPK on skin carcinogenesis. Previous studies suggested that TOPK contributed to p38 activation and the phosphorylation of JNKs during the UV-induced DNA damage response [6, 7]. Our recent study demonstrated that TOPK is an oncogenic factor in skin carcinogenesis. Furthermore, our recent clinical data showed that acute SUV irradiation increased epidermal thickness as a feature of inflammation and enhanced total protein and phosphorylation levels of TOPK in human skin tissues [8]. This suggests that TOPK plays an important role in SUV-induced skin carcinogenesis and can be a potential target for skin cancer prevention and therapy.
The activator protein 1 (AP-1) family of transcription factors has been generally characterized as a dominant promoter of skin cancer. Activated in SCC, the c-Jun is one of the core member of AP-1 which is a critical contributor to transformation and tumorigenesis. Co-expression of c-Jun with oncogenic Ras was sufficient to transform primary human epidermal cells into malignant cells in a regenerated human skin grafting model [9]. The previous study showed that TOPK promoted lung cancer resistance to EGFR tyrosine kinase inhibitors by phosphorylating and activating c-Jun, which consequently activated the transcription of AP-1 target genes [10]. However, the relationship between these two oncoproteins in mediating skin carcinogenesis is still unclear.
HI-TOPK-032 is a TOPK specific inhibitor, which is reported to inhibit TOPK kinase activity and effectively suppress colon cancer cell growth as well as increase colon cancer cell apoptosis. Administration of HI-TOPK-032 suppressed tumor growth in a colon cancer xenograft model [11].
Here, we elucidated TOPK’s regulation of c-Jun to better illustrate the mechanism of skin carcinogenesis. Moreover, we demonstrated that HI-TOPK-032 can suppress SUV-induced skin carcinogenesis through the TOPK-c-Jun axis.
Results
TOPK directly phosphorylates c-Jun at serine 63 and 73
To find the direct downstream target of TOPK on the AP-1 activation-associated signaling pathway in solar UV-induced skin carcinogenesis, we screened the potential candidate proteins including p38α, ATF2, and c-Jun by an in vitro kinase assay (Supplementary Fig. 1). Our in vitro kinase assay result indicated that TOPK can directly phosphorylate c-Jun (Fig. 1a). We next evaluated whether TOPK could phosphorylate serine residues within the N-terminal transactivation domain of the c-Jun protein at residues Ser63 and Ser73. Interestingly, TOPK can directly phosphorylate c-Jun at Ser63 and Ser73 (Fig. 1b). Next, we purified the mutant c-Jun proteins that both serine 63 and 73 residues were replaced with alanine (S63A and S73A) from BL21 competent Escherichia coli. In vitro kinase assay results indicated that TOPK phosphorylated wild-type (wt) c-Jun at Ser63 and Ser73 but could not phosphorylate mutant c-Jun (S63A and S73A) proteins (Fig. 1c). Our result indicated that TOPK could phosphorylate the serine residues within the N-terminal transactivation domain of the c-Jun protein at residues Ser63 and Ser73.
Fig. 1. The c-Jun is a substrate of TOPK.

a Kinase assay results with active TOPK and a purified His-tagged c-Jun protein detected by autoradiography. b Kinase assay results using active TOPK or JNKs and a purified His-tagged c-Jun protein detected by western blot analysis. Active JNK was used as a positive control. c Kinase assay results using active TOPK and purified GST-tagged c-Jun wild-type (wt) or mutant c-Jun (mut) detected by western blot analysis. Endogenous binding activity between TOPK and c-Jun in human precancerous HaCaT keratinocytes treated with SSL (60 kJ/m2 UVA and 2.9 kJ/m2 UVB). d Immunoprecipitation (IP) with TOPK or e p-c-Jun (Ser63) antibody and Protein G Sepharose 4 Fast Flow was performed and then TOPK, p-c-Jun (Ser63) or c-Jun was detected by western blot (WB) analysis. f Phosphorylated c-Jun levels in wild-type (TOPK+/+) control or TOPK knockout (TOPK−/−) SKH1 (Crl:SKH1-Hrhr) MEFs are stimulated by SSL (60 kJ/m2 UVA and 2.9 kJ/m2 UVB) in a time-dependent manner.
Our previous study showed that TOPK phosphorylation in HaCaT cells was increased by solar simulated light (SSL) irradiation in a dose- and time-dependent manner [8]. We then investigated the phosphorylation levels of c-Jun at Ser63 and Ser73 in SSL-stimulated HaCaT cells. SSL irradiation induced the phosphorylation of TOPK very early at 15–45 min [8] followed by the later phosphorylation of c-Jun at 60–120 min (Supplementary Fig. 2). We also examine the endogenous binding activity between TOPK and c-Jun in SSL-stimulated human precancerous HaCaT keratinocytes. SSL irradiation induced the protein interaction between TOPK and c-Jun in human precancerous HaCaT keratinocytes (Fig. 1d, e). To investigate whether c-Jun phosphorylation is dependent on the TOPK protein in SSL-stimulated cells, we isolated mouse embryonic fibroblasts (MEFs) from TOPK wt (TOPK+/+) or TOPK knockout (TOPK−/−)-SKH1 (Crl:SKH1-Hrhr) hairless mice. Phosphorylation of c-Jun at Ser63 and Ser73 was markedly increased after SSL irradiation in TOPK+/+ cells (Fig. 1f). However, although phosphorylation of c-Jun in TOPK−/− cells was slightly increased by SSL irradiation, the level of phosphorylated forms was extremely less than that observed in TOPK+/+ cells at the time point 180 min (Fig. 1f). Our results indicate that TOPK can directly phosphorylate c-Jun at Ser63 and Ser73 and the phosphorylation levels of c-Jun are dependent on the TOPK protein.
TOPK activates c-Jun independent of JNKs activation
According to a previous study, when activated by a MAPK cascade, JNKs specifically phosphorylate serine residues within the N-terminal transactivation domain of Jun at residues Ser63 and Ser73 and thereby enhances its transactivation activity [7, 12–14]. Therefore, to examine whether TOPK could phosphorylate the c-Jun on JNK1 absence, we used MEFs isolated from JNK1 knockout (JNK1−/−)-SKH1 hairless mice. To isolate the activate form of endogenous TOPK protein, the JNK1−/− cells were stimulated with solar stimulated light (SSL) and then subjected to an immunoprecipitation (IP)-kinase assay with a TOPK antibody. Purified His-c-Jun proteins as a substrate were reacted with the TOPK–bead complex. Our results showed that SSL-activated TOPK can phosphorylate the c-Jun at Ser63 and Ser73 in JNK1−/− cells (Fig. 2a).
Fig. 2. TOPK can phosphorylate c-Jun in the JNK-independent pathway.

a Immunoprecipitation (IP)-kinase assay results from lysates of JNK1−/− murine embryonic fibroblasts (MEFs) treated or not treated with SSL (60 kJ/m2 UVA and 2.9 kJ/m2 UVB) visualized by western blot analysis. b Immunoprecipitation (IP)-kinase assay results using the epidermal keratinocytes isolated from TOPK wild-type (TOPK+/+) or TOPK knockout (TOPK−/−)-SKH1 (Crl:SKH1-Hrhr) hairless mice. c Phosphorylation of c-Jun in wild-type (wt) and JNK1−/− (left panel), or JNK2−/− (right panel) MEFs transfected with shTOPK or shMock and treated or not treated with SSL irradiation.
To prove the requirement of TOPK kinase activity for c-Jun phosphorylation, we performed IP-kinase assay using the lysates isolated from the TOPK wild-type (TOPK+/+) or TOPK knockout (TOPK−/−)-SKH1 (Crl:SKH1-Hrhr) hairless mice. Briefly, we isolated the epidermal keratinocytes from TOPK+/+ or TOPK−/−-SKH1 hairless mice. And then, to isolate the active form of the endogenous TOPK protein, SSL (60 kJ/m2 UVA and 2.9 kJ/m2 UVB) was treated, and cells were incubated for 60 min at 37 °C. Then, cell extracts were incubated with an anti-TOPK antibody and Protein G Sepharose 4 Fast Flow (GE Healthcare). The purified His-c-Jun protein was used as a substrate of TOPK for the in vitro kinase assay. Phosphorylation levels of p-c-Jun at Ser63 or Ser73 were detected by western blot analysis. Our data showed that SSL-activated TOPK can phosphorylate the c-Jun at Ser63 and Ser73 (Fig. 2b).
Furthermore, we next investigated the effect of TOPK absence on c-Jun phosphorylation in JNK1−/− or JNK2−/− cells. We transfected shMock or shTOPK into JNK1−/− or JNK2−/− MEFs, and then cells were stimulated with SSL irradiation. The results indicated that phosphorylation of TOPK and c-Jun at Ser63 and Ser73 was increased in shMock-transfected wt cells, JNK1−/− (Fig. 2c, left panel) or JNK2−/− (Fig. 2c, right panel) MEFs stimulated with SSL irradiation. The wt cells expressing shTOPK decreased the phosphorylation of c-Jun at Ser63 and Ser73. Furthermore, SSL-induced c-Jun phosphorylation was decreased in JNK1−/−and JNK2−/− cells expressing shTOPK (Fig. 2c). Interestingly, all our results indicate that SSL irradiation-activated TOPK can directly phosphorylate c-Jun at both Ser63 and Ser73 through the JNK-independent mechanism.
TOPK inhibition attenuates colony formation and cell proliferation of skin cancer cells
Anchorage-independent growth is a frequent characteristic of transformed cells that correlates with tumorigenic potentials such as migration, colonization, and tumor growth in vivo [15]. Thus, to examine the effect of TOPK on colony formation and cell proliferation of human skin SCC cells, we established the SCC cell lines (A431 and SCC12) that TOPK is knocking down (Fig. 3a). Interestingly, the knocking down of TOPK decreased the anchorage-independent colony formation of both human skin epidermoid carcinoma A431 and human skin squamous cell carcinoma SCC12 cells (Fig. 3b). We then examined the effect of HI-TOPK-032 as a specific TOPK inhibitor on the anchorage-independent colony formation. HI-TOPK-032 significantly inhibited the epidermal growth factor (EGF)-induced colony formation in normal mouse skin epidermal JB6 Cl 41 cells (JB6) and immortal human precancerous HaCaT keratinocytes in a dose-dependent manner (Fig. 3c, d). Furthermore, HI-TOPK-032 attenuated the anchorage-independent colony formation of human skin SCC cell lines such as A431 (Fig. 3e) and SCC12 (Fig. 3f). Moreover, the TOPK absence suppressed the anchorage-dependent cell proliferation of A431 (Fig. 3g, left panel) and SCC12 (Fig. 3g, right panel). We confirmed that TOPK inhibition by treatment of HI-TOPK-032 can suppress the anchorage-dependent cell proliferation of A431 (Fig. 3h, left panel) and SCC12 (Fig. 3h, right panel) in a dose-dependent manner. Our results indicated that TOPK inhibition can attenuate colony formation and cell proliferation in skin cancer cells.
Fig. 3. TOPK inhibition suppresses anchorage-independent and -dependent growth of skin cancer cells.

a Human skin SCC cell lines (A431 and SCC12) expressing shMock or shTOPK. b The effect of shTOPK on anchorage-independent cell growth of A431 and SCC12. HI-TOPK-032 and EGF were treated in c JB6 Cl 41 or d HaCaT cells. HI-TOPK-032 was treated in e A431 and f SCC12 for colony formation assay. MTS assay was performed in g shTOPK-expressing or h HI-TOPK-032-treated A431 and SCC12 cells. Data represent mean values ± S.D. of three independent experiments. *P < 0.05 or **P < 0.01 vs. shMock (b, g), EGF only (c, d), or media only (e–h) group. #P < 0.01 vs. no EGF group (c, d).
Phosphorylated c-Jun levels are highly regulated in human AK and SCC
Next, we examined the protein levels of phosphorylated c-Jun at Ser63 and Ser73 in human cutaneous actinic keratosis (AK) and SCC. Normal skin, AK and SCC skin tissues in humans were supplied by the University of Arizona Cancer Center (Tucson, AZ) for immunofluorescence staining to check the phosphorylated c-Jun levels. Our data showed that phosphorylated c-Jun at Ser63 (Fig. 4a) and Ser73 (Fig. 4b) had levels that were markedly higher in AKs and SCCs from patients as compared with normal skin. We next asked whether TOPK inhibition by the TOPK specific inhibitor, HI-TOPK-032, can regulate the phosphorylation of c-Jun in human SCC cells. Thus, we treated HI-TOPK-032 to the human skin SCC cell line (A431) and examined the protein levels of phosphorylated c-Jun. Our results showed that TOPK inhibition attenuated the phosphorylation of c-Jun at both Ser63 and Ser73 (Fig. 4c).
Fig. 4. Effect of HI-TOPK-032 on c-Jun phosphorylation and AP-1 transactivation activity.

Immunofluorescence staining to check the phosphorylated c-Jun at a Ser63 or b Ser73 in human AKs (9 samples from 3 human subjects) and SCCs (15 samples from 5 human subjects) compared with normal skin (24 samples from 8 human subjects). Phosphorylated c-Jun levels are presented as the sum of relative intensity units. **P < 0.01 vs. normal tissue. Scale bar = 100 μm. c A431 cells were treated with HI-TOPK-032 for 24 h and analyzed by Western blot analysis. d AP-1 and β-galactosidase-transfected HaCaT cells were incubated with HI-TOPK-032 and then SSL (60 kJ/m2 UVA and 2.9 kJ/m2 UVB) were treated. AP-1 luciferase activity was measured and normalized against the β-galactosidase activity. #P < 0.01 vs. vehicle alone (without SSL). *P < 0.05 or **P < 0.01 vs. SSL alone (with vehicle).
Activated by a MAPK cascade, the JNKs translocated to the nucleus, where they phosphorylated Jun within its N-terminal transactivation domain including residues Ser63 and Ser73, thereby enhancing its transactivation potential [12]. Thus, we next asked whether TOPK inhibition can downregulate the AP-1 transactivation. HI-TOPK-032 decreased SSL-induced AP-1 transactivation activity (Fig. 4d). These results indicated that TOPK inhibition can downregulate the phosphorylation of c-Jun in SCC cells and SSL-induced AP-1 transactivation.
Potential preventive value of HI-TOPK-032 in chronic SSL-induced skin carcinogenesis
We next examined whether HI-TOPK-032 could have the preventive potential against SSL-induced skin carcinogenesis. Our previous study showed that chronic SSL irradiation induced the formation of cSCC and papilloma on SKH1 hairless mice [8, 16]. We used this cSCC mouse model as described in “Materials and methods”. We created an oil-in-water emulsion cream (patent pending) containing 0.1% or 1% (w/w) HI-TOPK-032, and then cream or HI-TOPK-032 cream was applied to the dorsal area of the SKH1 hairless mouse skin 1 h before SSL irradiation exposure on the same day. The time schedule of the cream treatment or SSL irradiation was illustrated in Supplementary Fig. 3. There are no studies that have examined gender differences in skin carcinogenesis following exposure to SSL in mouse models. Thus, we used both female and male mice and compared tumor volume and numbers. Topical application of 0.1% HI-TOPK-032 dramatically decreased tumor volume (Fig. 5a, left panel) and numbers (Fig. 5a, right panel) in chronic SSL-exposed skin in female SKH1 hairless mice (Fig. 5a, b). We also examine the efficacy of HI-TOPK-032 on chronic SSL-induced skin cancer development in male mice. Male mice showed a much larger volume and number of tumors by irradiation of chronic SSL (Fig. 5c, d) as compared with female mice. In fact, according to previous research, the overall estimated incidence rate in males was higher than in females in humans [17]. Topical application of HI-TOPK-032 decreased tumor volume (Fig. 5c, left panel) and numbers (Fig. 5c, right panel) that were induced by chronic SSL irradiation in male SKH1 hairless mice (Fig. 5c, d). We also examine the endogenous binding activity between TOPK and c-Jun in chronic SSL-stimulated SKH1 hairless mice. Protein binding activity between TOPK and c-Jun in chronic SSL-cSCC samples was increased by chronic SSL irradiation (Fig. 5e). Hematoxylin and eosin (H&E) staining results showed that chronic SSL irradiation induced large epidermal changes, dermal invasion, and the formation of abundant large keratin pearls as the histological features of SCC (Fig. 5f). Furthermore, the levels of phosphorylated c-Jun at Ser63 and Ser73 were decreased by TOPK inhibition in chronic SSL-exposed SKH1 hairless mouse skin (Fig. 5f). Our chronic SSL-exposed mouse models indicated that cSCC and papilloma development was prevented by blocking TOPK activity.
Fig. 5. The effect of HI-TOPK-032 as a TOPK inhibitor on chronic SSL-induced skin carcinogenesis in SKH1 (Crl:SKH1-Hrhr) hairless mice.
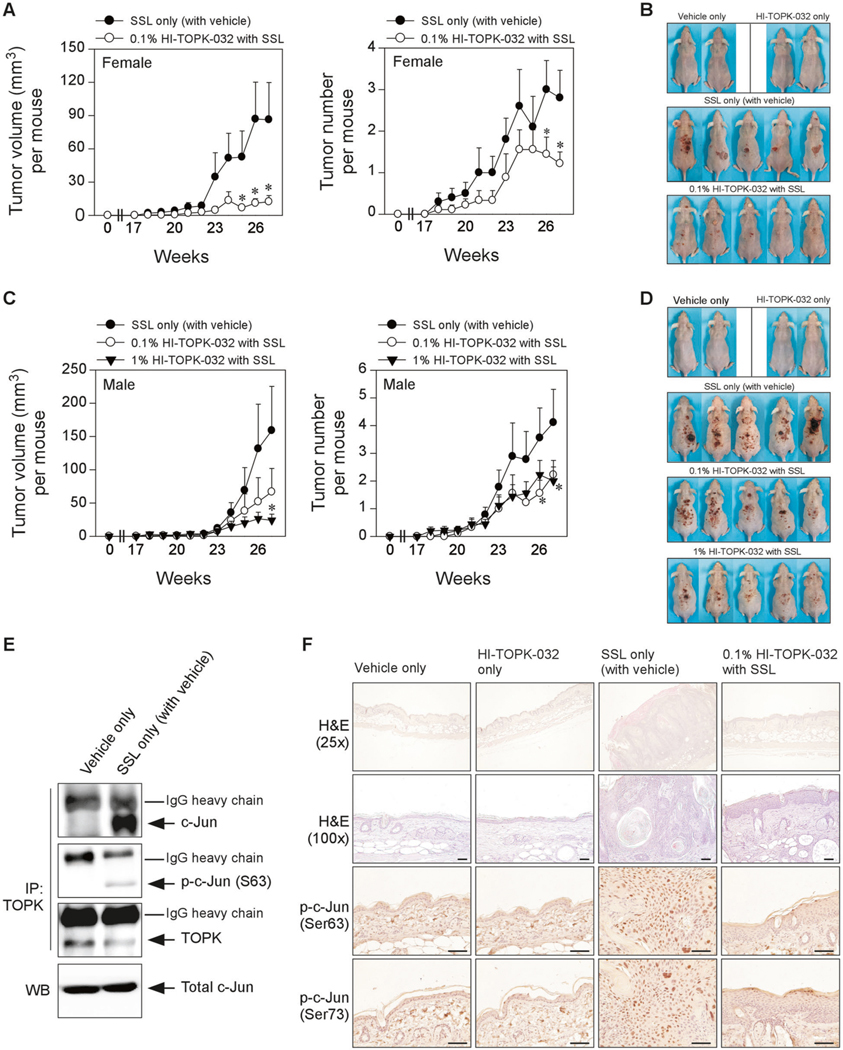
a Tumor volume and numbers and b skin photography at week 27 in female SKH1 hairless mice. c Tumor volume and numbers and d skin photography at week 27 in male SKH1 hairless mice. Data are shown as mean values ± S.D. Significant differences were determined by one-way ANOVA. The asterisk (*) indicates a significant (p < 0.1) difference compared with the groups treated with only SSL (with the vehicle). e Endogenous binding activity between TOPK and c-Jun in chronic SSL-treated SKH1 hairless mouse skin. Immunoprecipitation (IP) with TOPK antibody and Protein G Sepharose 4 Fast Flow was performed and then TOPK, p-c-Jun (Ser63) or c-Jun was detected by western blot (WB) analysis. f At week 27, tissues were collected for H&E staining (×25 and ×100 magnification) and IHC to detect phosphorylated c-Jun (Ser63 and Ser73). Scale bars = 1100 μm.
In addition, we examined the mutation levels of p53 protein in chronic SSL-exposed SKH1 hairless mice. Mutations of the p53 gene are usually detected in cSCC and solar UV irradiation is a pivotal factor to cause the mutation of p53 [18, 19]. Thus, we examined the mutation levels of p53 in chronic SSL-exposed mouse skin tissues. Importantly, chronic SSL irradiation increased the mutation of p53 (Fig. 6a, b). We observed the tissues with HI-TOPK-032 treatment and malignant lesions were not detected in mice treated with HI-TOPK-032. Notably, HI-TOPK-032 treatment groups showed less mutation of p53 on chronic SSL-stimulated female mouse skin tissues (Fig. 6a, b).
Fig. 6. The effect of HI-TOPK-032 on chronic SSL-induced p53 mutations in SKH1 hairless mice.

a Chronic SSL (60 kJ/m2 UVA and 2.9 kJ/m2 UVB) was irradiated to SKH1 hairless mice for 15 weeks and induced cSCC development. To examine the mutation of p53, the skin tissues from female mice were collected and IHC was performed. Scale bars = 1100 μm. b Mutant p53 levels are presented as the sum of integrated optical density (IOD) values. #P < 0.01 vs. vehicle only (without SSL). **P < 0.01 vs. SSL only (with vehicle).
Downregulation of phosphorylated c-Jun by inhibiting TOPK on acute SSL-exposed mouse skin
Increased epidermal thickness is a characteristic of solar UV-induced skin inflammation [20, 21]. Our previous clinical data with acute exposure of human skin to SSL supported the importance of the TOPK signaling pathway in solar UV-mediated inflammatory responses [8]. We next examined the effect of HI-TOPK-032 on epidermal thickness as an inflammation feature and c-Jun phosphorylation induced by acute SSL irradiation. Our results showed that the mouse skin exhibited increased epidermal thickness and this increase was attenuated by the topical application of HI-TOPK-032 cream in acute SSL-exposed SKH1 hairless mice (Fig. 7a). Furthermore, HI-TOPK-032 inhibited the protein levels of phosphorylated c-Jun at Ser63 and Ser73 (Fig. 7b).
Fig. 7. Effects of HI-TOPK-032 on acute SSL-induced inflammation in SKH1 hairless mice.
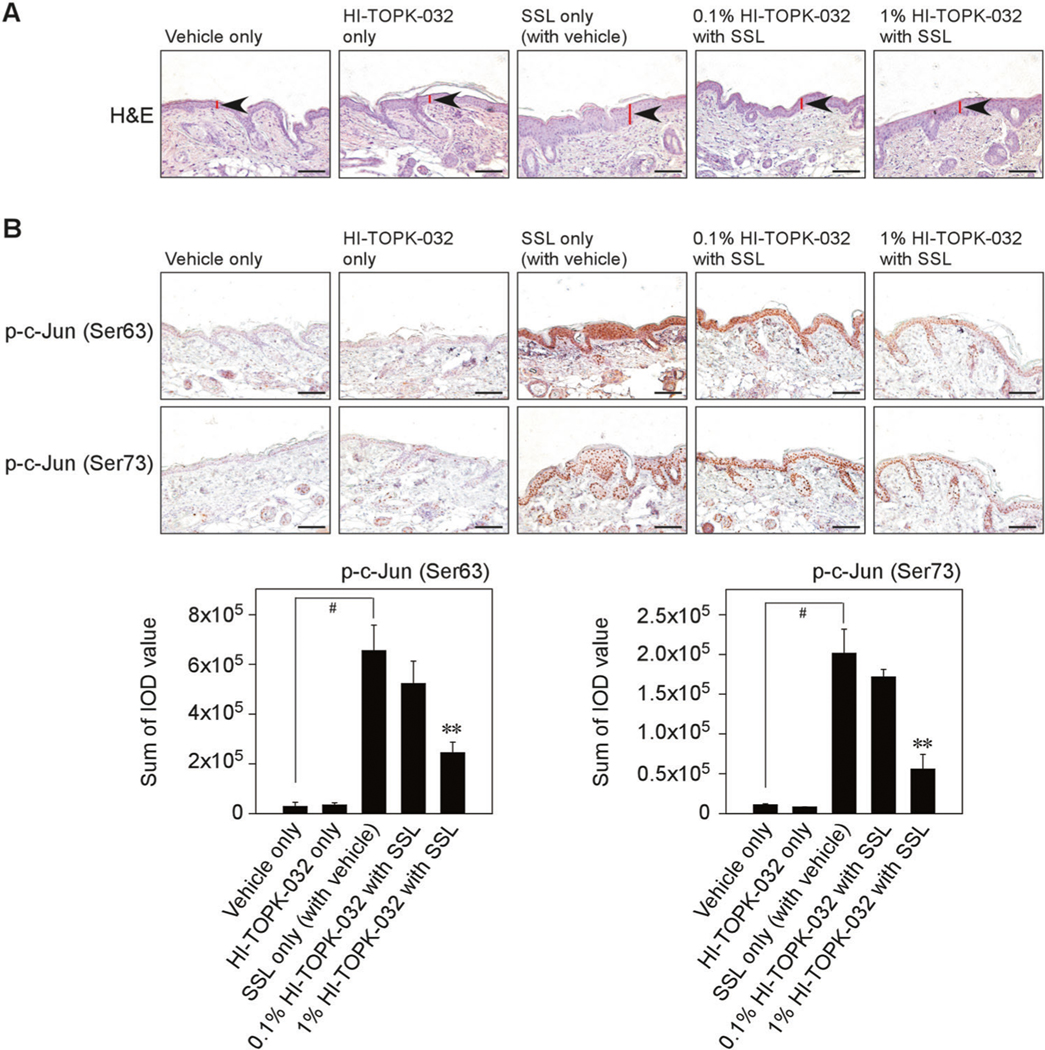
a At 24 h after SSL irradiation, mouse skin tissues were prepared for H&E staining to compare the epidermal thickness. b IHC was conducted to examine the effect of HI-TOPK-032 as a TOPK specific inhibitor on phosphorylated c-Jun levels in acute SSL-stimulated SKH1 hairless mouse skin. Phosphorylated c-Jun (Ser63 and Ser73) levels are presented as the sum of IOD values. #P < 0.01 vs. vehicle only (without SSL). **P < 0.01 vs. SSL only (with vehicle). Scale bar = 1100 μm.
The upregulated TOPK/c-Jun axis facilitates keratinocytes transformation
Next, to examine the effect of the TOPK/c-Jun axis during keratinocytes transformation, we used the pLVX-IRES-puro (pLVX) vector which is an HIV-1-based, lentiviral expression vector that allows the simultaneous expression of interest protein. Wt TOPK, wt c-Jun, or mutant c-Jun (S63A/S73A) gene was sub-cloned into the pLVX-IRES-puro vector. To establish the stable cell lines, the plasmids such as pLVX only, pLVX-TOPK, pLVX-c-Jun, pLVX-mutant c-Jun, pLVX-TOPK plus pLVX-c-Jun, or pLVX-TOPK plus pLVX-mutant c-Jun (S63A/S73A) were transfected into the human keratinocytes such as human precancerous HaCaT keratinocytes and human epidermal keratinocyte (HEKa). And then, keratinocytes transformation assay was performed with the treatment of 100 ng/ml human epidermal growth factor (hEGF) in HEKa or without hEGF in HaCaT. Colony transformation was successfully established from the single cell of human keratinocytes such as HaCaT and HEKa after 10 days and 3 weeks, respectively. The c-Jun overexpressing cells (pLVX-c-Jun) increased colony transformation in both human HaCaT keratinocytes (Fig. 8a, b) and HEKa (Fig. 8c, d). Interestingly, mutant c-Jun significantly increased colony transformation than wt c-Jun-overexpressing cells. Furthermore, TOPK overexpressing cells (pLVX-TOPK-transfected) contributed more high levels of colony transformation than wt c-Jun overexpressing cells (pLVX-c-Jun-transfected). Once both TOPK and c-Jun are overexpressed in HaCaT and HEKa, colony transformation was upregulated. Notably, TOPK and mutant c-Jun (S63A/S73A) overexpressing cells significantly upregulated keratinocytes transformation to the colony formation in both human HaCaT keratinocytes (Fig. 8a, b) and HEKa (Fig. 8c, d). Furthermore, both TOPK and wt c-Jun or mutant c-Jun overexpressing cells showed large colony formation in both human keratinocytes. Our data demonstrated that TOPK/c-Jun axis can contribute to the transformation of primary HEKa induced by EGF and human precancerous HaCaT keratinocytes.
Fig. 8. The effect of the TOPK/c-Jun axis during keratinocytes transformation.

TOPK, c-Jun, or mutant c-Jun (S63A/S73A) gene was sub-cloned to the pLVX-IRES-puro (pLVX) vector which is a lentiviral vector for bicistronic expression of a gene together with a puromycin-resistance marker. Stable cell lines that express the TOPK, c-Jun, mutant c-Jun (S63A/S73A), TOPK plus c-Jun, and TOPK plus mutant c-Jun (S63A/S73A) in human keratinocytes such as a, b HaCaT and c, d HEKa were established. And then, keratinocytes transformation assay was performed without human epidermal growth factor (hEGF) in HaCaT or with 100 ng/mL hEGF in HEKa. Colonies were observed and analyzed using a LEICA DM IRB microscope after 10 days (for HaCaT) or 3 weeks (for HEKa). Data represent mean values ± S.D. of three independent experiments. #P < 0.01 vs. pLVX vector only. *P < 0.05 or **P < 0.01 vs. pLVX-TOPK or pLVX-c-Jun.
Our previous study indicated that solar UV irradiation increased TOPK activation to induce skin carcinogenesis [8]. Therefore, understanding the TOPK molecular mechanism and identifying binding partners of TOPK that can help prevent skin carcinogenesis is an important task. In this study, we demonstrated that TOPK directly can bind and phosphorylate the c-Jun at Ser63 and Ser73 in a JNK-independent manner (representative TOPK-c-Jun pathway in skin carcinogenesis was illustrated in Supplementary Fig. 4). Furthermore, TOPK inhibition by HI-TOPK-032 as a TOPK specific inhibitor suppressed SSL-induced skin carcinogenesis and thus could be an effective clinical approach to prevent papilloma and cSCC development.
Discussion
The MAPKs belong to the family of serine/threonine protein kinases including JNKs, ERK1/2, and p38 MAPK to promote the various cellular processes including adaptation to stress, proliferation, differentiation, or apoptosis [22–25]. The TOPK could have an oncogenic potential by continuously maintaining the phosphorylation of oncogenic substrates [26]. In our recent study, we showed that TOPK could be a potential chemopreventive or therapeutic target against SUV-induced skin inflammation and carcinogenesis [8]. Thus, focusing on the inhibition of the promising TOPK protein could be a new potential strategy to prevent skin carcinogenesis. But, only a few potential TOPK inhibitors have been discovered and reported against SUV-induced skin inflammation and carcinogenesis [27–29]. Furthermore, although there are few studies about TOPK being highly overexpressed in solar dermatitis and melanoma, only one potential direct substrate of TOPK in skin carcinogenesis has been reported. Our previous paper showed that TOPK can directly bind and phosphorylate the p53-related protein kinase in skin carcinogenesis [8]. Thus, more research is necessary to identify potential downstream targets of TOPK and their specific phosphorylation site(s) associated with skin carcinogenesis.
JNKs are members of the MAPK superfamily and phosphorylate serine residues within the N-terminal transactivation domain of the Jun protein (as one of the core members of AP-1) at residues Ser63 and Ser73 [12, 30]. JNKs phosphorylate the c-Jun which is a central player for cell-cycle regulation and cell proliferation of fibroblasts and keratinocytes [31, 32]. Furthermore, the AP-1 family of transcription factors has been generally characterized as a dominant promoter of skin cancer. In the previous study, we provided evidence showing that TOPK could phosphorylate JNK1 and TOPK regulation of JNK1 activity enhanced the ability of JNKs to mediate H-Ras-induced cell transformation [7]. Interestingly, in the present study, our data indicated that TOPK directly can phosphorylate the c-Jun in a JNK-independent manner. Furthermore, the protein levels of phosphorylated c-Jun at Ser63 and Ser73 are higher in human cutaneous AK and cSCC compared with normal skin. Our finding could contribute to the additional strategies about the TOPK signaling mechanism. Thus, we expect that TOPK is a pivotal target that could control solar UV-induced skin carcinogenesis. Another paper showed that TOPK phosphorylated ERK2 in vitro and their interaction increased tumorigenesis of HCT116 colorectal cancer cells [26]. These studies clearly indicate that the TOPK is a potential central player of the MAPK cascade mechanism to regulate cell proliferation, colony formation, and cancer development.
A high-risk feature of cSCC includes immunosuppression and chronic SUV irradiation that induces immunosuppression by regulating interferon-γ, interleukin-12, and prostaglandin E2 (PGE2) [33–36]. The function of TOPK immunosuppression was not examined in the present study. Therefore, additional studies should be addressed to determine whether TOPK associates with SSL-induced immunosuppression in the future.
In conclusion, we demonstrated that TOPK can directly bind and phosphorylate the c-Jun at Ser63 and Ser73 on chronic SSL-exposed skin cells. Furthermore, HI-TOPK-032 as a specific TOPK inhibitor significantly attenuated the development of cSCC and papillomas by downregulating the TOPK-c-Jun pathway. Moreover, HI-TOPK-032 decreased the epidermal thickness and phosphorylation of c-Jun on acute SSL-stimulated mouse skin tissues. Thus, we propose that c-Jun is a new downstream target of TOPK in skin carcinogenesis and molecular targeting the TOPK-c-Jun axis could be an additional strategy to approach the clinical trials against skin cancer, especially cSCC.
Materials and methods
Human skin tissues
Normal tissues (eight human subjects), AKs (three human subjects) and SCCs (five human subjects) in humans were supplied by the University of Arizona Cancer Center (Tucson, AZ). The University of Arizona Institutional Review Board approved the study (the University of Arizona Human Subjects Protection Program) and written informed consent was obtained from all study participants. Methods were conducted in accordance with relevant regulations and guidelines from the University of Arizona Cancer Center Scientific Review Committee.
Animal care and maintenance
SKH1 (Crl:SKH1-Hrhr) hairless mice (strain code 477, Charles River Laboratories, Burlington, MA) were maintained in The Hormel Institute animal facilities according to the guidelines approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC). Mice were randomly grouped by age and body weight, and 10–15 mice per group are typically used and treated for criteria achievement of statistical significance without investigator blinding.
Solar simulated light (SSL) source for experiments
The SSL source comprised of UVA-340 lamps were purchased from Q-Lab Corporation (Cleveland, OH). This system of SSL irradiation mimics natural sunlight including both UVA and UVB. The UVA-340 lamps provide the best possible simulation of sunlight in the critical short wavelength region from 365 nm down to the solar cutoff of 295 nm, with peak emission at 340 nm. The percentage of UVA and UVB emitted from the UVA-340 lamps was measured by a UV radiometer and was 94.5% and 5.5%, respectively.
Generation of SKH1 (Crl:SKH1-Hrhr) TOPK knockout (TOPK−/−) mice
TOPK (PKB) heterozygous (TOPK+/−) female and male mice harboring the C57BL/6 × DBA hybrid genetic background were purchased from Texas Institute for Genomic Medicine (College Station, TX), and homozygous TOPK knockout (TOPK−/−) mice were produced by mating TOPK+/− female and male mice. The genetic background of homozygous TOPK−/− mice was filtered to the SKH1 (Crl: SKH1-Hrhr) hairless mouse (Charles River; Burlington, MA) background as described previously [8]. Mice were maintained in the animal facilities of The Hormel Institute following the University of Minnesota Animal Care and Use Committee (IACUC) guidelines.
Chronic SSL-induced skin carcinogenesis model in SKH1 hairless mice (prevention model)
Female and male SKH1 (Crl:SKH1-Hrhr) hairless mice (5–6 weeks old, body weight 22–24 g) were purchased from Charles River (Burlington, MA) and the mice were maintained in The Hormel Institute vivarium under ‘specific pathogen-free’ conditions following the University of Minnesota Animal Care and Use Committee (IACUC) guidelines. To establish the criteria for achieving statistical significance, 20 female and 20 male mice were randomly grouped. We created an oil-in-water emulsion cream (patent pending) containing 0.1% or 1% (w/w) of HI-TOPK-032 and the cream was applied to the dorsal area of the SKH1 (Crl:SKH1-Hrhr) hairless mice skin without investigator blinding, 1 h before SSL irradiation exposure on the same day. During week 1, the SSL dose was 37 kJ/m2 UVA and 1.8 kJ/m2 UVB applied three times per week. The doses of SSL irradiation were gradually increased by 10% each week. At week 6, the SSL dose was 60 kJ/m2 UVA and 2.9 kJ/m2 UVB and these doses were maintained until week 15. Only cream or cream containing HI-TOPK-032 was applied for 27 weeks and SSL irradiation was continued for 15 weeks as described in Supporting Information Fig. S3.
Statistical analyses
The GraphPad Prism 5.0 software (GraphPad Software; La Jolla, CA) was used for all the statistical analyses. To compare the statistical significance between three or more groups, one-way ANOVA was used, and the student’s t test was used to compare two groups. Statistical p values (*p < 0.05 or **p < 0.01) were statistically significant between groups.
Additional experimental procedures used in this study can be found in Supplementary Materials and Methods online.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Supplementary Material
Acknowledgements
We thank Dr Richard Flavell (Yale University School of Medicine, New Haven, CT) for providing JNK1−/− and JNK2−/− mice, and Dr Dirk Bohmann (European Molecular Biology Laboratory, Germany) for providing His6-tagged c-Jun expression plasmid. This work was supported by The Hormel Foundation and National institutes of Health grants (CA229112).
Footnotes
Compliance with ethical standards
Supplementary information The online version of this article (https://doi.org/10.1038/s41388-020-1286-4) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Rogers HW, Weinstock MA, Feldman SR, Coldiron BM. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the U.S. population, 2012. JAMA Dermatol. 2015;151:1081–6. [DOI] [PubMed] [Google Scholar]
- 2.Stern RS. Prevalence of a history of skin cancer in 2007: results of an incidence-based model. Arch Dermatol. 2010;146:279–82. [DOI] [PubMed] [Google Scholar]
- 3.Koh HK, Geller AC, Miller DR, Grossbart TA, Lew RA. Prevention and early detection strategies for melanoma and skin cancer. Curr Status Arch Dermatol. 1996;132:436–43. [PubMed] [Google Scholar]
- 4.Bode AM, Dong Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci STKE. 2003;2003:RE2. [DOI] [PubMed] [Google Scholar]
- 5.Gaudet S, Branton D, Lue RA. Characterization of PDZ-binding kinase, a mitotic kinase. Proc Natl Acad Sci USA. 2000;97: 5167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu K, Yu D, Cho YY, Bode AM, Ma W, Yao K, et al. Sunlight UV-induced skin cancer relies upon activation of the p38alpha signaling pathway. Cancer Res. 2013;73:2181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oh SM, Zhu F, Cho YY, Lee KW, Kang BS, Kim HG, et al. T-lymphokine-activated killer cell-originated protein kinase functions as a positive regulator of c-Jun-NH2-kinase 1 signaling and H-Ras-induced cell transformation. Cancer Res. 2007;67:5186–94. [DOI] [PubMed] [Google Scholar]
- 8.Roh E, Lee MH, Zykova TA, Zhu F, Nadas J, Kim HG, et al. Targeting PRPK and TOPK for skin cancer prevention and therapy. Oncogene. 2018;37:5633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin JY, Ke H, Hall RP, Zhang JY. c-Jun promotes whereas JunB inhibits epidermal neoplasia. J Investig Dermatol. 2011;131: 1149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Yang Z, Li W, Xu S, Wang T, Wang T, et al. TOPK promotes lung cancer resistance to EGFR tyrosine kinase inhibitors by phosphorylating and activating c-Jun. Oncotarget. 2016;7:6748–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim DJ, Li Y, Reddy K, Lee MH, Kim MO, Cho YY, et al. Novel TOPK inhibitor HI-TOPK-032 effectively suppresses colon cancer growth. Cancer Res. 2012;72:3060–8. [DOI] [PubMed] [Google Scholar]
- 12.Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117:5965–73. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Minemoto Y, Lin A. c-Jun N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for tumor necrosis factor alphainduced c-Jun kinase activation and apoptosis. Mol Cell Biol. 2004;24:10844–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell. 2004;15:713–25. [DOI] [PubMed] [Google Scholar]
- 15.Gassmann P, Haier J. The tumor cell-host organ interface in the early onset of metastatic organ colonisation. Clin Exp Metastasis. 2008;25:171–81. [DOI] [PubMed] [Google Scholar]
- 16.Kim JE, Roh E, Lee MH, Yu DH, Kim DJ, Lim TG, et al. Fyn is a redox sensor involved in solar ultraviolet light-induced signal transduction in skin carcinogenesis. Oncogene. 2016;35:4091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiser I, Scope A, Azriel D, Zloczower E, Carmel NN, Shalom A. Head and neck cutaneous squamous cell carcinoma clinicopathological risk factors according to age and gender: a population-based study. Isr Med Assoc J. 2016;18:275–8. [PubMed] [Google Scholar]
- 18.Edlund K, Larsson O, Ameur A, Bunikis I, Gyllensten U, Leroy B, et al. Data-driven unbiased curation of the TP53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proc Natl Acad Sci USA. 2012;109:9551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loyo M, Li RJ, Bettegowda C, Pickering CR, Frederick MJ, Myers JN, et al. Lessons learned from next-generation sequencing in head and neck cancer. Head Neck. 2013;35:454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Orazio J, Jarrett S, Amaro-Ortiz A, Scott T. UV radiation and the skin. Int J Mol Sci. 2013;14:12222–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumura Y, Ananthaswamy HN. Toxic effects of ultraviolet radiation on the skin. Toxicol Appl Pharmacol. 2004;195:298–308. [DOI] [PubMed] [Google Scholar]
- 22.Decean HP, Brie IC, Tatomir CB, Perde-Schrepler M, Fischer-Fodor E, Virag P. Targeting MAPK (p38, ERK, JNK) and inflammatory CK (GDF-15, GM-CSF) in UVB-activated human skin cells with Vitis vinifera seed extract. J Environ Pathol, Toxicol Oncol. 2018;37::261–72. [DOI] [PubMed] [Google Scholar]
- 23.Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, et al. Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell. 2018;34:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto H, Ryu J, Min E, Oi N, Bai R, Zykova TA, et al. TRAF1 is critical for DMBA/solar UVR-induced skin carcinogenesis. J Investig Dermatol. 2017;137:1322–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin Q, Han T, Fang B, Zhang G, Zhang C, Roberts ER, et al. K27-linked ubiquitination of BRAF by ITCH engages cytokine response to maintain MEK-ERK signaling. Nat Commun. 2019;10:1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu F, Zykova TA, Kang BS, Wang Z, Ebeling MC, Abe Y, et al. Bidirectional signals transduced by TOPK-ERK interaction increase tumorigenesis of HCT116 colorectal cancer cells. Gastroenterology. 2007;133:219–31. [DOI] [PubMed] [Google Scholar]
- 27.Gao G, Zhang T, Wang Q, Reddy K, Chen H, Yao K, et al. ADA-07 suppresses solar ultraviolet-induced skin carcinogenesis by directly inhibiting TOPK. Mol Cancer Ther. 2017;16:1843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Zhang Z, Ge R, Zhang J, Liu W, Mou K, et al. Gossypetin inhibits solar-uv induced cutaneous basal cell carcinoma through direct inhibiting PBK/TOPK protein kinase. Anti-cancer Agents Med Chem. 2019;19:1029–36. [DOI] [PubMed] [Google Scholar]
- 29.Xue P, Wang Y, Zeng F, Xiu R, Chen J, Guo J, et al. Paeonol suppresses solar ultraviolet-induced skin inflammation by targeting T-LAK cell-originated protein kinase. Oncotarget. 2017;8:27093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–68. [DOI] [PubMed] [Google Scholar]
- 31.Behrens A, Sibilia M, David JP, Mohle-Steinlein U, Tronche F, Schutz G, et al. Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. EMBO J. 2002;21:1782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li G, Gustafson-Brown C, Hanks SK, Nason K, Arbeit JM, Pogliano K, et al. c-Jun is essential for organization of the epidermal leading edge. Dev Cell. 2003;4:865–77. [DOI] [PubMed] [Google Scholar]
- 33.Burton KA, Ashack KA, Khachemoune A. Cutaneous squamous cell carcinoma: a review of high-risk and metastatic disease. Am J Clin Dermatol. 2016;17:491–508. [DOI] [PubMed] [Google Scholar]
- 34.Katiyar SK. Interleukin-12 and photocarcinogenesis. Toxicol Appl Pharmacol. 2007;224:220–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trifan OC, Hla T. Cyclooxygenase-2 modulates cellular growth and promotes tumorigenesis. J Cell Mol Med. 2003;7:207–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yokogawa M, Takaishi M, Nakajima K, Kamijima R, Digiovanni J, Sano S. Imiquimod attenuates the growth of UVB-induced SCC in mice through Th1/Th17 cells. Mol Carcinog. 2013;52:760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files].