

Abstract
Trichloroethene (TCE), a widely used industrial solvent, is associated with the development of autoimmune diseases (ADs), including systemic lupus erythematosus and autoimmune hepatitis. Increasing evidence support a linkage between altered gut microbiome composition and the onset of ADs. However, it is not clear how gut microbiome contributes to TCE-mediated autoimmunity, and initial triggers for microbiome-host interactions leading to systemic autoimmune responses remain unknown. To achieve this, female MRL+/+ mice were treated with 0.5 mg/ml TCE for 52 weeks and fecal samples were subjected to 16S rRNA sequencing to determine the microbiome composition. TCE exposure resulted in distinct bacterial community revealed by β-diversity analysis. Notably, we observed reduction in Lactobacillaceae, Rikenellaceae and Bifidobacteriaceae families, and enrichment of Akkermansiaceae and Lachnospiraceae families after TCE exposure. We also observed significantly increased colonic oxidative stress and inflammatory markers (CD14 and IL-1β), and decreased tight junction proteins (ZO-2, occludin and claudin-3). These changes were associated with increases in serum antinuclear and anti-smooth muscle antibodies and cytokines (IL-6 and IL-12), together with increased PD1+CD4+ T cells in TCE-exposed spleen and liver tissues. Importantly, fecal microbiota transplantation (FMT) using feces from TCE-treated mice to antibiotics-treated mice induced increased anti-dsDNA antibodies and hepatic CD4+ T cell infiltration in the recipient mice. Our studies thus delineate how imbalance in gut microbiome and mucosal redox status together with gut inflammatory response and permeability changes could be the key factors in contributing to TCE-mediated ADs. Furthermore, FMT studies provide a solid support to a causal role of microbiome in TCE-mediated autoimmunity.
Keywords: Environmental pollutant, microbiome, oxidative stress, permeability, inflammation, autoimmunity
Introduction
Autoimmune diseases (ADs) are chronic inflammatory diseases primarily mediated by autoreactive lymphocytes and circulating autoantibodies (Floreani et al., 2018; Doycheva et al., 2019). The etiology of ADs remains largely unclear but a number of factors, including genetic predisposition, hormonal and environmental exposures, can potentially contribute to disease progression (Cho and Gregersen, 2011). Trichloroethene (TCE), an organic solvent which is widely used as a cleaning and degreasing agent, is a ubiquitous environmental pollutant. TCE exposure via inhalation, ingestion or dermal contact results in of immune dysregulation in humans (Anagnostopoulos et al., 2004; Iavicoli et al., 2005; Kamijima et al., 2008). An estimated 3.5 million people are occupationally exposed to TCE, and more importantly, an estimated 14 million Americans are exposed to it via contaminated water (ATSDR, 2011). TCE exposure via drinking water in our established mouse model (MRL+/+) has been shown to result in increased oxidative stress (OS), impaired Kupffer cell function, hepatic inflammation/ inflammasome activation, glomerulonephritis and autoantibody production leading to autoimmune hepatitis (AIH) and systemic lupus erythematosus (SLE) (Khan et al., 1995; Cai et al., 2008; Wang et al., 2012b; Wang et al., 2019). A recent study showed that an early life TCE exposure is also associated with altered gut microbiome (Khare et al., 2019). However, whether TCE-induced gut microbiome dysbiosis is a causal factor or an outcome of the ADs remains unknown, and necessitates a clear understanding of how crosstalk between gut microbiota and host immune system promotes TCE-mediated systemic autoimmune responses.
Recent studies provide evidence that ADs are associated with an altered gut microbiome composition and impaired barrier function (Lin et al., 2015; Cai et al., 2017). AD patients with active disease have imbalances (dysbiosis) in the distribution/diversity of bacterial taxa in their intestinal microbiome with lower bacterial diversity and altered relative abundances (Hevia et al., 2014; DeGruttola et al., 2016; Luo et al., 2018). Increases in Ruminococcus gnavus of Lachnospiraceae family, and higher levels of fecal secretory IgA and calprotectin levels are reported in SLE patients (Azzouz et al., 2019). Also, enrichment in Veillonella genus correlated with AIH disease activity (Wei et al., 2020). Furthermore, impaired intestinal integrity as a result of decreased expression of tight junction proteins can lead to elevated plasma endotoxin lipopolysaccharide (LPS) levels, and chronic systemic microbial translocation can contribute to pathogenesis of SLE/AIH (Lin et al., 2015; Manfredo Vieira et al., 2018; Khan and Wang, 2019; Albillos et al., 2020). Even through, impaired gut epithelial integrity and barrier function are considered important predisposing factors to a number of inflammatory diseases, it is not known if TCE exposure also predisposes the host through a similar mechanism, resulting in autoimmune responses.
Increased activation of autoimmune effector T cells or inhibition of regulatory T cells are contributing factors in ADs (Skapenko et al., 2005). Our previous studies have shown that TCE exposure is associated with increased OS and substantial inflammatory T and B cell activation (Wang et al., 2008; Wang et al., 2019). Programmed cell death protein 1 (PD-1), an immune checkpoint, is mainly expressed by activated T and B cells, which infiltrate in tissues and contribute to AD progression (Liang et al., 2003; Salama et al., 2003). Intestinal microbiome can function as a trigger for autoreactive T and B cell responses that drive autoimmune responses in target organs (Lee and Kim, 2017; Ruff et al., 2019). Microbiome-derived short chain fatty acids (SCFAs) exhibit protective role in ADs through expansion of regulatory T cells (Zeng and Chi, 2015). Gut microbiome dysbiosis-mediated inflammation can also contribute to autoimmune responses by increasing membrane permeability and gut barrier dysfunction (Yu, 2018; Zhang et al., 2020). Although previous studies have shown that changes in gut microbiome and related intestinal modifications are possibly associated with ADs, there is a significant knowledge gap on the role of gut microbiome, especially chemical-host interactions and underlying mechanisms leading to ADs.
This study was primarily focused on determining whether the gut microbiome contributes to TCE-mediated SLE/AIH, and establishing a causal link between gut microbiome dysbiosis and autoimmune responses. To achieve these objectives, we first characterized the gut microbiome composition in female MRL+/+ mice after TCE treatment, and also analyzed various lymphocyte populations and autoantibodies, which are typical characteristics of SLE/AIH disease. Mechanistically, we evaluated intestinal OS, tight junction proteins (signs of leaky gut) and inflammatory markers to establish whether TCE induced microbiome dysbiosis can alter intestinal barrier function and mucosal immune responses, which could potentially promote systemic aberrant autoimmune response. Furthermore, fecal microbiota transplantation (FMT) from TCE-treated donor mice to antibiotic-treated recipient mice was performed to establish casual role of gut microbiome in TCE-mediated autoimmunity. This study thus identified changes in gut microbiome and unraveled potential mechanisms (intestinal integrity and mucosal immunity) that could contribute to systemic inflammatory immune disorders (SLE and AIH) following TCE exposure. Furthermore, FMT studies provide a firm support to a causal role of microbiome in TCE-mediated autoimmunity.
Method:
Animals and TCE treatment
Five-week old female MRL+/+ (Murphy Roths Large, MRL/MpJ; stock No: 000486) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained under specific pathogen-free facility and acclimatized for one week prior to any treatment. All experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch. Mice, randomly divided into groups of 8 each, were given TCE (Sigma, St. Louis, MO) at 0.5 mg/ml in drinking water ad libitum for 52 weeks. The MRL+/+ mouse spontaneously develops autoantibodies after several months and SLE late in the second year of life, which makes it an ideal model to study the effects of TCE in exacerbating ADs (Andrews et al., 1978; Khan et al., 1995; Gilbert et al., 1999). Female mice were chosen for these studies due to higher prevalence of ADs in females and also based on existing studies using this animal model, especially with TCE exposure (Andrews et al., 1978; Khan et al., 1995; Gilbert et al., 1999). Also, the choice of TCE dose (0.5 mg/ml) was based on earlier published studies, and this dose is lower than current 8-hour Permissible Exposure Limit [established by the Occupational Safety and Health Administration (OSHA)] for TCE of 100 ppm or approximately 76 mg/kg/day (Cai et al., 2008; Wang et al., 2012b; Gilbert et al., 2014). To circumvent the low water solubility of TCE, 1% Alkamuls EL-620, an emulsifier (Rhodia Chemicals, Cranbury, NJ), was used which has no adverse effects on mice or disease outcome. Control mice received drinking water containing 1% emulsifier only. After 52 weeks of TCE treatment, all the animals were euthanized. Major organs and feces were collected, and then stored at −80 °C for further analysis. Sera obtained from blood samples were stored in small aliquots at −80 °C until further analysis.
Fecal microbiota transplantation (FMT) studies
Four-week-old MRL-lpr mice were obtained from Jackson Laboratory. These mice can develop rapid and aggressive SLE disease, with autoantibody generation as early as 6 weeks and glomerulonephritis around 16 weeks (Chu et al., 1993). Mice were treated with broad-spectrum antibiotics cocktail (0.5 g/l vancomycin, 1.0 g/l ampicillin, 1.0 g/l metronidazole and 1.0 g/l neomycin) via drinking water for 4 days followed by 3 days of water only to clear up the antibiotics from the system (Mu et al., 2017). Mice were then orally fed (3 times/week) with fecal materials from 52-week control (CON) or TCE-treated MRL+/+ mice for two weeks. Feces (200 mg) collected from both control and TCE-treated mice were dissolved in 1 ml sterile PBS, centrifuged, and 200 μl supernatants were orally fed to the recipient MRL-lpr mice, essentially as described earlier (Mu et al., 2019). One week following the microbiome inoculation, mice were sacrificed, blood and organs collected, and the autoimmune parameters were assessed. Feces were collected for microbiome composition analysis.
Microbiome analysis (16S rRNA sequencing)
Taking fecal samples form control and TCE-exposed mice, DNA isolation and 16S rRNA sequencing were performed at the Alkek Center for Metagenomics and Microbiome Research at Baylor College of Medicine. Briefly, total bacterial genomic DNA was extracted using the MagAttract PowerSoil Kit (Qiagen, Redwood City, CA). The 16Sv4 region was amplified by PCR and sequenced on the MiSeq platform (Illumina, San Diego, CA) using a 2×250 bp paired-end protocol, yielding paired-end reads that overlap almost completely. Agile Toolkit for Incisive Microbial Analyses (ATIMA) was used to analyze microbiome data, which is a stand-alone tool for analyzing and visualizing trends in taxa abundance, alpha diversity, and beta diversity as they relate to sample metadata (Quast et al., 2013). Quantitative PCR was performed to further verify certain bacterial changes.
Enzyme-linked immunosorbent assays (ELISAs) and Bio-Plex assay
Serum autoantibodies were determined by using mouse-specific ELISA kits for antinuclear antibodies (ANA), anti-dsDNA antibodies (Alpha Diagnostic Int’l, San Antonio, TX) and anti-smooth muscle antibodies (ASMA) (Cusabio LLC, Houston, TX), respectively. Serum lipocalin-2 levels were measured by mouse lipocalin-2/NGAL quantikine ELISA kit (R&D Systems, Minneapolis, MN), according to the manufacturer’s instructions. MDA-protein adducts in the liver and colon tissues were analyzed according to our earlier published methods (Wang et al., 2008; Wang et al., 2012a; Wang et al., 2012b). Serum cytokines were determined using Cytokine 17-Plex Mouse ProcartaPlex Panel (Invitrogen, Carlsbad, CA) by following the manufacturer’s instructions.
Isolation of lymphocytes from spleen and liver tissues
Intrahepatic lymphocytes were isolated following our earlier published method (Wang et al., 2019). Briefly, the liver was perfused with PBS, minced and digested with RPMI-1640 containing 0.05% collagenase IV (Roche, Indianapolis, IN) at 37 °C for 30 min. Cell suspensions were passed through 70-μm cell strainers, followed by a enrichment centrifugation (400g for 30 min) over a 30/70% discontinuous Percoll density gradient (Sigma) at room temperature. The cells were then collected from the interphase, washed, and resuspended in complete RPMI-1640 containing 10% FBS. The spleens were gently mashed in RPMI-1640 medium through a cell strainer. Red blood cells were removed by using Red Cell Lysis buffer (Sigma). Cells were harvested by centrifugation and resuspended in complete RPMI-1640 containing 10% FBS.
Flow cytometry
For surface staining, cells were first incubated with FcγR blocker (CD16/32), followed by fluorochrome-labeled antibodies (Abs). For intracellular staining, cells were stimulated by phorbol myristate acetate (50 ng/ml) and ionomycin (750 ng/ml) for 5 hrs. After incubation, cells were stained for surface markers first, then fixed by using Foxp3/transcription factor staining set followed by intracellular staining. The specific antibodies and their corresponding isotype controls were purchased from Biolegend (San Diego, CA) and eBioscience (Waltham, MA). The following Abs were used in combinations: PE-Cy7 anti-mouse CD3, Pacific Blue anti-mouse CD4, APC-Cy7 anti-mouse CD8, APC anti-mouse CD11b, APC-Cy7 anti-mouse CD11c, AF700 anti-mouse CD19, Percp-cy5.5 anti-mouse CD45R/B220, FITC anti-mouse CD279 (PD-1) and PEAF610 anti-mouse IFN-γ Abs (BD Pharmingen, San Jose, CA). Flow cytometric analysis were done using an LSRII Fortessa (Becton Dickinson, San Jose, CA), and the data analyzed by using FlowJo software 10.0 (TreeStar, Ashland, OR).
Quantitative reverse transcriptase PCR (qRT-PCR) analysis
RNA was extracted from colon tissues using Trizol reagent (Sigma) followed by treatment with Qiagen DNase I (Qiagen, Hilden, Germany). cDNA was prepared using iScript reverse transcription supermix (Bio-Rad, Hercules, CA). qPCR was performed using universal SYBR green supermix kit (Bio-Rad) on a Bio-Rad CFX96 real time PCR machine. The mRNA expression of selected genes related to inflammation was determined. Mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the housekeeping gene. The primer sequences for the genes analyzed were according to our previous publication (Wang et al., 2019).
Western blot analysis
Total colon tissues were homogenized in T-PER lysis buffer containing 1% protease inhibitor cocktail (Sigma) and protein concentration was determined by Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA). Western blot analysis was done for tight junction proteins. Ten μg protein per lane was loaded onto 4–20% Trisglycine gel (Thermo Fisher Scientific) and transferred to PVDF membrane. The membrane was blotted with primary antibodies at 4°C overnight. Antibody detection was accomplished using horseradish peroxidase conjugated secondary antibodies and visualized with ECL. The signal intensity was quantified with Image Studio Lite Ver 5.2 (LI-COR).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software 7.0 (GraphPad, La Jolla, CA). Data are shown as mean ± SEM and were analyzed using the two-tailed Student’s t test when comparison was made between two groups. One-way analysis of variance (ANOVA) followed by Tukey-Kramer test was used for multiple group comparisons. The p values <0.05 were considered to be statistically significant. *p < 0.05; **p < 0.01. For microbiome data processed via ATIMA website, statistical tests used are Kruskall-Wallis and Mann-Whitney for alpha diversity and taxa abundance, and the PERMANOVA test for beta diversity.
Results:
Long-term TCE exposure induces autoimmune response in MRL +/+ mice
To demonstrate that TCE exposure induces autoimmune responses and to relate these responses with changes in microbiome, female MRL +/+ mice were treated with TCE via drinking water for 52 weeks. TCE treatment led to significant increases in ANA and ASMA, indicating that chronic low-dose TCE exposure exacerbates the autoimmune disease activities (Figure 1A–B). In addition, TCE exposure led to systemic inflammatory response, evidenced by increased serum levels of IL-6 and IL-12 (Figure 1C). The increases in ANA are consistent with our earlier findings showing that long-term TCE exposure can induce SLE-like disease in MRL+/+ mice (Wang et al., 2012b). The observed increases in ASMA along with ANA also suggest the onset of AIH disease in these mice after TCE exposure.
Figure 1.
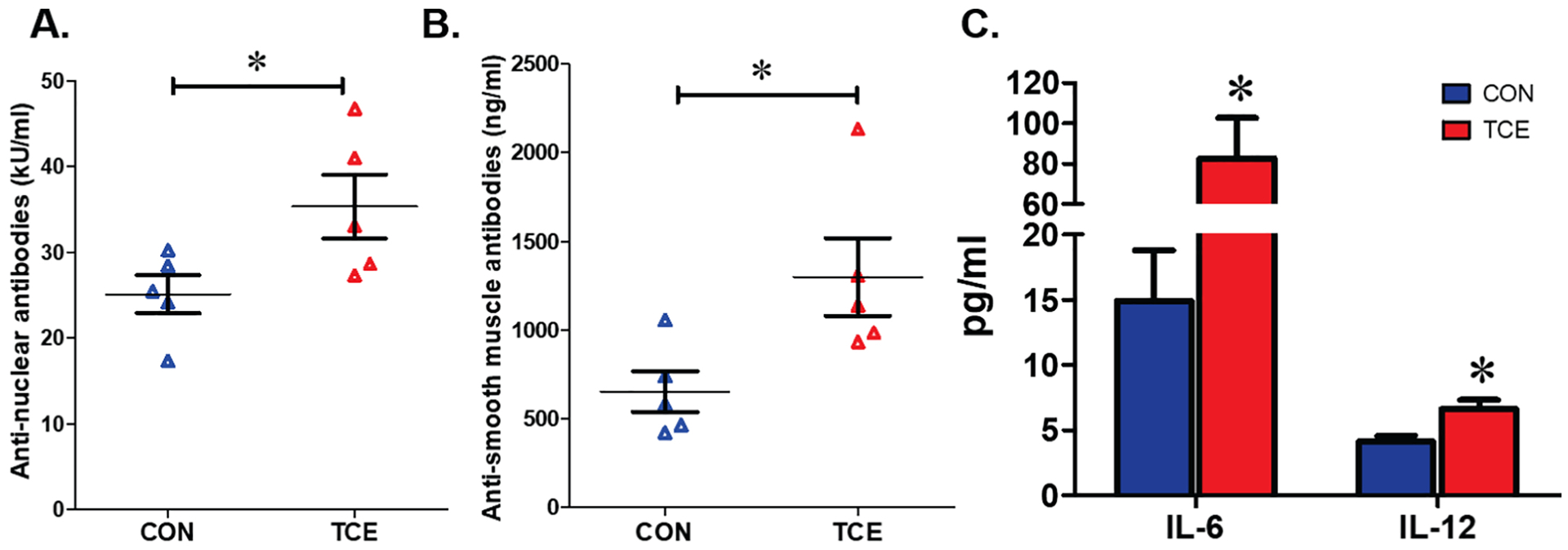
Chronic TCE exposure leads to autoimmune and inflammatory responses in MRL+/+ mice. Mice were exposed to 0.5 mg/ml TCE for 52 weeks. The autoantibodies and cytokines were measured to monitor autoimmune disease activities. Serum levels of ANA levels (A), ASMA levels (B) and pro-inflammatory IL-6 and IL-12 levels (C). Results are mean ± SEM. n=5 *p<0.05.
Splenic and hepatic immune responses after TCE exposure
To determine the contribution of immune cells in TCE-mediated SLE/AIH, various cell populations from spleen and liver were analyzed. TCE exposure resulted in significantly increased CD4+ T cells (Figure 2A) and a reducing trend for macrophages (CD11b+F4/80+) (Suppl. Figure 1) in the spleen. Although hepatic CD4+ T cells did not show any significant change after TCE exposure, reduced percentage of macrophages and dendritic cells were observed (Suppl. Figure 1). Furthermore, hepatic infiltrating CD4+ T cells from TCE-treated mice produced more INF-γ than those of controls (Figure 2E). Interestingly, we also observed significantly increased PD-1 expression in CD4+ T cells in the spleen and liver after TCE exposure (Figure 2C,F). Overall, our data show that TCE-mediated autoimmunity is associated with immune cell dysregulations, especially hepatic CD4+ T cell activation and reduced macrophages and dendritic cells.
Figure 2.
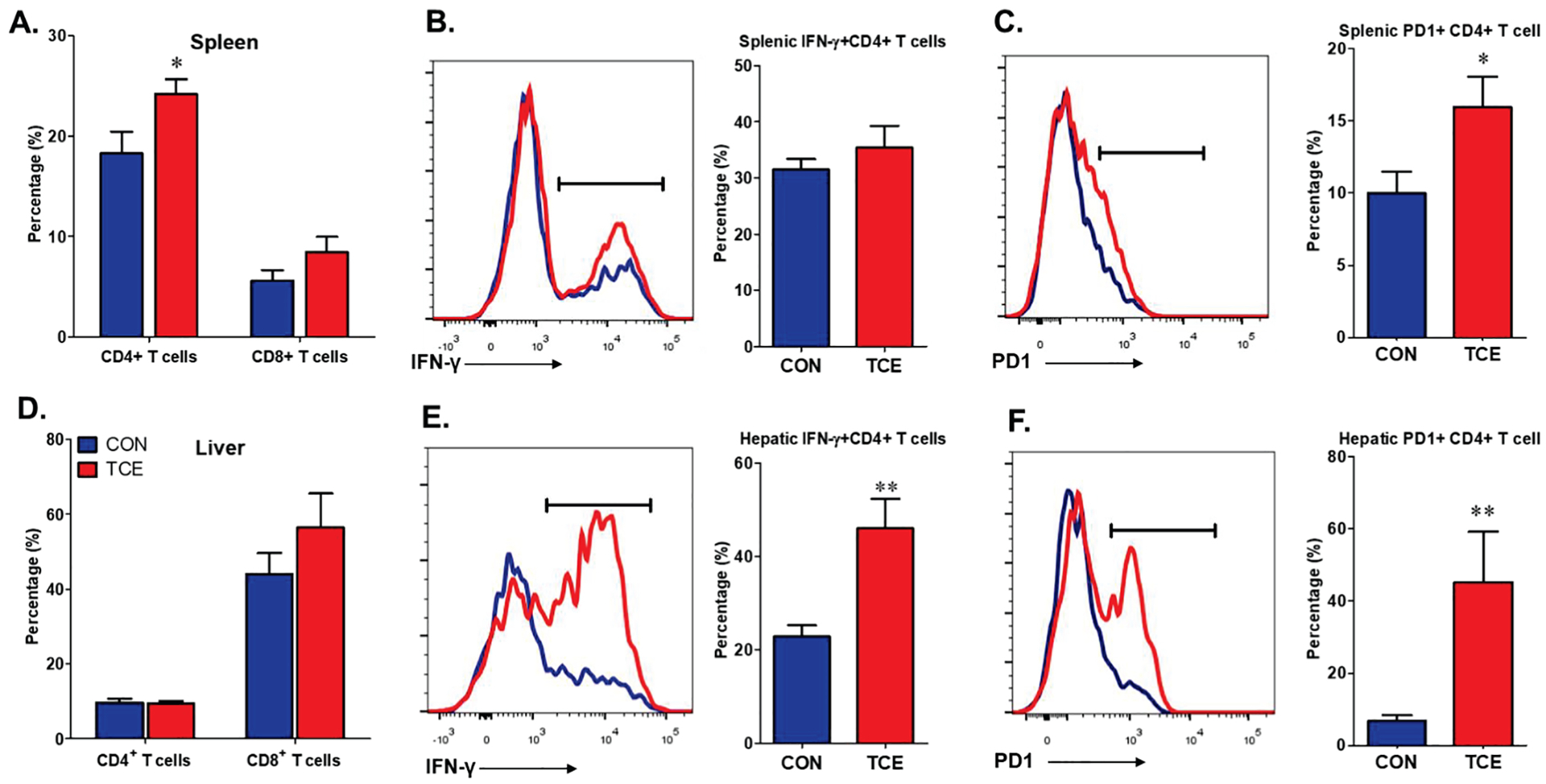
Splenic and hepatic T cell infiltration in CON and TCE-exposed MRL+/+ mice. Lymphocytes were harvested from the spleen and liver, then characterized for T cells. Quantification of flow cytometry analysis for CD4+ and CD8+ T cells (A), IFN-γ+ CD4+ T cells (B), PD1+ CD4+ T cells (C) in the spleen. Flow cytometry analysis for CD4+ and CD8+ T cells (D), IFN-γ+ CD4+ T cells (E), PD1+ CD4+ T cells (F) in intrahepatic lymphocytes. Results are mean ± SEM. n=5 *p<0.05; **p<0.01.
TCE exposure alters gut microbiome composition in MRL +/+ mice
Changes in gut microbiome composition (dysbiosis) can play a pivotal role in triggering an autoimmune response in susceptible individuals (Belkaid and Hand, 2014; Khan and Wang, 2019). To determine the potential of TCE in causing microbiome dysbiosis, fecal samples from TCE-treated and control mice were collected at 52 weeks and subjected to 16S rRNA sequencing. The α-diversity of bacterial communities was evaluated according to observed OTUs and Shannon’s diversity index. No significant change was observed in α-diversity of gut microbiome after TCE exposure (Figure 3A). As revealed by β-diversity analysis, TCE exposure resulted in distinct bacterial community clustering (Figure 3B). Notably, differences were observed for both unweighted and weighted measures, indicating that TCE treatment impacted not only the type of bacteria present but also the distribution of shared bacteria (Figure 3B). We subsequently identified significant TCE-induced changes at different taxonomical levels, including 6 bacterial families and 8 bacterial genera. Specifically, we observed reductions in the relative abundance of bacterial families Erysiopelotrichaceae and Lactobacillaceae under Firmicutes phylum and bacterial families Marinifilaceae and Rikenellaceae under Bacteroidetes phylum in TCE-treated animals as compared to controls (Figure 3C,D). In addition, significantly reduced Bifidobacteriaceae and Erysipelotrichaceae were observed in TCE-treated group (Figure 3D). At the lower taxonomic level, TCE exposure led to consistent decreasing trend in the genera Lactobacillus and Bifidobacterium (Figure 3E), which are established probiotics known to promote induction of Treg cells, and their reduced levels could thus promote ADs(Mu et al., 2017). TCE exposure, on the other hand, also showed enrichment of Akkermansia and Oscillibacter (Figure 3E). Other reductions at the genus level included Turicibacter and Faecalibaculum under Firmicutes phylum, and Alistipes and Odoribacter under Bacteroidetes phylum (Figure 3E). We further verified the enrichment of Akkermansia by RT-PCR with primers specific for Akkermansia muciniphila following TCE exposure. Consistently, TCE exposure induced significantly increased fecal Akkermansia muciniphila (Figure 3F). In addition, we also observed decreasing trend for Faecalibacterium prausnitzii after TCE exposure (Figure 3F). Our data thus show that TCE exposure causes significant alterations in the gut microbiome and suggest an association of gut dysbiosis with SLE disease pathogenesis.
Figure 3.
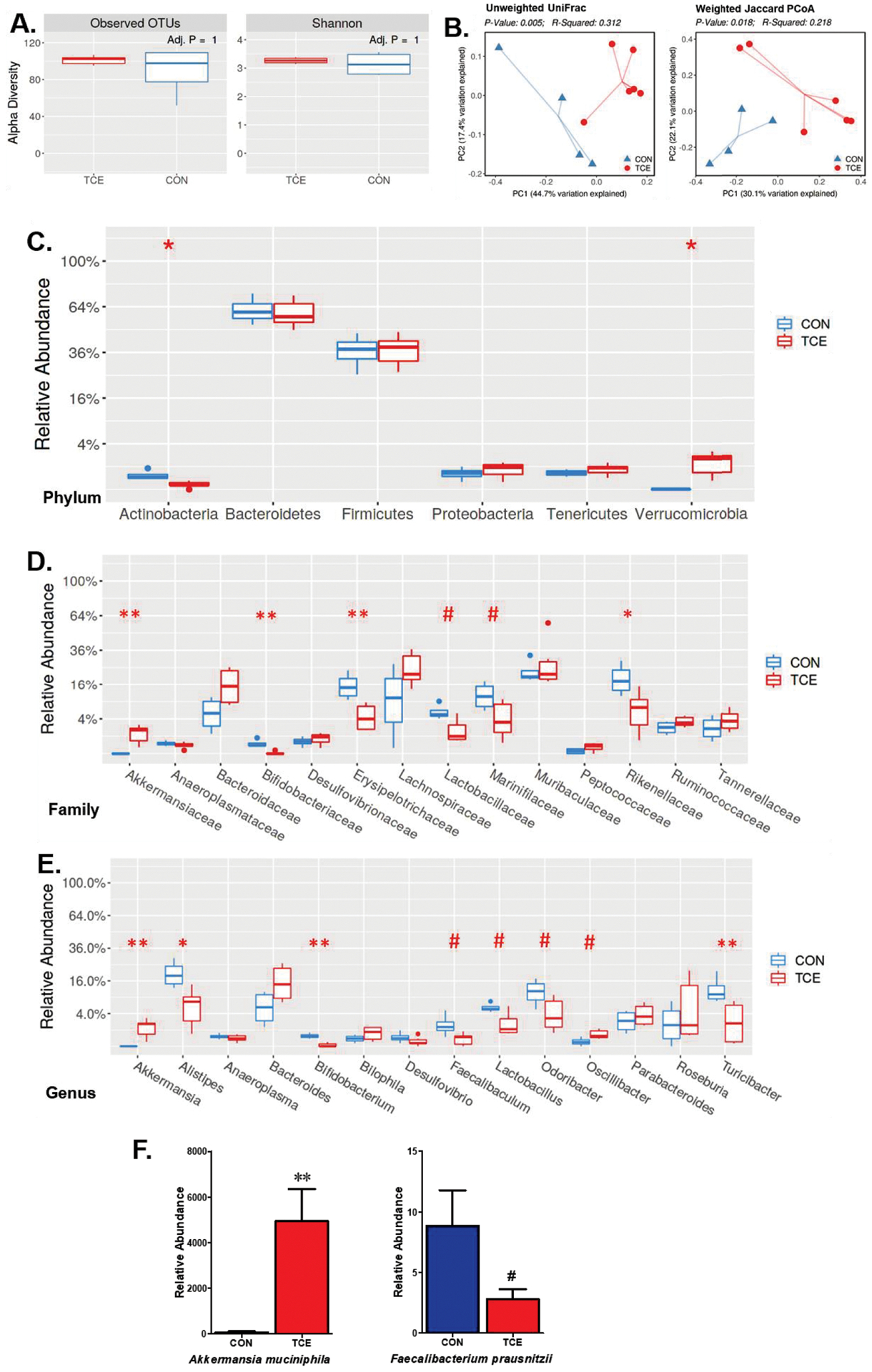
TCE exposure altered gut microbiome composition. Fecal pellets from CON and TCE-treated MRL+/+ mice were collected; bacterial DNA was isolated and subjected to 16s rDNA sequencing to evaluate the composition of microbiome. A. α-diversity of gut microbiota in each group. B. Distinct microbiome patterns revealed as β-diversity in each group. C. Average relative abundances of taxa at the phylum level. D. Relative abundance at taxonomic level of family. E. Relative abundance of major genera in fecal samples from CON and TCE-treated mice. F. Quantitative PCR analysis of the fecal samples from CON and TCE groups for Akkermansia muciniphila and Faecalibacterium prausnitzii. Results are mean ± SEM. CON: n=4; TCE: n=5 # p<0.1; *p<0.05; **p<0.01.
TCE exposure led to intestinal oxidative stress and barrier dysfunction
Our earlier studies have established that TCE exposure causes OS which contributes to TCE-mediated autoimmunity (Khan et al., 2001; Wang et al., 2008; Wang et al., 2012b; Wang et al., 2015; Wang et al., 2019). However, contribution of OS and its impact on intestinal barrier function are not known, which prompted us to evaluate the effect of TCE exposure on colonic OS and subsequent impact on intestinal permeability. TCE exposure led to significantly increased MDA-protein adducts, a conjugated form of lipid peroxidation product MDA with proteins and also a measure of OS (Khan et al., 2001; Wang et al., 2008; Wang et al., 2015), in the colon of TCE-treated mice (Figure 4A). To test whether TCE exposure alters gut barrier function-related proteins, we determined tight junction proteins (claudin1, claudin2, claudin3, ZO1, ZO2 and occludin). Among the tight junction proteins, the colon protein levels of ZO-2, occludin and claudin-3 were significantly decreased following TCE exposure (Figure 4B–C). More importantly, the decreases in tight junction proteins were associated with an increasing pattern for serum LPS in TCE-treated mice (data not shown). Our studies thus suggest that TCE-induced microbiome changes could potentially influence the host redox status and gut integrity, and suggest the importance of the gut microbiome in extra-intestinal immunomodulatory responses.
Figure 4.
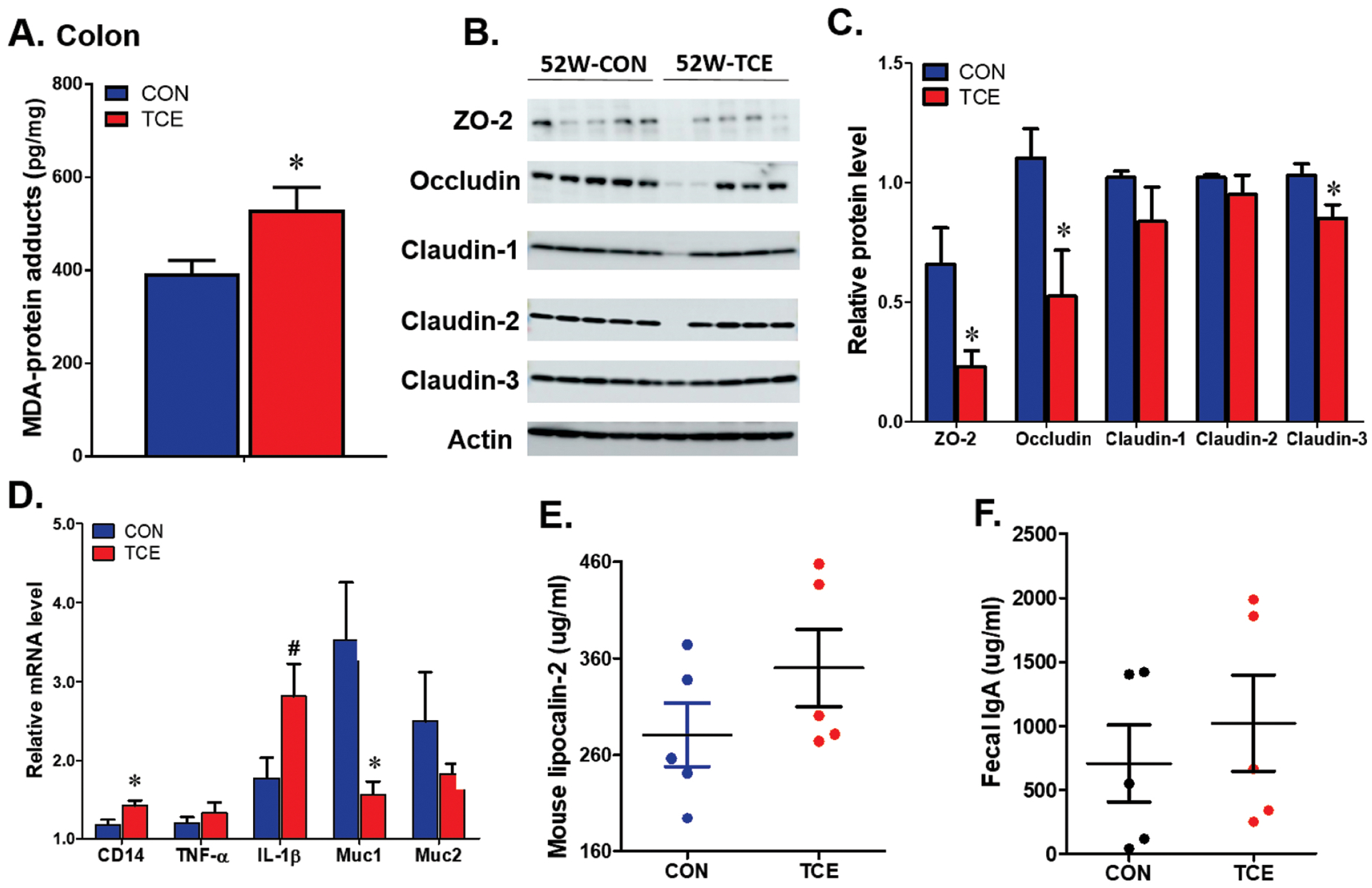
TCE exposure induced colonic oxidative stress, barrier dysfunction and inflammation. A. MDA-protein adducts in the colon of CON and TCE exposed mice at 52 weeks. B. Representative pictures for tight junction proteins in the colon. C. quantification of the signal intensity of tight junction proteins. D. Quantitative PCR analysis of inflammation related genes in the colon after TCE exposure. E. Serum Lipocalin-2 level. F. Fecal IgA levels. Results are mean ± SEM. n=5 # p<0.1; *p<0.05.
TCE exposure regulated intestinal inflammation
To determine if TCE-induced microbiome changes and their interaction with host result in intestinal inflammatory responses, we examined the inflammation markers (CD14, TNF-α, IL1-β, mucin-1, mucin-2 and lipocalin-2). The mRNA level of colonic CD14, a transmembrane co-receptor for TLRs, was significantly increased in TCE-exposed mice (Figure 4D). Moreover, TCE exposure showed an increasing trend for IL-1β and reduction in mucin1 at mRNA levels (Figure 4D). The serum lipocalin-2 and IgA levels, indicators of intestinal inflammation, also showed increasing trend following TCE exposure (Figure 4E–F). These data reveal that the gut microbiome host interactions can affect the intestinal and systemic immune responses.
Fecal microbiota transplantation from TCE-treated MRL+/+ mice generates autoimmune response in antibiotic-treated young MRL-lpr mice
To directly examine the potential contribution of TCE-mediated gut dysbiosis in SLE/AIH pathogenesis, FMT studies were conducted to assess if microbiome from TCE-exposed MRL +/+ mice could exacerbate the disease outcome in very young MRL-lpr mice (Figure 5A). FMT studies using fecal microbiome from TCE-treated mice (positive for SLE markers) and control mice to antibiotic-treated MRL-lpr mice were thus performed followed by evaluation of induction of inflammatory and autoimmune responses in the recipient mice. Feces from donor mice and recipient mice before or after FMT were subjected to 16S rRNA sequencing to monitor microbiome compositional changes. Generally, α- and β-diversity analysis revealed that microbiome compositions were significantly abated following antibiotics treatment in W0-CF and W0-TF mice (W0: before FMT), and recovered after FMT in W3-CF and W3-TF mice (W3: 3 weeks after FMT) (Figure 5B,C). Distinct microbiome compositions were further evaluated at the family level, which showed an increasing trend in Bacteroidaceae while a decreasing trend in Bifidobacteriaceae, Lactobacillaceae and Rikenellaceae (statistically not significant) in W3-TF groups compared to W3-CF(Figure 5D) – a pattern similar to microbiome of donor mice.
Figure 5.
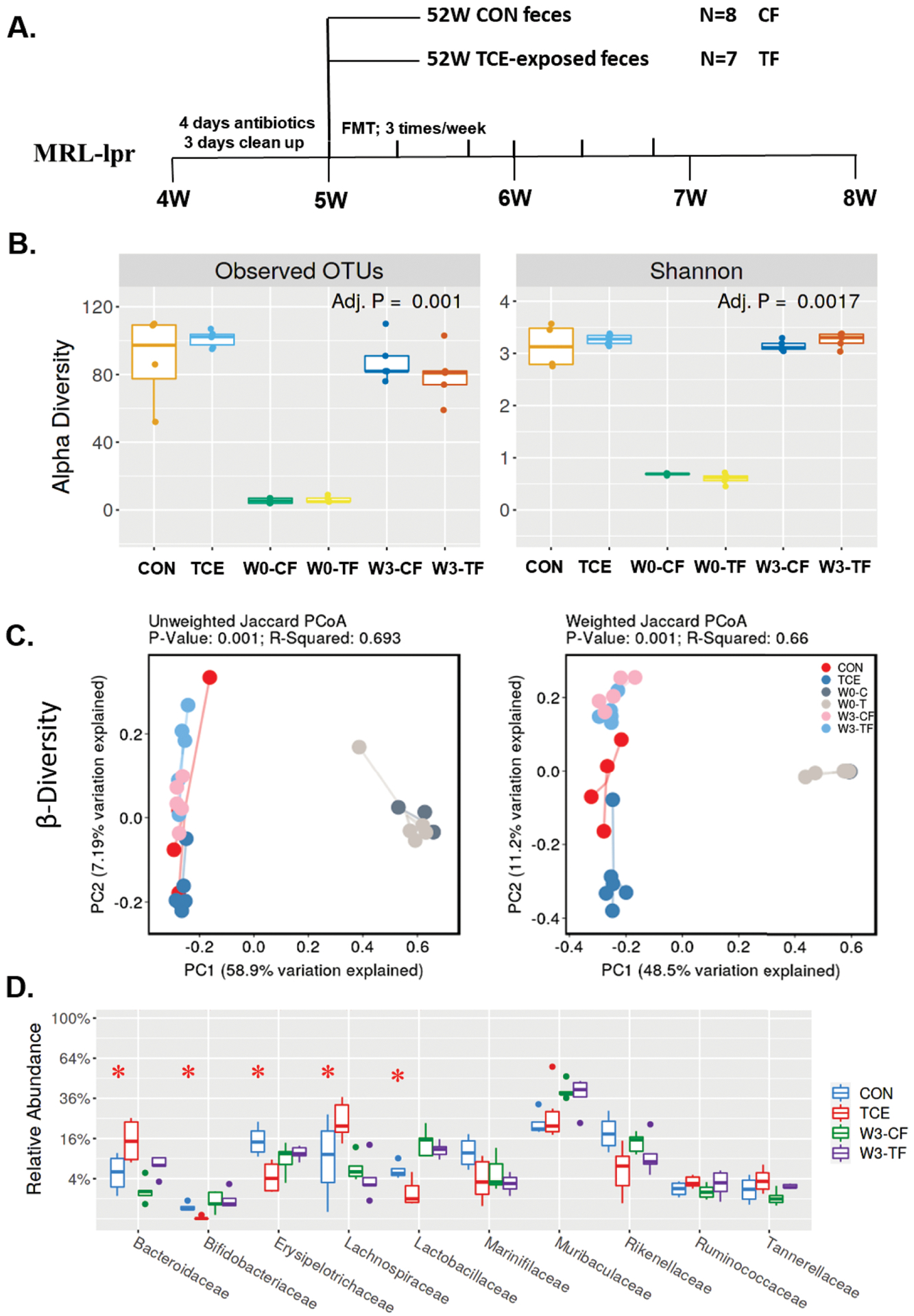
Fecal microbiota transplantation from CON and TCE-treated mice in microbiome-depleted young MRL-lpr mice. A. Study design of fecal transplantation from CON and TCE-exposed mice to MRL-lpr mice, designated as CF and TF. B. α-diversity of gut microbiota in donor (CON; TCE) and recipient mice before (W0-CF; W0-TF) and after FMT (W3-CF; W3-TF). C. β-diversity of gut microbiome in donor and recipient mice after FMT. D. Average relative abundances of taxa at the family level in donor and recipient mice after FMT. Results are mean ± SEM. CF: n=4; TF: n=5 *p<0.05.
Remarkably, oral feeding of antibiotic-pretreated MRL-lpr mice with fecal microbiota from TCE-treated mice resulted in significantly increased serum levels of anti-dsDNA autoantibodies (a prominent SLE disease marker) in TF mice, compared to CF group (Figure 6A). ANA levels also showed a similar increasing trend (Figure 6B). Interestingly, mice transplanted with microbiome from TCE-treated mice had increased total hepatic lymphocytes and CD4+ T cells than animals colonized with microbiome from controls, but no difference in the liver weight was noted (Figure 6C–D; Suppl. Figure 2A, E). Spleen was enlarged in TF group, however no significant changes were observed in the spleen lymphocyte or CD4+ T cells between TF and CF groups (Suppl. Figure 2B–E). Our data thus strongly support a causative role of microbiome in TCE-mediated autoimmunity.
Figure 6.
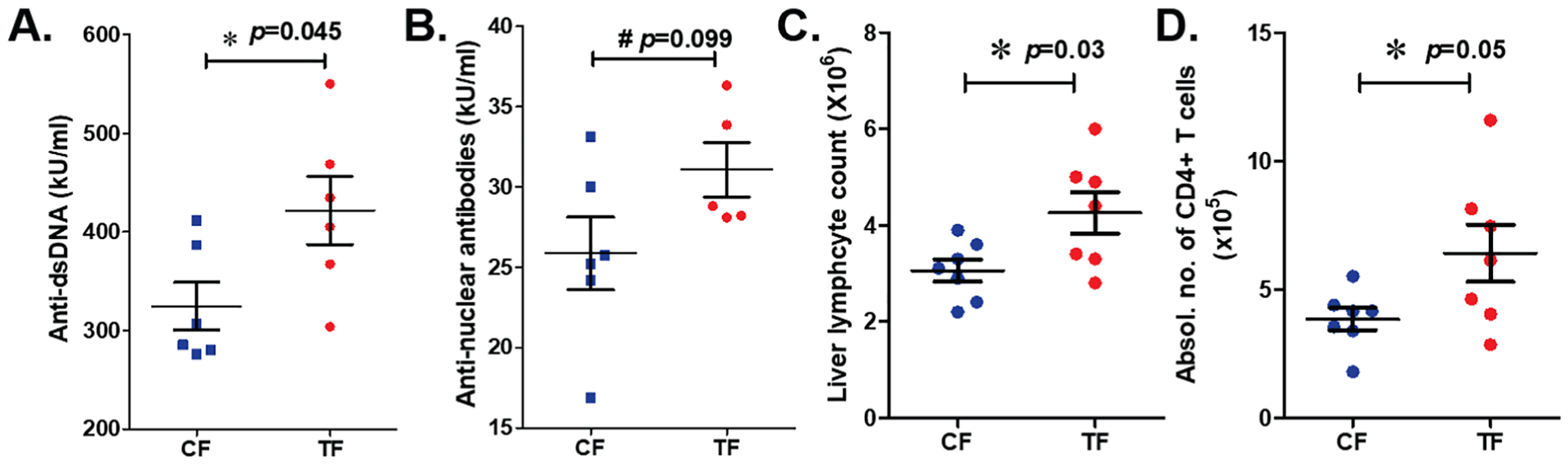
Effect of FMT from CON and TCE-treated mice in microbiome-depleted young MRL-lpr mice. Serum levels of anti-dsDNA (A) and ANA (B), total lymphocyte counts (C) and CD4+ T cell number (D) in the liver of recipient mice (CF and TF). Results are mean ± SEM. n=7 # p<0.1; *p<0.05.
Discussion
Increasing evidence suggest that alterations in the microbiome composition are critical players in numerous immune-mediated diseases in humans (Belkaid and Hand, 2014; Zheng et al., 2020). In fact, microbiome dysbiosis has also been associated with the pathogenesis of various ADs (Lin et al., 2015; De Luca and Shoenfeld, 2019; Zhang et al., 2020). However, efforts to establish a causal relationship between chemical-mediated microbiome dysbiosis and ADs, especially linking the microbes to SLE/AIH, are lacking. TCE exposure at an early developmental stage was shown to result in intestinal microbial dysbiosis (Blossom et al., 2020). However, there is no evidence to support the association of gut microbiota with SLE/AIH and their role in the pathogenesis of such ADs. Here, we report that chronic low dose TCE exposure causes aberrant gut microbiome, modulates intestinal redox potential, epithelial integrity and results in an inflammatory response. Furthermore, these changes were associated with significantly increased autoantibodies, representative of SLE/AIH. More importantly, microbiota transplantation using feces from TCE-exposed mice increased hepatic inflammation and systemic autoantibodies in recipient mice, further supporting a causal role of microbiome in such ADs.
Recent studies provide evidence to support that environmental chemicals can indeed affect the composition of gut microbiome and interferes with their metabolic activity, leading to dysbiosis of gut microbiome (Lu et al., 2015; Zhang et al., 2020), and contribute to intestinal and systemic inflammatory diseases (Bailey et al., 2020). To understand the impact of TCE in the modulation of gut microbiome and its casual effects in the pathogenesis of ADs, mice were exposed to 0.5 mg/ml TCE in drinking water for 52 weeks. The choice of TCE dose and the duration of exposure were based on findings that TCE exacerbates the development of both SLE (Cai et al., 2008; Wang et al., 2012b) and AIH (Gilbert et al., 1999; Wang et al., 2019) within this exposure period in MRL+/+ mice. It is clearly evident from our data that TCE exposure alters gut microbiome composition, particularly decreased relative abundance of Lactobacillus and Bifidobacterium, which have been implicated in the pathogenesis of multiple ADs (de Oliveira et al., 2017). Our findings are in agreement with and supported by the recent publication of Khare et al. (Khare et al., 2019) showing that gestational TCE exposure alters the gut microbiome with decreasing Bacteroides and Lactobacillus abundance. These findings are thus consistent with reported reductions in Lactobacillaceae and an increase of Lachnospiraceae in SLE patients (De Luca and Shoenfeld, 2019) and also in MRL-lpr mice with severe lupus symptoms (Zhang et al., 2014). Moreover, the observed TCE-induced decreasing trend for Faecalibacterium prausnitzii, which is predominant in the human microbiome, may contribute to increased intestinal inflammation (Miquel et al., 2013). Interestingly, Akkermansia muciniphila, which is associated with various metabolic disorders and ADs, especially multiple sclerosis (Berer et al., 2017; Cekanaviciute et al., 2018), was significantly increased in TCE-exposed mice. Akkermansia muciniphila, a mucin-degrading bacterium under the phylum Verrucomicrobia, was also significantly higher in mice exposed to ambient ultrafine particles (Li et al., 2017). A recent study has demonstrated that Akkermansia muciniphila can accelerate epithelial wound healing via intestinal ROS generation (Alam et al., 2016), suggesting that TCE-mediated enrichment of Akkermansia muciniphila may promote ROS production. Another important finding related to TCE exposure is enrichment in Oscillibacter under Firmicutes phylum, which is associated with increased gut permeability (Lam et al., 2012). Thus, our findings of aberrant microbiome profile following TCE exposure in MRL+/+ mice suggest a link and potential role of gut microbiome dysbiosis with inflammatory and autoimmune responses.
Another significant finding of this study was increased colonic OS as evident from higher levels of MDA-protein adducts, a marker of OS (Wang et al., 2012b), in TCE-treated mice. Levels of MDA-protein adducts are known to correlate with SLE-related autoantibodies and SLE disease activity in humans (Wang et al., 2010), and have also been shown to promote inflammatory cytokine production (IL17 and IL-21) in splenic cells form TCE-exposed mice (Wang et al., 2008), and thus could contribute to the pathogenesis of lupus. Studies depicting host-microbe interactions reveal that microorganisms can induce ROS generation from epithelium. In fact, certain intestinal epithelial cells can rapidly generate ROS in response to microbial signals, and supplementation of probiotics can indeed modulate gut redox status to restore homeostasis (Kumar et al., 2007); (Peterson et al., 2015). Furthermore, intestinal ROS is critical in enhancement of gut permeability through oxidative damage to epithelium (Aviello and Knaus, 2017; Bourgonje et al., 2020). OS can also alter tight junction proteins, thereby enhancing intestinal dysfunction (Rao, 2008). Tight junctions are intercellular junctions critical for intestinal integrity, and damage to tight junctions is accompanied by a leaky intestinal barrier, bacterial translocation and inflammation (Lee, 2015; Chelakkot et al., 2018). ZO-1, claudin-1 and occludin play pivotal roles in maintaining the integrity of tight junction between cells and the normal function of epithelial cell barriers. Mucin1, one of the transmembrane mucins, acts as a protective barrier underlying the epithelium (Grondin et al., 2020). It is clear from our results that TCE exposure leads to significant reductions in these tight junction proteins and decreasing mucin1 at mRNA level, and thus suggest a barrier dysfunction leading to increased permeability following TCE exposure. Impaired gut barrier function and microbial translocation are observed in AD patients and lupus-prone mice (Manfredo Vieira et al., 2018; Ruff et al., 2020). It is thus apparent that TCE-induced gut permeability might be the potential mechanism for hepatic or renal inflammation and eventually systemic autoimmune responses.
Oxidative stress is also an essential factor in the induction of host inflammation (Bhattacharyya et al., 2014; Mittal et al., 2014). Our previous studies have provided evidence that TCE exposure promotes oxidative responses and modulates redox-sensitive transcriptional factor NRF2 and microbial recognition receptor TLR4 in the spleen, contributing to autoimmunity (Banerjee et al., 2020; Wang et al., 2020). Here, our data show that TCE exposure induced transmembrane co-receptor CD14 and led to an increased trend for IL-1β in the colon tissues. In fact, higher levels of soluble CD14 are shown to correlate with SLE disease activity, also suggesting the involvement of free circulating LPS in SLE pathogenesis (Nockher et al., 1994). Excessive ROS generation, through microbial contact, can enhance IL-1β secretion via NLRP3 inflammasome activation (Bauer et al., 2010), and intestinal inflammation can enhance epithelial permeability (Ahmad et al., 2017). Subsequently, we compared systemic cytokine production and found that TCE also induced serum IL-6 and IL-12 levels, suggesting that the microbiome dysbiosis is a major determinant of TCE-mediated inflammatory responses. Our data thus provide evidence that TCE-mediated microbiome modifications can reshape the intestinal microenvironmental physiology and reveal that host-microbe interactions can promote intestinal and extraintestinal inflammation.
One of our most significant findings of this study was the demonstration that the transplantation of fecal microbiota from TCE-exposed mice with evident disease to antibiotic-treated mice induced relevant SLE/AIH phenotypes compared to those with FMT from control mice. Notably, we discovered that FMT of TCE-exposed feces led to increased autoantibodies and hepatic immune cell activation. These studies are thus critical first steps towards establishing a firm role of gut dysbiosis in the pathogenesis of chemical-mediated ADs, and warrant detailed characterization of microbiome during the disease initiation and progression. Extensive studies are needed to clarify the causal groups of bacterial involved in the pathogenesis of ADs.
In summary, the current study highlights the contribution of environmental chemical TCE-microbiome-host interactions in the development of autoimmunity and establishes a causal link between TCE-induced fecal microbiome dysbiosis and autoimmune disease manifestations. Mechanistically, our data provides evidence that TCE exposure induces microbiome dysbiosis along with excessive OS, impairing gut barrier integrity and inflammation, ultimately leading to ADs in autoimmune-prone mice. Our results not only provide insight into the molecular mechanisms of host-microbe interactions, but also reveal that manipulation of microbial compositions could be an effective therapeutic approach for ADs.
Supplementary Material
Supp. Figure 1. Splenic and hepatic immune cell infiltration in CON and TCE exposed mice at 52 weeks. Lymphocytes were harvested from the spleen and liver, profiled for different immune cells B cells (B220+), dendritic cells (CD11b+ CD11c+) and macrophages (CD11b+ F4/80+). Results are mean ± SEM. n=5 # p<0.1; **p<0.01.
Supp. Figure 2. Effects of fecal transplantation on liver weight, spleen weight, splenic lymphocyte number. A. Liver weight B. Spleen weight C. Splenic lymphocyte number D. Splenic CD4+ T cell number E. FACS analysis for splenic and hepatic CD4+ T cells. Results are mean ± SEM. n=7 **p<0.01.
Highlights.
TCE alters gut microbiome composition and mucosal inflammation in MRL +/+ mice.
TCE-induced microbiome dysbiosis is linked to altered gut redox status and integrity.
TCE-mediated autoimmunity is associated with hepatic immune cell dysregulations.
Fecal microbiota transplantation from TCE-treated mice generates autoimmune response.
Acknowledgements
This work was supported by RO1 grants [ES016302 and ES026887] from the National Institute of Environmental Health Sciences (NIEHS), NIH, and UTMB Institute for Human Infections and Immunity (IHII). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Ahmad R, Sorrell MF, Batra SK, Dhawan P, Singh AB, 2017. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunol 10, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A, Leoni G, Quiros M, Wu H, Desai C, Nishio H, Jones RM, Nusrat A, Neish AS, 2016. The microenvironment of injured murine gut elicits a local pro-restitutive microbiota. Nat Microbiol 1, 15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albillos A, de Gottardi A, Rescigno M, 2020. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol 72, 558–577. [DOI] [PubMed] [Google Scholar]
- Anagnostopoulos G, Sakorafas GH, Grigoriadis K, Margantinis G, Kostopoulos P, Tsiakos S, Arvanitidis D, 2004. Hepatitis caused by occupational chronic exposure to trichloroethylene. Acta Gastroenterol Belg 67, 355–357. [PubMed] [Google Scholar]
- Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB, Dixon FJ, 1978. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med 148, 1198–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATSDR, 2011. Toxicological profile for trichloroethylene. [PubMed]
- Aviello G, Knaus UG, 2017. ROS in gastrointestinal inflammation: Rescue Or Sabotage? Br J Pharmacol 174, 1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, Caricchio R, Buyon JP, Alekseyenko AV, Silverman GJ, 2019. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis 78, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MJ, Naik NN, Wild LE, Patterson WB, Alderete TL, 2020. Exposure to air pollutants and the gut microbiota: a potential link between exposure, obesity, and type 2 diabetes. Gut Microbes 11, 1188–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee N, Wang H, Wang G, Khan MF, 2020. Enhancing the Nrf2 antioxidant signaling provides protection against Trichloroethene-mediated inflammation and autoimmune response. Toxicol Sci 175, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, Tschopp J, Endres S, Latz E, Schnurr M, 2010. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW, 2014. Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, Liu C, Klotz L, Stauffer U, Baranzini SE, Kumpfel T, Hohlfeld R, Krishnamoorthy G, Wekerle H, 2017. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci U S A 114, 10719–10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE, 2014. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev 94, 329–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blossom SJ, Gokulan K, Arnold M, Khare S, 2020. Sex-dependent effects on liver inflammation and gut microbial dysbiosis after continuous developmental exposure to Trichloroethylene in autoimmune-prone mice. Front Pharmacol 11, 569008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgonje AR, Feelisch M, Faber KN, Pasch A, Dijkstra G, van Goor H, 2020. Oxidative stress and redox-modulating therapeutics in inflammatory bowel disease. Trends Mol Med 26, 1034–1046. [DOI] [PubMed] [Google Scholar]
- Cai P, Konig R, Boor PJ, Kondraganti S, Kaphalia BS, Khan MF, Ansari GA, 2008. Chronic exposure to trichloroethene causes early onset of SLE-like disease in female MRL +/+ mice. Toxicol Appl Pharmacol 228, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Ran Y, Li Y, Wang B, Zhou L, 2017. Intestinal microbiome and permeability in patients with autoimmune hepatitis. Best Pract Res Clin Gastroenterol 31, 669–673. [DOI] [PubMed] [Google Scholar]
- Cekanaviciute E, Probstel AK, Thomann A, Runia TF, Casaccia P, Katz Sand I, Crabtree E, Singh S, Morrissey J, Barba P, Gomez R, Knight R, Mazmanian S, Graves J, Cree BAC, Zamvil SS, Baranzini SE, 2018. Multiple sclerosis-associated changes in the composition and immune functions of spore-forming bacteria. mSystems 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelakkot C, Ghim J, Ryu SH, 2018. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med 50, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Gregersen PK, 2011. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med 365, 1612–1623. [DOI] [PubMed] [Google Scholar]
- Chu JL, Drappa J, Parnassa A, Elkon KB, 1993. The defect in Fas mRNA expression in MRL/lpr mice is associated with insertion of the retrotransposon, ETn. J Exp Med 178, 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca F, Shoenfeld Y, 2019. The microbiome in autoimmune diseases. Clin Exp Immunol 195, 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira GLV, Leite AZ, Higuchi BS, Gonzaga MI, Mariano VS, 2017. Intestinal dysbiosis and probiotic applications in autoimmune diseases. Immunology 152, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGruttola AK, Low D, Mizoguchi A, Mizoguchi E, 2016. Current understanding of dysbiosis in disease in human and animal models. Inflamm Bowel Dis 22, 1137–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doycheva I, Watt KD, Gulamhusein AF, 2019. Autoimmune hepatitis: Current and future therapeutic options. Liver Int 39, 1002–1013. [DOI] [PubMed] [Google Scholar]
- Floreani A, Restrepo-Jimenez P, Secchi MF, De Martin S, Leung PSC, Krawitt E, Bowlus CL, Gershwin ME, Anaya JM, 2018. Etiopathogenesis of autoimmune hepatitis. J Autoimmun 95, 133–143. [DOI] [PubMed] [Google Scholar]
- Gilbert KM, Griffin JM, Pumford NR, 1999. Trichloroethylene activates CD4+ T cells: potential role in an autoimmune response. Drug Metab Rev 31, 901–916. [DOI] [PubMed] [Google Scholar]
- Gilbert KM, Reisfeld B, Zurlinden TJ, Kreps MN, Erickson SW, Blossom SJ, 2014. Modeling toxicodynamic effects of trichloroethylene on liver in mouse model of autoimmune hepatitis. Toxicol Appl Pharmacol 279, 284–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grondin JA, Kwon YH, Far PM, Haq S, Khan WI, 2020. Mucins in intestinal mucosal defense and inflammation: learning from clinical and experimental studies. Front Immunol 11, 2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevia A, Milani C, Lopez P, Cuervo A, Arboleya S, Duranti S, Turroni F, Gonzalez S, Suarez A, Gueimonde M, Ventura M, Sanchez B, Margolles A, 2014. Intestinal dysbiosis associated with systemic lupus erythematosus. mBio 5, e01548–01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavicoli I, Marinaccio A, Carelli G, 2005. Effects of occupational trichloroethylene exposure on cytokine levels in workers. J Occup Environ Med 47, 453–457. [DOI] [PubMed] [Google Scholar]
- Kamijima M, Wang H, Huang H, Li L, Shibata E, Lin B, Sakai K, Liu H, Tsuchiyama F, Chen J, Okamura A, Huang X, Hisanaga N, Huang Z, Ito Y, Takeuchi Y, Nakajima T, 2008. Trichloroethylene causes generalized hypersensitivity skin disorders complicated by hepatitis. J Occup Health 50, 328–338. [DOI] [PubMed] [Google Scholar]
- Khan MF, Kaphalia BS, Prabhakar BS, Kanz MF, Ansari GA, 1995. Trichloroethene-induced autoimmune response in female MRL +/+ mice. Toxicol Appl Pharmacol 134, 155–160. [DOI] [PubMed] [Google Scholar]
- Khan MF, Wang H, 2019. Environmental Exposures and autoimmune diseases: contribution of gut microbiome. Front Immunol 10, 3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MF, Wu X, Ansari GA, 2001. Anti-malondialdehyde antibodies in MRL+/+ mice treated with trichloroethene and dichloroacetyl chloride: possible role of lipid peroxidation in autoimmunity. Toxicol Appl Pharmacol 170, 88–92. [DOI] [PubMed] [Google Scholar]
- Khare S, Gokulan K, Williams K, Bai S, Gilbert KM, Blossom SJ, 2019. Irreversible effects of trichloroethylene on the gut microbial community and gut-associated immune responses in autoimmune-prone mice. J Appl Toxicol 39, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Wu H, Collier-Hyams LS, Hansen JM, Li T, Yamoah K, Pan ZQ, Jones DP, Neish AS, 2007. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J 26, 4457–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YY, Ha CW, Campbell CR, Mitchell AJ, Dinudom A, Oscarsson J, Cook DI, Hunt NH, Caterson ID, Holmes AJ, Storlien LH, 2012. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS One 7, e34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Kim WU, 2017. Microbiota in T-cell homeostasis and inflammatory diseases. Exp Mol Med 49, e340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, 2015. Intestinal permeability regulation by tight junction: implication on inflammatory bowel diseases. Intest Res 13, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Yang J, Saffari A, Jacobs J, Baek KI, Hough G, Larauche MH, Ma J, Jen N, Moussaoui N, Zhou B, Kang H, Reddy S, Henning SM, Campen MJ, Pisegna J, Li Z, Fogelman AM, Sioutas C, Navab M, Hsiai TK, 2017. Ambient ultrafine particle ingestion alters gut microbiota in association with increased atherogenic lipid metabolites. Sci Rep 7, 42906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, Sharpe AH, 2003. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol 33, 2706–2716. [DOI] [PubMed] [Google Scholar]
- Lin R, Zhou L, Zhang J, Wang B, 2015. Abnormal intestinal permeability and microbiota in patients with autoimmune hepatitis. Int J Clin Exp Pathol 8, 5153–5160. [PMC free article] [PubMed] [Google Scholar]
- Lu K, Mahbub R, Fox JG, 2015. Xenobiotics: interaction with the intestinal microflora. ILAR J 56, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM, Ahmed SA, Bankole AA, 2018. Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Appl Environ Microbiol 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, Costa FRC, Tiniakou E, Greiling T, Ruff W, Barbieri A, Kriegel C, Mehta SS, Knight JR, Jain D, Goodman AL, Kriegel MA, 2018. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 359, 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquel S, Martin R, Rossi O, Bermudez-Humaran LG, Chatel JM, Sokol H, Thomas M, Wells JM, Langella P, 2013. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol 16, 255–261. [DOI] [PubMed] [Google Scholar]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB, 2014. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20, 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu H, Bai H, Sun F, Liu Y, Lu C, Qiu Y, Chen P, Yang Y, Kong L, Duan J, 2019. Pathogen-targeting glycovesicles as a therapy for salmonellosis. Nat Commun 10, 4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Q, Zhang H, Liao X, Lin K, Liu H, Edwards MR, Ahmed SA, Yuan R, Li L, Cecere TE, Branson DB, Kirby JL, Goswami P, Leeth CM, Read KA, Oestreich KJ, Vieson MD, Reilly CM, Luo XM, 2017. Control of lupus nephritis by changes of gut microbiota. Microbiome 5, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nockher WA, Wigand R, Schoeppe W, Scherberich JE, 1994. Elevated levels of soluble CD14 in serum of patients with systemic lupus erythematosus. Clin Exp Immunol 96, 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CT, Sharma V, Elmen L, Peterson SN, 2015. Immune homeostasis, dysbiosis and therapeutic modulation of the gut microbiota. Clin Exp Immunol 179, 363–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO, 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R, 2008. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci 13, 7210–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff WE, Dehner C, Kim WJ, Pagovich O, Aguiar CL, Yu AT, Roth AS, Vieira SM, Kriegel C, Adeniyi O, Mulla MJ, Abrahams VM, Kwok WW, Nussinov R, Erkan D, Goodman AL, Kriegel MA, 2019. Pathogenic autoreactive t and b cells cross-react with mimotopes expressed by a common human gut commensal to trigger autoimmunity. Cell Host Microbe 26, 100–113 e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff WE, Greiling TM, Kriegel MA, 2020. Host-microbiota interactions in immune-mediated diseases. Nat Rev Microbiol 18, 521–538. [DOI] [PubMed] [Google Scholar]
- Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, Azuma M, Yagita H, Sayegh MH, Khoury SJ, 2003. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med 198, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H, 2005. The role of the T cell in autoimmune inflammation. Arthritis Res Ther 7 Suppl 2, S4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Konig R, Ansari GA, Khan MF, 2008. Lipid peroxidation-derived aldehyde-protein adducts contribute to trichloroethene-mediated autoimmunity via activation of CD4+ T cells. Free Radic Biol Med 44, 1475–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Li H, Firoze Khan M, 2012a. Differential oxidative modification of proteins in MRL+/+ and MRL/lpr mice: Increased formation of lipid peroxidation-derived aldehyde-protein adducts may contribute to accelerated onset of autoimmune response. Free Radic Res 46, 1472–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Pierangeli SS, Papalardo E, Ansari GA, Khan MF, 2010. Markers of oxidative and nitrosative stress in systemic lupus erythematosus: correlation with disease activity. Arthritis Rheum 62, 2064–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wakamiya M, Wang J, Ansari GA, Firoze Khan M, 2015. iNOS null MRL+/+ mice show attenuation of trichloroethene-mediated autoimmunity: contribution of reactive nitrogen species and lipid-derived reactive aldehydes. Free Radic Biol Med 89, 770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang H, Banerjee N, Khan MF, 2020. Interplay and roles of oxidative stress, toll-like receptor 4 and Nrf2 in trichloroethene-mediated autoimmunity. Toxicol Appl Pharmacol 408, 115258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang J, Fan X, Ansari GA, Khan MF, 2012b. Protein adducts of malondialdehyde and 4-hydroxynonenal contribute to trichloroethene-mediated autoimmunity via activating Th17 cells: dose- and time-response studies in female MRL+/+ mice. Toxicology 292, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang G, Liang Y, Du X, Boor PJ, Sun J, Khan MF, 2019. Redox regulation of hepatic NLRP3 inflammasome activation and immune dysregulation in trichloroethene-mediated autoimmunity. Free Radic Biol Med 143, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Li Y, Yan L, Sun C, Miao Q, Wang Q, Xiao X, Lian M, Li B, Chen Y, Zhang J, Li Y, Huang B, Li Y, Cao Q, Fan Z, Chen X, Fang JY, Gershwin ME, Tang R, Ma X, 2020. Alterations of gut microbiome in autoimmune hepatitis. Gut 69, 569–577. [DOI] [PubMed] [Google Scholar]
- Yu LC, 2018. Microbiota dysbiosis and barrier dysfunction in inflammatory bowel disease and colorectal cancers: exploring a common ground hypothesis. J Biomed Sci 25, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Chi H, 2015. Metabolic control of regulatory T cell development and function. Trends Immunol 36, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liao X, Sparks JB, Luo XM, 2014. Dynamics of gut microbiota in autoimmune lupus. Appl Environ Microbiol 80, 7551–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen BD, Zhao LD, Li H, 2020. The gut microbiota: emerging evidence in autoimmune diseases. Trends Mol Med 26, 862–873. [DOI] [PubMed] [Google Scholar]
- Zheng D, Liwinski T, Elinav E, 2020. Interaction between microbiota and immunity in health and disease. Cell Res 30, 492–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp. Figure 1. Splenic and hepatic immune cell infiltration in CON and TCE exposed mice at 52 weeks. Lymphocytes were harvested from the spleen and liver, profiled for different immune cells B cells (B220+), dendritic cells (CD11b+ CD11c+) and macrophages (CD11b+ F4/80+). Results are mean ± SEM. n=5 # p<0.1; **p<0.01.
Supp. Figure 2. Effects of fecal transplantation on liver weight, spleen weight, splenic lymphocyte number. A. Liver weight B. Spleen weight C. Splenic lymphocyte number D. Splenic CD4+ T cell number E. FACS analysis for splenic and hepatic CD4+ T cells. Results are mean ± SEM. n=7 **p<0.01.