

Abstract
The healing of large bone defects represents a clinical challenge, often requiring some form of grafting. Three-dimensional (3D) nanofiber aerogels could be a promising bone graft due to their biomimetic morphology and controlled porous structures and composition. miR-26a has been reported to induce the differentiation of bone marrow-derived mesenchymal stem cells (BMSCs) and facilitate bone formation. Introducing miR-26a with a suitable polymeric vector targeting BMSCs could improve and enhance the functions of 3D nanofiber aerogels for bone regeneration. Herein, we first developed the comb-shaped polycation (HA-SS-PGEA) carrying a targeting component, biocleavable groups and short ethanolamine (EA)-decorated poly(glycidyl methacrylate) (PGMA) (abbreviated as PGEA) arms as miR-26a delivery vector. We then assessed the cytotoxicity and transfection efficiency of this polycation and cellular response to miR-26a-incorporated nanoparticles (NPs) in vitro. HA-SS-PGEA exhibited a stronger ability to transport miR-26a and exert its functions than the gold standard polyethyleneimine (PEI) and low-molecular-weight linear PGEA. We finally examined the efficacy of HA-SS-PGEA/miR-26a NPs loaded 3D hybrid nanofiber aerogels showing a positive effect on the cranial bone defect healing. Together, the combination of 3D nanofiber aerogels and functional NPs consisting of a biodegradable and targeting polycation and therapeutic miRNA could be a promising approach for bone regeneration.
Keywords: electrospun nanofibers, aerogel, miRNA delivery, poly(glycidyl methacrylate), bone regeneration
1. Introduction
Large bone defects, which commonly result from trauma, infection, and/or tumors, cause severe clinical issues and rapidly increased morbidity, and there is, therefore, huge demand for tissue-engineered bone.[1,2] The gold standard for treating critical-sized bone defects, which have no potent to heal spontaneously across the lifespan of patients if secondary intervention is not involved, are auto- and allo-grafts currently, but their applications in clinics are restricted by donor shortage, disease transmission, immune rejection, and persistent pain.[3–6] It is urgent and crucial to develop highly efficient, functionalized scaffolds for bone regeneration.
With advances in understanding of the molecular mechanisms of differentiation of mesenchymal stem cells (MSCs), more and more microRNAs (miRNAs) have been identified as regulators of osteoblastogenesis through governing various signaling pathways related to secretory molecules and transcription and growth factors including bone morphogenetic protein (BMP), Wnt protein, fibroblast growth factors (FGFs), vascular endothelial growth factor (VEGF), and transforming growth factor β (TGF-β).[7–14] Among the discovered miRNAs, miRNA-26a (miR-26a) has emerged as a promoter of bone marrow-derived MSC (BMSC) osteogenic differentiation by targeting on GSK-3β to activate Wnt signaling and translocate β-catenin from cytoplasm to nucleus for enhanced transcription.[15–18] miR-26a has also been proved to increase vascularization and coordinate the coupling of angiogenesis and osteogenesis, which is important for maturation of neo-bone.[15,19,20]
Cationic polymers which have been massively investigated and developed as gene delivery vectors are able to neutralize the negative charges of miRNAs and form nanoparticles (NPs) to protect miRNAs from degradation and enhance endocytosis, thereby improving their bioavailability and potential to exert function.[21–23] By now, several polycations such as polyethylenimine (PEI), polyamidoamine dendrimer (PAMAM), and polysacharide have exhibited certain abilities to introduce exogenous genes into MSCs.[24–27] However, these polycations are associated with drawbacks including low transfection efficiency, high toxicity, and/or non-degradability. Recently, some progress has been made to optimize polycationic gene delivery systems for MSCs through introducing specific ligands or reducible linkages.[28–30] There is a considerable difference in redox environment between extracellular and intracellular milieu, wherein glutathione (GSH) serves as an important indicator of reduction potential. The concentration of GSH in intracellular milieu is 50–1000 times higher than that in extracellular milieu, leading to ready cleavage of disulfide bonds in polymeric nanoparticles and further promoting release of DNA, RNA, and drugs.[31–34] Hyaluronic acid (HA) is an anionic glycoaminoglycan. As major component of extracellular matrix (ECM), HA possesses high biocompatibility and low immunogenicity, thereby becoming an attractive biomaterial for tissue engineering.[35–38] It has also been reported that HA and its derivatives specifically binds to cell surface receptors (like CD44 of MSCs) presenting in certain tissues and most tumor tissues, thus serving as targeted delivery carriers for peptides, nucleic acids, and antitumor drugs.[39–41]
Based on the aforementioned studies, a comb-shaped polycation (HA-SS-PGEA) consisting of a targeting component (HA), biocleavable groups (disulfide groups) and ethanolamine (EA)-functionalized poly(glycidyl methacrylate) (PGMA) (abbreviated as PGEA) could be useful as vehicle to deliver miR-26a into BMSCs for inducing osteoblastogenesis, where PGEA has been testified as reliable biomaterial in the previous work (Figure 1a).[42–44] In addition, a novel 3D hybrid nanofiber aerogel has been developed with fibrous morphology, controlled pore size, porosity, and composition for bone regeneration in our recent work.[45] In this study, we aimed to combine this novel functionalized polycation miR-26a delivery system with 3D hybrid nanofiber aerogels composed of poly(lactic-co-glycolic acid) (PLGA)-collagen-gelatin (PCG) and bioactive glass (BG) nanofiber segments for bone repair (Figure 1a).
Figure 1.
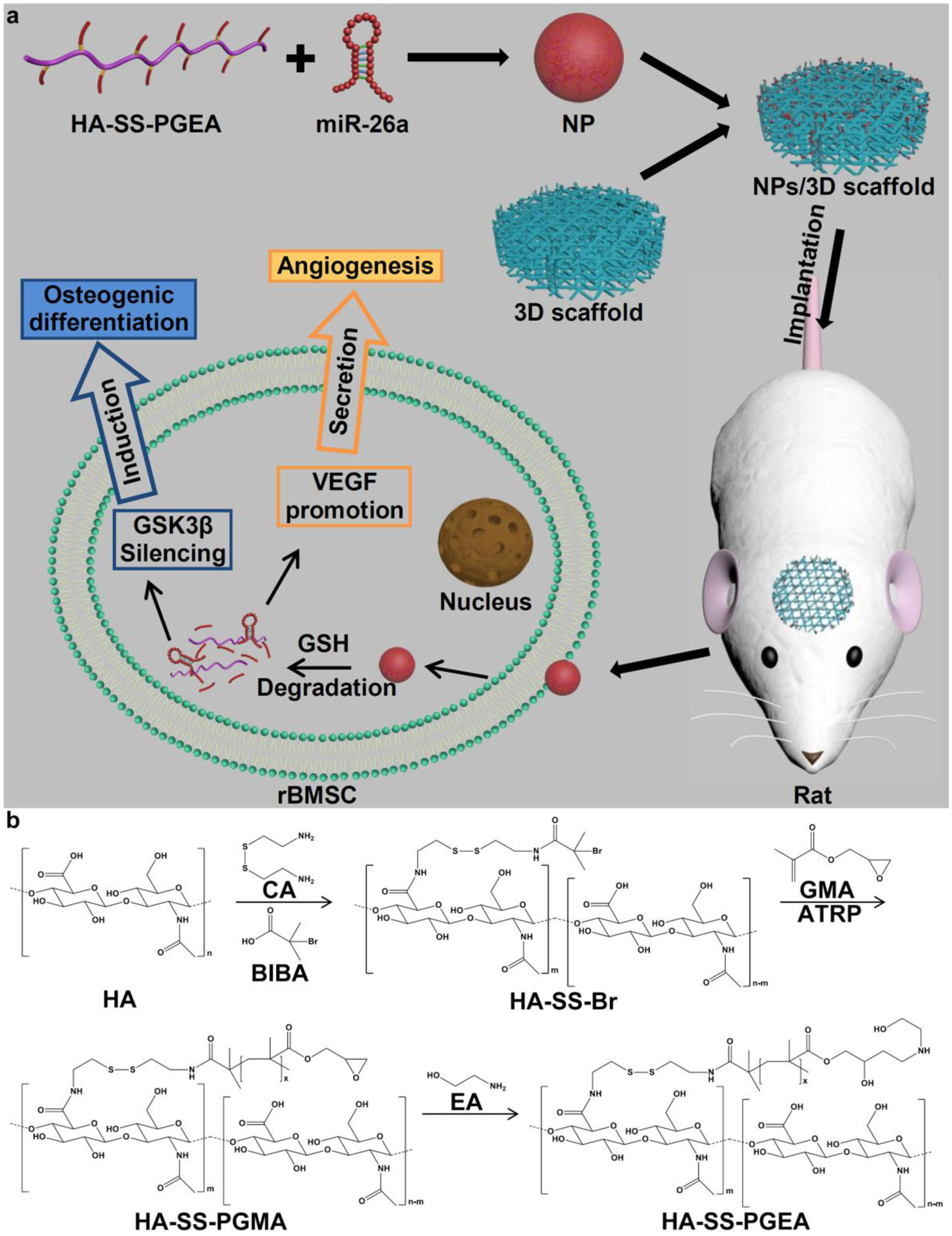
Schematic illustrating a) the combination of HA-SS-PGEA/miR-26a NPs and 3D nanofiber aerogels for cranial bone repair and b) the procedure of HA-SS-PGEA synthesis.
2. Results and Discussion
2.1. Preparation and Characterization of HA-SS-PGEA
For the preparation of miRNA-condensed NPs, the comb-like polycation HA-SS-PGEA consisting of a HA backbone, disulfide groups, and PGEA side chains was synthesized by combination of amidation, atom transfer radical polymerization (ATRP), and ring-opening reactions (Figure 1b). HA-SS-NH2 was first synthesized through amidation to introduce disulfide groups. The chemical structure of HA-SS-NH2 was determined by a 1H nuclear magnetic resonance (NMR). The peak in the scope of 4.36–4.63 ppm belonged to the protons of methylidyne group linked to two oxygen atoms (a, O-CH-O) (Figure S1a, Supporting Information). The peak at 2.02 ppm was attributed to the protons of methyl group adjacent to the amide group of HA (d, CH3-C(O)-NH). The signal at δ = 3.40 ppm was related to the protons of methylene group adjacent to the amide and amino groups (b, CH2-NH-C(O) and CH2-NH2). The peak at 3.02 ppm was associated with the protons of methylene group adjacent to disulfide bond (c, CH2-S-S). The other peaks between 3.2–4.1 ppm were attributed to the protons of glucose residue of HA. According to the area ratio of peak c to a (about 1.4:1), a total of 70% of carboxyl groups on HA were reacted with cystamine (CA) to generate amide groups.
HA-SS-Br was prepared as macromolecular initiator by transforming the amino groups to bromine terminals for the subsequent ATRP. In the typical 1H NMR spectrum of HA-SS-Br, the peak at 1.88 ppm belonged to methyl protons (e, -CH3) (Figure S1b). The area ratio of peak a and e was about 1:2, which suggested that 67% of the repeated units possessed terminal bromoisobutyryl groups.
HA-SS-PGMA was synthesized via ATRP to introduce side chains, and its chemical structure was determined by a 1H NMR. The peaks located at 4.31 and 3.74 ppm were related to the methylene protons adjacent to the oxygen moieties of the ester linkages (f, O=C-O-CH2-CH) (Figure S1c, Supporting Information). The signal δ = 3.21 ppm was associated with the methylidyne protons (g, CH2–CH(O)–CH2), and the peaks at 2.81 and 2.66 ppm belonged to the methylene protons (h, CH2–CH(O)–CH2 ) of the epoxy ring. The area ratio of peak f, g and h was about 2:1:2, indicating that the epoxy rings were not damaged during the ATRP process.
HA-SS-PGEA was finally synthesized via ring-opening reaction of epoxy groups with EA to provide hydrophilicity and cations. In the 1H NMR spectrum of HA-SS-PGEA, the signal at the scope of 3.82–4.36 ppm was mainly (two-thirds of the peak area) assigned to the methylene adjacent to the ester linkages (f, O=C-O-CH2-CH) (Figure S1d, Supporting Information). While, the residual partial scope (one-thirds of the peak area) was attributed to the methylidyne (g’, CH–OH). Besides, the characterized chemical shift at 3.73 ppm was associated with the methylene protons adjacent to the hydroxyl groups (g, CH2–OH). The signal δ = 2.77 ppm was related to the methylene protons adjacent to the secondary amine groups (i, CH2-NH). The area ratio of peak fg’, g and i was about 3:2:4. This result indicated that HA-SS-PGEA was successfully synthesized.
2.2. Preparation and Characterization of 3D Hybrid Nanofiber Aerogels
The 3D hybrid nanofiber aerogels composed of PCG and BG segments were prepared through electrospinning and control over the segment concentration and freezing conditions. The PCG nanofibers could mimic ECM and endow the scaffolds with biodegradability, and the BG short fibers were able to release silicon to promote osteogenesis.[46,47] The 3D aerogels had interconnected pores of about 100 μm in diameter (Figure 2a). Scaffolds with smaller pores (~50 μm) were ever fabricated in our previous work.[45] The pore size became larger with decreasing the segment concentration. It has been verified that larger pores contributed to better cell infiltration and vascularization with the tissue ingrowth.[48,49]
Figure 2.
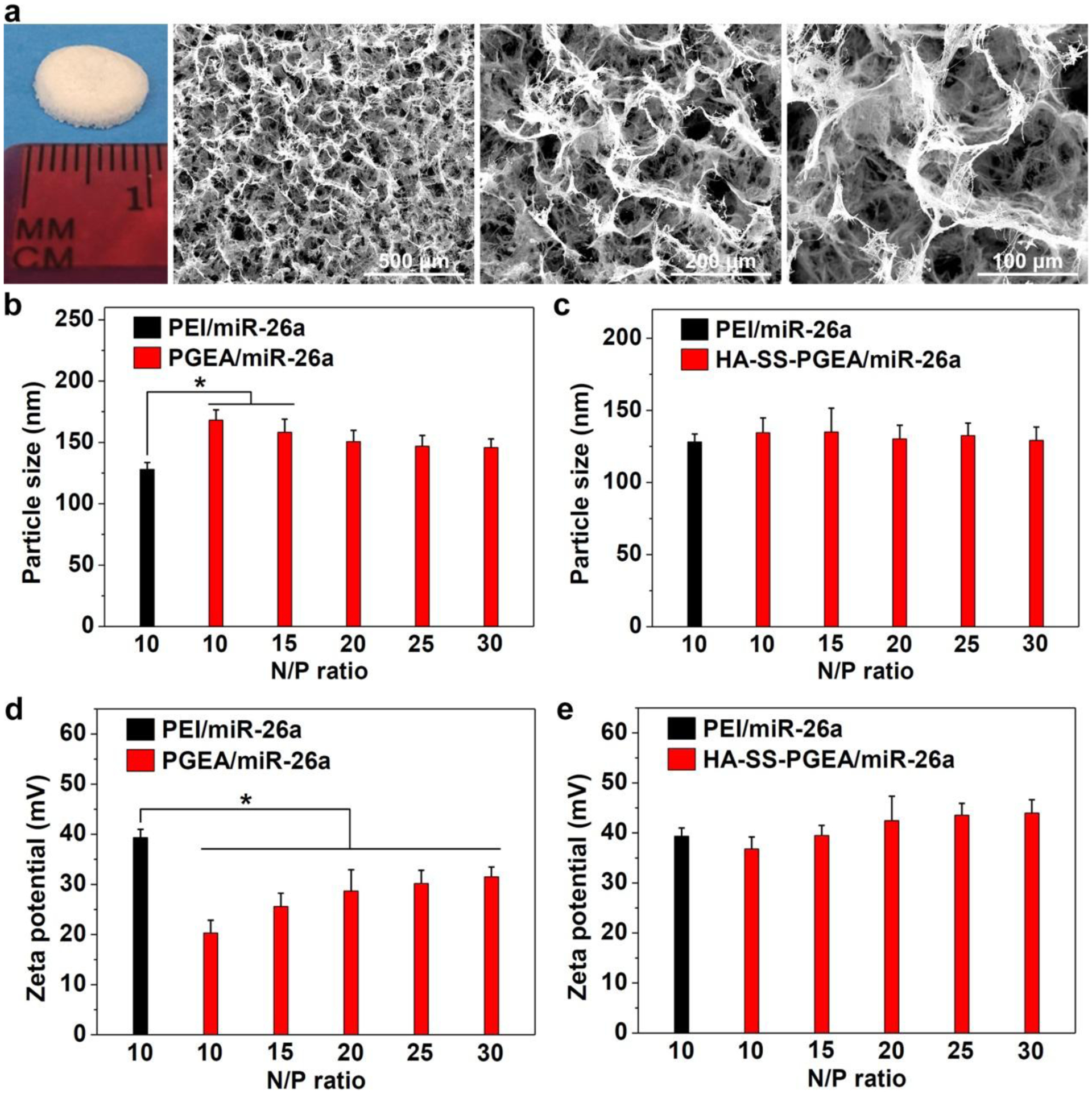
a) Photograph and SEM images of 3D hybrid nanofiber aerogels. b, c) The sizes of PEI/miR-26a, PGEA/miR-26a, and HA-SS-PGEA/miR-26a NPs. d, e) Zeta potentials of various NPs (*p < 0.05; n = 3 per group).
2.3. Formation and Characterization of NPs
For effective endocytosis of NPs, polycations should have strong ability to condense miRNA. The HA has carboxyl groups, and 67% of these groups were transformed into bromoisobutyryl groups which were used to graft PGEA chains. The amount of secondary amine groups on PGEA chains were much higher than that of the unreacted carboxyl groups on the HA backbone. Therefore, HA-SS-PGEA was positively charged and able to complex with negatively charged miRNA. Dynamic light scattering (DLS) assay was used to assess the miRNA binding performance of PGEA, HA-SS-PGEA, and PEI. Herein, all of the polycations could complex with negative-charged miRNA via electrostatic interaction to form NPs with sizes below 200 nm at N/P ratios of 10–30 (Figure 2b,c). Such size was suitable for undergoing NP diffusion and cellular internalization.[50,51] The particle size of PEI/miRNA NPs at N/P ratio of 10 was 128 nm. PGEA/miR-26a NPs had a gradual decline in particle size and then almost remained the same as the N/P ratio increased from 10 to 30, indicating that higher concentration of polycation contributed to making smaller NPs, but the low molecular weight of PGEA led to a restriction in compressing capability. In comparison with PEI/miR-26a NPs, PGEA/miR-26a NPs were larger, ranging from 138 nm to 177 nm. HA-SS-PGEA demonstrated a stronger ability to compress miRNA than PGEA. At low N/P ratios (10 and 15), HA-SS-PGEA/miR-26a NPs were similar to PEI-incorporated NPs in size and smaller than PGEA-incorporated NPs, which was probably attributed to the high molecular weight and comb-like structure of HA-SS-PGEA. The number of arms conjugated to one backbone likely led to mechanically induced denser packing compared to linear constructs.
The surface charge of complexes is also an important factor in modulating cellular internalization because positive surface charges provide NPs with affinity to negative-charged cell membrane. PEI/miR-26a NPs demonstrated +39.4 mV of zeta potential, while PGEA/miR-26a NPs possessed significantly less surface charges (Figure 2d,e). The zeta potential of PGEA/miR-26a NPs gradually increased from +20.3 mV to about +30 mV and then kept unchanged as increasing N/P ratio. In contrast, HA-SS-PGEA/miR-26a NPs demonstrated much higher zeta potential than PGEA-incorporated NPs at all N/P ratios, especially at low N/P ratios (10 and 15), probably attributed to the multi-armed structure of HA-SS-PGEA which provided more spatial positive charges to occupy larger surface area of NPs.
The abilities of PGEA and HA-SS-PGEA to condense miRNA were further confirmed through agarose gel electrophoresis assays. The PGEA and HA-SS-PGEA completely retarded miRNA migration at the N/P ratio of 1.5 and 1, respectively (Figure 3a), indicating stronger ability of HA-SS-PGEA to complex with miRNA than that of PGEA. Negatively charged heparin substituted the miRNA in the PGEA-based and HA-SS-PGEA-based NPs at the concentration of 120 and 300 μg/mL, respectively (Figure 3b), revealing that HA-SS-PGEA enabled NPs more stable to resist anionic macromolecules in the circulation to prevent premature failure of miRNA. In the presence of 10 mM of DL-dithiothreitol (DTT), which was analogous to the intracellular redox potential,[52] miRNA was released from HA-SS-PGEA-based NPs at a relatively low heparin concentration of 120 μg/mL as DTT induced disulfide bond cleavage, and the mechanical, spatial restriction of polycation side chains on miRNA was reduced, thus promoting miRNA expression. In contrast, DTT did not significantly alter the performance of PGEA-based NPs in binding miRNA. The results of agarose gel electrophoresis were consistent to those of DLS.
Figure 3.
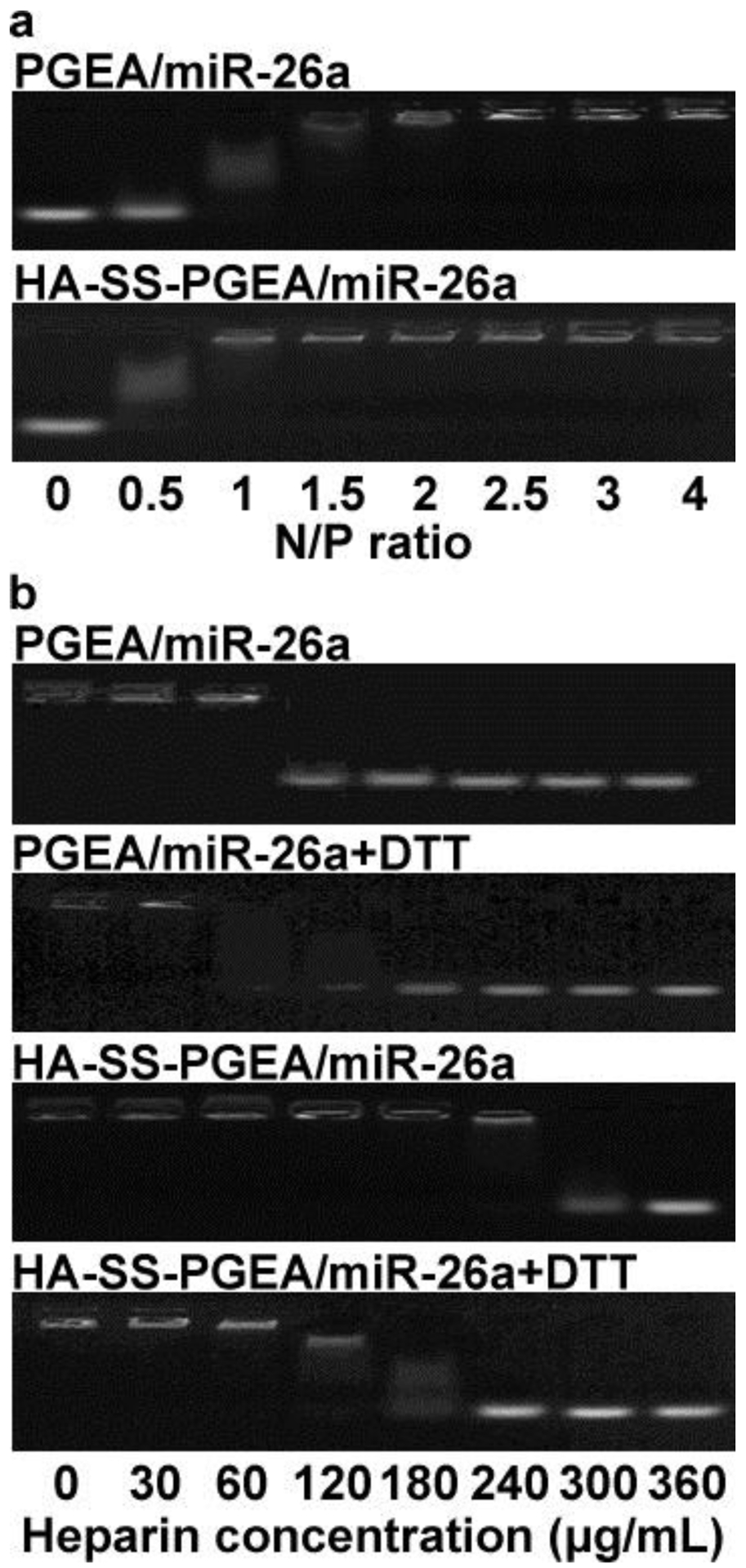
a) Agarose gel electrophoresis images of miR-26a migration retarded by PGEA and HA-SS-PGEA at various N/P ratios. b) Agarose gel electrophoresis images of miR-26a migration (at N/P ratio of 20) retarded by PGEA and HA-SS-PGEA with or without DTT treatment in the presence of heparin at series of concentrations.
2.4. MiRNA Release from 3D Hybrid Nanofiber Aerogels
The miR-26a and FITC-labeled HA-SS-PGEA/miR-26a NP release experiments over 30 days were carried out side-by-side. For both miRNA and FITC-labeled NPs, burst release from 3D aerogel scaffolds occurred during the first 3 days, and sustained release for one month was detected (Figure 4). The accumulative release percentages of miRNA and NPs were comparable at the same time point. The in vitro NP release from 3D aerogels could be attributed to the diffusion in the releasing medium. The analogous release profiles for miRNA and NPs indicated inapparent premature miRNA escape from NPs before they were internalized by cells. In addition, the HA-SS-PGEA/miR-26a polyplexes remained to be NPs, whose sizes were kept almost unchanged (Figure S2). Moreover, the released NPs became larger and looser after DTT treatment. Such NPs featuring good stability in the physiological conditions and degradability in the intracellular milieu probably contributed to potent protection of miRNA against RNase in the circulation and/or extracellular milieu and effective delivery and expression of miRNA inside cells.
Figure 4.
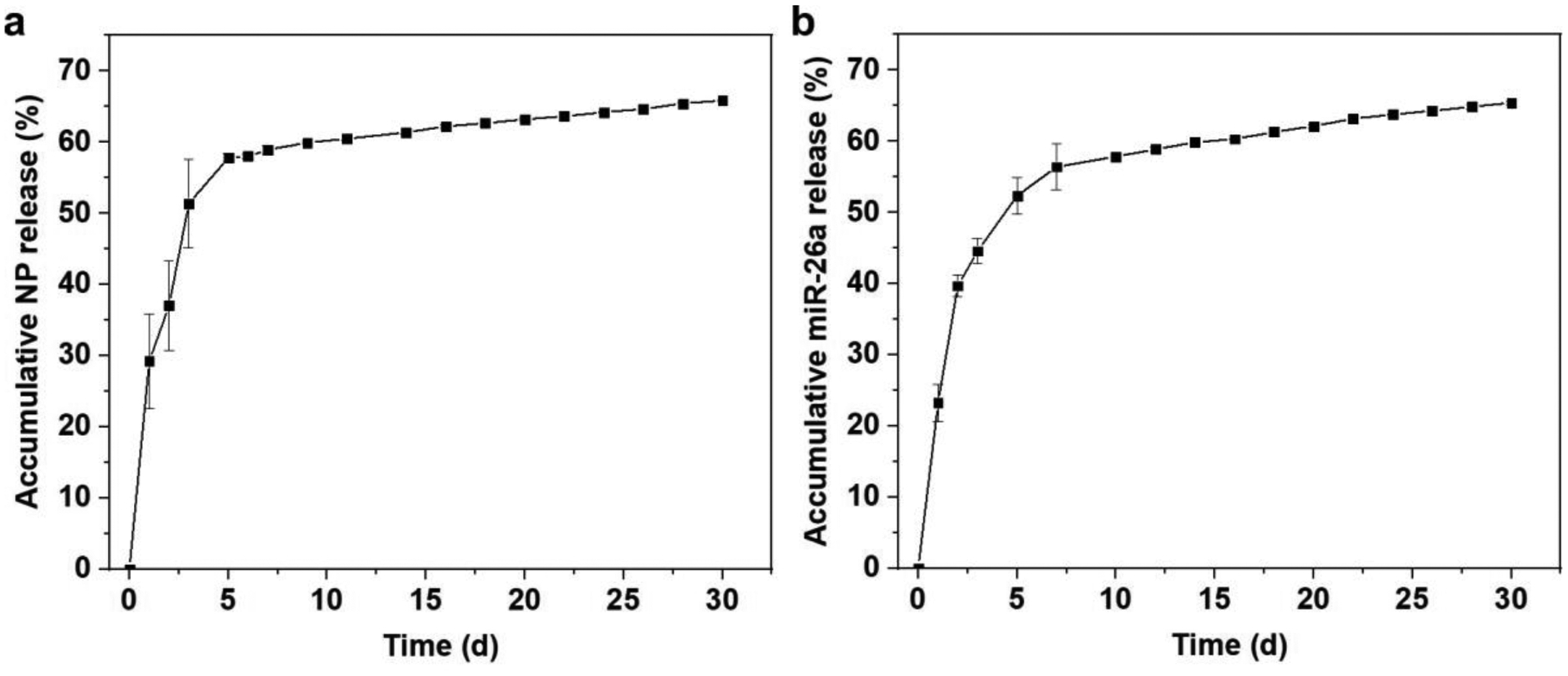
Release profiles of a) FITC-labeled HA-SS-PGEA/miR-26a NPs and b) miR-26a from 3D hybrid nanofiber aerogels over 30 days (data presented as mean ± standard deviation, n = 3).
2.5. Cell Viability
Low cytotoxicity is one of the most important factors for a satisfactory gene delivery system. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was first used to evaluate the cell viability of rBMSCs and MC3T3 cells treated with PEI/miRNA, PGEA/miRNA, and HA-SS-PGEA/miRNA NPs containing the same amounts of nitrogen (Figure 5a,b). PEI-based NPs caused death of around 50% of cells. In contrast, the cell viability mediated by PGEA-based NPs reached above 90%. Generally, the increased molecular weight may result in higher cytotoxicity. However, there was no obvious difference in the cytotoxicity between the PGEA/miRNA and HA-SS-PGEA/miRNA. The high biocompatibility of HA backbones was helpful for repressing the cytotoxicity, and the hydroxyl groups of PGEA could shield the deleterious surface charges.[42,43]
Figure 5.
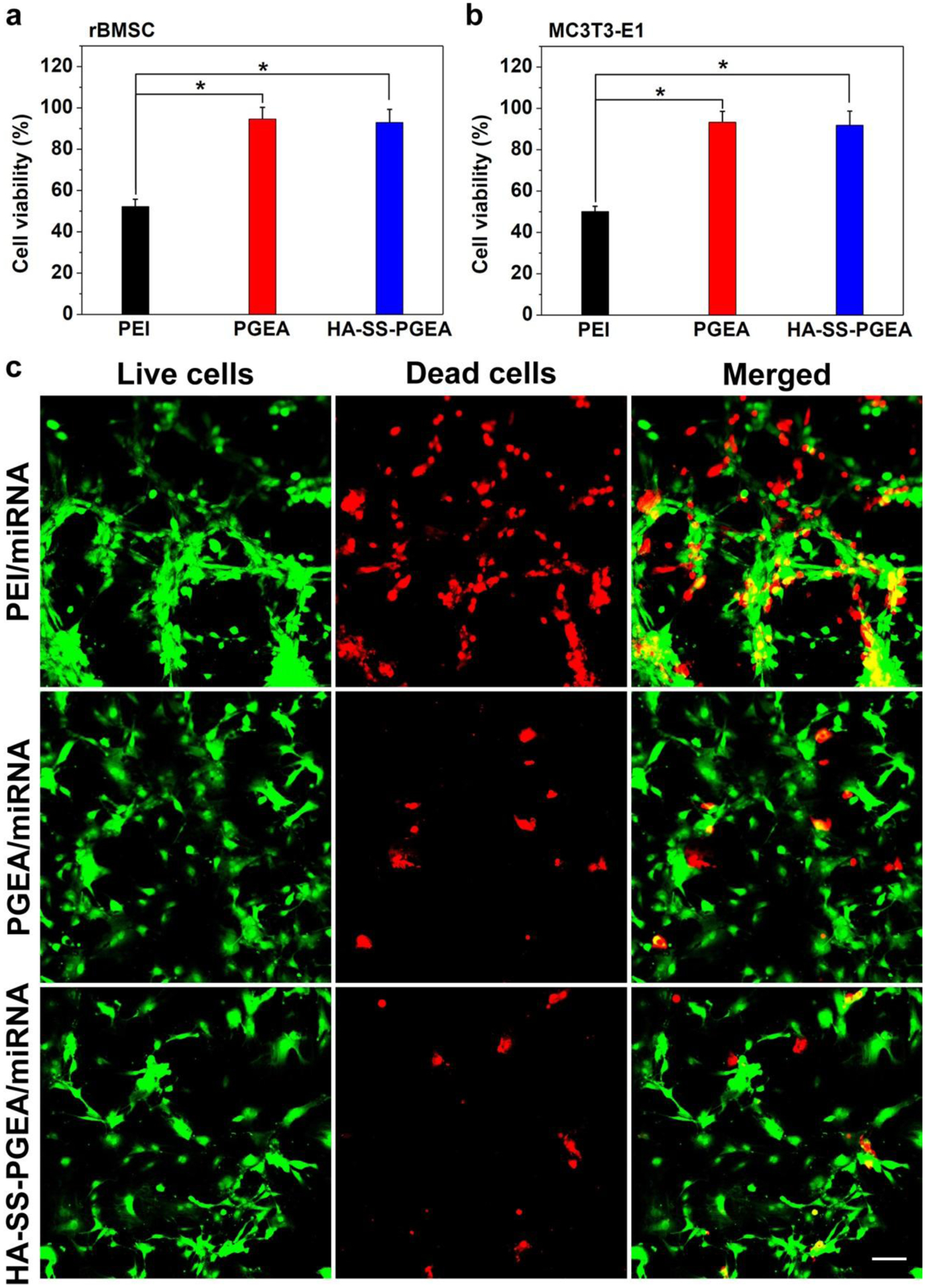
a, b) Cell viability of rBMSC and MC3T3-E1 mediated by PEI-, PGEA- and HA-SS-PGEA-containing NPs (*p < 0.05; n = 3 per group). The nitrogen concentration of polycation in medium was 2.6 mg/L. c) CLSM images of rBMSCs mediated by various miRNA-containing NPs. Live and dead cells were stained in green and red, respectively. Scale bar = 50 μm.
The live/dead assay kit was used to further confirm the cytotoxicity of different polycation/miRNA NPs in rBMSCs. Figure 5c shows the images of live cells stained in green and dead cells stained in red after treatments with PEI/miRNA, PGEA/miRNA, and HA-SS-PGEA/miRNA NPs. About half of cells were dead in the PEI/miRNA group, while sparse cells in red (less than 10% of cells) were observed in PGEA/miRNA and HA-SS-PGEA/miRNA groups, indicating that PGEA and HA-SS-PGEA exhibited lower cytotoxicity than PEI. The results of MTT and live/dead assays were consistent.
2.6. Cellular Internalization
Efficient cellular internalization is necessary for transfection. Figure 6a shows confocal laser scanning microscopy (CLSM) images of cellular internalization of fluorescein isothiocyanate (FITC)-labeled PEI/miRNA, PGEA/miRNA, and HA-SS-PGEA/miRNA NPs in rBMSCs over different incubation times. The amounts of fluorescent NPs significantly increased as the endocytosis time increased from 2 h to 4 h for each of the three types of polycation/miRNA NPs. In addition, more FITC-labeled NPs aggregated in rBMSCs treated by HA-SS-PGEA-FITC/miRNA than in rBMSCs mediated by PEI/miRNA and PGEA/miRNA after either 2 h or 4 h of cellular internalization. The stronger ability of HA-SS-PGEA to transport miRNA through cellular membrane than PEI and PGEA was partly attributed to the high affinity of HA to rBMSC membrane.[40] Moreover, HA-SS-PGEA was superior to PGEA in terms of structure and molecular weight, which led to smaller particle size and higher zeta potential of HA-SS-PGEA/miRNA than those of PGEA/miRNA and further enhancement of endocytosis.
Figure 6.
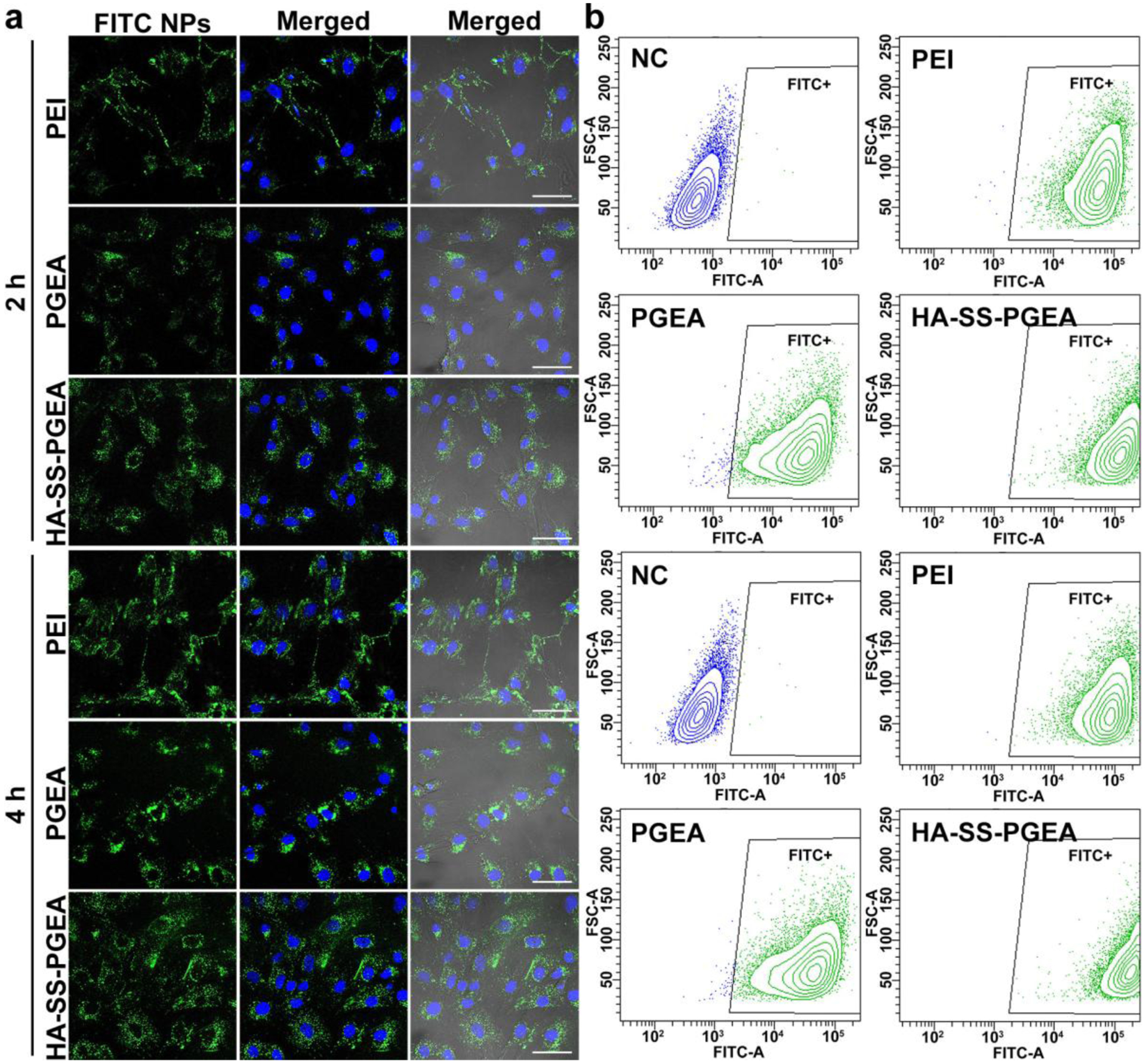
a) CLSM images of rBMSCs treated by PEI/miRNA, PGEA/miRNA, and HA-SS-PGEA/miRNA NPs for 2 h and 4 h. The NPs containing FITC-labeled polycations were shown in green, and the nuclei were stained by DAPI in blue. Scale bar = 50 μm. b) FCM analyses of FITC-positive rBMSCs treated by various NPs for 2 h and 4 h. NC is the negative control (rBMSCs were not treated by any NPs).
Flow cytometer (FCM) analyses were also carried out to further confirm the cellular internalization of various FITC-labeled polycation/miRNA NPs in rBMSCs (Figure 6b). Most or all of the rBMSCs were labeled by PEI/miR-26a, PGEA/miR-26a, and HA-SS-PGEA/miR-26a NPs after 2 h and 4 h of treatment (Table S2). The fluorescent intensity medians for PEI, PGEA, and HA-SS-PGEA groups were 6.00×104, 3.24×104, and 1.26×105, respectively, after 2 h of endocytosis, and increased to 8.55×104, 4.09×104, and 1.88×105, respectively, after 4 h of endocytosis (Table S2). In addition, the FITC-positive cells treated by HA-SS-PGEA/miRNA NPs distributed in a smaller region of fluorescent intensity than those mediated by PGEA/miRNA NPs. These results of FCM analyses revealed that HA-SS-PGEA promoted NP cellular internalization compared with PEI and PGEA, which were in line with the results displayed in CLSM images.
2.7. Transfection Affecting Cell Activities
Gene transfection can render specific gene expression of cells likely affecting cell activities. As shown in Figure 7a,b, miR-26a expression levels in rBMSCs mediated by PGEA/miR-26a and HA-SS-PGEA/miR-26a NPs at a series of N/P ratios and positive controls Lipofectamine 2000 (Lipo2000)- and PEI-based NPs were examined by quantitative reverse transcription polymerase chain reaction (qRT-PCR) relative to the internal reference U6 (RNU6–1). Both Lipo2000 and PEI demonstrated higher transfection efficiencies than the low-molecular-weight linear PGEA. There was also an apparent difference in transfection property between PGEA and HA-SS-PGEA at each N/P ratio, revealing that the chemical structure transformation and introduction of targeting property could dramatically affect miRNA delivery. The miR-26a expression level mediated by HA-SS-PGEA/miR-26a NPs first became higher and then decreased as the N/P ratio increased. In comparison with the Lipo2000 and PEI, HA-SS-PGEA exhibited significantly stronger ability to deliver miR-26a at the optimal N/P ratio of 20. The high affinity of HA to rBMSCs and enhanced NP endocytosis induced by HA-SS-PGEA could contribute to the improved transfection efficacy (Figure 6). Moreover, the miRNA was ready to escape from HA-SS-PGEA-based NPs upon exposure to highly reducing environment inside cells. Therefore, the HA-SS-PGEA/miR-26a NPs at the N/P ratio of 20 were selected for subsequent in vitro and in vivo studies.
Figure 7.
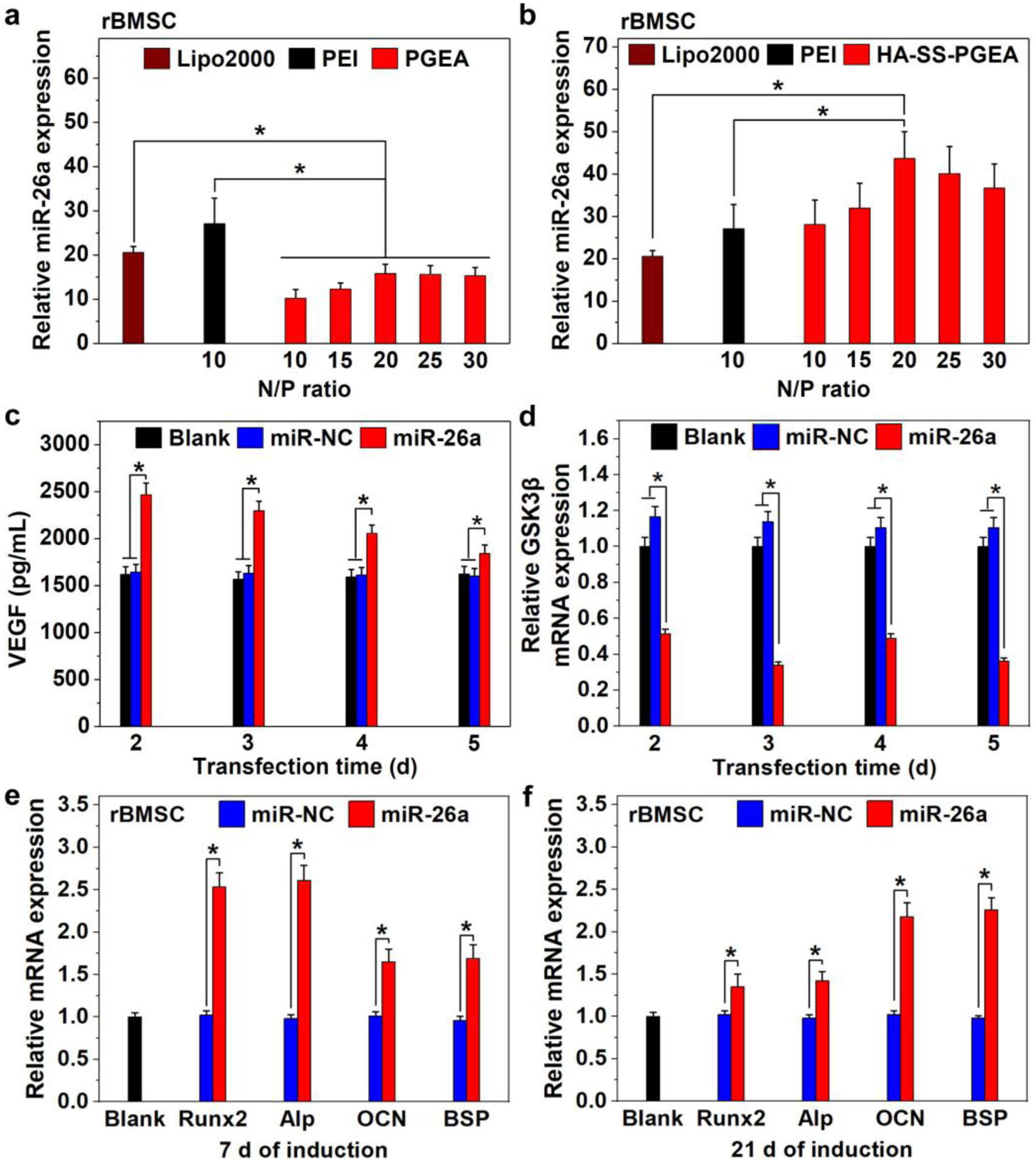
a, b) Relative miR-26a expression in rBMSCs mediated by Lipo2000-, PEI-, PGEA-, and HA-SS-PGEA-containing NPs, relative to the internal reference U6. c) VEGF expression in rBMSCs mediated by HA-SS-PGEA/miR-NC and HA-SS-PGEA/miR-26a at the N/P ratio of 20 after transfection for 2, 3, 4, and 5 days. d) Relative mRNA expression of GSK-3β in rBMSCs mediated by HA-SS-PGEA/miR-NC and HA-SS-PGEA/miR-26a at the N/P ratio of 20 after transfection for 2, 3, 4, and 5 days. The internal reference is GAPDH. e, f) Relative mRNA expressions of Runx2, Alp, OCN, and BSP in rBMSCs mediated by HA-SS-PGEA/miR-NC and HA-SS-PGEA/miR-26a at the N/P ratio of 20 after 7 d and 21 d of culture in osteo-differentiation medium (*p < 0.05; n = 3 per group). The internal reference is GAPDH.
Angiogenic growth factors can induce vascularization, which are crucial during entire bone formation.[15] A VEGF enzyme-linked immunosorbent assay (ELISA) kit was used to evaluate the angiogenic potential induced by delivered miR-26a because VEGF is a secreted mitogen related to angiogenesis.[53] Figure 7c shows VEGF expression levels in the media where rBMSCs were transfected by HA-SS-PGEA/miR-NC and HA-SS-PGEA/miR-26a NPs for different times. The rBMSCs, which were treated by HA-SS-PGEA/miR-NC NPs, secreted almost the same amount of VEGF to the cells without treatment of NPs at each transfection time point. In contrast, HA-SS-PGEA/miR-26a NPs resulted in a significant increase of VEGF secretion, indicating a likely positive effect of HA-SS-PGEA/miR-26a on angiogenesis.
miR-26a targets and inhibits GSK-3β, further promoting osteogenic differentiation.[17,18,54] Thus GSK-3β mRNA expression levels in rBMSCs were detected relative to the internal reference glyceraldehyde-3-phosphate dehydrogenase (GAPDH) after using HA-SS-PGEA/miR-NC and HA-SS-PGEA/miR-26a. As shown in Figure 7d, the GSK-3β expression in rBMSCs after HA-SS-PGEA/miR-26a treatment for 2–5 days was greatly suppressed, while rBMSCs with and without HA-SS-PGEA/miR-NC treatment showed no influence on the GSK-3β expression.
To investigate the effects of delivered miR-26a by HA-SS-PGEA on osteogenesis, the mRNA expression of runt-related transcription factor 2 (Runx2), alkaline phosphatase (Alp), osteocalcin (OCN), and bone sialoprotein (BSP) markers was determined via qRT-PCR after the rBMSCs were treated for 7 d and 21 d. For HA-SS-PGEA/miR-26a transfected rBMSCs, the Runx2 and Alp mRNA expression levels decreased as the induction time increased from 7 d to 21 d, while OCN and BSP mRNA expression levels increased during the same period of induction time. The different trends were because Runx2 and Alp are early-stage osteogenic markers and OCN and BSP are late-stage osteogenic markers. Moreover, a much higher mRNA expression of these four genes was detected in the HA-SS-PGEA/miR-26a group than in the HA-SS-PGEA/miR-NC and blank groups, revealing that HA-SS-PGEA/miR-26a effectively promoted osteogenesis of rBMSCs.
2.8. Radiographic Analysis
The rat cranial bone defect healing was quantified by micro computed tomography (micro-CT) after implantation of HA-SS-PGEA/miRNA-incorporated 3D hybrid nanofiber aerogels for 4 weeks. Figure 8a shows the representative micro-CT images of the new bone and defect in the groups of blank (without 3D aerogel or NPs), 3D aerogel with HA-SS-PGEA/miR-NC NPs (aerogel/miR-NC NPs), and HA-SS-PGEA/miR-26a NPs (aerogel/miR-26a NPs). Red circles were used to demarcate the newly formed bone from the pristine bone in the regions of interest. X-ray radiopacity induced by HA-SS-PGEA/miRNA-incorporated 3D aerogels increased remarkably in comparison with that in the blank group. Much larger defect healing area was observed in the aerogel/miR-26a NPs groups than in the aerogel/miR-NC NPs group after 4 weeks of implantation. The new bone volumes induced by blank, aerogel/miR-NC NPs, and aerogel/miR-26a NPs were 2.1 mm3, 7.5 mm3, and 21.8 mm3, respectively, and the corresponding closure percentages (new bone volume over defect volume) were 6.0%, 21.4%, and 62.2%, respectively (Figure 8b). Figure 8c shows 56.4% coverage (bone formation area over defect area) for aerogel/miR-26a NPs group, much higher than the coverage percentages in the unfilled defect (7.3%) and aerogel/miR-NC NPs (19.7%) groups.
Figure 8.
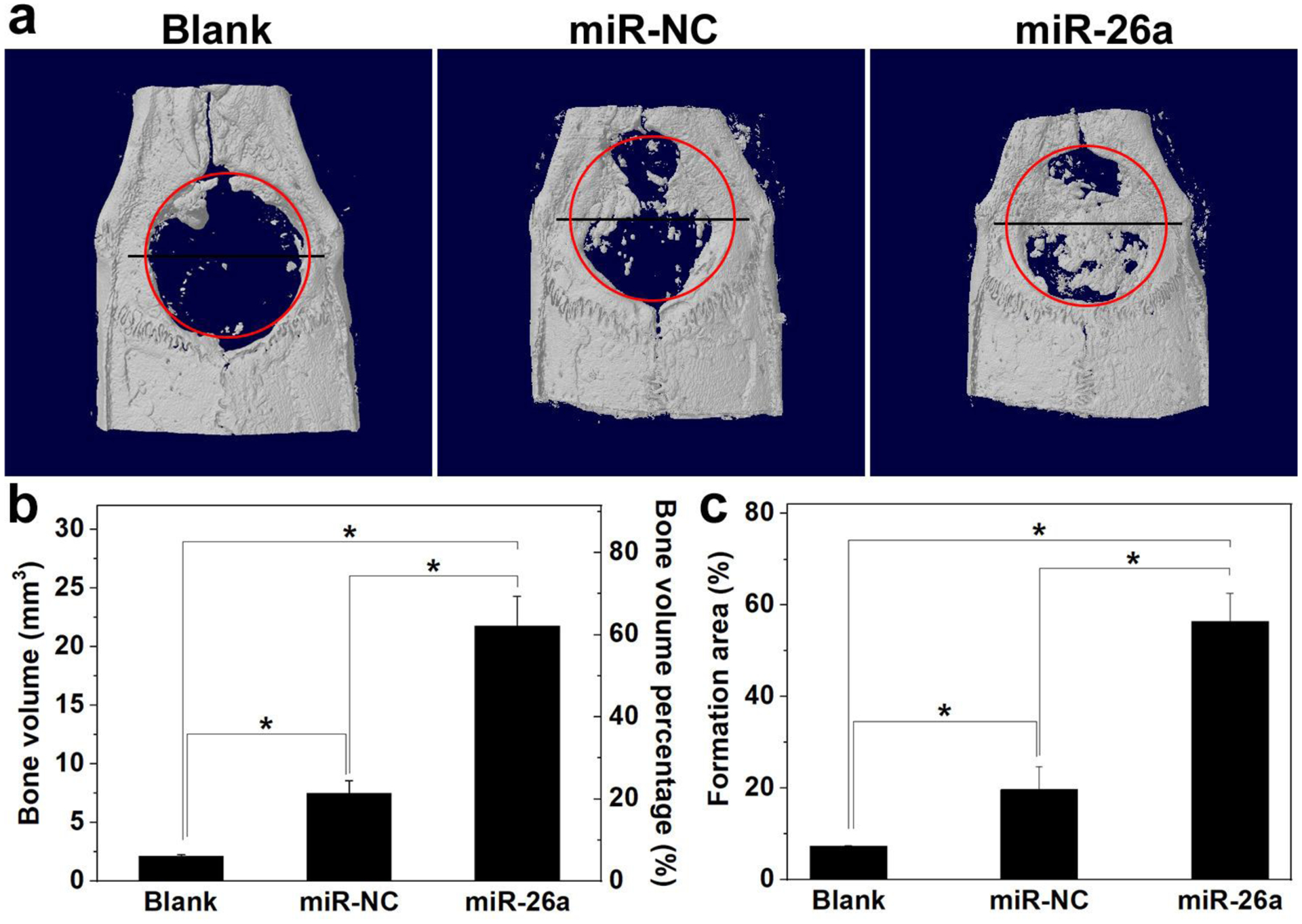
a) Representative planar radiographs of cranial bone defects at 4 weeks after implantation. b, c) Regenerated bone volume and bone formation area for various groups (*p < 0.05; n = 5 per group). Blank: without treatment. MiR-NC: 3D nanofiber aerogels with HA-SS-PGEA/miR-NC NPs. MiR-26a: 3D nanofiber aerogels with HA-SS-PGEA/miR-26a NPs.
2.9. Histopathological Analyses
The bone defect healing mediated by aerogel/miRNA NPs were further assessed via histomorphometric analysis. Figure 9a,b show the representative images of hematoxylin and eosin (H & E) and Masson’s trichrome (MTC) staining of slices sectioned from the positions of black straight lines as shown in Fig. 6a for all the groups. The absence of foreign body giant cells was the evidence that no inflammatory and/or foreign body reaction was induced by the implantation of 3D aerogels with or without HA-SS-PGEA/miR-26 NPs. In view of the inflammatory response induced by abruptly released acidic degradation products, bioactive glass possibly counteracted the acidic degradation to stabilize the local pH, thus remaining a mild physiological environment.[55–57] The new bone formation was badly retarded in the unfilled cranial defects as evident from the limited-size new bone formed at the periphery of the defects. The bone defect healing was found in the peripheral and middle regions of defects without integral bridging for the cranial defects of rats treated by 3D aerogels with miR-NC-containing NPs. In contrast, the new bone trabeculae were much thicker in case of 3D aerogel/miR-26a NPs implantation. As shown in Figure 9a,b, few tiny clumps of debris (indicated by black arrows) was observed in the 3D aerogel/miR-NC and 3D aerogel/miR-26a groups, indicating such 3D hybrid aerogels were able to degrade as the PLGA nanofiber were hydrolyzed and largely broke down after 4 weeks of implantation. PLGA nanofibers underwent a bulk erosion process and their degradation rate could exceed the defect healing rate, which may cause the incomplete healing of bone defects. PLGA with a higher ratio of lactide to glycolide or other polymers can be used in the future work to lower the scaffold degradation rate and provide a sufficiently long period for bone regeneration. Furthermore, more new blood vessels in the newly formed bone were observed in the 3D aerogel/miR-26a NPs group than in the other two groups. Figure 9c shows the neovascular density in rat cranial bone defects in various groups. The aerogel together with miR-26a-incorporated NPs induced the formation of higher density of blood vessels than the blank and aerogel/miR-NC NPs. The results of histopathological analyses further proved that 3D aerogels incorporated with HA-SS-PGEA/miR-26a NPs effectively facilitated new bone formation. According to the in vitro results, it could be speculated that transient dissociative rBMSCs interacted with the 3D aerogels and HA-SS-PGEA/miR-26a NPs and engulfed these NPs, eliciting VEGF secretion and further facilitating the recruitment of osteoblasts and their progenitors at the early stage of bone formation. MiR-26a could also regulate the coupling of angiogenesis and osteogenesis to promote bone repair during the late stage of bone formation. Nevertheless, HA-SS-PGEA/miR-26a NPs functionalized 3D aerogels demonstrated a great capacity for healing critical-sized cranial bone defects.
Figure 9.
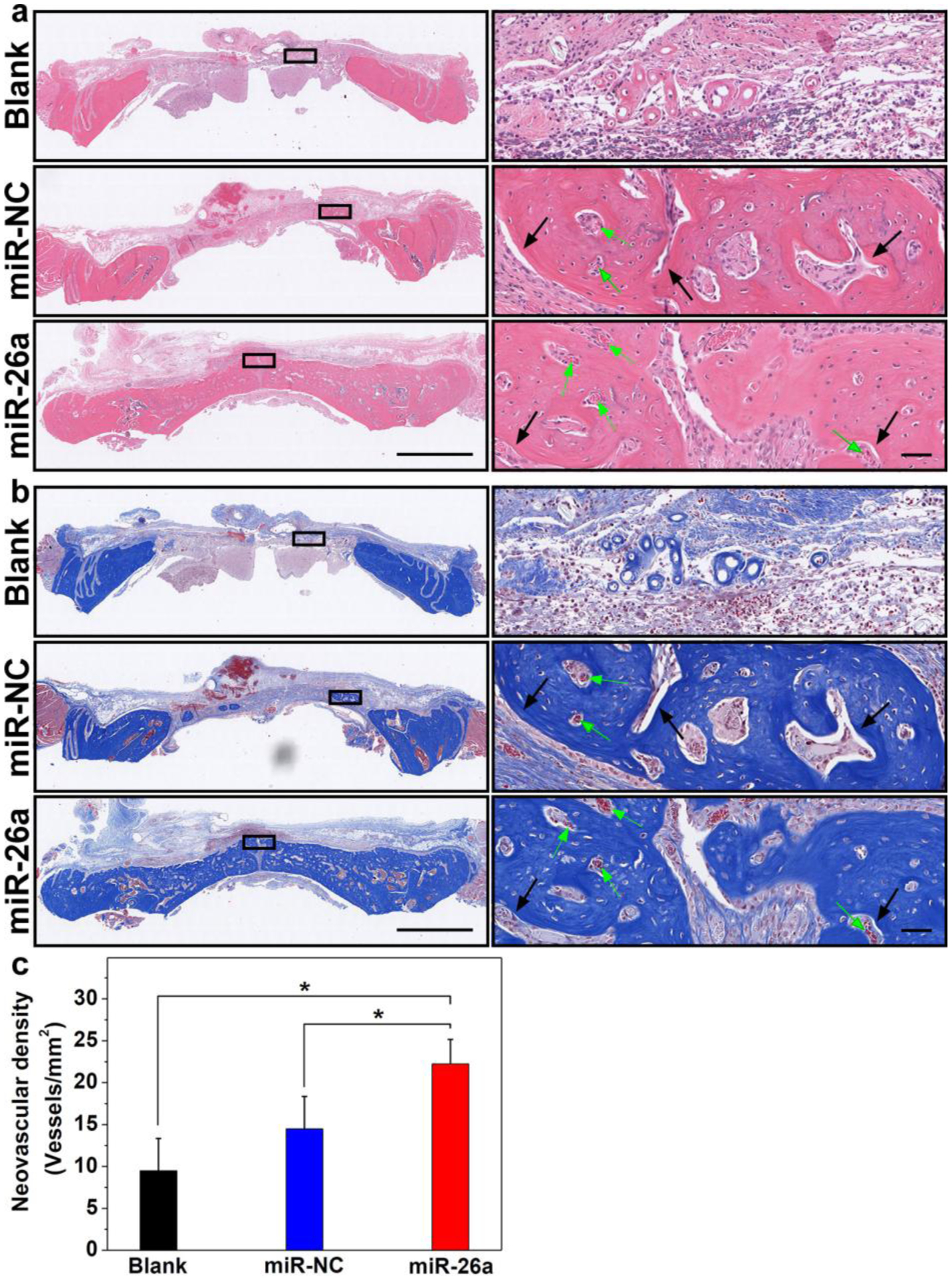
a) H & E staining and b) MTC staining of regenerated tissues after implantation for 4 weeks. The representative images presented in the left column were obtained by sectioning across the lines shown in Figure 6a. Scale bar in the left column = 2 mm. Scale bar in the right column = 50 μm. Green arrows indicate blood vessels. c) Neovascularization of the regenerated tissue in the calvarial bone defects in different groups after 4 weeks of implantation (*p < 0.05; n = 5 per group). Blank: without treatment. MiR-NC: 3D nanofiber aerogels with HA-SS-PGEA/miR-NC NPs. MiR-26a: 3D nanofiber aerogels with HA-SS-PGEA/miR-26a NPs.
3. Conclusions
In this study, the novel comb-like polycationic gene vector HA-SS-PGEA consisting of HA backbone, disulfide linkages, and short PGEA arms, was synthesized. HA-SS-PGEA demonstrated higher transfection efficiency than PEI and low-molecular-weight linear PGEA and lower cytotoxicity, likely attributed to the targeting content that induced receptor-mediated endocytosis, and biodegradability that increased potential to release incorporated miRNA and reduced toxicity. HA-SS-PGEA-mediated miR-26a delivery into rBMSCs suppressed GSK-3β signaling and enhanced VEGF secretion, coordinating the coupling between osteogenesis and angiogenesis. The implantation of 3D hybrid nanofiber aerogels loaded with HA-SS-PGEA/miR-26a NPs significantly promoted healing of rat cranial bone defects. The present work demonstrated that the combination of 3D aerogels and targeting, bioreducible polycation-mediated miRNA delivery is highly promising for tissue regeneration.
4. Experimental Section
Materials:
Sodium hyaluronate (Mw 15 kDa) was purchased from Lifecore Biomedical Inc., Chaska, MN. Polyvinylpyrrolidone (PVP, Mw 1300000), Dowex H+ 50W × 8–100 sulfonic resin, N-Hydroxysuccinimide (NHS, 98%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDAC, ≥99%), 2-(N-Morpholino)ethanesulfonic acid (MES, ≥99%), cystamine dihydrochloride (CADH, >98%), α-bromoisobutyric acid (BIBA, 98%), dimethyl sulfoxide (DMSO, ≥99.5%), trimethylamine (TEA, ≥99%), copper(I) bromide (CuBr, 99%), glycidyl methacrylate (GMA, 98%), ethanolamine (EA, 98%), polyethylenimine (PEI, 25 kDa), and fluorescein isothiocyanate isomer (FITC, 97.5%) were obtained from Sigma-Aldrich Chemical Co., St. Louis, MO. N,N,N’,N’,N”-pentamethyldiethylenetriamine (PMDETA, ≥99%) and ethylenediaminetetraacetic acid (EDTA, 99%) were obtained from Acros Organics N.V., New Jersey, US. GMA was used after removal of the inhibitors in a ready-to-use disposable inhibitor-removal column. Poly(lactic-co-glycolic acid) (PLGA, 50:50 lactide:glycolide) was purchased from Lactel, Birmingham, AL. Rno-miR-26a-5p (miRNA mimic) was purchased from ThermoFisher Scientific International Inc, Massachusetts, US.
Synthesis of HA-SS-PGEA:
To obtain HA, ion exchange was performed as follows: Dowex H+ sulfonic resin (500 mg) was added in 100 mL of deionized (DI) water containing 500 mg of sodium hyaluronate and stirred at RT for 1 h. When the pH reached 2.85, the resin was removed via filtration and the HA was harvested via lyophilization. HA (200 mg), NHS (43.7 mg), and EDAC (87.3 mg) were completely dissolved in 10 mL of MES buffer (pH 4.72) and the solution was stirred at RT for 4 h. Then, the activated HA solution was added dropwise into 10 mL of DI water containing 830.7 mg of CADH and the reaction was processed for 12 hours. A total of 160 mg of HA-SS-NH2 were harvest after being dialyzed against DI water with dialysis membrane (Molecular weight cut-off (MWCO) 3500–5000 Da) for 48 h and freeze-dried.
BIBA (30.2 mg) was first activated by NHS (25.0 mg) and EDAC (49.8 mg) in 500 μL of DMF at 30 °C for 4 h. Then, HA-SS-NH2 (100 mg) in 4.5 mL of MES buffer (pH 4.72) was added and the reaction solution was stirred at 37 ℃ for 48 h. A total of 70 mg of HA-SS-Br were collected after dialysis against DI water for 48 h and lyophilization.
ATRP was used to prepare HA-SS-PGMA. HA-SS-Br (30 mg) was dissolved in 4 mL of DMSO containing 1 mL of GMA and 15.4 μL of PMDETA. Then, CuBr (5.3 mg) was added and the ATRP was performed in an argon atmosphere for 20 min. Before dried in a vacuum, the product was precipitated with methanol and purified via redissolution in DMSO and reprecipitation in methanol. A total of 100 mg of HA-SS-PGMA were obtained. Linear PGMA was also synthesized as control via ATRP. BIBA was used as initiator and the polymerization was processed in DMSO at ratio [BIBA]:[GMA]:[CuBr]:[PMDETA] of 1:50:0.2:0.4 in an argon atmosphere for 1 h. Linear PGMA was harvested after methanol precipitation and drying in vacuum.
EA-functionalized HA-SS-PGMA (abbreviated as HA-SS-PGEA) and linear EA-functionalized PGMA (PGEA) as control were prepared via ring-opening reaction. HA-SS-PGMA or PGMA (80 mg) was dissolved in 4 mL of DMSO containing excess EA (680 μL) and 100 μL of TEA. The reaction solution was stirred at 37 °C for 5 days. Then, the reaction solution was diluted with 10 times volume of DI water and dialyzed against DI water with dialysis membrane (MWCO 3500–5000 Da) for 48 h. The yields of final product HA-SS-PGEA and PGEA were 82 mg and 90 mg, respectively, after lyophilization.
Fabrication of 3D Hybrid Nanofiber Aerogels:
The 3D hybrid nanofiber aerogel was prepared following the procedures in our previous work with some modifications.[45] PCG and BG nanofibers were first fabricated.[58–61] Briefly, PLGA, gelatin, and collagen were dissolved in hexafluoroisopropanol at a mass ratio of 2:1:1. To fabricate PCG nanofibers, 0.4 mL/h feeding rate, 15 kV applied direct-current voltage, and 15 cm distance between a spinneret and a collecting mandrel at a fast rotation speed were applied. The PCG nanofibers were then cross-linked with glutaraldehyde vapor for 12 h. For fabricating BG nanofibers, tetraethyl orthosilicate, triethyl phosphate, and Ca(NO3)2·4H2O were dissolved in acidic solution at a [Si]:[P]:[Ca] molar ratio of 80:10:10 and then mixed with an equal volume of 16.5% PVP solution in ethanol. The feeding rate of the mixed solution was 0.6 mL/h, and the mandrel rotated slowly to collect composite fibers with the same remaining conditions mentioned in the PCG nanofiber preparation. The PVP fibers were removed fibers through sintering the composite nanofibers in a muffle furnace at 600 °C for 5 h and BG nanofibers were harvested. The PCG and BG nanofibers were cut into segments and homogenized in DI water with a sonicator (Qsonica 500, Newtown, CT) at a weight ratio of 60:40. The final segment concentration was 26.7 mg/mL. The segment mixture was added in copper ring molds glued to an aluminum plate and frozen at −20 °C for 3 h and then −80 °C for 15 min. After lyophilization, the samples were immersed in prewarmed absolute ethanol at 48 °C for 10 min to thermally cross-link the segments. Subsequently, the ethanol was removed by washing the samples with DI water. The aerogels were shaped with 8 mm of diameter and 1 mm of thickness by using a cryotome and then freeze dried.
Characterization of Polymers and Aerogels:
The chemical structures of synthesized HA-SS-NH2, HA-SS-Br, HA-SS-PGMA, and HA-SS-PGEA were characterized by Bruker AVIII 500 1H NMR. The surface morphology of the 3D hybrid aerogels was characterized by a FEI Quanta 200 scanning electron microscope (SEM). The applied accelerating voltage was 25 kV. Charging was avoided by coating gold and palladium on the aerogels before acquiring the SEM images.
Formation and Characterization of Polyplex Nanoparticles:
PEI, PGEA, and HA-SS-PGEA were dissolved into sterilized diethyl pyrocarbonate (DEPC)-treated water to reach nitrogen concentration of 10 mM. HA-SS-PGEA/miRNA and PGEA/miRNA polyplex nanoparticles (NPs) at various N/P ratios (expressed as the ratio of nitrogen in the polycation to phosphorus in miRNA) were prepared via mixing polycation and miRNA solutions (in DEPC-treated water) on a vertex mixer and then being kept at room temperature for 30 min. The particle size and zeta potential of formed nanoparticles at N/P ratios of 10–30 were determined by using Zetasizer Nano ZS with a laser of wavelength 633 nm at a 173° scattering angle. The polycations were examined for their abilities to bind miRNA through agarose gel electrophoresis in a Sub-Cell system using the similar procedures described earlier.[62] For evaluation of the effect of DTT and heparin on NPs, the formed NPs at N/P ratio of 20 were treated by 10 mM of DTT for 1 h and a series of concentrations of heparin (60–360 μg/mL) for 1 h. MiRNA bands were visualized and photographed by a UV transilluminator and BioDco-It imaging system.
NP and miRNA Release:
3D aerogels loading FITC-labeled HA-SS-PGEA/miR-26a and unlabeled HA-SS-PGEA/miR-26a NPs at N/P ratio of 20 (60 pmol miRNA per aerogel) were immersed in 200 μL of phosphate-buffered saline (PBS, pH 7.4) shaken at 80 r.p.m. at 37 ℃. The release media was withdrawn and replaced with an equal volume of fresh PBS at predetermined time intervals. The fluorescent intensity of FITC-labeled NPs (100 μL of release medium in each well in a 96-well plate) was quantified by an Infinite M200PRO microplate reader at an excitation and emission wavelength of 490 nm and 525 nm, respectively. For unlabeled NPs, unknown, blank or standard sample (20 μL) was added to a spectrophotometer cell containing QuantiFluor RNA Dye working solution (200 μL) and incubated for 5 min in a dark room. The miRNA concentration of the sample was measured by a HORIBA Scientific FluoroMax-4 spectrofluorometer.
Cell Viability:
MTT assay was used to evaluate the cytotoxicity of HA-SS-PGEA, compared with that of PEI and PGEA, in rBMSC and MC3T3-E1 cell line. Dulbecco’s modified eagle medium (DMEM, low glucose concentration), replenished with 10% heat-inactivated fetal bovine serum (FBS), 100 units/mL of penicillin, and 100 μg/mL of streptomycin, was used to culture rBMSC. Alpha minimum essential medium (α-MEM, without ascorbic acid) with 10% FBS, 100 units/mL of penicillin, and 100 μg/mL of streptomycin was used to culture MC3T3-E1 cell line. Cells were seeded into 96-well plates at a density of 1×104/well and cultured for 24 hours. Then, the medium was replaced with fresh medium containing HA-SS-PGEA- or PEI-containing miRNA NPs (N concentration of 2.6 mg/L) to culture cells for 24 h. After washed with PBS three times, the cells were treated with sterile-filtered MTT solution in PBS (0.5 mg/mL) for 4 h. DMSO was added to the wells after the cells were washed with PBS. The absorbance was measured at a wavelength of 570 nm. For each sample, the final absorbance was the average of those measured from six wells in parallel. The formula [A]test/[A]control × 100% was used to calculate the cell viability (%), where [A]test and [A]control are defined as the absorbance values of the wells with the polycations and controls (without the polycations), respectively.
Live/dead assay kit (Invitrogen, Eugene, OR) was used to evaluate the viability of rBMSCs treated with PEI/miRNA, PGEA/miRNA, and HA-SS-PGEA/miRNA NPs. A total of 8×104 rBMSCs were seeded to the 3.5-mm dishes with glass bottom and cultured for 24 h. Then, PEI/miRNA, PGEA/miRNA, or HA-SS-PGEA/miRNA NPs were added to the media and incubated with cells for 12 h. The auld culture media were substituted with fresh media to culture the cells for additional 12 h. Afterwards, the cells were washed with PBS three times, and all the non-adherent cells were collected by centrifugation, resuspended in fresh PBS and added to the original dishes. Calcein AM and ethidium homodimer-1 stock solutions were added to the dishes, reaching final concentrations of 0.5 μM and 4 μM, respectively, for staining the cells at room temperature for 40 min. A Zeiss LSM 800 Airyscan CLSM was then used to acquire the images of live cells stained by calcein AM in green and dead cells stained by ethidium homodimer-1 in red.
Cellular Internalization:
A total of 16 mg of PEI and 2 mg of FITC were dissolved in 2 mL of ethanol and stirred in dark room at room temperature overnight. PGEA and HA-SS-PGEA were separately dissolved in ethanol containing FITC at the molar ratio [secondary amine group]:[FITC] of 14.5:1 and stirred away from light at room temperature for 24 h. The FITC-labeled PEI (PEI-FITC), PGEA (PGEA-FITC), and HA-SS-PGEA (HA-SS-PGEA-FITC) were then harvested via dialysis and lyophilization. These FITC-labeled polycations were separately dissolved in sterile DEPC-treated water to reach a final nitrogen concentration of 10 mM as working solutions used for the following endocytosis assay.
rBMSCs were seeded in 3.5-mm dishes with glass bottom and 12-well plates at the density of 8 ×104 cells/well and cultured in media for 24 h. The stale media was substituted with fresh media containing PEI-FITC/miRNA at the N/P ratio of 10 and PGEA-FITC/miRNA and HA-SS-PGEA/miRNA at the N/P ratios of 20. CLSM was used to collect images of cells in dishes at 2 h and 4 h after the FITC-labeled polycation/miRNA treatment. The nuclei were stained by immersing cells in 200 ng/mL of 4′,6-diamidino-2-phenylindole (DAPI) solution in PBS. The endocytosis was also analyzed by a FCM after the cells in plates were treated by various FITC-labeled polycation/miRNA NPs for 2 h and 4 h, digested in trypsin and resuspended in PBS with 2% FBS and 1 mM EDTA.
Transfection:
rBMSC cells were seeded in 12-well plates at the density of 1×105 cells/well. When the cell confluence was about 40%, polycation/miR-NC (negative control) and polycation/miR-26a nanoparticles (containing 120 pmol miRNA) were added and then cultured with cells for 12 h. Lipo2000 was used according to the manual. Then, the stale medium was substituted with fresh medium to culture cells for additional 48 h. The total RNA was extracted with PureLink RNA mini kit (Invitrogen) and reverse transcribed into cDNA by using a miScript II RT kit (Qiagene, Hilden, DE). The expression level of miR-26a was determined via SYBR-Green qRT-PCR and expressed as a relative value based on the U6 miRNA gene expression.
ELISA:
For secreted VEGF analysis, rBMSCs were transfected with HA-SS-PGEA/miRNA (miR-26a or miR-NC) as described above. After 12 h of transfection, the medium was substituted with fresh DMEM medium containing 5.0% serum substitute nu-Serum (Corning, Corning, NY). The supernatant was collected at 2 d, 3 d, 4 d, and 5 d. VEGF levels in the supernatant were detected with a VEGF ELISA kit (AVIVA System Biology, San Diego, US) according to the manual. A Synergy H1 hybrid reader was used to determine the absorbance at 450 nm.
qRT-PCR:
rBMSCs were transfected as described above. After 12 h of transfection, the medium was replaced with fresh medium every two days. The total mRNA was extracted from the cells by using RNeasy mini kit (Qiagene) at 2 d, 3 d, 4 d, and 5 d and reverse transcribed into cDNA by using cDNA synthesis kit (Quantabio, Beverly, MA). The GSK-3β mRNA expression levels were quantified via SYBR-Green qRT-PCR and expressed as a relative value based on the GAPDH mRNA gene expression.
Osteoblast differentiation was also determined by qRT-PCR. A total of 1×105 rBMSCs were seeded to each well of 12-well plates. HA-SS-PGEA/miR-NC and HA-SS-PGEA/miR-26a NPs were added to the medium after the confluence reached about 40%. The cells were cultured with the NPs for 12 h and then in fresh medium for additional 48 h. Subsequently, the medium was replaced with OsteoMAX-XF differentiation medium (ODM) (MilliporeSigma, Burlington, VT) and cultured with the cells for 7 d and 21 d. The total RNA was extracted with RNeasy mini kit (Qiagene) and generated cDNA using cDNA synthesis kit (Quantabio). The mRNA expression level of Runx2, Alp, OCN, and BSP were determined via SYBR-Green qRT-PCR and expressed as a relative value based on the GAPDH mRNA gene expression. The primers used are listed in Table S1.
Critical-Sized Cranial Bone Defect Model:
Care and use of laboratory animals were followed by the animal protocol (15–067-09-FC) approved by the Institutional Animal Care and Use committee at the University of Nebraska Medical Center. Twelve-week-old Sprague Dawley rats (male) were used to create critical-sized cranial bone defects for in vivo study. Linear incisions from the nasal bone to the occiput were created down to cranial periosteum. A trephine mounted on a dentist drill was used to create circular defects of 8 mm diameter centered on the parietal cranial bone. Sterile PBS was irrigated onto the wound to dissipate the heat generated while drilling. After removing the cranial disks, 3D hybrid aerogels loaded with miRNA-containing NPs were placed into the defects. Herein, HA-SS-PGEA/miRNA NP solution (5 μL) was dropped onto a 3D hybrid nanofiber aerogel (1 mm height × 8 mm diameter) five times to reach a total volume of 25 μL (containing 60 pmol miR-26a or miR-NC). The incisions were sutured with #6/0 suture. A total of 15 rats were divided into three groups (n = 5) as follows: (1) Blank group without aerogel implantation, (2) 3D aerogel with miR-NC group, and (3) 3D aerogel with miR-26a group. The rats were sacrificed after 4 weeks of implantation. The calvaria were harvested and fixed in 10% formalin for 3 d and then kept in 70% ethanol for further radiographic and histological analyses.
Radiographic Analysis:
A high-resolution micro-CT scanner (Skyscan 1172, Kontick, Belgium) was used to scan the formalin-fixed samples. Scan settings were as follows: X-ray tube voltage of 70 kV, current of 114 μA, and slice thickness/slice increment of 8.71 μm. The radiographic analysis was carried out by CT analyzer software (Bruker microCT). Multiple scan slices were used for a 3D reconstruction of the skull bone. New bone volume and formation area in the 8-mm round region of interest were determined through 3D analysis. The defect volume was calculated as πr2t, where radius of defect r = 4 mm and thickness t = 0.6 mm. The thickness of the rat skull has been reported to vary from 0.5–0.7 mm, depending on the body mass.[63,64]
Histopathological Analysis:
Following the radiographic analysis, the calvaria were decalcified with 10% EDTA solution for 30 d with solution replacement every 7 d, dehydrated in an increasing gradient series of ethanol (70–100%), and embedded in paraffin. Multiple sections with 5 μm of thickness were prepared per sample with a microtome and stained with H & E or MTC, following the manufacturer’s protocols. The stained sections were examined with a Ventana’s Coreo Au slide scanner. The blood vessel numbers were counted and normalized to the tissue area in the histology slice images by using a Ventana image viewer v. 3.1.3.
Statistical Analysis:
Each experiment was performed at least three replicates. The data was analyzed using the SPSS software. The statistical analysis was performed using one-way analysis of variance with Tukey test, and *p < 0.05 indicated a statistical significance.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institute of General Medical Science (NIGMS) at the NIH (5P30GM127200-03), National Institute of Dental and Craniofacial Research (NIDCR) at the NIH (1R21DE027516), NE LB606, and startup funds from the University of Nebraska Medical Center.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Primers, 1H NMR spectra, images of agarose gel electrophoresis, miRNA release profiles, NP sizes after release and DTT treatment, and quantified data of FCM can be found in the Supporting Information.
Contributor Information
Haiqing Song, Department of Chemical Engineering, University of Illinois at Chicago, Chicago, IL 60607, United States.
Matthew J. Teusink, Department of Orthopedic Surgery and Rehabilitation, University of Nebraska Medical Center, Omaha, NE 68198, United States
Jingwei Xie, Department of Surgery-Transplant and Holland Regenerative Medicine Program University of Nebraska Medical Center, Omaha, NE 68130, United States.
References
- [1].Kolambkar YM, Boerckel JD, Dupont KM, Bajin M, Huebsch N, Mooney DJ, Hutmacher DW, Guldberg RE, Bone 2011, 49, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liu Z, Chang H, Hou Y, Wang Y, Zhou Z, Wang M, Huang Z, Yu B, Mol. Med. Rep 2018, 18, 5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sanders DW, Bhandari M, Guyatt G, Heels-Ansdell D, Schemitsch EH, Swiontkowski M, Tornetta P, Walter S, Orthop J. Trauma 2014, 28, 632. [DOI] [PubMed] [Google Scholar]
- [4].Van Heest A, Swiontkowski M, Lancet 1999, 353, S28. [DOI] [PubMed] [Google Scholar]
- [5].Kolambkar YM, Dupont KM, Boerckel JD, Huebsch N, Mooney DJ, Hutmacher DW, Guldberg RE, Biomaterials 2011, 32, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li J, Hong J, Zheng Q, Guo X, Lan S, Cui F, Pan H, Zou Z, Chen C, J. Orthop. Res 2011, 29, 1745. [DOI] [PubMed] [Google Scholar]
- [7].Stadler BM, Ruohola-Baker H, Cell 2008, 132, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dirks PB, Cell 2009, 138, 423. [DOI] [PubMed] [Google Scholar]
- [9].Clark EA, Kalomoiris S, Nolta JA, Fierro FA, Stem Cells 2014, 32, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Karsenty G, Kronenberg HM, Settembre C, Annu. Rev. Cell Dev. Biol 2009, 25, 629. [DOI] [PubMed] [Google Scholar]
- [11].Karsenty G, Annu. Rev. Genomics. Hum. Genet 2008, 9, 183. [DOI] [PubMed] [Google Scholar]
- [12].Long F, Nat. Rev. Mol. Cell Biol 2012, 13, 27. [DOI] [PubMed] [Google Scholar]
- [13].Schipani E, Maes C, Carmeliet G, Semenza GL, Bone Miner J. Res. 2009, 24, 1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lin L, Shen Q, Leng H, Duan X, Fu X, Yu C, Mol. Ther 2011, 19, 1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Y, Fan L, Liu S, Liu W, Zhang H, Zhou T, Wu D, Yang P, Shen L, Chen J, Jin Y, Biomaterials 2013, 34, 5048. [DOI] [PubMed] [Google Scholar]
- [16].Su X, Liao L, Shuai Y, Jing H, Liu S, Zhou H, Liu Y, Jin Y, Cell Death Dis. 2015, 6, e1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vacik T, Stubbs JL, Lemke G, Genes Dev. 2011, 25, 1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rawadi G, Vayssiere B, Dunn F, Baron R, Roman-Roman S, Bone Miner J. Res. 2003, 18, 1842. [DOI] [PubMed] [Google Scholar]
- [19].Gerber HP, Ferrara N, Trends Cardiovasc. Med 2000, 10, 223. [DOI] [PubMed] [Google Scholar]
- [20].Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM, Development 2003, 130, 2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xie Y, Murray-Stewart T, Wang Y, Yu F, Li J, Marton LJ, Casero RA Jr., Oupický D, J. Control. Release 2017, 246, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huang S, Duan S, Wang J, Bao S, Qiu X, Li C, Liu Y, Yan L, Zhang Z, Hu Y, Adv. Funct. Mater 2016, 26, 2532. [Google Scholar]
- [23].Xu J, Sun J, Hoc PY, Luo Z, Ma W, Zhao W, Rathod SB, Fernandez CA, Venkataramanan R, Xie W, Yu A-M, Li S, Biomaterials 2019, 210, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wen Y, Guo Z, Du Z, Fang R, Wu H, Zeng X, Wang C, Feng M, Pan S, Biomaterials 2012, 33, 8111. [DOI] [PubMed] [Google Scholar]
- [25].Li W, Ma N, Ong LL, Nesselmann C, Klopsch C, Ladilov Y, Furlani D, Piechaczek C, Moebius JM, Lützow K, Lendlein A, Stem Cells 2007, 25, 2118. [DOI] [PubMed] [Google Scholar]
- [26].Kim HH, Choi HS, Yang JM, Shin S, Int. J. Pharm 2007, 335, 70. [DOI] [PubMed] [Google Scholar]
- [27].Bachrach U, Curr. Protein Pept. Sc 2005, 6, 559. [DOI] [PubMed] [Google Scholar]
- [28].Santos JL, Pandita D, Rodrigues J, Pego AP, Granja PL, Balian G, Tomas H, Mol. Pharmaceutics 2010, 7, 763. [DOI] [PubMed] [Google Scholar]
- [29].Liu S, Zhou D, Yang J, Zhou H, Chen J, Guo T. J. Am. Chem. Soc 2017, 139, 5102. [DOI] [PubMed] [Google Scholar]
- [30].Park JS, Park W, Park S-J, Larson AC, Kim D-H, Park K-H, Adv. Funct. Mater 2017, 27, 1700396. [Google Scholar]
- [31].Liu H, Wang H, Yang W, Cheng Y, J. Am. Chem. Soc 2012, 134, 17680. [DOI] [PubMed] [Google Scholar]
- [32].Meister A, Anderson ME, Annu. Rev. Biochem 1983, 52, 711. [DOI] [PubMed] [Google Scholar]
- [33].Lee H, Mok H, Lee S, Oh YK, Park TG, Control J. Release 2007, 119, 245. [DOI] [PubMed] [Google Scholar]
- [34].Li YL, Zhu L, Liu Z, Cheng R, Meng F, Cui JH, Ji SJ, Zhong Z, Angew. Chem. Int. Ed 2009, 48, 9914. [DOI] [PubMed] [Google Scholar]
- [35].Fraser JR, Laurent TC, Laurent UB, J. Int. Med 1997, 242, 27. [DOI] [PubMed] [Google Scholar]
- [36].Raia NR, Partlow BP, McGill M, Kimmerling EP, Ghezzi CE, Kaplan DL, Biomaterials 2017, 131, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ranga A, Lutolf MP, Hilborn J, Ossipov DA, Biomacromolecules 2016, 17, 1553. [DOI] [PubMed] [Google Scholar]
- [38].Lee D, Beack S, Yoo J, Kim S-K, Lee C, Kwon W, Hahn SK, Kim C, Adv. Funct. Mater 2018, 28, 1800941. [Google Scholar]
- [39].Venkatraman S, Howden T, Widjaja LK, U.S. Patent 2018, 9,987,367. [Google Scholar]
- [40].Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B, Cell 1990, 61, 1303. [DOI] [PubMed] [Google Scholar]
- [41].Sherman L, Sleeman J, Herrlich P, Ponta H, Curr. Opin. Cell Biol 1994, 6, 726. [DOI] [PubMed] [Google Scholar]
- [42].Ren Y, Li R-Q, Cai Y-R, Xia T, Yang M, Xu F-J, Adv. Funct. Mater 2016, 26, 7314. [Google Scholar]
- [43].Song L, Zhao N, Xu F-J, Adv. Funct. Mater 2017, 27, 1701255. [Google Scholar]
- [44].Yang Y, Qi Y, Zhu M, Zhao N, Xu F, Nano Res. 2016, 9, 2531. [Google Scholar]
- [45].Weng L, Boda SK, Wang H, Teusink MJ, Shuler FD, Xie J, Adv. Healthcare Mater 2018, 7, 1701415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xu T, Zhao W, Zhu JM, Albanna MZ, Yoo JJ, Atala A, Biomaterials 2013, 34, 130. [DOI] [PubMed] [Google Scholar]
- [47].Day RM, Tissue Eng. 2005, 11, 768. [DOI] [PubMed] [Google Scholar]
- [48].Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR, Biomaterials 2006, 27, 3413. [DOI] [PubMed] [Google Scholar]
- [49].Karageorgiou V, Kaplan D, Biomaterials 2005, 26, 5474. [DOI] [PubMed] [Google Scholar]
- [50].Mintzer MA, Simanek EE, Chem. Rev 2009, 109, 259. [DOI] [PubMed] [Google Scholar]
- [51].Van de Wetering P, Moret EE, Schuurmans-Nieuwenbroek NME, van Steenbergen MJ, Hennink WE, Bioconj. Chem 1999, 10, 589. [DOI] [PubMed] [Google Scholar]
- [52].Meng F, Hennink WE, Zhong Z, Biomaterials 2009, 30, 2180. [DOI] [PubMed] [Google Scholar]
- [53].Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, van Bruggen N, Chopp M, J. Clin. Invest 2000, 106, 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang X, Li Y, Chen YE, Chen J, Ma PX, Nat. Commun 2016, 7, 10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bergsma EJ, Rozema FR, Bos RRM, Debruijn WC, J. Oral. Maxillofac. Surg 1993, 51, 666. [DOI] [PubMed] [Google Scholar]
- [56].Martin C, Winet H, Bao JY, Biomaterials 1996, 17, 2373. [DOI] [PubMed] [Google Scholar]
- [57].Rich J, Jaakkola T, Tirri T, Narhi T, Yli-Urpo A, Seppala J, Biomaterials 2002, 23, 2143. [DOI] [PubMed] [Google Scholar]
- [58].Boda SK, Chen S, Chu K, Kim HJ, Xie J, ACS Appl. Mater. Interfaces 2018, 10, 25069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Boda SK, Almoshari Y, Wang H, Wang X, Reinhardt RA, Duan B, Wang D, Xie J, Acta Biomat. 2019, 85, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Xie J, Blough ER, Wang CH, Acta Biomater. 2012, 8, 811. [DOI] [PubMed] [Google Scholar]
- [61].Weng L, Boda SK, Teusink MJ, Shuler FD, Li X, Xie J, ACS Appl. Mater. Interfaces 2017, 9, 24484. [DOI] [PubMed] [Google Scholar]
- [62].Qi Y, Song H, Xiao H, Cheng G, Yu B, Xu F-J, Small 2018, 14, 1803061. [DOI] [PubMed] [Google Scholar]
- [63].Icli B, Wara AK, Moslehi J, Sun X, Plovie E, Cahill M, Marchini JF, Schissler A, Padera RF, Shi J, Cheng HW, Circ. Res 2013, 113, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kim K, Kim JH, Kim I, Lee J, Seong S, Park YW, Kim N, Mol. Cells 2015, 38, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.