

Abstract
Perturbation of tissue homeostasis accompanies a diversity of inflammatory pathologies elicits endoplasmic reticulum (ER) stress, protein misfolding, and cell death. ER stress triggers initiation of the unfolded protein response (UPR), which determines divergent cell fate decisions, either promoting recovery of ER proteostasis and cell survival or triggering leveraged single-cell RNA-Seq to define dynamic transcriptional states associated with the adaptive versus terminal UPR in the intestinal epithelium. We integrated these transcriptional programs with genome-scale CRISPR screening to functionally dissect the UPR pathway and identify QRICH1 as a key effector of the PERK-eIF2α axis. QRICH1 controls a transcriptional program associated with translation and secretory networks that are specifically upregulated in inflammatory pathologies. Thus, QRICH1 dictates cell fate in response to pathological ER stress.
Keywords: ER stress, UPR, transcriptional regulation, QRICH1
Summary
QRICH1 is a central regulator of cell stress.
The integrated stress response (ISR) is equipped to respond to a broad range of extrinsic and intrinsic stimuli, enabling maintenance of homeostasis under conditions of cellular stress (1). Tissue inflammation can alter cellular protein homeostasis by increasing the demands of protein synthesis or leading to the accumulation of unfolded proteins in the ER, consequently activating a network of pathways that converge upon the phosphorylation of EIF2S1 (eIF2α) and resulting in global reduction of protein synthesis. Protein homeostasis (proteostasis) in the ER is maintained by a system of interconnected, tightly regulated processes comprised of the unfolded protein response (UPR), autophagy, and proteasome-dependent ER-associated degradation (ERAD) (2). The UPR is induced by disruption of ER proteostasis and senses ER stress through three transmembrane proteins: Inositol-Requiring Enzyme 1 (IRE1/ERN1), Activating Transcription Factor 6 (ATF6), and protein kinase RNA-like ER kinase (PERK,EIF2AK3) (3, 4). However, if the adaptive UPR is unable to restore proteostasis, sustained ER stress results in terminal-UPR activity, which culminates in cell death.
ER stress can both elicit and amplify tissue pathology. Genetic variants that intrinsically impair protein folding can lead to the accumulation of misfolded protein aggregates that give rise to unresolvable ER stress. For example, a frameshift variant in the MUC1 gene that is associated with kidney disease induces misfolding, aggregation, and ER stress (5, 6). Similar mechanisms have been described in proteinopathies and neurodegenerative diseases, such as Huntington’s, Alzheimer’s, Parkinson’s, and prion diseases (7, 8). In addition to intrinsic mechanisms leading to protein misfolding and ER stress, extrinsic factors can elicit similar outcomes. For example, tissue pH dysregulation, oxidative stress, pyrexia, increased translational/secretory demands, metabolic constraints, and infections can all result in protein misfolding and impaired ER homeostasis (9-11). The capacity for inflammation to create a tissue microenvironment that triggers ER stress is particularly damaging at epithelial barrier surfaces, such as the lung and intestines, where a barrier breach can expose the host immune system to pathogens and commensal microorganisms that further amplify tissue inflammation and cellular ER stress (12-15).
Given the role of ER stress in the pathogenesis of disease, there has been increased interest in therapeutic intervention in this cycle of inflammation and ER stress. However, the mechanisms that dynamically modulate the UPR to elicit a beneficial adaptive response versus a terminal apoptotic response are not fully understood. To gain insight into these regulatory mechanisms, we defined UPR signature states, and identified QRICH1 as a terminal-UPR regulator and critical determinant of cell fate under conditions of cellular stress. Collectively, our study sheds light on cellular mechanisms that regulate ER proteostasis and identifies QRICH1 as a central regulator of proteotoxicity associated with inflammatory diseases in the intestines and liver.
Temporal scRNA-seq identifies dynamic transcriptional programs that reflect the outcome of ER stress
Cellular responses to ER stress vary temporally and between individual cells. Therefore, we sought to identify candidate terminal-UPR modulators by defining temporal gene expression dynamics during ER stress. We utilized single-cell RNA-Seq (scRNA-seq) to define transcriptional states at single-cell resolution in a primary mouse intestinal epithelial monolayer culture system that, in part, recapitulates the cellular composition of the polarized epithelium, including secretory goblet cells and absorptive enterocytes. We performed scRNA-seq of monolayer cells treated with Tunicamycin (Tm) to induce ER stress through inhibition of N-linked glycosylation. To select time points for analysis, we considered the fact that attenuation of Ern1 and Atf6 signaling under prolonged ER stress can induce cell death (16). We monitored Ern1 and Atf6 activity by measuring the aggregate population-level expression of Xbp1s (spliced Xbp1 transcript mediated by Ern1) and Hspa5 (an Atf6 target gene). Xbp1s and Hspa5 were maximally induced at 12 and 15 hours (h) after initiation of Tm treatment, and decreased substantially after 24 hours. The splicing pattern of Xbp1s, as assessed by gel electrophoresis, correlated with qPCR results (Fig. 1A). Thus, we harvested cells for scRNA-seq at 13 h after Tm treatment, when both Xbp1s and Hspa5 were highly induced, and at 25 hours, when their expression levels had abated. We hypothesized that these timepoints represent different modes of the UPR pathway in the epithelial monolayer (Fig. 1A). We recovered 5,122 high quality cell profiles (Methods), and annotated them by cell-type across different conditions by controlling for stimulation and replication as linear categorical covariates before clustering and comparing against known marker genes of intestinal epithelial cell-types (17) (fig. S1A and Methods). We confirmed the cell subsets by staining of the monolayers with cell type-specific markers (fig. S1B).
Fig. 1. Temporal scRNA-seq identifies gene expression signature of terminal-UPR.
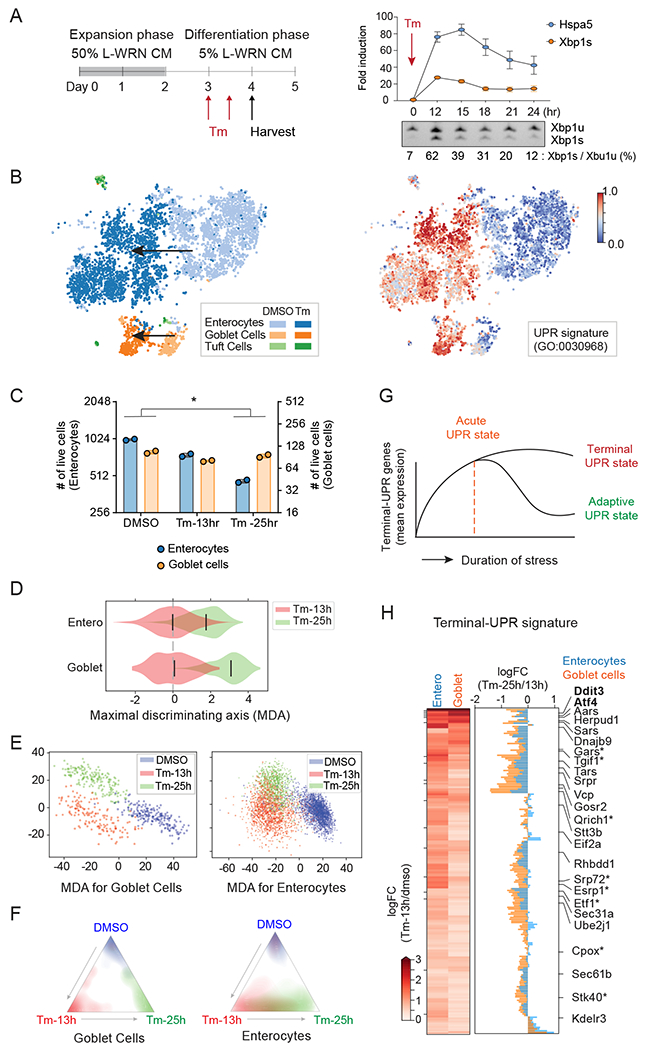
(A) Experimental timeline for differentiation of intestinal organoids into monolayers and treatment with Tunicamycin (Tm). Right upper panel shows a 24-hour time course of Hspa5 and Xbp1s expression levels upon treatment with 0.5ug/ml of Tm (n=3, error bars, mean +/− SD). Right lower panel shows the ratio of Xbp1s vs. Xbp1u transcripts (n=3). Xbp1u, unspliced transcript; Xbp1s, spliced transcript.
(B) ScRNA-seq tSNE plots recover known cell-types from primary intestinal monolayer cells (left) and identify transcriptomic signatures of UPR (right, colored by cell ranking). Arrows indicate the Tm-mediated positional shift.
(C) The number of enterocytes and goblet cells in each condition in scRNA-seq (n=2, unpaired student t-test; *p<0.05). Goblet cells exhibit stronger resistance to prolonged UPR stress than enterocytes.
(D and E) Violin (D) and scatter (E) plots of cells along the transcriptomic MDA between 13h and 25h (D) and MDAs among DMSO, 13 h, and 25 h (E). Goblet cells show more clearly distinct states between 13 h and 25 h Tm treatments compared to enterocytes. In D, bars indicate median values, and mean expression at 13 h is chosen as the baseline.
(F) Linear discriminant analysis predictions show distinct and overlapping states of goblet cells (left) and enterocytes (right). Colors and coordinates, respectively, indicate the ground truths and predicted probabilities of stimulation using scRNA-seq.
(G) The schematic diagram for the cellular state during ER stress. We hypothesized that terminal-UPR genes are up-regulated in the early phase of ER stress, and their persistent expression promotes UPR-mediated apoptosis under unresolved ER stress.
(H) Heatmap shows the logFC of terminal-UPR genes in Tm-13h vs DMSO. Barplot shows the logFCs between Tm-25 h and Tm-13 h in enterocytes and goblet cells. Our gating strategy identified 192 terminal-UPR genes. Known and novel UPR genes are shown (novel terminal-UPR regulators are indicated by an asterisk).
We tested for both cell intrinsic expression programs and cell subset proportions affected by Tm treatment before controlling for stimulation effects (Fig. 1 B,C). The cell profiles were clearly separated in response to Tm stimulation, with increased expression of known UPR genes (Fig. 1B and Methods). Upregulation of the UPR signature was similar between goblet cells and enterocytes. Importantly, however, we found a decrease in the relative proportion of enterocytes when compared with goblet cells at 25 h post Tm treatment but not at 13 h (Fig. 1C and fig. S1C). If this difference was due to an inherent resistance of goblet cells to apoptosis, we would expect population differences under short term ER stress as well, but no difference in these cell types was observed at 13 h, suggesting that goblet cells possess mechanisms that allow for adaptation to prolonged ER stress. This hypothesis is further supported by functional enrichment analysis of the upregulated genes in each cell type at 13 h, which shows a similar level of enrichment of UPR signature and apoptotic pathway genes (fig. S1D). To identify the set of genes related to different stress responses in each cell, we projected the transcriptomic profiles of all cells onto the cell-type-specific axis that maximally discriminates the 13 h and 25 h Tm treatments (i.e., maximal discriminating axis or MDA; Methods). Goblet cells showed more distinct transcriptome states between the 13 h and 25 h time points compared to enterocytes (Fig. 1D). This distinct state was even more pronounced after including dimethyl sulfoxide (DMSO)-treated cells in the Linear Discriminant Analysis (LDA) along two MDAs of the three stimulation conditions (Fig. 1E, F; Methods). Combined with the cell proportion changes, these data suggest that goblet cells successfully adapted to ER stress and resisted apoptosis by entering a unique adaptive state resembling the DMSO treatment state. Conversely, enterocytes underwent apoptosis during ER stress, without significant transcriptomic differences between short- and long-term ER stress. We distinguished cell state changes over time, classifying them into the following categories: (1) the acute-UPR, comprised of genes upregulated in both enterocytes and goblet cells after 13 h Tm, (2) the adaptive-UPR, associated with recovery from acute ER stress in goblet cells after 25 h Tm, and (3) the terminal-UPR, associated with unresolved ER stress in enterocytes after 25 h Tm treatment (Fig. 1G).
We performed differential gene expression analyses to define the UPR gene module associated with unresolved ER stress, which we refer to as the terminal-UPR signature. Defining regulators of the adaptive versus terminal UPR is challenging due to the many UPR responsive genes that have dual, context-dependent functions. For example, ATF4 is a well-known early acute UPR mediator and terminal-UPR mediator, which decreases expression upon resolution of ER stress (Fig. 1G). Based on this observation, we defined the gene signature associated with positive regulation of the terminal-UPR as follows: (1) genes that were up-regulated in both enterocytes and goblet cells at early time points (13 h Tm treatment) and (2) genes exhibiting reduced expression in goblet cells compared to enterocytes at later time points (25 h-vs-13 h Tm) (Fig. 1H and Table S1). Expression of terminal-UPR genes was inversely correlated with cell viability, and the relatively lower expression observed in goblet cells after prolonged ER stress reflects entry of these cells into the adaptive state. Using these criteria, we identified 192 terminal-UPR signature genes, including 18 known UPR genes, such as the UPR-mediated apoptotic regulators Atf4 and Ddit3 (Fig. 1H and Table S1). The signature also contained genes belonging to UPR and ER protein homeostasis-related signaling pathways, such as vesicle transport, protein targeting to ER, sterol metabolic process, and translation (fig. S1E). Taken together, the terminal-UPR signature contains key terminal-UPR pathway regulators that dictate adaptation versus programmed cell death in response to ER stress.
Functional dissection of the terminal-UPR program by genome-wide CRISPR screen
To discover regulators of the UPR pathway that influence the outcome of cellular ER stress responses, we performed genome-scale genetic perturbation screens. To closely monitor ER stress without interrupting the endogenous regulatory circuit, we generated an endogenous XBP1s-GFP reporter by CRISPR knock-in (Fig. 2A) and validated its dose-dependent response to Tm treatment (fig. S2A). The ERN1-XBP1 axis is the most conserved arm of the UPR signaling pathway, and all three UPR arms up-regulate the XBP1s transcript (18, 19). In this reporter system, ER stress induced splicing of XBP1 mRNA and subsequent expression of XBP1s and GFP proteins. Separation of the GFP protein from the XBP1s coding sequence by the P2A self-cleaving peptide ensured that GFP did not interfere with endogenous XBP1s signaling.
Fig. 2. Genome-wide CRISPR screens unveil gene functional proximities in, and identify novel regulators of, the UPR pathway.
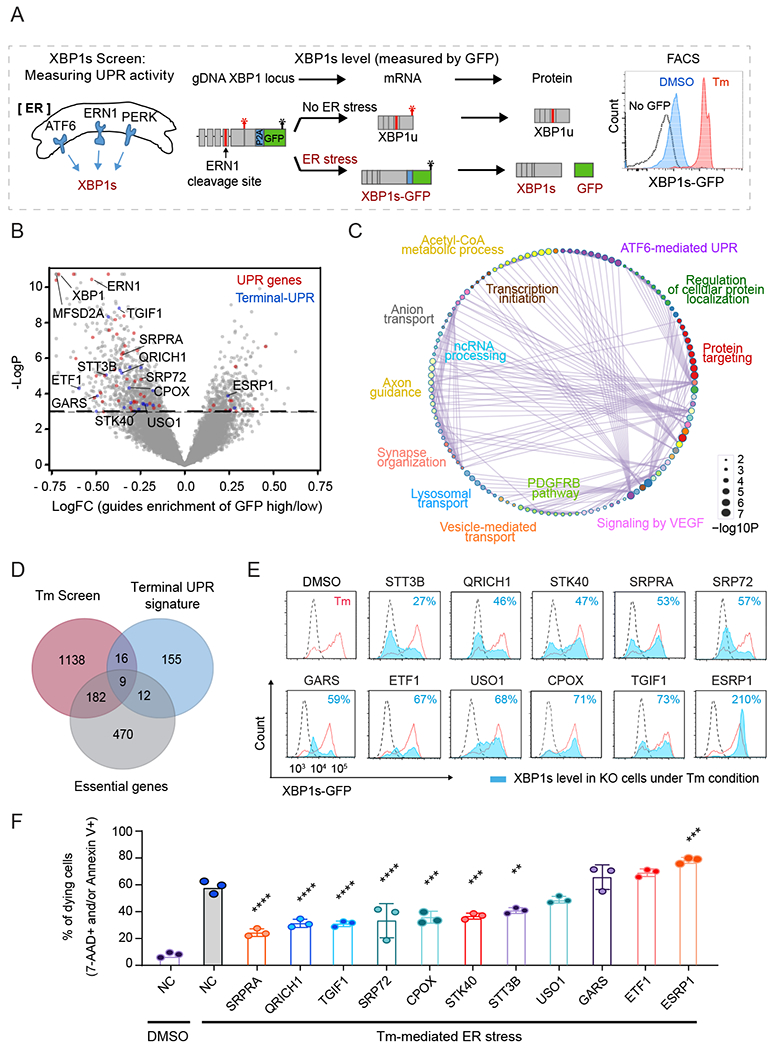
(A) Schematic overview of XBP1s-GFP reporter for CRISPR screen.
(B) Volcano plot of gRNA enrichment reveals putative regulators of the UPR pathway. Dashed line indicates the p-value cut-off (p<0.05). Known UPR genes and terminal-UPR genes are highlighted in red and blue, respectively.
(C) Pathway interaction networks of the screen hits obtained using Cytoscape. The color code indicates pathway nodes, and GO terms represent the most significant pathway in the node. The edges show the crosstalk between the pathways.
(D) Venn diagram of the CRISPR screen, single-cell analysis, and essential genes highlight 16 potent regulators of the terminal-UPR pathway.
(E) Assessment of XBP1s in wild-type (red) or knockout (filled blue) cells treated with Tm by measurement of GFP intensities. Symbol indicates the targeted gene. Dashed black line indicates the GFP intensity in DMSO condition.
(F) Measurement of dying (7-AAD or Annexin V positive) cells treated with Tm for 3 days (n=3, one-way ANOVA (indicated target gene values compared to Tm-treated negative control (NC)); error bars, mean +/− SD). **p<0.01, ***p<0.001, ****p<0.0001.
We reasoned that introducing genetic perturbations at baseline under non-stressed conditions would reveal genes involved in homeostatic ER functions, whereas analysis under ER stress would reveal genes that regulate UPR activity. Thus, we performed CRISPR screens in both DMSO and Tm treated HT-29-Cas9 cells to identify genetic perturbations that can induce ER stress and/or modulate UPR activity (Fig. 2A). We used a genome-wide CRISPR library consisting of 76,441 guides targeting 19,114 genes, as well as 1,000 non-targeting control guides (20). We used fluorescence-activated cell sorting (FACS) to separate the cells based on the top and bottom 15% of GFP expression (fig. S2B). Since XBP1s expression is low in DMSO conditions (Fig. 2A), perturbations that increase the expression of XBP1s can be detected most accurately in these conditions (fig. S2C and Table S2). We observed enrichment of genes involved in pathways that contribute to ER homeostasis, including protein folding, glycosylation, transport, and secretion (fig. S2D). These findings are consistent with genome-wide UPR screens reported previously (21-24).
Under Tm-induced ER stress conditions, knock-out (KO) of ERN1, XBP1, and the known Tm membrane transporter MFSD2A (25) were top hits for reducing XBP1s expression (Fig. 2B and Table S2). Importantly, the XBP1 screen performed in Tm-treated cells identified perturbations that caused significant expression changes in a larger number of known ER stress and UPR genes compared to DMSO-treated cells (Fig. 2B,C, and fig. S2C). Among the 428 hits that increased XBP1s in the Tm-treated condition, only 10 genes also increased XBP1s in the DMSO condition, and the enriched signaling pathways differ from those in the DMSO condition (fig. S2E), supporting our hypothesis that genes that maintain ER homeostasis are distinct from those that regulate UPR activity under stress. By comparing genes essential for cell viability (26) with hits from the ER stress XBP1 screen, we found that KO of almost all essential genes (177 of 182) identified in our screen resulted in a reduction of XBP1s reporter expression (fig. S2F and Table S2). This is expected based on the rationale that perturbation of essential genes might alter UPR activity by inducing a nonspecific cytotoxic response or an overall decrease in metabolic activity. Therefore, this criterion allowed us to prioritize UPR pathway-specific genes by excluding essential genes in subsequent analyses. Overall, our genome-wide screens represent expansive datasets that can be leveraged to elucidate signaling crosstalk during ER stress and to discover UPR regulators.
To identify regulatory nodes that control the terminal-UPR, we compared the screen hits from the ER stress condition with the terminal-UPR signature genes from our scRNA-seq analysis of primary intestinal epithelial cells. We identified 16 terminal-UPR signature genes whose perturbation modulated UPR activity only in the Tm-treated screens (Fig. 2D). To validate these potent UPR regulators, we tested two individual sgRNAs targeting each gene and measured XBP1s levels and cell viability under Tm-induced ER stress conditions. Overall, KO of 11 of the 16 genes potentiated or inhibited XBP1 splicing and cell viability in response to ER stress (Fig. 2E,F). Implicating these genes in regulation of the UPR pathway provides insights into connectivity with other signaling pathways. It is notable that KO of either SRP72 or SRPRA, which function in cotranslational protein targeting to the ER, lowered XBP1s activity and increased cell viability. Under prolonged stress, PERK-eIF2α may increase secreted protein synthesis, leading to cell death (27, 28). In the same sense, SRP72 or SRPRA KO can increase cell viability by reducing the production of misfolded proteins by reducing client proteins entering the ER. Overall, there was a negative correlation between the level of XBP1s and cell viability under ER stress across the perturbations, except in the case of perturbing genes necessary for ER homeostasis (Fig. 2E,F). KO of ETF1 (a subunit of the SURF complex) or GARS (Glycyl-tRNA synthetase 1) inhibited XBP1s upregulation, but not cell death, under ER stress (Fig. 2E,F), whereas these perturbations activated XBP1s expression in the absence of Tm (fig. S2G), suggesting that severe disruption of ER homeostasis can induce cell death regardless of UPR activation state. However, KO of several genes such as QRICH1 had no demonstrable effect on ER homeostasis and cell viability under non-stressed conditions but induced resistance to UPR-mediated cell death (Fig. 2F and fig. S2G), thus highlighting the crucial role for these genes specifically in UPR regulation.
QRICH1 promotes cell death under ER stress
Based on our CRISPR screens, we prioritized the previously uncharacterized protein QRICH1 for mechanistic investigation. QRICH1 is a member of the caspase recruitment domain (CARD)-containing gene family (fig. S3A), which encodes for proteins implicated in regulation of caspase activation in the context of apoptosis and inflammation, and in regulation of NF-κB signaling. Our findings indicated that QRICH1-deficient cells are more resilient to ER stress-mediated apoptosis (Fig. 2F and fig. S3B). Therefore, we hypothesized that QRICH1 acts as a regulator of the terminal-UPR response and sought to define the molecular mechanism underlying this regulation. First, we tested whether QRICH1 is sufficient for UPR-mediated apoptosis using CRISPR activation (CRISPRa) (29). CRISPRa_QRICH1 increased QRICH1 expression approximately 1.6-fold (fig. S3C), activated the UPR pathway, and potentiated induction of apoptosis after Tm treatment (Fig. 3A,B), indicating that QRICH1 expression is sufficient to potentiate UPR pathway activity and apoptosis under ER stress.
Fig. 3. QRICH1 promotes cell death and its translation is upregulated by the PERK-eIF2α axis under ER stress.
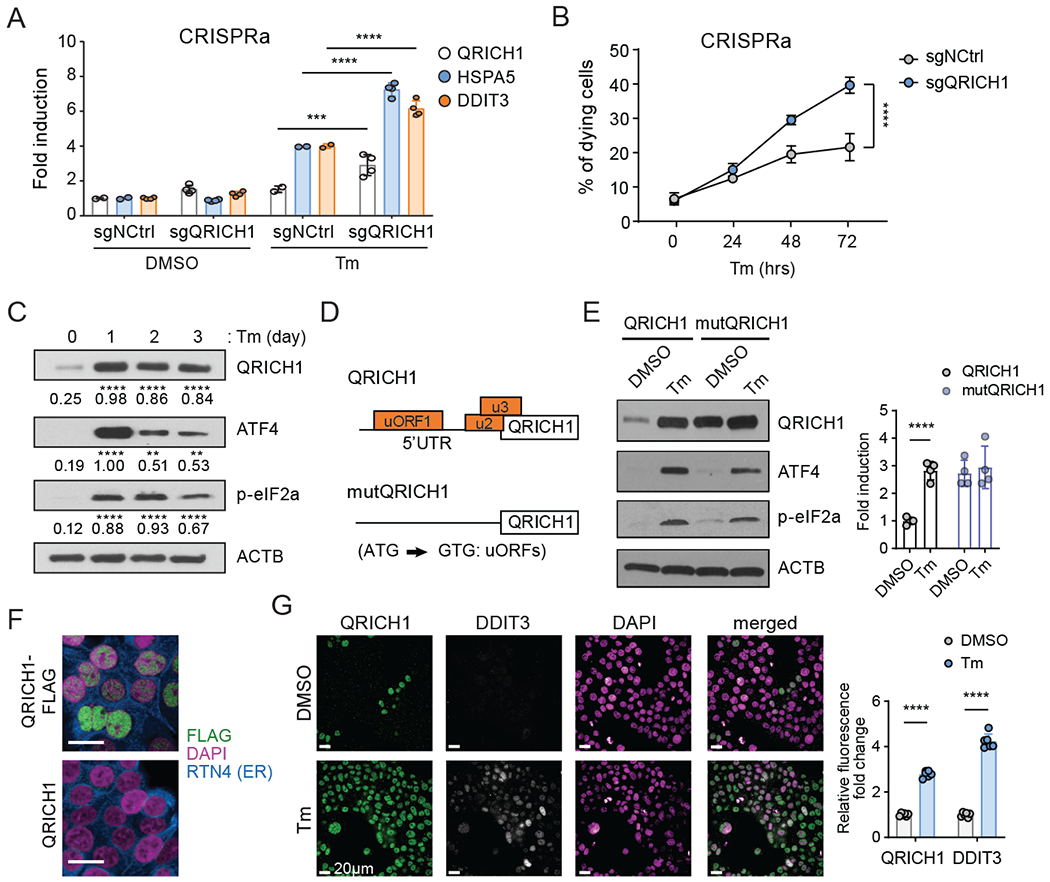
(A) Measuring the transcriptional activity of UPR pathway regulators in control (sgNCtrl) or CRISPRa QRICH1 cells in normal or Tm-mediated ER stress conditions (n=4, two-way ANOVA).
(B) The percentage of 7-AAD positive cells. sgNCtrl or sgQRICH1 cells treated with Tm for the indicated time. Three different guides (n=4, two-way ANOVA).
(C) Immunoblot shows the time-course expression pattern of QRICH1, ATF4, and p-eIF2α during prolonged Tm treatment. A representative blot is shown (n=3, two-way ANOVA, compared to DMSO).
(D) The structure of the wild type 5’UTR (containing three putative upstream ORFs) or mutated 5’UTR (substitution of A to G at the AUG translation start codon of three uORFs) of QRICH1 expression constructs.
(E) Immunoblot shows the protein expression patterns of QRICH1 constructs upon DMSO or Tm treatment. ATF4 and p-eIF2α are used as markers of ER stress (n=4, multiple t-test).
(F) Immunostaining for QRICH1-FLAG (green) or QRICH1 transduced cells. Nuclei and ER are stained with DAPI (magenta) and RTN4 (blue), respectively. Scale bars, 20 μm.
(G) Immunostaining of endogenous QRICH1 (green) and DDIT3 (gray) in cells with DMSO or Tm treatment. Nuclei are stained with DAPI (magenta). Scale bars, 20 μm. Right graph shows the normalized intensities of QRICH1 and DDIT3 (n=6, multiple t-test).
For all above panels, **p<0.01, ***p<0.005, ****p<0.0001; error bars, mean +/− SD.
PERK-eIF2α axis promotes the translation of QRICH1 during ER stress
Having demonstrated that QRICH1 functions as a determinant of cell survival in response to ER stress, we sought to define the mechanisms by which it is regulated by the UPR. First, we examined QRICH1 expression under ER stress. We found that QRICH1 exhibited a similar pattern of Tm-mediated upregulation as phospho-eIF2α and ATF4 (Fig. 3C). This upregulation of QRICH1, as well as ATF4, was suppressed by PERK KO (fig. S3D). We also quantified QRICH1 mRNA and protein levels by qPCR and western blotting after Tm treatment in HT-29 cells. ER stress significantly induced the expression of QRICH1 at the protein level, more so than at the mRNA level (fig. S3E). One well-characterized feature of stress-responsive genes is translational control by upstream open reading frames (uORFs) present in the 5’ untranslated regions of mRNA (30). Generally, uORFs reduce translation of the downstream protein-coding region by engaging with the 40S scanning ribosome. Transcripts containing uORFs exhibit decreased translation at baseline; however, when PERK phosphorylates eIF2α under ER stress, phospho-eIF2α slows the assembly of the 40S ribosome, allowing the ribosome to bypass the uORF and translate the protein-coding region (fig. S3F). We searched for short ORFs starting with ‘AUG’ in the 5’UTR of QRICH1 mRNA, and found that 7 of 8 QRICH1 transcripts contained putative uORFs (fig. S3G). To determine the effect of these putative uORFs on QRICH1 expression, we quantified protein expression from a mutated QRICH1 construct in which the first nucleotide of the start codons of the uORFs were changed from A to G (Fig. 3D,E). QRICH1 protein expression from the native uORF construct was lower at baseline compared to the mutated uORF construct, suggesting that translation of upstream ORFs inhibits translation of the coding sequence for QRICH1. In response to ER stress, QRICH1 protein expression from the native, but not the mutated uORF construct, was significantly upregulated (Fig. 3E). We also determined the subcellular localization of QRICH1 protein. First, we used Flag-tagged QRICH1 to assess localization of QRICH1 and observed that it is enriched exclusively in the nucleus (Fig. 3F). Immunostaining showed that endogenous QRICH1 is strictly localized to the nucleus under both ER stress and at baseline (Fig. 3G). Collectively, these data suggest that the PERK-eIF2α axis upregulates QRICH1 under ER stress and that QRICH1 in turn modulates the activity of the UPR in the nucleus.
QRICH1 promotes the expression of translation-related genes
During ER stress, the PERK-eIF2α axis modulates proteostasis through transcriptional programs mediated by ATF4 and DDIT3 (28). QRICH1 contains a DUF3504 domain (fig. S3A), which shares homology with DNA-binding proteins resembling transposons but lacking catalytic activity (31). Therefore, we sought to test the hypothesis that QRICH1 regulates cell viability by acting as a transcriptional regulator. To this end, we performed chromatin immunoprecipitation (ChIP), first confirming that the QRICH1 antibody was able to effectively capture endogenous QRICH1 following formaldehyde cross-linking (Fig. 4A). We sequenced the immunoprecipitated genomic DNA from WT or QRICH1 KO cells after Tm treatment. For comparison, we also performed ChIP-seq experiments with antibodies directed against ATF4. We identified a total of 70,565 QRICH1-bound peaks and 121,649 ATF4-bound peaks across the genome. Among these, 16.8% of QRICH1 and 9.5% of ATF4 peaks were located within gene promoter regions. Specifically, QRICH1 and ATF4 localized to the promoter regions of 2,626 and 1,875 genes, respectively (Fig. 4B,C).
Fig. 4. QRICH1 ChIP-seq identifies QRICH1 as a promoter of protein translation activity.
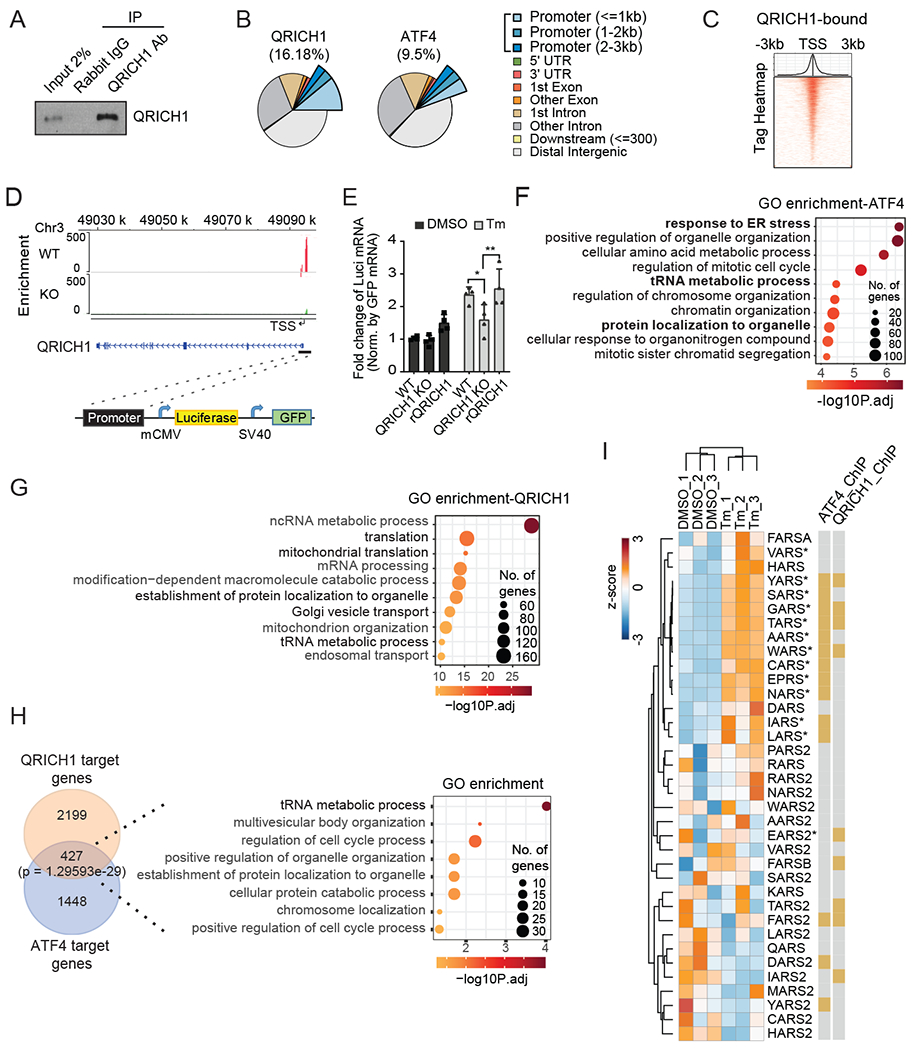
(A) WT HT29 cells were treated with Tm for 24 hrs and crosslinked by formaldehyde. The cells were immunoprecipitated (IP) by anti-QRICH1 or anti-rabbit IgG and detected by immunoblot analysis.
(B) The genomic annotation of QRICH1 or ATF4 ChIP-seq peaks. Promoter regions are defined as the indicated distance from the transcription start site (TSS).
(C) Peak distribution of QRICH1 ChIP-seq within 3kb from the TSS. Heatmap shows the read density for QRICH1 ChIP-seq.
(D) QRICH1 binding profiles on the QRICH1 promoter region in WT and QRICH1 KO cells. Black bar indicates the cloned genomic region in front of the minimal CMV promoter for the promoter activity reporter assay.
(E) Level of luciferase mRNAs in WT, QRICH1 KO, and QRICH1 reconstituted (rQRICH1) cells transduced with the promoter reporter-expressing lentivirus and treated with DMSO or Tm for 24 hrs. Luciferase mRNA was measured by normalizing GFP mRNA using qRT-PCR (n=3, two-way ANOVA; *p<0.05, **p<0.01; error bars, mean +/− SD).
(F and G) Functional enrichment analysis shows the top 10 enriched gene ontologies from ATF4-binding targets (F) and QRICH1-binding targets (G).
(H) Venn diagram illustrating overlap of QRICH1 and ATF4 targets from ChIP-seq. GO analysis shows that the tRNA metabolic process is the most significant biological process.
(I) RNA-seq shows that QRICH1 and/or ATF4-bounded tRNA synthetases are up-regulated during ER stress (n=3). An asterisk indicates the translationally upregulated genes during ER stress (28).
We found that QRICH1 bound its own promoter region, potentially establishing a self-regulation circuit that allows for stress-responsive QRICH1 regulation by increasing its transcription. To test this possibility, we cloned the 1.1 kb QRICH1 promoter identified in our analysis as a target binding region into a luciferase reporter construct (Fig. 4D). ER stress resulted in a stronger induction of luciferase mRNA, as measured by qRT-PCR, in WT cells compared with QRICH1 KO cells. Transcriptional repression in the QRICH1 KO cells was rescued with QRICH1 reconstitution (Fig. 4E). These findings support our hypothesis that QRICH1 activates its transcription to enhance its regulatory effects on the UPR.
Gene Ontology analysis of ChIP-seq associated genes showed that ATF4 is enriched at ER stress-responsive genes as well as translation-related genes, while QRICH1 preferentially binds promoters of genes related to protein production such regulators of ribosome biogenesis (ncRNA metabolic processes), translation, and mRNA processing (Fig. 4F,G and Table S3). Together, QRICH1 and ATF4 co-occupied 427 gene regions (p = 1.29*10−29, hypergeometric test) that are enriched for functional annotations associated with tRNA metabolism (Fig. 4H). It was recently reported that the PERK-eIF2α axis promotes synthesis of the secreted proteome through the translational regulation of amino acid biosynthesis, specifically tRNA synthetases associated with secreted and membrane-targeted proteins (27). Similarly, our combined ChIP-seq and RNA-seq analyses identified that QRICH1 and ATF4 were enriched at the promoters of these specific tRNA synthetases, and that ER stress positively regulated their transcription (Fig. 4I). Together, these findings suggest that QRICH1 and ATF4 modulate tRNA metabolic processes to promote secreted protein synthesis during ER stress.
QRICH1 regulates a transcriptional program that promotes protein secretion
Given our findings that QRICH1 occupies promoter regions of proteostasis-related genes, we sought to quantify its effect on putative target gene transcription. We compared transcriptomes from WT vs. QRICH1 KO cells after Tm treatment and identified 1,141 differentially expressed genes (DEGs) (633 genes decreased, and 508 genes increased in QRICH1 KO cells) (Fig. 5A and Table S4). Notably, 32% of upregulated genes and 15% of downregulated genes were directly bound by QRICH1 (Table S4). The most enriched annotations for QRICH1-activated targets were ‘translation’, ‘ER-Golgi transport’, and ‘protein localization to ER’, whereas cell cycle genes were enriched in QRICH1-suppressed targets (Fig. 5B). Since maintaining ER proteostasis is the key determinant for cell fate, it was notable that genes belonging to the ‘SRP-dependent cotranslational targeting to membrane’ pathway were reduced in QRICH1 KO cells. RNA-seq also showed that 28 of 30 DEGs, including SRP72 and SRPRA, belonging in the ‘SRP-dependent cotranslational targeting to membrane’ were down-regulated in QRICH1 KO cells (Fig. 5C,D). Moreover, the translocon components, SEC61B and TRAM1, are direct targets of QRICH1, and their upregulation under ER stress was inhibited in QRICH1 KO cells (fig. S4A). Therefore, we investigated whether the SRP-dependent secretion pathway is critical for cell viability during ER stress. We found that KO of SRP-dependent secretion pathway genes, including the QRICH1 targets TRAM1 and RPS7, increased cell viability within the context of ER stress (Fig. 5E). Consistent with this, KO of these genes did not disrupt ER homeostasis in the non-stressed condition, without Tm treatment (fig. S4B). Thus, our ChIP-seq and RNA-seq results indicate that QRICH1 is a critical mediator of the PERK-eIF2α axis for facilitating protein secretion and that these functions impact cell viability/fate during ER stress.
Fig. 5. QRICH1 upregulates protein synthesis and secretion during ER stress.
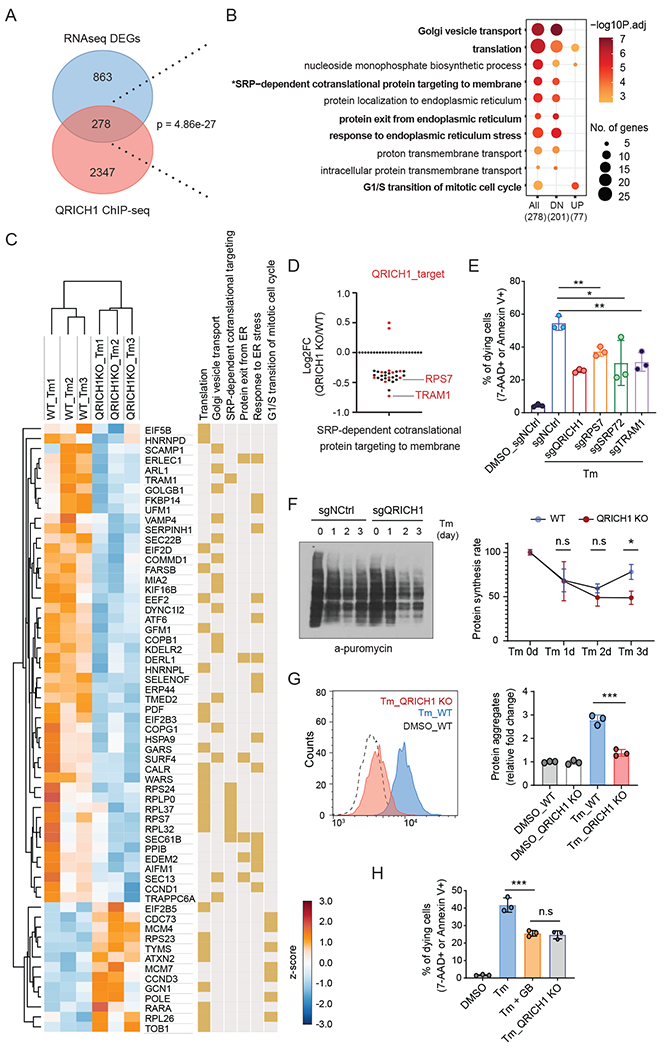
(A) Venn diagram illustrating the overlap of QRICH1 target genes from ChIP-seq and differentially expressed genes (DEGs) between WT and QRICH1 KO cells in response to Tm treatment.
(B) Functional enrichment analysis of the 278 overlapping gene set in A. All, 278 DEGs; DN, 201 down-regulated genes in QRICH1 KO cells; UP, 77 up-regulated genes in QRICH1 KO cells.
(C) RNA-seq performed with WT and QRICH1 KO cells to show response to Tm treatment. Heatmap shows selective DEGs belonging to specific biological processes (related to Fig. 5B). Yellow color indicates the involved biological processes of that gene.
(D) RNA-seq showed that 28 of 30 DEGs belonging to ‘SRP-mediated cotranslational ER targeting’ are down-regulated in the QRICH1 KO cells. Red dots indicate the QRICH1-target in ChIP-seq data.
(E) Measurement of dying (7-AAD or Annexin V positive) cells treated with Tm for 3 days (n=3, one-way ANOVA; error bars, mean +/− SD). X-axis labels indicate the target gene.
(F) Immunoblot shows the puromycin incorporation rate by anti-puromycin blot. WT and QRICH1 KO cells were pulse-labeled with puromycin after Tm treatment for the indicated times. A representative blot is shown. The graph shows the quantified intensities of anti-puromycin signals, compared to the signal of sgNCtrl-Tm 0hr (set to 100%) (n=3, two-way ANOVA; error bars, mean +/− SD, see Methods).
(G) FACS analysis of WT and QRICH1 KO cells after 72 hrs Tm treatment showing the intensity of the fluorescent dye which preferentially interacts with unfolded protein aggregates (n=3, one-way ANOVA, error bars, mean +/− SD).
(H) FACS analysis of cell viability in WT and QRICH1 KO cells treated with Tm or Tm plus guanabenz (GB) for 72 hrs (n=3, one-way ANOVA; error bars, mean +/− SD). For all above panels, *p<0.05, **p<0.01, ***p<0.001; n.s, not significant.
Translational recovery induces proteotoxicity-mediated cell death during ER stress
Increased production of misfolded proteins under ER stress is a key trigger for the induction of UPR-mediated apoptosis (28), and our data suggest that QRICH1 primarily regulates protein input (synthesis & targeting to the ER). Therefore, we hypothesized that QRICH1-regulated translational activation under ER stress may promote protein aggregate-mediated cell death by increasing the protein input to the ER. We directly monitored the requirement for QRICH1 in dynamic translational regulation during ER stress by measuring the protein synthesis rate in WT and QRICH1 KO cells using puromycin labeling. Translational activity was reduced in the early phase (48h) of ER stress in WT cells and increased in the later phase (72h) (Fig. 5F), which is consistent with previous reports (28). However, QRICH1 KO cells maintained reduced translational activity at later phases of ER stress (Fig. 5F). Next, we investigated the influx of the secretome into the ER during stress by using a subcellular fractionation approach to measure newly synthesized peptides in the ER/Golgi fraction. We found that under ER stress, puromycin-labeled proteins in the ER/Golgi fraction as well as the cytosolic fraction were reduced by approximately 25% (Fig. S4C). To determine the role of QRICH1 in accumulation of misfolded proteins under ER stress, we measured protein aggregates in the WT and QRICH1 KO cells under ER stress using red fluorescent molecular rotor dye, which preferentially interacts with denatured protein aggregates. QRICH1 KO cells showed reduced levels of protein aggregates compared to WT, which suggests that QRICH1 increases misfolded proteins by upregulation of protein synthesis (Fig. 5G). These data suggest that QRICH1-mediated translational recovery is a critical determinant balancing proteotoxicity versus cell viability. To test this possibility, we treated cells with Guanabenz, an inhibitor of phospho-eIF2α phosphatase (32). Guanabenz-treated cells showed increased cell viability under ER stress conditions (Fig. 5H), suggesting that resumption of translational activity can trigger terminal-UPR mediated cell death. Collectively, our data demonstrate that QRICH1 promotes protein synthesis and secretion during ER stress through transcriptional control of a functionally cohesive gene module regulating the terminal-UPR and especially translational activation and secretion.
QRICH1 sensitizes primary epithelial cells to ER stress
To validate the role of QRICH1 in primary intestinal epithelial cells, we employed CRISPR to target Qrich1 in mouse intestinal organoid cultures and subsequently differentiated these organoids into polarized monolayers (fig. S5A). To quantify apoptosis in response to ER stress, we measured cleaved caspase-3 (cCasp3) by Western blot after Tm treatment. Qrich1 KO cells showed reduced cCasp3 levels compared to WT (fig. S5B,C). We then measured cellular Caspase 3 activity by using a fluorogenic substrate, and observed reduced activity in Qrich1 KO monolayers consistent with the corresponding reduction in cCasp3 levels (fig. S5D). We also investigated whether reconstitution of QRICH1 could restore the sensitivity to ER stress. To address this, we transduced human QRICH1, which has 83% amino acid conservation with mouse Qrich1, into the mouse Qrich1 KO organoids. We confirmed that QRICH1 re-expression sensitized the mouse organoids, as measured by an increase in Caspase-3 activity (fig. S5E). These data suggest that QRICH1 expression promotes cell death in primary mouse epithelial cells under ER stress.
To further confirm these findings in primary human epithelial cells, we generated human intestinal organoids. We found that QRICH1 and the SRP-mediated pathway genes are upregulated under ER stress (Fig. 6A). We next sought to investigate whether cell type-specific translational activity is critical for cell viability in human organoids. Based on our scRNA-seq (Fig. 1) and our previously published scRNA-seq data for mouse and human intestinal epithelium (17, 33), we selected MUC2 and TMIGD1 as cell type-specific markers for goblet cells and enterocytes, respectively (fig. S6A). FACS analysis demonstrated that enterocytes, but not goblet cells, showed higher cell death and protein synthesis rate under ER stress (Fig. 6B and fig. S6B), which is consistent with our mouse scRNAseq results (Fig. 1C). These findings suggest that differential regulation of translation and proteotoxicity sensitizes enterocytes to apoptosis, whereas goblet cells are more resilient to ER stress.
Fig. 6. QRICH1 sensitizes primary intestinal epithelium to ER stress and is regulated during inflammatory conditions.
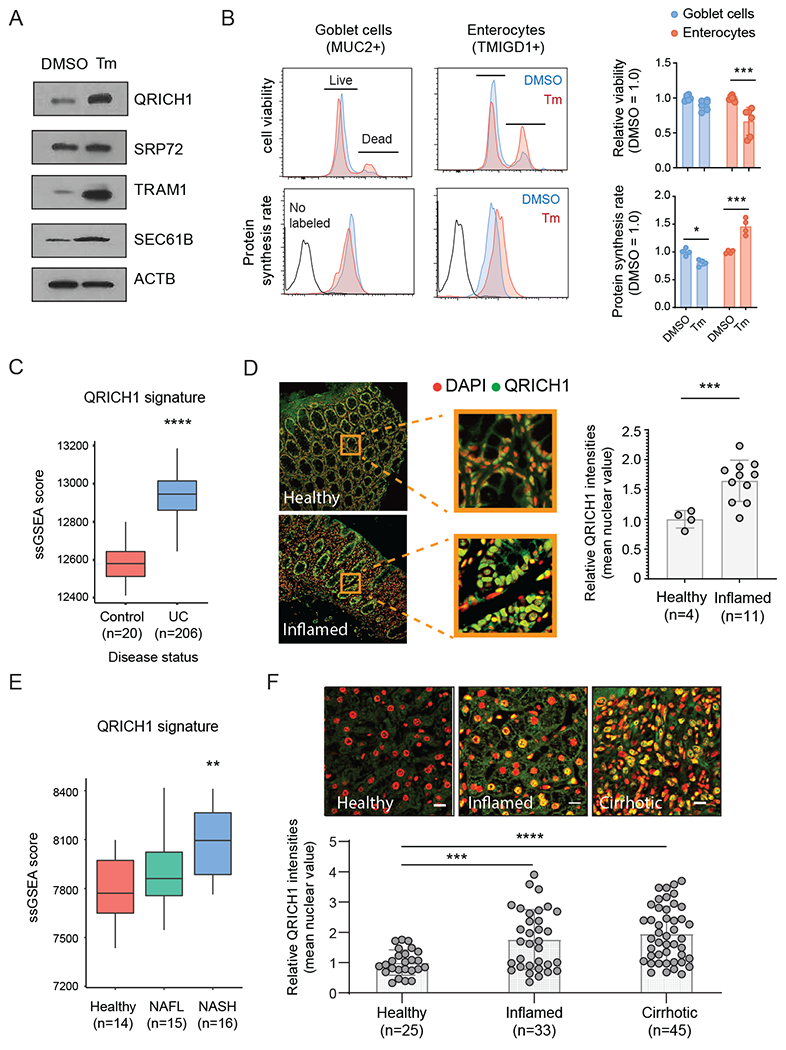
(A) Immunoblot shows the expression patterns of QRICH1 and SRP-mediated secretion pathway genes upon 0.5ug/ml of Tm treatment in human intestinal organoids for 24hrs. A representative blot is shown.
(B) FACS analysis of human intestinal organoids shows cell-type-specific protein synthesis rates and viability during ER stress. Cells were stained with non-permeable amine-reactive dye, and anti-puromycin antibodies to assess cell viability and protein synthesis rate in goblet cells (MUC2+) and enterocytes (TMIGD1+). The graphs show quantified signals from each FACS analysis (n=6 and 4 for viability and protein synthesis rate, respectively; two-way ANOVA, error bars, mean +/− SD).
(C) Single sample Gene Set Enrichment (ssGSEA) Scores for the QRICH1-signature were calculated in bulk RNA-seq data from the rectal biopsies of pediatric UC patients. Wilcoxon rank-sum test, error bars, mean +/− SD.
(D) Immunofluorescence assay of a colon tissue array stained for QRICH1 (green), and nuclei (red) in the healthy and inflamed colon (two-tailed unpaired t-test, error bars, mean +/− SD).
(E) ssGSEA Scores for the QRICH1-signature were calculated in bulk RNA-seq data from liver biopsy samples from healthy, NAFL, and NASH patients. Wilcoxon rank-sum test, error bars, mean +/− SD.
(F) Immunofluorescence assay of a liver tissue array stained for QRICH1 (green), and nuclei (red) in healthy, inflamed, and cirrhotic livers (one-way ANOVA, error bars, mean +/− SD).
For all above panels: **p<0.01, ***p<0.001, ****p<0.0001.
Given our findings that QRICH1 plays a central role in regulating proteotoxicity, we sought to identify conditions in which QRICH1 activity may contribute to human disease. Thus, we derived a QRICH1 transcriptional signature from our ChIP-seq and RNA-seq datasets (see Methods for details), and scored its expression in the context of inflammatory conditions such as inflammatory bowel disease (IBD) and liver disease. We found that the QRICH1 signature is increased in bulk RNA-seq of biopsies derived from treatment-naïve UC patients (Fig. 6C) (34). Moreover, analysis of scRNA-seq from UC patients revealed enrichment of the QRICH1 signature in inflamed biopsies, and in particular, in secretory epithelial cells and enterocytes (fig. S7A,B) (33). At the protein level, QRICH1 expression significantly increased in inflamed colonic biopsies relative to controls (Fig. 6D). We next sought to determine if upregulation of the QRICH1 signature is specific to intestinal inflammation or if it is a broadly conserved stress response that functions in other tissue-specific pathologies. Accordingly, we found that the QRICH1 signature increased in liver biopsies derived from nonalcoholic steatohepatitis (NASH) patients (Fig. 6E) (35, 36), and confirmed the upregulation of QRICH1 protein expression in inflamed and cirrhotic specimens using a liver tissue array (Fig. 6F). Taken together, these findings suggest a broadly conserved role for aQRICH1-regulated transcriptional program in managing cell stress responses across a diversity of tissue pathologies. Accordingly, QRICH1 controls a distinct arm of the PERK-eIF2α axis that modulates proteostasis and dictates cellular entry into the adaptive versus terminal UPR.
Discussion
Maintaining proteostasis in the ER requires a system of interconnected biological processes to orchestrate protein synthesis, folding, and degradation (2). When cells detect accumulation of misfolded proteins in the ER, they attempt to restore proteostasis by coordinately increasing protein folding capacity through the ATF6 and ERN1 axis, and eliminating misfolded proteins through induction of autophagy and ERAD. In parallel, transient translational suppression through activation of the PERK-eIF2α pathway can help restore ER proteostasis by reducing the flux of client proteins into the ER (4, 37). However, in the later phase of prolonged ER stress, the PERK axis promotes recovery of translational activity by upregulating amino acid biosynthesis and components of the secretory pathway (27). Therefore, dynamic regulation of the PERK-eIF2α axis is essential for coordinating translation and proteostasis under prolonged ER stress (27, 28). Here, we identify a distinct arm of the PERK-eIF2α axis mediated by the novel transcriptional regulator QRICH1 that dynamically responds to ER stress by modulating translation and transit through the secretory pathway.
Diverse intrinsic and extrinsic factors trigger eIF2α phosphorylation to initiate the integrated stress response (ISR), which is mediated in part through activation of ATF4 (1, 38). We demonstrated that QRICH1 is also a central mediator of the PERK-eIF2α axis that functions at the level of, and in parallel to, ATF4 (fig. S7C). Similar to ATF4, DDIT3, and PPP1R15A, we demonstrated that QRICH1 is post-transcriptionally regulated by the PERK-eIF2α axis through differential translation initiation at uORFs (1). Our ChIP-seq and RNA-seq results suggest that QRICH1 cooperates with ATF4 signaling to regulate ER proteostasis by promoting protein synthesis and secretory efficiency during cellular stress, which is critical for cell viability. Small molecule targeting of the PERK-eIF2α axis has shown mixed results for therapeutic modulation of UPR activity, with some evidence for beneficial effects in animal models of neurodegenerative disease or tumorigenesis (1, 39). Precise targeting of the PERK-eIF2α axis is further complicated by the dual functions of ATF4 signaling in promoting ER homeostasis as well as in inducing apoptosis (1, 40). By comparison, our results suggest that QRICH1 suppression specifically reduces protein flux into the ER, while preserving activity of the adaptive UPR mediated by the PERK-ATF4 axis. Thus, inhibition of QRICH1 activity may facilitate ER processing capacity and allow for restoration of ER homeostasis. Our work identifies a new modular sub-arm of the PERK pathway and sheds light on the mechanisms coordinating the UPR pathway arms that enable dynamic responses to stress.
While the UPR pathway is activated to restore ER homeostasis within the cells, it also aims to maintain tissue homeostasis by coping with the protein secretion demands to respond to diverse physiological conditions (8). Therefore, dysfunction of UPR activity and protein secretion are associated with developmental defects. Mouse mutagenesis studies highlighted an for Qrich1 in embryonic development, demonstrating that Qrich1 mutant mice exhibited renal agenesis as well as congenital heart defects (41). In humans, exome sequencing identified truncation or missense variants of QRICH1 that are associated with developmental disorders (DD) (42). In particular, de novo loss-of-function variants of QRICH1 have been associated with developmental delay, cognitive disability, autism, and chondrodysplasia (43, 44). These human phenotypes are consistent with our findings that QRICH1 controls ER stress responses and with prior studies demonstrating that loss of function mutations in the secretory pathway impair synapse formation or function in developing neural circuits (45-47). In this context, hippocampus-specific downregulation of ATF4 reduces dendritic spines and synaptic plasticity accompanying impairment of memory formation (48). From this perspective, our results support the hypothesis that QRICH1 truncation mutations associated with developmental delay may impair synaptic plasticity at the level of dysregulated ER stress responses.
Maladaptive ER stress responses are associated with a variety of pathological conditions. Disruption of epithelial barrier function accompanied by ER stress is a common feature of intestinal inflammation in IBD (49). Our CRISPR screen results add to and corroborate the notion that several genes located in IBD risk loci function within the ER stress pathway; including TMEM258, RFT1, and LMAN2 (12, 14, 49). Furthermore, we demonstrate that upregulation of QRICH1 promotes UPR-mediated proteotoxicity, and that the QRICH1 signature is significantly upregulated in inflamed intestinal epithelial cells from UC patients. Taken together, these findings highlight the importance of QRICH1 in maintaining proteostasis and suggest that dysregulation of QRICH1 transcriptional activity may sensitize epithelial cells to ER stress during inflammation. Furthermore, we provide evidence that QRICH1 plays a broader role in maintaining tissue homeostasis in the context of pathological conditions including inflammatory and metabolic diseases. Indeed, ER stress and cellular responses to stress are integral to maintaining tissue homeostasis in health and disease, and mechanistic characterization of QRICH1 within this context provides insight into how cells manage responses to stress. Specifically, QRICH1 controls a distinct transcriptional module that coordinates ISR activities to modulate cellular regulation of protein synthesis and secretion under homeostatic and pathological conditions (Summary Figure).
Supplementary Material
Acknowledgments
We thank Heather Kang for assisting with the writing and technical editing of the manuscript, and Caline Matar for help with optimization of the CRISPR experiments.
Funding:
Work was supported by NIH R01 DK097485, NIH P30 DK043351, NIH U19 AI109725, and the Helmsley Charitable Trust (to R.J.X.).
Footnotes
Competing Interests: R.J.X. is a consultant to Novartis and Nestle. A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and until August 31, 2020 was an SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and ThermoFisher Scientific. From August 1, 2020, A.R. is an employee of Genentech, a member of the Roche Group.
Data and materials availability:
Data used in the study have been deposited in the Gene Expression Omnibus (GEO), under accession number GSE146842.
References and Notes
- 1.Pakos-Zebrucka K et al. , The integrated stress response. EMBO Rep 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hetz C, Chevet E, Oakes SA, Proteostasis control by the unfolded protein response. Nat Cell Biol 17, 829–838 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang M, Kaufman RJ, Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Hetz C, Papa FR, The Unfolded Protein Response and Cell Fate Control. Mol Cell 69, 169–181 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Kirby A et al. , Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet 45, 299–303 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dvela-Levitt M et al. , Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell 178, 521–535 e523 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Mays CE, Soto C, The stress of prion disease. Brain Res 1648, 553–560 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Remondelli P, Renna M, The Endoplasmic Reticulum Unfolded Protein Response in Neurodegenerative Disorders and Its Potential Therapeutic Significance. Front Mol Neurosci 10, 187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frakes AE, Dillin A, The UPR(ER): Sensor and Coordinator of Organismal Homeostasis. Mol Cell 66, 761–771 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Arunagiri A et al. , Proinsulin misfolding is an early event in the progression to type 2 diabetes. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnston BP, McCormick C, Herpesviruses and the Unfolded Protein Response. Viruses 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaser A et al. , XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grootjans J, Kaser A, Kaufman RJ, Blumberg RS, The unfolded protein response in immunity and inflammation. Nat Rev Immunol 16, 469–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham DB et al. , TMEM258 Is a Component of the Oligosaccharyltransferase Complex Controlling ER Stress and Intestinal Inflammation. Cell Rep 17, 2955–2965 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kropski JA, Blackwell TS, Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest 128, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin JH et al. , IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haber AL et al. , A single-cell survey of the small intestinal epithelium. Nature 551, 333–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuru A, Imai Y, Saito M, Kohno K, Novel mechanism of enhancing IRE1alpha-XBP1 signalling via the PERK-ATF4 pathway. Sci Rep 6, 24217 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K, XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Doench JG et al. , Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamson B et al. , A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882 e1821 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leto DE et al. , Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol Cell 73, 377–389 e311 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schinzel RT et al. , The Hyaluronidase, TMEM2, Promotes ER Homeostasis and Longevity Independent of the UPR(ER). Cell 179, 1306–1318 e1318 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panganiban RA et al. , Genome-wide CRISPR screen identifies suppressors of endoplasmic reticulum stress-induced apoptosis. Proc Natl Acad Sci U S A 116, 13384–13393 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiling JH et al. , A haploid genetic screen identifies the major facilitator domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin. Proc Natl Acad Sci U S A 108, 11756–11765 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hart T et al. , Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 (Bethesda) 7, 2719–2727 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonen N, Meller A, Sabath N, Shalgi R, Amino Acid Biosynthesis Regulation during Endoplasmic Reticulum Stress Is Coupled to Protein Expression Demands. iScience 19, 204–213 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han J et al. , ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15, 481–490 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konermann S et al. , Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wek RC, Role of eIF2alpha Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb Perspect Biol 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kojima KK, Jurka J, Crypton transposons: identification of new diverse families and ancient domestication events. Mob DNA 2, 12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsaytler P, Harding HP, Ron D, Bertolotti A, Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91–94 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Smillie CS et al. , Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730 e722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyams JS et al. , Factors associated with early outcomes following standardised therapy in children with ulcerative colitis (PROTECT): a multicentre inception cohort study. Lancet Gastroenterol Hepatol 2, 855–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suppli MP et al. , Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am J Physiol Gastrointest Liver Physiol 316, G462–G472 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Lefebvre P et al. , Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARalpha-regulated dermatopontin. JCI Insight 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novoa I, Zeng H, Harding HP, Ron D, Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 153, 1011–1022 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harding HP et al. , An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11, 619–633 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Maas NL, Diehl JA, Molecular pathways: the PERKs and pitfalls of targeting the unfolded protein response in cancer. Clin Cancer Res 21, 675–679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hetz C, The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13, 89–102 (2012). [DOI] [PubMed] [Google Scholar]
- 41.San Agustin JT et al. , Genetic link between renal birth defects and congenital heart disease. Nat Commun 7, 11103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.S. Deciphering Developmental Disorders, Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ververi A, Splitt M, Dean JCS, Study DDD, Brady AF, Phenotypic spectrum associated with de novo mutations in QRICH1 gene. Clin Genet 93, 286–292 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Lui JC et al. , QRICH1 mutations cause a chondrodysplasia with developmental delay. Clin Genet 95, 160–164 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aziz A, Harrop SP, Bishop NE, Characterization of the deleted in autism 1 protein family: implications for studying cognitive disorders. PLoS One 6, e14547 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giannandrea M et al. , Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genet 86, 185–195 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez G, Khatiwada S, Costa-Mattioli M, Hetz C, ER Proteostasis Control of Neuronal Physiology and Synaptic Function. Trends Neurosci 41, 610–624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pasini S, Corona C, Liu J, Greene LA, Shelanski ML, Specific downregulation of hippocampal ATF4 reveals a necessary role in synaptic plasticity and memory. Cell Rep 11, 183–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adolph TE et al. , Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data used in the study have been deposited in the Gene Expression Omnibus (GEO), under accession number GSE146842.