

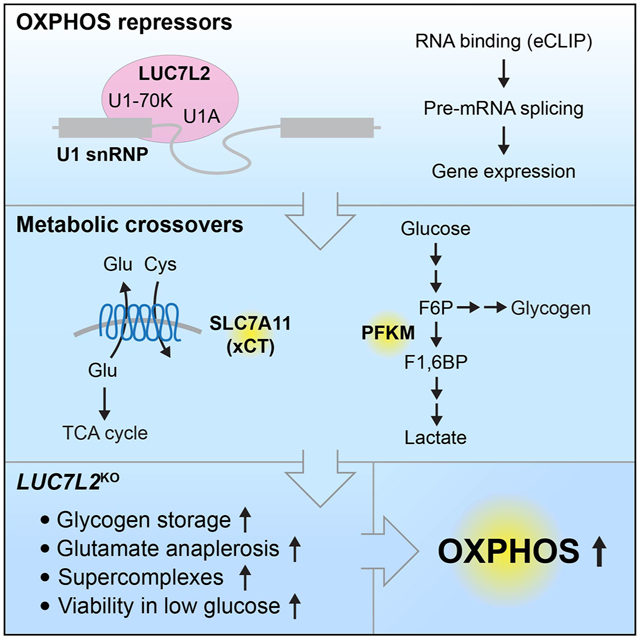
SUMMARY
Oxidative phosphorylation (OXPHOS) and glycolysis are the two major pathways for ATP production. The reliance on each varies across tissues and cell states, and can influence susceptibility to disease. At present, the full set of molecular mechanisms governing the relative expression and balance of these two pathways is unknown. Here, we focus on genes whose loss leads to an increase in OXPHOS activity. Unexpectedly, this class of genes is enriched for components of the pre-mRNA splicing machinery and in particular for subunits of the U1 snRNP. Among them, we show that LUC7L2 represses OXPHOS and promotes glycolysis by a coordinated mechanism, involving (1) splicing of the glycolytic enzyme PFKM to suppress glycogen synthesis, (2) splicing of the cystine/glutamate antiporter SLC7A11 (xCT) to suppress glutamate oxidation, and (3) secondary repression of mitochondrial respiratory supercomplex formation. Our results connect LUC7L2 and, more generally, the U1 snRNP to cellular energy metabolism.
Graphical Abstract
eTOC
Jourdain et al., report the identification of OXPHOS repressors: genes whose loss shifts metabolism from glycolysis to OXPHOS. Prominent in this set are members of the U1 snRNP, including LUC7L2, which the authors show is required for splicing of PFKM and SLC7A11 (xCT) and controls glycogen and glutamate metabolism.
INTRODUCTION
Human cells employ two chief pathways for generating ATP: glycolysis and oxidative phosphorylation (OXPHOS) (Fig. 1A). Use of these metabolic routes is associated with key tradeoffs, and while glycolysis tends to be kinetically favorable, the ATP yield from OXPHOS is higher (Pfeiffer et al., 2001). The relative balance of OXPHOS and glycolysis varies across tissues. For example, while cardiac tissue is rich in mitochondria and highly oxidative in its metabolism, proliferating cells from the thymus are highly glycolytic (Warburg, 1924). Balance between these two programs can vary during cellular differentiation and in response to environmental stimuli. Activation of immune cells is often accompanied by rewiring towards glycolysis, while stem cell differentiation results in increased OXPHOS (Pearce et al., 2013, Ito and Suda, 2014). Cells also acutely respond to nutrient and oxygen availability to adjust flux through these pathways within minutes (Crabtree, 1929, Pasteur, 1861), while oncogenic transformation promotes aerobic glycolysis (Warburg, 1924). Notably, differential reliance on OXPHOS versus glycolysis can be exploited for therapeutic benefit (Bonnet et al., 2007, Gohil et al., 2010).
Figure 1: Identification of Pre-mRNA Splicing Components as Repressors of OXPHOS. See also Figure S1 and Table S1.
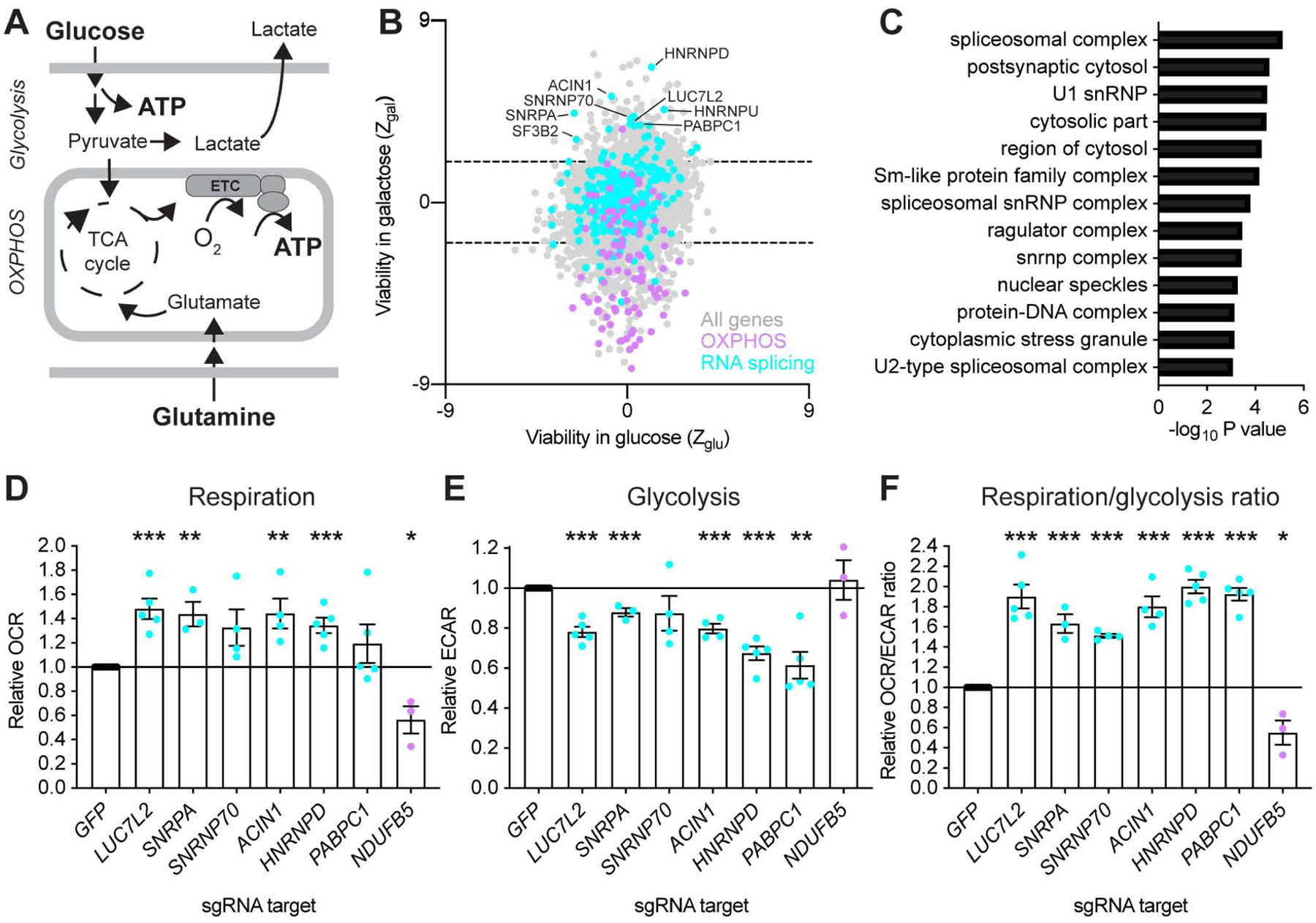
(A) Overview of the main ATP-generating pathways in human cells. OXPHOS: oxidative phosphorylation. ETC: electron transport chain. TCA: tricarboxylic acid cycle. (B) Gene-level analysis of a genome-wide CRISPR/Cas9 screen in glucose and galactose. Each dot represents an expressed, non-essential gene (n = 9,189). (C) Gene ontology analysis generated using a gene list ranked by viability in galactose against GO components. (D–F) Functional validation of the screening results. Basal whole-cell oxygen consumption rates (OCR), extracellular acidification rates (ECAR) and OCR/ECAR ratios were simultaneously measured after CRISPR/Cas9-mediated gene depletion in K562 cells grown in glucose-containing media. Data are shown as mean ± SEM (n≥3 independent experiments. *P<0.05, **P<0.01, ***P<0.001, t-test relative to control (GFP) sgRNA-treated cells. NDUFB5 is a control with known role in OXPHOS.
A small number of genomic programs have been identified that can influence the balance of cellular energy metabolism. For example, the transcriptional co-activator PGC-1α integrates nutrient levels and physiological inputs to orchestrate a genomic program that induces the expression of the OXPHOS machinery (Puigserver et al., 1998). Conversely, the transcription factor HIF-1α promotes expression of a set of genes, including glycolytic enzymes, in response to a decline in oxygen levels (Huang et al., 1998). Post-transcriptionally, RNA-binding proteins such as CLUH bind to a large number of mRNAs encoding mitochondrial proteins (Gao et al., 2014). A few instances of alternative splicing regulating individual metabolic enzymes have also been reported. For example, alternative splicing to generate the PKM2 isoform of the pyruvate kinase, re-routing lower glycolytic carbon flux, is believed to contribute to cancer progression (Christofk et al., 2008).
High-throughput approaches can provide insight into the regulation and patterning of cellular metabolic programs (Hillenmeyer et al., 2008, VanderSluis et al., 2014). We previously reported a nutrient-sensitized screen for small molecules that impact the fitness of cells in galactose, a poor substrate for glycolysis, and focused on dozens of small molecules that induce a shift from OXPHOS to glycolysis (Gohil et al., 2010). Recently, we reported the genome-wide identification of genes necessary to sustain OXPHOS (Arroyo et al., 2016). We systematically catalogued genes whose loss impaired OXPHOS in human cells, including 72 that underlie known OXPHOS diseases. However, that report did not explore the opposite side of the screen, which could in principle include pathways that tonically suppress OXPHOS.
Here, we report the genome-wide identification of “OXPHOS repressors”, defined as genes whose knockout promotes relative fitness in the absence of glucose as a fuel for glycolysis. We validate top-scoring genes and show that their depletion augments OXPHOS activity. OXPHOS repressors are enriched for components of the pre-mRNA splicing machinery, including subunits of the U1 snRNP. Among them, we show that LUC7L2 encodes a U1 snRNP subunit involved in pre-mRNA splicing and gene expression. Amongst LUC7L2 gene targets, we focus on two genes, PFKM and SLC7A11 (xCT), which we show represent two metabolic crossovers that influence the bioenergetic state of the cell in a LUC7L2-dependent manner.
RESULTS
Genome-Wide Search for Factors that Limit OXPHOS Identifies Components of the Pre-mRNA Splicing Machinery
To nominate genes whose depletion promotes OXPHOS, we reanalyzed the results of our genome-wide “death screen” that compared viability of CRISPR/Cas9 mutagenized K562 cells that are shifted to glucose or galactose conditions for 24 hours (Arroyo et al., 2016). Loss of genes required for OXPHOS is tolerated in the presence of glucose, whereas these genes are conditionally essential in galactose, as it is a poor substrate for glycolysis (Robinson et al., 1992). We reanalyzed 9,189 expressed non-essential genes in K562 cells, calculating a z-score of viability of the gene knockout under each condition (Fig. 1B, Table S1). In this analysis, sgRNAs targeting 3,726 unexpressed genes were used as negative controls. Gene Ontology (GO) analysis confirmed our previous result: that the depletion of genes encoding subunits of the mitochondrial respiratory chain caused loss of viability, as expected (Fig. S1A).
Unexpectedly, the genes whose depletion promotes relative viability in galactose were enriched in splicing-related GO terms, including “spliceosomal complex” (P<10−5) and “U1 snRNP” (P<10−4) (Fig. 1C, S1B). Hits with these GO terms included U1 snRNP-specific subunits (LUC7L2, SNRPA, SNRNP70), heterogeneous nuclear ribonucleoproteins (HNRNPD, HNRNPU), splicing factors (SF3B2, SFPQ), RNA helicases (DHX8, DDX47), an LSm-family protein (LSM1), an exon junction complex protein (ACIN1), and a polyadenylate-binding protein (PABPC1). Other gene expression pathways were not significantly enriched.
We used CRISPR/Cas9 and sgRNA sequences from the screening library to disrupt the expression of six representative genes identified in the screen that were not previously linked to energy metabolism, including LUC7L2, SNRPA (U1A), SNRNP70 (U1–70K), ACIN1, HNRNPD and PABPC1. We included a gene encoding a subunit of respiratory complex I (NDUFB5) as a control with known impact on metabolism, and measured oxygen consumption rates (OCR), a proxy for OXPHOS, and extracellular acidification rates (ECAR), a proxy for glycolysis, in transduced K562 cells. Notably, depletion of several of these genes significantly increased basal, maximal and ATP-linked OCR (Fig. 1D, S1C–D), while also decreasing ECAR (Fig. 1E), suggesting rewiring of metabolism from glycolysis to OXPHOS. In fact, the OCR/ECAR ratio was significantly increased upon depletion of all six selected genes (Fig. 1F), whereas it decreased in NDUFB5-depleted cells, as expected. Together, these results confirm that silencing certain pre-mRNA splicing genes boosts oxidative energy metabolism.
Expression of LUC7L2 Represses OXPHOS
Among the validated screening hits, LUC7L2 showed the most robust phenotype. This relatively unstudied gene belongs to the LUC7 family together with LUC7L and LUC7L3, which are all homologs of yeast LUC7, a U1 snRNP protein involved in pre-mRNA splicing (Fortes et al., 1999). Mutations, haploinsufficiency, and complete loss of LUC7L2 are all associated with poorer survival in myelodysplastic syndromes (MDS) (Singh et al., 2013), while Arabidopsis LUC7 genes contribute to development and stress responses (de Francisco Amorim et al., 2018). None of the LUC7 family members have been previously linked to energy metabolism.
To further investigate the function of LUC7L2, we used CRISPR/Cas9 to generate single-cell clones in which the expression of LUC7L2 was ablated (Fig. S2A–C). We observed that LUC7L2KO K562 cells grew more slowly relative to wild-type in standard 25mM glucose cell culture conditions (Fig. 2A). However, they grew comparatively faster than LUC7L2WT cells when glycolysis was limiting, either pharmacologically by treatment with 2-deoxyglucose (2-DG) or when glucose was replaced by galactose (Fig. 2B). In contrast, LUC7L2 depletion sensitized cells to killing by pharmacologic inhibition of OXPHOS (Fig. 2C).
Figure 2: LUC7L2 Impacts Metabolic State-Dependent Cell Growth and Bioenergetics. See also Figure S2.
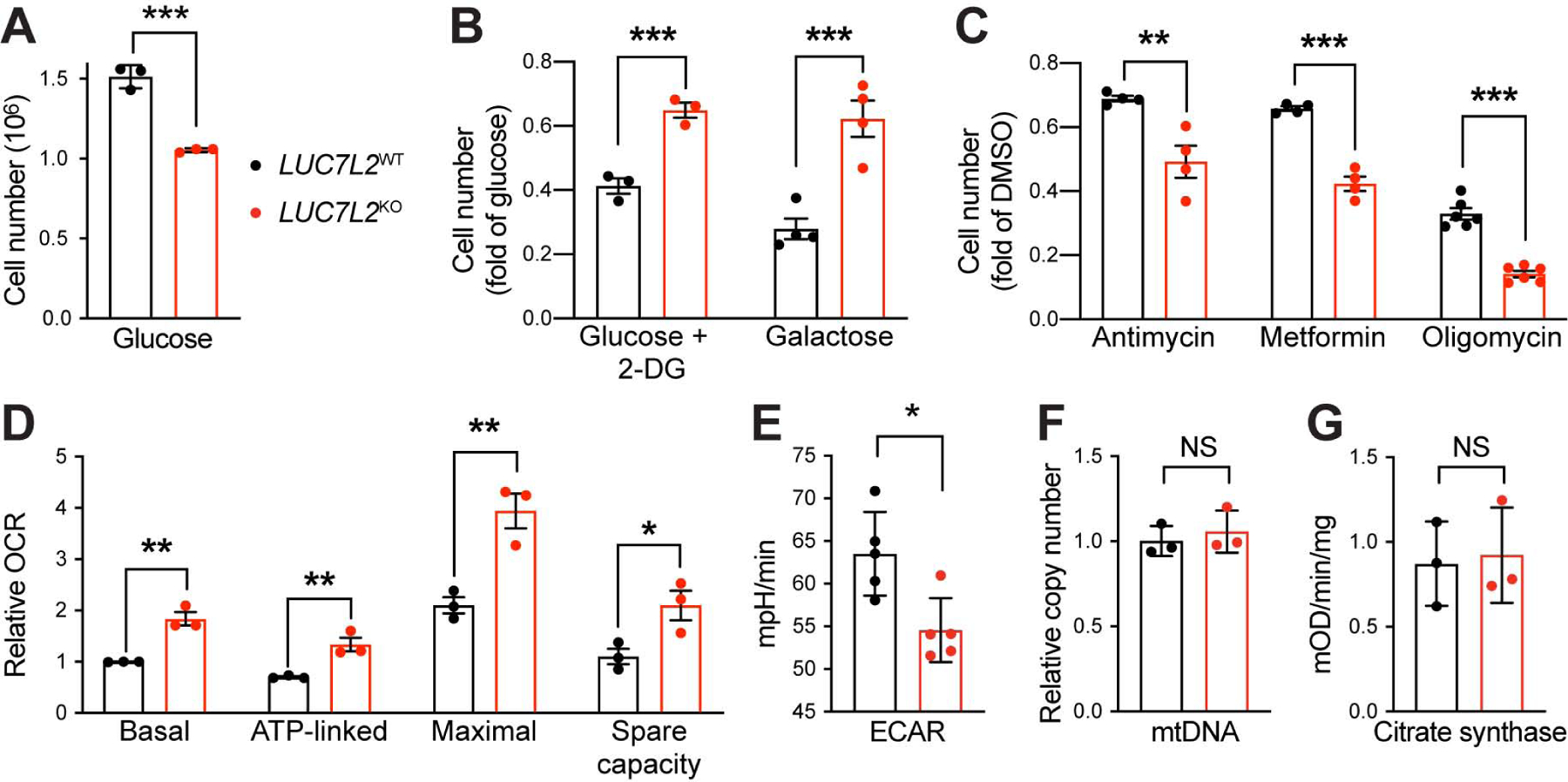
(A–C) Cell proliferation of LUC7L2KO K562 cells grown in (A) glucose and (B) treated with 2-deoxyglucose (2-DG) or when glucose was replaced by galactose or (C) glucose with OXPHOS inhibitors. (D) Respiratory parameters of LUC7L2KO cells as determined by oxygen consumption rate (OCR). (E) Basal glycolytic activity in LUC7L2KO cells as determined by extracellular acidification rate (ECAR). (F) Relative mtDNA abundance and (G) citrate synthase activity of LUC7L2KO cells. All data are shown as mean ±SEM (n≥3). *P< 0.05, **P<0.01, ***P<0.001, t-test relative to LUC7L2WT cells.
Next, we characterized the bioenergetic consequences of LUC7L2 depletion. OCR measurement confirmed our initial observation that LUC7L2 represses OXPHOS and, accordingly, all measured respiratory parameters in LUC7L2KO K562 and HAP1 cells were increased relative to controls (Fig. 2D, S2D–F). As expected from our initial validation, LUC7L2-depleted K562 clones exhibited less ECAR (Fig. 2E). To test whether the global abundance of mitochondria was affected by LUC7L2 depletion, we quantified mitochondrial DNA copy number and citrate synthase activity and observed no differences (Fig. 2F–G). Electron microscopy also confirmed the absence of gross differences in mitochondrial abundance or ultrastructure in these cells (Fig. S2G). Collectively, our results indicate that LUC7L2 impacts metabolic state-dependent cell growth and bioenergetics. While LUC7L2 loss does not appear to affect the gross abundance of mitochondria, it influences the balance between activity of glycolysis and OXPHOS.
Metabolic Basis of the Shift from Glycolysis to OXPHOS in LUC7L2-Depleted Cells
The rewiring of cellular bioenergetics upon LUC7L2 depletion prompted us to analyze the abundance of metabolic intermediates central to glycolysis and OXPHOS. We used LC-MS to quantify the relative steady-state levels of 122 metabolites in cell pellets as well as the absolute consumption/release rates of 22 media metabolites (Table S2–3). Consistent with our ECAR results, we observed reduced rates of glucose uptake, lactate secretion, and media acidification as well as a dramatically decreased media lactate/pyruvate ratio in LUC7L2KO K562 cells, all consistent with decreased glycolysis (Fig. 3A–C, S3C, S3F). In pellets of LUC7L2KO, we observed significant accumulation of glucose, glucose-6-phosphate and of the fructose-6-phosphate/glucose-1-phosphate isomers. Notably, levels of fructose-1,6-bisphosphate, the fourth intermediate of glycolysis and the product of phosphofructokinase (PFK), were significantly reduced in the absence of LUC7L2. Thus, we observed accumulation of the substrate of PFK and depletion of its product, identifying this enzyme as a metabolic crossover in LUC7L2KO cells. Accordingly, a loss of function mutation in PFKM leads to skeletal muscle glycogen accumulation (Tarui et al., 1965), and we observed significant accumulation of glycogen and its precursor UDP-glucose in these cells (Fig. 3A).
Figure 3: Metabolite Analysis in LUC7L2-Depleted Cells Reveals Crossovers at Phosphofructokinase and System Xc−. See also Figure S3 and Table S2–3.
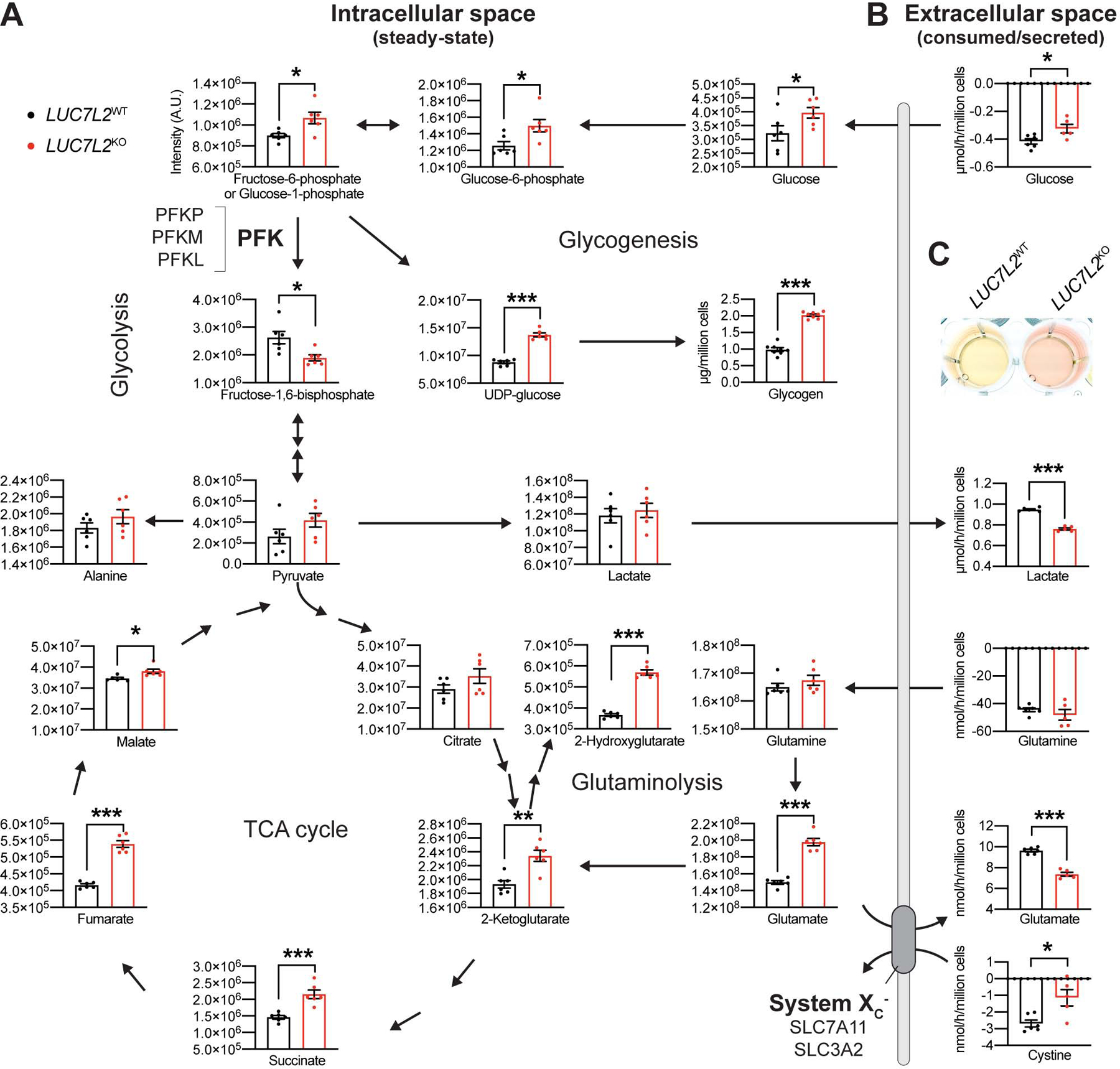
(A) Intracellular levels of metabolites in LUC7L2KO K562 cells as determined by LC-MS. (B) Extracellular levels of metabolites as determined by LC-MS analysis of the spent media from (A). Positive and negative values illustrate metabolite secretion and consumption by the cells, respectively. All data are shown as mean ±SEM (n = 5–8). *P<0.05, **P<0.01, ***P<0.001, t-test relative to LUC7L2WT. (C) Media acidification of LUC7L2KO K562 cells grown in glucose.
Our metabolite analysis also provided insight into how mitochondrial metabolism is rewired in LUC7L2KO cells. We observed accumulation of four of the five TCA metabolites analyzed (2-ketoglutarate, succinate, fumarate, malate) as well as two TCA-cycle derived metabolites (2-hydroxyglutarate, aspartate) (Fig. 3A, S3A). Glutamine is an important fuel that can contribute glutamate to the TCA cycle (Reitzer et al., 1979), but glutamine consumption and intracellular glutamine levels remained unchanged (Fig. 3A–B). In contrast, intracellular glutamate accumulated in LUC7L2KO cells. Intracellular glutamate may either be converted into 2-ketoglutarate to serve as an anaplerotic input into the TCA cycle or be exported out of the cell in exchange for cystine via the system Xc−, a plasma membrane antiporter encoded by two subunits, SLC7A11 (xCT) and SLC3A2 (4F2) (Sato et al., 1999). The system Xc− has previously been implicated in the survival of cells in low glucose conditions (Shin et al., 2017, Koppula et al., 2017). Importantly, we found that while intracellular glutamate accumulated in LUC7L2-depleted cells, its secretion to the culture media was significantly reduced (Fig. 3B). These observations pointed to a second crossover at the level of the system Xc−, as LUC7L2-depleted cells also consumed less media cystine (Fig. 3B, S3E).
Collectively, analysis of steady-state intracellular metabolites as well as consumption and release of media metabolites indicate that the loss of LUC7L2 modulates two key crossovers in energy metabolism: (1) upper glycolysis/glycogen storage at PFK, and (2) glutamate oxidation/secretion via the exchange of cystine and glutamate at the transporter system Xc−. The direction of these metabolic changes is concordant with the observed shift from glycolysis to OXPHOS in cells lacking LUC7L2.
LUC7L2 is a U1 snRNP Subunit Involved in Pre-mRNA Splicing
Next, we sought to determine how loss of LUC7L2 leads to remodeling of cellular energy metabolism. LUC7L2 is not a well-studied gene, but its yeast and plant homologs are components of the U1 snRNP. Hence, we predicted that LUC7L2 encodes a component of the mammalian snRNP, impacting splicing and expression of genes that might be influencing energy metabolism.
We used confocal microscopy to confirm the nuclear localization of LUC7L2 and observed that it localized in SRSF2-positive nuclear speckles, which are themselves enriched for the pre-mRNA splicing machinery (Rino et al., 2007) (Fig. 4A). We next immunoprecipitated LUC7L2 and discovered 29 interacting proteins using mass spectrometry, including our validated screening hits SNRPA and SNRNP70 and most other known U1 subunits (Fig. 4B–C, Table S4).
Figure 4: LUC7L2 Encodes a U1 snRNP Subunit Involved in Pre-mRNA Splicing. See also Figure S4 and Table S4–7.
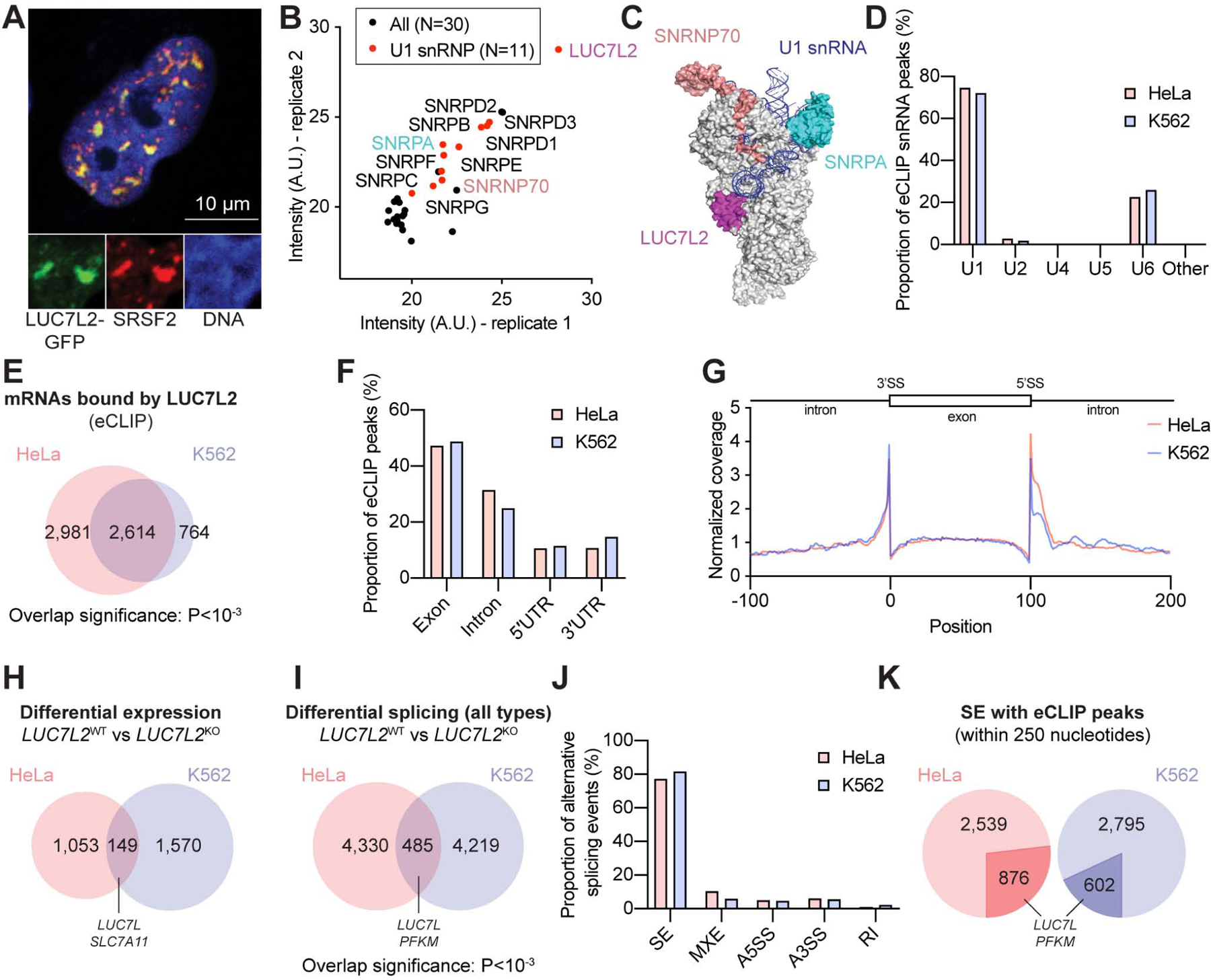
(A) Confocal microscopy of a single nucleus from a HeLa cell expressing LUC7L2-GFP and immunolabeled with antibodies to SRSF2. (B) LUC7L2-interacting proteins as determined by IP-MS (n = 2). (C) Representation of LUC7, SNRPA and SNRNP70 on the yeast U1 snRNP (PDB 5UZ5) (Li et al., 2017). (D) Proportion of eCLIP peaks mapping to splicing snRNAs in HeLa and K562 cells (eCLIP n = 2, each). (E) Representation of the genes bound by LUC7L2 at P<10−4. (F) Proportion of LUC7L2 eCLIP peaks in pre-mRNAs at P<10−4. (G) Meta-analysis of LUC7L2 binding sites across shared eCLIP peaks at P<10−4. (H) Differential gene expression in LUC7L2KO cells (n = 3 for each cell type and each genotype) as determined by RNA deep-sequencing at FDR<10−4 and >|1.5| fold change. (I) Alternative splicing events seen in LUC7L2KO cells as determined by rMATS at FDR<0.1 and |Δψ|>0.05 (n = 3 for each cell type and each genotype). (J) Types of alternative splicing in LUC7L2KO cells with SE: skipped exon; MXE: mutually-exclusive exons; A5SS: alternative 5′ splice site; A3SS: alternative 3′ splice site; RI: retained intron. (K) Alternative events presenting an eCLIP peak at a 250-nucleotide distance from splicing events at P<10−2 (in darker shade).
We next sought to identify the transcripts that are bound by endogenous LUC7L2 and used enhanced crosslinking and immunoprecipitation coupled to RNA deep-sequencing (eCLIP) (Van Nostrand et al., 2016). In agreement with the proposed role for LUC7L2 as a U1 snRNP subunit, we found that LUC7L2 crosslinks to the U1 snRNA, and to a lesser extent to the U6 snRNA, which comes in close proximity during the transfer of the 5′ splice site (5′SS) during spliceosome assembly (Plaschka et al., 2018) (Fig. 4D). We identified eCLIP peaks for LUC7L2 in 5,595 genes in HeLa cells and 3,378 genes in K562 cells. Of these, 2,614 were shared (expression-corrected overlap significance P<10−3, Poisson) (Fig. 4E, Table S5). Within pre-mRNAs, we found that LUC7L2 bound mainly to exons and introns (Fig. 4F). A meta-analysis revealed that LUC7L2 preferentially bound near splice sites (Fig. 4G). The pattern of binding near splice sites is consistent with association of LUC7L2 with U1 snRNP complexes, which recognize 5′SS motifs and also interact with U2 snRNP bound upstream of 3′ splice sites (De Conti et al., 2013)
To understand the impact of LUC7L2KO on gene expression, we profiled the transcriptomes of LUC7L2KO HeLa and K562 cells. Loss of LUC7L2 changed the expression of ~1000–1500 genes in each cell type (FDR<10−4 and >50% absolute fold change), 149 of which were shared (overlap not significant) (Fig. 4H, S4, Table S6). Splicing analysis using rMATS (Shen et al., 2014) identified 4,815 and 4,704 alternative splicing events in HeLa and K562 cells, respectively (FDR<0.1 and |Δψ|>0.05, where ψ represents “percent spliced in” and Δψ is the change in ψ following gene depletion) (Fig. 4I, Table S7). In all, 379 splicing changes were shared between both cell types (expression-corrected overlap significance P<10−3, Poisson) (Fig. 4I). Skipped exons (SE) comprised the majority of LUC7L2-induced alternative splicing events (Fig. 4J), and approximately 20–25% of these splicing events had a LUC7L2 binding site within a distance of 250 nucleotides from a splice site (Fig. 4K). The binding of LUC7L2 in close proximity to a SE event supports a direct effect of LUC7L2 on these exons, although absence of an adjacent CLIP peak does not imply that regulation is not direct, since CLIP does not detect all binding, and some regulation may occur across longer distances (Lovci et al., 2013, Van Nostrand et al., 2020).
Our RNA-seq and eCLIP analyses indicate that LUC7L2 impacts pre-mRNA splicing and gene expression. Importantly, among the genes most significantly impacted in the splicing analysis was PFKM (Fig. 4I), and amongst the most differentially expressed genes was SLC7A11 (Fig. 4H). Both of these correspond to metabolic crossovers identified above (Fig. 3). We next sought to validate these splicing and gene expression changes as being downstream of LUC7L2 to determine whether they might contribute to the observed metabolic phenotype.
LUC7L2 Promotes Expression of PFKM and Suppresses Glycogen Storage
We first investigated the changes in PFKM and other glycolytic enzymes following LUC7L2 depletion. Most transcripts encoding glycolytic enzymes were bound by LUC7L2 in eCLIP (FDR<10−4), and we found alternative splicing in PFKM, PKM, ALDOA and ENO3 (FDR<0.1, Table S5, 7). Of all glycolytic enzymes, PFKM was the most significantly alternatively spliced in both LUC7L2KO HeLa and K562 cells, with two altered splicing events detected: increased usage of an alternative 5′SS between exons 11 and 12 (Δψ = 0.29, FDR<10−11), and exon 12 skipping (Δψ = −0.19, FDR<10−12). LUC7L2 binding was observed near the 5′SS of both alternatively spliced exons (Fig. 5A). Notably, our unbiased metabolomics experiment suggested decreased PFKM activity in LUC7L2KO cells (Fig. 3A), and both alternative splicing events were indeed expected to reduce PFKM activity. First, the alternative 5′SS whose usage increases upon LUC7L2-depletion results in inclusion of a premature termination codon (PTC) (Fig. 5A). Second, skipping of exon 12 deletes 30 amino acids in the catalytic site of the enzyme (Fig. S5A). We validated the increased skipping of exon 12 in LUC7L2KO cells and observed a global decrease in the abundance of PFKM protein (Fig. 5B). To confirm the effects of this splicing change on PFKM levels, we designed an antisense oligonucleotide (ASO) targeting the 5′SS of this exon. Similar to LUC7L2 depletion, acute ASO treatment led to exon 12 skipping and decreased PFKM protein abundance, indicating that the skipping of PFKM exon 12 likely yields a less stable protein (Fig. 5B).
Figure 5: Role of LUC7L2-Mediated PFKM and SLC7A11 Alternative Splicing in Energy Metabolism. See also Figure S5.
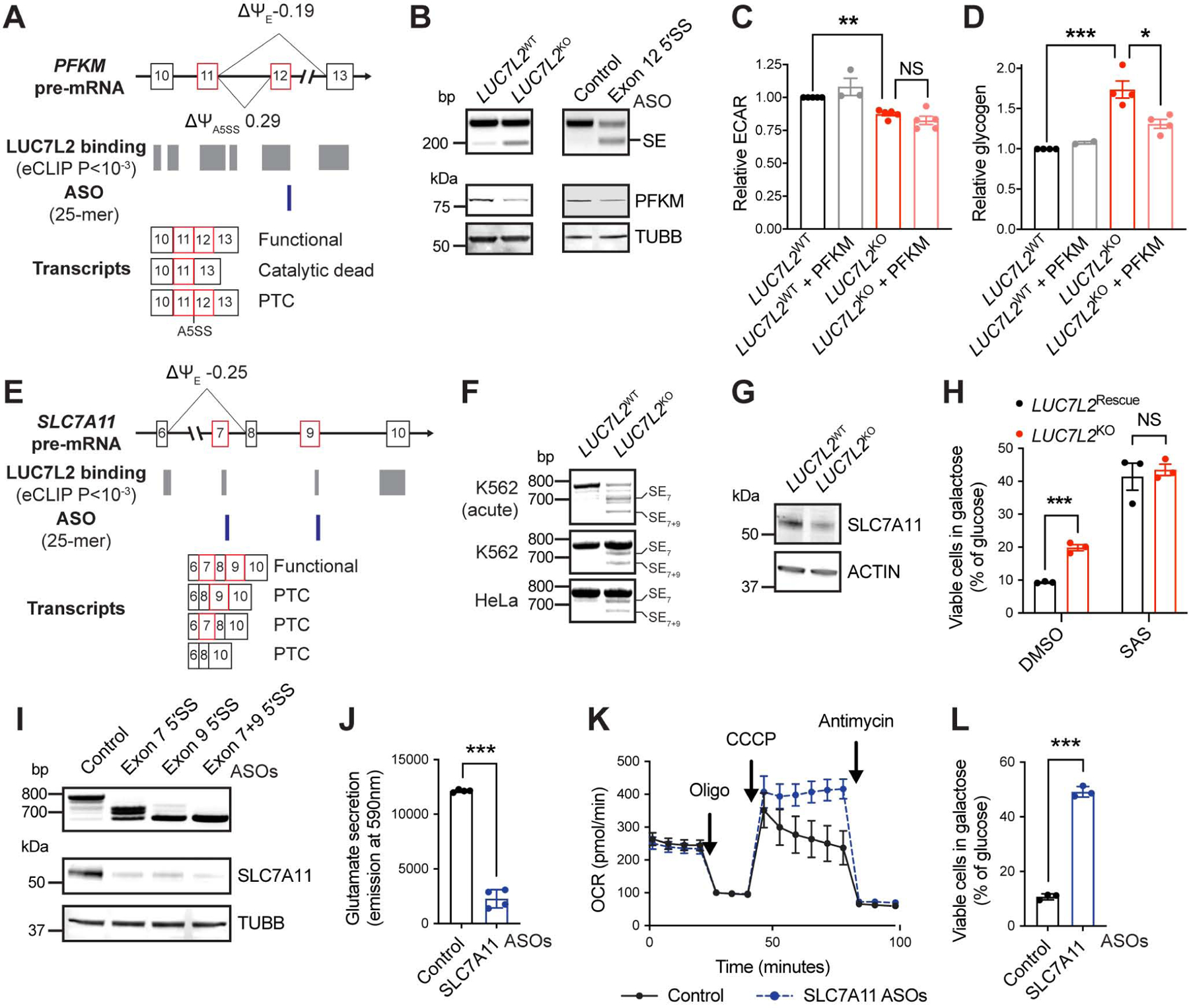
(A) Representation of PFKM exons 10–13, LUC7L2 binding sites as determined by eCLIP, antisense oligonucleotides (ASO) targeting sites and the expected transcripts. Ψ: percent spliced in reported by rMATS in K562 cells. E: exon. A negative Δψ value indicates exon skipping. PTC: premature termination codons. (B) RT-PCR (top) and immunoblot (bottom) of LUC7L2KO K562 cells (left) or HAP1 cells treated for 48h with ASO targeting the 5′SS of PFKM exon 12 (right). (C) Relative ECAR (n = 3–5) and (D) glycogen in LUC7L2KO K562 cells expressing control cDNAs (GFP) or PFKM cDNA (n = 2–4). (E) Representation of SLC7A11 exons 6–10 as in (A). (F) RT-PCR of LUC7L2KO K562 cells with primers amplifying transcripts corresponding to SLC7A11 exons 6–12. SE: skipped exon. (G) Immunoblot on LUC7L2KO K562 cells with antibodies to SLC7A11 and ACTIN. (H) Cell viability of LUC7L2Rescue (corresponds to LUC72L2KO expressing LUC7L2 cDNA) and LUC7L2KO HAP1 cells grown for 24h in galactose relative to glucose (n = 3). SAS: 500μM sulfasalazine. (I) RT-PCR (top) and immunoblot (bottom) of HAP1 cells treated for 48h with ASOs targeting the 5′SS of exon 7 and/or exon 9 of SLC7A11. (J) Media glutamate (n = 4), (K) representative seahorse trace (shown as mean ±SD), and (L) viability in galactose of HAP1 cells treated for 48h with the indicated ASOs (n = 3). All data are shown as mean ±SEM (unless otherwise stated) with * P<0.05, ** P<0.01, ***P<0.001, t-test relative to control.
The short time frame of the acute ASO treatment and the potential for long-term toxicity are not compatible with studying slow processes such as glycogen storage, so we opted for cDNA rescue to experimentally address the role of PFKM in mediating aspects of the phenotype of LUC7L2-depleted cells (Fig. S5B). We observed that overexpression of PFKM alone was insufficient to restore the bulk of glycolysis in a LUC7L2KO background, possibly due to the aforementioned consequences of LUC7L2 depletion on other glycolytic enzymes. However, it was sufficient to restore normal glycogen storage (Fig. 5C–D). Together, our data indicate that LUC7L2 is required for normal splicing and expression of full-length PFKM, and that its absence favors glycogenesis.
Expression of LUC7L2 is Limiting for Splicing of the Cystine/Glutamate Antiporter SLC7A11 (xCT)
Next, we addressed the genetic basis of the second crossover observed in LUC7L2-depleted cells at the level of the cystine/glutamate antiporter. Previous studies have shown that this antiporter controls survival in glucose-limiting conditions and OXPHOS activity (Shin et al., 2017, Koppula et al., 2017). Accordingly, we found that while inhibition with sulfasalazine prevented glutamate secretion and promoted maximal respiration, over-expression of the SLC7A11 subunit alone was sufficient to restore glutamate secretion in LUC7L2KO K562 cells (Fig. S5C–G), confirming that SLC7A11 is limiting for glutamate oxidation and OXPHOS. We next examined whether subunits of the system Xc− require LUC7L2 for their expression. SLC3A2 transcripts were not affected by LUC7L2 depletion but we found a significant reduction in transcript abundance of SLC7A11 in both LUC7L2KO HeLa and K562 cells (Fig. S5H). Reduced inclusion of SLC7A11 exon 7 was detected in these cells (Δψ = −0.25, FDR<6 × 10−11) and we noticed that exon 9 also showed reduced inclusion, which was previously annotated and which we confirmed by RT-PCR and Sanger sequencing (Fig. 5E–F, S5I). Skipping of exons 7 and/or 9 yield PTCs in SLC7A11 transcripts, likely reducing mRNA abundance via nonsense-mediated mRNA decay (NMD) (Fig. S5J). LUC7L2 binding was also observed at the 5′SS of these exons by eCLIP (Fig. 5E), suggesting a direct effect on their splicing. Accordingly, depletion of LUC7L2 led to a decrease in SLC7A11 expression in K562, HeLa and HAP1 cells (Fig. 5G, S5K). Reduced expression of SLC7A11 was also observed upon depletion of SNRPA and SNRNP70, the two other U1 snRNP subunits identified in our screen (Fig. S5L), and LUC7L2 over-expression was sufficient to stabilize SLC7A11 transcript and protein in K562 cells (Fig. S5M–N).
To test for a direct contribution of SLC7A11 splicing to the metabolic phenotypes observed in LUC7L2KO, we focused on LUC7L2KO HAP1 cells, a cell line in which the role of this antiporter in antagonizing viability in low glucose conditions is well characterized (Shin et al., 2017). As in K562 cells, we found that LUC7L2 depletion increased OXPHOS activity (Fig. S2F) and viability in galactose (Fig. 5H). We then treated wild-type HAP1 cells with ASOs targeting the 5′SS of SLC7A11 exons 7 and 9, where LUC7L2 and the U1 snRNP bind. Similar to the phenotype of LUC7L2KO cells, we found that acute ASO treatment led to skipping of these exons, prevented SLC7A11 expression and glutamate secretion, and boosted maximal respiration and viability in galactose (Fig. 5I–L), all resembling the phenotype observed upon LUC7L2 depletion. We conclude that splicing of SLC7A11 (xCT) is sensitive to perturbations of U1 snRNP components including LUC7L2, and that altered splicing of SLC7A11 induced by LUC7L2 depletion contributes to the metabolic and viability phenotypes observed in LUC7L2KO cells (Fig. 1, 3).
LUC7L2 Depletion Causes Secondary Accumulation of Respiratory Chain Complexes
To obtain a comprehensive view of proteome remodeling in LUC7L2-depleted K562 cells, we next performed global quantitative proteomics (Fig. 6A, S6A–D). As expected, we observed depletion of PFKM in LUC7L2KO cells as well as differential expression of multiple glycolytic enzymes, including rate-determining factors (Tanner et al., 2018) such as hexokinases HK1/2 (P<10−2), the glucose transporter GLUT1 (P<10−5) and the lactate transporter SLC16A1 (P<0.02) (Table S8). Transcripts of these factors were generally bound by LUC7L2 but splicing changes were not always observed in LUC7L2KO cells, suggesting regulation by both direct and indirect mechanisms. SLC7A11 was not detected in the proteomics analysis, possibly owing to its hydrophobicity. Importantly, while abundances of mitochondria and splicing-related proteins were not globally affected, GO analysis revealed a strong enrichment for OXPHOS proteins in LUC7L2KO cells, with “NADH dehydrogenase complex” as the top-scoring term associated with up-regulated proteins (P<10−4) (Fig. S6C).
Figure 6: Proteomic Analysis of LUC7L2KO and Galactose-Grown Cells Reveals Secondary Complexes I+III+IV Accumulation. See also Figure S6 and Table S8.
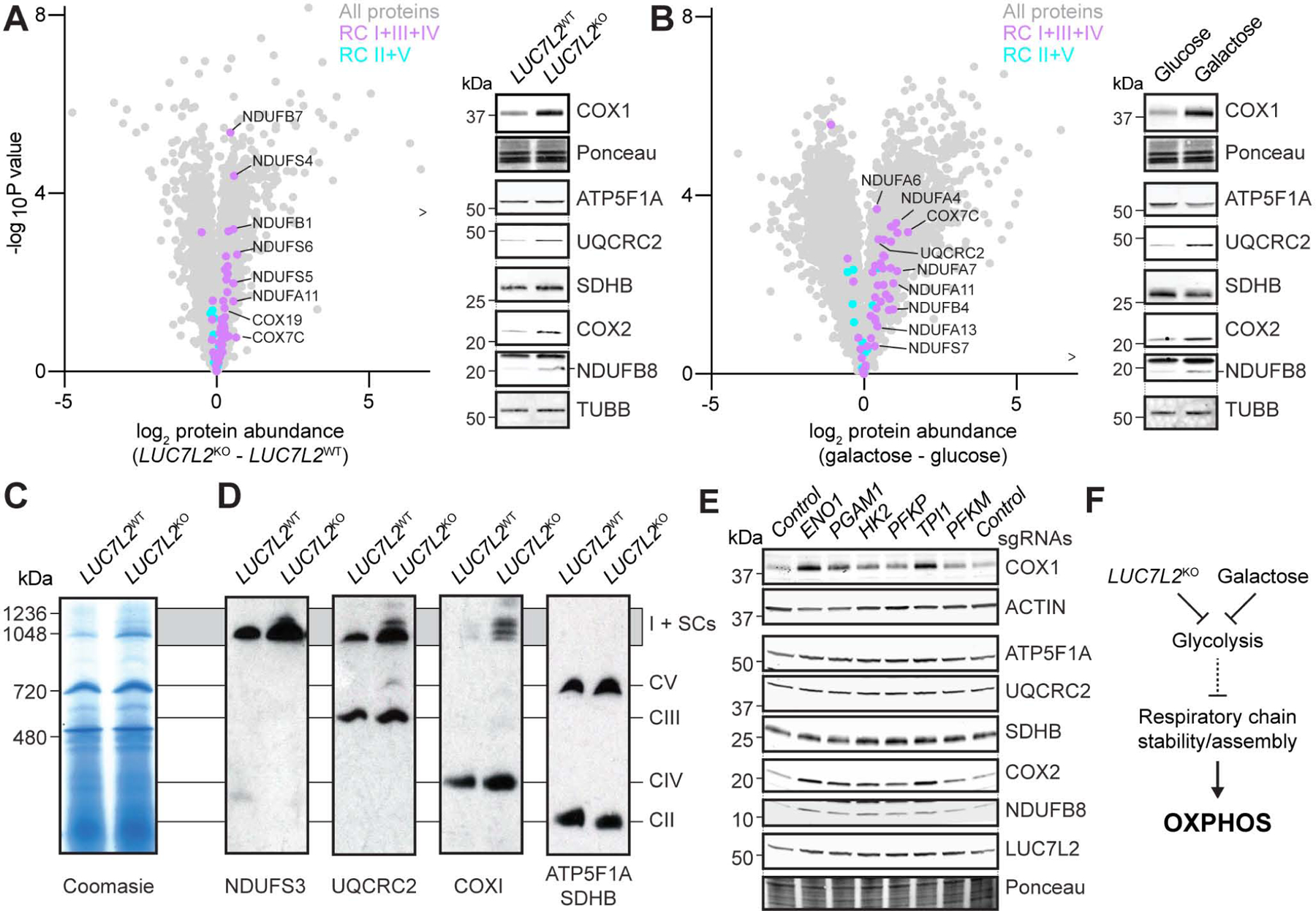
(A) Volcano plots and immunoblots of OXPHOS protein expression in LUC7L2KO and (B) in galactose-grown K562 cells. > indicates a protein not shown but reported in Table S8. (C) Blue-Native PAGE on a mitochondria-rich fraction isolated from LUC7L2KO K562 cells and stained with coomassie or (D) immunoblotted with the indicated antibodies. Parallel blots in which the same lysate was loaded were used to avoid antibodies cross-reactivity. SCs: supercomplexes. CI-V: complexes I to V. (E) Immunoblot on K562 cells expressing Cas9 and treated with sgRNAs targeting glycolytic enzymes with the indicated antibodies. (F) Model of the secondary regulation of the respiratory chain by LUC7L2 and galactose.
Given this inverse relationship between abundance of LUC7L2 and OXPHOS proteins, we analyzed their relative expression in published large-scale proteomics studies. Similar to our experimental observation in cells, LUC7L2 was significantly anti-correlated with OXPHOS protein expression across organs in two in vivo mouse tissue proteomic atlases (P<10−42, Wilcoxon) (Geiger et al., 2013, Huttlin et al., 2010) (Fig. S6E). A similar anti-correlation was observed in a proteomics study of brains from healthy subjects or from patients with neurodegenerative disease and showed that LUC7L2 protein accumulates in patients, whereas OXPHOS proteins are decreased in Parkinson’s disease (Fig. S6F) (Ping et al., 2018). OXPHOS protein abundance also decreases in skeletal muscle with aging, as recently confirmed by a proteomic study on 58 skeletal muscle biopsies (Ubaida-Mohien et al., 2019). In this study too, we observed significant up-regulation of LUC7L2 with age and anti-correlation with OXPHOS (P<10−26, Wilcoxon) (Fig. S6E). Thus, in these four in vivo datasets we observed inverse relationships between the abundance of LUC7L2 and OXPHOS proteins resembling LUC7L2KO depletion.
OXPHOS proteins also accumulate in glucose-limiting conditions (Rossignol et al., 2004). To compare this condition to LUC7L2KO, we performed proteomics analysis of galactose-grown cells and observed strong up-regulation, as expected (Fig. 6B). Importantly, we noticed a similar pattern in both LUC7L2KO cells and in galactose: the up-regulation of subunits of the respiratory chain (RC) complex I, III, IV. In contrast, protein subunits of RC complex II and the ATP synthase (V), as well as from other mitochondrial protein complexes and the gene expression machinery were not affected, or rather decreased (Fig. 6A–B, S6A–B,G). Transcripts of RC complexes I+III+IV did not accumulate in LUC7L2KO, suggesting an effect occurring after RNA processing (Fig. S6A). However, these same RC complexes I+III+IV have the ability to interact within the mitochondrial inner membrane to form “supercomplexes” (SCs), and their proposed roles in reinforcing RC complex stability (Acin-Perez et al., 2004) could explain their accumulation in LUC7L2KO cells. Accordingly, blue-native PAGE confirmed accumulation of higher molecular weight RC complexes in both K562 and HeLa LUC7L2KO and galactose-grown cells (Fig. 6C–D, S6H).
Galactose growth and LUC7L2 depletion both attenuate glycolysis, and we next directly tested whether lower glycolytic rates could explain increased RC abundance. We used CRISPR/Cas9 to acutely deplete six glycolytic enzymes and subsequently measured OXPHOS proteins (Fig. 6E, S6I). Importantly, we found that depletion of these genes generally led to the accumulation of the same RC subunits as LUC7L2KO and galactose, which was particularly apparent upon depletion of ENO1 and TPI1. While the mechanism by which attenuated glycolysis leads to RC complex accumulation was not investigated here, our observations indicate that the increased abundance of RC complexes in LUC7L2KO cells is likely secondary to the effect of this gene on glycolysis (Fig. 6F), and possibly involves the stabilization of mitochondrial SCs.
Cross-Regulation and Partial Redundancy Within the LUC7 Family
Finally, we investigated the role of LUC7L2 paralogs LUC7L and LUC7L3 in energy metabolism. All three LUC7 genes encode similar proteins (Tufarelli et al., 2001) (Fig. 7A). Analysis of LUC7 proteins across a mouse proteomics atlas (Geiger et al., 2013) revealed that although a general anti-correlation with OXPHOS was observed for all three proteins, the expression patterns of individual members of the LUC7 family were not identical (Fig. S7). For example, LUC7L was present at higher levels in the brain, while expression of LUC7L3 was not detectable in the spleen, suggesting tissue-specific roles in alternative splicing.
Figure 7: Pre-mRNA Splicing and Partial Redundancy Within the LUC7 Family. See also Figure S7.
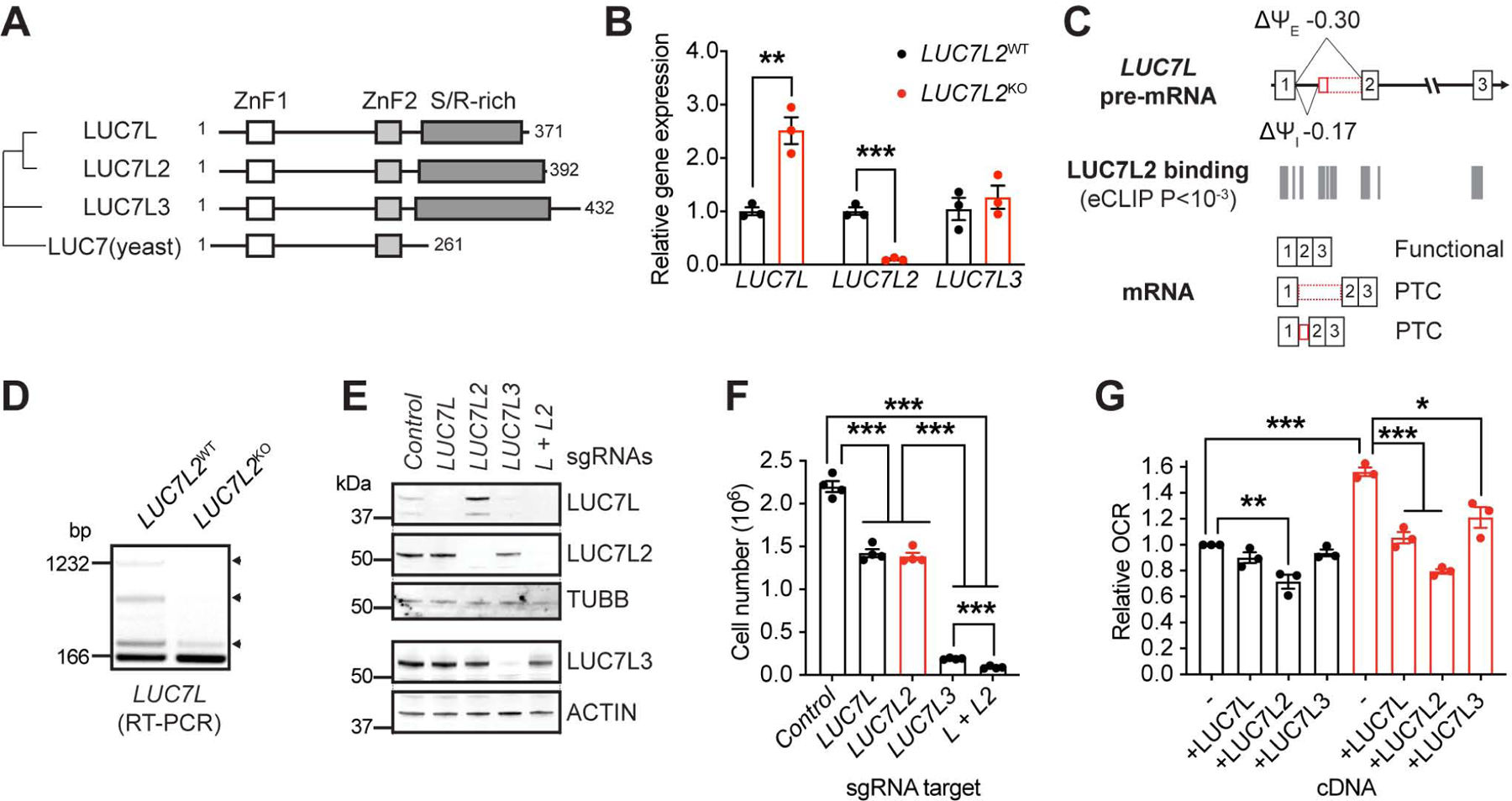
(A) Phylogenetic tree of the LUC7 protein family. LUC7 is from S. cerevisiae. ZnF: Zinc-finger domain. S/R-rich: Serine and arginine-rich domain. (B) Quantitative PCR detecting LUC7 family transcripts in LUC7L2KO K562 cells (n = 3). (C) Representation of LUC7L exons 1–3, LUC7L2 binding sites as determined by eCLIP, and the expected transcripts. Ψ: percent spliced in reported by rMATS in K562 cells. I: intron. E: exon. (D) RT-PCR amplifying LUC7L exon 1 to exon 2. Arrowheads: retained entities in LUC7L. (E) Immunoblot of LUC7 proteins in cell lines expressing Cas9 and sgRNAs targeting the indicated genes using the indicated antibodies and (F) number of cells after 4 days of growth in glucose-containing media. (G) Oxygen consumption analysis of LUC7L2KO K562 cells expressing cDNAs of LUC7 family members. All data are shown as mean ±SEM with * P<0.05, ** P<0.01, ***P<0.001, t-test relative to control.
With 85% protein identity, LUC7L is the closest homolog to LUC7L2. This gene was also one of the most impacted genes in LUC7L2KO (Fig. 7B) and we found decreased inclusion of an annotated exon located in an intron between canonical exons 1 and 2 of LUC7L as well as decreased retention of the associated intron, all within a region highly bound by LUC7L2 (Δψ = −0.30 and −0.17, respectively) (Fig. 7C,D). Importantly, inclusion of the exon with or without intron retention results in a PTC in LUC7L, which likely leads to transcript degradation by NMD. Accordingly, protein analysis revealed strong up-regulation of LUC7L in LUC7L2KO cells, indicating a repressive role for LUC7L2 (Fig. 7E, Table S8), an observation that is relatively common among paralogous RBPs (Spellman et al., 2007, Ni et al., 2007, Lareau et al., 2007)
To experimentally address the function of each of the LUC7 genes, we depleted each individually, as well as LUC7L and LUC7L2 together (Fig. 7E). We found that depletion of LUC7L or LUC7L2 individually led to a mild defect in cell proliferation (Fig. 7F) while depletion of LUC7L3 caused a strong growth defect. Simultaneous depletion of LUC7L and LUC7L2 showed a synthetic lethal phenotype, consistent with some degree of functional redundancy and cross-regulation. To directly address redundancy, we investigated whether members of the LUC7 family could rescue the metabolic phenotype observed in LUC7L2KO cells. For this purpose, we stably expressed LUC7L, LUC7L2 or LUC7L3 in these cells and measured oxygen consumption. Importantly, we found that while expression of LUC7L2 alone was able to decrease OCR in wild-type cells (Fig. 7G), re-expression of any of the members of the family could restore normal oxygen consumption in LUC7L2KO cells, to varying degrees (Fig. 5D). Together, our observations implicate all members of the LUC7 family in energy metabolism.
DISCUSSION
We have found that the expression of genes related to pre-mRNA splicing and the U1 snRNP impact the balance between glycolysis and OXPHOS. Genetic loss of any of three U1 snRNP-specific subunits (SNRPA, SNRNP70, LUC7L2) boosts cellular fitness when glycolysis is compromised. The U1 snRNP plays an essential role in pre-mRNA splicing and modulates other nuclear RNA-related processes such as cleavage and polyadenylation (Berg et al., 2012) and chromatin retention of non-coding RNAs (Yin et al., 2020). To our knowledge, our work is the first to establish a link between the expression of U1 snRNP components and the bioenergetic state of the cell.
In the current study, we were able to confirm that LUC7L2 is a genuine component of the U1 snRNP in humans, as has been observed for LUC7 homologs in yeast and plants (de Francisco Amorim et al., 2018, Fortes et al., 1999). Because LUC7 genes are expressed in a tissue-specific manner (Fig. S7), as are some of the transcripts of metabolic genes bound by LUC7L2, we speculate that the relative abundance of these paralogs in different cells may contribute to differences in levels of OXPHOS and glycolysis between different cell types.
We show that loss of LUC7L2 results in an activation of OXPHOS. The mechanisms linking LUC7L2 expression to energy metabolism are likely complex. Given that the U1 snRNP plays central roles in splicing and gene expression, we searched for changes in our transcriptome analysis that might help explain the observed metabolic phenotype. In the current study we have prioritized observations from our metabolic characterization of LUC7L2KO cells (Fig. 1–3) and investigated in detail PFKM and SLC7A11, which we show contribute to key aspects of the metabolic phenotype following depletion of LUC7L2 (Fig. 5).
First, we found that LUC7L2 binds to and contributes to splicing and expression of PFKM (Fig. 5A). This enzyme alone did not explain the global decrease in glycolysis in LUC7L2KO cells, as we found multiple instances of altered expression in glycolytic proteins in LUC7L2KO (Table S8), including in rate-limiting factors (Tanner et al., 2018). We propose that these changes may collectively lead to the decreased glycolytic activity in LUC7L2KO cells, and likely originate from both primary and secondary mechanisms. However, we show that altered splicing in PFKM is likely directly responsible for the glycogen storage phenotype in LUC7L2KO cells. The differential role of PFKM in glycogen storage versus glycolysis is not unexpected from observations of patients with Tarui disease, who present minimal effects on circulating lactate at rest (Piirila et al., 2016), possibly due to the complex interplay between PFKM, its paralogs and allostery.
Second, we have identified alternative splicing events in SLC7A11 whereby skipping of exons 7 and 9 leads to loss of SLC7A11 (xCT) protein, which has a direct impact on OXPHOS. xCT has emerged as a critical regulator of metabolism and cell viability, and while high expression of this cystine/glutamate antiporter blunts glutamine anaplerosis and creates a dependency on glycolysis for ATP production (Koppula et al., 2017, Shin et al., 2017), genetic ablation of this cancer-related gene in tumors induces death by lipid peroxidation (Badgley et al., 2020). Expression of SLC7A11 is highly regulated and is known to be transcriptionally activated by ATF4 (Sato et al., 2004) and NRF2 (Shin et al., 2017) and repressed by P53 (Jiang et al., 2015). SLC7A11 activity is also regulated by direct mTORC2 phosphorylation (Gu et al., 2017). Our work adds another layer of complexity to the regulation of this transporter. In the future, it will be interesting to determine whether these splicing events are regulated in disease states to influence the sensitivity to lipid peroxidation.
In addition, we report that, in general, shifts from glycolysis to OXPHOS appear to be associated with an accumulation of RC complexes (Fig. 6). When cells are grown in galactose, or in glucose-grown cells following genetic ablation of LUC7L2 or of glycolytic enzymes, we observed a characteristic pattern of RC I+III+IV accumulation which prompted us to investigate assembly of the SCs. Future studies are needed to determine how the reduction in glycolysis impacts RC assembly.
Our work predicts that the existence of shifts in energy metabolism will accompany human conditions associated with mis-expression of LUC7L2 and the U1 snRNP. U1 dysregulation and mutations have been reported to occur in cancer (Shuai et al., 2019, Suzuki et al., 2019, Oh et al., 2020). Similarly, mutations in LUC7L2 or haploinsufficiency through loss of chromosome 7q are associated with MDS (Singh et al., 2013). 7q- models recapitulate differentiation defects observed in MDS (Kotini et al., 2015) and reintroduction of LUC7L2 is sufficient to restore differentiation. How LUC7L2 loss impacts hematopoietic stem cell differentiation and MDS is not clear, but our work raises the possibility of a bioenergetics-related mechanism. It is notable that other splicing factors associated with MDS also participate in the splicing of metabolic enzymes; for example, MDS-associated mutations in SRSF2 give rise to the same splicing change in PFKM (Zhang et al., 2015) as we observe in LUC7L2KO. Our work has revealed a novel metabolic vulnerability of LUC7L2-depleted cells (Fig. 2C) and predicts that pharmacological blockade of OXPHOS using drugs such as metformin may be beneficial in these disorders.
While the current analysis has focused on the effects of LUC7L2 gene depletion, an exciting future direction is to determine whether metabolism is regulated by changes in expression of LUC7 family members in normal development or physiology. We observe a consistent, inverse correlation between the protein levels of LUC7L2 and OXPHOS across organs and during disease and aging (Fig. S6) Expression of LUC7L2 and its paralogs is known to respond to changes in the environment, including changes in oxygen tension (Kimura et al., 2004, Gao et al., 2011). It is notable that previous proteomics studies have established that LUC7L2 is post-translationally modified via phosphorylation and hydroxylation (Webby et al., 2009, Dephoure et al., 2008). In this context, it is possible that proteins of the LUC7 family, as well as other U1 snRNP components, might integrate signals such as oxygen and nutrients to balance the activity of major pathways in energy metabolism.
LIMITATION OF STUDY
Pre-mRNA splicing, gene expression and energy metabolism are cell type-specific processes. We report here that LUC7L2 impacts energy metabolism in three cellular models, but there are differences across cell types. For example, while we observed decreased levels of SLC7A11 in three LUC7L2KO cell lines investigated (Fig. 5G, S5K), it led to an increase in net oxygen consumption only in K562 and HAP1 cells, but not in HeLa cells (Fig. S2E). It is possible that cells with different metabolic programs, for instance, based on low SLC7A11 and PFKM expression or cells with high expression of LUC7L and LUC7L3 might be indifferent to LUC7L2 depletion. Furthermore, although we observed that LUC7L2 depletion results in altered splicing in PFKM, SLC7A11 and other genes, and that LUC7L2 crosslinks near the regulated exons in PFKM and SLC7A11, this does not formally prove that the effect on splicing is direct. Our profiling experiments of LUC7L2KO cells were likely biased towards abundant molecules, including in our sequencing analysis, as well as for the detection of peptides and metabolites by mass spectrometry. Thus, it is possible that additional low-abundance, splicing events in LUC7L2KO cells may have been missed and that those events may affect additional metabolic aspects in LUC7L2-depleted cells.
STAR METHOD
RESOURCE AVAILABILITY
Lead Contact
Vamsi K. Mootha (vamsi@hms.harvard.edu)
Material Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact.
Data and Code Availability
RNA sequencing data were deposited at GEO (GSE157917). Proteomics data were deposited at PRIDE (PXD021917). Plasmids were deposited at Addgene. Unedited gel scans are available in the supplemental information file (Data S1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
K562 (ATCC CCL-243), HeLa (ATCC CCL-2) and 293T (ATCC CRL-3216) were obtained from ATCC and were re-authenticated by STR profiling at ATCC prior submission of the manuscript. HAP1 cells were from Horizon Discovery (C631). Cells were periodically tested to ensure absence of mycoplasma.
METHOD DETAILS
CRISPR Screen Re-Analysis
CRISPR screen analysis was performed either using a normalized Z-score approach (To et al., 2019) or using MAGeCK (Li et al., 2014). Raw sgRNA read counts were normalized to reads per million and then log2 transformed using the following formula: Log2(reads from an individual sgRNA / total reads in the sample106 + 1) (To et al., 2019). Log2 fold-change of each sgRNA was determined relative to the pre-swap control. For each gene in each replicate, the mean log2 fold-change in the abundance of all 4 sgRNAs was calculated. Genes with low expression (log2 FPKM<0) according to publicly available K562 RNA-seq dataset (sample GSM854403 in GEO seriesGSE34740) and essential genes previously reported (Arroyo et al., 2016) were removed. Log2 fold-changes were averaged by taking the mean across replicates. For each treatment, a null distribution was defined by the 3,726 genes with lowest expression. To score each gene within each treatment, its mean log2 fold-change across replicates was Z-score transformed, using the statistics of the null distribution defined as above. To score each gene using MAGeCK, normalized sgRNA read counts from the duplicate in each condition were used as input for MAGeCK v0.5.3 to obtain a p-value and FDR for gene enrichment or depletion relative to the reference samples (pre-swap). MAGeCK was run with default parameters. For representation purpose, viability in galactose was defined as the - annexin V value.
Cell Culture and Cell Growth Assays
Unless otherwise specified, cells were maintained in DMEM containing 1mM sodium pyruvate (ThermoFisher Scientific) with 25 mM glucose, 10% fetal bovine serum (FBS, ThermoFisher Scientific), 50 μg/mL uridine (Sigma), and 100 U/mL penicillin/streptomycin (ThermoFisher Scientific) under 5% CO2 at 37°C. Cells were counted using a ViCell Counter (Beckman) and only viable cells were considered. Drugs were diluted in the same culture media for cell growth assays and compared to the solvent control (DMSO or water). For galactose growth assays, FBS was replaced by dialyzed FBS (Life Technologies) and glucose was replaced by an equivalent amount of galactose.
Gene-Specific CRISPR-Cas9 Knockouts
The two best sgRNAs from the Avana-library were ordered as complementary oligonucleotides (Integrated DNA Technologies) and cloned in pLentiCRISPRv2. An sgRNA targeting EGFP was used as a negative control. Lentiviruses were produced according to Addgene’s protocol (Sanjana et al., 2014) and 24h post-infection cells were selected with 2mg/mL puromycin (ThermoFisher Scientific) for 48h. Cells were then maintained in routine culture media for 10–20 addition days before analysis. Gene disruption efficiency was verified by qPCR and/or immunoblotting. For HAP1 cells, a LUC7L2KO cell line expressing LUC7L2 cDNA was used as control (LUC7L2Rescue). For acute treatment, K562 cells were transduced with a high titer of sgRNAs targeting LUC7L2 and analyzed after 7 days. Sequences of the sgRNAs used are in the Key Resources Table.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Actin (AC40) | Sigma | Cat # A4700; RRID: AB_476730 |
| Actin (EPR16769) | Abcam | Cat # ab179467; RRID: AB_2737344 |
| Anti-SC35 [SC-35] | Abcam | Cat # ab11826; RRID: AB_298608 |
| COX1 | Abcam | Cat # ab14705; RRID AB_2084810 |
| Enolase-1 | Cell Signaling Technology | Cat #3810; RRID AB_2246524 |
| FLAG M2 | Sigma | Cat # F1804; RRID: RRID: AB_262044 |
| Goat Anti-Mouse IgG H&L (Alexa Fluor® 488) | Abcam | Cat # ab150113; RRID AB_2576208 |
| Goat Anti-Rabbit IgG H&L (Alexa Fluor® 647) | Abcam | Cat # ab150079; RRID AB_2722623 |
| Hexokinase II (C64G5) Rabbit mAb | Cell Signaling Technology | Cat # 2867; RRID AB_2232946 |
| HRP-linked anti-mouse IgG | GE Healthcare | Cat # NA934; RRID: AB_772206 |
| HRP-linked anti-rabbit IgG | GE Healthcare | Cat # NXA931; RRID: AB_772209 |
| Human OXPHOS cocktail (ATP5A, SDHB, UQCRC2, COX2, NDUFB8) | Abcam | Cat # ab110411; RRID: AB_2756818 |
| IRDye 680RD Goat anti-Mouse IgG (H + L) | LI-COR Biosciences | Cat # 926–68070; RRID: RRID: AB_10956588 |
| IRDye 680RD Goat anti-Rabbit IgG (H + L) | LI-COR Biosciences | Cat # 926–68071; RRID: RRID: AB_10956166 |
| IRDye 800CW Goat anti-Mouse IgG (H + L) | LI-COR Biosciences | Cat # 926–32210; RRID: RRID: AB_621842 |
| IRDye 800CW Goat anti-Rabbit IgG (H + L) | LI-COR Biosciences | Cat # 926–32211; RRID: RRID: AB_621843 |
| LUC7L (43-I) | Santa Cruz | Cat # 101075; RRID: AB_2139473 |
| LUC7L2 | Sigma | Cat # HPA051631; RRID: AB_2681558 |
| LUC7L3 | Sigma | Cat # HPA018484; RRID: AB_1847248 |
| NDUFS3 | ThermoFisher Scientific | Cat # 15066–1-AP; RRID: AB_2151109 |
| PFKM | Abcam | Cat # ab97353; RRID: AB_10680060 |
| PFKP (D4B2) Rabbit mAb | Cell Signaling Technology | Cat # 8164; RRID AB_2713957 |
| PGAM1 (D3J9T) Rabbit mAb | Cell Signaling Technology | Cat # 12098; RRID AB_2736922 |
| SLC7A11 (D2M7A) | Cell signaling | Cat # 12691; RRID: AB_2687474 |
| TOMM20 | Santa Cruz | Cat # SC-11415; RRID: AB_2207533 |
| TUBB | ThermoFisher Scientific | Cat # MA5–16308; RRID: AB_2537819 |
| Chemicals, peptides, and recombinant proteins | ||
| 2-Deoxy-D-glucose | Sigma | Cat # D8375 |
| Agilent Seahorse XF Base Medium | Agilent | Cat #03334–100 |
| Ambion RNase I, cloned, 100 U/μL | Ambion | Cat # AM2295 |
| Anti-FLAG® M2 Magnetic Beads | Sigma | Cat # M8823 |
| Antimycin A | Sigma | Cat # A8674 |
| Carbonyl cyanide 3-chlorophenylhydrazone | Sigma | Cat # C2759 |
| Chloramphenicol | Sigma | Cat # C0378 |
| Coomassie Brilliant Blue R-250 Staining Solution | Biorad | Cat # 1610436 |
| Cycloheximide | Sigma | Cat # C4859 |
| DMEM, high glucose, pyruvate | ThermoFisher Scientific | Cat # 11995073 |
| DMEM, no glucose, no glutamine, no phenol red | ThermoFisher Scientific | Cat # A1443001 |
| Dulbecco’s PBS | Sigma | Cat # D8537 |
| Endo-Porter (PEG) | Gene Tools | Cat # OT-EP-PEG-1 |
| ExoSAP-IT PCR Product Cleanup Reagent | ThermoFisher Scientific | Cat # 78201.1.ML |
| EXPRESS [35S]-protein labeling mix | Perkin Elmer | Cat # NEG772 |
| Fetal Bovine Serum | ThermoFisher Scientific | Cat #26140079 |
| Fetal Bovine Serum, dialyzed | ThermoFisher Scientific | Cat # 26400044 |
| FLAG peptide | Sigma | Cat # F3290 |
| FluorSave | EMD Millipore | Cat # 345789 |
| G-418 solution | ThermoFisher Scientific | Cat # 10131035 |
| Galactose | Sigma | Cat # G5388 |
| Glucose | Sigma | Cat # G7021 |
| Hoescht 33342 | ThermoFisher Scientific | Cat # H1399 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | ThermoFisher Scientific | Cat # 28906 |
| L-Glutamine | ThermoFisher Scientific | Cat # 25030081 |
| M-MLV Reverse Transcriptase | Promega | Cat # M1701 |
| Meclizine dihydrochloride | Sigma | Cat # SML0950 |
| met/cys-free DMEM | ThermoFisher Scientific | Cat #21013024 |
| Metformin / 1,1-Dimethylbiguanide HCl | Sigma | Cat # D150959 |
| NEBNext® Ultra II Q5® Master Mix | New England BioLabs | Cat # M0544 |
| Odyssey Blocking Buffer | LI-COR Biosciences | Cat # 927–40000 |
| Odyssey® Blocking Buffer (PBS) | LI-COR Biosciences | Cat # 927–40100 |
| Oligomycin A | Sigma | Cat # 75351 |
| Penicillin-Streptomycin | ThermoFisher Scientific | Cat # 15140122 |
| Protease Inhibitor Cocktail (100X) | Cell Signaling | Cat # 5871 |
| Proteinase K, Molecular Biology Grade | New England BioLabs | Cat # P8107 |
| Puromycin Dihydrochloride | ThermoFisher Scientific | Cat # A1113803 |
| RNase-Free DNase Set | QIAGEN | Cat # 79254 |
| RNaseA | Takara | Cat # 740505 |
| RNaseOUT Recombinant Ribonuclease Inhibitor | ThermoFisher Scientific | Cat # 10777019 |
| Sulfasalasine | Sigma | Cat # S0883 |
| T4 Polynucleotide Kinase | New England BioLabs | Cat # M0201 |
| T4 RNA Ligase 1 (ssRNA Ligase) | New England BioLabs | Cat # M0204 |
| Universal Nuclease (Pierce) | ThermoFisher Scientific | Cat # 88702 |
| Uridine | Sigma | Cat # U3003 |
| XBridge BEH Amide column | Water | Cat # 186006091 |
| ZIC-philic column | Merck | Cat # 150460 |
| Critical commercial assays | ||
| Amplex red glutamic acid/glutamate oxidase assay kit | ThermoFisher Scientific | Cat # A22189 |
| Citrate Synthase Activity Assay Kit | Abcam | Cat # ab119692 |
| DC Protein Assay | Biorad | Cat # 5000112 |
| Glycogen Assay Kit II (Colorimetric) | Abcam | Cat # ab169558 |
| Hoechst 33342, Trihydrochloride, Trihydrate | ThermoFisher Scientific | Cat # H3570 |
| NativeMark Unstained Protein Standard | ThermoFisher Scientific | Cat # LC0725 |
| NativePAGE 3 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 10-well | ThermoFisher Scientific | Cat # BN1001BOX |
| NativePAGE Cathode Buffer Additive (20X) | ThermoFisher Scientific | Cat # BN2002 |
| NativePAGE Running Buffer (20X) | ThermoFisher Scientific | Cat # BN2001 |
| NativePAGE Sample Buffer (4X) | ThermoFisher Scientific | Cat # BN2003 |
| Novex 10–20% Tris-Glycine Mini Gels | ThermoFisher Scientific | Cat # XP10200BOX |
| Novex 4–12% Tris-Glycine Mini Gels | ThermoFisher Scientific | Cat # XP04122BOX |
| Precision Plus Protein Kaleidoscope Prestained Protein Standards | Biorad | Cat # 1610375 |
| RNeasy Mini Kit | QIAGEN | Cat # 74106 |
| Seahorse XFe96 FluxPaks | Agilent | Cat # 102416–100 |
| Trans-Blot Turbo Midi Nitrocellulose Transfer Packs | Biorad | Cat # 1704159 |
| LUC7L Taqman Assay | ThermoFisher Scientific | Hs00216077 |
| LUC7L2 Taqman Assay | ThermoFisher Scientific | Hs00255388 |
| LUC7L3 Taqman Assay | ThermoFisher Scientific | Hs00895240 |
| SLC7A11 Taqman Assay | ThermoFisher Scientific | Hs00921938 |
| TBP Taqman Assay | ThermoFisher Scientific | Hs00427620 |
| Deposited data | ||
| RNA-seq | GEO | GEO: GSE157917 |
| Proteomics | PRIDE | PRIDE: PXD021917 |
| Experimental models: cell lines | ||
| K562 | ATCC | CCL-243 |
| 293T | ATCC | CRL-3216 |
| HAP1 | Horizon Discovery | C631 |
| HeLa | ATCC | CCL-2 |
| Oligonucleotides | ||
| ACIN1_sg1_as | Integrated DNA Technologies | AAACCAGAGATGCGAGAGTCATCAC |
| ACIN1_sg1_s | Integrated DNA Technologies | CACCGTGATGACTCTCGCATCTCTG |
| ACIN1_sg2_as | Integrated DNA Technologies | AAACCCCTCTGTGTCACTGTTTCC |
| ACIN1_sg2_s | Integrated DNA Technologies | CACCGGAAACAGTGACACAGAGGG |
| AluYb8_as | Integrated DNA Technologies | GAGACGGAGTCTCGCTCTGTC |
| AluYb8_probe | Integrated DNA Technologies | VIC-ACTGCAGTCCGCAGTCCGGCCT-MGBNFQ |
| AluYb8_s | Integrated DNA Technologies | CTTGCAGTGAGCCGAGATT |
| ENO1_sg1_as | Integrated DNA Technologies | AAACTGAAGTTTGCTGCACCGACTC |
| ENO1_sg1_s | Integrated DNA Technologies | CACCGAGTCGGTGCAGCAAACTTCA |
| ENO1_sg2_as | Integrated DNA Technologies | AAACAGTGGTGCTTCAACTGGTATC |
| ENO1_sg2_s | Integrated DNA Technologies | CACCGATACCAGTTGAAGCACCACT |
| HK2_sg1_as | Integrated DNA Technologies | AAACCGCCTTTGTTCTCCTTGATGC |
| HK2_sg1_s | Integrated DNA Technologies | CACCGCATCAAGGAGAACAAAGGCG |
| HK2_sg2_as | Integrated DNA Technologies | AAACTGTGCTTTGGGTGAAAGTAAC |
| HK2_sg2_s | Integrated DNA Technologies | CACCGTTACTTTCACCCAAAGCACA |
| HNRNPD_sg1_as | Integrated DNA Technologies | AAACCGTTCTTACTGGCGTCAATC |
| HNRNPD_sg1_s | Integrated DNA Technologies | CACCGATTGACGCCAGTAAGAACG |
| HNRNPD_sg2_as | Integrated DNA Technologies | AAACCCGAACTGCTCCTCCGACATC |
| HNRNPD_sg2_s | Integrated DNA Technologies | CACCGATGTCGGAGGAGCAGTTCGG |
| LUC7L_sg1_as | Integrated DNA Technologies | AAACCAAGTGTAACTTGCCACCGAC |
| LUC7L_sg1_s | Integrated DNA Technologies | CACCGTCGGTGGCAAGTTACACTTG |
| LUC7L_sg2_as | Integrated DNA Technologies | AAACAGTCGTGGATTTTGGTACATC |
| LUC7L_sg2_s | Integrated DNA Technologies | CACCGATGTACCAAAATCCACGACT |
| LUC7L2_sg1_as | Integrated DNA Technologies | AAACCTCTTAAAGCCAGGTCATGGG |
| LUC7L2_sg1_s | Integrated DNA Technologies | CACCGCCATGACCTGGCTTTAAGAG |
| LUC7L2_sg2_as | Integrated DNA Technologies | AAACTTACTTTCTGGGATTCCTCC |
| LUC7L2_sg2_s | Integrated DNA Technologies | CACCGGAGGAATCCCAGAAAGTAA |
| LUC7L3_sg1_as | Integrated DNA Technologies | AAACCGTCTGATCCTACGTTCTAC |
| LUC7L3_sg1_s | Integrated DNA Technologies | CACCGTAGAACGTAGGATCAGACG |
| LUC7L3_sg2_as | Integrated DNA Technologies | AAACCGAGAGCGTAAGTCCCGCGGC |
| LUC7L3_sg2_s | Integrated DNA Technologies | CACCGCCGCGGGACTTACGCTCTCG |
| ND2_as | Integrated DNA Technologies | CCTGCAAAGATGGTAGAGTAGATGA |
| ND2_probe | Integrated DNA Technologies | FAM-CCCTGGCCCAACCC-MGBNFQ |
| ND2_s | Integrated DNA Technologies | TGTTGGTTATACCCTTCCCGTACTA |
| NDUFB5_sg1_as | Integrated DNA Technologies | AAACAAACTCTGGAATTTCTGCTAGTTC |
| NDUFB5_sg1_s | Integrated DNA Technologies | CACCGAACTAGCAGAAATTCCAGA |
| NDUFB5_sg2_as | Integrated DNA Technologies | AAACAAACTCTTGATATGGGATGCTACAC |
| NDUFB5_sg2_s | Integrated DNA Technologies | CACCGTGTAGCATCCCATATCAAGA |
| PABPC1_sg1_as | Integrated DNA Technologies | AAACCCTTTTTCTGAGCTCGACCAC |
| PABPC1_sg1_s | Integrated DNA Technologies | CACCGTGGTCGAGCTCAGAAAAAGG |
| PABPC1_sg2_as | Integrated DNA Technologies | AAACCTCCTTGGGCTACGCGTATGC |
| PABPC1_sg2_s | Integrated DNA Technologies | CACCGCATACGCGTAGCCCAAGGAG |
| PFKM_exon12_5SS Antisense Oligonucleotide | Gene Tools | GGCCACACAGCCCAGTGACTTACCA |
| PFKM_RT_as | Integrated DNA Technologies | TCATGGAATGTGTCCAGGTG |
| PFKM_RT_s | Integrated DNA Technologies | CATCATCATTGTGGCTGAGG |
| PFKM_RT2_as | Integrated DNA Technologies | ACACATTCCATGAGGGGCAG |
| PFKM_RT2_s | Integrated DNA Technologies | CCGTGGTTCTCGTCTCAACA |
| PFKP_sg1_as | Integrated DNA Technologies | AAACCCGAGAGTTTGACACACATC |
| PFKP_sg1_s | Integrated DNA Technologies | CACCGATGTGTGTCAAACTCTCGG |
| PFKP_sg2_as | Integrated DNA Technologies | AAACTTGGGATCTGATCATCCGGC |
| PFKP_sg2_s | Integrated DNA Technologies | CACCGCCGGATGATCAGATCCCAA |
| PGAM1_sg1_as | Integrated DNA Technologies | AAACACCTGAGCCCGGCGGGCCAC |
| PGAM1_sg1_s | Integrated DNA Technologies | CACCGTGGCCCGCCGGGCTCAGGT |
| PGAM1_sg2_as | Integrated DNA Technologies | AAACCTGAAGCGGTTCTCCAGGTTC |
| PGAM1_sg2_s | Integrated DNA Technologies | CACCGAACCTGGAGAACCGCTTCAG |
| Random Primers | ThermoFisher Scientific | Cat # 48190011 |
| SLC7A11_exon7_5SS Antisense Oligonucleotide | Gene Tools | CCAACTTGGACTTACCACTGCCACT |
| SLC7A11_exon9_5SS Antisense Oligonucleotide | Gene Tools | ATATACTTGTTAATATGCATTACCA |
| SLC7A11_RT_as | Integrated DNA Technologies | GGCAGATTGCCAAGATCTCAAG |
| SLC7A11_RT_s | Integrated DNA Technologies | TGCTGGCTGGTTTTACCTCAA |
| SNRNP70_sg1_as | Integrated DNA Technologies | AAACCCCGCTACGATGAGAGGTAAC |
| SNRNP70_sg1_s | Integrated DNA Technologies | CACCGTTACCTCTCATCGTAGCGGG |
| SNRNP70_sg2_as | Integrated DNA Technologies | AAACCTTACAAACACGCAGATGGC |
| SNRNP70_sg2_s | Integrated DNA Technologies | CACCGCCATCTGCGTGTTTGTAAG |
| SNRPA_sg1_as | Integrated DNA Technologies | AAACCCGCCTTGCACAGCCTTCTTC |
| SNRPA_sg1_s | Integrated DNA Technologies | CACCGAAGAAGGCTGTGCAAGGCGG |
| SNRPA_sg2_as | Integrated DNA Technologies | AAACGGCCTTTGTCATCTTCAAGG |
| SNRPA_sg2_s | Integrated DNA Technologies | CACCGGCCTTTGTCATCTTCAAGG |
| Standard Control Antisense Oligonucleotide | Gene Tools | CCTCTTACCTCAGTTACAATTTATA |
| TPI1_sg1_as | Integrated DNA Technologies | AAACCAGTCTTTGATCATGCCAGGC |
| TPI1_sg1_s | Integrated DNA Technologies | CACCGCCTGGCATGATCAAAGACTG |
| TPI1_sg2_as | Integrated DNA Technologies | AAACCGTGGGTGGTCCTGGGGCAC |
| TPI1_sg2_s | Integrated DNA Technologies | CACCGTGCCCCAGGACCACCCACG |
| Recombinant DNA | ||
| plentiCRISPR v2 | Addgene | Plasmid # 52961 |
| pLV-EF1a-IRES-Puro | Addgene | Plasmid # 85132 |
| pLV-EF1a-IRES-Puro GFP-3xFLAG | This study | N/A |
| pLV-EF1a-IRES-Puro LUC7L2–3xFLAG | This study | N/A |
| pLV-EF1a-IRES-Puro PFKM | This study | N/A |
| pMD2.G | Addgene | Plasmid # 12259 |
| psPAX2 | Addgene | Plasmid # 12260 |
| pWPI/Neo | Addgene | Plasmid # 35385 |
| pWPI/Neo GFP | This study | N/A |
| pWPI/Neo LUC7L-3xFLAG | This study | N/A |
| pWPI/Neo LUC7L2–3xFLAG | This study | N/A |
| pWPI/Neo LUC7L3–3xFLAG | This study | N/A |
| pWPI/Neo SLC7A11-HA | This study | N/A |
| pLENTICRISPR_v2_PFKP_sgRNA1 | To et al., 2019 | N/A |
| pLENTICRISPR_v2_PFKP_sgRNA1 | To et al., 2019 | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| CLIPper | Yeo lab | https://github.com/YeoLab/clipper/wiki/CLIPper-Home |
| Clustal Omega | EMBL-EBI | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| Gene Ontology GOrilla | Eden et al., 2009 | http://cbl-gorilla.cs.technion.ac.il/ |
| Gene set enrichment analysis (GSEA) | Broad Institute | https://www.gsea-msigdb.org/gsea/index.jsp |
| IGV | Broad Institute | https://software.broadinstitute.org/software/igv/ |
| MAGeCK | Li et al., 2014 | https://sourceforge.net/p/mageck/wiki/Home/ |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| rMATS | Shen et al., 2014 | http://rnaseq-mats.sourceforge.net/ |
| Seahorse Wave Desktop Software | Agilent | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-desktop-software-740897 |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Kallisto | Bray et al., 2016 | https://github.com/pachterlab/kallisto |
| R | The R Project | https://www.r-project.org/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
Antisense Oligonucleotides (ASOs) Treatment
2 × 105/mL HAP1 cells were seeded in a culture or seahorse plate. 24h later, the media was replaced by fresh media containing 10μM of the specific or control ASO, and 6μL/mL of PEG-based endoporter (GeneTools). Cells were analyzed 48h–72h later and recounter after the experiment.
Oxygen Consumption and Extracellular Acidification Rates by Seahorse XF Analyzer
1.25 × 105 K562 cells were plated on a Seahorse plate in Seahorse XF DMEM media (Agilent) containing 25mM glucose and 4mM glutamine (ThermoFisher Scientific). Oxygen consumption and extracellular acidification rates were simultaneously recorded by a Seahorse XFe96 Analyzer (Agilent) using the mito stress test protocol, in which cells were sequentially perturbed by 2mM oligomycin, 1μM CCCP and 0.5mM antimycin (Sigma). Data were analyzed using the Seahorse Wave Desktop Software (Agilent). Data were not corrected for carbonic acid derived from respiratory CO2. For seahorse in HeLa and HAP1 cells, 5 × 104 and 1 × 105 cells, respectively, were plated in a 96-well seahorse plate the day before the experiment. Cells were trypsinized after the experiment and recounted and the data was normalized to cell number.
Media Acidification
To visualize media acidification, 3 × 106 LUC7L2WT or LUC7L2KO K562 cells were collected by centrifugation, washed in PBS and incubate in 1mL of DMEM media (containing 25mM glucose and phenol red, a pH indicator) for 3h before imaging.
Mitochondrial and Nuclear DNA Determination
Mitochondrial and nuclear DNA determination was carried as previously described (Bao et al., 2016). LUC7L2WT and LUC7L2KO K562 cells were grown for 24h in fresh cell culture media and counted. 1 × 105 cells from each condition (n = 3) were harvested and lysed in 100uL mtDNA lysis buffer (25mM NaOH, 0.2mM EDTA) before incubation at 95°C for 15min. 100uL of 40mM Tris-HCl pH 7.5 was added to neutralize the reaction on ice. Samples were diluted 50x and the ratio between mitochondrial and nuclear DNA was determined using a custom Taqman based assay and qPCR using a CFx96 quantitative PCR machine (Biorad). Relative mtDNA abundance was determine using the ΔΔCt method.
RNA-Extraction, Reverse Transcription and qPCR
qPCR was performed using the TaqMan assays (ThermoFisher Scientific). RNA was extracted from total cells with an RNeasy kit (QIAGEN) and DNase-I digested before murine leukemia virus (MLV) reverse transcription using random primers (Promega) and a CFx96 quantitative PCR machine (rad). All data were normalized to TBP using ΔΔCt method.
Citrate Synthase Activity Determination
Citrate synthase activity was determined using a commercially available kit (Abcam). LUC7L2WT and LUC7L2KO K562 cells were grown for 48h in fresh cell culture media and counted. 5 × 106 cells from each condition (n = 3) were harvested, washed in PBS and resuspended in lysis buffer (provided by the kit) and completed with protease inhibitor. 100uL of lysate was used and the experiment was performed as described in the kit protocol. Protein abundance was determined using a DC protein assay (Biorad) and the citrate synthase activity signal was normalized to the protein abundance of each sample.
Glycogen Determination
Glycogen synthase activity was determined using a commercially available kit (Abcam). LUC7L2WT and LUC7L2KO K562 cells were grown for 24h in fresh cell culture media and counted. 1 × 106 cells from each condition (n = 8) were incubated in fresh media for another 6h before being harvested, washed in ice-cold PBS and resuspended in lysis buffer (provided by the kit). 25uL of lysate was used and the experiment was performed as described in the kit protocol and normalized to total protein abundance.
Glutamate Determination
Glutamate levels were determined using a commercially available AMPLEX kit (Life Technologies). LUC7L2WT, LUC7L2Rescue (LUC7L2KO cells were a LUC7L2 cDNA was stably expressed), LUC7L2KO and ASO treated K562 or HAP1 cells were grown for 24h in fresh culture media and counted. Cells were then washed with PBS, the media was replaced and the cells were incubated for another 3h. Medias were then collected and centrifuged at 2,000g for 3min, and glutamate concentrations were determined from the supernatant.
Cell Viability Assay in Galactose
To measure viability in galactose, cells were washed in PBS, counted and an equal number of cells was seeded in culture media containing 25mM glucose or 25mM galactose. 24h later, cells were collected and viable cells were determined using a Vi-Cell Counter (Beckman).
Electron Microscopy
5 × 106 LUC7L2WT and LUC7L2KO K562 cells were grown for 24h in fresh cell culture media and were fixed in 2.5% gluteraldehyde, 3% paraformaldehyde with 5% sucrose in 0.1M sodium cacodylate buffer (pH 7.4), pelleted, and post fixed in 1% OsO4 in veronal-acetate buffer. Cells were stained en block overnight with 0.5% uranyl acetate in veronal-acetate buffer (pH6.0), dehydrated and embedded in Embed-812 resin. Sections were cut on a Leica EM UC7 ultra microtome with a Diatome diamond knife at a thickness setting of 50 nm, stained with 2% uranyl acetate, and lead citrate. The sections were examined using a FEI Tecnai spirit at 80KV and photographed with an AMT CCD camera.
Confocal Microscopy and Immunofluorescence
HeLa cells were transduced with pWPI-LUC7L2-GFP at least 48h before the experiment and grown on coverslips until 30–50% confluent. Cells were successively fixed in 4% paraformaldehyde in cell culture media at room temperature for 30min, blocked/permeabilized for 30min in Abdil buffer (PBS + 0.1% Triton X-100 + 3% bovine serum albumin (w/v)), incubated with primary antibodies (1:200) in Abdil buffer for 1h, washed 3× 5min in PBS + 0.1% Triton X-100, incubated in fluorophore-coupled secondary antibodies (1:1000) and hoescht (1:10000) in Abdil for 30min, washed 3x in PBS + 0.1% Triton X-100 and mounted on a slide using FluorSave (EMD Millipore). Cells were imaged using a Zeiss LSM700 confocal microscope
Polyacrylamide Gel Electrophoresis and Immunoblotting
Cells were harvested, washed in PBS and lysed for 5min on ice in RIPA buffer (25mM Tris pH 7.5, 150mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% NP40 analog, 1x protease (Cell Signaling) and 1:500 Universal Nuclease (ThermoFisher Scientific). Protein concentration was determined from total cell lysates using DC protein assay (Biorad). Gel electrophoresis was done on Novex Tris-Glycine gels (ThermoFisher Scientific) before transfer using the Trans-Blot Turbo blotting system and nitrocellulose membranes (Biorad). All immunoblotting was performed in Intercept Protein blocking buffer (Licor). Washes were done in TBS + 0.1% Tween-20 (Sigma). Specific primary antibodies were diluted 1:100–1:5000 in blocking buffer. Fluorescent-coupled secondary antibodies were diluted 1:10,000 in blocking buffer. Membranes were imagined with an Odyssey CLx analyzer (Licor) or by chemiluminescence. In a few instances, the same lysates were loaded on parallel gels to avoid antibody cross-reactivity, or because the proteins of interest could not be resolved on the same percentage gels. In these cases, a loading control is provided for all gels, and panels from the same immunoblots are connected with a dotted line on the figure. All raw immunoblots pictures are provided in the supplemental information document.
Liquid Chromatography-Mass Spectrometry (LC-MS)
Intracellular Metabolite Profiling
LUC7L2WT and LUC7L2KO K562 cells were pre-incubated overnight in profiling media containing glucose-free DMEM media (ThermoFisher Scientific),10% dialyzed FBS (ThermoFisher Scientific), penicillin and streptomycin (ThermoFisher Scientific) and 25mM glucose (Sigma) but omitting supplemental pyruvate or uridine. On the day of the experiment, 2.5 × 106 cells were seeded in 3mL of profiling media in a 6-well plate (n = 6 replicate plates for each genotype). An additional well containing 3mL of media but no cells was included as control. After 8h of incubation, cells were centrifuged at 300g for 3min at room temperature and the culture media was saved and frozen at −80°C until further analysis (described below). The cell pellet was briefly washed in ice-cold 150mM NaCl and centrifuged again. 1mL dry ice-cold 80% methanol was then added to quench metabolism. Cells were incubated on ice for >20min, centrifuged at 20,000g (4°C) and the supernatant was saved and dried down in a speed vacuum concentrator (Savant SPD 1010, ThermoFisher Scientific) and stored at −80°C until analysis. On the day of analysis, samples were re-suspended in 120μL of 60/40 acetonitrile/water, vortexed, sonicated in ice-cold water for 1min, incubated on ice for 20min and the supernatant was collected in an autosampler vial after centrifugation at 21,000g for 20min at 4°C. Pooled quality control (PooledQC) samples were generated by combining ~20μL of each sample. Metabolite profiling was performed using a Dionex Ultimate 3000 UHPLC system coupled to a Q-Exactive Plus orbitrap mass spectrometer (ThermoFisher Scientific, Waltham, MA) with an Ion Max source and HESI II probe operating in polarity switching mode. A zwitterionic zic pHilic column (150 × 2.1mm, 5μm, Merck KGaA) was used for polar metabolite separation. Mobile phase A (MPA) was 20mM ammonium carbonate in water, pH9.6 (adjusted with ammonium hydroxide) and MPB was acetonitrile. The column was held at 27°C, with an injection volume of 5μL, and an autosampler temperature of 4°C. The LC conditions at a flow rate of 0.15 mL/min were: 0min: 80% B, 0.5min: 80% B, 20.5min: 20% B, 21.3min: 20%B, 21.5min: 80% B with 7.5min of column equilibration time. MS parameters were: sheath gas flow = 30, aux gas flow = 7, sweep gas flow = 2, spray voltage = 2.80 for negative & 3.80 for positive ion modes, capillary temperature = 310°C, S-lens RF level = 50 and aux gas heater temp 370°C. Data acquisition was performed using Xcalibur 4.1 (ThermoFisher Scientific) in full scan mode with a range of 70–1000m/z, a resolving power of 70,000, an AGC target of 1 × 106, and a maximum injection time of 80ms. Data analysis was done using Compound Discoverer 3.0. Samples were injected in a randomized order and pooled QC samples were injected regularly throughout the analytical batches. Metabolite annotation was based on accurate mass (±5ppm) and matching retention time (±0.3min) as well as MS/MS fragmentation pattern from the pooled QC samples against in-house retention time +MSMS library of reference chemical standards. Metabolites which had a pooled QC CV<20% were used for the statistical analysis.
Media Profiling
30μL of control or spent media was mixed with 120μL of ice-cold acetonitrile containing the metabolomics amino acid mix from Cambridge Isotope Labs (MSK-A2–1.2), 13C6 -glucose, 13C3 -pyruvate, and 13C3 -lactate as internal standards, was vortexed, incubated on ice for 20min, centrifuged at 21,000g for 20min at 4°C and the supernatant was transferred to an autosampler vial for LC-MS analysis. Calibration curves were prepared in water at varying concentration levels depending on the amino acid level in the DMEM media formulation. Metabolite separation was done using XBridge BEH amide (2.1 × 150mm, 1.7 μm, Waters Corporation, MA). Mobile phase A was 90/5/5 water/acetonitrile/methanol, 20mM ammonium acetate, 0.2% acetic acid and mobile phase B was 90/10 acetonitrile/water, 10mM ammonium acetate, 0.2% acetic acid. The column temperature was 40°C and flow rate was 0.3 mL/min. The chromatographic gradient was: 0min: 95% B, 9min : 70% B, 9.75min: 40% B, 12min: 40% B, 13min: 30% B, 14min : 30%B, 14.1min: 10% B,17min: 10% B, 17.5min: 95% B, 22min : 95% B. MS parameters were: sheath gas flow = 50, aux gas flow = 12, sweep gas flow = 2, spray voltage = 2.80 for negative (3.50 for positive), Capillary temperature = 320°C, S-lens RF level = 50 and aux gas heater temperature 380°C. Data acquisition was done using Xcalibur 4.1 (ThermoFisher Scientific) and performed in full scan mode with a range of 70–1000m/z, a resolving power of 70,000, an AGC target 106, and a maximum injection time of 100ms. Tracefinder 4.1 was used for quantitation analysis. One LUC7L2KO sample gave aberrant spectra and was excluded.
LUC7L2–3xFLAG Immunoprecipitation and Mass Spectrometry
For immunoprecipitation, a 3xFLAG-tagged version of LUC7L2 was cloned into pWPI-Neo (Addgene), and viruses were produced. pWPI-GFP served as control. 293T cells were infected and expanded for at least 48h. An equal number of cells from each condition in duplicate was lysed in IP lysis buffer (50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 1% NP-40, 0.1% sodium deoxycholate, 1× protease (Cell Signaling). Lysates were cleared by centrifugation at 20,000g for 20min and the supernatants were saved. Washed FLAG M2 magnetic beads (Sigma) were added to the lysate and incubated overnight at 4°C. Beads were recovered after extensive washing, and the protein/RNA complexes were eluted with 100 μg/mL 3xFLAG peptide (Sigma). For protein isolation, the eluate was run on an SDS-PAGE gel until the whole lysate entered the gel. Single bands containing all proteins from the sample were then cut and analyzed by mass spectrometry at the Whitehead proteomics facility. Peptides were identified and quantified using the Top 3 total ion current (TIC) method (Scaffold4). Interacting proteins were considered positive when they were enriched >2-fold over either control and identified only by unique peptides.
Enhanced Crosslinking and Immunoprecipitation (eCLIP)
Libraries were generated using standard eCLIP methods according to published protocols (Van Nostrand et al., 2017). In brief, K562 and Hela cells (2 × 107 for each replicate plate) were UV crosslinked (254 nm, 400 mJ/cm2), then lysed and sonicated (Bioruptor) in eCLIP lysis buffer (50 mM Tris–HCl pH 7.4, 100 mM NaCl, 1% NP-40 (Igepal CA630), 0.1% SDS, 0.5% sodium deoxycholate, 1:200 Protease Inhibitor Cocktail I, in RNase/DNase-free H2O). RNA fragments were created by incubating lysates with RNase I (Ambion) and LUC7L2:RNA complexes were immunoprecipitated for 2h at 4°C using Dynabeads bound to 4μg of LUC7L2-specific affinity-purified antibody. In parallel, libraries were generated from size-matched input (SMInput) samples containing RNAs present in the whole cell lysates, i.e. sans RBP-specific IP. For the IPs, a series of stringent washes (high salt wash buffer: 50 mM Tris–HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, in RNase/DNase-free H2O; wash buffer: 20 mM Tris–HCl pH 7.4, 10 mM MgCl2, 0.2% Tween-20, in RNase/DNase-free H2O) was followed by RNA dephosphorylation with FastAP (ThermoFisher Scientific) and T4 PNK (NEB) then ligation of an adaptor to the 3′ ends of the RNAs with T4 RNA ligase 1 (NEB). Protein:RNA complexes were separated on 4–12% polyacrylamide gels, transferred to a nitrocellulose membranes and RNA was extracted from the membranes using Proteinase K (NEB). Immunoprecipitation was confirmed by parallel western blotting of fractions of each sample with the antibody described previously. Following purification, SMInput RNA were dephosphorylated and 3′-ligated and all samples were reverse transcribed with Superscript III (Invitrogen). Free primers were removed with ExoSap-IT (Affymetrix) and a DNA adaptor was ligated to the 3′ ends of the cDNA with T4 RNA ligase 1. cDNA was quantified by qPCR and PCR amplified using Q5 Master Mix (NEB) and resulting libraries were purified prior to Illumina sequencing.
Blue-Native PAGE
For blue-native PAGE, a mitochondria-rich fraction (Jourdain et al., 2013) was isolated from LUC7L2WT and LUC7L2KO K562 and HeLa cells grown for two weeks in glucose or galactose-containing by differential centrifugation. An equal amount of mitochondria were resuspended in blue-native loading buffer containing 1% digitonin (Life Technologies) before electrophoresis on a 3 to 12% Native PAGE (Life Technologies) according to the manufacturer’s instruction. Gels were then fixed and stained with coomassie R-250, or transferred to PVDF membranes, denatured by 3% acetic acid treatment, destained with methanol, blocked and an immunodetection was performed with the indicated antibodies and secondary HRP-coupled antibodies. Samples were loaded on parallel gels to avoid cross-reactivity between antibodies.
Mitochondrial Translation
Determination of mitochondrial translation products in LUC7L2WT and LUC7L2KO cells was performed as previously described (Jourdain et al., 2013). K562s cells were incubated for 20 min in methionine/cysteine-free DMEM (Sigma) complemented with dialyzed serum and 2mM L-glutamine. Cells were then incubated for 1h in the same medium in the presence of 100μg/ml emetine and 100μCi/μL 35S-labeled methionine/cysteine (PerkinElmer). Total protein concentration of cell lysates was measured, and lysates were resolved on an acrylamide gel, transferred to a nitrocellulose membrane, and analyzed by autoradiography. A mitochondrial translation inhibitor (chloramphenicol) was used as a control.
Gene-Specific cDNA Cloning and Expression
cDNAs of interest were custom designed (Genewiz or IDT) and cloned into pWPI-Neo or pLV-lenti-puro (Visanji et al., 2011, Hayer et al., 2016) using BamHI/SpeI and BamHI/NotI (NEB), respectively. cDNA sequences were:
GFP-3xFLAG
atggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaaggattataaagatcatgatggcgattataaagatcatgatattgattataaagatgatgatgataaataatagtgagcggccgc
LUC7L-3xFLAG
atgtccgcccaggcgcagatgcgggccctgctggaccagctcatgggcacggctcgggacggagacgaaaccagacagagggtcaagtttacagatgaccgtgtctgcaagagtcaccttctggactgctgcccccatgacatcctggctgggacgcgcatggatttaggagaatgtaccaaaatccacgacttggccctccgagcagattatgagattgcaagtaaagaaagagacctgttttttgaattagatgcaatggatcacttggagtcctttattgctgaatgtgatcggagaactgagctcgccaagaagcggctggcagaaacacaggaggaaatcagtgcggaagtttctgcaaaggcagaaaaagtacatgagttaaatgaagaaataggaaaactccttgctaaagccgaacagctaggggctgaaggtaatgtggatgaatcccagaagattcttatggaagtggaaaaagttcgtgcgaagaaaaaagaagctgaggaagaatacagaaattccatgcctgcatccagttttcagcagcaaaagctgcgtgtctgcgaggtctgttcagcctaccttggtctccatgacaatgaccgtcgcctggcagaccacttcggtggcaagttacacttggggttcattcagatccgagagaagcttgatcagttgaggaaaactgtcgctgaaaagcaggagaagagaaatcaggatcgcttgaggaggagagaggagagggaacgggaggagcgtctgagcaggaggtcgggatcaagaaccagagatcgcaggaggtcacgctcccgggatcggcgtcggaggcggtcaagatctacctcccgagagcgacggaaattgtcccggtcccggtcccgagatagacatcggcgccaccgcagccgttcccggagccacagccggggacatcgtcgggcttcccgggaccgaagtgcgaaatacaagttctccagagagcgggcatccagagaggagtcctgggagagcgggcggagcgagcgagggcccccggactggaggcttgagagctccaacgggaagatggcttcacggaggtcagaagagaaggaggccggcgagatcgattataaagatcatgatggcgattataaagatcatgatattgattataaagatgatgatgataaataatagtgagcggccgc
LUC7L2–3xFLAG
atgtcggcgcaggcccagatgcgcgcgatgctggaccagttgatgggcacctcccgggacggagatacaactcgtcaacgaatcaaattcagtgatgacagagtatgcaagagtcaccttctcaactgttgtcctcatgatgtcctttctggaactagaatggatcttggagaatgtctgaaagtccatgacctggctttaagagcggattatgaaattgcatccaaagaacaagattttttctttgaacttgatgccatggatcatctgcagtcattcattgcagattgtgatcgtagaacagaagtggccaagaaaagattagcagaaactcaagaagagattagtgctgaagtagcagcaaaggcagaacgtgttcatgagttaaatgaagaaattggtaaattgttagccaaggtggaacaactaggagctgaagggaatgtggaggaatcccagaaagtaatggatgaagtagagaaagcacgggcaaagaaaagagaagcagaggaagtttatcggaattctatgccagcttccagttttcagcagcagaaacttcgagtctgtgaagtctgctctgcctatttaggacttcatgataatgacagacgactggctgatcattttgggggtaaactgcacctgggatttattgaaataagagagaagcttgaagaattaaagagagtcgtagctgagaagcaggagaaaagaaaccaggaacggctgaaacgaagagaagagagagagagagaagaaagggagaagctgaggaggtcccgatcacacagcaagaatccaaaaagatccaggtccagagagcatcgcagacatcgatctcgctccatgtcacgtgaacgcaagaggagaactcgatccaaatctcgggagaaacgccatcgccacaggtcccgctccagcagccgtagccgcagccgtagccaccagagaagtcggcacagttctagagataggagcagagaacgatccaagaggagatcctcaaaagaaagattcagagaccaagacttagcatcatgtgacagagacaggagttcaagagacagatcacctcgtgacagagatcggaaagataagaagcggtcctatgagagtgctaatggcagatcagaagacaggaggagctctgaagagcgcgaagcaggggagatcggagggggtgattataaagatcatgatggcgattataaagatcatgatattgattataaagatgatgatgataaataatagtga
LUC7L3–3xFLAG
atgatttcggccgcgcagttgttggatgagttaatgggccgggaccgaaacctagccccggacgagaagcgcagcaacgtgcggtgggaccacgagagcgtttgtaaatattatctctgtggtttttgtcctgcggaattgttcacaaatacacgttctgatcttggtccgtgtgaaaaaattcatgatgaaaatctacgaaaacagtatgagaagagctctcgtttcatgaaagttggctatgagagagattttttgcgatacttacagagcttacttgcagaagtagaacgtaggatcagacgaggccatgctcgtttggcattatctcaaaaccagcagtcttctggggccgctggcccaacaggcaaaaatgaagaaaaaattcaggttctaacagacaaaattgatgtacttctgcaacagattgaagaattagggtctgaaggaaaagtagaagaagcccaggggatgatgaaattagttgagcaattaaaagaagagagagaactgctaaggtccacaacgtcgacaattgaaagctttgctgcacaagaaaaacaaatggaagtttgtgaagtatgtggagcctttttaatagtaggagatgcccagtcccgggtagatgaccatttgatgggaaaacaacacatgggctatgccaaaattaaagctactgtagaagaattaaaagaaaagttaaggaaaagaaccgaagaacctgatcgtgatgagcgtctaaaaaaggagaagcaagaaagagaagaaagagaaaaagaacgggagagagaaagggaagaaagagaaaggaaaagacgaagggaagaggaagaaagagaaaaagaaagggctcgtgacagagaaagaagaaagagaagtcgttcacgaagtagacactcaagccgaacatcagacagaagatgcagcaggtctcgggaccacaaaaggtcacgaagtagagaaagaaggcggagcagaagtagagatcgacgaagaagcagaagccatgatcgatcagaaagaaaacacagatctcgaagtcgggatcgaagaagatcaaaaagccgggatcgaaagtcatataagcacaggagcaaaagtcgggacagagaacaagatagaaaatccaaggagaaagaaaagaggggatctgatgataaaaaaagtagtgtgaagtccggtagtcgagaaaagcagagtgaagacacaaacactgaatcgaaggaaagtgatactaagaatgaggtcaatgggaccagtgaagacattaaatctgaaggtgacactcagtccaatgattataaagatcatgatggcgattataaagatcatgatattgattataaagatgatgatgataaataatagtgagcggccgc
SLC7A11-HA
atggtcagaaagcctgttgtgtccaccatctccaaaggaggttacctgcagggaaatgttaacgggaggctgccttccctgggcaacaaggagccacctgggcaggagaaagtgcagctgaagaggaaagtcactttactgaggggagtctccattatcattggcaccatcattggagcaggaatcttcatctctcctaagggcgtgctccagaacacgggcagcgtgggcatgtctctgaccatctggacggtgtgtggggtcctgtcactatttggagctttgtcttatgctgaattgggaacaactataaagaaatctggaggtcattacacatatattttggaagtctttggtccattaccagcttttgtacgagtctgggtggaactcctcataatacgccctgcagctactgctgtgatatccctggcatttggacgctacattctggaaccattttttattcaatgtgaaatccctgaacttgcgatcaagctcattacagctgtgggcataactgtagtgatggtcctaaatagcatgagtgtcagctggagcgcccgaatccagattttcttaaccttttgcaagctcacagcaattctgataattatagtccctggagttatgcagctaattaaaggtcaaacgcagaactttaaagacgccttttcaggaagagattcaagtattacgcggttgccactggctttttattatggaatgtatgcatatgctggctggttttacctcaactttgttactgaagaagtagaaaaccctgaaaaaaccattccccttgcaatatgtatatccatggccattgtcaccattggctatgtgctgacaaatgtggcctactttacgaccattaatgctgaggagctgctgctttcaaatgcagtggcagtgaccttttctgagcggctactgggaaatttctcattagcagttccgatctttgttgccctctcctgctttggctccatgaacggtggtgtgtttgctgtctccaggttattctatgttgcgtctcgagagggtcaccttccagaaatcctctccatgattcatgtccgcaagcacactcctctaccagctgttattgttttgcaccctttgacaatgataatgctcttctctggagacctcgacagtcttttgaatttcctcagttttgccaggtggctttttattgggctggcagttgctgggctgatttatcttcgatacaaatgcccagatatgcatcgtcctttcaaggtgccactgttcatcccagctttgttttccttcacatgcctcttcatggttgccctttccctctattcggacccatttagtacagggattggcttcgtcatcactctgactggagtccctgcgtattatctctttattatatgggacaagaaacccaggtggtttagaataatgtcagagaaaataaccagaacattacaaataatactggaagttgtaccagaagaagataagttatatccatatgatgttccagattatgcttaatagtga
PFKM
atgacccatgaagagcaccatgcagccaaaaccctggggattggcaaagccattgctgtcttaacctctggtggagatgcccaaggtatgaatgctgctgtcagggctgtggttcgagttggtatcttcaccggtgcccgtgtcttctttgtccatgagggttatcaaggcctggtggatggtggagatcacatcaaggaagccacctgggagagcgtttcgatgatgcttcagctgggaggcacggtgattggaagtgcccggtgcaaggactttcgggaacgagaaggacgactccgagctgcctacaacctggtgaagcgtgggatcaccaatctctgtgtcattgggggtgatggcagcctcactggggctgacaccttccgttctgagtggagtgacttgttgagtgacctccagaaagcaggtaagatcacagatgaggaggctacgaagtccagctacctgaacattgtgggcctggttgggtcaattgacaatgacttctgtggcaccgatatgaccattggcactgactctgccctgcatcggatcatggaaattgtagatgccatcactaccactgcccagagccaccagaggacatttgtgttagaagtaatgggccgccactgtggatacctggcccttgtcacctctctgtcctgtggggccgactgggtttttattcctgaatgtccaccagatgacgactgggaggaacacctttgtcgccgactcagcgagacaaggacccgtggttctcgtctcaacatcatcattgtggctgagggtgcaattgacaagaatggaaaaccaatcacctcagaagacatcaagaatctggtggttaagcgtctgggatatgacacccgggttactgtcttggggcatgtgcagaggggtgggacgccatcagcctttgacagaattctgggcagcaggatgggtgtggaagcagtgatggcacttttggaggggaccccagataccccagcctgtgtagtgagcctctctggtaaccaggctgtgcgcctgcccctcatggaatgtgtccaggtgaccaaagatgtgaccaaggccatggatgagaagaaatttgacgaagccctgaagctgagaggccggagcttcatgaacaactgggaggtgtacaagcttctagctcatgtcagacccccggtatctaagagtggttcgcacacagtggctgtgatgaacgtgggggctccggctgcaggcatgaatgctgctgttcgctccactgtgaggattggccttatccagggcaaccgagtgctcgttgtccatgatggtttcgagggcctggccaaggggcagatagaggaagctggctggagctatgttgggggctggactggccaaggtggctctaaacttgggactaaaaggactctacccaagaagagctttgaacagatcagtgccaatataactaagtttaacattcagggccttgtcatcattgggggctttgaggcttacacagggggcctggaactgatggagggcaggaagcagtttgatgagctctgcatcccatttgtggtcattcctgctacagtctccaacaatgtccctggctcagacttcagcgttggggctgacacagcactcaatactatctgcacaacctgtgaccgcatcaagcagtcagcagctggcaccaagcgtcgggtgtttatcattgagactatgggtggctactgtggctacctggctaccatggctggactggcagctggggccgatgctgcctacatttttgaggagcccttcaccattcgagacctgcaggcaaatgttgaacatctggtgcaaaagatgaaaacaactgtgaaaaggggcttggtgttaaggaatgaaaagtgcaatgagaactataccactgacttcattttcaacctgtactctgaggaggggaagggcatcttcgacagcaggaagaatgtgcttggtcacatgcagcagggtgggagcccaaccccatttgataggaattttgccactaagatgggcgccaaggctatgaactggatgtctgggaaaatcaaagagagttaccgtaatgggcggatctttgccaatactccagattcgggctgtgttctggggatgcgtaagagggctctggtcttccaaccagtggctgagctgaaggaccagacagattttgagcatcgaatccccaaggaacagtggtggctgaaactgaggcccatcctcaaaatcctagccaagtacgagattgacttggacacttcagaccatgcccacctggagcacatcacccggaagcggtccggggaagcggccgtctaatagtga
Quantitative Proteomics
LUC7L2WT and LUC7L2KO cells were grown in n = 3 replicate plates in DMEM media containing 25mM glucose or galactose for 2 weeks. Quantitative proteomics was performed at the Thermofisher for Multiplexed Proteomics (Harvard). In short, cells were harvested and total protein quantification was performed using micro-BCA assay (Pierce). Samples were reduced with DTT and alkylated with iodoacetamide before protein precipitation in methanol/chloroform. Pellets were resuspended in 200 mM EPPS, pH 8.0 and a digestion was performed sequentially using LysC (1:50) and Trypsin (1:100) based on protease to protein ratio. ~50μg peptide per sample was labeled with TMTpro 16 reagents. A small aliquot of each sample was then combined and analyzed by LC-MS3 to verify labeling efficiency and mixing ratios. Samples were combined, desalted, and dried by speedvac. 14 fractions from the total proteome HPRP set were analyzed on an Orbitrap Eclipse mass spectrometer using a 180-minute method MS3 method with real-time search. Peptides were detected (MS1) and quantified (MS3) in the Orbitrap. Peptides were sequenced (MS2) in the ion trap. MS2 spectra were searched using the COMET algorithm against a custom protein database containing only one protein per gene (referenced as the canonical isoform). Peptide spectral matches were filtered to a 1% false discovery rate (FDR) using the target-decoy strategy combined with linear discriminant analysis. The proteins from the 14 runs were filtered to a <1% FDR. Proteins were quantified only from peptides with a summed SN threshold of >100. Only unique peptides were considered for downstream analysis. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD021917 and 10.6019/PXD021917″.
Next-Generation RNA Sequencing
Total RNA from LUC7L2WT and LUC7L2KO K562 and HeLa cells (n = 3, replicate plates each) were isolated using a RNeasy kit (QIAGEN). RNA sequencing libraries were prepared by the Genomics Platform at the Broad Institute based on the True-Seq protocol (Illumina), which selects for polyadenylated RNA and preserves strand specificity. Libraries were sequenced using a NovaSeq 6000 instrument, generating 2 × 101bp paired-end reads with a minimum of 40M pairs per sample. The RNA sequencing data have been deposited to the Gene Expression Omnibus (GEO) with the dataset identifier GSE157917.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests and sample sizes are reported in the legends associated with each figure. Methods for quantification and statistical analysis of large-scale datasets are described below.
eCLIP
Raw reads were mapped using STAR (Dobin et al., 2013) to the hg19 genome following standard ENCODE guidelines (https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf, page 9). Read length was relaxed to accommodate the slightly shorter average eCLIP read length (--outFilterMatchNminOverLread 0.33). Duplicate PCR reads were removed from the mapped reads to generate final reads. Mapped reads were then processed into peaks using CLIPper (Lovci et al., 2013) with standard specifications. For each dataset, only peaks shared between two replicates and not appearing in the input controls were considered in subsequent analyses. Reads were then filtered to a robust p-value of P<10−4 or as indicated in the manuscript. Using these highest-confidence peaks, a metaplot centered on the exon was created, showing an additional 500 bases upstream and 500 bases downstream of the flanking introns. For each region (exonic or intronic), the relative positions of each single base of a CLIP peak were summed and normalized to the mean base coverage in that region.
Gene Expression
The reads were aligned with STAR (Dobin et al., 2013) to the human genome hg19 using default parameters and a two-pass approach. Following a first-pass alignment of each sample, novel splice junctions were pooled across all samples from the same cell type and incorporated into the genome annotation for a second-pass alignment. Second-pass gene counts derived from uniquely mapping pairs with the expected strandedness were output by STAR. Differential gene expression analysis between LUC7L2WT and LUC7L2KO samples was performed in R using the package DESeq2 (Love et al., 2014). Differentially-expressed genes were considered significant below FDR<10−4 and with an absolute fold change value greater than 50%.
Splicing
Splicing events were analyzed with rMATS.4.0.2 (three replicates per condition per cell line) using standard specifications and the hg19 genome. Events were considered significant below FDR<0.1 and with an absolute Δψ value greater than 0.05. Only events in genes with total TPM>1 in the WT cell line of interest as called by kallisto (Bray et al., 2016) according to standard parameters were considered.
Overlap
Overlap analyses were normalized for gene expression as in (Friedman et al., 2008). Briefly, ten bins (percentiles) of gene expression were established for HeLa and K562 cells using the average gene expression from three replicates determined by kallisto (Bray et al., 2016) according to standard parameters. Averaged TPMs were filtered for TPM>1 as above. For each bin, the percentage of genes within that bin appearing in corresponding eCLIP or rMATS was calculated. A 10 × 10 matrix was then created by multiplying these frequencies together for each cell. A second 10 × 10 matrix was then populated by the actual counts of total overlapping genes in each cell (of 100). A third 10 × 10 matrix of expected counts of eCLIP/rMATS was subsequently generated from these two matrices and the total sum of this matrix represented the “expected” value of the overlap based on gene expression alone. Finally, the significance of the observed count of overlapping genes per query was estimated by Poisson (R 1.3.1093, poisson.test).
Correlation
Published protein expression datasets (Geiger et al., 2013, Huttlin et al., 2010, Ping et al., 2018, Ubaida-Mohien et al., 2019) were filtered to exclude genes with null values in >20% of samples and remaining missing values were replaced with half the minimum observed for that protein. Each protein was normalized via Z-score and then Pearson correlations were calculated between LUC7L2 profiles and each protein. Anti-correlation with OXPHOS was assessed via a Wilcoxon Rank Sum test.
Gene Ontology
Gene ontology analysis was performed using GOrilla with default settings and using a ranked gene list as input (Eden et al., 2009). Only GO categories with <500 genes and represented by >2 significant genes were considered. Highlighted genes in figures correspond to the GO categories “RNA splicing” (GO:0008380), “U1 snRNP” (GO:0005685) and “OXPHOS” (manually curated) and GO gene lists are reported in Table S1.
Supplementary Material
Table S4: LUC7L2 Immunoprecipitation and Mass Spectrometry (IP-MS), Related to Figure 4 and S4.
Table S6: Differential Gene Expression Analysis in LUC7L2KO K562 Cells, Related to Figure 4 and S4.
Table S5: LUC7L2 Enhanced Crosslinking and Immunoprecipitation (eCLIP), Related to Figure 4 and S4.
Table S8: Global Quantitative Proteomics in LUC7L2KO And Galactose-Grown K562 Cells Relative to Glucose-Grown LUC7L2WT Cells, Related to Figure 6 and S6.
Table S7: Differential Splicing Analysis in LUC7L2KO K562 Cells, Related to Figure 4 and S4.
Table S1: Genome-Wide CRISPR/Cas9 Screening Results and Gene Ontology, Related to Figure 1 and S1.
HIGHLIGHTS.
Expression of LUC7L2 and the U1 snRNP represses OXPHOS
Pre-mRNA splicing and expression of PFKM and SLC7A11 (xCT) requires LUC7L2
Loss of LUC7L2 and glycolysis promotes respiratory chain (super)complex assembly
Cross-regulation and role in energy metabolism for the LUC7 family
ACKNOWLEDGEMENTS
We thank J. Wengrod, T.L. To and the VKM laboratory for advice and feedback on the manuscript; M. Shadpour for technical help; D. Steensma, B. Ebert, M. Blower, A. Kotini and E. Papapetrou for fruitful discussion. This work was supported by NIH grants R35GM122455 (VKM), R01GM085319 (CBB), HG004659 and U41HG009889 (GWY), EMBO long-term ALTF 554–2015 and SNF Advanced Postdoc.Mobility P300PA_171514 fellowships (AAJ) and a F32 Fellowship from the National Institute of General Medical Sciences 1F32GM133047–01 (OSS). VKM is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
VKM is a paid scientific advisor to 5AM Ventures and Janssen Pharmaceuticals. OSS is a paid consultant for Proteinaceous. RS holds equity in BlueBird Bio. GWY is co-founder, member of the Board of Directors, on the scientific advisory board, equity holder, and paid consultant for Locanabio and Eclipse Bioinnovations. GWY is a visiting professor at the National University of Singapore. GWY’s interest(s) have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies. AAJ and VKM are co-inventors on a US provisional patent application related to the work in this manuscript. The authors declare no other competing financial interests.
INCLUSION AND DIVERSITY STATEMENT
One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
REFERENCES
- ACIN-PEREZ R, BAYONA-BAFALUY MP, FERNANDEZ-SILVA P, MORENOLOSHUERTOS R, PEREZ-MARTOS A, BRUNO C, MORAES CT & ENRIQUEZ JA 2004. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell, 13, 805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARROYO JD, JOURDAIN AA, CALVO SE, BALLARANO CA, DOENCH JG, ROOT DE & MOOTHA VK 2016. A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab, 24, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BADGLEY MA, KREMER DM, MAURER HC, DELGIORNO KE, LEE HJ, PUROHIT V, SAGALOVSKIY IR, MA A, KAPILIAN J, FIRL CEM, DECKER AR, SASTRA SA, PALERMO CF, ANDRADE LR, SAJJAKULNUKIT P, ZHANG L, TOLSTYKA ZP, HIRSCHHORN T, LAMB C, LIU T, GU W, SEELEY ES, STONE E, GEORGIOU G, MANOR U, IUGA A, WAHL GM, STOCKWELL BR, LYSSIOTIS CA & OLIVE KP 2020. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science, 368, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAO XR, ONG SE, GOLDBERGER O, PENG J, SHARMA R, THOMPSON DA, VAFAI SB, COX AG, MARUTANI E, ICHINOSE F, GOESSLING W, REGEV A, CARR SA, CLISH CB & MOOTHA VK 2016. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERG MG, SINGH LN, YOUNIS I, LIU Q, PINTO AM, KAIDA D, ZHANG Z, CHO S, SHERRILL-MIX S, WAN L & DREYFUSS G 2012. U1 snRNP determines mRNA length and regulates isoform expression. Cell, 150, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONNET S, ARCHER SL, ALLALUNIS-TURNER J, HAROMY A, BEAULIEU C, THOMPSON R, LEE CT, LOPASCHUK GD, PUTTAGUNTA L, BONNET S, HARRY G, HASHIMOTO K, PORTER CJ, ANDRADE MA, THEBAUD B & MICHELAKIS ED 2007. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell, 11, 37–51. [DOI] [PubMed] [Google Scholar]
- BRAY NL, PIMENTEL H, MELSTED P & PACHTER L 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol, 34, 525–7. [DOI] [PubMed] [Google Scholar]
- CHRISTOFK HR, VANDER HEIDEN MG, HARRIS MH, RAMANATHAN A, GERSZTEN RE, WEI R, FLEMING MD, SCHREIBER SL & CANTLEY LC 2008. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature, 452, 230–3. [DOI] [PubMed] [Google Scholar]
- CRABTREE HG 1929. Observations on the carbohydrate metabolism of tumours. Biochem J, 23, 536–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE CONTI L, BARALLE M & BURATTI E 2013. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip Rev RNA, 4, 49–60. [DOI] [PubMed] [Google Scholar]
- DE FRANCISCO AMORIM M, WILLING EM, SZABO EX, FRANCISCO-MANGILET AG, DROSTE-BOREL I, MACEK B, SCHNEEBERGER K & LAUBINGER S 2018. The U1 snRNP Subunit LUC7 Modulates Plant Development and Stress Responses via Regulation of Alternative Splicing. Plant Cell, 30, 2838–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEPHOURE N, ZHOU C, VILLEN J, BEAUSOLEIL SA, BAKALARSKI CE, ELLEDGE SJ & GYGI SP 2008. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A, 105, 10762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOBIN A, DAVIS CA, SCHLESINGER F, DRENKOW J, ZALESKI C, JHA S, BATUT P, CHAISSON M & GINGERAS TR 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDEN E, NAVON R, STEINFELD I, LIPSON D & YAKHINI Z 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics, 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORTES P, BILBAO-CORTES D, FORNEROD M, RIGAUT G, RAYMOND W, SERAPHIN B & MATTAJ IW 1999. Luc7p, a novel yeast U1 snRNP protein with a role in 5’ splice site recognition. Genes Dev, 13, 2425–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRIEDMAN BA, STADLER MB, SHOMRON N, DING Y & BURGE CB 2008. Ab initio identification of functionally interacting pairs of cis-regulatory elements. Genome Res, 18, 1643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO G, XIE A, HUANG SC, ZHOU A, ZHANG J, HERMAN AM, GHASSEMZADEH S, JEONG EM, KASTURIRANGAN S, RAICU M, SOBIESKI MA 2ND, BHAT G, TATOOLES A, BENZ EJ JR., KAMP TJ & DUDLEY SC JR. 2011. Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure. Circulation, 124, 1124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO J, SCHATTON D, MARTINELLI P, HANSEN H, PLA-MARTIN D, BARTH E, BECKER C, ALTMUELLER J, FROMMOLT P, SARDIELLO M & RUGARLI EI 2014. CLUH regulates mitochondrial biogenesis by binding mRNAs of nuclear-encoded mitochondrial proteins. J Cell Biol, 207, 213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEIGER T, VELIC A, MACEK B, LUNDBERG E, KAMPF C, NAGARAJ N, UHLEN M, COX J & MANN M 2013. Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol Cell Proteomics, 12, 1709–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOHIL VM, SHETH SA, NILSSON R, WOJTOVICH AP, LEE JH, PEROCCHI F, CHEN W, CLISH CB, AYATA C, BROOKES PS & MOOTHA VK 2010. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol, 28, 249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GU Y, ALBUQUERQUE CP, BRAAS D, ZHANG W, VILLA GR, BI J, IKEGAMI S, MASUI K, GINI B, YANG H, GAHMAN TC, SHIAU AK, CLOUGHESY TF, CHRISTOFK HR, ZHOU H, GUAN KL & MISCHEL PS 2017. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol Cell, 67, 128–138 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAYER A, SHAO L, CHUNG M, JOUBERT LM, YANG HW, TSAI FC, BISARIA A, BETZIG E & MEYER T 2016. Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat Cell Biol, 18, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILLENMEYER ME, FUNG E, WILDENHAIN J, PIERCE SE, HOON S, LEE W, PROCTOR M, ST ONGE RP, TYERS M, KOLLER D, ALTMAN RB, DAVIS RW, NISLOW C & GIAEVER G 2008. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science, 320, 362–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG LE, GU J, SCHAU M & BUNN HF 1998. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A, 95, 7987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUTTLIN EL, JEDRYCHOWSKI MP, ELIAS JE, GOSWAMI T, RAD R, BEAUSOLEIL SA, VILLEN J, HAAS W, SOWA ME & GYGI SP 2010. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell, 143, 1174–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITO K & SUDA T 2014. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol, 15, 243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIANG L, KON N, LI T, WANG SJ, SU T, HIBSHOOSH H, BAER R & GU W 2015. Ferroptosis as a p53-mediated activity during tumour suppression. Nature, 520, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOURDAIN AA, KOPPEN M, WYDRO M, RODLEY CD, LIGHTOWLERS RN, CHRZANOWSKA-LIGHTOWLERS ZM & MARTINOU JC 2013. GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab, 17, 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIMURA E, HIDAKA K, KIDA Y, MORISAKI H, SHIRAI M, ARAKI K, SUZUKI M, YAMAMURA KI & MORISAKI T 2004. Serine-arginine-rich nuclear protein Luc7l regulates myogenesis in mice. Gene, 341, 41–7. [DOI] [PubMed] [Google Scholar]
- KOPPULA P, ZHANG Y, SHI J, LI W & GAN B 2017. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J Biol Chem, 292, 14240–14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOTINI AG, CHANG CJ, BOUSSAAD I, DELROW JJ, DOLEZAL EK, NAGULAPALLY AB, PERNA F, FISHBEIN GA, KLIMEK VM, HAWKINS RD, HUANGFU D, MURRY CE, GRAUBERT T, NIMER SD & PAPAPETROU EP 2015. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat Biotechnol, 33, 646–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAREAU LF, INADA M, GREEN RE, WENGROD JC & BRENNER SE 2007. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature, 446, 926–9. [DOI] [PubMed] [Google Scholar]
- LI W, XU H, XIAO T, CONG L, LOVE MI, ZHANG F, IRIZARRY RA, LIU JS, BROWN M & LIU XS 2014. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol, 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI X, LIU S, JIANG J, ZHANG L, ESPINOSA S, HILL RC, HANSEN KC, ZHOU ZH & ZHAO R 2017. CryoEM structure of Saccharomyces cerevisiae U1 snRNP offers insight into alternative splicing. Nat Commun, 8, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVCI MT, GHANEM D, MARR H, ARNOLD J, GEE S, PARRA M, LIANG TY, STARK TJ, GEHMAN LT, HOON S, MASSIRER KB, PRATT GA, BLACK DL, GRAY JW, CONBOY JG & YEO GW 2013. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol, 20, 1434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVE MI, HUBER W & ANDERS S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NI JZ, GRATE L, DONOHUE JP, PRESTON C, NOBIDA N, O’BRIEN G, SHIUE L, CLARK TA, BLUME JE & ARES M JR. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev, 21, 708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OH JM, VENTERS CC, DI C, PINTO AM, WAN L, YOUNIS I, CAI Z, ARAI C, SO BR, DUAN J & DREYFUSS G 2020. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat Commun, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PASTEUR L 1861. Animalcules infusoires vivant sans gaz oxygène libre et déterminant des fermentations. Comptes rendus - Académie des sciences, 52, 344–347. [Google Scholar]
- PEARCE EL, POFFENBERGER MC, CHANG CH & JONES RG 2013. Fueling immunity: insights into metabolism and lymphocyte function. Science, 342, 1242454.24115444 [Google Scholar]
- PEREZ-RIVEROL Y, CSORDAS A, BAI J, BERNAL-LLINARES M, HEWAPATHIRANA S, KUNDU DJ, INUGANTI A, GRISS J, MAYER G, EISENACHER M, PEREZ E, USZKOREIT J, PFEUFFER J, SACHSENBERG T, YILMAZ S, TIWARY S, COX J, AUDAIN E, WALZER M, JARNUCZAK AF, TERNENT T, BRAZMA A & VIZCAINO JA 2019. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PFEIFFER T, SCHUSTER S & BONHOEFFER S 2001. Cooperation and competition in the evolution of ATP-producing pathways. Science, 292, 504–7. [DOI] [PubMed] [Google Scholar]
- PIIRILA P, SIMILA ME, PALMIO J, WUORIMAA T, YLIKALLIO E, SANDELL S, HAAPALAHTI P, UOTILA L, TYYNISMAA H, UDD B & AURANEN M 2016. Unique Exercise Lactate Profile in Muscle Phosphofructokinase Deficiency (Tarui Disease); Difference Compared with McArdle Disease. Front Neurol, 7, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PING L, DUONG DM, YIN L, GEARING M, LAH JJ, LEVEY AI & SEYFRIED NT 2018. Global quantitative analysis of the human brain proteome in Alzheimer’s and Parkinson’s Disease. Sci Data, 5, 180036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLASCHKA C, LIN PC, CHARENTON C & NAGAI K 2018. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature, 559, 419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUIGSERVER P, WU Z, PARK CW, GRAVES R, WRIGHT M & SPIEGELMAN BM 1998. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell, 92, 829–39. [DOI] [PubMed] [Google Scholar]
- REITZER LJ, WICE BM & KENNELL D 1979. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem, 254, 2669–76. [PubMed] [Google Scholar]
- RINO J, CARVALHO T, BRAGA J, DESTERRO JM, LUHRMANN R & CARMO-FONSECA M 2007. A stochastic view of spliceosome assembly and recycling in the nucleus. PLoS Comput Biol, 3, 2019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBINSON BH, PETROVA-BENEDICT R, BUNCIC JR & WALLACE DC 1992. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem Med Metab Biol, 48, 122–6. [DOI] [PubMed] [Google Scholar]
- ROSSIGNOL R, GILKERSON R, AGGELER R, YAMAGATA K, REMINGTON SJ & CAPALDI RA 2004. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res, 64, 985–93. [DOI] [PubMed] [Google Scholar]
- SATO H, NOMURA S, MAEBARA K, SATO K, TAMBA M & BANNAI S 2004. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun, 325, 109–16. [DOI] [PubMed] [Google Scholar]
- SATO H, TAMBA M, ISHII T & BANNAI S 1999. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem, 274, 11455–8. [DOI] [PubMed] [Google Scholar]
- SHEN S, PARK JW, LU ZX, LIN L, HENRY MD, WU YN, ZHOU Q & XING Y 2014. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A, 111, E5593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIN CS, MISHRA P, WATROUS JD, CARELLI V, D’AURELIO M, JAIN M & CHAN DC 2017. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat Commun, 8, 15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHUAI S, SUZUKI H, DIAZ-NAVARRO A, NADEU F, KUMAR SA, GUTIERREZ-FERNANDEZ A, DELGADO J, PINYOL M, LOPEZ-OTIN C, PUENTE XS, TAYLOR MD, CAMPO E & STEIN LD 2019. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature, 574, 712–716. [DOI] [PubMed] [Google Scholar]
- SPELLMAN R, LLORIAN M & SMITH CW 2007. Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol Cell, 27, 420–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUZUKI H, KUMAR SA, SHUAI S, DIAZ-NAVARRO A, GUTIERREZ-FERNANDEZ A, DE ANTONELLIS P, CAVALLI FMG, JURASCHKA K, FAROOQ H, SHIBAHARA I, VLADOIU MC, ZHANG J, ABEYSUNDARA N, PRZELICKI D, SKOWRON P, GAUER N, LUU B, DANIELS C, WU X, FORGET A, MOMIN A, WANG J, DONG W, KIM SK, GRAJKOWSKA WA, JOUVET A, FEVRE-MONTANGE M, GARRE ML, RAO AAN, GIANNINI C, KROS JM, FRENCH PJ, JABADO N, NG HK, POON WS, EBERHART CG, POLLACK IF, OLSON JM, WEISS WA, KUMABE T, LOPEZ-AGUILAR E, LACH B, MASSIMINO M, VAN MEIR EG, RUBIN JB, VIBHAKAR R, CHAMBLESS LB, KIJIMA N, KLEKNER A, BOGNAR L, CHAN JA, FARIA CC, RAGOUSSIS J, PFISTER SM, GOLDENBERG A, WECHSLERREYA RJ, BAILEY SD, GARZIA L, MORRISSY AS, MARRA MA, HUANG X, MALKIN D, AYRAULT O, RAMASWAMY V, PUENTE XS, CALARCO JA, STEIN L & TAYLOR MD 2019. Recurrent non-coding U1-snRNA mutations drive cryptic splicing in Shh medulloblastoma. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANNER LB, GOGLIA AG, WEI MH, SEHGAL T, PARSONS LR, PARK JO, WHITE E, TOETTCHER JE & RABINOWITZ JD 2018. Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst, 7, 49–62 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TARUI S, OKUNO G, IKURA Y, TANAKA T, SUDA M & NISHIKAWA M 1965. Phosphofructokinase Deficiency in Skeletal Muscle. A New Type of Glycogenosis. Biochem Biophys Res Commun, 19, 517–23. [DOI] [PubMed] [Google Scholar]
- TO TL, CUADROS AM, SHAH H, HUNG WHW, LI Y, KIM SH, RUBIN DHF, BOE RH, RATH S, EATON JK, PICCIONI F, GOODALE A, KALANI Z, DOENCH JG, ROOT DE, SCHREIBER SL, VAFAI SB & MOOTHA VK 2019. A Compendium of Genetic Modifiers of Mitochondrial Dysfunction Reveals Intraorganelle Buffering. Cell, 179, 1222–1238 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUFARELLI C, FRISCHAUF AM, HARDISON R, FLINT J & HIGGS DR 2001. Characterization of a widely expressed gene (LUC7-LIKE; LUC7L) defining the centromeric boundary of the human alpha-globin domain. Genomics, 71, 307–14. [DOI] [PubMed] [Google Scholar]
- UBAIDA-MOHIEN C, LYASHKOV A, GONZALEZ-FREIRE M, THARAKAN R, SHARDELL M, MOADDEL R, SEMBA RD, CHIA CW, GOROSPE M, SEN R & FERRUCCI L 2019. Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. Elife, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN NOSTRAND EL, FREESE P, PRATT GA, WANG X, WEI X, XIAO R, BLUE SM, CHEN JY, CODY NAL, DOMINGUEZ D, OLSON S, SUNDARARAMAN B, ZHAN L, BAZILE C, BOUVRETTE LPB, BERGALET J, DUFF MO, GARCIA KE, GELBOIN-BURKHART C, HOCHMAN M, LAMBERT NJ, LI H, MCGURK MP, NGUYEN TB, PALDEN T, RABANO I, SATHE S, STANTON R, SU A, WANG R, YEE BA, ZHOU B, LOUIE AL, AIGNER S, FU XD, LECUYER E, BURGE CB, GRAVELEY BR & YEO GW 2020. A large-scale binding and functional map of human RNA-binding proteins. Nature, 583, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN NOSTRAND EL, NGUYEN TB, GELBOIN-BURKHART C, WANG R, BLUE SM, PRATT GA, LOUIE AL & YEO GW 2017. Robust, Cost-Effective Profiling of RNA Binding Protein Targets with Single-end Enhanced Crosslinking and Immunoprecipitation (seCLIP). Methods Mol Biol, 1648, 177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN NOSTRAND EL, PRATT GA, SHISHKIN AA, GELBOIN-BURKHART C, FANG MY, SUNDARARAMAN B, BLUE SM, NGUYEN TB, SURKA C, ELKINS K, STANTON R, RIGO F, GUTTMAN M & YEO GW 2016. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods, 13, 508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANDERSLUIS B, HESS DC, PESYNA C, KRUMHOLZ EW, SYED T, SZAPPANOS B, NISLOW C, PAPP B, TROYANSKAYA OG, MYERS CL & CAUDY AA 2014. Broad metabolic sensitivity profiling of a prototrophic yeast deletion collection. Genome Biol, 15, R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VISANJI NP, WISLET-GENDEBIEN S, OSCHIPOK LW, ZHANG G, AUBERT I, FRASER PE & TANDON A 2011. Effect of Ser-129 phosphorylation on interaction of alpha-synuclein with synaptic and cellular membranes. J Biol Chem, 286, 35863–73. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- WARBURG O 1924. Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften, 12, 1131–1137. [Google Scholar]
- WEBBY CJ, WOLF A, GROMAK N, DREGER M, KRAMER H, KESSLER B, NIELSEN ML, SCHMITZ C, BUTLER DS, YATES JR 3RD, DELAHUNTY CM, HAHN P, LENGELING A, MANN M, PROUDFOOT NJ, SCHOFIELD CJ & BOTTGER A 2009. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science, 325, 90–3. [DOI] [PubMed] [Google Scholar]
- YIN Y, LU JY, ZHANG X, SHAO W, XU Y, LI P, HONG Y, CUI L, SHAN G, TIAN B, ZHANG QC & SHEN X 2020. U1 snRNP regulates chromatin retention of noncoding RNAs. Nature, 580, 147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG J, LIEU YK, ALI AM, PENSON A, REGGIO KS, RABADAN R, RAZA A, MUKHERJEE S & MANLEY JL 2015. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci U S A, 112, E4726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4: LUC7L2 Immunoprecipitation and Mass Spectrometry (IP-MS), Related to Figure 4 and S4.
Table S6: Differential Gene Expression Analysis in LUC7L2KO K562 Cells, Related to Figure 4 and S4.
Table S5: LUC7L2 Enhanced Crosslinking and Immunoprecipitation (eCLIP), Related to Figure 4 and S4.
Table S8: Global Quantitative Proteomics in LUC7L2KO And Galactose-Grown K562 Cells Relative to Glucose-Grown LUC7L2WT Cells, Related to Figure 6 and S6.
Table S7: Differential Splicing Analysis in LUC7L2KO K562 Cells, Related to Figure 4 and S4.
Table S1: Genome-Wide CRISPR/Cas9 Screening Results and Gene Ontology, Related to Figure 1 and S1.
Data Availability Statement
RNA sequencing data were deposited at GEO (GSE157917). Proteomics data were deposited at PRIDE (PXD021917). Plasmids were deposited at Addgene. Unedited gel scans are available in the supplemental information file (Data S1).