

Abstract
Background
To examine immune-epithelial interactions and their impact on epithelial transformation in primary sclerosing cholangitis–associated ulcerative colitis (PSC-UC) using patient-derived colonic epithelial organoid cultures (EpOCs).
Methods
The EpOCs were originated from colonic biopsies from patients with PSC-UC (n = 12), patients with UC (n = 14), and control patients (n = 10) and stimulated with cytokines previously associated with intestinal inflammation (interferon (IFN) γ and interleukin (IL)-22). Markers of cytokine downstream pathways, stemness, and pluripotency were analyzed by real-time quantitative polymerase chain reaction and immunofluorescence. The OLFM4 expression in situ was assessed by RNAscope and immunohistochemistry.
Results
A distinct expression of stem cell–associated genes was observed in EpOCs derived from patients with PSC-UC, with lower expression of the classical stem-cell marker LGR5 and overexpression of OLFM4, previously associated with pluripotency and early stages of neoplastic transformation in the gastrointestinal and biliary tracts. High levels of OLFM4 were also found ex vivo in colonic biopsies from patients with PSC-UC. In addition, IFNγ stimulation resulted in the downregulation of LGR5 in EpOCs, whereas higher expression of OLFM4 was observed after IL-22 stimulation. Interestingly, expression of the IL-22 receptor, IL22RA1, was induced by IFNγ, suggesting that a complex interplay between these cytokines may contribute to carcinogenesis in PSC-UC.
Conclusions
Higher expression of OLFM4, a cancer stemness gene induced by IL-22, is present in PSC-UC, suggesting that IL-22 responses may result in alterations of the intestinal stem-cell niche in these patients.
Keywords: primary sclerosing cholangitis, inflammatory bowel disease, organoids, epithelium
INTRODUCTION
Primary sclerosing cholangitis (PSC) is a chronic progressive disorder of the hepatobiliary system characterized by inflammation, fibrosis, and stricturing of the intrahepatic and extrahepatic bile ducts, leading to liver cirrhosis. There is no cure for PSC, and prognosis remains poor. It is associated with inflammatory bowel disease (IBD) in 60% to 80% of patients, of whom 80% are diagnosed with ulcerative colitis (UC).1 However, it is now recognized that colitis in PSC-IBD has distinct features, including more frequent pancolonic, right-sided patchy inflammation, rectal sparing and “backwash” ileitis, and an usually quiescent course.2 Most important, patients with primary sclerosing cholangitis–associated ulcerative colitis (PSC-UC) have a 4-fold increased risk of developing colorectal cancer (CRC) compared with patients with UC alone, and 10 times more than the general population.3 These cancers more frequently arise from the right colon in PSC, suggesting distinct pathogenetic mechanisms and a possible link with the underlying inflammatory process. Patients with PSC are also at increased risk of hepatobiliary malignancies, particularly cholangiocarcinoma (cumulative lifetime incidence, 10% to 30%), gall bladder adenoma, dysplasia, and carcinoma. Patient prognosis remains poor, with mortality rates as high as 30% at 6 years from diagnosis, with cancer as the main cause (40% to 50% of deaths), followed by liver failure (30% to 40%).4 Unfortunately, no medical therapy has proven to be effective, and liver transplant remains the only treatment available.
The pathogenesis of PSC remains unknown. Several hypotheses have been put forward, such as an aberrant homing of gut-derived memory T cells to the liver and bacterial translocation secondary to increased intestinal permeability (the “leaky gut” hypothesis) contributing to the pathogenesis of the disease.5, 6 Genetic studies have identified multiple loci associated with disease susceptibility with genome-wide significance and other suggestive associations.5, 7 In particular, a strong HLA antigen association suggests that antigen-driven adaptive immune responses, possibly mediated by CD4+ T cells, contribute to disease pathogenesis.8 The intestinal microbiome has emerged as a key player in modulating mucosal immune responses, and dysbiosis has been linked to multiple immune disorders, including PSC.9 In a recent study, gnotobiotic mice inoculated with microbiota derived from patients with PSC exhibited T helper (Th) 17 cell responses in the liver and increased susceptibility to hepatobiliary injuries.10 These observations suggest that altered host-microbial interactions may result in immune dysregulation in PSC, leading to chronic inflammation and increased carcinogenesis.
Previous research observed distinct chemokine receptor profiles on circulating T cells in PSC-UC, confirming that specific changes in T-cell trafficking properties are involved in the pathogenesis of intestinal and liver inflammation. Studies have also found an accumulation of innate lymphoid cells (ILC) and increased Th1 responses in the colon of these patients, even in the presence of quiescent or mild activity.11 Furthermore, Th17 responses have been described in PSC,12 and increased levels of interleukin (IL)-17A and IL-22 in the colonic mucosa of patients with PSC-UC have been observed compared to levels in patients with UC alone (unpublished data). This information suggests that overactive type 1 and type 17 T-cell and unconventional lymphocyte responses may play a role in promoting intestinal inflammation in PSC-UC. However, nothing is known about the epithelial signature during chronic inflammation in PSC-UC and how this may impact neoplastic transformation.
The development of human organoids represents an invaluable tool to evaluate intestinal pathophysiology in vitro.13 In particular, human epithelial organoid cultures (EpOCs) can be derived ex vivo from intestinal crypts isolated from biopsies and surgical specimens and have been shown to retain the specificity of the intestinal segment of origin.14 Interestingly, a distinct transcriptional profile has been described in colonic EpOCs derived from patients with IBD, suggesting the presence of sustained changes in the epithelial stem cell compartment in these patients.15
This is the first study in which colonic organoids have been obtained from patients with PSC-UC. In particular, we cultured EpOCs derived from patients with PSC-UC, patients with UC alone, and healthy control patients to characterize the epithelial signature and evaluate the role of specific immune-epithelial interactions in the pathogenesis of increased risk of CRC in patients with PSC-UC.
MATERIALS AND METHODS
Patient Recruitment and Sample Collection
All patients and control patients were recruited from the Department of Gastroenterology, and samples were collected as part of the Translational Gastroenterology Unit biobank at the John Radcliffe Hospital in Oxford (UK) and part of the National Institute for Health Research Oxford Biomedical Research Centre IBD Cohort (REC 09/H1204/30) and the Oxford Biomedical Research Centre GI Illness Biobank (REC 16/YH/0247). Patients with a previous formal diagnosis of PSC-UC or UC according to clinical, radiological, endoscopic, and histological criteria were identified from the databases of our center and approached at outpatient clinic or endoscopy appointments to confirm consent and obtain colonic samples. Colonic biopsies were obtained from patients with PSC-UC and patients with UC alone undergoing colonoscopy for surveillance or disease activity and extension assessment and from healthy control patients undergoing colonoscopy for CRC screening or chronic abdominal pain/diarrhea with a macroscopically and microscopically normal colon. Endoscopic activity of PSC-UC and UC was defined by the Ulcerative Colitis Endoscopic Index of Severity score,16 and microscopic inflammation was defined by the Nancy score.17 Other clinical and demographic information, including current medications, were obtained from our clinical patient database.
Crypt Isolation and Culture of EpOCs
The EpOCs were obtained as previously described.13, 18 Detailed protocols for human organoid culture were generously provided by Marc van de Wetering (Hubrecht Institute, Netherlands). In brief, colonic biopsies (n = 8-10) were washed in chelating buffer,18 incubated on ice for 20 minutes in 0.5M EDTA and 1mM dithiothreitol solution, vortexed and washed in Advanced Dulbecco’s Modified Eagle’s Medium with Nutrient Mixture Ham’s F-12 (Ad-DF; Gibco), and supplemented with 1% penicillin-streptomycin (Invitrogen), 1M Hepes (Invitrogen), and L-glutamine (Invitrogen; Ad-DF+++). Supernatants containing the epithelial crypts were collected and resuspended in reduced growth factor basement membrane extract (BME) type 2 (AMSBIO) to allow for the seeding of 100 to 300 crypts per 10 μL of BME in 48 well plates. Next, 200 μL of prewarmed organoid media18 was added to each well and kept at 37°C, 5% CO2 in a humidified incubator. Organoids were allowed to grow for 3 to 4 days before the first passage, then passaged once weekly for 2 weeks. The EpOCs were stimulated 2 days after second passage with IL-22 and interferon (IFN) γ (10 ng/mL; R&D Bio-Techne) for 24 hours (gene expression studies) and 15 minutes (confocal microscopy). The EpOCs were collected after 2 weeks of culture, embedded in paraffin to create a formalin fixed paraffin embedded (FFPE) block and cut at 5 μm for hematoxylin and eosin staining.
RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction Analysis
Total RNA was isolated from organoids using the RNeasy Minikit (Qiagen), including an on-column DNase digestion step (Qiagen). The cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s protocol. Gene expression was analyzed by real-time quantitative polymerase chain reaction (qPCR; TaqMan real-time PCR) and the Precision Fast qPCR Mastermix with ROX at a lower level with inert blue dye (Primer Design) and run on a ViiA7 Real-Time PCR System (Applied Biosystems). Data were analyzed using the ΔCT method [2-ΔCT where ΔCT = CT(target gene) – CT(endogenous control)]. RPLP0 was used as the endogenous reference gene for all qPCR analyses. The following TaqMan probes (Applied Biosystems) were used: RPLP0 (Hs99999902_ m1), FUT2 (Hs00382834_m1), SOCS3 (Hs02330328_s1), CIITA (Hs00172106_m1), CXCL2 (Hs00601975_m1), OLFM4 (Hs00197437_m1), LGR5 (Hs00969422_m1), POU5F1 (Hs04260367_gh), NANOG (Hs04260366_g1), STAT1 (Hs01014002_m1), STAT3 (Hs01047580_m1), MUC1 (Hs00159357_m1), MUC2 (Hs00159374_m1), IL22RA1 (Hs00222035_s1), IRF1 (Hs00971965_m1), IDO1 (Hs00984148_m1), IFNGR1 (Hs00988304_m1), IFNGR2 (Hs001942264_m1), S100A9 (Hs00610058_m1), CDH1 (Hs01023894_m1), CDH2 (Hs00983056_m1), SOCS1 (Hs00705164_s1), PDL1 (Hs00204257_m1), and PDL2 (Hs00228839_m1). For RNA expression analysis in colonic tissue, biopsies were collected in RNAprotect Tissue Tubes (Qiagen) and homogenized using Soft Tissue Homogenizing CK14 Kit vials (Stretton Scientific) in a Precellys 24 (Bertin Instruments) homogenizer (30 seconds at 3500 rpm). The RNA extraction, cDNA synthesis, and qPCR analysis were performed as described above.
Immunofluorescence Imaging
Organoid samples were fixed with 4% paraformaldehyde, permeabilized using ice-cold methanol, blocked with 10% goat serum for 1 hour at room temperatire, and stained with the following primary antibodies overnight at 4°C: anti-E-cadherin clone 36/E-cadherin, (610181, BD Biosciences), anti-Phospho-Stat3 (Tyr705) clone D3A7, (9145, Cell Signalling), and anti-LGR5 (21833-1-AP, Proteintech). Goat anti-mouse IgG conjugated with Alexa Fluor 488 (A11001, Life Technologies) and goat anti-rabbit IgG conjugated with Alexa Fluor 555 (A32732, Life Technologies) were used as secondary antibodies. Nuclei were stained with Hoechst 33258 (Life Technologies). Images were acquired on an Olympus FV1200 IX83 Confocal System.
RNA In Situ Hybridization
In situ hybridization for OLFM4 was performed on colonic tissue sections using an RNAscope FFPE assay kit (Advanced Cell Diagnostics Inc., Hayward, CA). Following manufacturer protocol, 5 μm FFPE tissue sections were pretreated with H2O2 for blocking, antigen retrieval buffer, and protease digestion followed by hybridization with an OLFM4 probe. Next, a horseradish peroxidase–based signal amplification system was hybridized before color development with 3,3’-di-aminobenzidine tetrahydrochloride. Samples were counterstained with hematoxylin and eosin and mounted with a mixture of distyrene (a polystyrene), a plasticiser (tricresyl phosphate), and xylene, called DPX. Positive staining was indicated by brown punctate dots in the nucleus and/or cytoplasm. Images were visualized and captured using the NanoZoomer S210 (Hamamatsu).
Immunohistochemistry
Formalin-fixed paraffin-embedded tissue was sectioned at 5 microns and collected onto Superfrost glass slides. Tissue sections were dewaxed in xylene and rehydrated through alcohol to water. Endogenous peroxidase activity was blocked with 3% (v/v) H2O2 before masked antigens were retrieved by microwaving the tissue sections in target retrieval solution (Dako). Endogenous avidin and biotin were blocked (Vector Laboratories) before additional blocking of endogenous enzymes was performed using BloXall (Vector Laboratories). Tissue sections were further blocked with 10% (v/v) normal goat serum (Sigma-Aldrich) before being incubated overnight at 4°C in a humidified environment with anti-OLFM4 XP rabbit monoclonal antibody (Clone D1E4M; Cell Signaling Technology) or normal rabbit immunoglobulin fraction (Dako).
After incubation, primary antibody labeling was detected with a biotinylated goat anti-rabbit IgG secondary antibody (Vector Laboratories). Tissue sections were then incubated with streptavidin horseradish peroxidase (Vector Laboratories) and signal-detected using diaminobenzidine (Vector Laboratories). Tissue sections were counterstained with Mayer’s Haematoxylin (Sigma-Aldrich) before being dehydrated through alcohol to xylene and mounted with DPX (Sigma-Aldrich) and coverslips applied. Images were collected on a Zeiss AxioScope A1 with ZEN Blue software (Carl Zeiss).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA) version 8. Differences between 2 groups were determined using the nonparametric Mann-Whitney U test: patients with PSC vs patients with UC, patients with PSC vs control patients, and patients with UC vs control patients. An ordinary 1-way analysis of variance (Tukey test as recommended) was used to show differences in cytokine stimulation between groups. For significance level, *P < 0.05 and **P < 0.01. Error bars represented the standard error of the mean.
RESULTS
Patient Characteristics
The characteristics of patients included in this study are shown in Table 1. The majority of patients with PSC-UC and those with UC only were male, whereas control individuals were equally distributed between male and female. Patients with PSC-UC were significantly younger than patients with UC (P < 0.04; Mann-Whitney U test), possibly reflecting the clinical indication for surveillance colonoscopy from diagnosis in patients with PSC-UC, whereas surveillance was indicated for long-standing colitis in patients with UC. No significant age difference was observed when comparing patients with colitis with control patients. As expected, the majority of patients with PSC-UC had extensive colitis, whereas disease distribution in patients with UC included both extensive and distal colitis. Colitis endoscopic activity was measured by the Ulcerative Colitis Endoscopic Index of Severity score and was comparable between patients with UC and patients with PSC-UC. The majority of intestinal samples showed quiescent or mild to moderate inflammation at microscopic examination, as measured by the Nancy score.
TABLE 1.
Patient Characteristics
| PSC-UC | UC | Control Patients | |
|---|---|---|---|
| Colonic samples, n | 12 | 14 | 10 |
| Sex, male, n (%) | 8 (66) | 10 (71) | 5 (50) |
| Age, y, median (range) | 31 (21-64) | 58 (26-80) | 52 (36-78) |
| Small-duct PSC, n (%) | 1 | — | — |
| Liver cirrhosis | 0 | — | — |
| PSC-UC | 12 | 14 | — |
| UC duration, y, median (range) | 13.5 (5-33) | 11 (1-47) | — |
| UC extent | |||
| Extensive, n (%) | 12 | 7 | — |
| Left-sided, n (%) | 0 | 6 | — |
| Proctitis, n (%) | 0 | 1 | — |
| UCEIS score | — | ||
| 0-2, n | 9 | 13 | — |
| 3-5, n | 3 | 1 | — |
| 6-8, n | 0 | 0 | — |
| Nancy score, average (range) | 1.3 (0-4) | 0.8 (0-4) | — |
| Treatment at time of sample collection | |||
| 5-aminosalicylates, n (%) | 11 | 9 | — |
| Immunosuppressants, n (%) | 4 | 5 | — |
| Corticosteroids, n (%) | 3 | 0 | — |
| Anti-TNFs, (%) | 2 | 1 | — |
| Anti-integrin therapy, n (%) | 1 | 2 | — |
| Ursodeoxycholic acid, n (%) | 5 | 0 | — |
TNF indicates tumor necrosis factor drugs; UCEIS, Ulcerative Colitis Endoscopic Index of Severity.
In terms of liver disease, all except 1 patient with PSC-UC presented with large-duct PSC, and none had liver cirrhosis. At the time of sample collection, the majority of the patients with PSC-UC and UC were on aminosalicylates, and similar proportions were on immunosuppressants (ie, azathioprine) or biologic therapies. In addition, 42% of the patients with PSC-UC were also on ursodeoxycholic acid. Other concomitant treatments are noted in Table 1.
EpOCs Showed Similar Expression of Genes Associated With Proliferation and Intestinal Cell Differentiation
We successfully grew EpOCs from colonic biopsies obtained at colonoscopy from control patients, patients with UC, and patients with PSC-UC (Fig. 1). Colonic crypts isolated from biopsies were seeded in BME-conditioned medium containing multiple growth factors, and the formation of 3-dimensional epithelial structures, or spheroids, was observed within 48 hours of culture (Fig. 1A). Once a week, spheroids were trypsinized to single-cell suspension, and cells were reseeded in BME. After 2 passages, we were able to obtain large numbers of EpOCs (Figs. 1B, C), which were characterized by a single layer of epithelial cells as confirmed by immunofluorescence (Fig. 1D).
FIGURE 1.

EpOCs show similar expression of genes associated with proliferation and intestinal cell differentiation. A-D, Morphological characterization of EpOCs derived from patient biopsies. A, Bright-field microscopy 48 hours after feeding. B, Two days after second passage, in BME and 24 well plates. C, Hematoxylin and eosin staining 2 days after second passage. and D, Immunofluorescent staining 2 days after second passage, with nuclei in blue and E-cadherin in green. Scale bar, 100 μm. E, mRNA expression of MKI67, CDH1, MUC1, and MUC2 in EpOCs derived from control patients, patients with UC, and patients with PSC-UC. *P < 0.05. Error bars represent standard error of the mean.
To further characterize the EpOCs, we analyzed the expression of genes involved in proliferation and intestinal cell differentiation (Fig. 1E). We observed comparable expression of MKI67, a gene encoding for the nuclear protein Ki67, in EpOCs derived from all 3 patient groups, suggesting similar proliferative activity. The gene CDH1, encoding for the epithelial marker E-cadherin, was also equally expressed in EpOCs derived from the different groups. We observed no difference in the expression of the membrane-bound mucin MUC1, whereas MUC2, encoding for the main gel-forming mucin secreted by goblet cells in the mucus layers, was significantly overexpressed in PSC-UC derived EpOCs compared with those derived from control patients but not compared with those derived from patients with UC (Fig. 1E).
Distinct Expression of Stemness-Associated Genes Observed in EpOCs Derived From Patients With PSC-UC
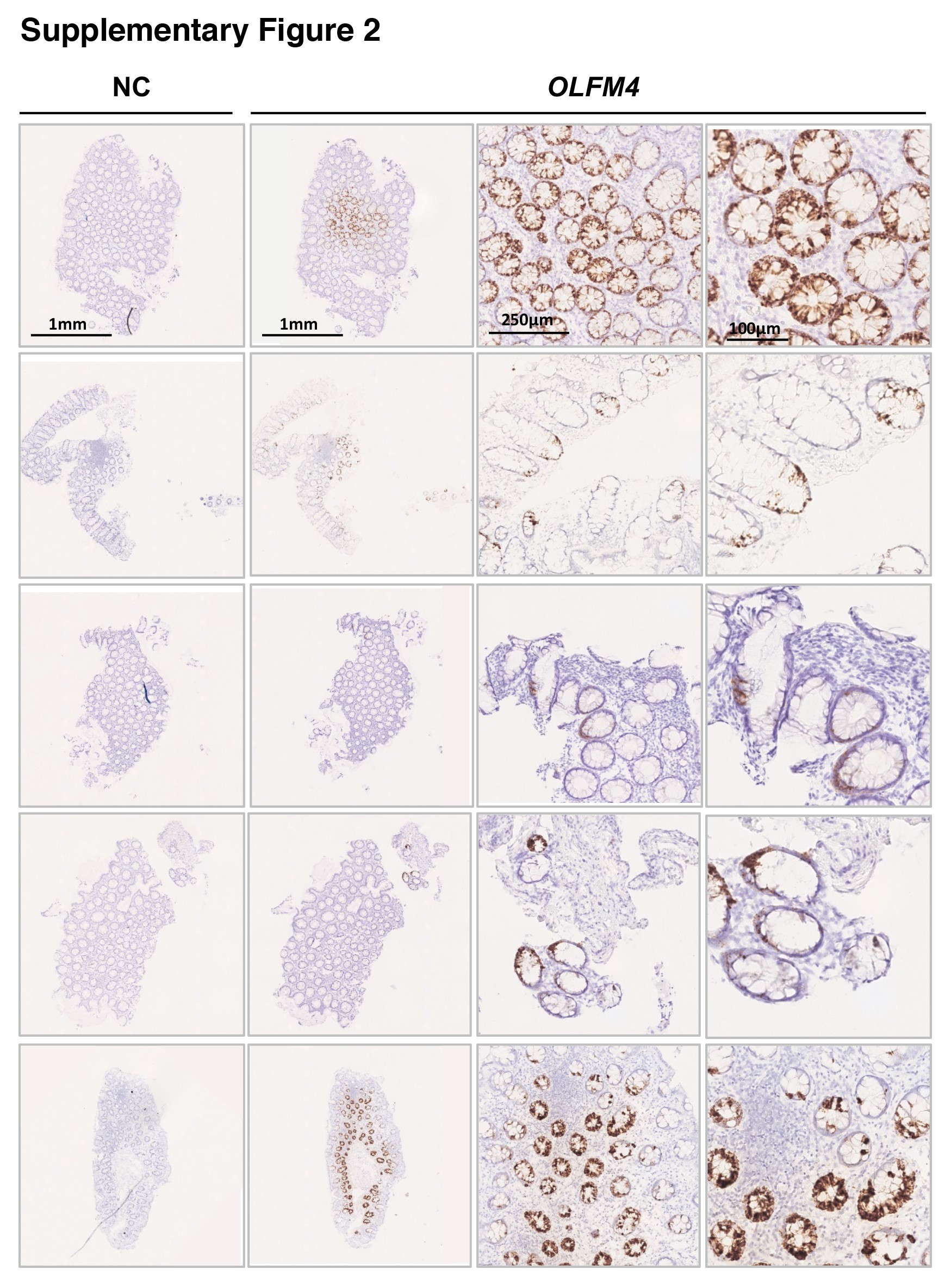
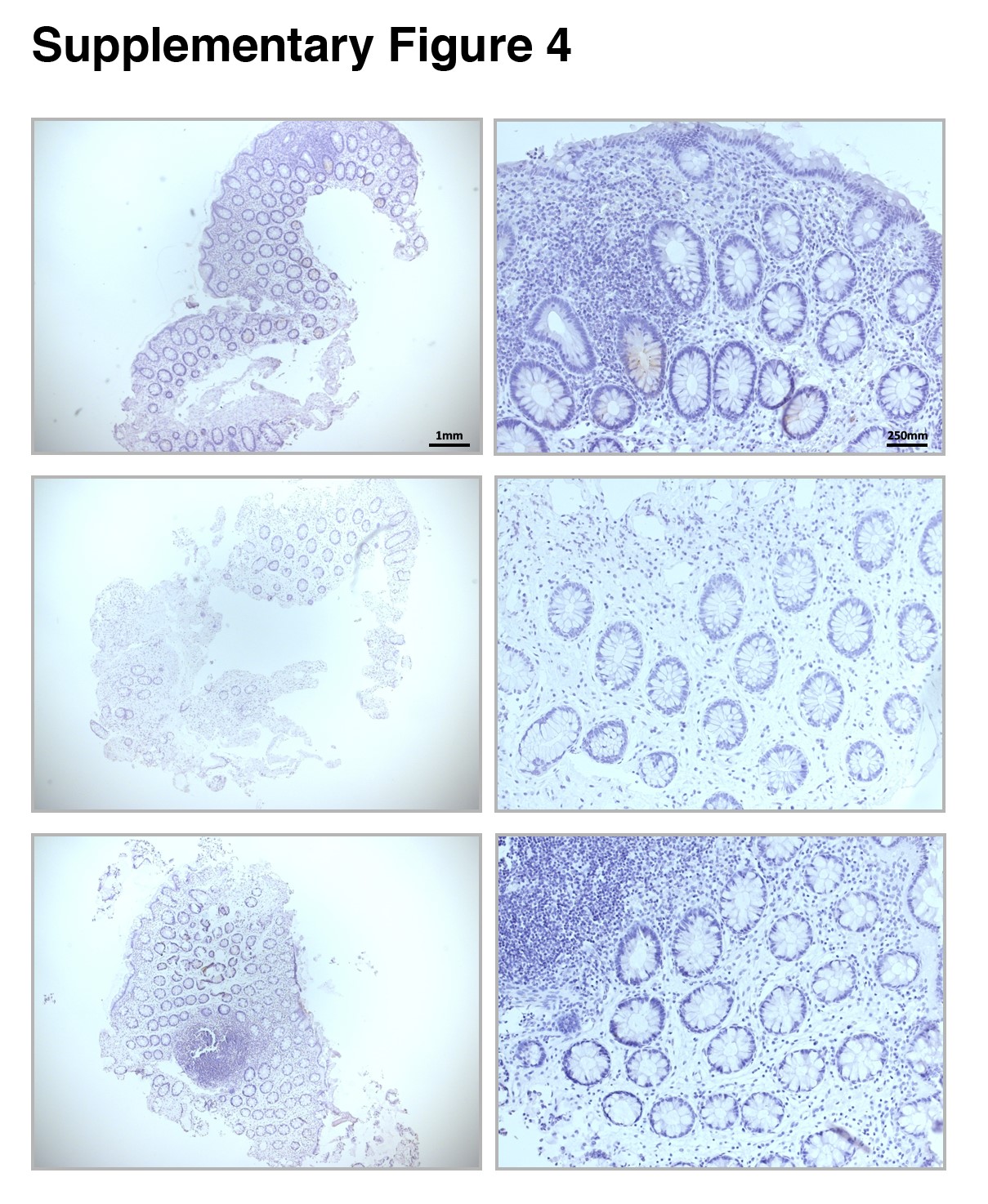
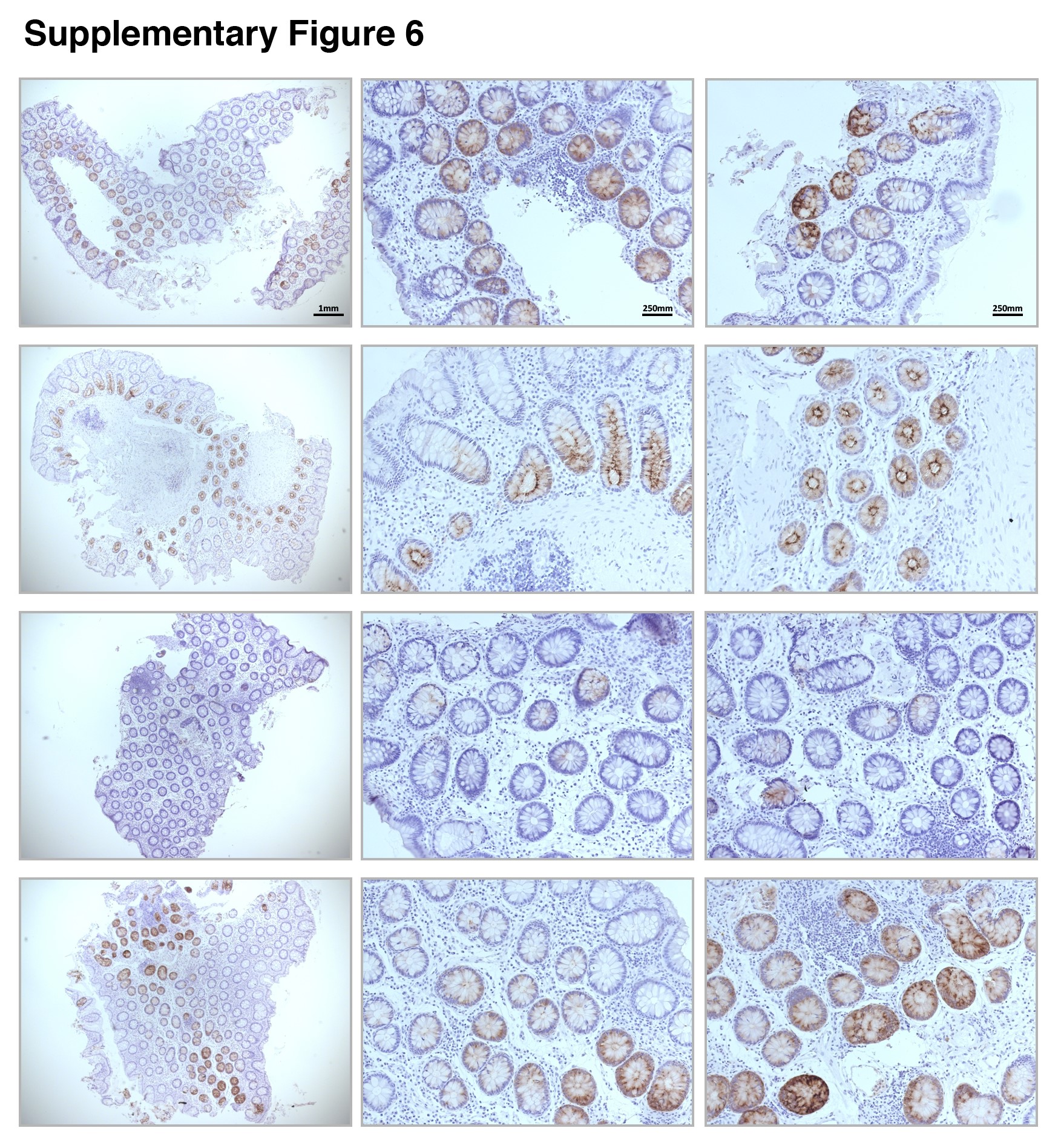
To assess epithelial stemness function, we evaluated markers of stem cell proliferation, self-renewal, and pluripotency in patient-derived EpOCs. The classical stem cell marker LGR5 was expressed by EpOCs derived from patients with PSC-UC, UC, and control individuals, as observed by immunofluorescence staining (Fig. 2A). A significant decrease in the mRNA expression of LGR5 was found in PSC-UC-derived EpOCs compared with those derived from patients with UC. No difference was observed in the expression of other stemness-associated genes, such as NANOG, SOX9, and POU5F1, between the 3 groups. Interestingly, a significantly increased expression of OLFM4, a member of the olfactomedin domain-containing protein family typically expressed in the stem cell niche, was found in EpOCs derived from patients with PSC-UC compared with both patients with UC and control patients (Fig. 2B). High levels of OLFM4 expression were also observed by in situ hybridization (Fig. 2CSupplementary Figs. 1-3) and at the protein level by immunohistochemistry (Fig. 2DSupplementary Figs. 4-6) in colonic tissue from patients with PSC-UC, even in the presence of quiescent disease.
FIGURE 2.

Distinct expression of stemness genes in EpOCs derived from patients with PSC-UC. A, Representative images of immunofluorescent staining of colonic organoids derived from patients with PSC-UC, patients with UC, and control patients with nuclei in blue, E-cadherin in green, and LGR5 in red. Scale bar 100 μm. B, mRNA expression of LGR5, NANOG, SOX9, POU5F1, and OLFM4 in EpOCs derived from 3 patient groups. *P < 0.05, **P < 0.01. Error bars represent standard error of the mean. C, Representative images of OLFM4 transcript staining of noninflamed colon tissues derived from 3 patient groups. D, Representative images of OLFM4 immunohistochemistry staining of noninflamed colon tissues derived from 3 patient groups. Scale bar 250 μm.
Patient-Derived EpOCs Respond to Cytokine Stimulation Through STAT Phosphorylation and Gene Regulation
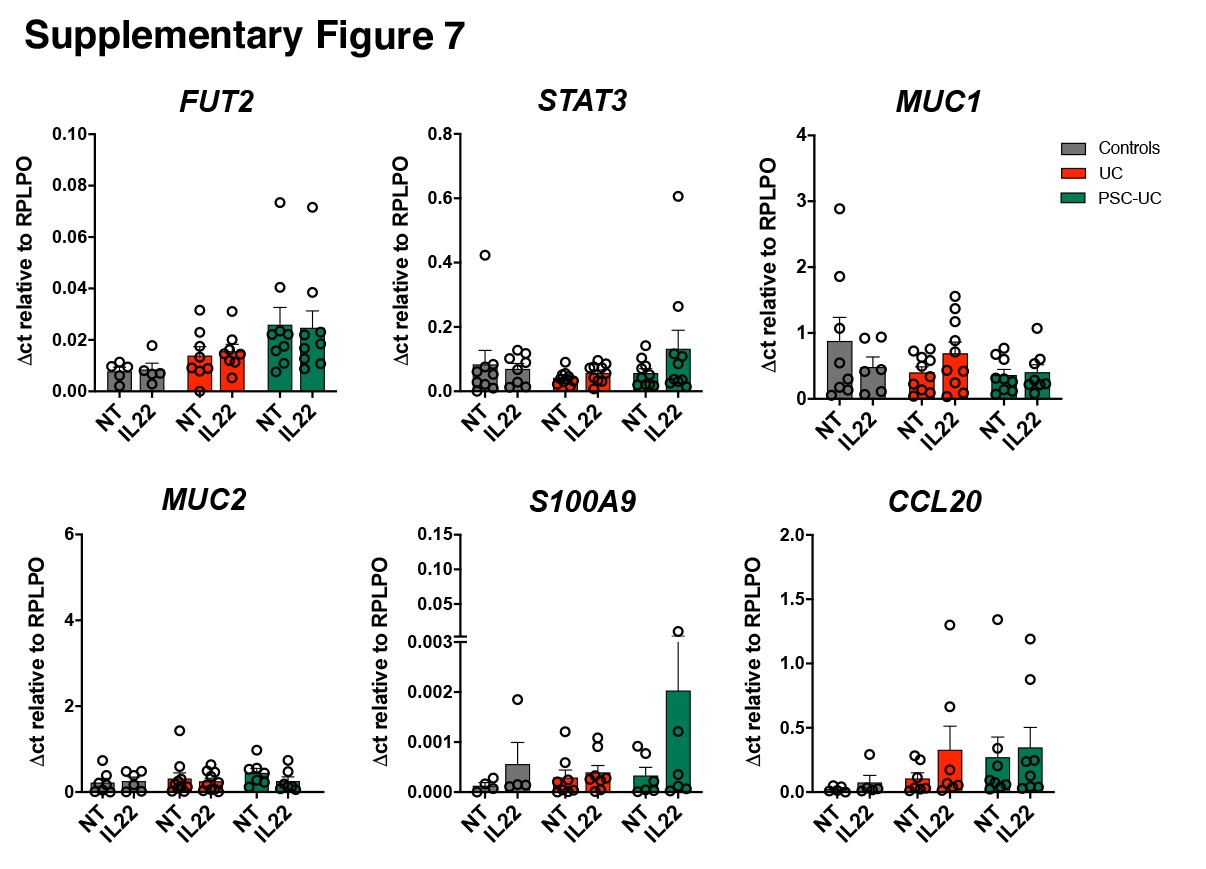
Previous research has shown an increased frequency of IFNγ-secreting T cells and an accumulation of ILC in the colon of patients with PSC-UC compared with patients with UC,11 and unpublished data have shown increased expression of Th1 and Th17 cytokines in the colon of these patients. To assess how inflammation can affect colonic epithelial function, we stimulated patient-derived EpOCs with IFNγ and IL-22. As expected, after IFNγ stimulation, we observed upregulation of IFNγ-induced genes, such as STAT1, IRF1, IDO, SOCS1, PDL1, PDL2, and CIITA (Fig. 3A) in the EpOCs derived from patients with UC/PSC-UC and control patients, with no difference observed between the groups. Stimulation with IL-22 resulted in STAT3 phosphorylation (Fig. 3B) and the induction of IL-22-regulated genes, such as SOCS3 and CXCL2 (Fig. 3C). In addition, SOCS3 expression after IL-22 stimulation was significantly higher in EpOCs from patients with PSC-UC than in EpOCS from patients with UC. However, no induction was observed for other IL-22-induced genes (Supplementary Fig. 7). These results suggest that EpOCs respond to IFNγ and IL-22 stimulation, providing a model to investigate immune-epithelial interactions.
FIGURE 3.

EpOCs respond to stimulation with cytokines. A, mRNA expression of STAT1, IRF1, IDO, SOCS1, PDL1, PDL2, and CIITA in EpOCs from control patients (grey bars), patients with UC (red bars), and patients with PSC-UC (green bars), with no treatment (NT) and after 24 hours of stimulation with IFNγ (10 ng/mL). B, Immunofluorescent staining, with nuclei in blue, E-cadherin in green, and phospho-STAT3 (p-S3) in red in EpOCs untreated and stimulated with IL-22 for 15 minutes. Scale bar 100 μm. C, mRNA expression of SOCS3 and CXCL2 in EpOCs from control patients (grey bars), patients with UC (red bars), and patients with PSC-UC (green bars), with no treatment (NT) and after 24 hours of stimulation with IL-22 (10 ng/mL). Ordinary 1-way analysis of variance (Tukey test as recommended) was performed in the cytokine stimulation experiments. *P < 0.05, **P < 0.005. Error bars represent standard error of the mean.
Differential Regulation of Stem Cell Gene Expression by Cytokine Stimulation in Patient-Derived EpOCs
When we looked at the expression of stemness-associated genes, we observed that IFNγ stimulation induced a significant downregulation of LGR5 in EpOCs derived from control individuals and noted a similar trend in UC-derived EpOCs. The expression of LGR5 in response to IL-22 stimulation was significantly higher in EpOCs derived from control patients when compared with PSC-UC derived EpOCS (Fig. 4A). The expression of POU5F1 was significantly higher after IFNγ stimulation in PSC-UC-derived EpOCs compared with those of control patients (Fig. 4B). There was a trend toward a reduction of OLFM4 expression after IFNγ stimulation in PSC-UC-derived organoids, but it remained higher than in EpOCs derived from patients with UC and from control individuals. Although a trend toward an induction of OLFM4 was observed after IL-22 stimulation in all EpOCs, higher OLFM4 expression was observed after IL-22 stimulation in EpOCs derived from patients with PSC-UC than in EpOCs derived from patients with UC and control patients (Fig. 4C). Cytokine stimulation with either IFNγ or IL-22 did not have any significant effect on the expression of NANOG and SOX9 (Figs. 4D, E). These results indicate that immune responses involved in intestinal inflammation can affect the expression of genes associated to stemness and pluripotency in the intestinal epithelium. In particular, our data suggest that IFNγ induces the downregulation of LGR5 and higher POU5F1 expression in PSC-UC, whereas IL-22 results in higher levels of OLFM4 in PSC-UC.
FIGURE 4.

Cytokine stimulation regulating stem-cell gene expression in patient-derived EpOCs. mRNA expression of the stemness genes (A) LGR5, (B) POU5F1, (C) OLFM4, (D) NANOG, and (E) SOX9 in EpOCs from control patients (grey bars), patients with UC (red bars), and patients with PSC-UC (green bars), with no treatment (NT) and after 24 hours of stimulation with IL-22 (10 ng/mL) and IFNγ (10 ng/ml). *P < 0.05, **P < 0.01. Error bars represent standard error of the mean.
IFNγ Stimulation Induces Increased IL-22 Responsiveness in Patient-Derived EpOCs
To assess epithelial responsiveness to the inflammatory milieu, we analyzed the expression of IFNγ and IL-22 cytokine receptor subunits on the EpOCs derived from the 3 groups. We found no difference in the expression of IFNGR1 and IFNGR2, encoding for the 2 chains of the IFNγ receptor, between EpOCs derived from all 3 groups, and their expression was not affected by IFNγ or IL-22 stimulation (Fig. 5A). On the other hand, IL22RA1 was significantly induced by IFNγ stimulation in the EpOCs from patients with PSC-UC and from control individuals, and a similar trend was also observed in UC-derived EpOCs (Fig. 5B). Moreover, the IL22RA1 expression levels after IFNγ stimulation were significantly higher in the PSC-UC vs the control EpOCs, and a trend toward increased expression of IL22RA1 was also found in colonic biopsies in PSC-UC (Supplementary Fig. 8). These results suggest that the high levels of IFNγ observed in PSC-UC19 may increase the epithelial responsiveness to IL-22, which in turn can induce OLFM4 expression.
FIGURE 5.

IFNγ stimulation inducing IL-22 responsiveness in EpOCs. mRNA expression of (A) IFNGR1 and IFNGR2 and (B) IL22RA1 in EpOCs from control patients (grey bars), patients with UC (red bars), and patients with PSC-UC (green bars), with no treatment (NT) and after 24 hours of stimulation with IL-22 and IFNγ (10 ng/mL). *P < 0.05, **P < 0.00. Error bars represent standard error of the mean.
DISCUSSION
Research has shown that PSC is a rare and severe hepatobiliary disease, leading to cirrhosis and liver failure, which is associated with IBD in 80% of patients, mostly diagnosed as UC. In addition, PSC-IBD is linked to a high risk of developing colorectal and hepatobiliary cancers, including cholangiocarcinoma, which together with liver failure are responsible for a high mortality rate in these patients. Unfortunately, the cause of PSC is unknown and there is no treatment available.
Colitis is often right-sided in PSC, and CRC more frequently arises from the right colon in these patients, suggesting that the underlying chronic inflammation may contribute to cancer initiation. Previous research noted an accumulation of Th1 and ILC in the colon of patients with PSC-UC,11 and other studies have implicated Th17 responses in the pathogenesis of PSC-IBD.10, 19, 20 In this study, we aimed to evaluate the role of immune-epithelial interactions in the pathogenesis of PSC-UC and the increased risk of CRC using patient-derived EpOCs.
We obtained EpOCs from colonic biopsies from control patients, patients with PSC-UC, and patients with UC with comparable disease activity, according to endoscopic and histological criteria. Organoids are 3-dimensional cell culture models or “mini organs” that are derived from stem cells.14 In intestinal organoids, LGR5+ stem cells can differentiate into enterocytes, goblet cells, Paneth cells, enteroendocrine cells, and enterochromaffin cells, rendering them representative of the original tissue and an invaluable tool for in vitro studies. Distinct transcriptomic profiles have been described in EpOCs derived from patients with UC compared with those derived from control patients, with a differential expression of genes involved in secretory and absorptive functions, antimicrobial defense, and inflammatory and carcinogenic pathways.15, 21 However, this is the first study to evaluate colonic EpOCs from patients with PSC-UC.
In this work, we observed a similar expression of genes associated with proliferation and differentiation in EpOCs derived from patients with UC, patients with PSC-UC, and control patients. However, a significantly higher expression of MUC2 was found in EpOCs derived from patients with PSC-UC than in patients with UC and control patients. Studies have shown that MUC2 is a mucin synthesized and secreted by goblet cells to form the inner mucous layer, a thick and dense barrier between the luminal microbiome and the epithelial cells, which contributes to mucosal defense.22 Mucin secretion is known to be regulated by bacterial products such as lipopolysaccharide and flagellin and by various cytokines.23 Dysbiosis has been previously described in PSC,9 and alterations of the mucous layer, through its effect on microbial-epithelial interactions, could contribute to the pathogenesis of intestinal inflammation in these patients. Interestingly, IL-22, a cytokine secreted mainly by Th17, Th22, and type 3 ILC, has been shown to play a central role in controlling mucin production.24, 25 IL-22-/- mice show a significant reduction in MUC1 and MUC2 expression, and IL-22 stimulation can induce MUC1 and MUC2 expression in intestinal epithelial cells in mice.26 The increased Th17 and ILC responses in PSC may therefore contribute to mucosal barrier alterations in these patients.
We next evaluated the expression of genes associated with a stem-cell phenotype in EpOCs. Interestingly, we observed a significant downregulation of the classical stem-cell marker LGR5 and an upregulation of OLFM4, which was highly expressed at the base of the crypts in patients with PSC-UC. OLFM4 is a secreted glycoprotein identified as a signature gene for LGR5+ stem cells and is considered a robust marker of murine and human intestinal stem cells.27 Interestingly, OLFM4 is also overexpressed in active IBD, and bacteria-led induction of OLFM4 through Notch signaling has been previously reported in colon adenocarcinoma and mouse epithelial cell lines.28, 29 Furthermore, OLFM4 is upregulated in colorectal adenomas30 and cancers of the gastrointestinal and hepatobiliary systems such as CRC, gastric cancer, and gall bladder cancer.31 Conversely, other studies have shown that OLFM4 deletion in mice results in dextran sodium sulfate–induced intestinal inflammation and colon adenocarcinoma,32 and its downregulation has been observed in poorly differentiated and metastatic cancers.31 This disparity clearly indicates that sufficient levels of OLFM4 are required to play a regulatory role in the intestine to maintain homeostasis and prevent dysbiosis.
To study immune-epithelial interactions, we stimulated EpOCs with cytokines previously associated with PSC-UC and with colitis-associated cancer, such as IFNγ and IL-22.11, 33 We confirmed that EpOCs respond to cytokine stimulation with STAT phosphorylation and gene induction. Interestingly, LGR5 was downregulated in EpOCs derived from control patients after IFNγ stimulation, whereas higher POU5F1 and OLFM4 expression was observed in EpOCs derived from patients with PSC-UC after stimulation with IFNγ and IL-22, respectively.
Previously, IL-22 was shown to induce OLFM4 in colon adenocarcinoma cell lines29; however, the opposite effects have also been reported, with IL-22 dampening the expression of LGR5 and OLFM4 in murine small bowel organoids.34, 35 Moreover, Th22 cells can promote CRC through the activation of STAT3 and induction of the DOT1L complex, which regulates the expression of stem cell–associated genes, such as NANOG, SOX2, and POU5F1.36 However, in our study, NANOG and POU5F1 were not significantly affected by IL-22 stimulation.
The cytokine IL-22 is considered as a double-edged sword in inflammation, with protective and pathogenic roles depending on the microenvironment. It is clearly involved in epithelial regeneration, and IL22RA1 is upregulated in the transit-amplifying region of the intestinal crypts.24, 36 However, continuous IL-22 exposure in chronic inflammation may induce permanent changes in the stem niche that could contribute to epithelial transformation. Interestingly, we observed amplifying activity of Th1 and Th17 responses in the colonic epithelium, with IFNγ stimulation resulting in the upregulation of IL22RA1. A similar synergism between IFNγ and IL-22 receptors was previously found to play a central role in protection against viral infections in the intestine,37 highlighting the complex amplifying interaction between these evolution-related cytokines.
CONCLUSIONS
Our study suggests that increased Th1 and Th17 responses in PSC may impact the intestinal epithelium, leading to alteration of the stem cell niche. The observed expansion of OLFM4+ and POU5F1+ transient amplifying cells may contribute to disease progression and increased cancer risk. Further studies are needed to characterize the stem cell niche in the intestinal and biliary epithelium in PSC and to assess whether targeting IL-12/IL-23 or more downstream cytokines, such as IFNγ or IL-22, may be beneficial for the treatment of these patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
The authors thank Dr. Matthias Friedrich at the Kennedy Institute of Rheumatology for providing protocols and helping to set up initial organoid cultures; James Chivenga, Roxanne Williams, and Krishnageetha Manoharan for patient sample collection; and Ida Parisi at the Kennedy Institute of Rheumatology for her technical support. The authors thank all volunteers and patients who took part in this study. The views expressed are those of the authors and not necessarily those of the National Health Service, the National Institute for Health Research, or the Department of Health. We thank the Oxford Biomedical Research Centre GI Biobank, which is funded by the National Institute for Health Research Oxford Biomedical Research Centre.
Glossary
Abbreviations
- BME
basement membrane extract
- CRC
colorectal cancer
- EpOCs
epithelial organoid cultures
- IBD
inflammatory bowel disease
- IFN
interferon
- IL
interleukin
- ILC
innate lymphoid cells
- PSC
primary sclerosing cholangitis
- PSC-UC
primary sclerosing cholangitis–associated ulcerative colitis
- Th
T helper
- UC
ulcerative colitis
Author contributions: M.N. and S.S.B. designed, performed, and analyzed experiments and wrote the manuscript. Z.V. and S.B. performed experiments and provided technical support. S.T., C.V.A-C., and F.P. contributed to data interpretation and revised the manuscript. A.G. conceived and designed the project and contributed to data interpretation and revision of the manuscript.
Supported by: This work was supported by the Wellcome Trust (101734/Z/13/Z to A.G., 212240/Z/18/Z to F. P.), the National Institute for Health Research Research Capability Fund (to A.G.), the National Institute for Health Research Oxford Biomedical Research Centre (Gastroenterology and Mucosal Immunity Theme; to C.V.A-C., F.P., and S.T.), and Versus Arthritis (to S.B.).
Conflicts of interest: A.G. is currently an employee of UCB Pharma, but this work was completed before she took up this position.
OXFORD IBD COHORT INVESTIGATORS
Phil Allan, Tim Ambrose, Adam Bailey, Ellie Barnes, Elizabeth Bird-Lieberman, Barbara Braden, Oliver Brain, Jane Collier, Emma Culver, James East, Lucy Howarth, Satish Keshav, Paul Klenerman, Simon Leedham, Rebecca Palmer, Astor Rodrigues, Jack Satsangi, Alison Simmons, Peter Sullivan, Holm Uhlig, and Alissa Walsh.
REFERENCES
- 1. Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med. 2016;375:2501–2502. [DOI] [PubMed] [Google Scholar]
- 2. Loftus EV Jr, Harewood GC, Loftus CG, et al. . PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut. 2005;54:91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ananthakrishnan AN, Cagan A, Gainer VS, et al. . Mortality and extraintestinal cancers in patients with primary sclerosing cholangitis and inflammatory bowel disease. J Crohns Colitis. 2014;8:956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Williamson KD, Chapman RW. Primary sclerosing cholangitis: a clinical update. Br Med Bull. 2015;114:53–64. [DOI] [PubMed] [Google Scholar]
- 5. Eksteen B, Grant AJ, Miles A, et al. . Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med. 2004;200:1511–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grant AJ, Lalor PF, Salmi M, et al. . Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet. 2002;359:150–157. [DOI] [PubMed] [Google Scholar]
- 7. Jiang X, Karlsen TH. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat Rev Gastroenterol Hepatol. 2017;14:279–295. [DOI] [PubMed] [Google Scholar]
- 8. Karlsen TH. A lecture on the genetics of primary sclerosing cholangitis. Dig Dis. 2012;30(Suppl 1):32–38. [DOI] [PubMed] [Google Scholar]
- 9. Hov JR, Karlsen TH. The microbiome in primary sclerosing cholangitis: current evidence and potential concepts. Semin Liver Dis. 2017;37:314–331. [DOI] [PubMed] [Google Scholar]
- 10. Nakamoto N, Sasaki N, Aoki R, et al. . Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol. 2019;4:492–503. [DOI] [PubMed] [Google Scholar]
- 11. Gwela A, Siddhanathi P, Chapman RW, et al. . Th1 and innate lymphoid cells accumulate in primary sclerosing cholangitis-associated inflammatory bowel disease. J Crohns Colitis. 2017;11:1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quraishi MN, Acharjee A, Beggs AD, et al. . A pilot integrative analysis of colonic gene expression, gut microbiota, and immune infiltration in primary sclerosing cholangitis-inflammatory bowel disease: association of disease with bile acid pathways. J Crohns Colitis. 2020;14:935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sato T, Vries RG, Snippert HJ, et al. . Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. [DOI] [PubMed] [Google Scholar]
- 14. Sato T, Stange DE, Ferrante M, et al. . Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. [DOI] [PubMed] [Google Scholar]
- 15. Dotti I, Mora-Buch R, Ferrer-Picón E, et al. . Alterations in the epithelial stem cell compartment could contribute to permanent changes in the mucosa of patients with ulcerative colitis. Gut. 2017;66:2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Travis SP, Schnell D, Krzeski P, et al. . Reliability and initial validation of the Ulcerative Colitis Endoscopic Index of Severity. Gastroenterology. 2013;145:987–995. [DOI] [PubMed] [Google Scholar]
- 17. Marchal-Bressenot A, Salleron J, Boulagnon-Rombi C, et al. . Development and validation of the Nancy histological index for UC. Gut. 2017;66:43–49. [DOI] [PubMed] [Google Scholar]
- 18. McCuaig S, Barras D, Mann EH, et al. . The interleukin 22 pathway interacts with mutant KRAS to promote poor prognosis in colon cancer. Clin Cancer Res. 2020;26:4313–4325. [DOI] [PubMed] [Google Scholar]
- 19. Katt J, Schwinge D, Schoknecht T, et al. . Increased T helper type 17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology. 2013;58:1084–1093. [DOI] [PubMed] [Google Scholar]
- 20. Kunzmann LK, Schoknecht T, Poch T, et al. . Monocytes as potential mediators of pathogen-induced T-helper 17 differentiation in patients with primary sclerosing cholangitis (PSC). Hepatology. 2020;72:1310–1326. [DOI] [PubMed] [Google Scholar]
- 21. Sarvestani SK, Signs SA, Lefebvre V, et al. . Cancer-predicting transcriptomic and epigenetic signatures revealed for ulcerative colitis in patient-derived epithelial organoids. Oncotarget. 2018;9:28717–28730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johansson ME, Phillipson M, Petersson J, et al. . The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cornick S, Tawiah A, Chadee K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers. 2015;3:e982426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mizoguchi A. Healing of intestinal inflammation by IL-22. Inflamm Bowel Dis. 2012;18:1777–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sugimoto K, Ogawa A, Mizoguchi E, et al. . IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Turner JE, Stockinger B, Helmby H. IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLoS Pathog. 2013;9:e1003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van der Flier LG, Haegebarth A, Stange DE, et al. . OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology. 2009;137:15–17. [DOI] [PubMed] [Google Scholar]
- 28. Liu W, Yan M, Liu Y, et al. . Olfactomedin 4 down-regulates innate immunity against Helicobacter pylori infection. Proc Natl Acad Sci U S A. 2010;107:11056–11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gersemann M, Becker S, Nuding S, et al. . Olfactomedin-4 is a glycoprotein secreted into mucus in active IBD. J Crohns Colitis. 2012;6:425–434. [DOI] [PubMed] [Google Scholar]
- 30. Besson D, Pavageau AH, Valo I, et al. . A quantitative proteomic approach of the different stages of colorectal cancer establishes OLFM4 as a new nonmetastatic tumor marker. Mol Cell Proteomics. 2011;10:M111.009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang XY, Chen SH, Zhang YN, et al. . Olfactomedin-4 in digestive diseases: a mini-review. World J Gastroenterol. 2018;24:1881–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu W, Li H, Hong SH, et al. . Olfactomedin 4 deletion induces colon adenocarcinoma in ApcMin/+ mice. Oncogene. 2016;35:5237–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kirchberger S, Royston DJ, Boulard O, et al. . Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. 2013;210:917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zha JM, Li HS, Lin Q, et al. . Interleukin 22 expands transit-amplifying cells while depleting Lgr5+ stem cells via inhibition of wnt and notch signaling. Cell Mol Gastroenterol Hepatol. 2019;7:255–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zwarycz B, Gracz AD, Rivera KR, et al. . IL22 inhibits epithelial stem cell expansion in an ileal organoid model. Cell Mol Gastroenterol Hepatol. 2019;7:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kryczek I, Lin Y, Nagarsheth N, et al. . IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity. 2014;40:772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hernández PP, Mahlakoiv T, Yang I, et al. . Interferon-λ and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat Immunol. 2015;16:698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.