

Abstract
In this paper, we report the synthesis of hitherto unknown 5-haloethynyl and 5-(1,2-dihalo)vinyluracil nucleoside analogues of the anti-HIV AZT, and FLT drugs. The key step of those syntheses is a Pd(0) cross-coupling at C5 position under Sonogashira conditions. Finally, based on their in vitro anti-HIV activities and their cytotoxicity on PBM, CEM, and VERO cell lines, the best compounds were the 2′,3′-dideoxy-3′-fluoro-5-(bromo-2-iodo)vinyluridine (10b, EC50 of 0.6 μM), and the 3′-azido-2′,3′-dideoxy-5-(bromo-2-iodo)vinyluridine (16b, EC50 of 1.1 μM).
1. Introduction
The search for new drugs against the human immunodeficiency virus (HIV), the causative agent of AIDS is intense.1 There are now more than 20 anti-HIV drugs approved for clinical use and more than 30 anti-HIV compounds under (pre)clinical development. The nucleoside or nucleotide analogue reverse transcriptase inhibitors (NRTIs or NtRTIs) are integral components of nearly all anti-retroviral treatment regimens. They are part of several families of nucleosides from which the thymidine analog 3′-azido-3′-dideoxythymidine (AZT)2 or 2′,3′-dideoxy-3′-fluorothymidine (3′-FddThd, FLT)3 is particularly potent and selective anti-HIV agents.
These drugs, as the other anti-HIV nucleosides, which lack a 3′-hydroxyl group, cannot support continued synthesis of the newly made DNA strand and thus terminate or abort the polymerization process catalyzed by the HIV reverse transcriptase. Moreover, a number of 5-substituted pyrimidine nucleosides have quite potent antiviral activity.4
Current agents have been found to have some limitations, including short- and long-term adverse effects, mitochondrial toxicity and as with all anti-retroviral drugs, the development of drug-resistant or multi-drug-resistant The intense development of novel NRTIs with an excellent safety profile and limited or no cross-resistance with currently available drugs is still the goal for new agents.
Thus, based on our ongoing research program, we report herein a full account of the synthesis of 10 C5-haloethynyl and C5-(1,2-dihalovinyl) analogues of the well-known anti-HIVAZT and FLT (Fig. 1). The key step is a cross-coupling reaction under Sonogashira conditions. All final compounds were tested on HIV-1 using different cell lines and their cytotoxicities evaluated in lymphocytic CEM, African Green monkey (VERO), and activated human peripheral blood mononuclear (PBM) cells.
Figure 1.
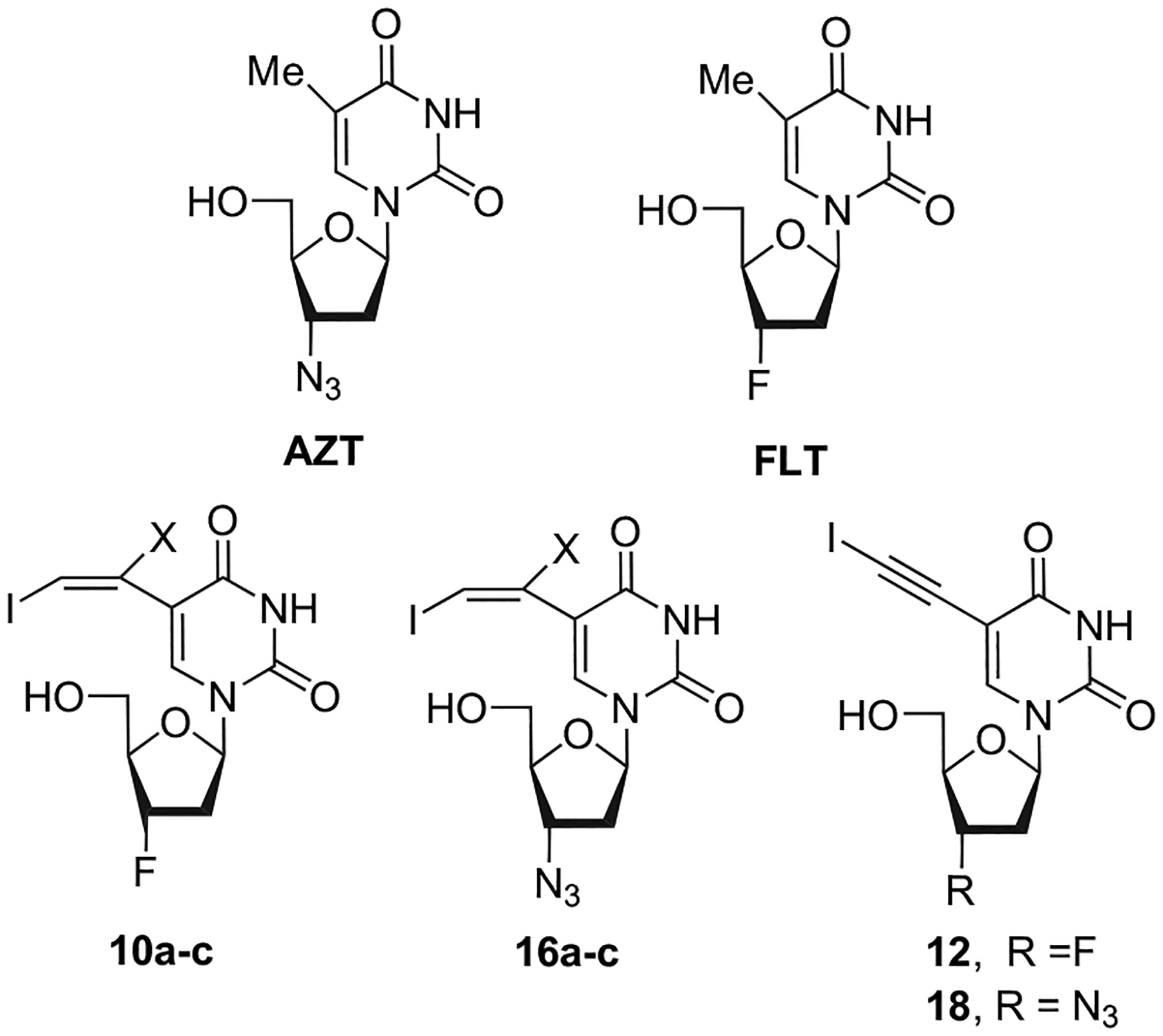
AZT, FLT, and their newly synthesized analogues.
2. Results and discussion
Starting from 2′-deoxyuridine (1), the synthesis of the key intermediate 6, which can lead to either fluorinated or azido compounds, is outlined in Scheme 1.
Scheme 1.
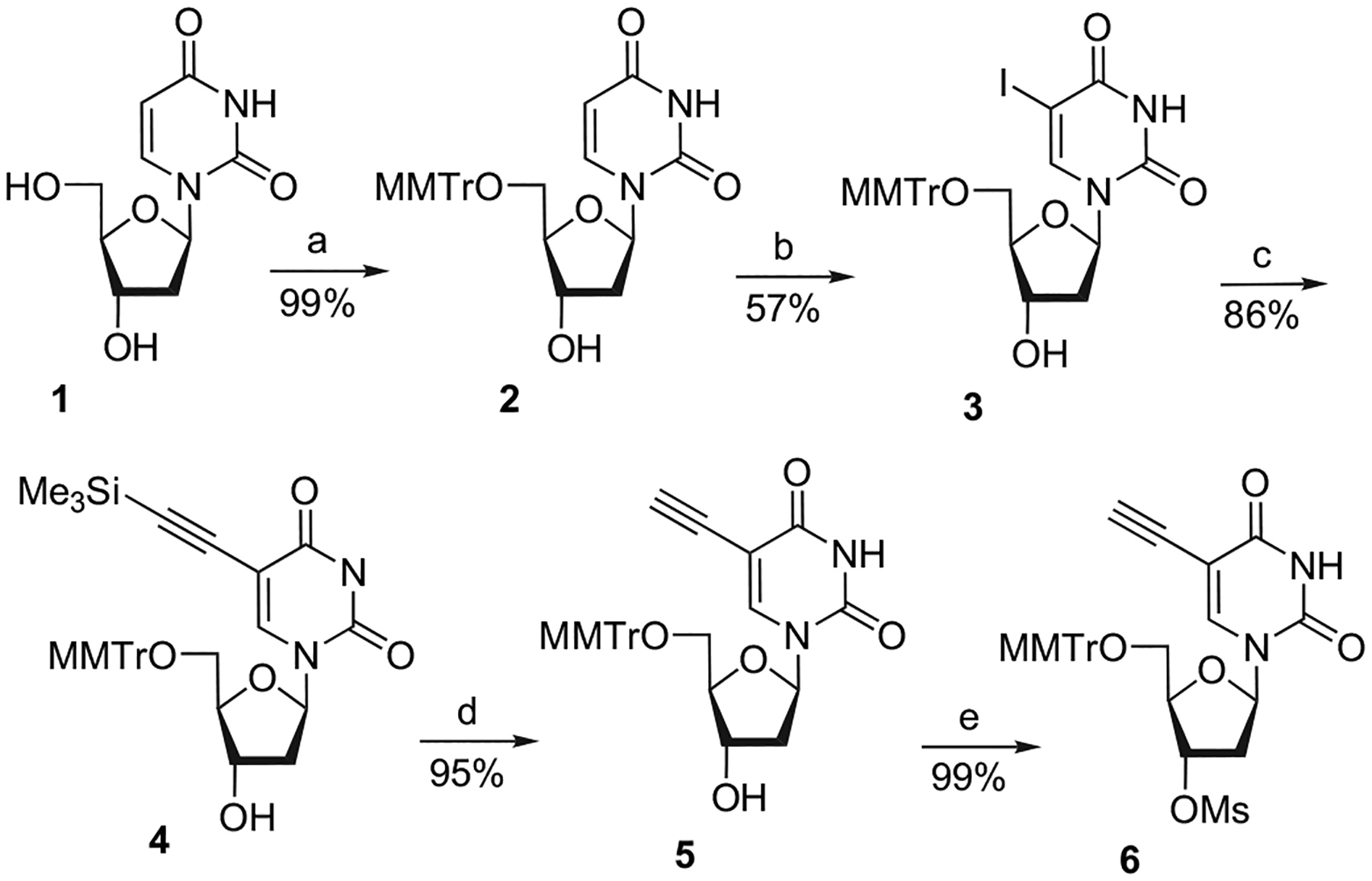
Reagents and conditions: (a) MMTrCl, pyridine, reflux; (b) I2, CAN, NaHCO3, CH3CN, reflux, 4 days; (c) TMSC≡CH, CuI, PdCl2(PPh3)2, Et3N, DMF, rt; (d) TBAF, CH3CN, rt; (e) MsCl, pyridine, 0 °C.
After a selective protection of the 2′-deoxyuridine (1) with monomethoxytritylchloride (MMTrCl), the resulting compound 2 was directly iodinated at C5 position following the Asakura and Robins methodology5 with elemental iodine and ceric(IV) ammonium nitrate (CAN) at 80 °C. The corresponding protected 5-iodouracil analogue 3 was isolated in 57% yield. The introduction of a C-5-alkynyl group was performed by a Pd(0)-mediated reaction, using the Sonogashira cross-coupling reaction.4g,6 Thus, reaction of the resulting pure 5′-protected-5-iodouracil 3 with trimethylsilylacetylene (TMS) in the presence of Et3N (base), CuI (co-catalyst), and PdCl2(PPh3)2 (catalyst) in anhydrous dimethylformamide (DMF) at rt afforded 4 in 86% yield. The removal of the trimethylsilyl group in 4 with n-Bu4NF yielded the corresponding 5-ethynyluracil 5 (95%), which was activated at 3′-OH by mesylation. The key intermediate 6 was isolated in 99% yield.
To reach the analogues of FLT, the mesylate 6 was treated with aqueous 1 M NaOH in dioxane at reflux for 2 h to give 7 having the 3′-OH with a β configuration. Fluorination of 7 was performed in dichloromethane at 0 °C with diethylaminosulfur trifluoride (DAST)7,8 to give 8, from which mono- or dihalogenation can be processed (Scheme 2).
Scheme 2.
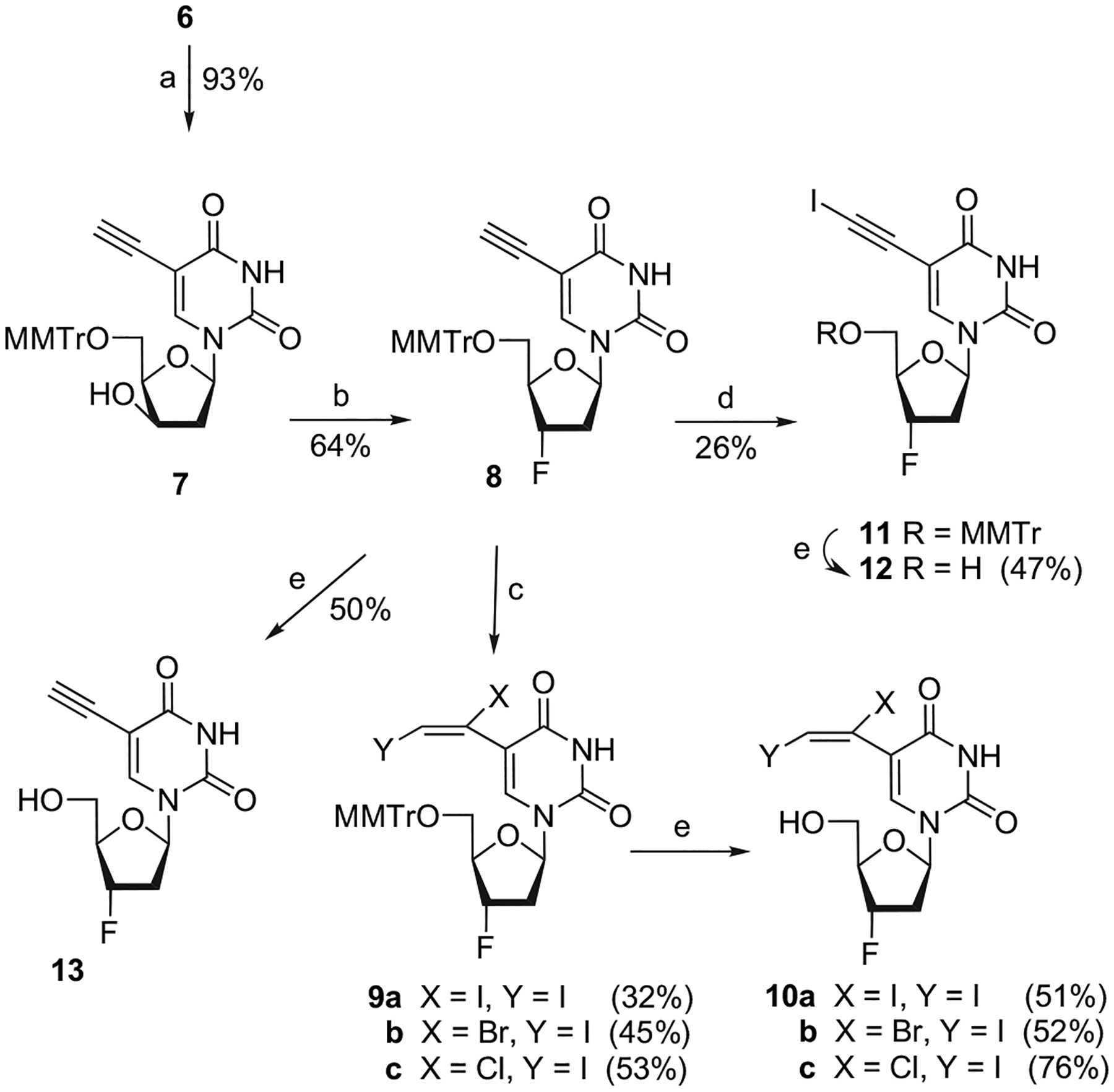
Reagents and conditions: (a) 1 M NaOH, dioxane, reflux; (b) DAST, CH2Cl2, 0 °C; (c) XY (i.e., I2, IBr, and ICl), CH3CN, 0 °C; (d) I(coll)2ClO4 (IDCP), Ag(coll)2ClO4, CH3CN, rt; (e) AcOH 80%, rt.
We first turned our attention to the dihalogenation of 5-ethynyl nucleoside 8 to reach 5-(1,2-dihalogenated)vinyl analogues 9a–c. Several halogenating reagents (i.e., I2, IBr, ICl) were used to obtain the corresponding 1,2-dihaloderivatives.
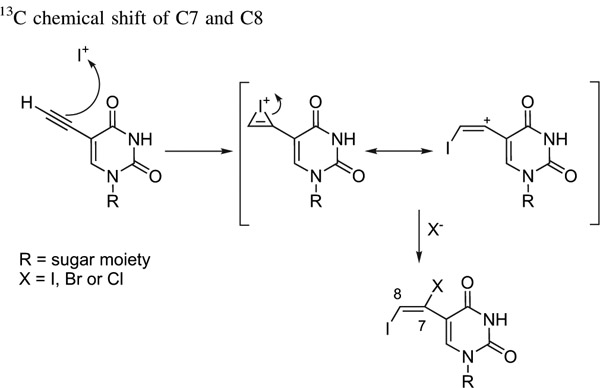
In all the cases of halo-iodination, the formation of vicinal halo-iodoalkenes occurred with high anti-stereospecificity, implicating the intermediary of an iodonium ion in the reaction sequence, and a unique regioselectivity, implicating a predominantly Markovnikov addition (Table 1). The regioselectivity of this dihalogenation has been checked by 13C NMR, in agreement with some recent papers,9,10 taking into account the different effects exerted by chlorine and iodine atoms on the chemical shift of the carbon to which they were directly bonded. In fact, a chlorine (and bromine) atom induces a downfield shift of the carbon resonance (C7) due to its high electronegativity, whereas an iodine atom leads to an upfield shift of the carbon resonance (C8) due to the ‘heavy atom effect’11 (see Table 1).
Table 1.
13C chemical shift of C7 and C8
 | |||
|---|---|---|---|
| Substrate | Substituent X | δ | |
| C7 | C8 | ||
| Alkyne | None | 87.5 | 74.5 |
| Alkene | I | 85.3 | 87.5 |
| Alkene | Br | 113.8 | 82.4 |
| Alkene | Cl | 114.2 | 80.6 |
Finally, when 8 was treated with the iodonium di-syn-collidine perchlorate (IDCP), acting as very reactive electrophile (I+), in anhydrous CH3CN, the desired 5-iodoethynyluracil nucleoside 11 was obtained in a low yield (26%). The final detritylation of 8, 9a–c, and 11 under smooth acidic conditions (acetic acid 80%) afforded 13, 10a–c, and 12, respectively, in good to moderate yields.
Figure 2 represents a zoom of the 1H NMR spectra of new 5-ethynyl-3′-fluoro-3′-deoxyuridine (13) in CD3OD. The chemical shift and multiplicity of H3′ and H4′ of 13 are in agreement with those found in FLT 1H NMR spectra and with the α-position of the fluorine.
Figure 2.
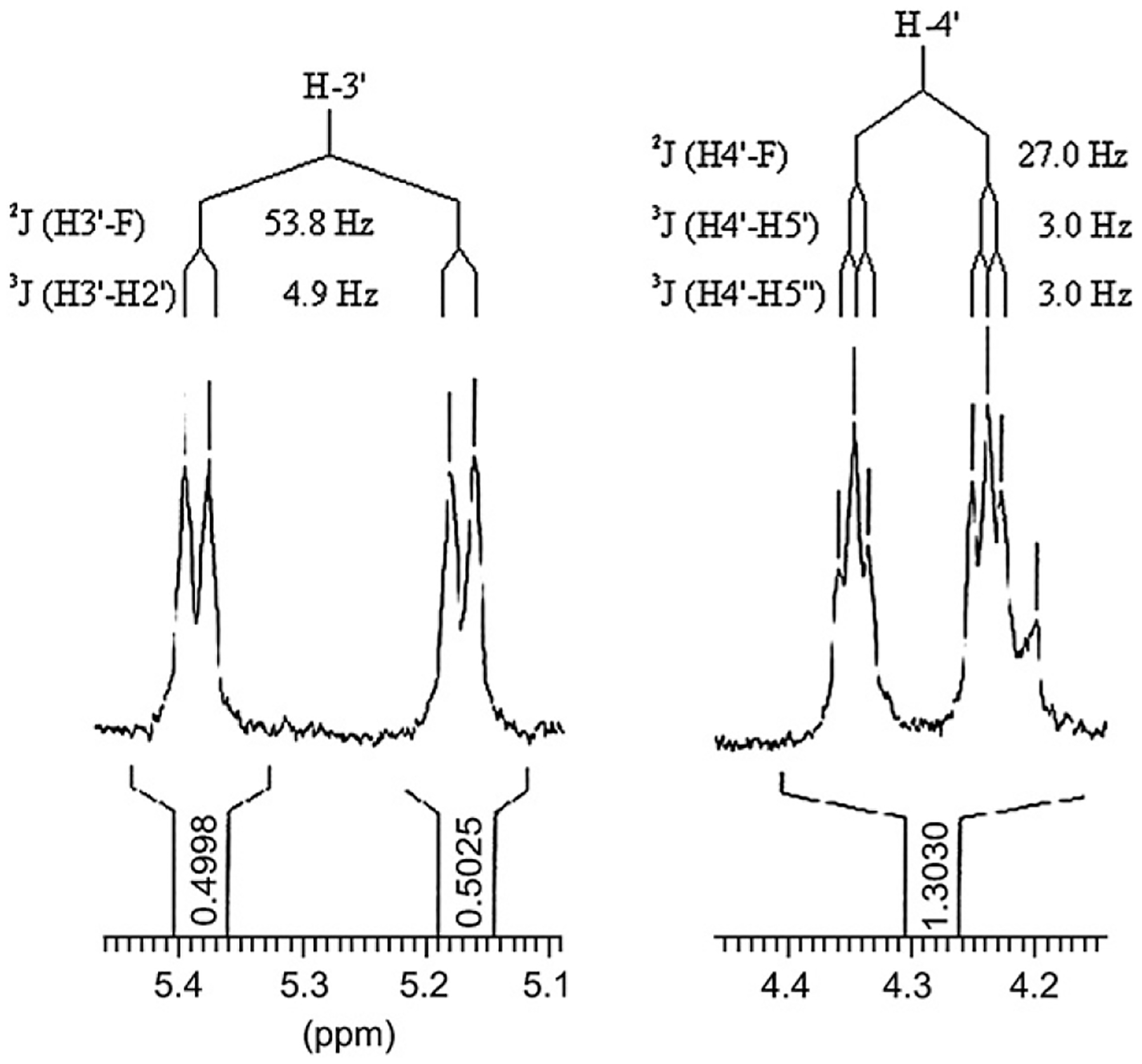
Zoom of the 1H NMR spectra of 13.
The intermediate 6 was also used for the synthesis of the AZT derivatives 16a–c and 18 (Scheme 3), following a procedure from Matsuda et al.12 Treatment of 6 with NaN3 in DMF at reflux for 16 h led to the formation of 14, via (1) the formation of the anhydro O2,3′ nucleoside and (2) its opening at C3′ by NaN3. The stereochemistry at C3′ was determined by NMR. The iodination of the C5-ethynyl and the 1,2-dihalogenation of the C5-vinyl were performed as described above and the desired compounds 15a–c and 17 were isolated in good to moderate yields, respectively. The final detritylation of 14, 15a–c, and 17 under the same acidic conditions (acetic acid 80%) afforded 18, 19, and 16a–c, respectively, in good to moderate yields.
Scheme 3.
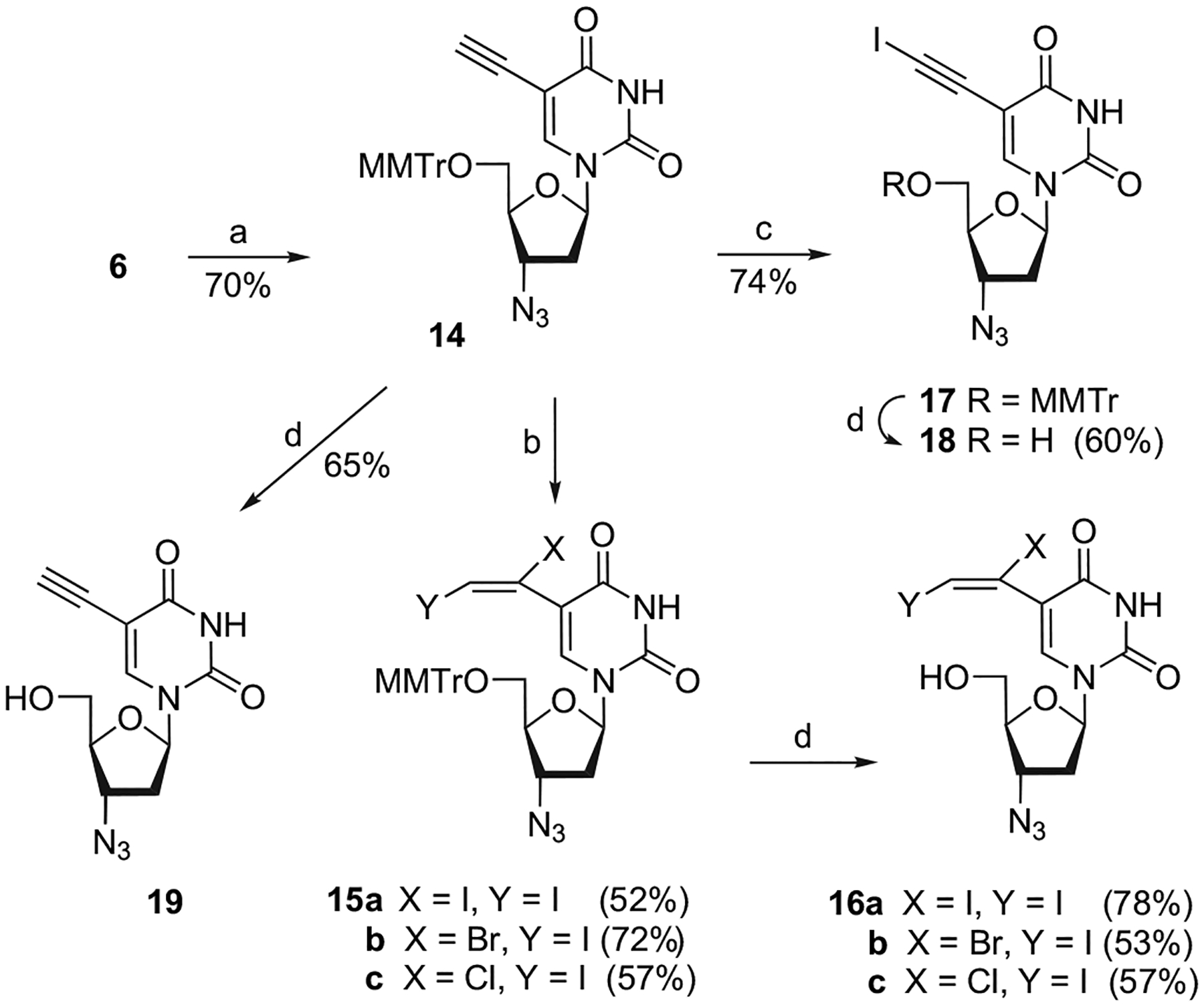
Reagents and conditions: (a) NaN3, DMF, reflux; (b) XY (i.e., I2, IBr, ICl), CH3CN, 0 °C; (c) I(coll)2ClO4 (IDCP), Ag(coll)2ClO4, CH3CN, rt; (d) AcOH 80%, rt.
3. Biological results
The synthesized compounds 10a–c, 12, 16a–c, and 18, along with the known antiviral compound (AZT), were tested for their anti-HIV activity in vitro, and the results are shown in Table 2.
Table 2.
Evaluation of the antiviral activity against human immunodeficiency virus (HIV) and cytotoxicity against PBM, CEM, and VERO cells in vitro, expressed in μM, of synthesized AZT and FLT analogues
| Compound | Anti-HIV-1 activity in PBMCs (EC50) | Toxicity (IC50) | ||
|---|---|---|---|---|
| PBM | CEM | VERO | ||
| AZTa | 0.016 | >100 | 14.0 | 29.0 |
| 12 | 6.9 | 25.6 | 15.1 | 22.9 |
| 10a | 3.1 | 43.0 | 100 | 82.7 |
| 10b | 0.6 | 26.8 | 31.6 | 29.5 |
| 10c | 4.9 | 18.4 | 33.1 | 18.5 |
| 18 | 2.3 | 19.7 | 31.6 | 14.4 |
| 16a | 3.4 | 18.6 | 30.7 | 52.3 |
| 16b | 1.1 | 26.2 | 30.6 | 27.7 |
| 16c | 2.7 | 23.8 | 48.1 | 9.0 |
Reference compound.
Among these nucleoside analogues, only compounds 10b and 16b were found to exhibit moderate anti-HIV activity, with an EC50 of 0.6 and 1.1 μM, respectively. Nevertheless, those compounds showed also toxicity against PBM (26.8 and 26.2 μM, respectively). Their toxicity against CEM (31.6 and 30.6 μM, respectively) or VERO cells (29.5 and 27.7 μM, respectively) is comparable to that of AZT. The antiviral13 and cytotoxicity14 assays were done as previously described.
4. Conclusion
An efficient route to various derivatives of FLT (10a–c, 12, and 13) or of AZT (18, 19, and 16a–c) has been developed using a combination of Pd(0) under Sonogashira conditions, and regio- and stereoselective (di)halogenation methodologies. The antiviral activities of all final compounds have been determined.
5. Experimental
5.1. General
Commercially available chemicals were of reagent grade and used as received. Dry pyridine was obtained from distillation over Na, and N,N-dimethylformamide and ethanol over CaH2. Dimethylamine was dried by passage through a KOH filled tower. Methanol was dried with CaH2 and triethylamine with KOH. The reactions were monitored by thin layer chromatography (TLC) analysis using silica gel plates (Kieselgel 60 F254, E. Merck). Compounds were visualized by UV irradiation and/or spraying with 20% H2SO4 in EtOH, followed by charring at 150 °C. Column chromatography was performed on silica gel 60 M (0.040–0.063 mm, E. Merck). The 1H and 13C NMR spectra were recorded on a Brucker AVANCE DPX 250 Fourier Transform spectrometer at 250 MHz and 62.9 MHz , respectively, in chloroform (d), methanol (d4), and DMSO-(d6), and shift values in parts per million relative to SiMe4 as internal reference, unless otherwise stated; signals are reported as s (singlet), d (doublet), t (triplet), m (multiplet); J in hertz. High Resolution Mass spectra (HRMS) were performed using FAB (Fast Atom Bombardment) or ESI (Electron Spray Ionization). The nomenclature of the obtained compounds is in accordance with the IUPAC rules and was checked with autonome. The numbering and assignment of the chemical shifts for all described compounds are related to the corresponding ribose derivatives. Evidence of purity has been obtained from a proton-decoupled 13C NMR spectrum with a signal-to-noise ratio sufficient to permit seeing peak with 5% of the intensity of the strongest peak.
5.1.1. 1-{4-Hydroxy-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-1H-pyrimidine-2,4-dione (2)
A solution of 2′-deoxyuridine (10.0 g, 43.9 mmol) and para-anisylchlorodiphenylmethane (16.3 g, 52.6 mmol) in dry pyridine (235 mL) was refluxed for 2 h. After cooling at rt, pyridine was evaporated under reduced pressure, then dichloromethane (400 mL) was added and the solution was washed with water (50 mL×3), dried over MgSO4, filtered through a fritted glass funnel, and concentrated under reduced pressure to afford the crude product, which was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 5:5 to EtOAc/MeOH, 9:1) to give the pure protected nucleoside 2 (21.9 g, 99%) as a white solid (CAS number: 70255-96-8).
5.1.2. 1-{4-Hydroxy-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-5-iodo-1H-pyrimidine-2,4-dione (3)
A solution of 2 (21.9 g, 43.8 mmol), I2 (17.0 g, 66.9 mmol), CAN (17.7 g, 32.3 mmol), and NaHCO3 (1.0 g) in dry acetonitrile (480 mL) was refluxed for 4 days. After cooling at rt, a solution of saturated Na2SO3 (800 mL) and EtOAc (200 mL) were added, aqueous layer was extracted with EtOAc (300 mL×2), then organic layer was washed with saturated Na2SO3 (300 mL), dried over MgSO4, filtered through a fritted glass funnel, and concentrated under reduced pressure to afford the crude product, which was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 6:4 to EtOAc) to give the pure iodinated nucleoside 3 (12.8 g, 57%) as a white solid. The physico-chemical data of the compound are fully related with those previously published.15
5.1.3. 1-{4-Hydroxy-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-5-trimethylsilanyl-ethynyl-1H-pyrimidine-2,4-dione (4)
Compound 3 (5.00 g, 8.0 mmol) was dissolved in a mixture of dry DMF (62.5 mL), dry Et3N (3.3 mL, 24.0 mmol), and trimethylsilylacetylene (3.4 mL, 24.0 mmol). CuI (300.0 mg, 1.6 mmol) and PdCl2(PPh3)2 (563 mg) were then added and the reaction mixture was stirred at rt under argon for 16 h. Solvents were evaporated under reduced pressure. The crude product was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 5:5 to 2:8) to give 4 (4.10 g, 86%) as a pale yellow solid. (c 0.5, CHCl3); mp 104 °C; 1H NMR (CDCl3) δ: 2.08–2.33 (m, 1H, H-2′a), 2.42–2.64 (m, 1H, H-2′b), 3.20–3.52 (m, 2H, H-5′), 3.78 (s, 3H, OCH3), 4.05–4.25 (m, 1H, H-4′), 4.40–4.53 (m, 1H, H-3′), 6.31 (dd, J=5.6, 8.1 Hz, 1H, H-1′), 6.78–7.55 (m, 14H, MMTr), 8.05 (s, 1H, H-6); 13C NMR (CDCl3) δ: 41.5 (C-2′), 55.3 (OCH3), 63.7 (C-5′), 72.5 (C-3′), 85.9 (C-1′), 86.6 (C-4′), 87.3 (C), 94.9 (C), 99.9 (C), 100.7 (C), 113.5 (CH), 127.2 (CH), 128.2 (CH), 128.3 (C), 130.5 (CH), 135.1 (C), 142.8 (C-6), 144.0 (C), 144.1 (C), 149.5 (C=O), 158.8 (C), 161.5 (C); HRMS: C34H36N2O6NaSi, calcd m/z 619.2240; found m/z 619.2245.
5.1.4. 5-Ethynyl-1-{4-hydroxy-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-1H-pyrimidine-2,4-dione (5)
A solution of 4 (616.3 mg, 1.03 mmol) and TBAF (303.4 mg, 1.16 mmol) in dry acetonitrile (24.0 mL) was stirred at rt for 2 h. Solvent was removed under reduced pressure and the crude product was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 5:5 to 2:8) to give 5 (516.8 mg, 95%) as a pale yellow solid. (c 1.0, CHCl3); mp 110 °C; 1H NMR (CDCl3) δ: 2.22–2.40 (m, 1H, H-2′a), 2.45–2.60 (m, 1H, H-2′b), 2.90 (s, 1H, CH alkyne), 3.41 (t, J=3.6 Hz, 2H, H-5′), 3.81 (s, 3H, OCH3), 4.15–4.20 (m, 1H, H-4′), 4.51–4.62 (m, 1H, H-3′), 6.30 (t, J=8.2 Hz, 1H, H-1′), 6.70–7.50 (m, 14H, MMTr), 8.10 (s, 1H, H-6); 13C NMR (CDCl3) δ: 41.6 (C-2′), 55.4 (OCH3), 63.7 (C-5′), 72.4 (C-3′), 74.2 (C), 82.2 (C), 85.9 (C-1′), 86.6 (C-4′), 87.5 (C), 99.4 (C), 113.5 (CH), 127.3 (CH), 128.2 (CH), 128.3 (C), 128.4 (C), 130.5 (CH), 135.1 (C), 143.8 (C-6), 143.9 (C), 144.1 (C), 149.4 (C=O), 158.9 (C), 161.7 (C); HRMS: C31H28N2O6Na, calcd m/z 547.1845; found m/z 547.1844.
5.1.5. Methanesulfonic acid 5-(5-ethynyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-2-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-3-yl ester (6)
A solution of 5 (500.0 mg, 0.95 mmol) and methanesulfonylchloride (0.73 mL, 9.50 mmol) in dry pyridine (30 mL) at 0 °C was allowed to warm to rt and stirred for 16 h. After cooling at 0 °C, ice, water (100 mL), and EtOAc (100 mL) were added, the aqueous layer was extracted with EtOAc (50 mL×3), the organic layers were washed with water (50 mL×3), dried over MgSO4, filtered through a fritted glass funnel, and concentrated under reduced pressure to afford the crude product, which was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 5:5) to give 6 (587.0 mg, 99%) as an orange solid. (c 1.0, CHCl3); mp 99 °C; 1H NMR (CDCl3) δ: 2.30–2.50 (m, 1H, H-2′a), 2.70–2.82 (m, 1H, H-2′b), 2.91 (s, 1H, CH alkyne), 3.02 (s, 3H, SCH3), 3.33–3.57 (m, 2H, H-5′), 3.80 (s, 3H, OCH3), 4.35 (br s, 1H, H-4′), 5.31 (se, 1H, H-3′), 6.29 (t, J=8.2 Hz, 1H, H-1′), 6.75–7.50 (m, 14H, MMTr), 8.07 (s, 1H, H-6), 8.58 (br s, 1H, NH); 13C NMR (CDCl3) δ: 38.8 (SCH3), 39.3 (C-2′), 55.4 (OCH3), 63.0 (C-5′), 74.0 (C-3′), 79.8 (C), 82.4 (C), 84.5 (C-1′), 85.4 (C-4′), 88.0 (C), 99.9 (C), 113.6 (CH), 127.5 (CH), 128.3 (CH), 128.3 (CH), 129.2 (C), 130.5 (CH), 134.6 (C), 143.2 (C-6), 143.6 (C), 143.7 (C), 149.2 (C=O), 159.0 (C), 161.2 (C); HRMS: C32H30N2O8NaS, calcd m/z 625.1621; found m/z 625.1632.
5.1.6. 5-Ethynyl-1-{4-hydroxy-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-1H-pyrimidine-2,4-dione (7)
A solution of 6 (50.0 mg, 0.08 mmol) in dioxane (2.0 mL) and aqueous 1 M NaOH (168 μL, 0.16 mmol) was refluxed for 2 h. After cooling at rt, solvents were removed in vacuo and the crude product was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 5:5 to EtOAc) to give 7 (41.0 mg, 93%) as a pale yellow solid. (c 1.0, CHCl3); mp 124 °C; 1H NMR (CDCl3) δ: 2.08–2.25 (m, 1H, H-2′a), 2.47–2.66 (m, 1H, H-2′b), 3.07 (s, 1H, CH alkyne), 3.43–3.72 (m, 2H, H-5′), 3.79 (s, 3H, OCH3), 4.00–4.12 (m, 1H, H-4′), 4.38–4.50 (m, 1H, H-3′), 6.15 (t, J=6.1 Hz, 1H, H-1′), 6.80–7.55 (m, 14H, MMTr), 8.15 (s, 1H, H-6); 13C NMR (CDCl3) δ: 41.1 (C-2′), 55.4 (OCH3), 61.9 (C-5′), 70.9 (C-3′), 75.0 (C), 81.7 (CH alkyne), 83.4 (C-1′), 85.9 (C-4′), 87.5 (C), 98.5 (C), 113.6 (CH), 127.4 (CH), 128.2 (CH), 130.4 (CH), 134.9 (C), 143.8 (C), 143.8 (C), 145.3 (C-6), 149.5 (C=O), 159.0 (C), 161.8 (C). HRMS: C31H28N2O6Na, calcd m/z 547.5617; found m/z 547.5612.
5.1.7. 5-Ethynyl-1-{4-fluoro-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-1H-pyrimidine-2,4-dione (8)
To a solution of 7 (1.26 g, 2.4 mmol) in dry dichloromethane (77.0 mL) at 0 °C was added dropwise diethylaminosulfur trifluoride (0.6 mL, 4.8 mmol), and the reaction mixture was stirred for 15 min. Then a saturated NaHCO3 solution (50.0 mL) was added, the aqueous layer was extracted with CH2Cl2 (50 mL×3), the organic layers were washed with water (50 mL), dried over MgSO4, filtered through a fritted glass funnel, and concentrated under reduced pressure to afford the crude product, which was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 5:5) to give 8 (805.0 mg, 64%) as a pale yellow solid. (c 1.0, CHCl3); mp 54 °C; 1H NMR (CDCl3) δ: 2.12–2.46 (m, 1H, H-2′a), 2.68–2.90 (m, 1H, H-2′b), 2.88 (s, 1H, CH alkyne), 3.35–3.48 (m, 2H, H-5′), 3.80 (s, 3H, OCH3), 4.37 (d, J=27.4 Hz, 1H, H-4′), 5.28 (dd, J=4.6, 53.6 Hz, 1H, H-3′), 6.34 (dd, J=5.3, 9.0 Hz, 1H, H-1′), 6.80–7.50 (m, 14H, MMTr), 8.10 (s, 1H, H-6); 13C NMR (CDCl3) δ: 39.7 (d, J=84.2 Hz, C-2′), 55.4 (OCH3), 63.4 (d, J=40.9 Hz, C-5′), 82.2 (CH alkyne), 84.8 (d, J=101.1 Hz, C-4′), 85.7 (C-1′), 87.8 (C), 94.5 (d, J=710.0 Hz, C-3′), 99.7 (C), 113.6 (CH), 127.4 (CH), 128.3 (CH), 128.4 (CH), 130.5 (CH), 134.6 (C), 143.4 (C-6), 143.6 (C), 143.7 (C), 149.2 (C=O), 159.0 (C×2); HRMS: C31H27N2O5FNa, calcd m/z 549.1802; found m/z 549.1806.
5.2. General procedure for dihalogenation of 5-ethynyl compound
5-Ethynyl nucleoside (0.5 mmol) was dissolved in dry acetonitrile (5.0 mL) and the reaction mixture was cooled in an ice bath. A solution of the halogenating reagent (i.e., I2, IBr, or ICl; 0.6 mmol) in dry CH3CN (1.0 mL) was added dropwise at 0 °C and the reaction mixture was stirred until completion (typically for 15 min to 2 h, checked by TLC). The reaction mixture was quenched at 0 °C by a saturated Na2SO3 solution (2 mL) and then extracted with EtOAc (10 mL×3). The organic layer was washed with water (5 mL×2) and brine (10 mL), then dried over MgSO4, filtered through a fritted glass funnel, and concentrated under reduced pressure to afford the crude product, which was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 8:2 to 3:7) to give the desired 1,2-dihalogenated compound.
5.2.1. 5-(1,2-Diiodo-vinyl)-1-{4-fluoro-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-1H-pyrimidine-2,4-dione (9a)
Prepared from compound 8 using I2 with the typical procedure described before to give 9a (32%) as an orange solid. (c 1.0, CHCl3); mp 58 °C; 1H NMR (CDCl3) δ: 2.05–2.35 (m, 1H, H-2′a), 2.67–2.86 (m, 1H, H-2′b), 3.43 (d, J=3.8 Hz, 2H, H-5′), 3.81 (s, 3H, OCH3), 4.34 (td, J=3.5, 27.0 Hz, 1H, H-4′), 5.23 (dd, J=4.7, 53.7 Hz, 1H, H-3′), 6.33 (dd, J=5.0, 9.1 Hz, 1H, H-1′), 6.75–7.50 (m, 15H, CH alkene, MMTr), 7.63 (s, 1H, H-6), 9.00 (br s, 1H, NH); 13C NMR (CDCl3) δ: 39.4 (d, J=84.2 Hz, C-2′), 55.5 (OCH3), 63.1 (d, J=38.5 Hz, C-5′), 84.5 (d, J=101.0 Hz, C-4′), 85.3 (C), 85.5 (CH alkene), 87.5 (C), 87.7 (C-1′), 94.1 (d, J=710.0 Hz, C-3′), 113.6 (CH), 118.1 (C), 127.4 (CH), 128.3 (CH), 128.4 (CH), 130.5 (CH), 134.8 (C), 139.2 (C-6), 143.7 (C), 143.8 (C), 149.5 (C=O), 158.9 (C), 159.0 (C); HRMS: C31H27N2O5-FI2Na, calcd m/z 802.9891; found m/z 802.9902.
5.2.2. 5-(1-Bromo-2-iodo-vinyl)-1-{4-fluoro-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-1H-pyrimidine-2,4-dione (9b)
Prepared from compound 8 using IBr with the typical procedure described before to give 9b (45%) as a yellow oil. (c 1.0, CHCl3); 1H NMR (CDCl3) δ: 2.10–2.40 (m, 1H, H-2′a), 2.70–2.92 (m, 1H, H-2′b), 3.35–3.52 (m, 2H, H-5′), 3.81 (s, 3H, OCH3), 4.35 (td, J=3.2, 27.6 Hz, 1H, H-4′), 5.24 (dd, J=4.6, 53.6 Hz, 1H, H-3′), 6.33 (dd, J=5.1, 9.2 Hz, 1H, H-1′), 6.96 (s, 1H, CH alkene), 6.80–7.50 (m, 14H, MMTr), 7.72 (s, 1H, H-6), 8.68 (br s, 1H, NH); 13C NMR (CDCl3) δ: 39.5 (d, J=81.8 Hz, C-2′), 55.4 (OCH3), 63.1 (d, J=43.3 Hz, C-5′), 82.7 (C-1′), 84.5 (d, J=101.1 Hz, C-4′), 85.6 (CH alkene), 87.4 (C), 87.5 (C), 94.1 (d, J=707.6 Hz, C-3′), 113.6 (CH), 113.7 (CH), 127.3 (C), 127.4 (CH), 128.2 (CH), 128.4 (CH), 130.6 (CH), 134.7 (C), 140.7 (C-6), 143.7 (C), 143.8 (C), 149.6 (C=O), 159.0 (C×2); HRMS: C31H27N2O5FBrINa, calcd m/z 755.0030; found m/z 755.0004.
5.2.3. 5-(1-Chloro-2-iodo-vinyl)-1-{4-fluoro-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-1H-pyrimidine-2,4-dione (9c)
Prepared from compound 8, using ICl with the typical procedure described before to give 9c (53%) as a pale yellow solid. (c 1.0, CHCl3); mp 91 °C; 1H NMR (CDCl3) δ: 2.05–2.35 (m, 1H, H-2′a), 2.65–2.90 (m, 1H, H-2′b), 3.30–3.55 (m, 2H, H-5′), 3.80 (s, 3H, OCH3), 4.35 (td, J=3.0, 27.3 Hz, 1H, H-4′), 5.23 (dd, J=4.7, 53.7 Hz, 1H, H-3′), 6.34 (dd, J=5.3, 9.1 Hz, 1H, H-1′), 6.72 (s, 1H, CH alkene), 6.75–7.50 (m, 14H, MMTr), 7.78 (s, 1H, H-6), 8.85 (br s, 1H, NH); 13C NMR (CDCl3) δ: 39.5 (d, J=84.3 Hz, C-2′), 55.4 (OCH3), 63.1 (d, J=40.9 Hz, C-5′), 80.9 (CH alkene), 84.6 (d, J=101.0 Hz, C-4′), 85.6 (C-1′), 87.6 (C), 94.1 (d, J=707.6 Hz, C-3′), 113.6 (CH), 113.9 (CH), 127.2 (C), 127.4 (CH), 128.3 (CH), 128.4 (CH), 130.5 (CH), 134.7 (C), 141.1 (C-6), 143.7 (C), 143.8 (C), 149.8 (C=O), 164.2 (C×2); HRMS: C31H27N2O5FClINa, calcd m/z 711.0535; found m/z 711.0531.
5.2.4. 1-{4-Azido-5-[(4-methoxy-phenyl)-diphenyl-methoxy-methyl]-tetrahydrofuran-2-yl}-5-(1,2-diiodo-vinyl)-1H-pyrimidine-2,4-dione (15a)
Prepared from compound 14, using I2 with the typical procedure described before to give 15a (52%) as a pale yellow solid. (c 1.0, CH3OH); mp 139 °C; 1H NMR (CDCl3) δ: 2.23–2.34 (m, 1H, H-2′a), 2.51–2.61 (m, 1H, H-2′b); 3.36–3.49 (m, 2H, H-5′), 3.81 (s, 3H, OCH3), 3.98–4.05 (m, 1H, H-4′), 4.16–4.26 (m, 1H, H-3′), 6.16 (t, J=6.6, 1H, H-1′), 6.87 (d, J=8.8 Hz, 2H, MMTr), 7.21–7.43 (m, 13H, MMTr, CH alkene), 7.58 (s, 1H, H-6); 13C NMR (CDCl3) δ: 38.1 (C-2′), 55.2 (OCH3), 60.4 (C-3′), 62.8 (C-5′), 83.5 (C-4′), 85.1 (C-1′), 87.1 (C), 87.2 (CH alkene), 113.3 (CH), 117.6 (C), 127.1 (CH), 128.0 (CH), 128.1 (CH), 130.2 (CH), 135.6 (C), 139.0 (C-6), 143.5 (C), 143.6 (C), 149.1 (C=O), 158.6 (C), 158.7 (C); HRMS: C31H27N5O5I2Na, calcd m/z 826.0093; found m/z 825.9999.
5.2.5. 1-{4-Azido-5-[(4-methoxy-phenyl)-diphenyl-methoxy-methyl]-tetrahydrofuran-2-yl)}-5-(1-bromo-2-iodo-vinyl)-1H-pyrimidine-2,4-dione (15b)
Prepared from compound 14, using IBr with the typical procedure described before to give 15b (72%) as a pale yellow solid. (c 0.9, CH3OH); mp 89 °C; UV (MeOH) λmax 227, 279 nm; 1H NMR (CDCl3) δ: 2.25–2.36 (m, 1H, H-2′a), 2.51–2.61 (m, 1H, H-2′b), 3.35–3.50 (m, 2H, H-5′), 3.80 (s, 3H, OCH3), 3.98–4.03 (m, 1H, H-4′), 4.17–4.23 (m, 1H, H-3′), 6.17 (t, J=6.3 Hz, H-1′), 6.86 (d, J=9.1 Hz, 2H, MMTr), 6.95 (s, 1H, CH alkene), 7.21–7.42 (m, 12H, MMTr), 7.67 (s, 1H, H-6), 8.85 (s, 1H, NH); 13C NMR (CDCl3) δ: 38.5 (C-2′), 55.4 (OCH3), 60.7 (C-3′), 63.11 (C-5′), 82.5 (CH), 83.9 (CH), 85.6 (CH), 87.4 (C), 113.5 (CH), 113.8 (C), 115.0 (C), 127.4 (CH), 128.3 (CH), 128.4 (CH), 130.5 (CH), 138.9 (C), 140.7 (C-6), 143.8 (C), 149.3 (C=O), 159.8 (C), 159.9 (C); HRMS: C31H27N5O5BrINa, calcd m/z 778.0138; found m/z 778.0142.
5.2.6. 1-{4-Azido-5-[(4-methoxy-phenyl)-diphenyl-methoxy-methyl]-tetrahydrofuran-2-yl}-5-(1-chloro-2-iodo-vinyl)-1H-pyrimidine-2,4-dione (15c)
Prepared from compound 14, using ICl with the typical procedure described before to give 15c (57%) as a pale yellow gum. (c 0.9, CH3OH); 1H NMR (CDCl3) δ: 2.27–2.39 (m, 1H, H-2′a), 2.53–2.63 (m, 1H, H-2′b), 3.36–3.53 (m, 2H, H-5′), 3.98–4.05 (m, 1H, H-4′), 4.18–4.27 (m, 1H, H-3′), 6.20 (t, J=6.5 Hz, 1H, H-1′), 6.73 (s, 1H, CH alkene), 6.88 (d, J=8.8 Hz, 2H, MMTr), 7.25–7.48 (m, 12H, MMTr), 7.74 (s, 1H, H-6), 8.94 (s, 1H, NH); 13C NMR (CDCl3) δ: 38.5 (C-2′), 55.4 (OCH3), 60.7 (C-3′), 63.1 (C-5′), 80.7 (CH alkene), 83.9 (C-4′), 85.4 (C-1′), 87.4 (C), 113.6 (CH), 127.2 (CH), 127.3 (CH), 127.9 (CH), 127.97 (CH), 128 (CH), 128.1 (CH), 128.2 (CH), 128.3 (CH), 129.3 (CH), 130.4 (CH), 134.9 (C), 141.2 (C-6), 143.8 (C), 149.4 (C=O), 158.9 (C), 159.1 (C); HRMS: C31H27N5O5ClINa, calcd m/z 734.0643; found m/z 734.0646.
5.3. General procedure for monohalogenation of 5-ethynyl compound
5-Ethynyl nucleoside (0.5 mmol) was dissolved in dry acetonitrile (5.0 mL) and the reaction mixture was cooled in an ice bath. A solution of the halogenating reagent (i.e., IDCP; 0.7 mmol) and Ag(coll)2ClO4 (11.0 mg) were added and the reaction mixture was stirred in dark at rt (typically 2–20 h, checked by TLC). The reaction mixture was quenched at 0 °C by a saturated Na2SO3 solution (2 mL) and then extracted with EtOAc (10 mL×4). The organic layer was washed with water (10 mL), a 1 M HCl solution (5 mL×3), water (5 mL×2), and brine (10 mL), then dried over MgSO4, filtered through a fritted glass funnel, and concentrated under reduced pressure to afford the crude product, which was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 8:2 to 3:7) to give the desired mono-halogenated compound.
5.3.1. 1-{4-Fluoro-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-5-iodoethynyl-1H-pyrimidine-2,4-dione (11)
Prepared from compound 8, using the typical procedure described before to give 11 (26%) as yellow solid. (c 1.0, CHCl3); mp 73 °C; 1H NMR (CDCl3) δ: 2.15–2.48 (m, 1H, H-2′a), 2.68–2.93 (m, 1H, H-2′b), 3.30–3.56 (m, 2H, H-5′), 3.81 (s, 3H, OCH3), 4.38 (d, J=27.0 Hz, 1H, H-4′), 5.32 (dd, J=4.4, 53.4 Hz, 1H, H-3′), 6.33 (dd, J=5.3, 9.1 Hz, 1H, H-1′), 6.75–7.50 (m, 14H, MMTr), 8.01 (s, 1H, H-6), 9.20 (br s, 1H, NH); 13C NMR (CDCl3) δ: 12.1 (CCI), 39.8 (d, J=86.6 Hz, C-2′), 55.4 (OCH3), 63.4 (d, J=40.9 Hz, C-5′), 84.2 (C), 84.9 (d, J=101.1 Hz, C-4′), 86.0 (C-1′), 87.8 (C), 94.5 (d, J=707.6 Hz, C-3′), 101.0 (C), 113.6 (CH), 127.5 (CH), 127.9 (C), 128.1 (CH), 128.2 (CH), 130.4 (CH), 134.6 (C), 143.5 (C), 143.7 (C-6), 143.8 (C), 149.2 (C=O), 159.0 (C), 161.6 (C); HRMS: C31H26N2O5FINa, calcd m/z 675.0768; found m/z 675.0771.
5.3.2. 1-{4-Azido-5-[(4-methoxy-phenyl)-diphenyl-methoxymethyl]-tetrahydrofuran-2-yl}-5-iodoethynyl-1H-pyrimidine-2,4-dione (17)
Prepared from compound 14 using the typical procedure described before to give 17 (74%) as a pale yellow solid. (c 1.0, CH3OH); mp 95 °C; 1H NMR (CDCl3) δ: 2.37–2.47 (m, 1H, H-2′a), 2.52–2.62 (m, 1H, H-2′b), 3.36–3.48 (m, 2H, H-5′), 3.81 (s, 3H, OCH3), 4.01–4.05 (m, 1H, H-4′), 4.28–4.38 (m, 1H, H-3′), 6.15 (t, J=6.1 Hz, 1H, H-1′), 6.85–6.89 (m, 2H, MMTr), 7.22–7.47 (m, 12H, MMTr), 8.02 (s, 1H, H-6); 13C NMR (CDCl3) δ: 12.9 (C), 39.3 (C-2′), 56.0 (OCH3), 61.2 (C-3′), 63.5 (C-5′), 84.8 (C-4′), 86.4 (C-1′), 88.2 (C), 101.4 (C), 114.2 (CH), 128.0 (CH), 128.8 (CH), 128.9 (CH), 131.0 (CH), 135.4 (C), 144.3 (C-6), 149.6 (C), 159.5 (C), 162.1 (C); HRMS: C31H26N5O5INa, calcd m/z 698.0876; found m/z 698.0871.
5.4. General procedure for deprotection
A solution of the protected nucleoside (0.3 mmol) in acetic acid (80%, 30 mL) is stirred at rt until completion (checked by TLC, typically 18 h). After evaporation of the solvent, the crude product was submitted to a flash silica gel column chromatography (eluent: hexanes/EtOAc, 2:8) to give the desired deprotected nucleoside.
5.4.1. 5-(1,2-Diiodo-vinyl)-1-(4-fluoro-5-hydroxymethyl-tetrahydrofuran-2-yl)-1H-pyrimidine-2,4-dione (10a)
Prepared from compound 9a, using the typical procedure described before to give 10a (51%) as a yellow solid. (c 1.0, CH3OH); mp 112 °C; UV (MeOH) λmax 232, 274 nm; 1H NMR (CD3OD) δ: 2.13–2.47 (m, 1H, H-2′a), 2.50–2.77 (m, 1H, H-2′b), 3.80 (d, J=2.8 Hz, 2H, H-5′), 4.32 (td, J=2.9, 27.0 Hz, 1H, H-4′), 5.30 (dd, J=4.6, 53.7 Hz, 1H, H-3′), 6.36 (dd, J=5.6, 9.0 Hz, 1H, H-1′), 7.53 (s, 1H, CH alkene), 8.28 (s, 1H, H-6); 13C NMR (CD3OD) δ: 40.3 (d, J=81.9 Hz, C-2′), 62.7 (d, J=43.3 Hz, C-5′), 87.0 (C-1′), 87.3 (d, J=96.3 Hz, C-4′), 87.3 (C), 88.1 (CH alkene), 96.0 (d, J=698.0 Hz, C-3′), 119.2 (C), 142.2 (C-6), 151.5 (C=O), 161.8 (C=O); HRMS: C11H11N2O4FI2Na, calcd m/z 530.8690; found m/z 530.8687.
5.4.2. 5-(1-Bromo-2-iodo-vinyl)-1-(4-fluoro-5-hydroxy-methyl-tetrahydrofuran-2-yl)-1H-pyrimidine-2,4-dione (10b)
Prepared from compound 9b, using the typical procedure described before to give 10b (52%) as a pale yellow solid. (c 0.6, CH3OH); mp 107 °C; UV (MeOH) λmax 296 nm; 1H NMR (CD3OD) δ: 2.14–2.47 (m, 1H, H-2′a), 2.52–2.78 (m, 1H, H-2′b), 3.80 (d, J=3.0 Hz, 2H, H-5′), 4.31 (td, J=2.9, 27.0 Hz, 1H, H-4′), 5.30 (dd, J=4.7, 53.7 Hz, 1H, H-3′), 6.36 (dd, J=5.6, 9.0 Hz, 1H, H-1′), 7.30 (s, 1H, CH alkene), 8.36 (s, 1H, H-6); 13C NMR (CD3OD) δ: 40.3 (d, C-2′), 62.7 (d, C-5′), 83.2 (CH alkene), 87.0 (C-1′), 87.3 (d, C-4′), 87.3 (C), 96.0 (d, C-3′), 115.5 (C), 116.0 (C), 143.9 (C-6), 151.5 (C=O), 161.8 (C=O); HRMS: C11H11N2O4FBrINa, calcd m/z 482.8829; found m/z 482.8838.
5.4.3. 5-(1-Chloro-2-iodo-vinyl)-1-(4-fluoro-5-hydroxy-methyl-tetrahydrofuran-2-yl)-1H-pyrimidine-2,4-dione (10c)
Prepared from compound 9c, using the typical procedure described before to give 10c (76%) as an orange solid. (c 0.7, CH3OH); mp 87 °C; UV (MeOH) λmax 274 nm; 1H NMR (CD3OD) δ: 2.13–2.48 (m, 1H, H-2′a), 2.52–2.75 (m, 1H, H-2′b), 3.80 (d, J = 2.8 Hz, 2H, H-5′), 4.30 (td, J=2.8, 26.8 Hz, 1H, H-4′), 5.30 (dd, J=4.6, 53.5 Hz, 1H, H-3′), 6.37 (dd, J=5.7, 8.8 Hz, 1H, H-1′), 7.06 (s, 1H, CH alkene), 8.38 (s, 1H, H-6); 13C NMR (CD3OD) δ: 40.2 (d, J=84.2 Hz, C-2′), 62.7 (d, J=43.3 Hz, C-5′), 81.1 (CH alkene), 87.0 (C-1′), 87.4 (d, J=93.9 Hz, C-4′), 96.2 (d, J=698.0 Hz, C-3′), 114.5 (C), 128.9 (C), 146.0 (C-6), 151.2 (C=O), 164.2 (C=O); HRMS: C11H11N2O4FClINa, calcd m/z 438.9334; found m/z 438.9340.
5.4.4. 1-(4-Fluoro-5-hydroxymethyl-tetrahydrofuran-2-yl)-5-iodoethynyl-1H-pyrimidine-2,4-dione (12)
Prepared from compound 11, using the typical procedure described before to give 12 (47%) as a yellow solid. (c 1.0, CH3OH); mp 140 °C; UV (MeOH) λmax 233, 298 nm; 1H NMR (CD3OD) δ: 2.11–2.45 (m, 1H, H-2′a), 2.46–2.75 (m, 1H, H-2′b), 3.79 (d, J=3.2 Hz, 2H, H-5′), 4.29 (td, J=2.9, 27.0 Hz, 1H, H-4′), 5.27 (dd, J=4.9, 53.9 Hz, 1H, H-3′), 6.28 (dd, J=5.5, 8.9 Hz, 1H, H-1′), 8.30 (s, 1H, H-6); 13C NMR (CD3OD) δ: 15.8 (CCI), 39.8 (d, J=81.8 Hz, C-2′), 62.5 (d, J=43.3 Hz, C-5′), 85.2 (C), 87.0 (C-1′), 87.3 (d, J=96.3 Hz, C-4′), 96.0 (d, J=700.4 Hz, C-3′), 101.5 (C), 146.0 (C-6), 151.1 (C=O), 164.4 (C=O); HRMS: C11H11N2O4FBrINa, calcd m/z 402.9567; found m/z 402.9571.
5.4.5. 5-Ethynyl-1-(4-fluoro-5-hydroxymethyl-tetrahydro-furan-2-yl)-1H-pyrimidine-2,4-dione (13)
Prepared from compound 8, using the typical procedure described before to give 13 (50%) as a pale yellow solid. (c 1.0, CH3OH); mp 209 °C; UV (MeOH) λmax 287 nm; 1H NMR (CD3OD) δ: 2.13–2.45 (m, 1H, H-2′a), 2.47–2.71 (m, 1H, H-2′b), 3.56 (s, 1H, H alkyne), 3.79 (d, J=3.2 Hz, 2H, H-5′), 4.30 (td, J=3.0, 27.0 Hz, 1H, H-4′), 5.27 (dd, J=4.9, 53.8 Hz, 1H, H-3′), 6.29 (dd, J=5.5, 8.9 Hz, 1H, H-1′), 8.38 (s, 1H, H-6); 13C NMR (CD3OD) δ: 39.8 (d, J=84.2 Hz, C-2′), 62.6 (d, J=43.3 Hz, C-5′), 75.9 (CH alkyne), 82.9 (C), 87.0 (C-1′), 87.4 (d, J=96.3 Hz, C-4′), 96.0 (d, J=698 Hz, C-3′), 100.0 (C), 146.0 (C-6), 151.2 (C=O), 164.2 (C=O); HRMS: C11H11FN2O4Na, calcd m/z 277.2071; found m/z 277.2065.
5.4.6. 1-{4-Azido-5-[(4-methoxy-phenyl)-diphenyl-methoxy-methyl]-tetrahydrofuran-2-yl}-5-ethynyl-1H-pyrimidine-2,4-dione (14)
A solution of 6 (100 mg, 0.16 mmol) and NaN3 (35 mg, 0.54 mmol) in DMF (5 mL) was refluxed overnight. After evaporation of volatiles, the crude product was submitted to a flash silica gel column chromatography (eluent: hexanes/ EtOAc, 2:8) to give 14 (61.0 mg, 70%) as a pale yellow solid. (c 0.8, CH3OH); mp 110 °C; 1H NMR (CDCl3) δ: 2.35–2.46 (m, 1H, H-2′a), 2.52–2.62 (m, 1H, H-2′b), 2.89 (s, 1H, CH alkyne), 3.37–3.52 (m, 2H, H-5′), 3.82 (s, 3H, OCH3), 4.00–4.04 (m, 1H, H-4′), 4.28–4.34 (m, 1H, H-3′), 6.15 (t, J=6.1 Hz, 1H, H-1′), 6.88 (d, J=8.9 Hz, 2H, MMTr), 7.21–7.51 (m, 14H, MMTr), 8.08 (s, =1H, H-6); 13C NMR (CDCl3) δ: 38.6 (C-2′), 55.3 (OCH3), 60.4 (C-3′), 62.7 (C-5′), 82.3 (CH alkyne), 84.0 (C-4′), 85.5 (C-1′), 87.6 (C), 99.4 (C), 113.6 (CH 2), 127 .3 (CH), 128.1 (CH), 128.2 (CH), 130.4 (CH), 134.7 (C), 143.7 (C), 149.2 (C), 158.9 (C), 161.6 (C); HRMS: C31H27N5O5Na, calcd m/z 572.5745; found m/z 572.5741.
5.4.7. 1-(4-Azido-5-hydroxymethyl-tetrahydrofuran-2-yl)-5-(1,2-diiodo-vinyl)-1H-pyrimidine-2,4-dione (16a)
Prepared from compound 15a using the typical procedure described before to give 16a (78%) as a pale yellow gum. (c 1.0, CH3OH); UV (MeOH) λmax 225, 284 nm; 1H NMR (DMSO-d6) δ: 2.34–2.43 (m, 2H, H-2′), 3.56–3.71 (m, 2H, H-5′), 3.85–3.90 (m, 1H, H-4′), 4.35–4.41 (m, 1H, H-3′), 5.26 (t, J=4.9 Hz, 1H, OH), 6.09 (t, J=6.3 Hz, 1H, H-1′), 7.61 (s, 1H, CH alkene), 8.06 (s, 1H, H-6′),=11.68 (s, 1H, NH); 13C NMR (DMSO-d6) δ: 37.5 (C-2′), 60.4 (C-3′), 61.4 (C-5′), 85.0 (C-1′, C-4′), 88.9 (C-I), 90.2 (CH alkene), 117.7 (C-5), 140.5 (C-6), 150.1 (C=O), 159.9 (C=O); HRMS: C11H11N5O4I2Na, calcd m/z 553.8798; found m/z 553.8803.
5.4.8. 1-(4-Azido-5-hydroxymethyl-tetrahydrofuran-2-yl)-5-(1-bromo-2-iodo-vinyl)-1H-pyrimidine-2,4-dione (16b)
Prepared from compound 15b using the typical procedure described before to give 16b (53%) as a pale yellow gum. (c 1.0, CH3OH); UV (MeOH) λmax 229, 276 nm; 1H NMR (DMSO-d6) δ: 2.36–2.43 (m, 2H, H-2′), 3.57–3.69 (m, 2H, H-5′), 3.86–3.91 (m, 1H, H-4′), 4.35–4.42 (m, 1H, H-3′), 5.28 (br s, 1H, OH), 6.09 (t, J=5.9 Hz, 1H, H-1′), 7.45 (s, 1H, CH alkene), 8.18 (s, 1H, H-6′), 11.7 (s, 1H, NH); 13C NMR (DMSO-d6) δ: 37.0 (C-2′), 60.0 (C-3′), 60.7 (C-5′), 84.6 (C-1′, C-4′), 85.6 (CH alkene), 113.9 (C), 114.2 (C), 141.9 (C-6), 149.6 (C=O), 159.4 (C=O); HRMS: C11H11N5O4BrINa, calcd m/z 505.8937; found m/z 505.8939.
5.4.9. 1-(4-Azido-5-hydroxymethyl-tetrahydrofuran-2-yl)-5-(1-chloro-2-iodo-vinyl)-1H-pyrimidine-2,4-dione (16c)
Prepared from compound 15c using the typical procedure described before to give 16c (57%) as a pale yellow gum. (c 1.0, CH3OH); UV (MeOH) λmax 214, 273 nm; 1H NMR (DMSO-d6) δ: 2.33–2.44 (m, 2H, H-2′), 3.33–3.71 (m, 1H, H-5′), 3.86–3.91 (m, 1H, H-4′), 4.35–4.42 (m, 1H, H-3′), 5.28 (t, J=4.9 Hz, 1H, OH), 6.09 (t, J=6.2 Hz, 1H, H-1′), 7.23 (s, 1H, CH alkene), 8.22 (s, 1H, H-6), 11.73 (s, 1H, NH); 13C NMR (DMSO-d6) δ: 37.7 (C-2′), 60.7 (C-3′), 61.3 (C-5′), 84.5 (CH), 85.2 (CH 2), 113.0 (C), 127.4 (C), 143.1 (C-6), 150.3 (C=O), 160.1 (C=O); C11H11N5O4ClINa, calcd m/z 461.9442; found m/z 461.9453.
5.4.10. 1-(4-Azido-5-hydroxymethyl-tetrahydrofuran-2-yl)-5-iodoethynyl-1H-pyrimidine-2,4-dione (18)
Prepared from compound 17 using the typical procedure described before to give 18 (60%) as a pale yellow gum. (c 1.0, CH3OH); UV (MeOH) λmax 236, 295 nm; 1H NMR (DMSO-d6) δ: 2.24–2.34 (m, 1H, H-2′a), 2.39–2.46 (m, 1H, H-2′b), 3.54–3.72 (m, 2H, H-5′), 3.79–3.84 (m, 1H, H-4′), 4.34–4.41 (m, 1H, H-3′), 5.30 (t, J=5.3 Hz, 1H, OH), 6.01 (t, J=6.0 Hz, 1H, H-1′), 8.22 (s, 1H, H-6), 11.66 (s, 1H, NH); 13C NMR (DMSO-d6) δ: 19.8 (C), 36.8 (C-2′), 59.2 (C-3′), 60.2 (C-5′), 84.3 (C-1′, C-4′), 84.8 (C), 99.0 (C), 144.6 (C-6), 149.3 (C=O), 161.7 (C=O); HRMS: C11H10N5O4INa, calcd m/z 425.9682; found m/z 425.9675.
5.4.11. 1-(4-Azido-5-hydroxymethyl-tetrahydrofuran-2-yl)-5-ethynyl-1H-pyrimidine-2,4-dione (19)
Prepared from compound 14 using the typical procedure described before to give 19 (65%) as a pale yellow gum. (c 1.0, CH3OH); UV (MeOH) λmax 220, 290 nm; 1H NMR (DMSO-d6) δ: 3.36–2.50 (m, 2H, H-2′), 3.53–3.62 (m, 2H, H-5′), 3.79–3.88 (m, 1H, H-4′), 4.32–4.43 (m, 1H, H-3′), 5.33 (t, J=4.7 Hz, 1H, OH), 6.01 (t, J=5.9 Hz, 1H, H-1′), 8.28 (s, 1H, H-6′), 11.65 (br s, 1H, NH); 13C NMR (DMSO-d6) δ: 36.9 (C-2′), 59.2 (C-3′), 60.2 (C-5′), 76.4 (CH alkyne), 83.6 (C), 84.5 (C-1′, C-4′), 97.6 (C), 144.5 (C-6), 149.4 (C=O), 161.7 (C=O); HRMS: C11H11N5O4Na, calcd m/z 300.2287; found m/z 300.2285.
Acknowledgements
The authors would like to acknowledge the University of Orléans, Région Centre, and the CNRS for funding. R.F.S. is supported by NIH grant 1RO37-AI-41980, the Emory University Center for AIDS and the Department of Veterans Affairs.
References and notes
- 1.For reviews on anti-HIV nucleosides, see:; (a) Vittori S; Dal Ben D; Lambertucci C; Marucci G; Volpini R; Cristalli G. Curr. Med. Chem 2006, 13, 3529–3552; [DOI] [PubMed] [Google Scholar]; (b) De Clercq E. Antiviral Res. 2007, 75, 1–13; [DOI] [PubMed] [Google Scholar]; (c) De Clercq E. Biochem. Pharmacol 2007, 73, 911–922; [DOI] [PubMed] [Google Scholar]; (d) Khandazhinskaya A; Yasko M; Shirokova E Curr. Med. Chem 2006, 13, 2953–2980; [DOI] [PubMed] [Google Scholar]; (e) Oberg B. Antiviral Res. 2006, 71, 90–95; [DOI] [PubMed] [Google Scholar]; (f) Mathé C; Gosselin G Antiviral Res. 2006, 71, 276–281; [DOI] [PubMed] [Google Scholar]; (g) Meanwell NA; Belema M; Carini DJ; D’Andrea SV; Kadow JF; Krystal M; Naidu BN; Regueiro-Ren A; Scola PM; Sit SY; Walker MA; Wang T; Yeung KS Curr. Drug Targets Infect. Disord 2005, 5, 307–400; [DOI] [PubMed] [Google Scholar]; (h) Agrofoglio LA; Nolan SP Curr. Top. Med. Chem 2005, 5, 1541–1558; [DOI] [PubMed] [Google Scholar]; (i) Agrofoglio LA; Suhas E; Farese A; Condom R; Challand SR; Earl RA; Guedj R Tetrahedron 1994, 50, 10611–10670; [Google Scholar]; (j) Agrofoglio LA; Challand SR Acyclic, Carbocyclic and l-Nucleosides; Kluwer Academic: Dordrecht, Boston, London, 1998; (English, 385 pp). [Google Scholar]
- 2.(a) Horwitz JP; Chua J; Noel M. J. Org. Chem 1964, 29, 2076–2078; [Google Scholar]; (b) Mitsuya H; Weinhold KJ; Furman PA; St Clair MH; Lehrman SN; Gallo RC; Bolognesi D; Barry DW; Broder S Proc. Natl. Acad. Sci. U.S.A 1985, 82, 7096–7100; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kong X-B; Zhu Q-Y; Vidal PM; Watanabe KA; Polsky B; Armstrong D; Ostrander M; Lang SA Jr.; Muchmore E; Chou T-C Antimicrob. Agents Chemother 1992, 36, 808–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Langen P; Etzold G; Hintsche R; Kowollik G Acta Biol. Med. Ger 1969, 23, 759–766; [PubMed] [Google Scholar]; (b) Matthes E; Lehmann C; Scholz D; Rosenthal HA; Langen P Biochem. Biophys. Res. Commun 1988, 153, 825–831; [DOI] [PubMed] [Google Scholar]; (c) Etzold G; Hintsche R; Kowollik G; Langen P Tetrahedron 1971, 27, 2463–2472; [Google Scholar]; (d) Hartmann HM; Vogt W; Durno AG; Hirsch MS; Hunsmann G; Eckstein F AIDS Res. Hum. Retroviruses 1988, 4, 457–466. [DOI] [PubMed] [Google Scholar]
- 4.(a) De Clercq E Clin. Microbiol. Rev 2001, 14, 382–397; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bradshaw TK; Hutchinson DW Chem. Soc. Rev 1977, 6, 43–62; [Google Scholar]; Kundu NG; Das B; Spears CP; Majumdar A; Kang S-I. J. Med. Chem 1990, 33, 1975–1979; [DOI] [PubMed] [Google Scholar]; (c) Kundu NG; Dasgupta SK; Chaudhuri LN; Mahanty JS; Spears CP; Shahinian AH Eur. J. Med. Chem 1993, 28, 473–479; [Google Scholar]; (d) Kundu NG; Mahanty JS; Spears CP Bioorg. Med. Chem. Lett 1996, 6, 1497–1502; [Google Scholar]; (e) Kumar R; Nath M; Tyrrell J. J. Med. Chem 2002, 45, 2032–2040; [DOI] [PubMed] [Google Scholar]; (f) De Clercq E; Descamps J; De Somer P; Barr PJ; Jones AS; Walker RT Proc. Natl. Acad. Sci. U.S.A 1979, 76, 2947–2951; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Agrofoglio LA; Gillaizeau I; Saito Y Chem. Rev 2003, 103, 1875–1916. [DOI] [PubMed] [Google Scholar]
- 5.Asakura J-I; Robins MJ J. Org. Chem 1990, 55, 4928–4933. [Google Scholar]
- 6.For reviews on Pd(0) cross-coupling reactions, see:; (a) Sonogashira K; Tohda Y; Hagihara N. Tetrahedron Lett. 1975, 16, 4467–4770; [Google Scholar]; (b) Sonogashira K Comprehensive Organic Synthesis; Pergamon: New York, NY, 1991; p 521; [Google Scholar]; (c) Takahashi S; Kuroyama Y; Sonogashira K; Hagihara N Synthesis 1980, 627–630; [Google Scholar]; (d) Agrofoglio LA Curr. Org. Chem 2006, 10, 333–362; [Google Scholar]; (e) See Ref 4g.
- 7.(a) Middleton WJ J. Org. Chem 1975, 40, 574–578; [Google Scholar]; (b) Markovskij LN; Pashinnik VE; Kirsanov AV Synthesis 1973, 787–789. [Google Scholar]
- 8.von Janta-Lipinski M; Costisella B; Ochs H; Hübscher U; Hafkemeyer P; Matthes E. J. Med. Chem 1998, 41, 2040–2046. [DOI] [PubMed] [Google Scholar]
- 9.Bellina F; Colzi F; Mannina L; Rossi R; Viel S. J. Org. Chem 2003, 68, 10175–10177. [DOI] [PubMed] [Google Scholar]
- 10.Heasly VL; Buczala DM; Chappell AE; Hill DJ; Whisenand JM; Shellhamer DF J. Org. Chem 2002, 67, 2183–2187. [DOI] [PubMed] [Google Scholar]
- 11.Friebolin H Basic One- and Two-Dimensional NMR Spectroscopy; VCH: New York, NY, 1991; p 56. [Google Scholar]
- 12.Matsuda A; Watanabe KA; Fox JJ J. Org. Chem 1980, 45, 3274–3278. [Google Scholar]
- 13.Schinazi RF; Sommadossi J-P; Saalmann V; Cannon DL; Xie M-W; Hart GC; Smith GA; Hahn EF Antimicrob. Agents Chemother 1990, 34, 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stuyver LJ; Lostia S; Adams M; Mathew J; Pai BS; Grier J; Thamish P; Choi Y; Chong Y; Choo H; Chu CK; Otto MJ; Schinazi RF Antimicrob. Agents Chemother 2002, 46, 3854–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aucagne V; Escuret V; Zoulim F; Durantel D; Trepo C; Agrofoglio LA; Joubert N; Amblard F EP 2004–356207 (CAN 145:63122), December 24, 2004. [Google Scholar]