

Abstract
Each day, the retina converts an immense number of photons into chemical signals that are then transported to higher order neural centers for interpretation. This process of photo transduction requires large quantities of cellular energy and anabolic precursors, making the retina one of the most metabolically active tissues in the body. With such a large metabolic demand, the retina is understandably sensitive to perturbations in perfusion and hypoxia. Indeed, retinal ischemia underlies many prevalent retinal disorders including diabetic retinopathy (DR), retinal vein occlusion (RVO), and retinopathy of prematurity (ROP). Retinal ischemia leads to the expression of growth factors, cytokines, and other cellular mediators which promote inflammation, vascular dysfunction, and ultimately, vision loss. This review aims to highlight the most recent and compelling findings that have advanced our understanding of the molecular mechanisms underlying retinal ischemias.
Metabolism and retinal ischemia
An important cellular response to ischemia is activation of hypoxia inducible factors (HIFs). While stabilization of HIFs is most often attributed to reduced intracellular oxygen concentrations, HIF activation is also highly dependent upon the metabolic status of the cell. In both PHD2-mediated and FIH-1-mediated hydroxylation of HIFs, one oxygen atom from O2 is inserted into the respective amino acid residue while the remaining oxygen atom is bound to the citric acid cycle (TCA) intermediate α-ketoglutarate [1,2]. When metabolism is compromised and α-ketoglutarate levels are low, PDH2 and FIH-1 hydroxylation is inhibited, promoting HIF stabilization [3]. HIF activation is also highly dependent upon the presence of increased reactive oxygen species (ROS) generated by mitochondrial respiration [4], and in the absence of mitochondrial-derived ROS, hypoxia-induced HIF stabilization may be blunted [5]. Metabolism and redox homeostasis are tightly balanced in the normal state but may become misaligned in response to ischemia such that perturbations in metabolism result in increased ROS [6]. Hence, HIF activation is tightly coupled to the metabolic status of the cell (Figure 1).
Figure 1.
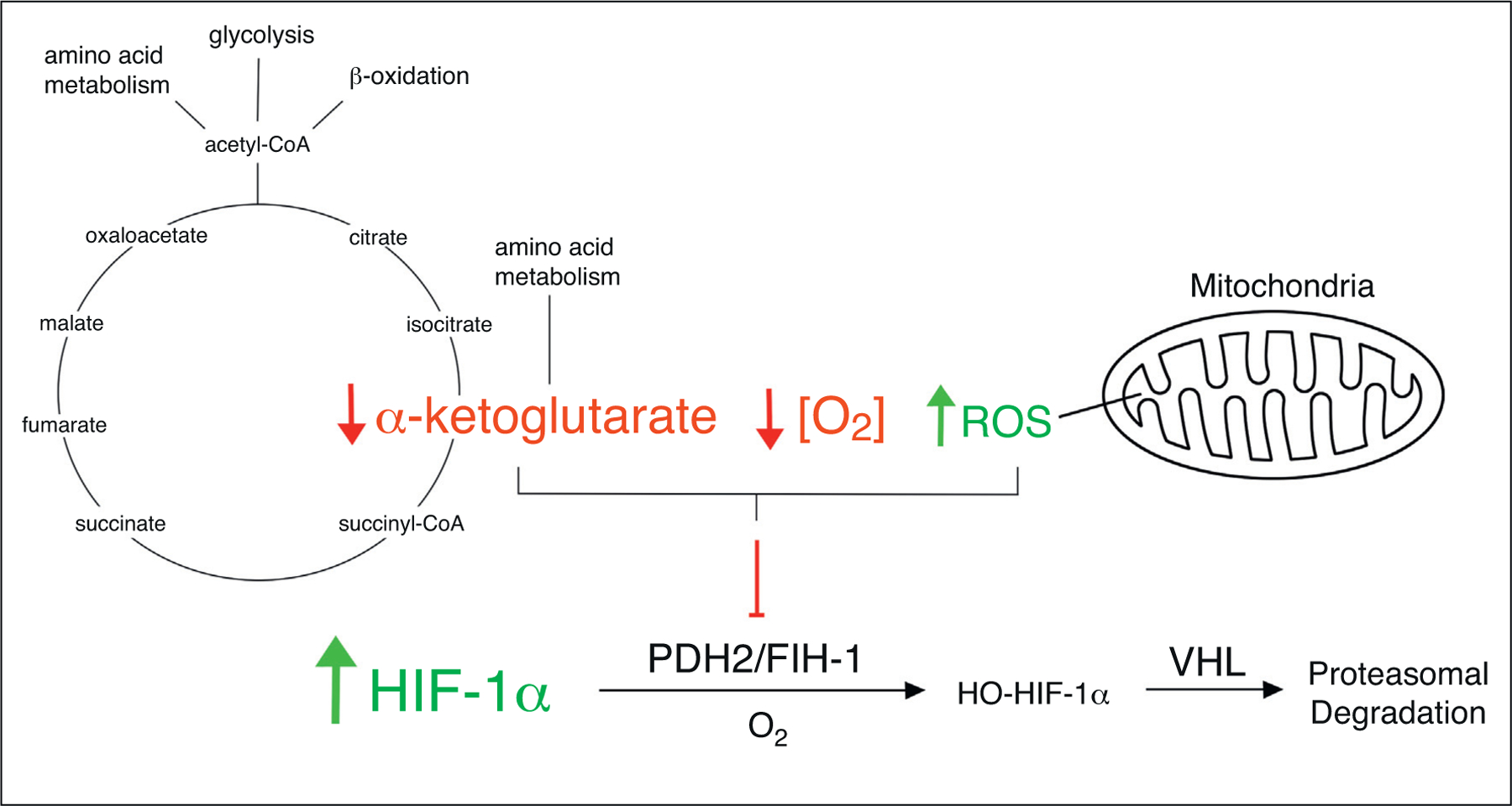
Molecular mechanisms regulating HIF activation. All metazoans share an evolutionarily conserved heterodimer system known as hypoxia-inducible factor 1 (HIF-1) which indirectly senses intracellular oxygen concentrations and acts as a transcription factor to initiate a cellular response to hypoxia. HIF-1 consists of two subunits, HIF-1α and HIF-1β, both of which are constitutively expressed at the transcriptional and translational levels. Regulation of the HIF-1 heterodimer system primarily occurs through post-translational modifications of HIF-1α which is hydroxylated by the prolyl hydroxylase PHD2 and the asparaginyl hydroxylase FIH-1. Hydroxylated HIF-1α binds von Hippel-Lindau (VHL) which recruits ubiquitin ligases and targets HIF-1α for proteasomal degradation. PHD2 and FIH-1-mediated hydroxylation of HIF-1α utilizes O2 as a substrate and is thus limited by the availability of oxygen, providing a real-time sensor of intracellular O2 concentrations. During hypoxia, HIF-1α hydroxylation is inhibited, preventing its degradation and allowing heterodimerization with HIF-1β. This unit directly interacts with DNA at hypoxia response elements (HREs) and induces the transcription of a diverse set of genes including those involved in angiogenesis, metabolism, and inflammation [7]. In PHD2 and FIH-1-mediated hydroxylation of HIF-1α, one oxygen atom is inserted into HIF-1α while the remaining oxygen atom is inserted into α-ketoglutarate, splitting it into CO2 and succinate. Thus, independent of hypoxia, reduced α-ketoglutarate levels can act as a limiting reagent in the hydroxylation of HIF-1α, leading to its activation. In addition, increased mitochondrial-derived ROS can inhibit hydroxylation of HIF-1α through an unknown mechanism, leading to its activation.
The retina is one of the most metabolically active tissues in the body [8,9], and until recently, it was assumed that the retina satisfied its vast metabolic needs through oxidative glycolysis. However, Smith et al. recently demonstrated that photoreceptors rely heavily on β-oxidation for ATP production [10●●]. Remarkably, 87% of glucose in ex vivo mouse retinas was converted to lactate rather than being used for oxidative phosphorylation, suggesting that aerobic glycolysis may occur in the retina. This conclusion is further supported by two recent studies which showed that photoreceptors rely on aerobic glycolysis to accumulate anabolic precursors for outer segment biogenesis [11] and that aerobic glycolysis in photoreceptors is important for adaptation to metabolic stress [12]. Moreover, Joyal et al. demonstrated coupled regulation of lipid and glucose metabolism in photoreceptors and showed that simultaneous impairment of lipid and glucose metabolism in the photoreceptors of very-low-density lipoprotein receptor knockout mice (Vldlr−/−) resulted in decreased α-ketogluterate, HIF-1α stabilization, and subsequent formation of neovascularization in the deep capillary plexus (Figure 2a). These vascular lesions highly resemble those seen in age-related macular degeneration (AMD) patients with retinal angiomatous proliferation (RAP) and suggest that HIF-1α stabilization in photoreceptors may be the underlying mechanism in this AMD subpopulation. Interestingly, mice that lack the lipid metabolism regulator PPARα develop retinal degeneration and vascular dysfunction [13]. These mice demonstrate impaired oxidative phosphorylation of lipids, diminished mitochondrial function, and compensatory increases in oxidative glycolysis, further underscoring the importance of β-oxidation in the retina. In patients, two large clinical trials have confirmed that fenofibrates, PPARα agonists, significantly reduce the progression of DR [14,15], and animal studies using novel PPARα agonists have shown decreased vascular dysfunction in diabetic rats [16] and reduced vascular lesion size in mice with laser-induced choroidal neovascularization (CNV) [17].
Figure 2.
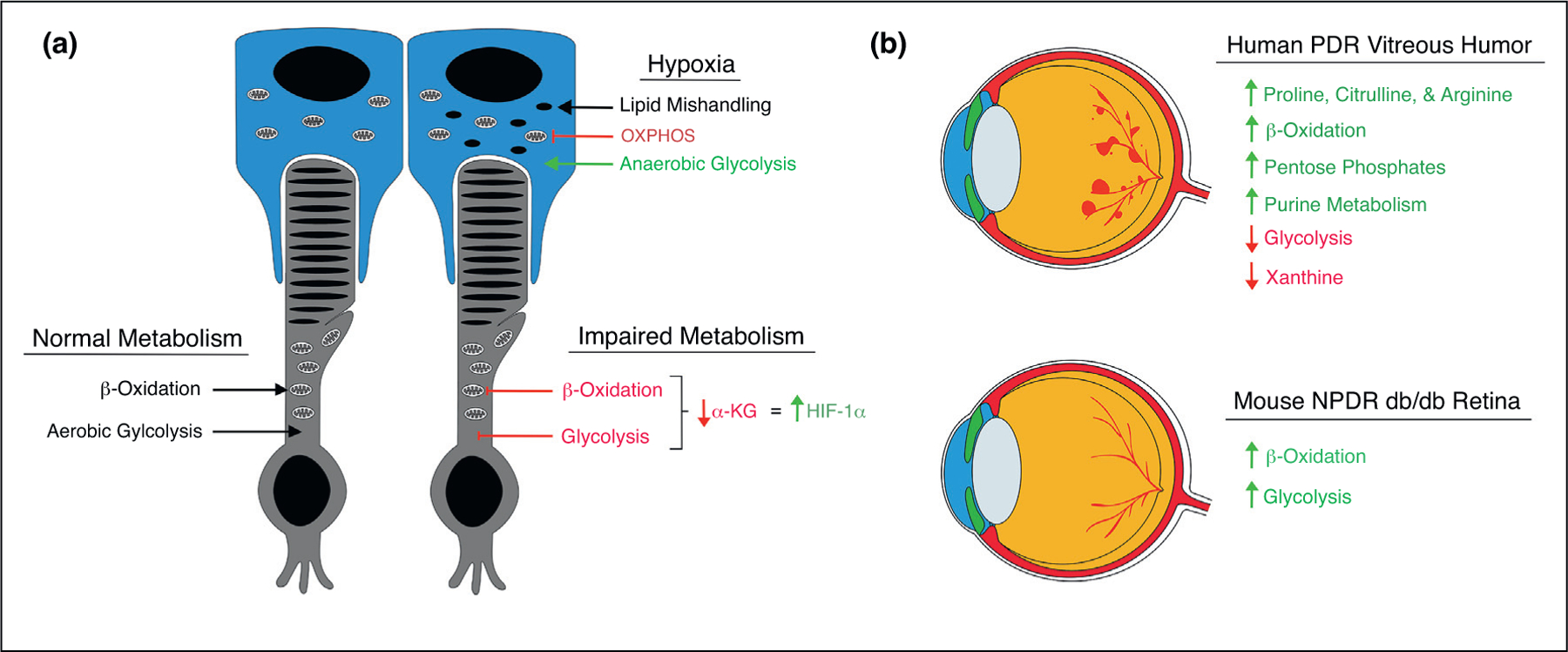
Novel insights into the metabolic alterations in retinal ischemias. (a) In the normal state, photoreceptors (depicted here in grey) rely on aerobic glycolysis for synthesis of anabolic precursors and β-oxidation for production of ATP. When both β-oxidation and glycolysis are impaired, as in Vldlr−/− mice, α-ketogluterate levels are reduced leading to activation of HIF-1α and subsequent neovascularization. In pseudohypoxic RPE cells (depicted here in blue), β-oxidation regulatory proteins are reduced and long-chain acylcarnitines aggregate, suggesting impaired lipid handling. In addition, TCA cycle enzymes are downregulated and glycolytic enzymes are increased, suggesting a shift to anaerobic glycolysis. (b) Metabolomic studies on vitreous humor from PDR patients (depicted here as upper eye) revealed several metabolic alterations including increased proline, citrulline, arginine, acylcarnitines, pentose phosphates, and purine intermediates, and decreased glycolytic intermediates and xanthine. Metabolomic analysis of retinas from diabetic db/db mice (depicted here as lower eye) revealed increased acylcarnitines and glycolytic metabolites.
In another recent report, Friedlander et al. demonstrated that genetically induced pseudohypoxia in retinal pigmented epithelial (RPE) cells, via conditional VHL knockdown, resulted in rapid metabolic stress in RPE cells and led to photoreceptor degeneration [18●]. In these mice, accumulation of glycogen and lipid droplets in RPE cells were observed after only three days post-induction of VHL deletion. Interestingly, gene profiling experiments demonstrated downregulation of fatty acid metabolism regulatory genes while mass spectrometry revealed increased levels of several acylcarnitines, suggesting impaired lipid handling in pseudohypoxic RPE. Lastly, Kurihara et al. demonstrated that pseudohypoxic RPE upregulates glycolytic regulatory enzymes while simultaneously downregulating those involved in the TCA cycle, seemingly shifting to anaerobic glycolysis (Figure 2a). Interestingly, the pathological changes observed in pseudohypoxic RPE cells were dependent upon HIF-2α but not HIF-1α. However, HIF-1α inhibition in rodent models of laser-induced choroidal neovascularization (CNV), using a small molecule inhibitor [19] and RPE-specific genetic ablation [20], significantly reduced vascular lesion size, demonstrating the complex context-dependent effects of HIFs in hypoxic RPE.
Other studies have supported the notion of a metabolic shift in response to retinal ischemia. In a metabolomics study on vitreous humor from patients with proliferative diabetic retinopathy (PDR), proline, citrulline, arginine, and β-oxidation products were found to be significantly increased [21] (Figure 2b). Eyes from mice with oxygen-induced retinopathy (OIR) showed similar increases in metabolites, supporting the notion that OIR mice recapitulate aspects of PDR. In a more recent metabolomics study on PDR vitreous, researchers found a downregulation of glycolytic intermediates and an upregulation of β-oxidation, purine metabolism, and pentose phosphate pathway (PPP) intermediates [22]. In addition, the group found increased levels of citrulline and proline and decreased levels of xanthine (Figure 2b). Similarly, pentose phosphates were found to be elevated in the plasma of patients with DR and may be suggestive of a cellular response to oxidative stress because the PPP is the main source of NADPH [23]. In a metabolomics study in rodents, the retinas from diabetic db/db mice showed significant increases in glycolytic metabolites and long-chain acylcarnitines, suggesting an increase in glycolysis and β-oxidation in this model of nonproliferative diabetic retinopathy (NPDR) [24]. Interestingly, Vancura et al. found that transcripts of β-oxidation regulatory proteins in mouse whole retina and isolated photoreceptors follow a dopamine-dependent circadian cycle and that this transcript pattern is misaligned in diabetic db/db mice [25]. Consistent with this observation, recent reports have identified HIF-1α as a regulator of circadian clock expression during pathological hypoxia [26,27].
Painting a cohesive picture of the metabolic alterations observed in retinal ischemia is a challenging proposition. The retina is composed of many different cell types, and each has a unique context-dependent response to hypoxia and energy deprivation; this creates a mosaic when viewed as a whole. For example, endothelial cells (ECs) rely upon increased aerobic glycolysis for EC rearrangement during vessel sprouting [28] but decreased glycolytic intermediates were observed in PDR vitreous, suggesting significant contributions from other cell types. Moreover, the setting in which retinal ischemia occurs is different for each disease, further complicating our ability to draw broad conclusions. For example, diabetic retinopathy occurs in the backdrop of hyperglycemia and hyperlipidemia and unraveling the coordinated metabolic responses to these additional variables is difficult. While the study of metabolomics in retinal ischemia is in its infancy, a clear trend is that β-oxidation appears to be critically important in both normal and pathological physiology of the retina. Further studies are needed to determine how and if metabolic alterations play a causal role in the progression of ischemic retinopathies.
Inflammation and retinal ischemia
The retina, like its parent organ system the CNS, was historically viewed as an immunologically privileged tissue because of its inability to mount an adaptive immune response. The retina maintains a highly controlled blood-retina barrier (BRB) which uses tight junctions to limit the extravasation of leukocytes and leakage of intravascular fluid [29]. However, the notion of immunological privilege in the retina has been challenged by accumulating evidence demonstrating the central role of inflammation in ischemic retinopathies.
In a novel mouse model of DR where pericyte coverage of retinal vessels is reduced by treatment with anti-PDGFRβ, Uemura et al. observed irreversible BRB breakdown secondary to sustained leukocyte infiltration [30●]. The group found a >10-fold increase in retinal macrophages which persisted throughout the life of the animal and were associated with growth factor release. Remarkably, macrophage depletion using clodronate liposomes dramatically suppressed retinal edema and restored vessel integrity. Thus, in DR patients with pericyte-free vessels, infiltrating macrophages may exacerbate BRB dysfunction via paracrine growth factor release (Figure 3a). However, in laser-induced CNV mice, macrophage deletion has shown inconsistent results. Local partial depletion of CD11b+ macrophages and microglia via drug-induced knockdown was not sufficient to reduce vascular lesion size [31], while splenic-denervated and splenectomized mice resulted in decreased infiltrating macrophages and concomitant reductions in vascular lesion size [32].
Figure 3.
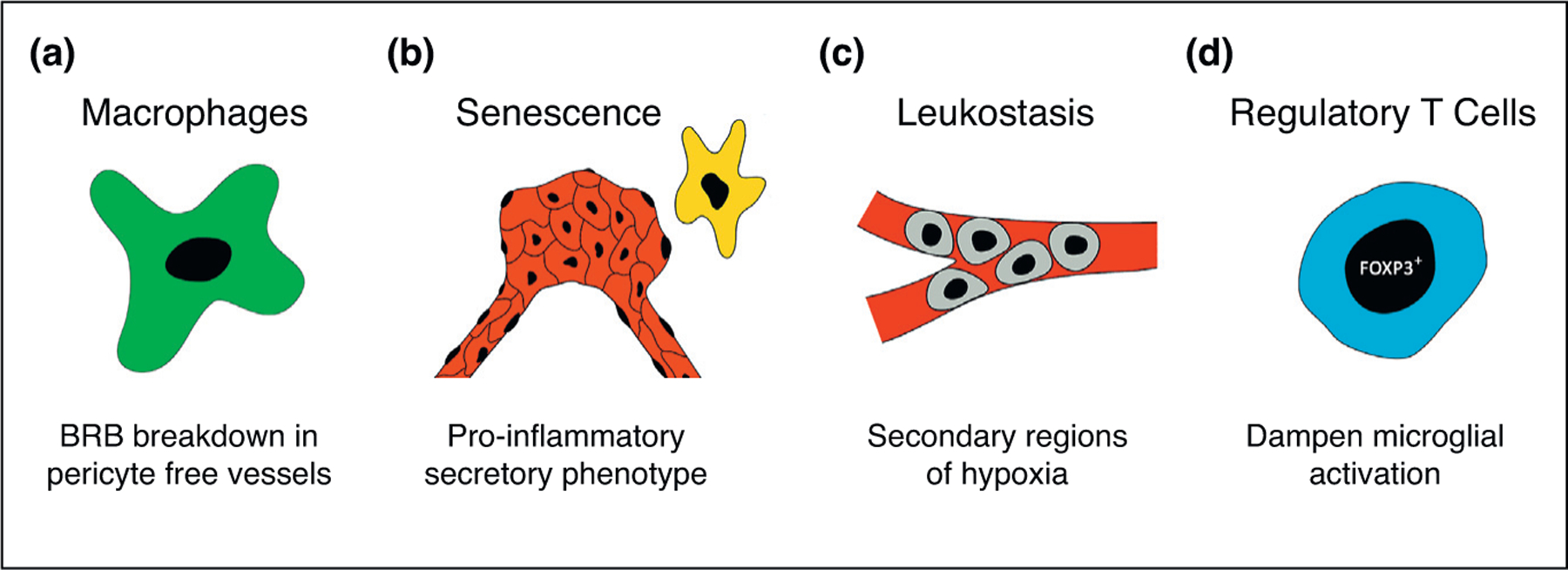
Novel insights into the inflammatory responses to retinal ischemias. (a) CD45hi CD11b+ macrophages are increased in mouse retinas with pericyte free vessels and promote vascular dysfunction through paracrine growth factor release. (b) Neovascular endothelial cells and microglia enter a senescent-like state in hypoxic mouse retinas and develop a pro-inflammatory secretory phenotype. (c) Leukostasis-mediated vascular plugging occurs in response to increased retinal VEGF and results in secondary regions of retinal hypoxia. (d) FOXP3+ Tregs improve vascular dysfunction in hypoxic mouse retinas by decreasing microglial activation via direct cell contact.
In another recent report, Sapieha et al. demonstrated that hypoxic retinal cells enter senescence and take on a pro-inflammatory secretory phenotype [33●●]. In OIR mice, the group observed a dynamically evolving pattern of cellular senescence that commenced in the ganglion cell layer (GCL) of avascular regions and propagated to ECs of neovascular tufts and microglia (Figure 3b). Senescent cells demonstrated semaphorin 3A (SEMA3A) dependent expression of several pro-inflammatory mediators, and a similar panel of cytokines was found to be elevated in the vitreous of PDR patients. Likewise, SEMA3A was found to be elevated in the plasma of NPDR and PDR patients and compared between the two, was significantly higher in the PDR group [34]. Interestingly, in OIR mice, administration of metformin significantly reduced senescence, cytokine secretion, and total neovascular area. This report suggests a potential model in which the ischemic neural retina enters a senescence-like state as a form of protection from hypoxia-associated cell death but with the unintended consequence of promoting inflammatory factors that spread senescence to microglia and endothelial cells, ultimately leading to vascular dysfunction. Subsequent reports have demonstrated premature senescence of retinal ECs in microaneurysms of elderly patients [35], ganglion cells of mice with IOP-elevation-induced ischemia [36], and retinal capillaries of streptozotocin-induced type 1 diabetic mice [37]. Further studies are needed to fully understand the different stimuli that trigger and mediate senescence in the retina.
In patients with RVO, clinicians observed that treatment with anti-VEGF agents promoted reperfusion of previously occluded retinal vessels [38]. Campochiaro et al. helped to elucidate the mechanism underlying this observation by showing that increased retinal secretion of VEGF stimulated marked leukocyte trafficking to the retina [39●●]. These leukocytes physically plugged the capillary lumen and led to secondary regions of hypoxia in the downstream tissue (Figure 3c). Moreover, reducing retinal VEGF levels by either therapeutic or genetic means was sufficient to restore perfusion in previously occluded vessels similar to what was observed in RVO patients treated with anti-VEGF injections. Thus, a single ischemic event in the retina can lead to a cycle of increasing hypoxia through VEGF-dependent leukostasis. Interestingly, leukostasis was similarly observed in the retinal vessels of streptozotocin-induced type 1 diabetic mice and was abolished with viral overexpression of angiotensin-converting enzyme (ACE)-2 [40]. Leukostasis is likely triggered by a number of different stimuli, and further research is needed to elucidate the mechanisms.
While FOXP3+ T-regulatory (Treg) cells are known to modulate inflammation in a variety of tissues [41], their role in the ischemic retina has remained largely unexplored. In a paradigm-shifting report, Wilkinson-Berka et al. displayed the beneficial properties of FOXP3+ Treg cells in OIR eyes by demonstrating their ability to inhibit the formation of pathological neovessels and dampen microglial activation [42●●]. The group showed that FOXP3+ Tregs are able to penetrate the BRB and function to reduce microglial activation through direct cell contact, leading to reduced expression of pro-inflammatory mediators (Figure 3d). Moreover, interventions that increased FOXP3+ Tregs significantly reduced vasobliteration and neovascularization and decreased VEGF expression. These findings support the notion that FOXP3+ Tregs are an endogenous cellular mechanism by which retinal inflammation can be controlled. Similar findings were reported in OIR mice lacking toll-like receptor 2 and 4 which mediates the balance between IL-17A-expressing Th17 cells and FOXP3+ Tregs [43]. The retinas of these mice had significantly increased FOXP3+ Treg cells and displayed decreased vasobliteration, neovascularization, microglial activation, and pro-inflammatory cytokines. Interestingly, in zebrafish, FOXP3+ Tregs are necessary for retinal regeneration and function to secrete regenerative factors and dampen excessive inflammation [44]. Thus, treatments that increase FOXP3+ Tregs may be beneficial in patients with ischemic retinopathies.
Retinal inflammation is now understood to be a central player in the progression of ischemic retinopathies. Indeed, intraocular treatment with corticosteroids is known to slow the development of PDR [45] and abrogate the expression of vasoactive proteins in DR patients [46]. Compelling cases have been put forth that implicate both innate and adaptive immune cells in the inflammatory response to retinal ischemia. However, a lack of animal models that fully recapitulate all aspects of the human disease has prevented a unified conclusion regarding the causal cell types and factors involved. Further research is needed to fully understand the molecular mechanisms underlying inflammation in retinal ischemias.
Vascular dysfunction and retinal ischemia
The retinal vasculature is unique in that it is readily visualized in living animals and isolated tissue. As such, the retina has served as an invaluable tool for studying EC biology. A well characterized consequence of retinal ischemia is aberrant angiogenesis referred to as neovascularization [47]. Neovessels lack proper cell adhesion proteins, leading to leakage of intravascular fluid and subsequent vision loss, if left untreated [48]. Vascular dysfunction is central to ischemic retinopathies, and recent studies have expanded our knowledge of the molecular mechanisms underlying its pathogenesis.
Neovessels are composed of highly proliferative ECs, but the exact population of cells giving rise to neovessels has remained elusive. Recent work by Dimmeler et al. has helped to elucidate the underlying mechanism of neovascular proliferation by showing that the majority of neovessels (≤69%) arise due to clonal expansion of previously quiescent ECs [49●●]. Using Confetti mice, which tags mature ECs with either GFP, YFP, or RFP following inducible cre-recombinase expression, the authors demonstrated that normal retinal vasculature is lined with a heterogeneous assortment of ECs. However, in OIR mice, neovessels were characterized by homogenous cell populations, suggesting that neovascularization is largely due to clonal expansion of formerly quiescent ECs (Figure 4a). Moreover, the group found that clonally expanded ECs demonstrate hypoxia and TGFβ-dependent expression of gene products involved in endothelial-to-mesenchymal transition (EndMT), suggesting that clonal expansion of ECs may involve EndMT. In agreement, expression of microRNA-20a in human umbilical vein ECs was shown to inhibit EndMT through repression of canonical TGFβ signaling [50]. Moreover, expression of the EndMT transcription factor SNAI1 was found to be increased in the vascular lesions of OIR and CNV mice [51]. Interestingly, epiretinal membranes from PDR patients demonstrated colocalization of the endothelial marker CD31 and the fibroblastic marker S100A4, providing evidence that EndMT occurs in patients [52]. Taken together, these findings suggest that retinal neovascularization may be due to EndMT-mediated clonal expansion of ECs, and that suppression of EndMT mediators may be of therapeutic value in patients with ischemic retinopathies.
Figure 4.
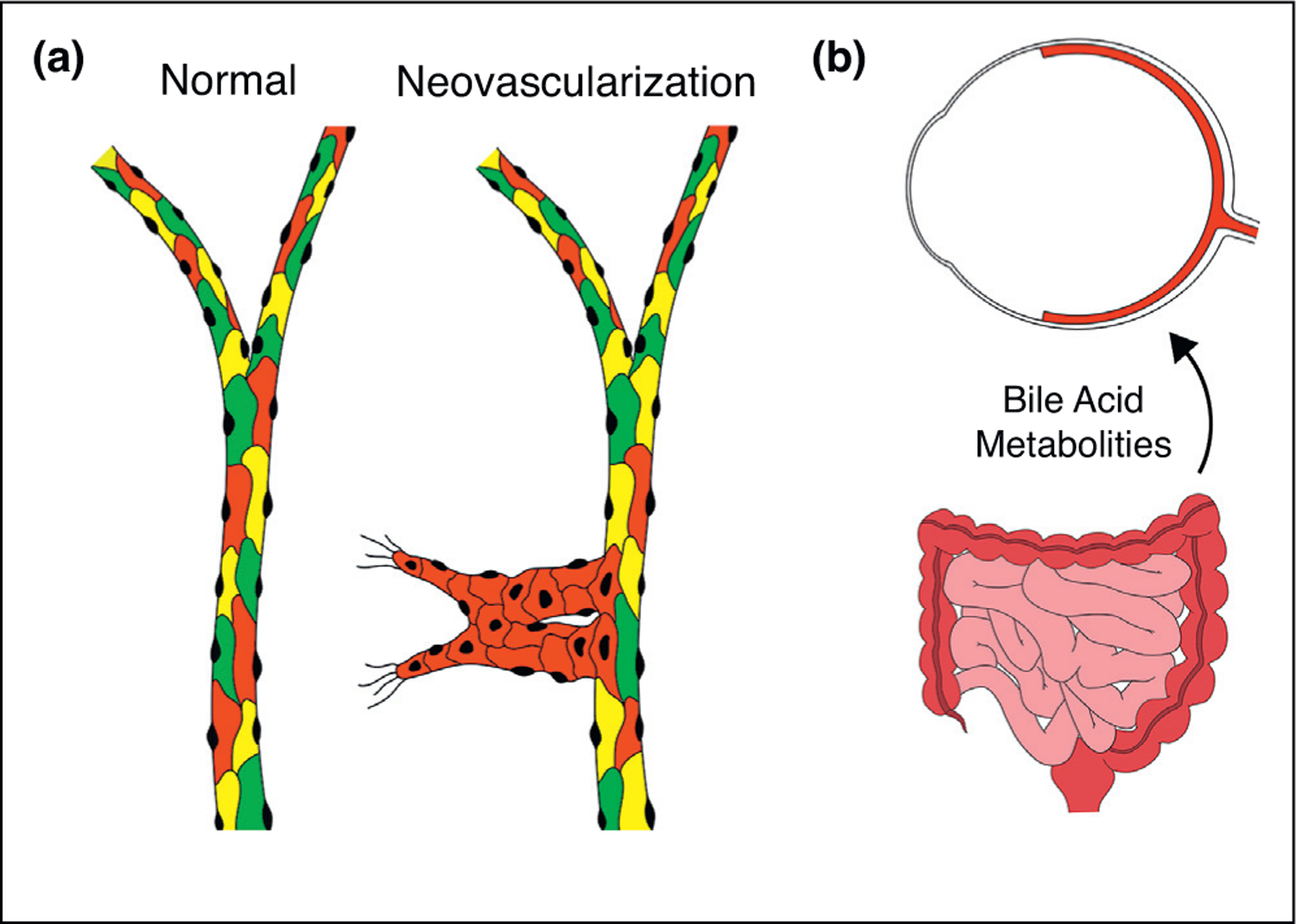
Novel insights into vascular dysfunction in retinal ischemias. (a) Normal retinal vasculature is composed of a heterogeneous assortment of ECs while neovascularization is homogenous and composed of clonally expanded ECs originating from a previously quiescent EC. (b) Changes in gut flora, induced by intermittent fasting, alters BA metabolism, leading to increased TUDCA. TUDCA travels via the blood to the neural retina where it signals through its cognate receptor and protects the retinal vasculature.
In another recent report, Beli et al. identified a novel function of the retina-gut axis by showing that shifts in the gut microbiome attenuate retinal vascular dysfunction in diabetic mice through altered bile acid (BA) metabolism [53●]. In two separate mouse models of diabetic retinopathy, intermittent fasting (IF) was shown to alter the bacterial populations of the gut microbiome and decrease numbers of acellular retinal capillaries. The authors found increased circulating levels of the BA metabolite tauroursodeoxycholate (TUDCA) and showed that ganglion cells express TGR-5, the cognate TUDCA receptor. Transcripts of TNF-α, the negatively-regulated downstream target of TGR-5 activation, were significantly decreased in the retinas of IF treated mice and resulted in decreased leukocyte infiltration. Importantly, the protective effects of IF were replicated with pharmacological activation of TGR5, confirming the beneficial effects of altered BA metabolism on the retinal vasculature of diabetic mice (Figure 4b).
In wet AMD patients, gut metagenome sequencing revealed altered bacterial populations compared to non-AMD controls [54]. Interestingly, in mice fed a high-fat diet, CNV vascular lesions were found to be significantly larger, and this increase was abrogated by fecal transplant from control mice, suggesting complicity of the gut flora [55]. Thus, alterations in the retina-gut axis accentuate vascular dysfunction in rodents and suggest that dietary restrictions may be of therapeutic value in patients with retinal ischemias. Further studies are needed to elucidate the molecular signals that govern the retina-gut axis.
Conclusion
The retina is particularly susceptible to ischemia given its immense metabolic needs. The molecular responses to retinal ischemia are multifaceted, and include pathways involved in metabolism, inflammation, and vascular homeostasis, among others. While these pathways are designed to protect the retina from hypoxia and nutrient deprivation, they often lead to exudation, pathological vessel growth, and vision loss. A current focus in the field is to determine which of these molecular responses are causative versus consequential, so that clinical interventions may be developed that effectively prevent disease progression. The discovery of VEGF as a central mediator in the progression of retinal ischemias and the subsequent development of anti-VEGF agents has preserved vision in millions of patients [56,57]. Even with the remarkable benefits of anti-VEGF agents, some patients still experience disease progression, highlighting the need for new treatments that target ischemic retinopathies from different perspectives[58]. The recent studies highlighted in this review represent significant advances in understanding the molecular mechanisms responsible for retinal ischemia and may be influential in the development of novel treatments.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
● of special interest
●● of outstanding interest
- 1.Epstein Andrew CR, Gleadle Jonathan M, McNeill Luke A, Hewitson Kirsty S, O’Rourke John, Mole David R, Mukherji Mridul et al. : C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107:43–54 10.1016/S0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 2.Kaelin WG: Cancer and altered metabolism: potential importance of hypoxia-inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harbor Symp Quant Biol 2011, 76:335–345 10.1101/sqb.2011.76.010975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W et al. : Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1. Science 2009, 324:261–265 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tormos Kathryn V, Chandel Navdeep S: Inter-connection between mitochondria and HIFs. J Cell Mol Med 2010, 14:795–804 10.1111/j.1582-4934.2010.01031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamanaka Robert B, Weinberg Samuel E, Reczek Colleen R, Chandel Navdeep S: The mitochondrial respiratory chain is required for organismal adaptation to hypoxia. Cell Rep 2016, 15:451–459 10.1016/j.celrep.2016.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Celia Quijano, Madia Trujillo, Laura Castro, Andrés Trostchansky: Interplay between oxidant species and energy metabolism. Redox Biol 2016, 8:28–42 10.1016/j.redox.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenza Gregg L: Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148:399–408 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Margaret Wong-Riley: Energy metabolism of the visual system. Eye Brain 2010, 99 10.2147/EB.S9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keshav Kooragayala, Norimoto Gotoh, Tiziana Cogliati, Jacob Nellissery, Kaden Talia R, French Stephanie, Robert Balaban, Wei Li, Raul Covian, Anand Swaroop: Quantification of oxygen consumption in retina ex vivo demonstrates limited reserve capacity of photoreceptor mitochondria. Invest Opthalmol Vis Sci 2015, 56:8428 10.1167/iovs.15-17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.●●.Sébastien Joyal Jean, Sun Ye, Gantner Marin L, Shao Zhuo, Evans Lucy P, Saba Nicholas, Fredrick Thomas et al. : Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat Med 2016, 22:439–445 10.1038/nm.4059.This work demonstrates that photoreceptors rely on β-oxidation for ATP production and that impaired metabolism in photoreceptors leads to HIF-1α activation and subsequent development of neovascularization.
- 11.Yashodhan Chinchore, Tedi Begaj, David Wu, Eugene Drokhlyansky, Cepko Constance L: Glycolytic reliance promotes anabolism in photoreceptors. eLife 2017, 6:1–22 10.7554/eLife.25946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lolita Petit, Shan Ma, Joris Cipi, Cheng Shun-Yun, Zieger Marina, Nissim Hay, Claudio Punzo: Aerobic glycolysis is essential for normal rod function and controls secondary cone death in retinitis pigmentosa. Cell Rep 2018, 23:2629–2642 10.1016/j.celrep.2018.04.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearsall Elizabeth A, Cheng Rui, Zhou Kelu, Takahashi Yusuke, Greg Matlock H, Vadvalkar Shraddha S, Shin Younghwa et al. : PPARα is essential for retinal lipid metabolism and neuronal survival. BMC Biol 2017, 15 10.1186/s12915-017-0451-x. [DOI] [PMC free article] [PubMed]
- 14.Keech AC, Mitchell P, Summanen PA, O’Day J, Davis TME, Moffitt MS, Taskinen M-R et al. : Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (field study): a randomised controlled trial. Lancet 2007, 370:1687–1697 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- 15.The ACCORD Study Group and ACCORD Eye Study Group: Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med 2010, 363:233–244 10.1056/NEJMoa1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guotao Deng, Moran Elizabeth P,Cheng Rui, Greg Matlock, Kelu Zhou, David Moran, Danyang Chen, Qiang Yu, Jian-Xing Ma: Therapeutic effects of a novel agonist of peroxisome proliferator-activated receptor alpha for the treatment of diabetic retinopathy. Invest Opthalmol Vis Sci 2017, 58:5030 10.1167/iovs.16-21402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fangfang Qiu, Greg Matlock, Qian Chen, Kelu Zhou, Yanhong Du, Xiang Wang, Jian-Xing Ma : Therapeutic effects of PPARα agonist on ocular neovascularization in models recapitulating neovascular age-related macular degeneration. Invest Opthalmol Vis Sci 2017, 58:5065 10.1167/iovs.17-22091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.●.Kurihara Toshihide, Westenskow Peter D, Gantner Marin L, Usui Yoshihiko, Schultz Andrew, Bravo Stephen, Aguilar Edith et al. : Hypoxia-induced metabolic stress in retinal pigment epithelial cells is sufficient to induce photoreceptor degeneration. eLife 2016, 5:1–22 10.7554/eLife.14319.This work demonstrates that pseudohypoxia in RPE leads to the development of metabolic stress that includes impaired lipid handling and a shift towards anaerobic glycolysis.
- 19.Mingbing Zeng, Jikui Shen, Yuanyuan Liu, Lucy Yang Lu, Ding Kun, Fortmann Seth D, Khan Mahmood et al. : The HIF-1 antagonist acriflavine: visualization in retina and suppression of ocular neovascularization. J Mol Med 2017, 95:417–429 10.1007/s00109-016-1498-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taeyoung Koo, Wook Park Sung, Hyun Jo Dong, Daesik Kim, Hyoung Kim Jin, Yeon Cho Hee, Jeungeun Kim, JeongHun Kim, Soo Kim Jin: CRISPR-LbCpf1 prevents choroidal neovascularization in a mouse model of age-related macular degeneration. Nat Commun 2018, 9 10.1038/s41467-018-04175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paris Liliana P, Johnson Caroline H, Aguilar Edith, Usui Yoshihiko, Cho Kevin, Hoang Lihn T, Feitelberg Daniel et al. : Global metabolomics reveals metabolic dysregulation in ischemic retinopathy. Metabolomics 2016, 12:1–10 10.1007/s11306-015-0877-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haines Nathan R, Manoharan Niranjan, Olson Jeffrey L, D’Alessandro Angelo, Reisz Julie A: Metabolomics analysis of human vitreous in diabetic retinopathy and rhegmatogenous retinal detachment. J Proteome Res 2018, 17:2421–2427 10.1021/acs.jproteome.8b00169. [DOI] [PubMed] [Google Scholar]
- 23.Liyan Chen, Cheng Ching Yu, Choi Hyungwon, Kamran Ikram Mohammad, Sabanayagam Charumathi, Tan Gavin SW, Tian Dechao et al. : Plasma metabonomic profiling of diabetic retinopathy. Diabetes 2016, 65:1099–1108 10.2337/db15-0661. [DOI] [PubMed] [Google Scholar]
- 24.Sas Kelli M, Kayampilly Pradeep, Byun Jaeman, Nair Viji, Hinder Lucy M, Hur Junguk, Zhang Hongyu et al. : Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1 10.1172/jci.insight.86976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patrick Vancura, Tanja Wolloscheck, Kenkichi Baba, Gianluca Tosini, Michael Iuvone P, Spessert Rainer: Circadian and dopaminergic regulation of fatty acid oxidation pathway genes in retina and photoreceptor cells. PLoS One 2016, 11:1–17 10.1371/journal.pone.0164665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walton Zandra E, Patel Chirag H, Brooks Rebekah C, Yongjun Yu, Ibrahim-Hashim Arig, Riddle Malini, Porcu Alessandra et al. : Acid suspends the circadian clock in hypoxia through inhibition of MTOR. Cell 2018, 174:72–87.e32 10.1016/j.cell.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yaling Wu, Dingbin Tang, Na Liu, Wei Xiong, Huanwei Huang, Yang Li, Zhixiong Ma et al. : Reciprocal regulation between the circadian clock and hypoxia signaling at the genome level in mammals. Cell Metab 2017, 25:73–85 10.1016/j.cmet.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Bert Cruys, Wong Brian W, Kuchnio Anna, Verdegem Dries, Rita Cantelmo Anna, Conradi Lena-Christin, Vandekeere Saar et al. : Glycolytic regulation of cell rearrangement in angiogenesis. Nat Commun 2016, 7:12240 10.1038/ncomms12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaur C, Foulds W, Ling E: Blood–retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retinal Eye Res 2008, 27:622–647 10.1016/j.preteyeres.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 30.●.Ogura Shuntaro, Kurata Kaori, Hattori Yuki, Takase Hiroshi, Ishiguro-Oonuma Toshina, Hwang Yoonha, Ahn Soyeon et al. : Sustained inflammation after pericyte depletion induces irreversible blood-retina barrier breakdown. JCI Insight 2017, 2:1–22 10.1172/jci.insight.90905.This work presents a novel DR mouse model with pericyte-free retinal vessels and demonstrates that sustained infiltration of macrophages promotes vascular dysfunction through growth factor release.
- 31.Brockmann Claudia, Kociok Norbert, Dege Sabrina, Davids Anja Maria, Brockmann Tobias, Miller Kelly R, Joussen Antonia M: Local partial depletion of CD11b+cells and their influence on choroidal neovascularization using the CD11b-HSVTK mouse model. Acta Ophthalmol 2018:1–8 10.1111/aos.13716. [DOI] [PubMed]
- 32.Xue Tan, Katsuhito Fujiu, Ichiro Manabe, Junko Nishida, Reiko Yamagishi, Yuya Terashima, Kouji Matsushima, Toshikatsu Kaburaki, Ryozo Nagai, Yasuo Yanagi: Choroidal neovascularization is inhibited in splenic-denervated or splenectomized mice with a concomitant decrease in intraocular macrophage. PLoS One 2016, 11:1–22 10.1371/journal.pone.0160985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.●●.Oubaha Malika, Miloudi Khalil, Dejda Agnieszka, Guber Vera, Mawambo Gaëlle, Germain Marie Anne, Bourdel Guillaume et al. : Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med 2016, 8 10.1126/scitranslmed.aaf9440.This work demonstrates that hypoxic neural retina develops a dynamically evolving pattern of senescence that spreads to neovessels and microglia and promotes a pro-inflammatory phenotype.
- 34.Kwon Soo Hyun, Shin JaePil, Taek Kim, Park Dong Ho: Association of plasma semaphorin 3a with phenotypes of diabetic retinopathy and nephropathy. Invest Ophthalmol Vis Sci 2016, 57:2983–2989 10.1167/iovs.16-19468. [DOI] [PubMed] [Google Scholar]
- 35.Mariana López-Luppo, Joana Catita, David Ramos, Marc Navarro, Ana Carretero, Luísa Mendes-Jorge, Pura Muñoz-Cánoves, Alfonso Rodriguez-Baeza, Victor Nacher, Jesus Ruberte: Cellular senescence is associated with human retinal microaneurysm formation during aging. Invest Ophthalmol Vis Sci 2017, 58:2832–2842 10.1167/iovs.16-20312. [DOI] [PubMed] [Google Scholar]
- 36.Li LU, Zhao Yin, Zhang Hong: P16INK4a upregulation mediated by TBK1 induces retinal ganglion cell senescence in ischemic injury. Cell Death Dis 2017, 8:1–12 10.1038/cddis.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Modesto Rojas, Tahira Lemtalsi, Haroldo Toque, Zhimin Xu, David Fulton, Robert Caldwell, Ruth Caldwell: NOX2-induced activation of arginase and diabetes-induced retinal endothelial cell senescence. Antioxidants 2017, 6:43 10.3390/antiox6020043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mir Tahreem A, Kherani Saleema, Hafiz Gulnar, Scott Adrienne W, Zimmer-Galler Ingrid, Wenick Adam S, Solomon Sharon et al. : Changes in retinal nonperfusion associated with suppression of vascular endothelial growth factor in retinal vein occlusion. Ophthalmology 2016, 123:625–634.e1 10.1016/j.ophtha.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.●●.Liu Yuanyuan, Shen Jikui, Fortmann Seth D, Wang Jiangxia, Vestweber Dietmar, Campochiaro Peter A: Reversible retinal vessel closure from VEGF-induced leukocyte plugging. JCI Insight 2017, 2 10.1172/jci.insight.95530.This work demonstrates that increased retinal VEGF recruits peripheral leukocytes, leadings to leukostasis and secondary regions of retinal hypoxia.
- 40.Dominguez James M, Hu Ping, Caballero Sergio, Moldovan Leni, Verma Amrisha, Oudit Gavin Y, Li Qiuhong, Grant Maria B: Adeno-associated virus overexpression of angiotensin-converting enzyme-2 reverses diabetic retinopathy in type 1 diabetes in mice. Am J Pathol 2016, 186:1688–1700 10.1016/j.ajpath.2016.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ling Lu, Joseph Barbi, Fan Pan: The regulation of immune tolerance by FOXP3. Nat Rev Immunol 2017, 17:703–717 10.1038/nri.2017.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.●●.Deliyanti Devy, Talia Dean M, Zhu Tong, Maxwell Mhairi J, Agrotis Alex, Jerome Jack R, Hargreaves Emily M et al. : Foxp3+ tregs are recruited to the retina to repair pathological angiogenesis. Nat Commun 2017, 8:1–12 10.1038/s41467-017-00751-w.This work demonstrates that FOXP3+ T-regulatory cells dampen microglial activation and inhibit retinal vascular dysfunction.
- 43.Chang He, Peilong Lai, Jing Wang, Tian Zhou, Zijing Huang, Lingli Zhou, Xialin Liu: TLR2/4 deficiency prevents oxygen-induced vascular degeneration and promotes revascularization by downregulating IL-17 in the retina. Sci Rep 2016, 6 10.1038/srep27739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hui Subhra P, Delicia Z, Sheng Kotaro, Sugimoto Alvaro, Gonzalez-Rajal Shinichi Nakagawa, Hesselson Daniel, Kikuchi Kazu: Zebrafish regulatory T cells mediate organ-specific regenerative programs. Dev Cell 2017, 43:659–672.e5 10.1016/j.devcel.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 45.Wykoff Charles C, Chakravarthy Usha, Campochiaro Peter A, Bailey Clare, Green Ken, Cunha-Vaz Jose: Long-term effects of intravitreal 0.19 Mg fluocinolone acetonide implant on progression and regression of diabetic retinopathy. Ophthalmology 2017, 124:440–449 10.1016/j.ophtha.2016.11.034. [DOI] [PubMed] [Google Scholar]
- 46.Campochiaro Peter A, Hafiz Gulnar, Mir Tahreem A, Scott Adrienne W, Zimmer-Galler Ingrid, Shah Syed M, Wenick Adam S et al. : Pro-permeability factors in diabetic macular edema; the diabetic macular edema treated with ozurdex trial. Am J Ophthalmol 2016, 168:13–23 10.1016/j.ajo.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campochiaro Peter A: Molecular pathogenesis of retinal and choroidal vascular diseases. Progr Retin Eye Res 2015, 49:67–81 10.1016/j.preteyeres.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campochiaro Peter A: Ocular neovascularization. J Mol Med 2013, 91:311–321 10.1007/s00109-013-0993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.●●.Manavski Yosif, Lucas Tina, Glaser Simone F, Dorsheimer Lena, Günther Stefan, Braun Thomas, Rieger Michael A, Zeiher Andreas M, Boon Reinier A, Dimmeler Stefanie: Clonal expansion of endothelial cells contributes to ischemia-induced neovascularizationnovelty and significance. Circ Res 2018, 122:670–677 10.1161/CIRCRESAHA.117.312310.This work demonstrates that retinal neovascularization is largely the result of clonal expansion of previously quiescent ECs and that clonal expansion of ECs involves hypoxia and TGFβ-dependent endothelial-to-mesenchymal transition.
- 50.Correia Ana CP, Moonen Jan-Renier, Brinker Marja G, Krenning Guido: FGF2 inhibits endothelial–mesenchymal transition through microRNA-20a-mediated repression of canonical TGF-03B2 signaling. J Cell Sci 2016, 129:569–579 10.1242/jcs.176248. [DOI] [PubMed] [Google Scholar]
- 51.Sun Jia-Xing, Chang Tian-Fang, Li Man-Hong, Sun Li-Juan, Yan Xian-Chun, Yang Zi-Yan, Yuan Liu et al. : SNAI1, an endothelial–mesenchymal transition transcription factor, promotes the early phase of ocular neovascularization. Angiogenesis 2018, 21:635–652 10.1007/s10456-018-9614-9. [DOI] [PubMed] [Google Scholar]
- 52.William Chang, Michelle Lajko, Amani A Fawzi: Endothelin-1 is associated with fibrosis in proliferative diabetic retinopathy membranes. Edited by Ljubimov Alexander V. PLoS One 2018, 13:e0191285 10.1371/journal.pone.0191285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.●.Beli Eleni, Yan Yuanqing, Moldovan Leni, Vieira Cristiano P, Gao Ruli, Duan Yaqian, Prasad Ram et al. : Restructuring of the gut microbiome by intermittent fasting prevents retinopathy and prolongs survival in Db/Db mice. Diabetes 2018. 10.2337/db18-0158.db180158.This work demonstrates that the retina-gut axis is capable of abrogating retinal vascular dysfunction through a protective bile acid metabolite pathway.
- 54.Zinkernagel Martin S, Zysset-Burri Denise C, Keller Irene, Berger Lieselotte E, Leichtle Alexander B, Largiadér Carlo R, Fiedler Georg M, Wolf Sebastian: Association of the intestinal microbiome with the development of neovascular age-related macular degeneration. Sci Rep 2017, 7 10.1038/srep40826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andriessen Elisabeth M, Wilson Ariel M, Mawambo Gaelle, Dejda Agnieszka, Miloudi Khalil, Sennlaub Florian, Sapieha Przemyslaw: Gut microbiota influences pathological angiogenesis in obesity-driven choroidal neovascularization. EMBO Mol Med 2016, 8:1366–1379 10.15252/emmm.201606531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campochiaro Peter A, Aiello Lloyd Paul, Rosenfeld Philip J: Anti-vascular endothelial growth factor agents in the treatment of retinal disease. Ophthalmology 2016, 123:S78–88 10.1016/j.ophtha.2016.04.056. [DOI] [PubMed] [Google Scholar]
- 57.Ferrara Napoleone, Adamis Anthony P: Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov 2016, 15:385–403 10.1038/nrd.2015.17. [DOI] [PubMed] [Google Scholar]
- 58.Nguyen Quan Dong, Brown David M, Marcus Dennis M, Boyer David S, Patel Sunil, Feiner Leonard, Gibson Andrea et al. : Ranibizumab for diabetic macular edema. Ophthalmology 2012, 119:789–801 10.1016/j.ophtha.2011.12.039. [DOI] [PubMed] [Google Scholar]