

Abstract
Son of sevenless homologue 1 (SOS1) is a guanine nucleotide exchange factor that catalyzes the exchange of GDP for GTP on RAS. In its active form, GTP-bound RAS is responsible for numerous critical cellular processes. Aberrant RAS activity is involved in ~30% of all human cancers; hence, SOS1 is an attractive therapeutic target for its role in modulating RAS activation. Here, we describe a new series of benzimidazole-derived SOS1 agonists. Using structure-guided design, we discovered small molecules that increase nucleotide exchange on RAS in vitro at sub-micromolar concentrations, bind to SOS1 with low double digit nanomolar affinity, rapidly enhance cellular RAS-GTP levels, and invoke biphasic signaling changes in phosphorylation of ERK 1/2. These compounds represent the most potent series of SOS1 agonists reported to date.
Keywords: RAS, SOS1, Agonist, Cancer
Graphical Abstract
INTRODUCTION
RAS proteins act as molecular switches, cycling between active (GTP-bound) and inactive (GDP-bound) states in order to regulate multiple intracellular signaling pathways that control cell proliferation, growth, differentiation, and survival.1,2 Deregulation of these pathways via oncogenic RAS mutations can result in cells displaying many of the hallmarks of cancer and is implicated in the oncogenesis and progression of ~30% of all malignant tumors.3–7 Therefore, therapeutic targeting of oncogenic RAS has been an important and long-standing goal in modern drug discovery. Unfortunately, only limited success has been realized when directly targeting RAS due to the lack of chemically tractable pockets on the protein surface.8–10 The recalcitrant nature of these proteins toward traditional drug discovery strategies has inspired the pursuit of alternative approaches to inhibit RAS signaling.11–19 One strategy is to indirectly target RAS through therapeutic modulation of the regulatory proteins that control its activity. The conversion of RAS-GDP to RAS-GTP is the rate-limiting step in the activation of RAS signaling and is controlled by guanine nucleotide exchange factors (GEFs) such as son of sevenless homologue 1 (SOS1).7,20,21 Due to its importance in RAS signaling, SOS1 has emerged as an attractive therapeutic target for the treatment of RAS-driven cancers.22,23
Our group recently reported two unrelated chemical series that bind to the same hydrophobic pocket on SOS1 as part of a RAS:SOS1:RAS complex.24–26 We have previously shown that these compounds activate nucleotide exchange on RAS, resulting in increased levels of cellular RAS-GTP and, at low concentrations, enhance phosphorylation of extracellular regulared kinase (ERK) 1/2 via the RAS-MAPK signaling pathway.27 At higher doses of compound, phosphorylated ERK (pERK) phosphorylates SOS1 leading to disruption of the SOS1-GRB2 complex and dissociation of RAS from SOS1, thereby achieving a compound-mediated negative feedback loop that results in reduced ERK phosphorylation. Although seemingly counterintuitive, a growing body of evidence suggest that activation of RAS may be a viable therapeutic strategy.28–30 The “Goldilocks principle” suggests that cells can only tolerate changes in signaling that stay within certain parameters, which do not go either above, or below a certain cellular threshold.31 For example, enhancement of RAS-GTP levels in the cell by ectopic overexpression of mutant RAS can lead to pronounced anti-tumoral responses.32 Furthermore, naturally occurring concomitant expression of mutant RAS with either mutant BRAF or mutant EGFR is very rare, and overexpression of RAS is synthetically lethal in the context of an EGFR or BRAF mutation.33–35 Thus, the RAS activation that we observe with SOS1 agonists may represent a promising approach for targeting cells with underlying oncogenic RAS, BRAF, or EGFR mutations. Here, we describe the structure-guided design, synthesis, and structure–activity relationships (SAR) of a new benzimidazole-derived series of SOS1 ligands. These efforts led to the discovery of molecules that robustly activate SOS1-mediated nucleotide exchange in vitro, bind with high affinity to SOS1, rapidly increase cellular RAS-GTP levels, and modulate ERK phosphorylation in cells, resulting in the most potent SOS1 agonists yet reported.
RESULTS AND DISCUSSION
Discovery of Benzimidazole-Derived SOS1 Compounds.
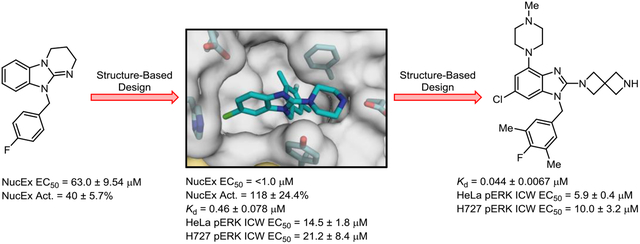
In order to identify compounds that bind to SOS1 and affect SOS1-mediated nucleotide exchange in vitro, we conducted a high throughput screen (HTS) of the Vanderbilt Compound Collection.36 This screen revealed multiple molecules in different chemical series that elicited robust and reproducible SOS1-mediated activation of nucleotide exchange on RAS. Similar to our previously reported work,24–26,36 compound efficacy is described by relative percent activation (Act.), which is the rate of nucleotide exchange at the maximum compound dose of 100 μM, relative to the previously reported indole-derived control compound at the same concentration in the same run of the assay.25 The corresponding potency is described as the half maximal effective concentration (EC50) determined from the dose response curve (0–100 μM of compound) generated from the nucleotide exchange assay. From the HTS hits, several benzimidazole-containing molecules emerged as potentially useful starting points. For example, compounds 1 and 2 were found to activate nucleotide exchange with EC50 values of 63.0 μM and 23.2 μM, respectively (Figure 1A). An early follow-up analog, 3, was synthesized that removed the ring constraints from the 2-position nitrogen atom and introduced a substitution pattern on the benzyl group similar to that of an aniline ring system in a previously reported anilinoquinazoline series.26 These structural modifications afforded a 3-fold improvement in nucleotide exchange activation EC50 when comparing derivative 3 to 2. Furthermore, an X-ray co-crystal structure obtained of 3 bound to the RAS:SOS1:RAS ternary complex confirmed that it binds in the same hydrophobic pocket of the CDC25 domain of SOS1, adjacent to the Switch II region of RAS, that is utilized by previously reported series of SOS1 ligands (Figure 1B).23–26,36
Figure 1.
(A) Early hit benzimidazole-derived activators of SOS1-mediated nucleotide exchange. (B) X-ray co-crystal structure of 3 (yellow; PDB ID code 6D5V) bound to SOS1 in the RAS:SOS1:RAS ternary complex. The RAS and SOS1 protein surfaces are colored yellow and grey, respectively. Areas identified for SAR development are highlighted, including: (i) a water-mediated interaction between the 2-amine functionality with Asp887 (blue oval), (ii) occupation of a hydrophobic subpocket beneath Phe890 (red arrows), (iii) occupation of a hydrophobic region along the wall of the CDC25 domain (pink rectangle), (iv) occupation of a hydrophobic subpocket behind Glu902 and adjacent to Phe890 (green circle), and (v) interaction with proximal residues of SOS1 from the 4-position of the benzimidazole scaffold (white arrows).
C-2 Substituent SAR.
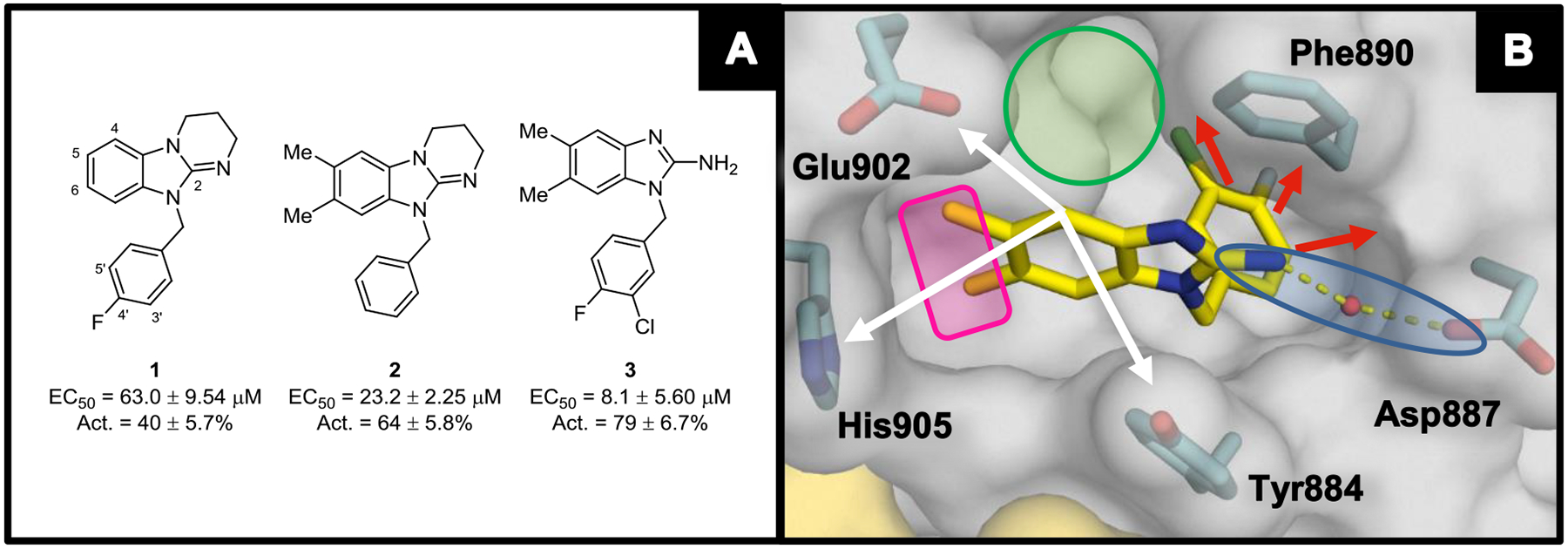
Analysis of the X-ray co-crystal structure of compound 3 revealed several opportunities to improve binding to SOS1 (Figure 1B). We initiated our SAR studies by focusing on the 2-position of the benzimidazole core with the goal of deploying a Lewis base closer to Asp887 in an attempt to establish a direct, rather than water-mediated, interaction (Table 1). Notably, improved potency and efficacy were observed with several 2-amine variants. This was highlighted by 2-piperazino 10, which demonstrated a ~4-fold and ~3-fold improvement in nucleotide exchange EC50 and relative percent activation, respectively. Conversely, negligible nucleotide exchange activation was observed when the Lewis base was removed, as exemplified by 2H-benzimidazole 4. The X-ray co-crystal structure of compound 10 (Figure 2A) revealed that the piperazine moiety occluded the water molecule that facilitated the hydrogen bonding interaction between Asp887 and 3. As a result, the piperazine nitrogen atom of 10 achieved a direct charge–charge interaction with the carboxylate side chain of Asp887. Based on its combination of potent and efficacious nucleotide exchange activity and limited number of hydrogen bond donors,37 piperazino-analog 10 was selected as a template for continued structural modification.
Table 1.
2-Amino Substituents
 |
|||
|---|---|---|---|
| Compd | R2 | EC50 (μM) | Act. (%)a |
| 3 | NH2 | 8.1 ± 6.83 | 79 ± 8.0 |
| 4 | H | --b | -- |
| 5 | 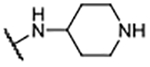 |
37.1 ± 12.57 | 129 ± 1.8 |
| 6 | 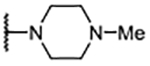 |
26.0 ± 0.80 | 143 ± 18.9 |
| 7 | 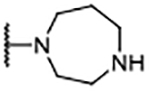 |
8.5 ± 2.06 | 176 ± 10.1 |
| 8 | 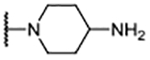 |
6.6 ± 0.05 | 114 ± 6.3 |
| 9 | 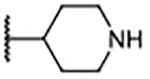 |
5.6 ± 0.11 | 128 ± 0.6 |
| 10 | 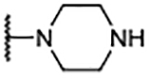 |
2.2 ± 0.52 | 211 ± 20.5 |
| 11 | 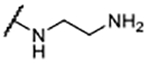 |
1.9 ± 0.62 | 114 ± 12.2 |
Measurements are reported as the mean ± SD of two or more independent experiments, each conducted in duplicate.
Activation values represent percentage relative to control.25
“--“ denotes an EC50 value of >100 μM, an EC50 value that was not calculated due to low efficacy, or an activation value that was not determined due to low efficacy.
Figure 2.
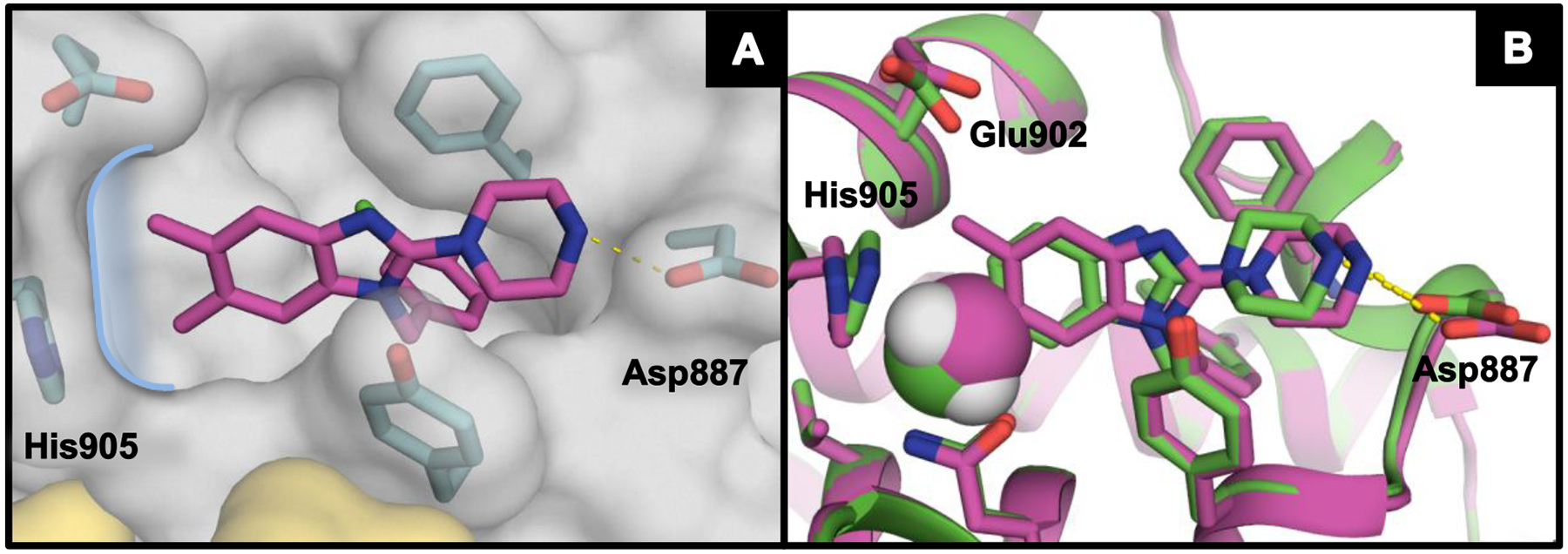
(A) X-ray co-crystal structure of 10 (magenta; PDB ID code 6D5M) bound to SOS1 in the RAS:SOS1:RAS ternary complex. The curved blue line highlights the subpocket near His905. The RAS and SOS1 protein surfaces are colored yellow and grey, respectively. (B) 28 (green; PDB ID code 6D5L) overlaid with 10. The green sphere represents space filling of the 6-chlorine atom in 28, whereas the magenta and white spheres represent space filling of the 6-methyl group in 10.
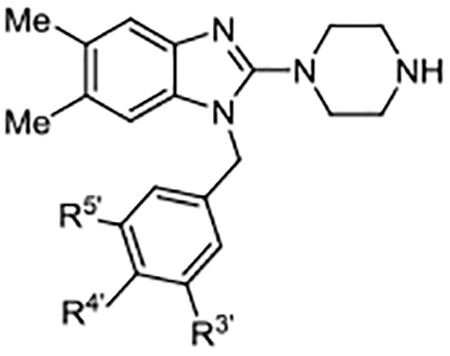
Benzyl Substituent SAR.
With improvements in potency and efficacy achieved via the inclusion of the 2-piperazine moiety, we turned our focus to investigating the chemical space around the benzyl group (Table 2). While ortho-substitutions were generally poorly tolerated (not shown), the contribution of the aromatic halogen atoms in other positions was quite evident, as unsubstituted benzyl analog 12 showed an EC50 that was ~4-fold worse than 10. Interestingly, of the 4’-monosubstituted derivatives, only the 4’-fluoro compound 13 demonstrated improved nucleotide exchange potency relative to 12, and the activity dropped off sharply with larger 4’-moieties. The benefit of meta-substitution also appeared to be limited by size as evidenced by the drop in potency observed for compounds featuring a 3’-trifluoromethyl (19) or a 3’-cyclopropyl (20) group relative to 10. Together, these data suggested that limited steric bulk at the para- and meta-positions provided improved potency. We noted that compound potency (EC50) was observed to correlate poorly with compound efficacy (Act.). Empirically, it had been previously determined that SOS1 agonists achieving Act. values greater than ~50% elicited more robust increases in cellular RAS–GTP levels.36 Beyond this perceived minimum threshold, the origin and consequences of varying degrees of percent relative activation is not well understood and is currently under further investigation.
Table 2.
Benzyl Substituents
 |
|||||
|---|---|---|---|---|---|
| Compd | R3’ | R4’ | R5’ | EC50 (μM) | Act. (%)a |
| 10 | Cl | F | H | 2.2 ± 0.52 | 211 ± 20.5 |
| 12 | H | H | H | 8.6 ± 1.64 | 122 ± 17.8 |
| 13 | H | F | H | 5.9 ± 4.88 | 130 ± 28.1 |
| 14 | H | Cl | H | 12.4 ± 2.70 | 164 ± 15.0 |
| 15 | H | Me | H | 36.6 ± 17.26 | 93 ± 13.0 |
| 16 | H | CF3 | H | 19.6 ± 5.01 | 148 ± 30.7 |
| 17 | F | F | H | 5.9 ± 1.33 | 183 ± 25.3 |
| 18 | Me | F | H | 4.1 ± 2.00 | 190 ± 20.6 |
| 19 | CF3 | F | H | 8.1 ± 0.52 | 263 ± 52.5 |
| 20 |  |
F | H | 11.7 ± 2.74 | 151 ± 5.0 |
| 21 | Me | F | Me | 3.7 ± 0.29 | 270 ± 73.8 |
Measurements are reported as the mean ± SD of two or more independent experiments, each conducted in duplicate.
Activation values represent percentage relative to control.25
Arene 5,6-Substitution SAR.
Having learned much about the tolerance of substitution around the benzyl group, we transitioned to examining various hydrophobic substitutions at the 5- and 6-positions of the benzimidazole core to optimize the occupancy of the small hydrophobic subpocket near His905 (Figure 2A). For this, the 2-piperazino and 3’-chloro-4’-fluoro benzyl moieties were held constant to isolate the effects of the 5- and 6-positions of the benzimidazole nucleus (Table 3). Removal of the 5-position substituent had a relatively minor effect on compound potency. Compare, for example, the 5H-6-methyl derivative, 24, and the 5H-6-chloro derivative, 28, to their respective 5-methyl analogs 10 and 22. It is interesting to note however, that 5H-substitution resulted in a strong decrease in relative percent activation. The X-ray co-crystal structure of 28 overlaid with that of 10 (Figure 2B) showed that both ligands rest in the hydrophobic subpocket near His905 and effected minimal differences in the positioning of the SOS1 binding pocket residues. This suggested that the differences in observed Act. levels between analogs 10 and 28 are not due to a significant discrepancy in compound binding mode. From these same co-crystal structures, it is apparent that there is little room in the hydrophobic subpocket near His905 for 6-position moieties much larger than a methyl group. This observation is supported by the substantial loss in potency observed for the 6-cyclopropyl compound 26. Notably, removal of both 5- and 6-substitution, exemplified by compound 23, resulted in a ~5-fold reduction in potency relative to 28. These data suggested that an appropriately sized 6-substituent was the primary contributor to potency in this region of the binding pocket.
Table 3.
5,6-Substituents
 |
||||
|---|---|---|---|---|
| Compd | R5 | R6 | EC50 (μM) | Act. (%)a |
| 10 | Me | Me | 2.2 ± 0.52 | 211 ± 20.5 |
| 22 | Me | Cl | 3.0 ± 0.56 | 200 ± 13.3 |
| 23 | H | H | 5.4 ± 1.22 | 109 ± 4.2 |
| 24 | H | Me | 2.4 ± 0.33 | 106 ± 13.1 |
| 25 | H | CF3 | 4.4 ± 0.54 | 78 ± 2.2 |
| 26 | H |  |
24.3 ± 5.33 | 65 ± 2.2 |
| 27 | H | Br | 1.3 ± 0.3 | 115 ± 5.4 |
| 28 | H | Cl | 1.0 ± 0.16 | 109 ± 1.0 |
Measurements are reported as the mean ± SD of two or more independent experiments, each conducted in duplicate.
Activation values represent percentage relative to control.25
The SAR developed in Tables 2 and 3 prompted the synthesis of several matched pairs of analogs to investigate the interplay between substituents on the aromatic ring of the benzyl moiety and the substituents at the 5- and 6-positions of the benzimidazole nucleus. From this effort, crossover analog 29—containing the 3’,5’-dimethyl-4’-fluorobenzyl moiety and a 6-position chlorine atom—was found to exhibit an EC50 value below 1 μM in the nucleotide exchange assay (Figure 3A). As compounds continued to improve, we observed a loss of SAR texture for derivatives with EC50 values in this range. To effectively differentiate more potent compounds, a fluorescence polarization anisotropy (FPA) assay, based on the displacement of a labeled probe from SOS1 (Supplementary Figure S2), was developed to provide a measurement of binding affinity (Kd) to SOS1.38,39 Several earlier compounds were re-evaluated using this FPA assay, showing a general agreement between Kd and nucleotide exchange EC50 for previously investigated SAR trends (Supplementary Figure S3 and Table S1). This reaffirmed our established SAR and prompted a switch to the FPA assay as the primary tool for evaluating analogs. Moving forward with a new biochemical assay to drive optimization, compound 29 was selected as a template upon which to conduct further work.
Figure 3.
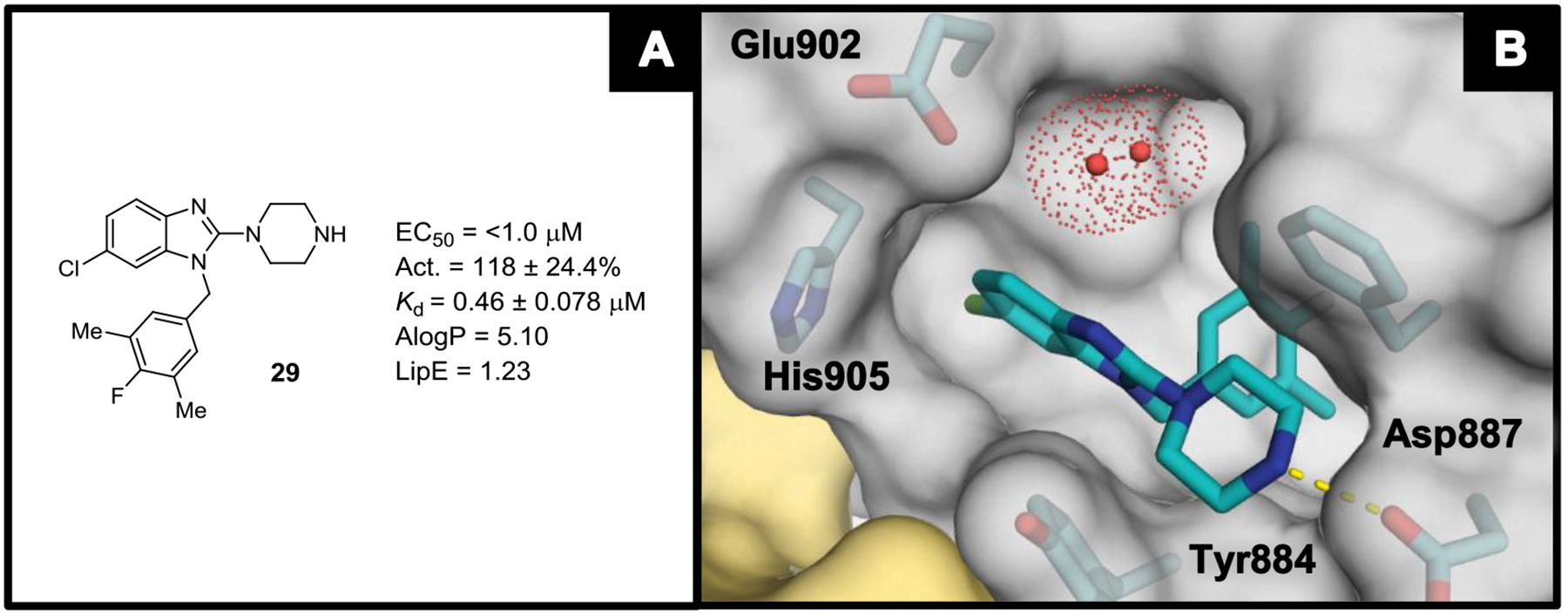
(A) Nucleotide exchange and binding affinity data for compound 29. (B) X-ray co-crystal structure of compound 29 (teal; PDB ID code 6D5J) bound to SOS1 in the RAS:SOS1:RAS ternary complex. The red dotted spheres represent molecules of water and the space they occupy. The RAS and SOS1 protein surfaces are colored yellow and grey, respectively.
C-4 Substituent SAR.
The binding mode of compound 29 (Figure 3B) highlighted several opportunities for continued optimization. First, a largely hydrophobic space proximal to the 4-position of the benzimidazole remained unoccupied, as demonstrated by the two molecules of water occupying the back of the binding pocket. Additionally, several residues at the lip of the binding pocket, primarily Glu902, His905, and Tyr884, seemed accessible for interaction. As these regions are largely solvent-exposed, a considerable tolerance for groups that may offer the ability to tune the overall molecular properties was expected. To exploit these opportunities, we explored a wide range of chemical diversity at the 4-position. A selection of these results is shown in Table 4.
Table 4.
C-4 SAR
 |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
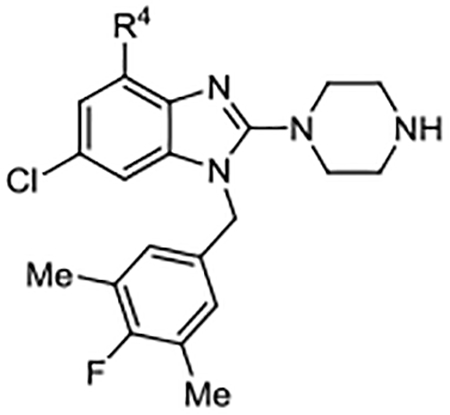
| Compd | R4 | Kd (μM)a | AlogP | LipE | Compd | R4 | Kd (μM)b | AlogP | LipE | Compd | R4 | Kd (μM)b | AlogP | LipE |
| 29 | H | 0.46 ± 0.078 | 5.10 | 1.23 | 39 | 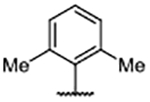 |
0.30 ± 0.078 | 7.59 | −1.06 | 49 | 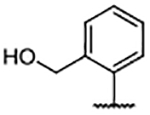 |
0.14 ± 0.009 | 6.02 | 0.85 |
| 30 |  |
0.27 ± 0.005 | 6.62 | −0.06 | 40 | 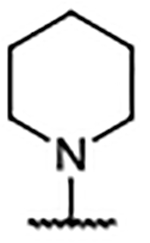 |
0.20 ± 0.003 | 6.18 | 0.51 | 50 | 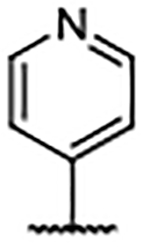 |
0.24 ± 0.056 | 5.47 | 1.16 |
| 31 | 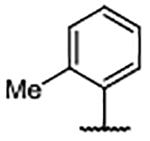 |
0.21 ± 0.010b | 7.11 | −0.44 | 41 | 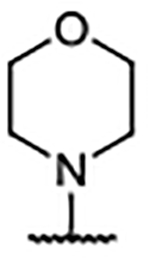 |
0.13 ± 0.030 | 4.95 | 1.95 | 51 | 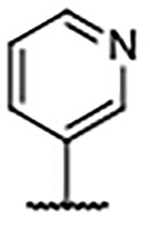 |
0.33 ± 0.027a | 5.47 | 1.01 |
| 32 | 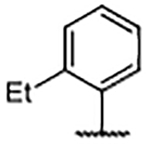 |
0.21 ± 0.029 | 7.56 | −0.88 | 42 | 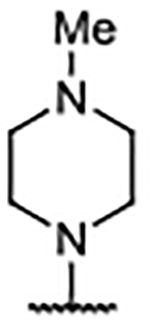 |
0.14 ± 0.013 | 5.22 | 1.65 | 52 | 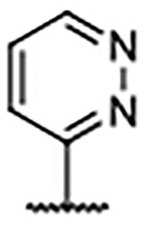 |
0.45 ± 0.104 | 5.43 | 0.92 |
| 33 | 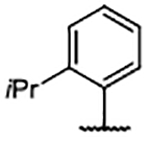 |
0.39 ± 0.093 | 7.82 | −1.41 | 43 | 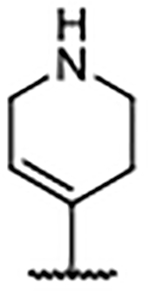 |
0.25 ± 0.057a | 5.14 | 1.46 | 53 | 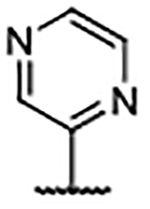 |
0.39 ± 0.064 | 4.75 | 1.66 |
| 34 | 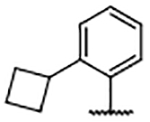 |
0.44 ± 0.139 | 7.90 | −1.54 | 44 | 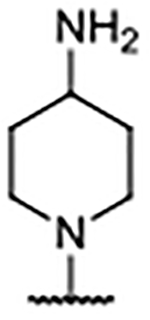 |
0.24 ± 0.083a | 4.41 | 2.21 | 54 | 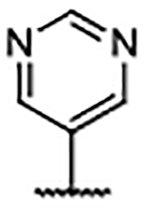 |
0.35 ± 0.001 | 4.84 | 1.61 |
| 35 | 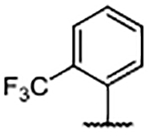 |
0.15 ± 0.048 | 7.56 | −0.74 | 45 | 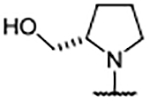 |
0.31 ± 0.083 | 5.22 | 1.30 | 55 | 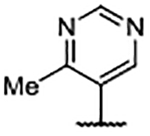 |
0.11 ± 0.018 | 5.12 | 1.82 |
| 36 |  |
0.67 ± 0.007 | 7.56 | −1.39 | 46 |  |
0.38 ± 0.025 | 5.22 | 1.20 | 56 | 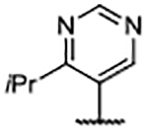 |
0.10 ± 0.011 | 6.25 | 0.74 |
| 37 | 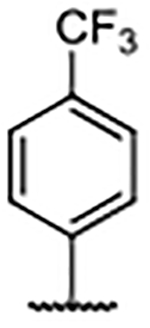 |
1.05 ± 0.056 | 7.56 | −1.59 | 47 | 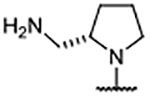 |
0.14 ± 0.009 | 4.93 | 1.92 | 57 | 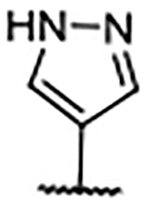 |
0.18 ± 0.081 | 5.06 | 1.68 |
| 38 | 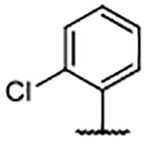 |
0.12 ± 0.034 | 7.29 | −0.43 | 48 |  |
0.24 ± 0.036 | 4.93 | 1.69 | 58 |  |
<0.10 | 5.49 | --c |
Measurements are reported as the mean ± SD of two or more independent experiments, each conducted in duplicate.
Evaluated through displacement of probe S1 (Supplementary Figure S2).
Evaluated through displacement of probe S2 (Supplementary Figure S2).
LipE was not determined.
In an effort to fill the neighboring hydrophobic subpocket, several biaryl derivatives with lipophilic substitutions were synthesized and evaluated by FPA (Table 4). Multiple 4-phenyl analogs demonstrated improved binding affinity relative to the 4H-parent 29. In some instances, ortho-substitution of the 4-phenyl ring offered further enhancement—best exemplified by the ortho-trifluoromethyl analog 35 and the ortho-chloro compound 38 —relative to the unsubstituted phenyl derivative 30. Increasing the size of the ortho-alkyl groups resulted in a decline in binding affinity from 30, suggesting that the nearby pocket has a limited size tolerance. Notably, biaryl analogs, such as 36 and 37, bearing meta- or para-substituents exhibited diminished affinity when compared to their ortho-substituted or unsubstituted comparators.
Analysis of the X-ray co-crystal structure of 38 revealed that the aryl group is rotated out of plane from the benzimidazole scaffold (Figure 4A). Further, when the co-crystal structure of 38 was overlaid with that of 29 (Figure 4D), it was evident that 38 leaned over into the SOS1 binding pocket to drive the ortho-chlorine atom down into the available lipophilic space, thus displacing the previously bound waters from this subpocket. The presence of the hydrophobic aryl group in 38 disrupted the positioning of Glu902, rotating the carboxylic acid side chain upward toward solvent compared to the downward pose observed in the co-crystal structure of 29. The observed gains in binding affinity from the addition of phenyl groups at the 4-position corresponded with an increase in compound lipophilicity as evidenced by a rise in ALogP and correspondingly poor lipophilic ligand efficiency (LipE)40 when compared with the 4H-compound, 29.
Figure 4.
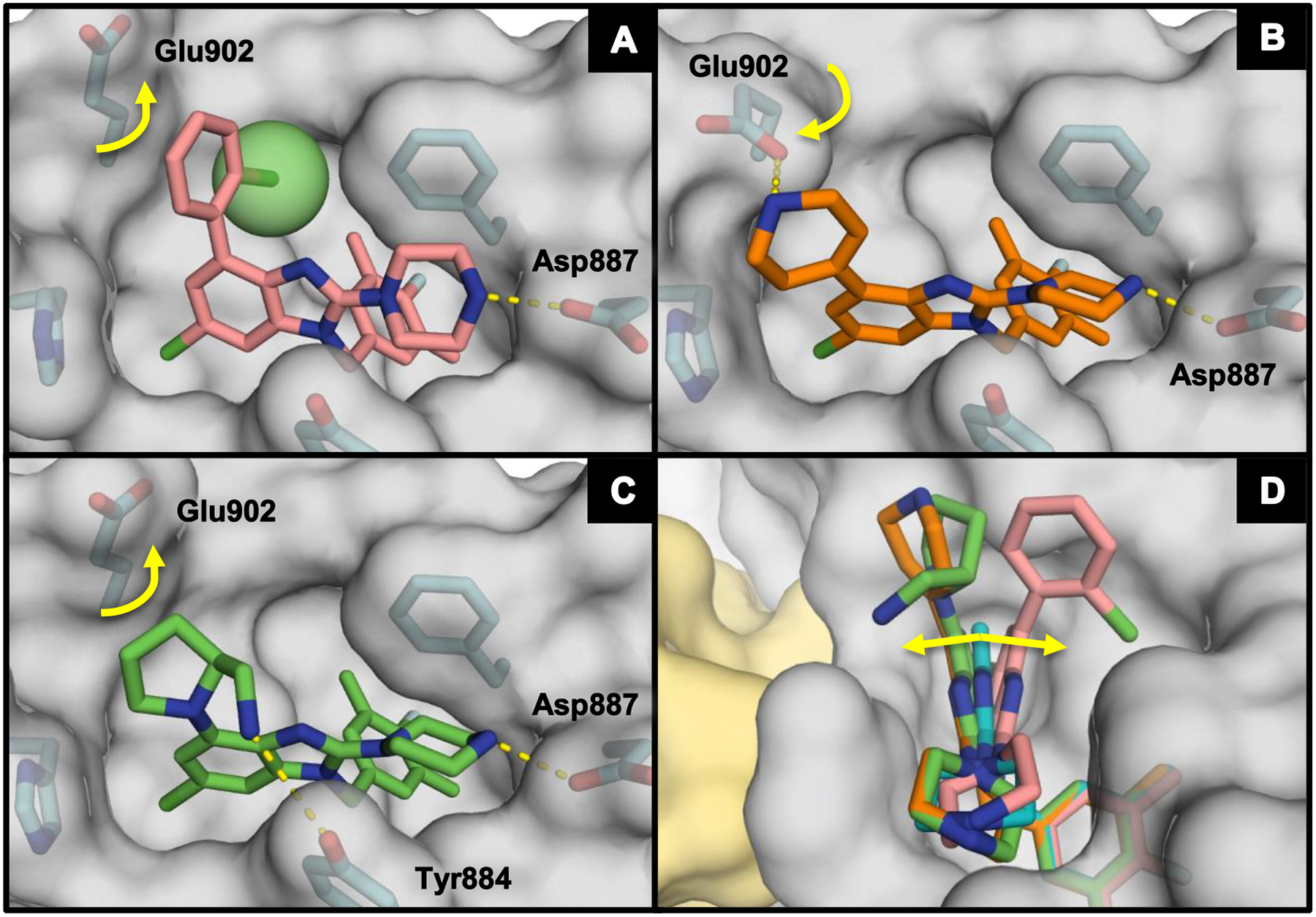
X-ray co-crystal structures of (A) 38 (salmon; PDB ID code 6D5H), (B) 43 (orange; PDB ID code 6D5G), (C) 47 (green; PDB ID code 6D5E), and (D) 38, 43, and 47 overlaid with 29 (teal) bound to SOS1 in the RAS:SOS1:RAS ternary complex. The RAS and SOS1 protein surfaces are colored yellow and grey, respectively.
We hypothesized that directly engaging residues Glu902, His905, and Tyr884 with heteroatoms might provide significant affinity enhancements. Furthermore, successful incorporation of polar functionality at the 4-position was expected to result in improved solubility and lower lipophilicity relative to the biaryl compounds. We thus explored the use of a protonatable nitrogen atom to target the carboxylate of Glu902 with a potential charge–charge interaction. As the data in Table 4 illustrate, amine substitutions at the 4-position were well-tolerated and generally demonstrated lower lipophilicity. This is best exemplified by the N-methylpiperazine analog, 42, which demonstrated a greater than 3-fold improvement over the 4H-benzimidazole predecessor 29. Notably, 42 had a similar binding affinity to that of 38, but with much improved AlogP and LipE values. The X-ray co-crystal structure of the tetrahydropyridine analog 43 (Figure 4B) revealed that Glu902 was rotated down toward the nitrogen atom of 43 compared to the position of the same residue in the X-ray co-crystal structure of 38 (Figure 4A). In contrast to 29 and 38, compound 43 leaned toward the RAS:SOS1 interface to accommodate the positioning of Glu902 (Figure 4D), confirming that the tetrahydropyridine moiety of 43 did indeed interact with the carboxylate of Glu902. However, replacement of the basic nitrogen atom in the piperazine moiety with a methylene group (40) or oxygen atom (41) proved to have little effect on binding affinity, suggesting that the charge–charge interaction with Glu902 offered minimal benefit to binding.
Since engaging Glu902 from the 4-position of the benzimidazole scaffold improved lipophilicity, but provided only limited binding affinity improvements over 4-aryl analogs, the focus turned toward creating a vector for a heteroatom to engage His905 or Tyr884 via a hydrogen bond, as exemplified by compounds 45–49 (Table 4). These entities also proved to be widely tolerated, affording binding affinities similar to that of other 4-substituents highlighted thus far. The X-ray co-crystal structure for 47 revealed that the extended nitrogen atom did not engage His905, but was orientated toward Tyr884 instead (Figure 4C). Interaction with the phenol of Tyr884 caused compound 47 to lean toward the RAS:SOS1 interface, in similar fashion to 43 (Figure 4D). Notably, Glu902 was rotated upward toward solvent—as it was in the X-ray co-crystal structure of 38—to accommodate the hydrocarbon portion of the pyrrolidine ring.
Importantly, the examples incorporating polar functionality at the 4-position of the benzimidazole scaffold were widely tolerated, and resulted in compounds with greatly improved physical metrics relative to the highly lipophilic biaryl analogs. It was hypothesized that heteroaromatic substituents at the 4-position might merge the observed tolerance of polar functionality with occupation of the proximal hydrophobic subpocket and generate compounds with a favorable combination of potency, physiochemical properties, and a reduced number of hydrogen bond donors (Table 4). Unsubstituted heterocyclic compounds generally showed a marginal decrease in binding affinity relative to unsubstituted phenyl congener 30, but exhibited improved AlogP and LipE values. As was the case in the 4-phenyl series, ortho-substitution provided a marked boost in ligand binding affinity, exemplified by the 3-fold decrease in Kd from pyrimidine 54 to ortho-methyl pyrimidine 55 and ortho-isopropyl pyrimidine 56. Furthermore, disubstituted pyrazole 58 generated an affinity below the reliable detection limit (Kd <0.10 μM) afforded by the assay using the fluorescent probe S2 (Supplementary Figure S2).
Inspection of the X-ray co-crystal structure of 58 revealed that the disubstituted pyrazole functionality was rotated out of plane from the benzimidazole scaffold (Figure 5A). This oriented the lipophilic methyl group toward the back of the hydrophobic pocket in a comparable fashion to the ortho-chloro derivative 38. This caused the ligand to lean away from the RAS:SOS1 interface (Figure 5D) and overrode a potential interaction between the pyrazole nitrogen atoms and Glu902, as evidenced by the rotation of Glu902 up toward solvent. The ortho-substituted heterocycle subset of compounds consistently offered the best SOS1 affinities measured to this point. In addition, these compounds possessed improved physical properties relative to their more lipophilic phenyl counterparts.
Figure 5.
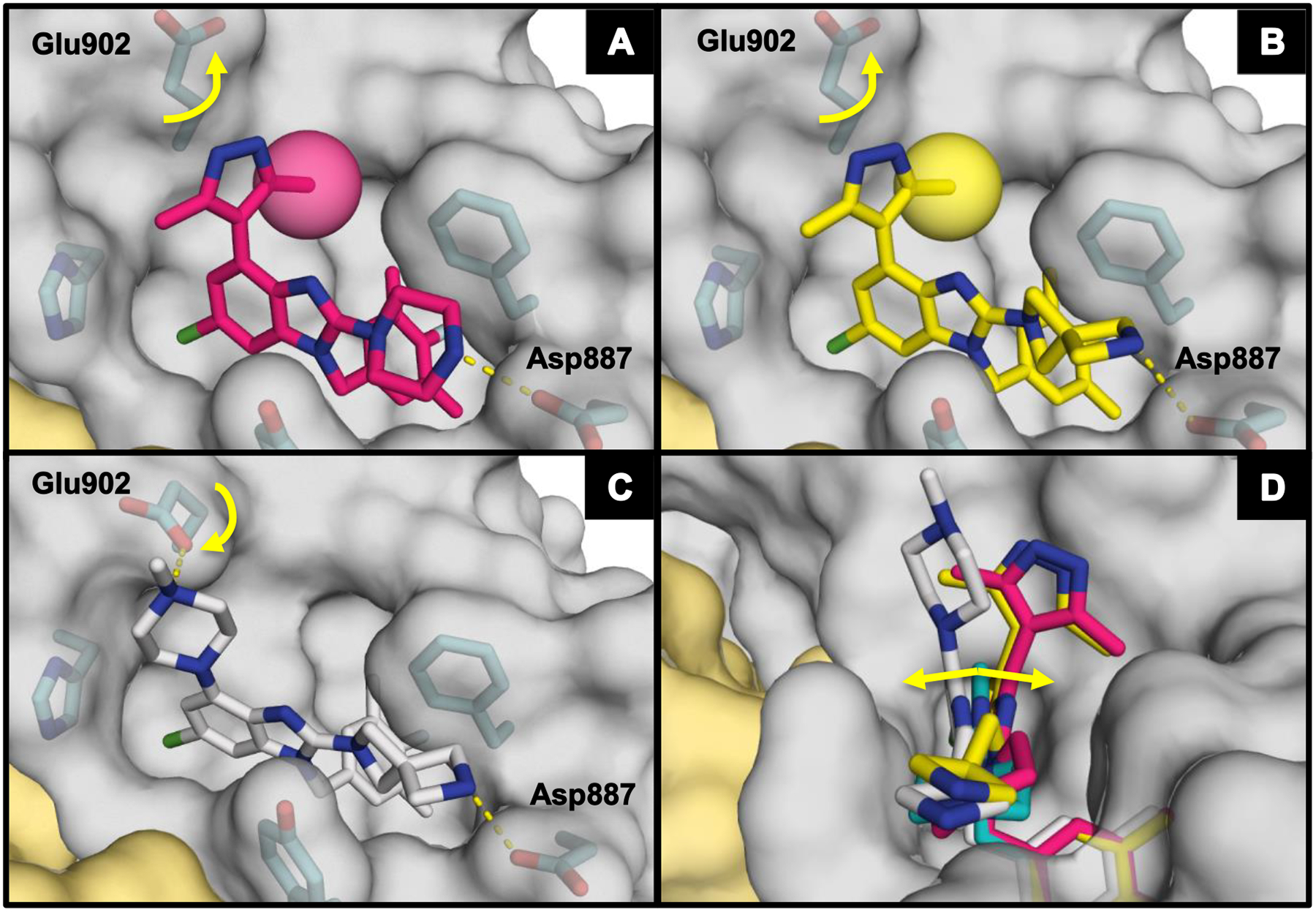
X-ray co-crystal structure of (A) 58 (pink; PDB ID code 6D59), (B) 65 (yellow; PDB ID code 6D56), (C) 64 (white; PDB ID code 6D55), and (D) 58, 64, and 65 overlaid with 29 (teal) bound to SOS1 in the RAS:SOS1:RAS ternary complex. The RAS and SOS1 protein surfaces are colored yellow and grey, respectively.
C-2 Diazaspiro[3.3]heptane Analogs.
After a thorough structure-guided SAR study, several compounds were discovered to bind to SOS1 with binding affinities near or below 0.10 μM (e.g. 58). We postulated that the numerous modifications to the benzimidazole scaffold may have affected earlier investigated SAR. Thus, the 2-position was re-evaluated using several amines in the context of our more advanced benzimidazole derivatives. From these efforts, a multitude of C-2 diazaspiro[3.3]heptane derivatives were found to have binding affinities below 0.10 μM,41–43 leading to the synthesis of a new fluorescent probe (S3, Supplementary Figure S2) bearing this moiety. The FPA competition assay with this probe proved capable of reproducible measurement of affinities well below 0.10 μM. Indeed, data for these analogs (Table 5) showed up to an order of magnitude improvement in affinity from their 2-piperazino counterparts.
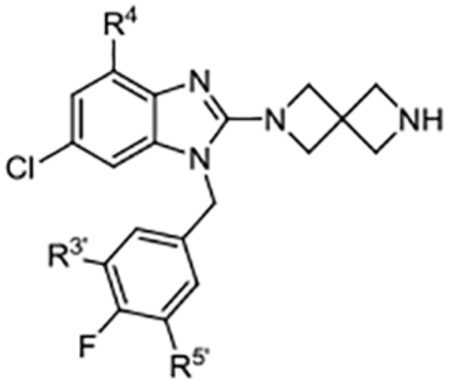
Table 5.
C-2 Diazaspiro[3.3]heptane Derivatives
 |
|||||
|---|---|---|---|---|---|
| Compd | R4 | R3’, R5’ | Kd (μM)a | ALogP | LipE |
| 59 | 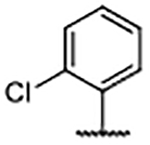 |
Me | 0.012 ± 0.0020 | 6.92 | 1.02 |
| 60 | 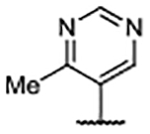 |
Me | 0.016 ± 0.0037 | 4.76 | 3.05 |
| 61 | 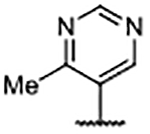 |
H | 0.18 ± 0.003b | 3.78 | 2.98 |
| 62 | 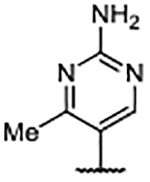 |
Me | 0.012 ± 0.0008 | 4.53 | 3.38 |
| 63 | 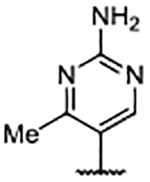 |
H | 0.036 ± 0.0055 | 3.56 | 3.88 |
| 64 | 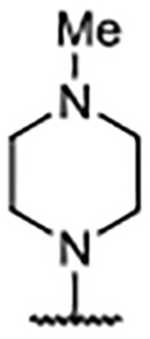 |
Me | 0.044 ± 0.0067 | 4.85 | 2.50 |
| 65 | 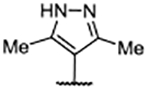 |
Me | 0.009 ± 0.0017 | 5.12 | 2.91 |
Measurements are reported as the mean ± SD of two or more independent experiments, each conducted in duplicate.
Evaluated through displacement of fluorescent probe S3 (Supplementary Figure S2).
Evaluated through displacement of fluorescent probe S2 (Supplementary Figure S2).
Analysis of the X-ray co-crystal structure for pyrazole 65 showed a very similar binding pose to that demonstrated by 58, with Glu902 rotated up and the ligand leaning in the pocket to place the methyl group of 65 into the hydrophobic subpocket (Figure 5B, 5D). The X-ray co-crystal structure for 64 (Figure 5C) showed a similar binding pose to 4-tetrahydropyridine 43, with Glu902 rotated down to establish a charge–charge interaction with the piperazine nitrogen atom and causing the ligand to lean toward the RAS:SOS1 interface (Figure 5D). Since little difference in the binding mode for either 64 or 65 from comparable 2-piperazine variants was evident, it was speculated that the more rigid C-2-diazaspiro[3.3]heptane moiety benefited from a lower entropic penalty as a result of binding compared with the more flexible C-2 piperazine functionality. Additionally, the diazaspiro[3.3]heptane functionality is more basic than its piperazine counterpart,43 which might potentiate a stronger charge–charge interaction with Asp887.
In addition to improved binding affinities, compounds featuring the diazaspiro[3.3]heptane moiety demonstrated improved ALogP and LipE values compared to previously examined SOS1 ligands. Compound lipophilicity was further reduced by appending an amine to the para-position of the pyrimidine, as exemplified by 62. Removal of the 3’- and 5’-methyl substituents, as in 63, generated the best ALogP and LipE metrics for this series of SOS1 agonists, albeit with a binding affinity ~3-fold worse than the parent molecule 62, as might be expected from the decreased occupancy of the hydrophobic pocket beneath Phe890. As a result of this extensive SAR campaign, these analogs represent the best binders to SOS1 reported to date, highlighted by pyrazole 65 with a binding affinity of 9 nM.
Cellular Analysis.
With compounds that bind tightly to SOS1 in hand, we sought to assess whether this series of benzimidazole-derived molecules also elicited the previously observed, characteristic biphasic ERK modulation and RAS-GTP activation in cancer cells.25,26 To assess the effects on pERK signaling, an In-Cell Western (ICW) assay was used to evaluate cellular pERK1/2T202/Y204 levels relative to total ERK1/2 levels in response to treatment with varying concentrations of compounds (Figure 6). Two different cell lines—HeLa (WT KRAS) and NCI-H727 (G12V KRAS)—were treated with compounds at concentrations ranging from 1.5 to 100 μM for 30 min. The levels of pERK1/2T202/Y204 were normalized to the levels of total ERK1/2 protein and the DMSO vehicle control. Accordingly, the ICW EC50 values reported in Table 6 represent the half maximal concentration of compound required to elicit peak induction of pERK1/2T202/Y204 levels. Thus, a lower value is considered indicative of a more potent SOS1 agonist. Guided by our previous mechanistic observations,27 we hypothesized that potent SOS1 agonists would typically demonstrate biphasic modulation of pERK1/2T202/Y204. This is characterized by an increase in pERK1/2T202/Y204 levels at lower doses of compound followed by a decrease below baseline in pERK1/2T202/Y204 levels at higher doses (Figure 6A). Observing an increase in pERK1/2T202/Y204 levels with no corresponding decrease over the concentration range of compound tested (Figure 6B) was considered to indicate an insufficiently potent SOS1 agonist. Alongside EC50 values, Table 6 also indicates whether biphasic modulation of pERK1/2T202/Y204 was elicited by each derivative. A selection of molecules featuring diverse chemical motifs and binding affinities, spanning five orders of magnitude, were examined by ICW to evaluate whether any inferences could be drawn between structural features, binding affinities, and phenotypic cellular responses.
Figure 6.
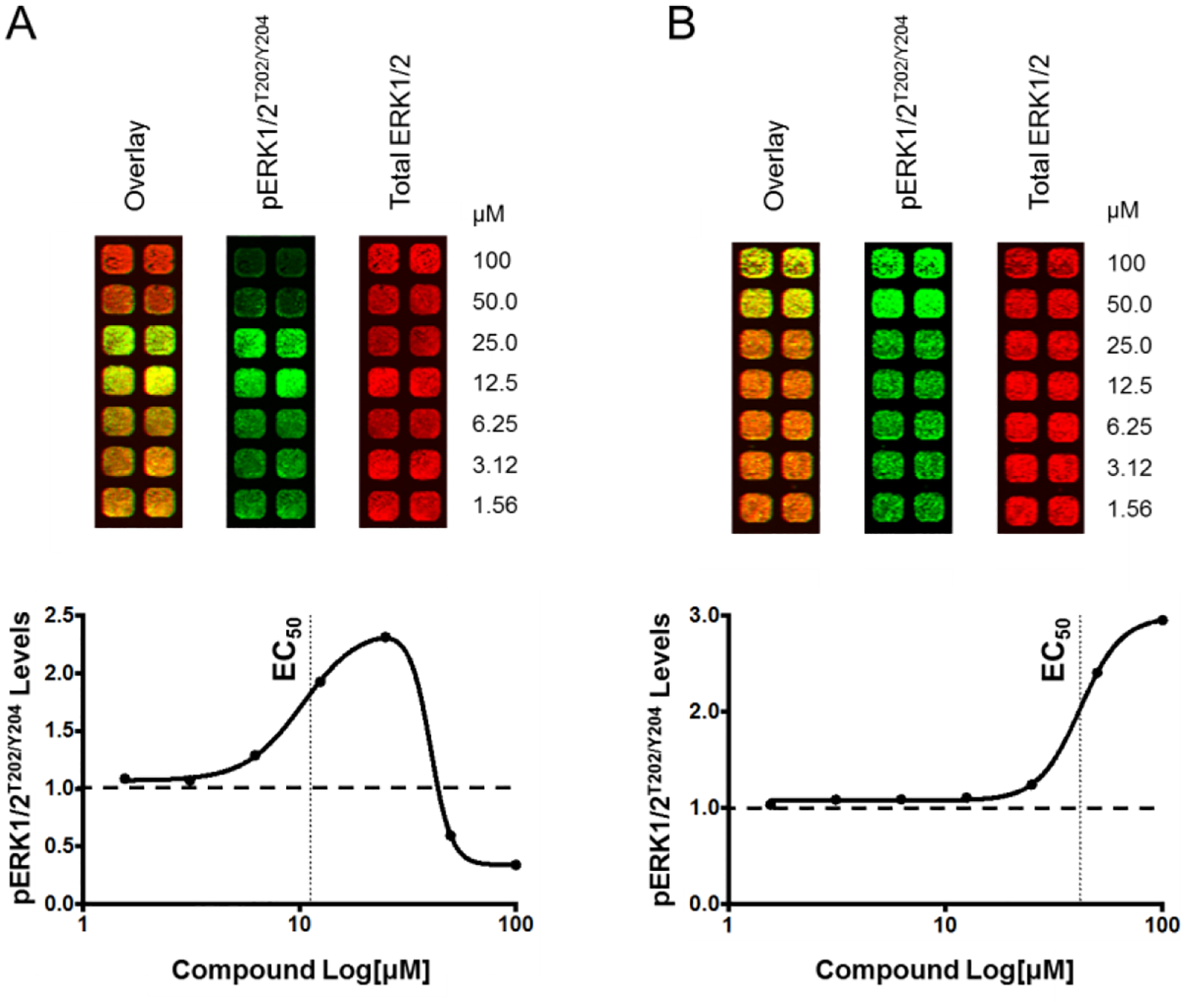
Representative ICW data for benzimidazole compounds that elicit (A) biphasic modulation of pERK1/2T202/Y204 protein levels, or (B) only an increase in pERK1/2T202/Y204 protein levels. The green channel of the LI-COR infrared scanner was used to detect pERK1/2T202/Y204 protein levels. In the same well, the red channel of the LI-COR infrared scanner was used to detect total ERK1/2 protein levels. The graphs represent the quantified immunofluorescence data, where pERK1/2T202/Y204 levels were normalized to total ERK1/2 levels from the same well and to the vehicle control treated well. The vertical black dotted line indicates how EC50 values were determined, and the horizontal black dotted line indicates baseline pERK1/2T202/Y204 signal. Each ICW experiment was conducted three independent times, and in duplicate on each plate.
Table 6.
In-Cell Western Analysis of pERK1/2T202/Y204 Levels
| Compd | 47 | 64 | 42 | 62 | 56 | 58 | 55 | 38 | 60 | 59 | 63 | 65 | 29 | 23 | 10 | 3 | 61 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HeLa ICW EC50 (μM) | 3.2 ± 0.1 | 5.9 ± 0.4 | 6.0 ± 1.2 | 7.8 ± 1.9 | 8.8 ± 2.9 | 9.0 ± 2.4 | 10.2 ± 4.1 | 11.0 ± 5.0 | 11.4 ± 3.8 | 14.0 ± 1.5 | 14.0 ± 0.8 | 14.1 ± 1.3 | 14.5 ± 1.8 | 21.0 ± 7.6 | 21.3 ± 10.0 | 32.2 ± 11.0 | 33.2 ± 10.9 |
| H727 ICW EC50 (μM) | 7.3 ± 3.1 | 10.0 ± 3.2 | 7.3 ± 1.5 | 13.4 ± 2.6 | 13.9 ± 0.2 | 15.6 ± 5.5 | 14.5 ± 1.0 | 36.8 ± 4.7 | 15.7 ± 2.2 | 19.7 ± 6.4 | 27.5 ± 2.8 | 21.1 ± 6.1 | 21.2 ± 8.4 | 51.4 ± 16.8 | 29.1 ± 1.8 | 18.7 ± 5.8 | 42.5 ± 15.1 |
| Kd (μM) | 0.14 | 0.049 | 0.14 | 0.012 | 0.10 | <0.10 | 0.11 | 0.12 | 0.013 | 0.012 | 0.036 | 0.009 | 0.46 | 14.8 | 2.59 | 55.0 | 0.18 |
| HeLa biphasic | + | + | + | + | + | + | + | + | + | + | + | + | + | − | + | − | − |
| H727 biphasic | + | + | + | + | + | + | + | − | + | + | − | + | + | − | + | + | − |
ICW EC50 measurements are reported as the mean ± SD of three individual experiments, each conducted in duplicate. Kd measurements are reported as the mean of two or more independent experiments, each conducted in duplicate. A “+” is used to indicate biphasic modulation of pERK1/2T202/Y204, as characterized by an increase in pERK1/2T202/Y204 levels at lower concentrations and a decrease in pERK1/2T202/Y204 levels below baseline at higher concentrations. A “–” sign indicates that compound treatment caused an increase in pERK1/2T202/Y204 levels, but no subsequent decrease below baseline over the concentrations tested. All pERK1/2T202/Y204 data are normalized to the DMSO vehicle control and to the total ERK1/2 protein levels from the same well.
The majority of compounds examined by ICW exhibited the characteristic biphasic modulation that we have come to expect from potent activators of SOS1-mediated nucleotide exchange, based on prior work.25–27 On the other hand, less potent compounds with higher ICW EC50 values were less likely to elicit biphasic modulation of ERK1/2 phosphorylation within the concentration range tested. We hypothesized that compounds with higher binding affinities would modulate pERK1/2T202/Y204 signaling with greater potency. Indeed, analogs with weaker binding affinities, such as 3, 23, and 10, all modulated pERKT202/Y204 signaling with low potency. However, compounds with higher binding affinities did not necessarily modulate pERK1/2T202/Y204 signaling with the greatest potencies. This is best illustrated by comparing the data for 47 and 65. Although the Kd value for 47 is an order of magnitude higher than that of 65, the ICW EC50 values for 47 were 3–4-fold better than those of 65 in both cell lines. These data suggest that other factors beyond only SOS1 binding affinity can influence pERK1/2T202/Y204 modulation in cells. These factors may include the physical properties of the compounds such as lipophilicity and solubility or issues with membrane permeability and/or plasma protein binding.44,45 Indeed, 4-N-methylpiperazino 64 displayed superior solubility at pH 6.8 (>110 μg/mL) relative to its direct comparator 4-pyrimidino 60 (3 μg/mL). Furthermore, several compounds, while capable of showing adequate passive permeability, appear to suffer from excessive efflux (Supplementary Table S2). In addition to these characteristics, cellular considerations such as protein localization,46 post-translational modifications,27,47 and additional layers of allosteric regulation48 that are not accounted for in a biochemical system could play a role in a disconnect between binding affinity to SOS1 and cellular activity.
Finally, to assess whether compounds from the benzimidazole series activate RAS signaling in cells we measured RAS-GTP levels and accompanying pERKT202/Y204 levels in HeLa cells (Figure 7). We chose to test matched sets of compounds that featured the same substituents at the 4-position of the benzimidazole scaffold, but between these sets, differed in their ability to modulate pERK1/2T202/Y204 signaling as measured by ICW. Regardless of the increase in binding affinity afforded by the diazaspiro[3.3]heptane substituent at the 2-position, an N-methylpiperazine substituent at the 4-position, as in 42 and 64, resulted in enhanced potency by pERK1/2T202/Y204 ICW, relative to the ortho-methylpyrimidine of 55, 60, and 61, which modulated pERK1/2T202/Y204 signaling less potently.
Figure 7.
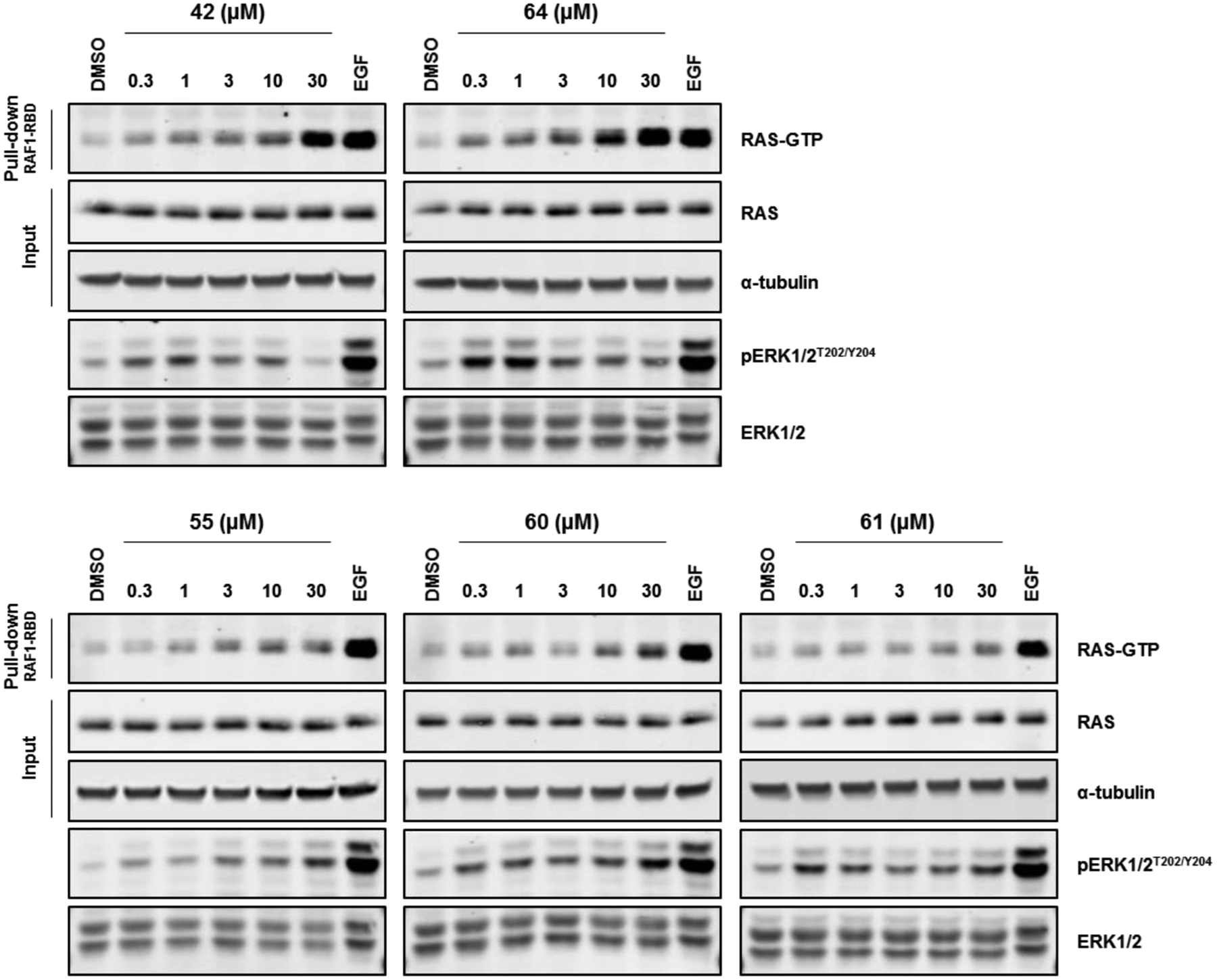
RAS-GTP levels and corresponding pERK1/2T202/Y204 levels from HeLa cells that were treated for 10 min with up to 30 μM of compound 42, 64, 55, 60, or 61. EGF (50 ng/mL for 5 min) was used as a positive control for pathway stimulation. Data are representative of two independent experiments.
Treatment with compounds 42 or 64 caused substantial increases in RAS-GTP levels at 10–30 μM, and induced small increases in RAS-GTP at concentrations as low as 0.3 μM. On the other hand, only modest increases in RAS-GTP levels were observed with 55, 60, and 61 over the same range of dosage concentrations. These findings are consistent with ICW pERK1/2T202/Y204 EC50 data, which identified compounds 42 and 64 as amongst the most potent compounds tested in cells. Our laboratory has typically assessed RAS-GTP levels after 30 min of compound treatment with all previous chemical series of SOS1 agonists that we have discovered, and generally, treatment concentrations up to 100 μM were required to observe notable changes in RAS-GTP levels.25,26 However, compounds 42 and 64 activated RAS-GTP and caused biphasic modulation of pERK1/2T202/Y204 signaling with such high potencies that a lower maximum concentration of 30 μM and only 10 min of treatment were required to sufficiently visualize their effects. Furthermore, 64 elicited robust activation of RAS-GTP at lower concentrations than 42, which may be a result of the higher SOS1 binding affinity of 64 compared to 42. Overall, compared to the other series of SOS1 agonists discovered in our lab, advanced examples from this series of benzimidazole-derived compounds modulate RAS-GTP and associated downstream signaling with the greatest potency that we have observed, to date.
Compound Synthesis.
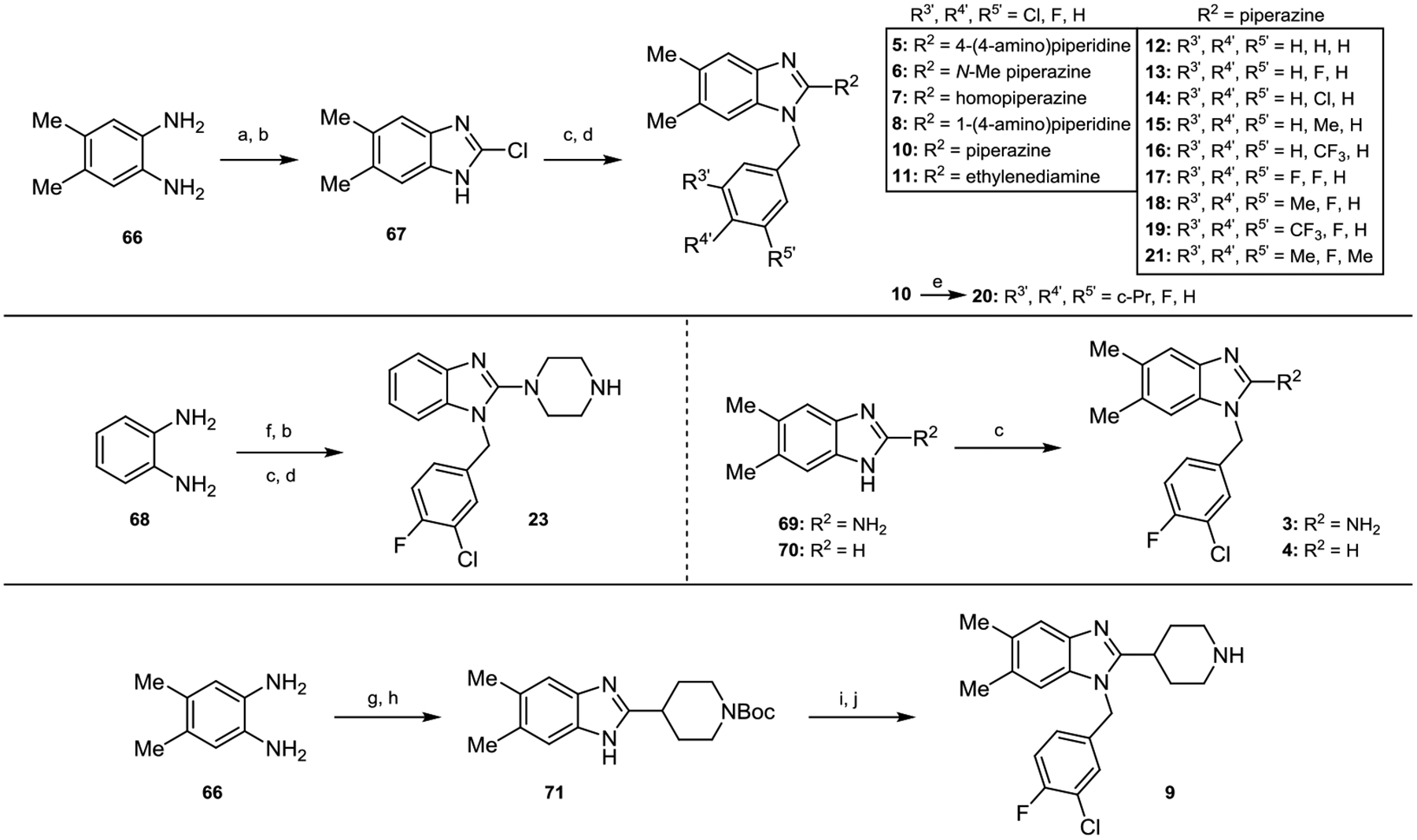
To access various benzimidazoles with 5,6-dimethyl moieties, commercially available bisaniline derivative 66 was acylated with 1,1-carbonyldiimidazole (CDI) and then chlorinated in refluxing POCl3 to afford 2-chloro benzimidazole 67 (Scheme 1). From there, alkylation of the benzimidazole nitrogen atom with a variety of substituted benzyl bromides and subsequent displacement of the 2-chlorine atom with an array of amines via an SNAr-type reaction furnished analogs 5–8, 10–19 and 21. The 5,6H-derivative 23 was constructed in a similar manner, beginning from commercially available benzene-1,2-diamine (68). Compound 20 was accessed via a Suzuki coupling between 10 and cyclopropylboronic acid. 2-Amino and 2H-derivatives 3 and 4 were prepared by alkylation of commercially available compounds 69 and 70, respectively, with 3-chloro-4-fluorobenzyl bromide. Lastly, 66 was condensed with piperidine-4-carboxylic acid followed by Boc-protection of the piperidine nitrogen atom to afford intermediate 71. Subsequent benzylation of the benzimidazole nitrogen atom and Boc-removal furnished analog 9.
Scheme 1.
Synthesis of Various Benzimidazole Derivatives.a
aReaction conditions: (a) CDI, DMAP, CH2Cl2, rt. (b) POCl3, 90 °C. (c) benzyl bromide derivative, K2CO3, DMF, rt. (d) diamine, DMA, 165 °C in microwave; TFA, CH2Cl2, rt when Boc removal necessary. (e) K3PO4, cyclopropylboronic acid, Pd(OAc)2, PCy3·HBF4, DME/H2O, 90 °C. (f) urea, xylenes, 130 °C. (g) piperidine-4-carboxylic acid, 4 M HCl(aq), 105 °C. (h) Boc2O, Et3N, THF/DMF, 35 °C. (i) NaH, 3-chloro-4-fluorobenzyl bromide, DMF, rt. (j) TFA, CH2Cl2, rt.
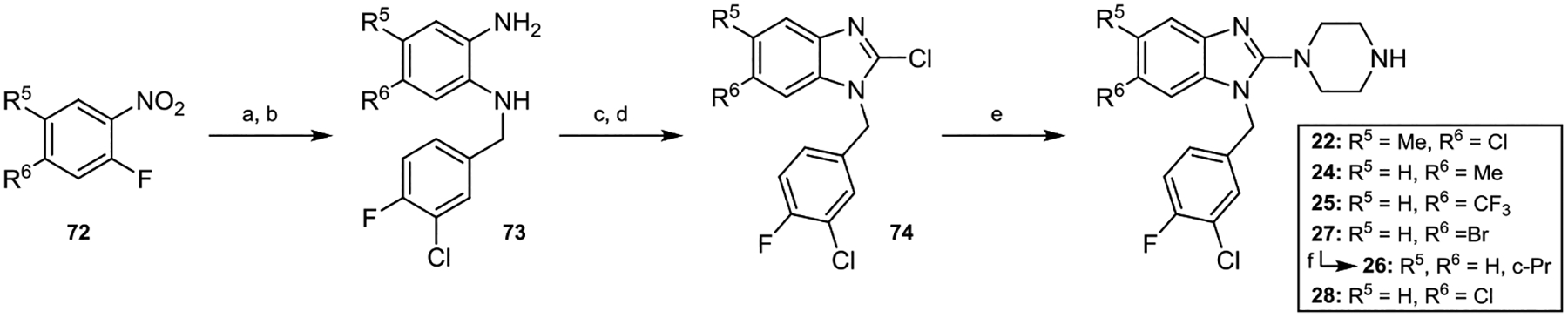
In order to synthesize derivatives with varying 5,6-substitution patterns, commercially available 1-fluoro-2-nitrobenzenes, represented by 72, were treated with 3-chloro-4-fluorobenzylamine via an SNAr-type reaction and then reduced using Zn/HCl to provide anilines 73 (Scheme 2). These intermediates were acylated using CDI and then chlorinated by heating with POCl3 to form 2-chlorobenzimidazoles 74. Substitution of the 2-chlorine atom with piperazine afforded derivatives 22, 24, 25, 27, and 28. Compound 26 was accessed via a Suzuki coupling between 27 and cyclopropylboronic acid.
Scheme 2.
Synthesis of 5,6-Substituted Benzimidazole Derivatives.a
aReaction conditions: (a) i-Pr2NEt, 3-chloro-4-fluorobenzylamine, NMP, rt. (b) Zn°, 12 M HCl, EtOH/CH2Cl2, rt. (c) CDI, DMAP, CH2Cl2, rt. (d) POCl3, 100 °C. (e) piperazine, DMA, 165 °C in microwave. (f) K3PO4, PdCl2dppf·CH2Cl2, cyclopropylboronic acid, dioxane, 100 °C.
Compounds with various 4-position substituents were constructed by treating 3-bromo-5-chlorobenzene-1,2-diamine 75 with CDI and then POCl3 to furnish 2-chlorobenzimidazole intermediate 76 (Scheme 3). Benzylation with 4-fluoro-3,5-dimethylbenzyl bromide or 4-fluorobenzyl bromide and successive displacement of the 2-chlorine atom with piperazine, 1-Boc-piperazine or 1-Boc-2,6-diazaspiro[3.3]heptane provided 4-bromo compounds 77, 78, 79, or 80. These intermediates were functionalized at C-4 by various metal catalyzed C–C and C–N bond forming reactions, or reduced via palladium catalyzed hydrogenolysis to afford analogs 29–51 and 54–65 after exposure to trifluoroacetic acid to effect N-Boc deprotection.
Scheme 3.
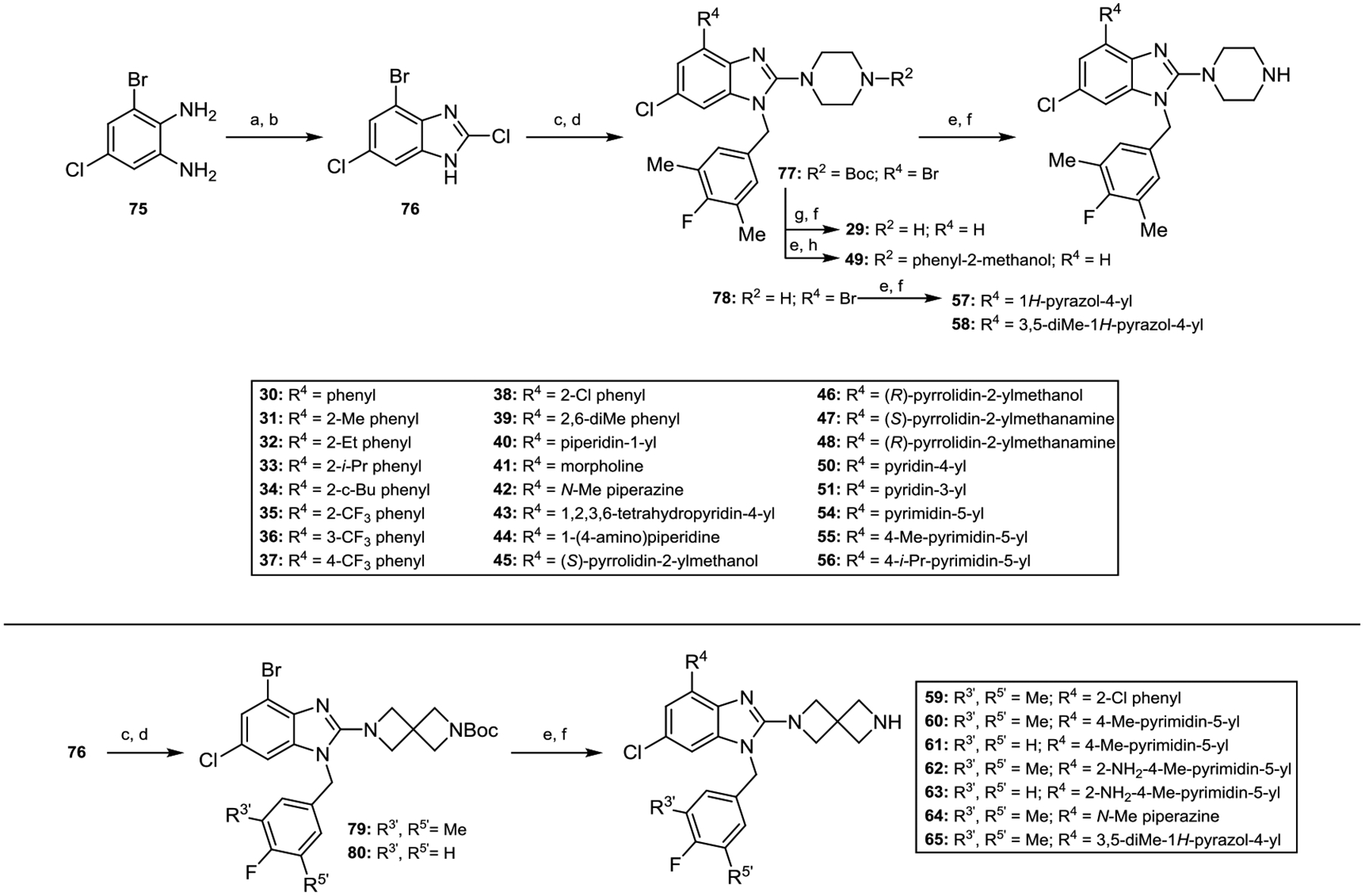
Synthesis of 4-Substituted Benzimidazole Derivatives.a
aReaction conditions: (a) CDI, DMAP, CH2Cl2, rt. (b) POCl3, 110 °C. (c) 4-fluoro-3,5-dimethylbenzyl bromide or 4-fluorobenzyl bromide, K2CO3, DMF, rt. (d) diamine, K2CO3 or i-Pr2NEt, DMF or DMSO, 80–100 °C. (e) Pd cat. C–C or C–N coupling. (f) TFA, CH2Cl2, rt. (g) 10% Pd/C, H2, AcOH, i-PrOH, rt. (h) NaBH4, MeOH, rt; 4 M HCl in dioxane.
CONCLUSION
A set of early hit molecules based on a benzimidazole scaffold were found to activate SOS1-mediated nucleotide exchange on RAS. The SAR of these compounds was systematically investigated using two different biochemical assays and X-ray co-crystal structures of select SOS1 ligands bound to the RAS:SOS1:RAS complex were used as guides for analog design. Through these studies, several novel derivatives were found to exhibit low double-digit nanomolar affinity for SOS1, which represent the tightest binders of SOS1 reported to date. These compounds also elicited increases of RAS-GTP levels in cells with the highest potency observed from any SOS1 ligands yet tested, while inducing a robust biphasic response in downstream pERK1/2T202/Y204 signaling that is characteristic of SOS1 agonists. These observations are in agreement with our proposed mechanism that compound-mediated agonism of SOS1 can modulate RAS signaling via negative feedback on SOS1 by active pERK1/2T202/Y204. The benzimidazole-derived compounds described here and future derivatives may serve as chemical tools for studying modulation of RAS signaling via binding to SOS1 and could inform future discoveries regarding small molecule therapeutics for the treatment of RAS-driven malignancies.
EXPERIMENTAL
General Procedures.
All chemical reagents and reaction solvents were purchased from commercial suppliers and used as received. Proton nuclear magnetic resonance (1H NMR) spectra were recorded at either 400 MHz or 600 MHz on a Bruker spectrometer. For 1H NMR spectra, chemical shifts are reported in parts per million (ppm) and are reported relative to residual non-deuterated solvent signals. Coupling constants are reported in hertz (Hz). The following abbreviations (or a combination, thereof) are used to describe splitting patterns: s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; comp, overlapping multiplets of non-magnetically equivalent protons; br, broad. All final compounds were of 95% purity or higher, unless otherwise noted, as measured by analytical reversed-phase HPLC. Analytical HPLC was performed on an Agilent 1200 series system with UV detection at 214 and 254 nm, along with evaporative light scattering detection (ELSD). Low-resolution mass spectra were obtained on an Agilent 6140 mass spectrometer with electrospray ionization (ESI). LC-MS experiments were performed with the following parameters: Phenomenex Kinetex 2.6 μm XB-C18 100 Å, LC column 50 × 2.1 mm; 2 min gradient, 5%–95% MeCN in H2O, and 0.1% TFA or 0.1% formic acid. Analytical thin layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel. TLC plates were visualized with UV light and iodine. Silica gel chromatography was performed using a Teledyne ISCO Combiflash® Rf system, eluting with varying concentrations of EtOAc in hexanes or MeOH in CH2Cl2. Preparative reversed-phase HPLC was performed on a Gilson instrument equipped with a Phenomenex Kinetex C18 column, using varying concentrations of MeCN in H2O, and 0.1% TFA. Compounds 1 and 2 were purchased from ChemDiv and used as received. Compounds 52 and 53 were purchased from Viva Biotech (Shanghai, China) and used as received. Starting materials 66, 68, 69, 70, and 1-fluoro-2-nitrobenzene derivatives, represented by 72, were purchased from commercial suppliers and used as received. Compound 75 was prepared according to a previously reported protocol.49
General Procedure A: Synthesis of 5–8, 10–19, 21, and 23.
Step 1: A mixture of 67 (1.0 equiv), an appropriate benzyl bromide derivative (1.1 equiv), and K2CO3 (2.0 equiv) in DMF (0.25 M) was stirred at rt for 16 h. The mixture was diluted with EtOAc, poured into a 50% sat. brine solution, and extracted with EtOAc (3x). The combined organics were washed with a 50% sat. brine solution, a sat. brine solution, dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography. Step 2: The material obtained from step 1 (1.0 equiv) was mixed with an appropriate diamine (10.0 equiv) in N,N-dimethylacetamide (0.2 M) and heated to 165 °C in a microwave reactor for 45 min. The mixture was cooled to rt, diluted with EtOAc, poured into a 50% sat. brine solution, and extracted with EtOAc (3x). The combined organics were washed with a 50% sat. brine solution, a sat. brine solution, dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by reversed-phase preparative HPLC and the pure fractions were concentrated under a stream of warm air to afford the target compound as a TFA salt. Alternatively, the free-base was obtained by neutralizing the pure HPLC fractions with solid Na2CO3, extraction with CH2Cl2 (3x), drying over Na2SO4, and concentrating under reduced pressure.
General Procedure B: Synthesis of 22 and 24–28.
Step 1: A mixture of an appropriately substituted 1-fluoro-2-nitrobenzene derivative (72, 1.0 equiv) and N,N-diisopropylethylamine (1.5 equiv) in N-methyl-2-pyrrolidinone (0.5 M) was charged with 3-chloro-4-fluorobenzylamine (1.2 equiv) and stirred at rt for 16 h. The reaction mixture was poured into ice cold brine and stirred for 5 min. The resultant yellow precipitate was collected via filtration, rinsed with water, and dried under reduced pressure. Step 2: The crude material obtained from step 1 was suspended in a mixture of EtOH/CH2Cl2/12 M HCl (3:1:1 v/v/v, 0.2 M) and Zn dust (10.0 equiv) was added. After the reaction was complete, as indicated by LC-MS, the reaction mixture was filtered. Water was added to the filtrate and the suspension was extracted with CH2Cl2 (3x), dried (Na2SO4), and concentrated. Step 3: The crude residue obtained from step 2 was re-suspended in CH2Cl2 (0.3 M), charged with DMAP (0.1 eq) and CDI (2.0 eq), and stirred at rt. After 2 h, 1 M HCl was added and the mixture was stirred rapidly. The resultant solid was collected via filtration, rinsed with water, rinsed with CH2Cl2, and dried under reduced pressure. Step 4: The crude solid obtained from step 3 was suspended in POCl3 (1.0 M) and heated to 100 °C. After the reaction was complete, as indicated by LC-MS, the mixture was cooled to 0 °C and poured slowly into water. After all the POCl3 was quenched, the mixture was slowly neutralized with solid NaOH. The resulting precipitate was collected via filtration, rinsed with water, and dried under reduced pressure. Step 5: The crude solid obtained from step 4 and piperazine (10.0 equiv) were dissolved in a mixture of N,N-dimethylacetamide (0.2 M) and heated to 165 °C in a microwave reactor for 45 min. The mixture was cooled to rt, diluted with EtOAc, poured into a 50% sat. brine solution, and extracted with EtOAc (3x). The combined organics were washed with a 50% sat. brine solution, a sat. brine solution, dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by reversed-phase preparative HPLC and the pure fractions were concentrated under a stream of warm air to afford the target compound as a TFA salt.
General Procedure C: Synthesis of 30–39, 43, 54–63, and 65.
Step 1: A mixture of an appropriate boronic acid or boronic acid pinacol ester (1.5 equiv), PdCl2dppf·CH2Cl2 (0.1 equiv), K2CO3 (3.0 equiv), and 77, 78, 79, or 80 (1.0 equiv) under Ar(g) was charged with degassed DMF/ethanol (4:1 v/v, 0.35 M) and heated to a temperature of 90 °C. After 18 h, the reaction mixture was cooled to rt, diluted with sat. aqueous ammonium chloride, and extracted with CH2Cl2 (3x). The organic phases were combined, dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography. Step 2: The compound obtained from step 1 (1.0 equiv) was dissolved in CH2Cl2 (0.05 M) and stirred at rt. TFA (0.2 M) was added and the mixture was stirred for 2 h before being concentrated under reduced pressure. The residue was purified by reversed-phase preparative HPLC and the pure fractions were concentrated under a stream of warm air to afford the target compound as a TFA salt. Alternatively, the free-base was obtained by neutralizing the pure HPLC fractions with solid Na2CO3, extraction with CH2Cl2 (3x), drying over Na2SO4, and concentrating under reduced pressure.
General procedure D: Synthesis of 40–42, 44–48, and 64.
A mixture of amine (2.0 equiv), Pd(OAc)2 (0.1 equiv), BINAP (0.2 equiv), and K3PO4 (3.0 equiv) under Ar(g) was charged with a solution of either 77 or 79 (1.0 equiv) in degassed toluene (0.1 M) and heated to 100 °C with vigorous stirring. Once the starting material was consumed, as indicated by LC-MS, the mixture was diluted with EtOAc, filtered over diatomaceous earth, and concentrated under reduced pressure. The crude residue was dissolved in a mixture of TFA and CH2Cl2 (1:2 v/v, 0.05 M) and stirred at rt for 1 h. The reaction mixture was concentrated under reduced pressure and purified by reversed-phase preparative HPLC. The pure fractions were concentrated under a stream of warm air to afford the target compound as a TFA salt. Alternatively, the free-base was obtained by neutralizing the pure HPLC fractions with solid Na2CO3, extraction with CH2Cl2 (3x), drying over Na2SO4, and concentrating under reduced pressure.
General procedure E: Synthesis of 50 and 51.
Step 1: A mixture of 77 (1.0 equiv), an appropriate potassium pyridine trifluoroborate (1.1 equiv), Pd(OAc)2 (0.1 equiv), RuPhos (0.2 equiv), and Na2CO3 (2.0 equiv) under Ar(g) was charged with degassed ethanol (0.2 M). The reaction mixture was heated to 80 °C for 48 h. The reaction mixture was cooled to rt, diluted with sat. aqueous ammonium chloride, and extracted with dichloromethane (3x). The organic phases were combined, dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by silica gel chromatography. Step 2: The compound obtained from step 1 (1.0 equiv) was dissolved in CH2Cl2 (0.05 M) and TFA (0.2 M) was added. The mixture was stirred for 2 h before being concentrated under reduced pressure. The crude residue was purified by reversed-phase preparative HPLC and the pure fractions were concentrated under a stream of warm air to afford the target compound as a TFA salt.
1-(3-Chloro-4-fluorobenzyl)-5,6-dimethyl-1H-benzo[d]imidazol-2-amine (3 as TFA salt).
A mixture of 69 (50 mg, 0.31 mmol), 3-chloro-4-fluorobenzyl bromide (50 μL, 0.37 mmol), and NaH (60% dispersion in mineral oil, 25 mg, 0.63 mmol) in EtOH (4 mL) was stirred at 60 °C until starting material was completely consumed, as indicated by LC-MS. The mixture was cooled to rt, acidified with a 10% HCl solution, extracted witch CH2Cl2 (3x), dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by reversed-phase preparative HPLC and the pure fractions were concentrated under a stream of warm air to afford 8 mg of the title compound as the corresponding TFA salt (6% yield). 1H NMR (400 MHz, CD3OD): δ 7.41 (dd, J = 6.8, 2.2 Hz, 1H), 7.25 (t, J = 8.8 Hz, 1H), 7.22–7.16 (comp, 2H), 7.14 (s, 1H), 5.35 (s, 2H), 2.34 (s, 3H), 2.32 (s, 3H). MS (ESI) m/z = 304.0 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-5,6-dimethyl-1H-benzo[d]imidazole (4 as TFA salt).
A mixture of 70 (50 mg, 0.34 mmol), 3-chloro-4-fluorobenzyl bromide (50 μL, 0.38 mmol), and K2CO3 (71 mg, 0.51 mmol) in DMF (3 mL) was stirred at rt for 1 h. The mixture was acidified with a 10% HCl solution, diluted with EtOAc, poured into a 50% sat. brine solution, and extracted with EtOAc (3x). The combined organics were washed with a 50% sat. brine solution, a sat. brine solution, dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by reversed-phase preparative HPLC and the pure fractions were concentrated under a stream of warm air to afford 15 mg of the title compound as the corresponding TFA salt (11%). 1H NMR (400 MHz, CD3OD): δ 9.28 (s, 1H), 7.64–7.59 (comp, 2H), 7.58 (br s, 1H), 7.40 (dq, J = 8.5, 2.2 Hz, 1H), 7.29 (t, J = 8.8 Hz, 1H), 5.66 (s, 2H), 2.44 (s, 3H), 2.43 (s, 3H). MS (ESI) m/z = 289.0 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-5,6-dimethyl-N-(piperidin-4-yl)-1H-benzo[d]imidazol-2-amine (5 as TFA salt).
The title compound was prepared from 3-chloro-4-fluorobenzyl bromide and tert-butyl 4-aminopiperidine-1-carboxylate in similar fashion to general procedure A with a slight modification. Prior to purification as described in step 2 of general procedure A, the crude residue obtained was dissolved in a solution of TFA (0.2 M) in CH2Cl2 and stirred at ambient temperature for 2 h before being concentrated under reduced pressure. Purification as described in step 2 of general procedure A afforded the title compound as the corresponding TFA salt in 7% yield over three steps. 1H NMR (400 MHz, CD3OD): δ 7.44 (dd, J = 6.8, 2.2 Hz, 1H), 7.35 (s, 1H), 7.29 (t, J = 8.7 Hz, 1H), 7.20 (ddd, J = 8.5, 4.4, 2.3 Hz, 1H), 7.14 (s, 1H), 5.41 (s, 2H), 3.79 (app d, J = 13.0 Hz, 2H), 3.46–3.33 (comp, 3H), 2.38 (s, 3H), 2.33 (s, 3H), 2.13 (app d, J = 12.6 Hz, 2H), 1.81 (dq, J = 12.3, 4.2 Hz, 2H). MS (ESI) m/z = 387.0 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-5,6-dimethyl-2-(4-methylpiperazin-1-yl)-1H-benzo[d]imidazole (6 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 3-chloro-4-fluorobenzyl bromide and N-methyl piperazine in similar fashion to general procedure A in 38% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.38 (s, 1H), 7.25 (m, 2H), 7.14 (m, 1H), 7.09 (s, 1H), 5.40 (s, 2H), 3.59–3.52 (comp, 4H), 3.51–3.45 (comp, 4H), 2.99 (s, 3H), 2.38 (s, 3H), 2.34 (s, 3H). MS (ESI) m/z = 387.1 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-2-(1,4-diazepan-1-yl)-5,6-dimethyl-1H-benzo[d]imidazole (7 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 3-chloro-4-fluorobenzyl bromide and homopiperazine in similar fashion to general procedure A in 47% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.48 (d, J = 6.6 Hz, 1H), 7.36 (s, 1H), 7.32 (t, J = 8.7 Hz, 1H), 7.26–7.21 (m, 1H), 7.19 (s, 1H), 5.51 (s, 2H), 3.99 (t, J = 2.3 Hz, 2H), 3.83 (t, J = 2.3 Hz, 2H), 3.55 (t, J = 4.8 Hz, 2H), 3.43 (t, J = 5.4 Hz, 2H), 2.41 (s, 3H), 2.36 (s, 3H), 2.26 (t, J = 4.9 Hz, 2H). MS (ESI) m/z = 387.1 [M+H]+.
1-(1-(3-Chloro-4-fluorobenzyl)-5,6-dimethyl-1H-benzo[d]imidazol-2-yl)piperidin-4-amine (8 as free-base).
The title compound was prepared from 3-chloro-4-fluorobenzyl bromide and tert-butyl piperidin-4-ylcarbamate in similar fashion to general procedure A with a slight modification. Prior to purification as described in step 2 of general procedure A, the crude residue obtained was dissolved in a solution of TFA (0.2 M) in CH2Cl2 and stirred at ambient temperature for 2 h before being concentrated under reduced pressure. Purification as described in step 2 of general procedure A afforded the title compound as the free-base in 56% yield over three steps. 1H NMR (400 MHz, DMSO-d6): δ 7.41–7.31 (comp, 2H), 7.20 (s, 1H), 7.09–7.04 (m, 1H), 6.99 (s, 1H), 5.20 (s, 2H), 3.17 (s, 1H), 2.87 (t, J = 11.1 Hz, 2H), 2.76–2.66 (m, 1H), 2.24 (s, 3H), 2.21 (s, 3H), 1.78–1.30 (comp, 5H). MS (ESI) m/z = 387.1 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-5,6-dimethyl-2-(piperidin-4-yl)-1H-benzo[d]imidazole (9 as TFA salt).
A mixture of compound 66 (375 mg, 2.75 mmol) and piperidine-4-carboxylic acid (1.04 g, 8.26 mmol) in HCl(aq) (4 M, 10 mL), was heated at 105 °C for 18 h. The reaction mixture was cooled to rt and neutralized by slowly adding solid NaOH. The resulting precipitate was filtered, washed with water, and dried by pulling air through the solid to afford 400 mg of crude 5,6-dimethyl-2-(piperidin-4-yl)-1H-benzo[d]imidazole. This material (200 mg, 0.873 mmol) was then dissolved in a THF/DMF mixture (1:1 v/v, 10 mL), charged with Boc2O (335 mg, 1.53 mmol) and Et3N (160 μL, 1.15 mmol), and heated at 35 °C for 2 h. The reaction mixture was poured into 50% sat. brine (20 mL) and extracted with EtOAc (3×10 mL). The combined extracts were washed with 50% sat. brine (10 mL) followed by sat. brine (10 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography to afford tert-butyl 4-(5,6-dimethyl-1H-benzo[d]imidazol-2-yl)piperidine-1-carboxylate (80 mg, 0.24 mmol). This material (80 mg, 0.24 mmol) was subsequently dissolved in DMF (4 mL) and charged with NaH (60% dispersion in mineral oil, 61 mg, 1.5 mmol) followed by 3-chloro-4-fluorobenzyl bromide (154 uL, 1.14 mmol). After 1 h, the mixture was poured into 50% sat. brine (20 mL) and extracted with EtOAc (3x). The combined extracts were washed with 50% sat. brine (10 mL) followed by sat. brine (10 mL), dried (Na2SO4), and concentrated. The crude residue was re-dissolved in a mixture of CH2Cl2 (1 mL) and TFA (2 mL). Upon complete conversion of starting material, as indicated by LC-MS, the mixture was concentrated and purified via reversed-phase preparative HPLC. The resultant pure fractions were concentrated under a stream of warm air to afford 37 mg of the title compound as the corresponding TFA salt (41% yield). 1H NMR (400 MHz, CDCl3): δ 7.60 (s, 1H), 7.46 (s, 1H), 7.46–7.43 (comp, 1H), 7.28 (t, J = 8.8 Hz, 1H), 7.17–7.12 (m, 1H), 5.76 (s, 2H), 3.79–3.67 (m, 1H), 3.59 (d, J = 13.2 Hz, 2H), 3.25–3.13 (m, 2H), 2.46 (s, 3H), 2.43 (s, 3H), 2.24–2.16 (m, 4H). MS (ESI) m/z = 372.1 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-5,6-dimethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (10 as free-base).
The title compound was prepared as the free-base from 3-chloro-4-fluorobenzyl bromide and piperazine in similar fashion to general procedure A in 27% yield over two steps. 1H NMR (400 MHz, DMSO-d6): δ 7.40 (dd, J = 7.1, 2.0 Hz, 1H), 7.36 (t, J = 9.0 Hz, 1H), 7.22 (s, 1H), 7.06 (ddd, J = 8.6, 4.5, 2.1 Hz, 1H), 7.00 (s, 1H), 5.22 (s, 2H), 3.04–3.02 (comp, 4H), 2.82–2.79 (comp, 4H), 2.24 (s, 3H), 2.22 (s, 3H). MS (ESI) m/z = 372.9 [M+H]+.
N1-(1-(3-Chloro-4-fluorobenzyl)-5,6-dimethyl-1H-benzo[d]imidazol-2-yl)ethane-1,2-diamine (11 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 3-chloro-4-fluorobenzyl bromide and ethane-1,2-diamine in similar fashion to general procedure A in 21% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.43 (dd, J = 7.0, 1.9 Hz, 1H), 7.29 (s, 1H), 7.27–7.17 (comp, 2H), 7.13 (s, 1H), 5.39 (s, 2H), 3.85 (t, J = 6.0 Hz, 2H), 3.33 (m, 2H), 2.36 (s, 3H), 2.32 (s, 3H). MS (ESI) m/z = 347.2 [M+H]+.
1-Benzyl-5,6-dimethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (12 as TFA salt).
The title compound was prepared as the corresponding TFA salt from benzyl bromide and piperazine in similar fashion to general procedure A in 39% yield over two steps. 1H NMR (400 MHz, DMSO-d6): δ 8.97 (s, 2H), 7.39–7.27 (comp, 4H), 7.23–7.18 (comp, 2H), 7.09 (s, 1H), 5.39 (s, 2H), 3.52–3.46 (comp, 4H), 3.30–3.24 (comp, 4H), 2.29 (s, 3H), 2.23 (s, 3H). MS (ESI) m/z = 321.0 [M+H]+.
1-(4-Fluorobenzyl)-5,6-dimethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (13 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 4-fluorobenzyl bromide and piperazine in similar fashion to general procedure A in 32% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.40 (s, 1H), 7.30 (dd, J = 8.6, 5.2 Hz, 2H), 7.18 (s, 1H), 7.18–7.11 (comp, 2H), 5.49 (s, 2H), 3.74–3.69 (comp, 4H), 3.46–3.40 (comp, 4H), 2.39 (s, 3H), 2.33 (s, 3H). MS (ESI) m/z = 339.1 [M+H]+.
1-(4-Chlorobenzyl)-5,6-dimethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (14 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 4-chlorobenzyl bromide and piperazine in similar fashion to general procedure A in 29% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.44–7.39 (comp, 3H), 7.26 (d, J = 8.4 Hz, 2H), 7.17 (s, 1H), 5.49 (s, 2H), 3.71–3.67 (comp, 4H), 3.45–3.41 (comp, 4H), 2.39 (s, 3H), 2.33 (s, 3H). MS (ESI) m/z = 355.0 [M+H]+.
5,6-Dimethyl-1-(4-methylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (15 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 4-methylbenzyl bromide and piperazine in similar fashion to general procedure A in 14% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.39 (s, 1H), 7.22 (d, J = 7.8 Hz, 2H), 7.17 (s, 1H), 7.13 (d, J = 8.2 Hz, 2H), 5.44 (s, 2H), 3.71–3.66 (comp, 4H), 3.43–3.38 (comp, 4H), 2.39 (s, 3H), 2.33 (s, 6H). MS (ESI) m/z = 335.1 [M+H]+.
5,6-Dimethyl-2-(piperazin-1-yl)-1-(4-(trifluoromethyl)benzyl)-1H-benzo[d]imidazole (16 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 4-trifluoromethylbenzyl bromide and piperazine in similar fashion to general procedure A in 15% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.70 (d, J = 8.5 Hz, 2H), 7.42 (d, J = 8.1 Hz, 2H), 7.39 (s, 1H), 7.10 (s, 1H), 5.55 (s, 2H), 3.64–3.59 (comp, 4H), 3.44–3.39 (comp, 4H), 2.38 (s, 3H), 2.32 (s, 3H). MS (ESI) m/z = 389.1 [M+H]+.
1-(3,4-Difluorobenzyl)-5,6-dimethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (17 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 3,4-difluorobenzyl bromide and piperazine in similar fashion to general procedure A in 25% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.41 (s, 1H), 7.36–7.21 (comp, 2H), 7.16 (s, 1H), 7.12–7.06 (m, 1H), 5.49 (s, 2H), 3.76–3.70 (comp, 4H), 3.48–3.43 (comp, 4H), 2.39 (s, 3H), 2.34 (s, 3H). MS (ESI) m/z = 357.1 [M+H]+.
1-(4-Fluoro-3-methylbenzyl)-5,6-dimethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (18 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 4-fluoro-3-methylbenzyl bromide and piperazine in similar fashion to general procedure A in 33% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.39 (s, 1H), 7.19–7.15 (comp, 2H), 7.07 (d, J = 1.3 Hz, 1H), 7.06–7.04 (m, 1H), 5.44 (s, 2H), 3.73–3.67 (comp, 4H), 3.45–3.39 (comp, 4H), 2.39 (s, 3H), 2.33 (s, 3H), 2.25 (d, J = 1.6 Hz, 3H). MS (ESI) m/z = 353.1 [M+H]+.
1-(4-Fluoro-3-(trifluoromethyl)benzyl)-5,6-dimethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (19 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 4-fluoro-3-(trifluoromethyl)benzyl bromide and piperazine in similar fashion to general procedure A in 1.2% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.74 (s, 1H) 7.53 (s, 1H), 7.43 (s, 1H), 7.39 (t, J = 9.5 Hz, 1H), 7.18 (s, 1H), 5.58 (s, 2H), 3.49 (d, J = 5.1 Hz, 4H), 3.49 (d, J = 5.1 Hz, 4H), 2.41 (s, 3H), 2.35 (s, 3H). MS (ESI) m/z = 407.1 [M+H]+.
1-(3-Cyclopropyl-4-fluorobenzyl)-5,6-dimethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (20 as TFA salt).
A mixture of 10 (36 mg, 0.097 mmol), K3PO4 (62 mg, 0.29 mmol), cyclopropylboronic acid (13 mg, 0.15 mmol), Pd(OAc)2 (2.2 mg, 0.0097 mmol), and PCy3·HBF4 (7.0 mg, 0.019 mmol) under Ar(g) was charged with a degassed mixture of 1,2-dimethoxyethane and water (3:1 v/v, 1 mL) and stirred at 90 °C for 16 h. The mixture was cooled to rt, poured into water (10 mL), extracted with CH2Cl2 (3x), dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by reversed-phase preparative HPLC and the pure fractions were concentrated under a stream of warm air to afford 7 mg of the title compound as the corresponding TFA salt (15%). 1H NMR (400 MHz, CD3OD): δ 7.41 (s, 1H), 7.17 (s, 1H), 7.08–6.95 (comp, 3H), 5.44 (s, 2H), 3.76–3.70 (comp, 4H), 3.46–3.40 (comp, 4H), 2.40 (s, 3H), 2.34 (s, 3H), 2.12–2.04 (m, 1H),1.01 (ddd, J = 6.4, 4.5, 2.0 Hz, 2H). 0.70 (ddd, J = 5.0, 4.5, 1.6 Hz, 2H). MS (ESI) m/z = 379.2 [M+H]+.
1-(4-Fluoro-3,5-dimethylbenzyl)-5,6-dimethyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (21 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 3,5-dimethyl-4-fluorobenzyl bromide and piperazine in similar fashion to general procedure A in 13% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.39 (s, 1H), 7.14 (s, 1H), 6.94 (s, 1H), 6.93 (s, 1H), 5.37 (s, 2H), 3.70–3.64 (comp, 4H), 3.45–3.39 (comp, 4H), 2.39 (s, 3H), 2.33 (s, 3H), 2.21 (d, J = 2.1 Hz, 6H). MS (ESI) m/z = 367.0 [M+H]+.
6-Chloro-1-(3-chloro-4-fluorobenzyl)-5-methyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (22 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 1-chloro-5-fluoro-2-methyl-4-nitrobenzene in similar fashion to general procedure B in 5% yield over five steps. 1H NMR (400 MHz, CD3OD): δ 7.56 (s, 1H), 7.49 (dd, J = 6.8, 2.2 Hz, 1H), 7.41 (s, 1H), 7.30 (t, J = 8.8 Hz, 1H), 7.26–7.21 (m, 1H), 5.49 (s, 2H), 3.76 (t, J = 5.2 Hz, 4H), 3.47 (t, J = 5.2 Hz, 4H), 2.49 (s, 3H). MS (ESI) m/z = 393.0 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (23 as TFA salt).
A mixture of 1,2-diaminobenzene (5.0 g, 46 mmol) and urea (3.11 g, 51.8 mmol) in xylenes (100 mL) was heated at 130 °C in a flask equipped with a reflux condenser. The mixture became a solution at elevated temperatures. After 18 h, the reaction was cooled to rt and the solid product was collected via filtration and dried over air to afford crude 1,3-dihydro-2H-benzo[d]imidazol-2-one. This crude material was suspended in POCl3 (100 mL) and heated to 100 °C. After complete conversion of starting material, as indicated by LC-MS, the reaction mixture was cooled to 0 °C, slowly added to ice water, stirred at 0 °C until all POCl3 was quenched, and then neutralized with solid NaOH. The resulting solid was collected via filtration, washed with water, and dried under reduced pressure to afford 5.5 g of crude 2-chlorobenzimidazole as a white solid. The title compound was prepared as the corresponding TFA salt from crude 2-chlorobenzimidazole used in place of 67 with 3-chloro-4-fluorobenzyl bromide, and piperazine in similar fashion to general procedure A in 32% yield over two steps. 1H NMR (400 MHz, CD3OD): δ 7.62 (d, J = 7.8 Hz, 1H), 7.43 (dd, J = 6.9, 2.2 Hz, 1H), 7.40–7.34 (m, 1H), 7.33–7.30 (comp, 2H), 7.27 (t, J = 8.8 Hz, 1H), 7.20–7.16 (m, 1H), 5.47 (s, 2H), 3.64 (t, J = 5.2 Hz, 4H), 3.46 (t, J = 5.2 Hz, 4H). MS (ESI) m/z = 345.1 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-6-methyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (24 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 2-fluoro-4-methyl-1-nitrobenzene in similar fashion to general procedure B in 3% yield over five steps. 1H NMR (400 MHz, CD3OD): δ 7.49 (d, J = 8.3 Hz, 1H), 7.43 (dd, J = 6.8, 2.1 Hz, 1H), 7.30–7.22 (comp, 2H), 7.18 (ddd, J = 8.6, 4.4, 2.3 Hz, 1H), 7.15 (s, 1H), 5.45 (s, 2H), 3.67–3.62 (comp, 4H), 3.47–3.41 (comp, 4H), 2.42 (s, 3H). MS (ESI) m/z = 359.1 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-2-(piperazin-1-yl)-6-(trifluoromethyl)-1H-benzo[d]imidazole (25 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 2-fluoro-1-nitro-4-(trifluoromethyl)benzene in similar fashion to general procedure B in 6% yield over five steps. 1H NMR (400 MHz, DMSO-d6): δ 8.82 (br s, 2H), 7.70 (s, 1H), 7.66 (d, J = 8.6 Hz, 1H), 7.51 (dd, J = 7.2, 1.6 Hz, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.37 (t, J = 8.8 Hz, 1H), 7.14–7.09 (m, 1H), 5.47 (s, 2H), 3.46 (br s, 4H), 3.29 (br s, 4H). MS (ESI) m/z = 413.1 [M+H]+.
1-(3-Chloro-4-fluorobenzyl)-6-cyclopropyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (26 as free-base).
A mixture of 27 (50 mg, 0.093 mmol), K3PO4 (79 mg, 0.37 mmol), PdCl2dppf·CH2Cl2 (7.6 mg, 0.009 mmol), and cyclopropylboronic acid (16 mg, 0.19 mmol) in degassed dioxane (1.0 mL) was heated to 100 °C under Ar(g) for 2 h. The reaction mixture was cooled to rt, diluted with EtOAc, filtered through diatomaceous earth, and concentrated under reduced pressure. The crude residue was purified by reversed-phase preparative HPLC, the pure fractions were neutralized with solid Na2CO3, and extracted with CH2Cl2 (3x). The combined organics were dried (Na2SO4) and concentrated under reduced pressure to afford 11 mg of the title compound as the free-base (31% yield). 1H NMR (400 MHz, CD3OD): δ 7.38 (d, J = 8.0 Hz, 1H), 7.31 (dd, J = 6.8, 2.0 Hz, 1H), 7.19 (t, J = 8.8 Hz, 1H), 7.06 (dq, J = 8.6, 2.2 Hz, 1H), 6.93 (dd, J = 8.2, 1.6 Hz, 1H), 6.89 (d, J = 1.3 Hz, 1H), 5.28 (s, 2H), 3.21–3.14 (comp, 4H), 3.00–2.93 (comp, 4H), 1.98–1.90 (m, 1H), 0.94–0.88 (comp, 2H), 0.63–0.57 (comp, 2H). MS (ESI) m/z = 385.0 [M+H]+.
6-Bromo-1-(3-chloro-4-fluorobenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (27 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 4-bromo-2-fluoro-1-nitrobenzene in similar fashion to general procedure B in 24% yield over five steps. 1H NMR (400 MHz, CD3OD): δ 7.48 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 1.7 Hz, 1H), 7.38 (dd, J = 8.5, 1.9 Hz, 1H), 7.36 (dd, J = 6.9, 2.2 Hz, 1H), 7.24 (t, J = 8.8 Hz, 1H), 7.08 (dq, J = 8.5, 2.2 Hz, 1H), 5.38 (s, 2H), 3.54–3.49 (comp, 4H), 3.43–3.38 (comp, 4H). MS (ESI) m/z = 422.8 [M+H]+.
6-Chloro-1-(3-chloro-4-fluorobenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (28 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 4-chloro-2-fluoro-1-nitrobenzene in similar fashion to general procedure B in 20% yield over five steps. 1H NMR (400 MHz, CD3OD): δ 7.54 (d, J = 8.5 Hz, 1H), 7.38 (dd, J = 6.8, 2.0 Hz, 1H), 7.32 (d, J = 1.8 Hz, 1H), 7.28 (dd, J = 8.0, 2.0 Hz, 1H), 7.23 (d, J = 8.8 Hz, 1H), 7.11 (ddd, J = 8.5, 4.4, 2.3 Hz, 1H), 5.40 (s, 2H), 3.57–3.52 (comp, 4H), 3.44–3.40 (comp, 4H). MS (ESI) m/z = 379.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (29 as TFA salt).
Intermediate 77 (20 mg, 0.36 mmol) was dissolved in 2-propanol (1 mL) and acetic acid (3 drops) was added. The vial was sealed, evacuated, and refilled with Ar(g). Palladium/carbon (10% w/w, 10 mg) was added. The vial was resealed, evacuated, and refilled with H2(g) via balloon. After 18 h, the vial was purged with Ar(g) and the reaction mixture was filtered over silica gel with the aid of EtOAc. The filtrate was concentrated under reduced pressure. The residue was purified by reversed-phase preparative HPLC. The resultant material was dissolved in CH2Cl2 (2 mL) and stirred at rt. TFA (0.5 mL) was added and the mixture was stirred for 2 h and then concentrated under reduced pressure. The residue was purified by reversed-phase preparative HPLC and the pure fractions were concentrated under a stream of warm air to afford 7 mg of the title compound as the corresponding TFA salt as a white solid (38% yield, two steps). 1H NMR (400 MHz, CD3OD): δ 7.52 (dd, J = 8.3, 0.7 Hz, 1H), 7.28–7.25 (comp, 2H), 6.87 (d, J = 6.6 Hz, 2H), 5.30 (s, 2H), 3.55–3.52 (comp, 4H), 3.41–3.39 (comp, 4H), 2.20 (d, J = 2.1 Hz, 6H). MS (ESI) m/z = 373.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-4-phenyl-2-(piperazin-1-yl)-1H-benzo[d]imidazole (30 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and phenylboronic acid in similar fashion to general procedure C in 75% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.94–7.91 (comp, 2H), 7.50–7.46 (comp, 2H), 7.42–7.38 (m, 1H), 7.34 (d, J = 2.0 Hz, 1H), 7.21 (d, J = 2.0 Hz, 1H), 6.88 (d, J = 6.7 Hz, 2H), 5.30 (s, 2H), 3.52–3.49 (comp, 4H), 3.40–3.38 (comp, 4H), 2.21 (d, J = 2.1 Hz, 6H). MS (ESI) m/z = 449.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(o-tolyl)-1H-benzo[d]imidazole (31 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and ortho-tolylboronic acid in similar fashion to general procedure C in 73% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.35–7.28 (comp, 5H), 7.12 (d, J = 1.9 Hz, 1H), 6.93 (d, J = 6.6 Hz, 2H), 5.35 (s, 2H), 3.53–3.50 (comp, 4H), 3.37–3.34 (comp, 4H), 2.22 (d, J = 2.0 Hz, 6H), 2.20 (s, 3H). MS (ESI) m/z = 463.0 [M+H]+.
6-Chloro-4-(2-ethylphenyl)-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (32 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and (2-ethylphenyl)boronic acid in similar fashion to general procedure C in 24% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.39–7.38 (comp, 2H), 7.30–7.24 (comp, 3H), 7.10–7.09 (m, 1H), 6.90 (d, J = 6.4 Hz, 2H), 5.33 (s, 2H), 3.49–3.45 (comp, 4H), 3.35–3.33 (comp, 4H), 2.54 (q, J = 7.6 Hz, 2H), 2.22 (d, J = 2.0 Hz, 6H), 1.04 (t, J = 7.6 Hz, 3H). MS (ESI) m/z = 477.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-4-(2-isopropylphenyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (33 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and (2-isopropylphenyl)boronic acid in similar fashion to general procedure C in 19% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.47 (d, J = 7.6 Hz, 1H), 7.41 (td, J = 8.0, 1.7 Hz, 1H), 7.29–7.21 (comp, 3H), 7.06 (d, J = 1.9 Hz, 1H), 6.89 (d, J = 6.6 Hz, 2H), 5.32 (s, 2H), 3.44–3.42 (comp, 4H), 3.35–3.33 (comp, 4H), 2.89–2.79 (m, 1H), 2.22 (d, J = 2.1 Hz, 6H), 1.16 (br s, 6H). MS (ESI) m/z = 490.9 [M+H]+.
6-Chloro-4-(2-cyclobutylphenyl)-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (34 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and (2-cyclobutylphenyl)boronic acid in similar fashion to general procedure C in 8% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.47 (d, J = 7.8 Hz, 1H), 7.41 (dt, J = 7.9, 2.0 Hz, 1H), 7.29–7.23 (comp, 3H), 7.01 (d, J = 2.0 Hz, 1H), 6.85 (d, J = 6.6 Hz, 2H), 5.32 (s, 2H), 3.66–3.57 (m, 1H), 3.43–3.40 (comp, 4H), 3.36–3.34 (comp, 4H), 2.21 (d, J = 2.0 Hz, 6H), 2.04–1.99 (comp, 2H), 1.85–1.65 (comp, 4H). MS (ESI) m/z = 502.9 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(2-(trifluoromethyl)phenyl)-1H-benzo[d]imidazole (35 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and (2-(trifluoromethyl)phenyl)boronic acid in similar fashion to general procedure C in 67% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.85 (d, J = 7.7 Hz, 1H), 7.70 (t, J = 7.2 Hz, 1H), 7.63 (t, J = 7.6 Hz, 1H), 7.48 (d, J = 7.4 Hz, 1H), 7.32 (d, J = 1.8 Hz, 1H), 7.08 (s, 1H), 6.86 (d, J = 6.6 Hz, 2H), 5.31 (s, 2H), 3.44–3.39 (comp, 4H), 3.35–3.33 (comp, 4H), 2.22 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 516.9 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(3-(trifluoromethyl)phenyl)-1H-benzo[d]imidazole (36 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and (3-(trifluoromethyl)phenyl)boronic acid in similar fashion to general procedure C in 24% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 8.46 (s, 1H), 8.18–8.15 (m, 1H), 7.70–7.65 (comp, 2H), 7.43 (d, J = 1.9 Hz, 1H), 7.25 (d, J = 1.9 Hz, 1H), 6.88 (d, J = 6.6 Hz, 2H), 5.31 (s, 2H), 3.54–3.52 (comp, 4H), 3.42–3.39 (comp, 4H), 2.21 (d, J = 1.9 Hz, 6H). MS (ESI) m/z = 516.9 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(4-(trifluoromethyl)phenyl)-1H-benzo[d]imidazole (37 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and (4-(trifluoromethyl)phenyl)boronic acid in similar fashion to general procedure C in 43% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 8.18 (d, J = 8.3 Hz, 2H), 7.78 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 1.2 Hz, 1H), 7.27 (d, J = 1.8 Hz, 1H), 6.88 (d, J = 6.6 Hz, 2H), 5.31 (s, 2H), 3.53–3.51 (comp, 4H), 3.41–3.38 (comp, 4H), 2.21 (d, J = 1.9 Hz, 6H). MS (ESI) m/z = 516.9 [M+H]+.
6-Chloro-4-(2-chlorophenyl)-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (38 as free-base).
The title compound was prepared as the free-base from 77 and (2-chlorophenyl)boronic acid in similar fashion to general procedure C in 43% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.56–7.51 (m, 1H), 7.49–7.45 (m, 1H), 7.42–7.36 (m, 2H), 7.19 (d, J = 1.9 Hz, 1H), 7.08 (d, J = 1.9 Hz, 1H), 6.87 (d, J = 6.7 Hz, 2H), 5.22 (s, 2H), 3.19–3.14 (m, 4H), 2.93–2.86 (m, 4H), 2.20 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 483.0 [M+H]+.
6-Chloro-4-(2,6-dimethylphenyl)-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (39 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and (2,6-dimethylphenyl)boronic acid in similar fashion to general procedure C in 14% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.31 (d, J = 2.0 Hz, 1H), 7.23 (dd, J = 8.6, 6.5 Hz, 1H), 7.15 (d, J = 7.5 Hz, 2H), 7.00–6.99 (m, 1H), 6.89 (d, J = 6.7 Hz, 2H), 5.33 (s, 2H), 3.47–3.45 (comp, 4H), 3.36–3.32 (comp, 4H), 2.22 (d, J = 2.1 Hz, 6H), 2.02 (s, 6H). MS (ESI) m/z = 477.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(piperidin-1-yl)-1H-benzo[d]imidazole (40 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and piperidine in similar fashion to general procedure D in 45% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.21 (d, J = 1.7 Hz, 1H), 7.17 (d, J = 1.7 Hz, 1H), 6.86 (d, J = 6.8 Hz, 2H), 5.28 (s, 2H), 3.81 (t, J = 5.5 Hz, 4H), 3.56 (dd, J = 6.8, 5.0 Hz, 4H), 3.41 (dd, J = 5.5, 3.8 Hz, 4H), 2.20 (d, J = 2.0 Hz, 6H), 2.02–1.95 (m, 4H), 1.82–1.73 (m, 2H). MS (ESI) m/z = 456.0 [M+H]+.
4-(6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazol-4-yl)morpholine (41 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and morpholine in similar fashion to general procedure D in 37% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 6.84–6.81 (comp, 3H), 6.61 (d, J = 1.8 Hz, 1H), 5.21 (s, 2H), 3.90 (dd, J = 5.4, 4.7 Hz, 4H), 3.50–3.42 (comp, 8H), 3.40–3.36 (comp, 4H), 2.19 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 458.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-4-(4-methylpiperazin-1-yl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (42 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and N-methylpiperazine in similar fashion to general procedure D in 35% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 6.90 (d, J = 1.6 Hz, 1H), 6.83 (d, J = 6.8 Hz, 2H), 6.71 (d, J = 1.6 Hz, 1H), 5.24 (s, 2H), 4.40 (d, J = 13.6 Hz, 2H), 3.65 (d, J = 13.6 Hz, 2H), 3.59–3.34 (comp, 10H), 3.23–3.14 (m, 2H), 3.00 (s, 3H), 2.19 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 471.3 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(1,2,3,6-tetrahydropyridin-4-yl)-1H-benzo[d]imidazole (43 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate in similar fashion to general procedure C in 36% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.20 (s, 2H), 6.84 (d, J = 6.7 Hz, 2H), 6.84–6.79 (m, 1H), 5.27 (s, 2H), 3.95–3.91 (comp, 2H), 3.56–3.48 (comp, 6H), 3.42–3.48 (comp, 4H), 3.08–3.01 (comp, 2H), 2.19 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 454.0 [M+H]+.
1-(6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazol-4-yl)piperidin-4-amine (44 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and tert-butyl piperidin-4-ylcarbamate in similar fashion to general procedure D in 8% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 6.84–6.82 (comp, 3H), 6.69 (d, J = 1.7 Hz, 1H), 5.23 (s, 2H), 4.26 (br d, J = 12.7 Hz, 2H), 3.47 (dd, J = 9.7, 4.5 Hz, 4H), 3.40 (dd, J = 8.1, 3.1 Hz, 4H), 3.34–3.28 (m, 1H), 2.91 (td, J = 12.2, 1.7 Hz, 2H), 2.19 (d, J = 2.0 Hz, 6H), 2.12 (br d, J = 12.2 Hz, 2H), 1.95–1.85 (m, 2H). MS (ESI) m/z = 471.0 [M+H]+.
(S)-(1-(6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazol-4-yl)pyrrolidin-2-yl)methanol (45 as free-base).
The title compound was prepared as the free-base from 77 and (S)-pyrrolidin-2-ylmethanol in similar fashion to general procedure D in 57% yield over two steps as a white solid. 1H NMR (400 MHz, CDCl3): δ 6.75 (d, J = 6.6 Hz, 2H), 6.40–6.33 (comp, 2H), 5.06–4.95 (comp, 3H), 3.80–3.59 (comp, 2H), 3.56–3.47 (m, 1H), 3.37–3.01 (comp, 8H), 2.68–2.50 (m, 1H), 2.20 (d, J = 1.7 Hz, 6H), 2.07–1.72 (comp, 4H). MS (ESI) m/z = 472.0 [M+H]+.
(R)-(1-(6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazol-4-yl)pyrrolidin-2-yl)methanol (46 as free-base).
The title compound was prepared as the free-base from 77 and (R)-pyrrolidin-2-ylmethanol in similar fashion to general procedure D in 74% yield over two steps as a white solid. 1H NMR (400 MHz, CDCl3): δ 6.75 (d, J = 6.7 Hz, 2H), 6.40–6.32 (comp, 2H), 5.08–4.95 (comp, 3H), 3.80–3.61 (comp, 2H), 3.57–3.48 (m, 1H), 3.37–3.01 (comp, 8H), 2.67–2.51 (m, 1H), 2.20 (d, J = 1.8 Hz, 6H), 2.08–1.73 (comp, 4H). MS (ESI) m/z = 472.0 [M+H]+.
(S)-(1-(6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazol-4-yl)pyrrolidin-2-yl)methanamine (47 as TFA salt).
The title compound was prepared as the free-base from 77 and tert-butyl (S)-(pyrrolidin-2-ylmethyl)carbamate in similar fashion to general procedure D in 65% yield over two steps as a white solid. 1H NMR (400 MHz, CDCl3) δ 6.76 (d, J = 6.5 Hz, 2H), 6.34 (d, J = 1.3 Hz, 1H), 6.24 (d, J = 1.3 Hz, 1H), 5.04–4.94 (comp, 2H), 4.90–4.81 (m, 1H), 3.64–3.53 (m, 1H), 3.37–3.26 (m, 1H), 3.21–3.05 (comp, 5H), 3.04–2.92 (comp, 4H), 2.69–2.56 (m, 1H), 2.20 (d, J = 1.6 Hz, 6H), 2.07–1.96 (comp, 4H). MS (ESI) m/z = 471.0 [M+H]+.
(R)-(1-(6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazol-4-yl)pyrrolidin-2-yl)methanamine (48 as free-base).
The title compound was prepared as the free-base from 77 and tert-butyl (R)-(pyrrolidin-2-ylmethyl)carbamate in similar fashion to general procedure D in 75% yield over two steps as a white solid. 1H NMR (400 MHz, CDCl3) δ 6.78 (d, J = 6.5 Hz, 2H), 6.34 (d, J = 1.6 Hz, 1H), 6.24 (d, J = 1.6 Hz, 1H), 5.04–4.95 (comp, 2H), 4.90–4.81 (m, 1H), 3.64–3.55 (m, 1H), 3.38–3.27 (m, 1H), 3.19–3.03 (comp, 5H), 3.03–2.90 (comp, 4H), 2.68–2.55 (m, 1H), 2.20 (d, J = 1.7 Hz, 6H), 2.08–1.96 (comp, 4H). MS (ESI) m/z = 471.0 [M+H]+.
(2-(6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazol-4-yl)phenyl)methanol (49 as free-base).
A mixture of 77 (75 mg, 0.14 mmol), (2-formylphenyl)boronic acid (41 mg, 0.27 mmol), PdCl2dppf·CH2Cl2 (11 mg, 0.014 mmol), and K3PO4 (87 mg, 0.41 mmol) was charged with degassed dioxane (1 mL) and heated to a temperature of 100 °C under Ar(g). After 2 h, the reaction mixture was cooled to rt, diluted with EtOAc, filtered through diatomaceous earth, and concentrated. The crude residue was dissolved in MeOH (2 mL), charged with NaBH4 (26 mg, 0.68 mmol), and stirred for 1 h at rt. A solution of HCl in dioxane (4 M, 1 mL) was added and the reaction mixture was stirred for 1 h at rt. The mixture was concentrated under reduced pressure and purified by reversed-phase preparative HPLC. The pure HPLC fractions were neutralized with solid Na2CO3, extracted with CH2Cl2 (3x), dried (Na2SO4), and concentrated under reduced pressure to afford 44 mg of the title compound (68%) as a white solid. 1H NMR (600 MHz, CD3OD): δ 7.59 (d, J = 7.7 Hz, 1H), 7.45 (td, J = 7.6, 1.2 Hz, 1H), 7.39 (td, J = 7.6, 1.2 Hz, 1H), 7.30 (dd, J = 7.6, 1.2 Hz, 1H), 7.19 (d, J = 1.8 Hz, 1H), 7.07 (d, J = 1.8 Hz, 1H), 6.88 (d, J = 6.6 Hz, 2H), 5.24 (s, 2H), 4.44 (s, 2H), 3.20–3.16 (m, 4H), 2.94–2.91 (m, 4H), 2.21 (d, J = 1.8 Hz, 6H)). MS (ESI) m/z = 478.9 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(pyridin-4-yl)-1H-benzo[d]imidazole (50 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and potassium pyridine-4-trifluoroborate in similar fashion to general procedure E in 27% yield over two steps as a yellow solid. 1H NMR (400 MHz, CD3OD): δ 8.94 (d, J = 6.9 Hz, 2H), 8.86 (d, J = 6.9 Hz, 2H), 7.85 (d, J = 1.8 Hz, 1H), 7.47 (d, J = 1.8 Hz, 1H), 6.88 (d, J = 6.7 Hz, 2H), 5.35 (s, 2H), 3.64–3.61 (comp, 4H), 3.43–3.41 (comp, 4H), 2.20 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 450.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(pyridin-3-yl)-1H-benzo[d]imidazole (51 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and potassium pyridine-3-trifluoroborate in similar fashion to general procedure E in 39% yield over two steps as a yellow solid. 1H NMR (400 MHz, CD3OD): δ 9.82 (d, J = 2.0 Hz, 1H), 9.23 (dt, J = 8.4, 1.5 Hz, 1H), 8.82 (d, J = 5.2 Hz, 1H), 8.14 (dd, J = 8.2, 5.9 Hz, 1H), 7.71 (d, J = 1.8 Hz, 1H), 7.39 (d, J = 1.9 Hz, 1H), 6.88 (d, J = 6.6 Hz, 2H), 5.34 (s, 2H), 3.61–3.58 (comp, 4H), 3.43–3.40 (comp, 4H), 2.20 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 450.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(pyrimidin-5-yl)-1H-benzo[d]imidazole (54 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and pyrimidin-5-ylboronic acid in similar fashion to general procedure C in 79% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 9.47 (s, 2H), 9.15 (s, 1H), 7.55 (t, J = 1.8 Hz, 1H), 7.32 (d, J = 1.8 Hz, 1H), 6.88 (d, J = 6.7 Hz, 2H), 5.32 (s, 2H), 3.56–3.54 (comp, 4H), 3.42–3.40 (comp, 4H), 2.20 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 451.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-4-(4-methylpyrimidin-5-yl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (55 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and 4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine in similar fashion to general procedure C in 93% yield over two steps as a yellow solid. 1H NMR (400 MHz, CD3OD): δ 9.07 (s, 1H), 8.70 (s, 1H), 7.37 (d, J = 1.9 Hz, 1H), 7.24 (d, J = 1.9 Hz, 1H), 6.89 (d, J = 6.6 Hz, 2H), 5.32 (s, 2H), 3.45 (br s, 4H), 3.37–3.35 (comp, 4H), 2.49 (s, 3H), 2.22 (d, J = 2.2 Hz, 6H). MS (ESI) m/z = 465.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-4-(4-isopropylpyrimidin-5-yl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (56 as TFA salt).
The title compound was prepared as the corresponding TFA salt from 77 and (4-isopropylpyrimidin-5-yl)boronic acid in similar fashion to general procedure C in 58% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 9.14 (s, 1H), 8.63 (s, 1H), 7.37 (d, J = 1.9 Hz, 1H), 7.19 (d, J = 1.9 Hz, 1H), 6.89 (d, J = 6.7 Hz, 2H), 5.32 (s, 2H), 3.45–3.42 (comp, 4H), 3.37–3.34 (comp, 4H), 3.11–3.04 (m, 1H), 2.22 (d, J = 2.0 Hz, 6H), 1.23 (d, J = 6.9 Hz, 6H). MS (ESI) m/z = 493.0 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-4-(1H-pyrazol-4-yl)-1H-benzo[d]imidazole (57 as free-base).
The title compound was prepared as the free-base from 78 and tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylate in dioxane in similar fashion to general procedure C in 31% yield over two steps as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 12.97 (br s, 1H), 8.60 (br s, 1H), 8.38 (br s, 1H), 7.46 (d, J = 1.9 Hz, 1H), 7.05 (d, J = 2.0 Hz, 1H), 6.92 (d, J = 6.7 Hz, 2H), 5.20 (s, 2H), 3.22–3.15 (comp, 4H), 2.89–2.80 (comp, 4H), 2.16 (d, J = 1.6 Hz, 6H). MS (ESI) m/z = 439.0 [M+H]+.
6-Chloro-4-(3,5-dimethyl-1H-pyrazol-4-yl)-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (58 as free-base).
The title compound was prepared as the free-base from 78 and tert-butyl 3,5-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylate in dioxane in similar fashion to general procedure C in 31% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.10 (d, J = 2.0 Hz, 1H), 7.00 (d, J = 1.9 Hz, 1H), 6.86 (d, J = 6.7 Hz, 2H), 5.21 (s, 2H), 3.23–3.17 (m, 4H), 2.98–2.93 (m, 4H), 2.32 (s, 6H), 2.19 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 467.0 [M+H]+.
6-Chloro-4-(2-chlorophenyl)-1-(4-fluoro-3,5-dimethylbenzyl)-2-(2,6-diazaspiro[3.3]heptan-2-yl)-1H-benzo[d]imidazole (59 as free-base).
The title compound was prepared as the free-base from 79 and (2-chlorophenyl)boronic acid in dioxane in similar fashion to general procedure C in 46% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.55–7.50 (m, 1H), 7.48–7.44 (m, 1H), 7.41–7.36 (m, 2H), 7.13 (d, J = 2.0 Hz, 1H), 7.01 (d, J = 2.0 Hz, 1H), 6.83 (d, J = 6.9 Hz, 2H), 5.17 (s, 2H), 4.25 (s, 4H), 3.82 (s, 4H), 2.20 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 494.9 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-4-(4-methylpyrimidin-5-yl)-2-(2,6-diazaspiro[3.3]heptan-2-yl)-1H-benzo[d]imidazole (60 as free-base).
The title compound was prepared as the free-base from 79 and 4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine in dioxane in similar fashion to general procedure C in 91% yield over two steps as a white solid. 1H NMR (600 MHz, CD3OD): δ 9.03 (s, 1H), 8.63 (s, 1H), 7.22 (d, J = 1.9 Hz, 1H), 7.06 (d, J = 1.9 Hz, 1H), 6.83 (d, J = 6.7 Hz, 2H), 5.18 (s, 2H), 4.27 (s, 4H), 3.84 (s, 4H), 2.46 (s, 3H), 2.19 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 477.0 [M+H]+.
6-Chloro-1-(4-fluorobenzyl)-4-(4-methylpyrimidin-5-yl)-2-(2,6-diazaspiro[3.3]heptan-2-yl)-1H-benzo[d]imidazole (61 as free-base).
The title compound was prepared as the free-base from 80 and 4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine in dioxane/water (5:1 v/v) in similar fashion to general procedure C in 78% yield over two steps as a white solid. 1H NMR (400 MHz, CDCl3): δ 9.14 (s, 1H), 8.67 (s, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.36–7.32 (m, 3H), 7.21–7.16 (m, 2H), 5.45 (s, 2H), 4.63 (s, 4H), 4.30 (s, 4H), 2.45 (s, 3H). MS (ESI) m/z = 449.0 [M+H]+.
5-(6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(2,6-diazaspiro[3.3]heptan-2-yl)-1H-benzo[d]imidazol-4-yl)-4-methylpyrimidin-2-amine (62 as free-base).
The title compound was prepared as the free-base from 79 and 4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidin-2-amine in dioxane in similar fashion to general procedure C in 27% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 8.13 (s, 1H), 7.13 (d, J = 2.0 Hz, 1H), 6.98 (d, J = 2.0 Hz, 1H), 6.82 (d, J = 6.4 Hz, 2H), 5.16 (s, 2H), 4.25 (s, 4H), 3.77 (s, 4H), 2.26 (s, 3H), 2.19 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 492.0 [M+H]+.
5-(6-Chloro-1-(4-fluorobenzyl)-2-(2,6-diazaspiro[3.3]heptan-2-yl)-1H-benzo[d]imidazol-4-yl)-4-methylpyrimidin-2-amine (63 as free-base).
The title compound was prepared as the free-base from 80 and 4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine in dimethoxyethane/water (5:1 v/v) in similar fashion to general procedure C in 73% yield over two steps as a white solid. 1H NMR (400 MHz, CDCl3): δ 8.02 (s, 1H), 7.09–7.05 (m, 3H), 7.00–6.96 (m, 2H), 6.87 (s, 1H), 5.14 (s, 2H), 4.74 (s, 4H), 4.21 (s, 4H), 2.15 (s, 3H). MS (ESI) m/z = 464.1 [M+H]+.
6-Chloro-1-(4-fluoro-3,5-dimethylbenzyl)-4-(4-methylpiperazin-1-yl)-2-(2,6-diazaspiro[3.3]heptan-2-yl)-1H-benzo[d]imidazole (64 as free-base).
The title compound was prepared as the free-base from 79 and N-methylpiperazine in similar fashion to general procedure D in 99% yield over two steps as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 6.81 (d, J = 6.8 Hz, 2H), 6.75 (d, J = 1.8 Hz, 1H), 6.40 (d, J = 1.8 Hz, 1H), 5.06 (s, 2H), 4.13 (s, 4H), 3.58 (s, 4H), 3.45 (br s, 4H), 2.47–2.44 (comp, 4H), 2.22 (s, 3H), 2.15 (d, J = 1.8 Hz, 6H). MS (ESI) m/z = 483.0 [M+H]+.
6-Chloro-4-(3,5-dimethyl-1H-pyrazol-4-yl)-1-(4-fluoro-3,5-dimethylbenzyl)-2-(2,6-diazaspiro[3.3]heptan-2-yl)-1H-benzo[d]imidazole (65 as free-base).
The title compound was prepared as the free-base from 79 and tert-butyl 3,5-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylate in dioxane in similar fashion to general procedure C in 38% yield over two steps as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.05 (d, J = 1.9 Hz, 1H), 6.93 (d, J = 1.9 Hz, 1H), 6.83 (d, J = 6.4 Hz, 2H), 5.14 (s, 2H), 4.24 (s, 4H), 3.75 (s, 4H), 2.22 (s, 6H), 2.20 (d, J = 2.0 Hz, 6H)). MS (ESI) m/z = 479.0 [M+H]+.
2-Chloro-5,6-dimethyl-1H-benzo[d]imidazole (67).
A mixture of compound 66 (2.0 g, 15 mmol), DMAP (183 mg, 1.5 mmol), and CDI (4.86 g, 30 mmol) in CH2Cl2 (50 mL) was stirred for 16 h at rt. The resulting precipitate was collected via filtration, rinsed with CH2Cl2 (30 mL), and dried under reduced pressure. The crude solid was suspended in POCl3 (15 mL) and heated to 90 °C. After 6 h, the suspension had become a clear solution and the reaction was complete, as indicated by LC-MS. The reaction mixture was cooled to 0 °C and chilled water was added slowly with rapid stirring to avoid excessive exotherm. Solid pellets of KOH were then added portionwise until the mixture was neutral, at which time a solid precipitate formed. The solid was collected via filtration, rinsed with water, and dried under reduced pressure to provide 1.8 g of the title compound (67%) as an off-white powder. 1H NMR (400 MHz, CD3OD): δ 7.26 (s, 2H), 2.35 (s, 6H). MS (ESI) m/z = 181.1 [M+H]+.
4-Bromo-2,6-dichloro-1H-benzo[d]imidazole (76).
A mixture of compound 75 (8.92 g, 40.3 mmol), DMAP (0.49 g, 4.0 mmol), and CDI (13.1 g, 80.5 mmol) in CH2Cl2 (400 mL) was stirred for 16 h at rt. The resulting precipitate was collected via filtration, rinsed with CH2Cl2 (100 mL), and dried under reduced pressure. The crude solid was suspended in POCl3 (23 mL) and heated to 110 °C. After 3 h, the suspension had become a clear solution and the reaction was complete, as indicated by LC-MS. The mixture was cooled to rt and added slowly to rapidly stirred water (200 mL), taking care to avoid excessive exotherm. The mixture was cooled in an ice bath and solid pellets of KOH were then added portionwise until the mixture was neutral, at which time a solid precipitate formed. The solid was collected via filtration, rinsed with water, and dried under reduced pressure to provide 1.62 g of the title compound (75%) as an off-white solid. 1H NMR (400 MHz, CD3OD): δ 7.52 (d, J = 1.8 Hz, 1H), 7.48 (d, J = 1.8 Hz, 1H). MS (ESI) m/z = 266.8 [M+H]+.
tert-Butyl-4-(4-bromo-6-chloro-1-(4-fluoro-3,5-dimethylbenzyl)-1H-benzo[d]imidazol-2-yl)piperazine-1-carboxylate (77).
A mixture of 76 (5.08 g, 19.1 mmol), 4-fluoro-3,5-dimethylbenzyl bromide (4.35 g, 20.1 mmol), and K2CO3 (5.28 g, 38.2 mmol) in DMF (38 mL) was stirred at rt for 16 h. The mixture was diluted with EtOAc, poured into a 50% sat. brine solution, and extracted with EtOAc (3x). The combined organics were washed with a 50% sat. brine solution, a sat. brine solution, dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography to provide 4.1 g of 4-bromo-2,6-dichloro-1-(4-fluoro-3,5-dimethylbenzyl)-1H-benzo[d]imidazole. A portion of this material (800 mg, 1.99 mmol) was mixed with 1-Boc-piperazine (1110 mg, 5.97 mmol) and N,N-diisopropylethylamine (514 mg, 3.98 mmol) in DMF (0.69 mL) and heated to 80 °C for 16 h. The mixture was cooled to rt and poured into brine. The resulting precipitate was collected via filtration and dried under reduced pressure. The crude solid was purified by silica gel chromatography to afford 1.09 g of the title compound (99%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.38 (d, J = 1.8 Hz, 1H), 6.93 (d, J = 1.8 Hz, 1H), 6.72 (d, J = 6.5 Hz, 2H), 5.06 (s, 2H), 3.55–3.53 (m, 4H), 3.27–3.25 (m, 4H), 2.21 (d, J = 1.9 Hz, 6H), 1.46 (s, 9H). MS (ESI) m/z = 552.9 [M+H]+.
4-Bromo-6-chloro-1-(4-fluoro-3,5-dimethylbenzyl)-2-(piperazin-1-yl)-1H-benzo[d]imidazole (78).
A mixture of 76 (5.08 g, 19.1 mmol), 4-fluoro-3,5-dimethylbenzyl bromide (4.35 g, 20.1 mmol), and K2CO3 (5.28 g, 38.2 mmol) in DMF (38 mL) was stirred at rt for 16 h. The mixture was diluted with EtOAc, poured into a 50% sat. brine solution, and extracted with EtOAc (3x). The combined organics were washed with a 50% sat. brine solution, a sat. brine solution, dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography to provide 4.1 g of 4-bromo-2,6-dichloro-1-(4-fluoro-3,5-dimethylbenzyl)-1H-benzo[d]imidazole. A portion of this material (1.00 g, 2.49 mmol) was mixed with piperazine (1.29 g, 14.9 mmol) and K2CO3 (1.03 g, 7.46 mmol) in DMSO (5.0 mL) and heated to 100 °C for 16 h. The mixture was cooled to rt, poured into brine, and the precipitate was collected via filtration. The solid was washed with water and then dried under reduced pressure to afford 1.06 g of the title compound (95%) as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.34 (d, J = 1.8 Hz, 1H), 7.14 (d, J = 1.8 Hz, 1H), 6.82 (d, J = 6.8 Hz, 2H), 5.19 (s, 2H), 3.28–3.24 (comp, 4H), 2.96–2.91 (comp, 4H), 2.19 (d, J = 2.0 Hz, 6H). MS (ESI) m/z = 450.9 [M+H]+.
tert-Butyl-6-(4-bromo-6-chloro-1-(4-fluoro-3,5-dimethylbenzyl)-1H-benzo[d]imidazol-2-yl)-2,6-diazaspiro[3.3]heptane-2-carboxylate (79).
A mixture of 76 (5.08 g, 19.1 mmol), 4-fluoro-3,5-dimethylbenzyl bromide (4.35 g, 20.1 mmol), and K2CO3 (5.28 g, 38.2 mmol) in DMF (38 mL) was stirred at rt for 16 h. The mixture was diluted with EtOAc, poured into a 50% sat. brine solution, and extracted with EtOAc (3x). The combined organics were washed with a 50% sat. brine solution, a sat. brine solution, dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography to provide 4.1 g of 4-bromo-2,6-dichloro-1-(4-fluoro-3,5-dimethylbenzyl)-1H-benzo[d]imidazole. A portion of this material (400 mg, 1 mmol) was mixed with 2,6-diazaspiro[3.3]heptane-2-carboxylic acid tert-butyl ester hemioxylate (290 mg, 1.19 mmol) and K2CO3 (412 mg, 2.98 mmol) in DMSO (2 mL) and heated to 100 °C for 3 h. The mixture was cooled to rt and poured into brine. The resulting solid precipitate was collected via filtration, washed with water, and dried under reduced pressure to afford 554 mg of the title compound (99%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.28 (d, J = 1.9 Hz, 1H), 7.10 (d, J = 1.9 Hz, 1H), 6.78 (d, J = 6.6 Hz, 2H), 5.13 (s, 2H), 4.36 (s, 4H), 4.08 (s, 4H), 2.19 (d, J = 2.0 Hz, 6H), 1.43 (s, 9H)). MS (ESI) m/z = 562.8 [M+H]+.
tert-Butyl-6-(4-bromo-6-chloro-1-(4-fluorobenzyl)-1H-benzo[d]imidazol-2-yl)-2,6-diazaspiro[3.3]heptane-2-carboxylate (80).
A mixture of 76 (1.06 g, 4 mmol), 4-fluorobenzyl bromide (907 mg, 4.8 mmol), and K2CO3 (829 mg, 6 mmol) in DMF (14 mL) was stirred at rt for 16 h. The mixture was quenched with 10% HCl, extracted with EtOAc (3x), and washed with water. The combined organics were dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography to afford 1.07 g of 4-bromo-2,6-dichloro-1-(4-fluorobenzyl)-1H-benzo[d]imidazole. A portion of this material (374 mg, 1 mmol) was mixed with 2,6-diazaspiro[3.3]heptane-2-carboxylic acid tert-butyl ester hemioxylate (486 mg, 1.5 mmol) and K2CO3 in DMSO (3 mL) and heated to 100 °C for 12 h. The mixture was cooled to rt, diluted with EtOAc, poured into a 50% sat. brine solution, and extracted with EtOAc (3x). The combined organics were washed with a 50% sat. brine solution, a sat. brine solution, dried (Na2SO4), and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography to afford 386 mg of the title compound (72%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.46 (d, J = 2.0 Hz, 2H), 7.11 (d, J = 2.0 Hz, 2H), 6.92 (br s, 1H), 6.81 (br s, 1H), 5.22 (s, 2H), 4.96 (s, 4H), 4.25 (s, 4H), 1.49 (s, 9H). MS (ESI) m/z = 536.8 [M+H]+.
Supplementary Material
ACKNOWLEDGEMENTS
Funding for this work came from the following sources: U. S. National Institutes of Health, NIH Director’s Pioneer Award (DP1OD006933/DP1CA174419) to S. W. Fesik; Lustgarten Foundation Research Investigator Grant to S. W. Fesik; National Cancer Institute SPORE Grant in GI Cancer (5P50A095103-09) to R. J. Coffey.
The authors would like to thank the Vanderbilt High Throughput Screening (HTS) Core Facility, with assistance provided by C. David Weaver, Paige Vinson, Chris Farmer, and Corbin Whitwell. This facility receives support from the Vanderbilt Institute of Chemical Biology and the Vanderbilt Ingram Cancer Center (P30 CA68485). The authors would also like to thank the Vanderbilt University Biomolecular NMR Facility, with assistance provided by Donald Stec. This facility receives support from an NIH SIG Grant (1S-10RR025677-01) and Vanderbilt University matching funds. Finally, the authors would like to thank the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences for use of the Advanced Photon Source (Contract: DE-AC02-06CH11357).
ABBREVIATIONS USED
- CDI
1,1’-carbonyldiimidazole
- EGF
epidermal growth factor
- ERK1/2
extracellular regulated kinases 1 and 2
- GEF
guanine nucleotide exchange factor
- LipE
lipophilic ligand efficiency
- MAPK
mitogen-activated protein kinase
- pERK1/2
phosphorylated ERK1/2
- Act.
relative percent activation
- SOS1
son of sevenless homologue 1
Footnotes
Supporting Information. The Supporting Information associated with this manuscript is available free of charge on the ACS Publications website. Files include:
Biochemical and cellular assay conditions; saturation binding curves for FPA probe molecules S1, S2, and S3; additional biochemical data; preliminary in vitro solubility and permeability data; experimental details for the syntheses of S1, S2, and S3; details regarding protein expression and purification; details regarding crystallization, X-ray data collection, structure solution, and refinement; and a table containing X-ray data collection and refinement statistics (PDF)
Molecular formula strings (CSV)
Accession Codes.
Compound 3 (PDB ID code 6D5V)
Compound 10 (PDB ID code 6D5M)
Compound 28 (PDB ID code 6D5L)
Compound 29 (PDB ID code 6D5J)
Compound 38 (PDB ID code 6D5H)
Compound 43 (PDB ID code 6D5G)
Compound 47 (PDB ID code 6D5E)
Compound 58 (PDB ID code 6D59)
Compound 64 (PDB ID code 6D55)
Compound 65 (PDB ID code 6D56)
Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare the following competing financial interest(s): RAS activator compounds have been licensed to Boehringer Ingelheim.
REFERENCES
- (1).Vetter IR The Guanine Nucleotide-Binding Switch in Three Dimensions. Science 2001, 294 (5545), 1299–1304. [DOI] [PubMed] [Google Scholar]
- (2).Schubbert S; Shannon K; Bollag G Hyperactive Ras in Developmental Disorders and Cancer. Nat. Rev. Cancer 2007, 7 (4), 295–308. [DOI] [PubMed] [Google Scholar]
- (3).Hanahan D; Weinberg RA The Hallmarks of Cancer. Cell 2000, 100 (1), 57–70. [DOI] [PubMed] [Google Scholar]
- (4).Hanahan D; Weinberg RA Hallmarks of Cancer: The Next Generation. Cell 2011, 144 (5), 646–674. [DOI] [PubMed] [Google Scholar]
- (5).Pylayeva-Gupta Y; Grabocka E; Bar-Sagi D RAS Oncogenes: Weaving a Tumorigenic Web. Nat. Rev. Cancer 2011, 11 (11), 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chin L; Tam A; Pomerantz J; Wong M; Holash J; Bardeesy N; Shen Q; O’Hagan R; Pantginis J; Zhou H; Horner JW; Cordon-Cardo C; Yancopoulos GD; DePinho RA Essential Role for Oncogenic Ras in Tumour Maintenance. Nature 1999, 400 (6743), 468–472. [DOI] [PubMed] [Google Scholar]
- (7).Vigil D; Cherfils J; Rossman KL; Der CJ Ras Superfamily GEFs and GAPs : Validated and Tractable Targets for Cancer Therapy ? Nat Rev Cancer 2010, 10, 842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rao S; Cunningham D; de Gramont A; Scheithauer W; Smakal M; Humblet Y; Kourteva G; Iveson T; Andre T; Dostalova J; Illes A; Belly R; Perez-Ruixo JJ; Park YC; Palmer PA Phase III Double-Blind Placebo-Controlled Study of Farnesyl Transferase Inhibitor R115777 in Patients With Refractory Advanced Colorectal Cancer. J. Clin. Oncol 2004, 22 (19), 3950–3957. [DOI] [PubMed] [Google Scholar]
- (9).Maurer T; Garrenton LS; Oh A; Pitts K; Anderson DJ; Skelton NJ; Fauber BP; Pan B; Malek S; Stokoe D; Ludlam MJC; Bowman KK; Wu J; Giannetti AM; Starovasnik MA; Mellman I; Jackson PK; Rudolph J; Wang W; Fang G Small-Molecule Ligands Bind to a Distinct Pocket in Ras and Inhibit SOS-Mediated Nucleotide Exchange Activity. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (14), 5299–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sun Q; Burke JP; Phan J; Burns MC; Olejniczak ET; Waterson AG; Lee T; Rossanese OW; Fesik SW Discovery of Small Molecules That Bind to K-Ras and Inhibit Sos-Mediated Activation. Angew. Chemie Int. Ed 2012, 51 (25), 6140–6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Waldmann H; Karaguni I-M; Carpintero M; Gourzoulidou E; Herrmann C; Brockmann C; Oschkinat H; Müller O Sulindac-Derived Ras Pathway Inhibitors Target the Ras–Raf Interaction and Downstream Effectors in the Ras Pathway. Angew. Chemie Int. Ed 2004, 43 (4), 454–458. [DOI] [PubMed] [Google Scholar]
- (12).Rosnizeck IC; Graf T; Spoerner M; Tränkle J; Filchtinski D; Herrmann C; Gremer L; Vetter IR; Wittinghofer A; König B; Kalbitzer HR Stabilizing a Weak Binding State for Effectors in the Human Ras Protein by Cyclen Complexes. Angew. Chemie Int. Ed 2010, 49 (22), 3830–3833. [DOI] [PubMed] [Google Scholar]
- (13).Wang W; Fang G; Rudolph J Ras Inhibition via Direct Ras Binding—is There a Path Forward? Bioorg. Med. Chem. Lett 2012, 22 (18), 5766–5776. [DOI] [PubMed] [Google Scholar]
- (14).Hocker HJ; Cho K-J; Chen C-YK; Rambahal N; Sagineedu SR; Shaari K; Stanslas J; Hancock JF; Gorfe AA Andrographolide Derivatives Inhibit Guanine Nucleotide Exchange and Abrogate Oncogenic Ras Function. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (25), 10201–10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ostrem JM; Peters U; Sos ML; Wells JA; Shokat KM K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503 (7477), 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shima F; Yoshikawa Y; Ye M; Araki M; Matsumoto S; Liao J; Hu L; Sugimoto T; Ijiri Y; Takeda A; Nishiyama Y; Sato C; Muraoka S; Tamura A; Osoda T; Tsuda K.-i.; Miyakawa T; Fukunishi H; Shimada J; Kumasaka T; Yamamoto M; Kataoka T In Silico Discovery of Small-Molecule Ras Inhibitors That Display Antitumor Activity by Blocking the Ras-Effector Interaction. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (20), 8182–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Spiegel J; Cromm PM; Zimmermann G; Grossmann TN; Waldmann H Small-Molecule Modulation of Ras Signaling. Nat. Chem. Biol 2014, 10 (8), 613–622. [DOI] [PubMed] [Google Scholar]
- (18).Leshchiner ES; Parkhitko A; Bird GH; Luccarelli J; Bellairs JA; Escudero S; Opoku-Nsiah K; Godes M; Perrimon N; Walensky LD Direct Inhibition of Oncogenic KRAS by Hydrocarbon-Stapled SOS1 Helices. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (6), 1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ostrem JML; Shokat KM Direct Small-Molecule Inhibitors of KRAS: From Structural Insights to Mechanism-Based Design. Nat. Rev. Drug Discov 2016, 15 (11), 771–785. [DOI] [PubMed] [Google Scholar]
- (20).Bourne HR; Sanders DA; McCormick F The GTPase Superfamily: A Conserved Switch for Diverse Cell Functions. Nature 1990, 348 (6297), 125–132. [DOI] [PubMed] [Google Scholar]
- (21).Konstantinopoulos PA; Karamouzis MV; Papavassiliou AG Post-Translational Modifications and Regulation of the RAS Superfamily of GTPases as Anticancer Targets. Nat. Rev. Drug Discov 2007, 6 (7), 541–555. [DOI] [PubMed] [Google Scholar]
- (22).Jeng H-H; Taylor LJ; Bar-Sagi D Sos-Mediated Cross-Activation of Wild-Type Ras by Oncogenic Ras Is Essential for Tumorigenesis. Nat. Commun 2012, 3 (1), 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Winter JJG; Anderson M; Blades K; Brassington C; Breeze AL; Chresta C; Embrey K; Fairley G; Faulder P; Finlay MRV; Kettle JG; Nowak T; Overman R; Patel SJ; Perkins P; Spadola L; Tart J; Tucker JA; Wrigley G Small Molecule Binding Sites on the Ras:SOS Complex Can Be Exploited for Inhibition of Ras Activation. J. Med. Chem 2015, 58 (5), 2265–2274. [DOI] [PubMed] [Google Scholar]
- (24).Burns MC; Sun Q; Daniels RN; Camper D; Kennedy JP; Phan J; Olejniczak ET; Lee T; Waterson AG; Rossanese OW; Fesik SW Approach for Targeting Ras with Small Molecules That Activate SOS-Mediated Nucleotide Exchange. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 3401–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Abbott JR; Hodges TR; Daniels RN; Patel PA; Kennedy JP; Howes JE; Akan DT; Burns MC; Sai J; Sobolik T; Beesetty Y; Lee T; Rossanese OW; Phan J; Waterson AG; Fesik SW Discovery of Aminopiperidine Indoles That Activate the Guanine Nucleotide Exchange Factor SOS1 and Modulate RAS Signaling. J. Med. Chem 2018, 61 (14), 6002–6017. [DOI] [PubMed] [Google Scholar]
- (26).Abbott JR; Patel PA; Howes JE; Akan DT; Kennedy JP; Burns MC; Browning CF; Sun Q; Rossanese OW; Phan J; Waterson AG; Fesik SW Discovery of Quinazolines That Activate SOS1-Mediated Nucleotide Exchange on RAS. ACS Med. Chem. Lett [Online early-access]. DOI: 10.1021/acsmedchemlett.8b00296. Published Online: August 8, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Howes JE; Akan DT; Burns MC; Rossanese OW; Waterson AG; Fesik SW Small Molecule–Mediated Activation of RAS Elicits Biphasic Modulation of Phospho-ERK Levels That Are Regulated through Negative Feedback on SOS1. Mol. Cancer Ther 2018, 17 (5), 1051–1060. [DOI] [PubMed] [Google Scholar]
- (28).Chi S; Kitanaka C; Noguchi K; Mochizuki T; Nagashima Y; Shirouzu M; Fujita H; Yoshida M; Chen W; Asai A; Himeno M; Yokoyama S; Kuchino Y Oncogenic Ras Triggers Cell Suicide through the Activation of a Caspase-Independent Cell Death Program in Human Cancer Cells. Oncogene 1999, 18 (13), 2281–2290. [DOI] [PubMed] [Google Scholar]
- (29).Overmeyer JH; Maltese WA Death Pathways Triggered by Activated Ras in Cancer Cells. Front. Biosci 2011, 16, 1693–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lv C; Hong Y; Miao L; Li C; Xu G; Wei S; Wang B; Huang C; Jiao B Wentilactone A as a Novel Potential Antitumor Agent Induces Apoptosis and G2/M Arrest of Human Lung Carcinoma Cells and Is Mediated by HRas-GTP Accumulation to Excessively Activate the Ras/Raf/ERK/P53-P21 Pathway. Cell Death Dis 2013, 4 (12), e952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Martin SJ Oncogene-Induced Autophagy and the Goldilocks Principle. Autophagy 2011, 7 (8), 922–923. [DOI] [PubMed] [Google Scholar]
- (32).Overmeyer JH; Kaul A; Johnson EE; Maltese WA Active Ras Triggers Death in Glioblastoma Cells through Hyperstimulation of Macropinocytosis. Mol. Cancer Res 2008, 6 (6), 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Unni AM; Lockwood WW; Zejnullahu K; Lee-Lin S-Q; Varmus H Evidence That Synthetic Lethality Underlies the Mutual Exclusivity of Oncogenic KRAS and EGFR Mutations in Lung Adenocarcinoma. Elife 2015, 4, e06907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ambrogio C; Barbacid M; Santamaría D In Vivo Oncogenic Conflict Triggered by Co-Existing KRAS and EGFR Activating Mutations in Lung Adenocarcinoma. Oncogene 2017, 36 (16), 2309–2318. [DOI] [PubMed] [Google Scholar]
- (35).Petti C; Molla A; Vegetti C; Ferrone S; Anichini A; Sensi M Coexpression of NRAS Q61R and BRAF V600E in Human Melanoma Cells Activates Senescence and Increases Susceptibility to Cell-Mediated Cytotoxicity. Cancer Res 2006, 66 (13), 6503–6511. [DOI] [PubMed] [Google Scholar]
- (36).Burns MC; Howes JE; Sun Q; Little AJ; Camper DV; Abbott JR; Phan J; Lee T; Waterson AG; Rossanese OW; Fesik SW High-Throughput Screening Identifies Small Molecules That Bind to the RAS:SOS:RAS Complex and Perturb RAS Signaling. Anal. Biochem 2018, 548, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Veber DF; Johnson SR; Cheng H-Y; Smith BR; Ward KW; Kopple KD Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem 2002, 45 (12), 2615–2623. [DOI] [PubMed] [Google Scholar]
- (38).Souza-Fagundes EM; Frank AO; Feldkamp MD; Dorset DC; Chazin WJ; Rossanese OW; Olejniczak ET; Fesik SW A High-Throughput Fluorescence Polarization Anisotropy Assay for the 70N Domain of Replication Protein A. Anal. Biochem 2012, 421 (2), 742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Nikolovska-Coleska Z; Wang R; Fang X; Pan H; Tomita Y; Li P; Roller PP; Krajewski K; Saito NG; Stuckey JA; Wang S Development and Optimization of a Binding Assay for the XIAP BIR3 Domain Using Fluorescence Polarization. Anal. Biochem 2004, 332 (2), 261–273. [DOI] [PubMed] [Google Scholar]
- (40).Leeson PD; Springthorpe B The Influence of Drug-like Concepts on Decision-Making in Medicinal Chemistry. Nat. Rev. Drug Discov 2007, 6 (11), 881–890. [DOI] [PubMed] [Google Scholar]
- (41).Zheng Y; Tice CM; Singh SB The Use of Spirocyclic Scaffolds in Drug Discovery. Bioorg. Med. Chem. Lett 2014, 24 (16), 3673–3682. [DOI] [PubMed] [Google Scholar]
- (42).Burkhard J; Carreira EM 2,6-Diazaspiro[3.3]Heptanes: Synthesis and Application in Pd-Catalyzed Aryl Amination Reactions. Org. Lett 2008, 10 (16), 3525–3526. [DOI] [PubMed] [Google Scholar]
- (43).Burkhard J. a; Wagner B; Fischer H; Schuler F; Müller K; Carreira EM Synthesis of Azaspirocycles and Their Evaluation in Drug Discovery. Angew. Chem. Int. Ed. Engl 2010, 49 (20), 3524–3527. [DOI] [PubMed] [Google Scholar]
- (44).Goodwin JT; Conradi RA; Ho NFH; Burton PS Physicochemical Determinants of Passive Membrane Permeability: Role of Solute Hydrogen-Bonding Potential and Volume. J. Med. Chem 2001, 44 (22), 3721–3729. [DOI] [PubMed] [Google Scholar]
- (45).Liu X; Wright M; Hop CECA Rational Use of Plasma Protein and Tissue Binding Data in Drug Design. J. Med. Chem 2014, 57 (20), 8238–8248. [DOI] [PubMed] [Google Scholar]
- (46).Aronheim A; Engelberg D; Li N; Al-Alawi N; Schlessinger J; Karin M Membrane Targeting of the Nucleotide Exchange Factor Sos Is Sufficient for Activating the Ras Signaling Pathway. Cell 1994, 78 (6), 949–961. [DOI] [PubMed] [Google Scholar]
- (47).Kamioka Y; Yasuda S; Fujita Y; Aoki K; Matsuda M Multiple Decisive Phosphorylation Sites for the Negative Feedback Regulation of SOS1 via ERK. J. Biol. Chem 2010, 285 (43), 33540–33548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Iversen L; Tu H-L; Lin W-C; Christensen SM; Abel SM; Iwig J; Wu H-J; Gureasko J; Rhodes C; Petit RS; Hansen SD; Thill P; Yu C-H; Stamou D; Chakraborty AK; Kuriyan J; Groves JT Ras Activation by SOS: Allosteric Regulation by Altered Fluctuation Dynamics. Science 2014, 345 (6192), 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Soll M; Le Hir De Fallois L; Huber S; Lee H; Douglas E; Jacobs R; Beck B Preparation of Enantiomerically Enriched Aryloazol-2-Yl Cyanoethylamino Parasiticidal Compounds. WO 2010056999, 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.