

Abstract
The SARS-CoV-2 coronavirus is an enveloped, positive-sense single-stranded RNA virus that is responsible for the COVID-19 pandemic. The spike is a class I viral fusion glycoprotein that extends from the viral surface and is responsible for viral entry into the host cell, and is the primary target of neutralizing antibodies. The receptor binding domain (RBD) of the spike samples multiple conformations in a compromise between evading immune recognition and searching for the host-cell surface receptor. Using atomistic simulations of the glycosylated wild-type spike in the closed and 1-up RBD conformations, we map the free energy landscape for RBD opening and identify interactions in an allosteric pocket that influence RBD dynamics. The results provide an explanation for experimental observation of increased antibody binding for a clinical variant with a substitution in this pocket. Our results also suggest the possibility of allosteric targeting of the RBD equilibrium to favor open states via binding of small molecules to the hinge pocket. In addition to potential value as experimental probes to quantify RBD conformational heterogeneity, small molecules that modulate the RBD equilibrium could help explore the relationship between RBD opening and S1 shedding.
Graphical Abstract
Introduction
The emergence of COVID-19 in late 2019 sparked a global pandemic, causing > 3 million deaths as of 20211, and crippling the international economy. The disease is caused by the SARS-CoV-2 coronavirus, a positive-sense single-stranded RNA virus that can cause respiratory distress, pneumonia, and death. The severity of the pandemic, coupled with the globe’s past history with coronavirus outbreaks, ignited a massive effort to develop effective therapeutics. A particularly promising target in the viral life cycle for therapeutic design is the spike glycoprotein, a class I membrane fusion protein2–5 that decorates the surface of the virus.6–7 The spike is the dominant antigen for immune response,8 and the goal of COVID-19 vaccines is to expose the human immune system to the spike prior to viral infection9–10.
The SARS-CoV-2 spike is a homotrimeric glycoprotein consisting of two subunits, S1 and S2 (Figure 1A), and is cleaved by host cell proteases at two distinct sites.2, 11–14 Both the S1 and S2 subunits are heavily decorated with glycans.15 The N-terminal S1 subunits sit atop the spike and are responsible for recognizing and binding the host cell receptor angiotensin converting enzyme 2 (ACE2) and stabilizing the S2 core.12, 14, 16–21 While the S1 subunit is responsible for receptor binding, the S2 subunit contains the fusion machinery of the spike.3 At some point after the S1 region binds to ACE2, the S1 subunits dissociate to expose the S2 core, which undergoes dramatic conformational changes to initiate membrane fusion.3, 22–24 Each S1 subunit consists of an N-terminal domain (NTD), a receptor binding domain (RBD), and two C-terminal domains (CTD1 and CTD2); the S1/S2 interface lies at the C-terminal end of CTD2 (Figure 1B–C).25–27
Figure 1.
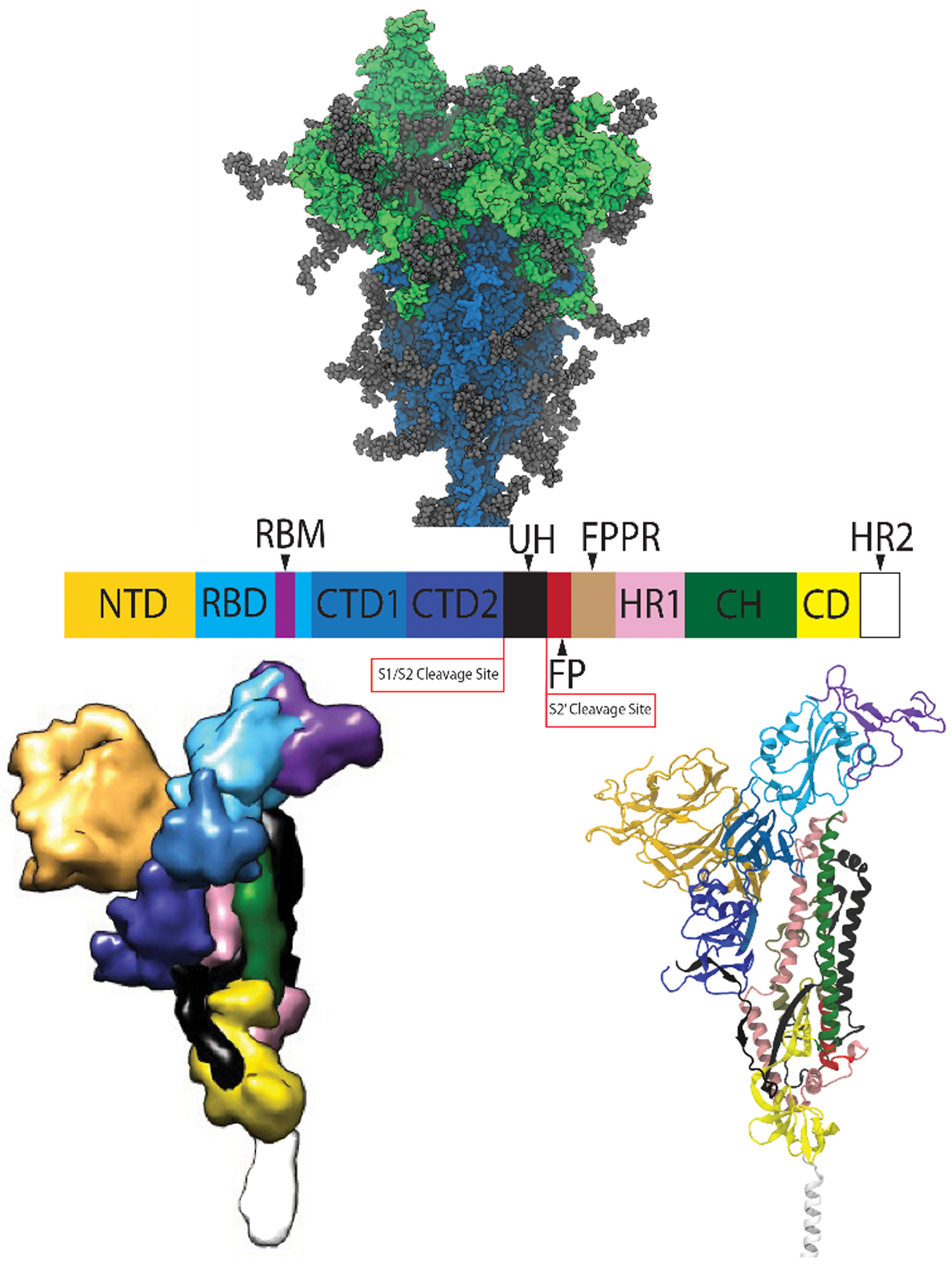
(A, upper) structure of the trimeric SARS-CoV-2 spike glycoprotein ectodomain fromMD, with the S1 subunit shown in green the S2 subunit shown in blue, and the glycans in gray. (B, middle) domain organization of the spike protomer along the protein sequence. (C, bottom) protomer structure, with colors matching the domain organization in the middle image (space-filling on the left, ribbon diagram on right).
In SARS-CoV-2, the RBD in the S1 subunit is responsible for recognizing and binding ACE2.17–21, 28 The RBD alternates between two distinct conformational states relative to the remainder of the spike: ‘open’ and ‘closed’ (Figure 2A,B).6, 26–27, 29 A two-stranded hinge region links the RBD and CTD1, and allows the RBD to shift between the two geometries.30 An open RBD is a prerequisite for ACE2 binding; in the closed state binding of ACE2 is precluded by a steric clash with the RBDs of other protomers.27, 29–31 Another key feature of the closed state is that the RBD is shielded by the extensive glycans decorating the surface; only upon opening of the RBD does the receptor binding motif RBM) protrude out of the glycan shield (Figure 3).32 This allows it to recognize and contact the ACE2 receptor (Figure 2C) -- but also makes the critical RBD residues vulnerable to neutralization by antibody binding.32–34
Figure 2.
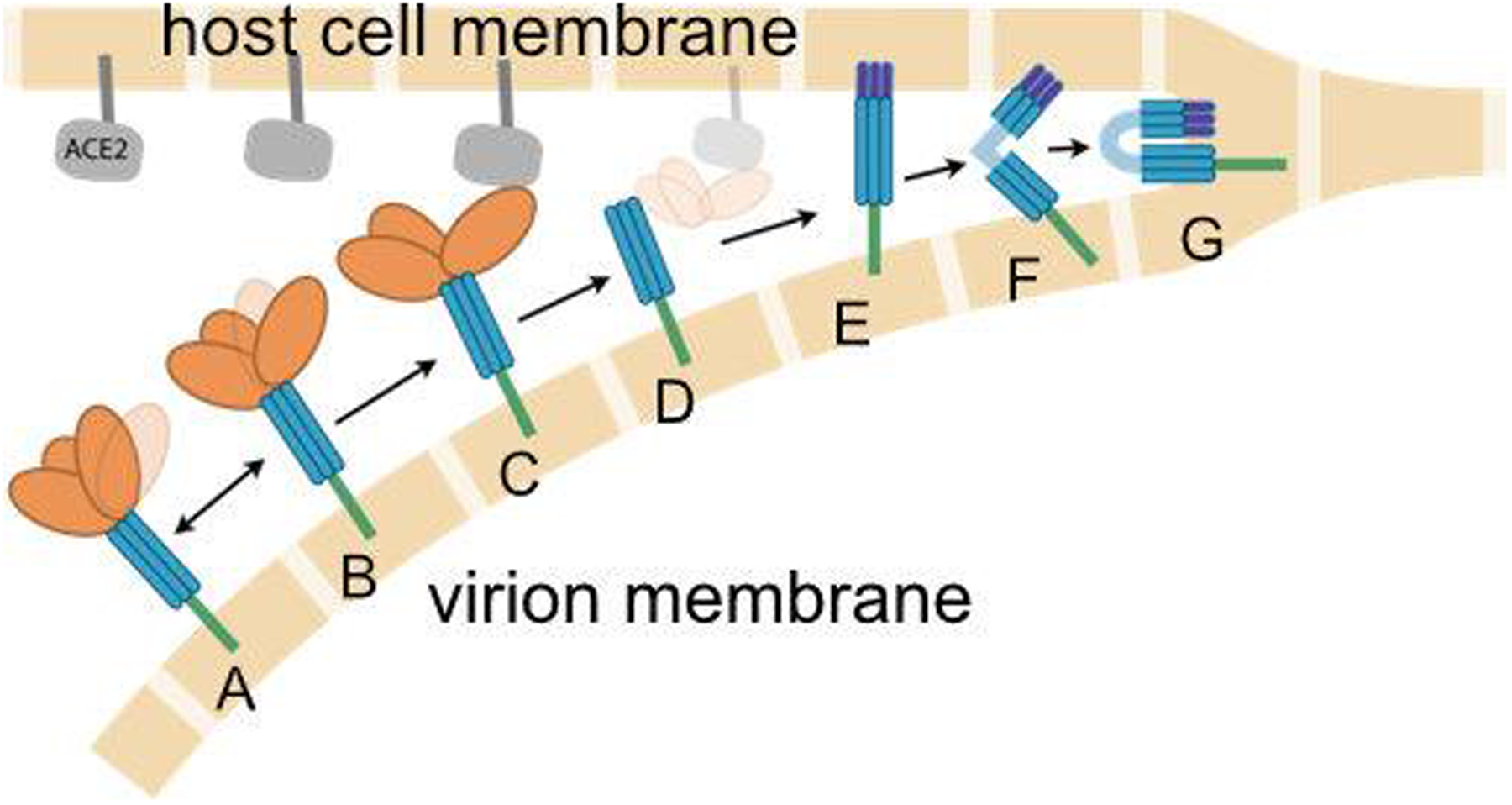
Cartoon illustration of the presumed role of the spike in fusion of the viral (lower beige blocks) and host cell (upper blocks) membranes. The RBD on the S1 subunit (orange) is attached to the S2 subunit (blue), and fluctuates between (A) closed and (B) open states. When the spike approaches the ACE2 receptor (gray), the open RBD is capable of binding to ACE2 (C), leading to shedding of the S1 subunit (D), insertion of fusion peptides into the host membrane (E), additional conformational changes to co-localize the membranes (F) and eventual membrane fusion (G). Double arrows indicate reversible dynamics, while single arrows indicate presumably irreversible events. Experimental structures for states D, E and F have not been reported. Image credit: Sarina Bromberg and Carlos Simmerling
Figure 3.
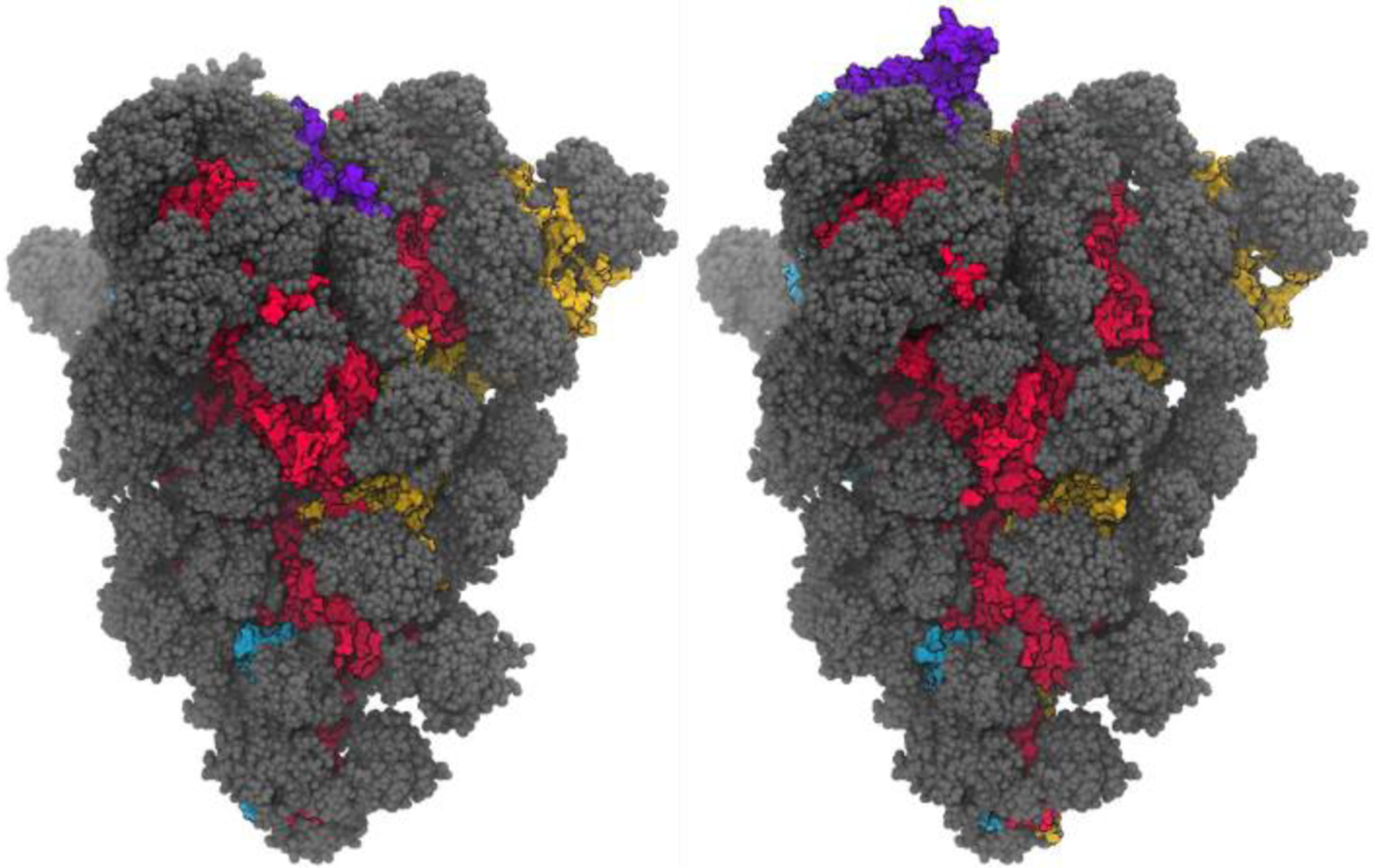
Structures (from MD simulations reported here) of the spike glycoprotein with all 3 RBD domains closed (left), and with a single RBD in the open position (right, 1-up). A single snapshot of the protein is shown in space-filling model, with red/yellow/blue for the 3 protomers, with the RBM region of the opening RBD shown in purple. Glycans are shown in gray, with multiple MD snapshots shown to indicate the region of the protein covered by the dynamic glycan shield.
Although the SARS and SARS-CoV-2 S proteins bind to the same host cell receptor using the same RBD region, the residues responsible for ACE2 recognition and binding are substantially different. The isolated RBD of the SARS-CoV-2 S protein binds ACE2 more strongly than that of SARS, but the complete S proteins bind with nearly the same affinity.14, 17, 19, 23, 35 SARS-CoV-2 S is thought to occupy the active, open RBD state less often than in the SARS S, but is compensated by the greater affinity of the RBD for ACE2.23 Since the RBD is largely obscured by the glycan shield in the closed state32, the shifted equilibrium allows the SARS-CoV-2 spike to spend less time in the vulnerable open conformation without sacrificing net affinity for ACE2.
Our focus here is on the dynamics of the RBD, and how it transitions from the hidden and binding-incompetent closed position, to the open conformation in which it can bind ACE2 but is also susceptible to immune surveillance. Locking the spike in either the open or closed conformation could potentially interfere with the viral entry into host cells. Sealing it in the closed state would eliminate its ability to bind ACE2, preventing infection. This approach has been demonstrated through introduction of disulfide bonds that lock the RBD closed, preventing ACE2 binding.36–39 Small molecules may also be able to reduce RBD opening for wild-type spike, as suggested by the identification of a linoleic acid binding pocket in the RBD of the SARS-CoV-2 spike.40–41 Conversely, substitutions that increase population of the open RBD39 may more frequently expose antibody epitopes, potentially facilitating a neutralizing immune response42–43.
Shifting the distribution to the open state also could weaken the S1-S2 interface and help to prematurely trigger S1 shedding, irreversibly neutralizing the spike6, 18, 23. Studies of the wild-type (WT) cleaved spike for both SARS-CoV and SARS-CoV-2, either isolated or on viral particles, observe detectable amounts of post-fusion S2 structures even in the absence of ACE2.23, 29–30, 44–45 The CR3022 antibody also has been shown to bind the pre-fusion spike and induce large conformational changes.46 These results suggest that the S1 subunit can spontaneously shed and prematurely trigger the irreversible S2 transition to the post-fusion state, though no reports have yet quantified the coupling of RBD dynamics to S1 shedding.
Rational design of small molecules that can penetrate the glycan shield and serve as probes or modulators of RBD positioning would be facilitated by a complete, atomically-detailed model of the RBD opening mechanism. Here, we use an array of computational methods to model a conformational transition pathway and free energy landscape between the all-closed and 1-up RBD states in the fully glycosylated, solvated spike models based on prior simulations32, cryo-EM26–27 and crystal21 structures, and mass spectroscopy experiments15. The data provide a detailed description of RBD opening, indicating specific interactions that may stabilize the closed, hidden state, and provide opportunities to control RBD positioning. The results are briefly summarized here.
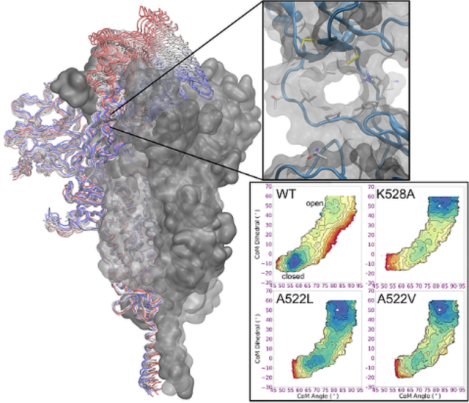
Analysis of the RBD transition profile identified formation of a transient pocket at the hinge region beneath the RBD (Figure 4). This pocket is present only in the open RBD state, and is formed by the bottom of the RBD, the top of the CTD1 domain, and the inner surface of the two protein strands connecting these domains. Importantly, the amino acids composing the hinge-pocket are well-conserved across other betacoronaviruses, despite substantial variations in the RBD and rest of S1. This is consistent with an important mechanistic role for the hinge region.
Figure 4.
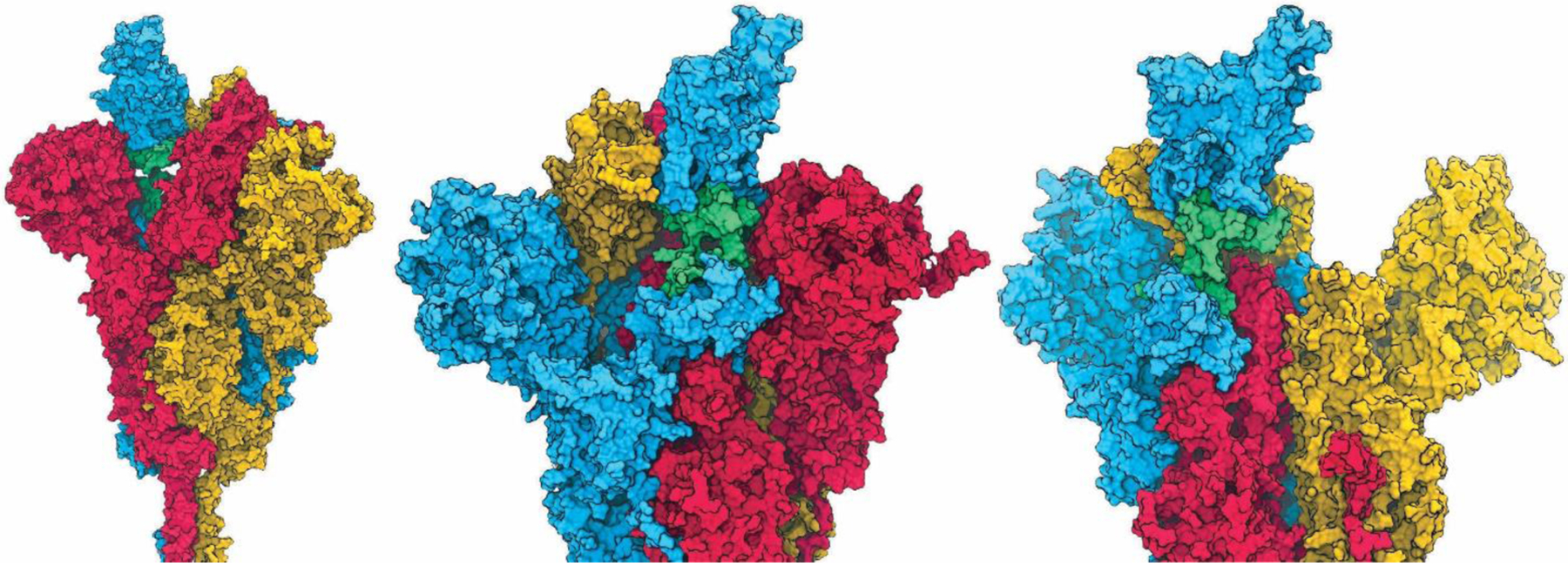
Space-filling model of the 1-up spike trimer, colored blue for protomer with open RBD, and red and yellow for the counterclockwise and clockwise protomers with closed RBD, respectively. Glycans and water are not shown. The RBD hinge region is shown in green. Three different views are shown: (left) pocket accessibility through gap left by opened RBD; (middle) narrow tunnel between hinge connectors; and (right, red S1 subunit not shown) concave pocket between RBD and CTD1 domains.
We calculated free energy landscapes to quantify the role in RBD positioning of specific interactions near the hinge region. Consistent with experiments,7, 23 the closed RBD is favored on free energy landscapes of the wild-type spike. The conserved K528 forms a salt bridge with D389 only when the RBD is closed27, and simulation of K528A shifts the RBD equilibrium toward the open, exposed state. The pocket is tightly packed when the RBD is closed; substitution of A522 with bulkier Val or Leu also remodels the simulated free energy landscape to favor the open, exposed RBD. These results help rationalize experimental observations47–48 for the A522V clinical variant, and also suggest that binding of a small molecule that makes appropriate contacts in the allosteric hinge pocket could preferentially stabilize the spike in a more open conformation. Such hinge pocket binders could avoid problems with low yields37 that often accompany spike variants that modulate RBD dynamics. In particular, hinge pocket binders could serve as valuable probes to characterize a stable spike construct over a range of RBD opening angles, providing insight into the coupling of RBD positioning to spike activation.
Methods
General Protocols
Simulations described here used the ff14Sbonlysc,49 GLYCAM,50 and OPC351 force fields for the protein, glycans, and water, respectively, with salt described by the Joung and Cheatham monovalent ion set52–53. Unless otherwise specified, all simulations used default settings in Amber v2054, with a 4 fs timestep via hydrogen mass repartitioning55, an 8.0 Å direct space cutoff with particle mesh Ewald56 for long-range electrostatics, a Langevin thermostat with collision frequency of 1.0 ps−1, a Berendsen barostat with pressure coupling constant of 0.5 ps, and SHAKE on all bonds involving hydrogen atoms with 0.00001 A tolerance. Simulations were carried out using the sander and pmemd.CUDA modules of Amber2054. Structure visualization and salt bridge identification were performed with VMD57. RMSD values, CoM angle and dihedral values, and other structural measurements were calculated using the cpptraj58 module of Amber. Backbone RMSD values used Cα, C, and N atoms. The imshow() and contour() functions in the pyplot module of matplotlib library59 were used to visualize the free energy landscapes. All amino acid position numbers refer to those in the full-length spike sequence.
Glycosylated Spike Model Building and Simulation Protocols
The full-length, glycosylated wild-type (WT) spike ectodomain model was described by Casalino et al.32, in both the closed and 1-up RBD states (the “initial models” below). These models are based on cryo-EM structures (the “experimental structures” below) of the soluble ectodomain of the spike (6VXX, closed;27 6VSB, chain A open26), which included amino acid substitutions (e.g. in 6VSB: R682S, R683G, R685S, K986P, and V987P) to stabilize the spike in the pre-fusion state. These variants are easier to image and handle experimentally, but these substitutions also potentially alter23, 36 the structure and dynamics of the spike. Thus, our models were converted back to the WT sequence, as well as cleaved at the S1/S2 interface furin site (R685|S686) to better mimic the expected state in situ. The models from Casalino et al.32 include both N-linked and O-linked glycans on each protomer,15, 33, 60 with a glycosylation profile congruent with cryo-EM and MS reports on the spike. Each protomer has 22 N-linked glycans and 1 (chains B and C) or 2 (chain A) O-linked glycans (Table S1). The initial models had the full-length stalk embedded into a membrane.32 Since our focus was on RBD dynamics, we simplified the initial model by removing the membrane and truncating the stalk at V1164 in each protomer.
The experimental models, particularly the 1-up structure, were missing density for the spike stalk, loops and several sidechains, including a portion of the RBM. Starting from the glycosylated structures from Casalino et al.32, we replaced the RBD of each protomer in both initial models with RBD coordinates obtained from the crystal structure of the RBD:ACE2 complex (PDB 6M0J21). The non-RBM region from 6M0J was first aligned with the non-RBM backbone atoms of each protomer. The atoms between A334 and G526 from the initial model were replaced with those from the crystallographic structure 6M0J. This resolved missing loops, as well as repositioned two cysteine residues (C480 and C488) close enough to introduce a disulfide bond that was absent from the cryo-EM structures and our initial models. The remaining disulfides not resolved in the cryo-EM structures were included based on their distance and sequence conservation against the consensus sequences of SARS coronavirus; all of the involved cysteines were conserved in the SARS spike protein. A complete list of all the disulfides is provided in Table S2. This protocol resulted in the “amended models” used in the remainder of this work.
The amended models were solvated with a 20.0 Å minimum distance to the box edge, yielding 403,743 molecules of explicit water, as well as a 200 mM NaCl buffer. A large minimum distance was used to ensure an adequate box to enclose the flexible surface glycans. The 1-up and closed systems each consisted of the same 1,298,646 atoms. Each was equilibrated using a 10-step protocol. First, the water molecules were minimized for 1,000 steps using steepest descent, and then for an additional 9,000 steps with conjugate gradient, while the rest of the system was restrained with 1 kcal/(mol·Å2) Cartesian positional restraints. The systems were then heated to 310 K at constant volume over 0.5 ns, again with 100 kcal/(mol·Å2) positional restraints applied to all atoms except hydrogens and waters. The box size and density were then equilibrated over 1 ns with constant pressure of 1 bar, with positional restraints maintained. The restraints were then lowered to 10 kcal/(mol·Å2) for an additional 1 ns of equilibration, before a second minimization of 10,000 steps of conjugate gradient with only backbone atoms restrained using a force constant of 10 kcal/(mol·Å2). The next three steps of equilibration were done for 1 ns each at constant NPT with positional restraints on protein backbone atoms at 10, 1, and 0.1 kcal/(mol·Å2), respectively. This was followed by a final 1 ns of unrestrained MD at constant NPT. These equilibration steps used a 1 fs integration time step. Production simulations of the closed and 1-up spike systems were both conducted in triplicate, each for ~ 0.3 μs at 310 K and constant NPT.
Building models for sequence substitutions
The 1-up structure used for building the WT model before equilibration was used as initial structure for building the sequence variants A522L, A522V, and K528A. The sidechain atoms of residues to be substituted were deleted, and the new sidechains were introduced using the tleap module of Amber20. The protocols used to equilibrate the WT system were repeated for the substituted spike systems.
Reference models
The cryo-EM structures 6VYB and 6VXX27 were used as reference structures for RMSD analysis on the closed and 1-up structures, respectively. Since the experimental structures were missing coordinates for some regions, we built complete models to facilitate comparison by substituting the coordinates of backbone Cɑ, C, N, and O atoms of each experimental structure into our closed and 1-up models, and retaining coordinates for all other atoms. These are denoted as the “reference models”. RMSD calculations using the reference models were limited to amino acids that were resolved in the 6VSB 1-up cryo-EM structure; these are listed for each domain in Table S3.
Collective variables used to quantify the RBD open/closed transition
We defined center-of-mass (COM) variables to quantify RBD motion during MD, the RBD-opening pathway from NEB, and also as the collective variables (CVs) for the umbrella sampling simulations. Figure S1 gives a visual representation of both CV definitions and COM groups are provided in Table S4. Gui et al.29 used the angle between the long axis of RBD and the plane vertical to the symmetry axis of S protein to quantify the state of RBD. Here, to facilitate use as restraints during umbrella sampling, a COM angle was defined to measure the open/closed movement of the RBD. The first point of the angle was chosen to be the center of the opening RBD. The second point is in CTD1 on the same protomer, and the third is in the center of a short helical region in the upstream helix. The second group was chosen at approximately the fulcrum of RBD opening, and the third group is roughly at the same distance along the CH vector as the second group. These were chosen to make this COM angle definition similar to that used by Gui et al.29 As the RBD opens and becomes less constrained by packing against neighboring RBDs, we also observed RBD rotation along its own long axis. We added a second CV, the COM dihedral, to improve sampling of this rotation and speed convergence in the open state. The first two points of the COM dihedral are in CTD1, while the other 2 are in the RBD.
Nudged Elastic Band Pathway Optimization
We used a GPU implementation61 of a large-system variant62 of the nudged elastic band (NEB) method63 to explore a pathway of the spike transition between the closed and 1-up RBD states for the WT spike. We have previously used this protocol on several systems to obtain pathways for biomolecular recognition dynamics that were subsequently validated through experiments.64–69 NEB does not require definition of collective variables, but maps the pathway in full Cartesian space. The equilibrated closed and 1-up structures were used as the two pathway endpoints, which in NEB are held fixed. An additional 30 intermediate simulations (beads) were placed to map the transition pathway, giving a total of 32 simulations run simultaneously, with bead 0 corresponding to the equilibrated closed state, and bead 32 corresponding to the equilibrated 1-up state. NEB springs were applied to the backbone heavy atoms of all residues with a 1 kcal/(mol·Å2) force constant. NEB was run in four stages: heating, equilibration, annealing, and production. The set of 32 simulations were first heated from 100 to 300 K over 0.5 ns at constant volume, then the box size was allowed to equilibrate for 1 ns at constant pressure and temperature. Annealing was done over 5 ns at constant volume: 1 ns of heating from 300 to 400 K, 1 ns at 400 K, 1 ns heating from 400 to 500 K, 1 ns at 500 K, then cooling to 300 K over 0.5 ns and a final 0.5 ns at 300 K. The final annealed structure from each bead was used to initiate 15 ns of NEB at 300 K and constant P to locally explore the annealed pathway.
Steered molecular dynamics
The NEB calculations applied springs only to the protein backbone, leading to potential discontinuities in other degrees of freedom along the optimized path that could hinder calculation of free energy landscapes. Therefore, steered molecular dynamics (SMD70) using the Amber NFE module was used to generate initial structures for umbrella sampling. The structure first was steered to match the 1-up endpoint of NEB by reducing RMSD from the initial value (1.95 Å) to zero, using a force constant of 50000 kcal/(mol· Å2) during 0.5 ns at 310 K and constant volume. The RMSD region included the Cα atoms of the RBD (residues 338–517), CTD1 (residues 324–327,538–585) and helices in S2 (residues 747–782, 946–966, 987–1034) of only one protomer. The 1-up spike was then closed gradually along the final NEB pathway in a stepwise fashion (31 steps, one for each subsequent NEB bead) until the RBD was fully closed. At each step, the last frame from the neighboring bead was used as the new reference, and the RMSD was reduced to zero as in the previous step. The simulation conditions and lengths for each step were the same as the first step. This strategy was applied to both wild-type and substituted full spike models.
Umbrella sampling and free energy landscapes
To map the energy landscape for the open-to-closed RBD transition in each system, we performed 2-dimensional umbrella sampling (US). A 2-D grid was generated, ranging from 45° to 95° in CoM angle and −30° to 70° in CoM dihedral, spaced evenly by 2° in both dimensions. The range of values for the grid was selected based on the range of values sampled during the NEB simulations with additional space for expanding the grid beyond minima. The SMD trajectories for WT and substituted spike systems were mapped onto the 2D grid, and the median potential energy structure occupying each grid point was selected to initiate US simulations. This resulted in a total of 102 grid blocks for the WT, 114 grid blocks for A522L and A522V, and 102 grid blocks for K528A S-protein. After visual inspection of the initial PMF, additional points were added to expand the sampled grid beyond local minima. Initial structures for expanded grid points were obtained from the final structure sampled in the US run of the grid left or right in CoM angle space; if not available, the grid above or below in CoM dihedral space was selected. The US protocol described above was repeated for the newly added grid points. This brought the total number of grid points to 328 for WT, 280 for K528A and 308 for A522L and A522V (Figure S2).
Umbrella sampling was carried out with 1 window (simulation) for each selected grid point, simulated in parallel. For each window, the CoM angle and CoM dihedral of each initial structure were restrained to their respective grid center using a harmonic potential with force constants of 1600 and 1400 kcal/(mol·rad2) respectively. Each grid structure was equilibrated for 2 ns at 310 K under NPT conditions, followed by a production run of 16 ns under the same conditions, saving structures every 20 ps and values of the CVs every 0.4 ps. PMFs for each system were calculated using the weighted histogram analysis method71 (WHAM) in WHAM v2.0.1072, with a convergence tolerance of 0.00001 kcal/mol and histogram boundaries of 45° and 95° for the CoM angle and −30° and 70° for the CoM dihedral reaction coordinates, respectively. Bin widths of 0.25° for CoM angle and 0.35° for CoM dihedral were optimized using the Shimazaki-Shinomoto algorithm73
Fpocket analysis
Fpocket 3.0.474 was used to extract solvent accessible surface area (SASA) and volume descriptors of the hinge pocket. The algorithm requires a selection of grid points to define the pocket. Therefore, we used mdpocket75 to obtain the grid points for the hinge pocket, using the trajectory from the NEB bead at 75% progress on the opening path. The structures in the NEB trajectory were aligned along the lower portion of the central helices (S1006-V1036). Since the hinge pocket is contained inside a protomer, the other two protomers were deleted to reduce the calculation time. The analysis yielded the same results whether glycans were retained or removed. Default fpocket settings were used to select and cluster the α-spheres. The grid points that corresponded to the cluster of α-spheres located in the hinge pocket were extracted using Chimera76 v1.14. All 395 extracted grid points were used to define the pocket to extract descriptors using a second run of mdpocket, which calculated descriptors using trajectories from the entire RBD opening pathway (all 32 beads).
Sequence conservation analysis
A total of 28 sequences belonging to a variety of coronaviruses were utilized to quantify amino acid conservation at various locations of the spike. Lineages were chosen based on previous work done by Cagliani et al.77 who noted the clustering of SARS-CoV-2 with SARS-CoV in pangolins and bats. The authors used knowledge of previous zoonotic spillover events that resulted in SARS and MERS outbreak to determine the relevance of pangolin and bat coronaviruses. Sequences were obtained using the genbank78 and GISAID79 databases. Lineages were chosen based on previous work done by Cagliani et al.77 who note the clustering of SARS-CoV-2 with SARS-CoV in pangolins and bats. The authors used knowledge of previous zoonotic spillover events that resulted in SARS and MERS outbreak to determine the relevance of pangolin and bat coronaviruses. Differing from Cagliani, the GISAID79 reference sequence was utilized and two common human coronaviruses were included: 229E and OC43. Table S5 details the specific accession numbers and hosts of coronaviruses chosen. Alignment of sequences was performed using default alignment parameters on the T-Coffee80 online server. Following the alignment, the sequences were processed to remove gaps that were present in the SARS-CoV-2 sequence. Table S6 shows the calculated percent identity for the hinge pocket region alongside the sequence alignment. Lastly, sequence logos were generated utilizing the WebLogo server81.
Results and Discussion
MD simulations of the spike in closed and 1-up states
Following addition of missing loops and glycans, followed by solvation, the complete spike ectodomain system consisted of ~ 1.3 million atoms (see Methods for details on construction of the initial structures for each state). We performed three independent ~ 300 ns MD simulations of the spike with all RBDs closed, as well as three independent runs of the 1-up spike (with only one RBD in the open position; in this work we refer to an individual RBD as being open or closed, while the spike trimer can be in the closed, 1-up, 2-up or 3-up state).
We first confirmed that the spike was reasonably stable in our simulation model. We show in Figure S3 the best-fit backbone atom RMSD of the S1 and S2 subunits, during our 1-up and closed simulations, against their respective cryo-EM reference structures (see “Reference Models” in Methods). The simulations of the closed spike were largely stable, with each S2 subunit showing low deviations of ~ 2 Å, and the more flexible S1 subunit sampling RMSD values of ~ 3 Å. Although the 1-up simulations are also stable, with similar S2 RMSD values, the additional space from reduced packing of the RBD leads to a more flexible S1 subunit with larger fluctuations and average RMSD values near 4–5 Å. These observations are consistent with prior simulations reports and lower resolution in the cryo-EM datasets for open RBDs.26, 32 Overall, the spike behavior appears reasonable in our model.
We next examined the behavior of individual regions of the spike in the closed and 1-up simulations (Figures S4, S5). For the closed system, the domains tended to be quite stable with most sampling RMSD values between 1–2 Å. Similar domain stability is seen in the 1-up simulations, with larger changes in the NTD domains that become more loosely constrained when the packing at the top of the spike is reduced. The CTD2 domain in one protomer shows higher deviation in both systems, likely due to an inaccurately modeled surface loop in the initial structures. The cleaved S1/S2 site is highly mobile in both the closed and 1-up simulations (data not shown), consistent with it typically being unresolved in cryo-EM experiments. In contrast, the uncleaved S2’ site is exceptionally stable and samples RMSD values of only ~ 0.5 Å in all simulations, with R815 in the KRSF segment remaining partially buried and presumably inaccessible to proteases. This is consistent with experimental evidence for inaccessibility of S2’ to proteases in pre-fusion SARS spikes.82 The fusion peptide regions proximal to the S2’ site are similarly stable, with small changes occurring in some trials, but the results are anecdotal and were not subjected to detailed analysis given our focus here on the RBD.
Quantifying RBD positioning
In addition to RMSD values, we characterized the extent of RBD opening using two collective variables (CVs) that describe the opening angle of the RBD and rotation of the RBD in the plane roughly perpendicular to the central helices (see Methods and Figure S1 for CV definitions). In the 1-up system, the two closed RBDs sample somewhat different angle ranges due to their different positions relative to the cavity left by the open RBD. All RBDs in the 1-up and closed states remained in their respective conformations for the entirety of the 0.3 μs simulations, with no spontaneous transitions between open and closed RBDs (Figure 5, with 2 additional independent runs shown in Figure S6). This metastable RBD behavior on the sub-μsec timescale here is consistent with the behavior observed by Casalino et al. in their 4 μsec simulations.32
Figure 5.
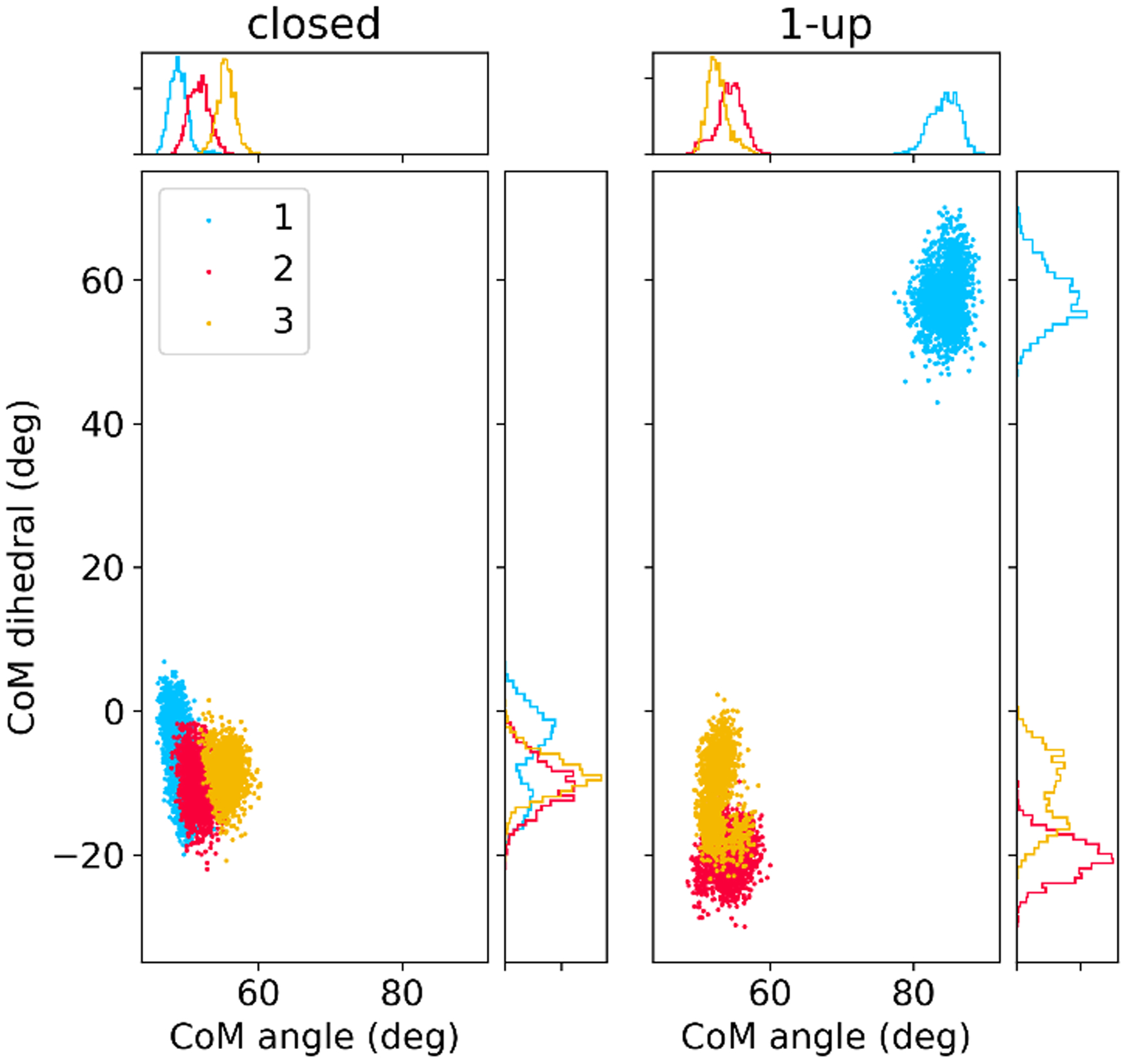
Sampling of collective variables quantifying RBD position in MD simulations of closed (left) and 1-up (right) spike systems. Dots represent MD snapshots of 310 ns runs, and each protomer is shown in a different color corresponding to Figure 4. Collective variables are defined in Figure S1; the CoM angle measures motion of the RBD away from the S2 core, with the CoM dihedral measures rotation of the RBD relative to the CTD1 domain.
Glycans
Behavior of the glycans in SARS-CoV-2 spike simulations was previously described by Casalino et al., who proposed that the N-glycans at N165 and N234 in the NTD play an essential structural role in RBD conformational transitions.32 In our closed simulations, the glycans on N234 point towards the solvent, rather than the core of the protein. In the 1-up simulations, the glycans on N234 of the adjacent NTD direct inward, occupying the vacancy left by the RBD and interacting with the closed, opposing RBD. During the same simulations, the glycans at N165 interacted with various residues on the open RBD and inserted into the vacancy along with the glycans at with those at N234. These observations are consistent with those reported by Casalino et al. using independent force fields and MD software packages.32
Mapping the RBD opening pathway
Since brute-force MD simulations were unable to capture spontaneous opening or closing of the RBD during the 300 ns runs, we used a nudged elastic band (NEB) approach62–63 to optimize a low-energy pathway between the spike in the closed and 1-up RBD states (see Methods). Briefly, NEB uses a set of multiple simulations (“beads”) that connect two fixed conformational endpoints (here, the closed and 1-up states). Virtual springs ensure that the beads remain spaced apart along the path, but the intermediate beads are otherwise free to explore changes in the positions of all atoms. These coupled simulations are run concurrently, and an annealing optimization provides a model for a low-energy pathway connecting the defined conformations of the endpoints.
The NEB pathway maps the RBD transition in the full Cartesian space of all atoms, unlike many other approaches where one or more collective variables that describe the transition must be defined in advance of pathway mapping, leading to dependence of the path on the choice of collective variables. Importantly, projecting the NEB pathway onto different collective variables during post-processing can facilitate the interpretation of the pathway, but the NEB-optimized pathway itself is robust to such choices.62–63
Projections of the NEB pathway onto the two CVs that quantify the RBD position (Figure S1) are shown as a function of progress along the closed-to-open RBD transition in Figure S7, along with overlaid structure snapshots of a single protomer during the transition shown in Figure 6. At roughly 15% progress along the pathway, the COM angle rises, indicating the start of RBD opening. Changes earlier in the path correspond to modest adjustments in the NTD as it shifts away from the RBD to provide clearance for opening. Next, the RBD lifts up out of the cavity formed by the NTD and RBD of the counterclockwise protomer, giving rise to a sharper increase in the CoM angle value until ~ 60% progress along the path. As the RBD clears the cavity it rotates outward, quantified by the increase in the CoM dihedral starting at roughly 30% progress.
Figure 6.
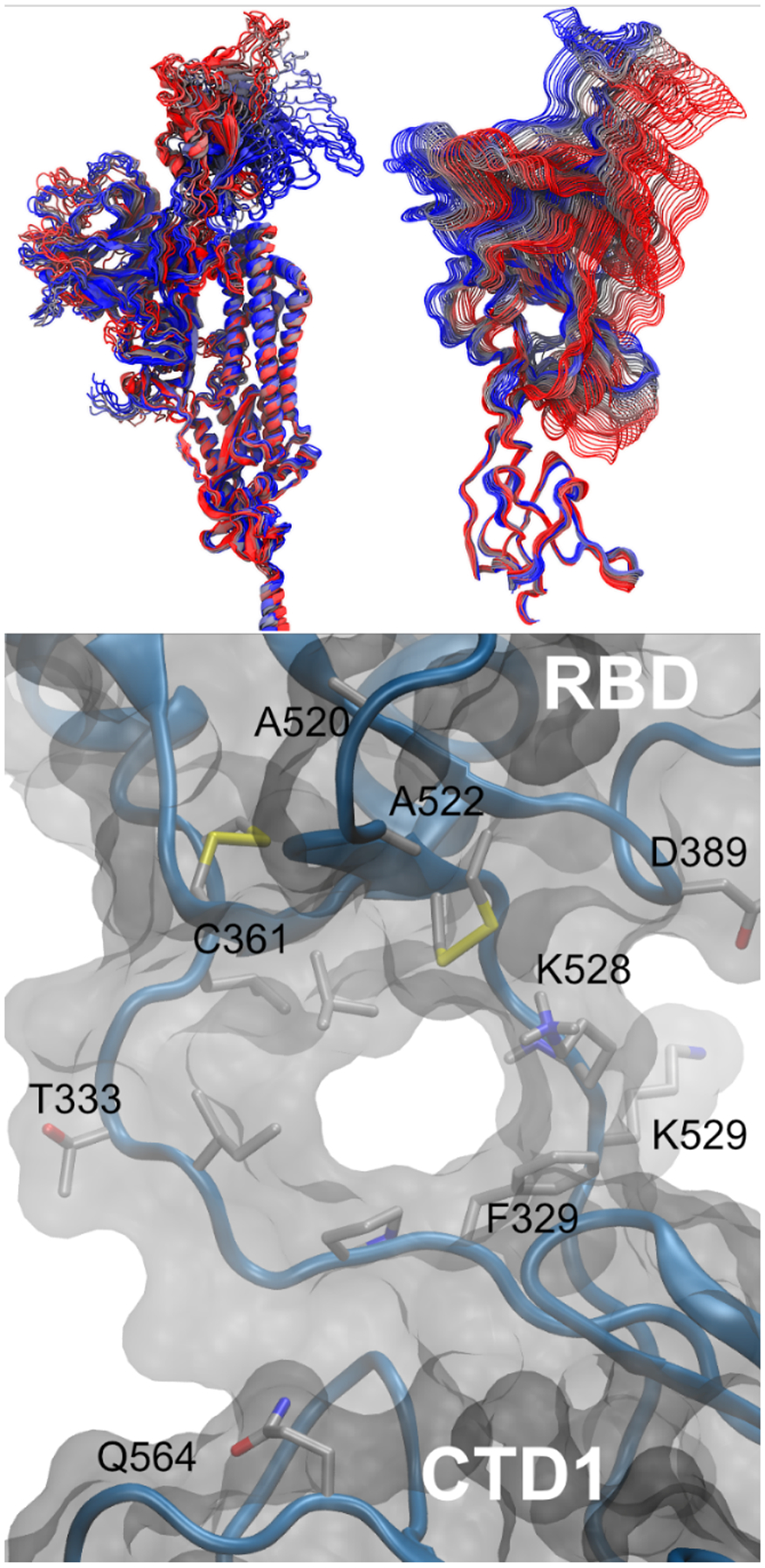
Spike structure data from the RBD opening pathway. (Top, A) structures of a single protomer colored by opening pathway progress from blue to red, with the entire protomer shown on the left (best-fit to central helices to emphasize the larger change in RBD as compared to CTD1 domain) and close-up of the pocket formed between the RBD and CTD1 shown on the right (best-fit to CTD1 to emphasize the hinge motion between these domains). (Bottom, B) hinge pocket and tunnel in simulated open RBD structure, viewed from inside the spike. Sidechains discussed in the text are labeled. For clarity, the solvent, most sidechains and hydrogen atoms are not shown.
RBD opening is achieved through a hinge
As the RBD lifts, it moves away from the rest of the spike, leading to an RMSD increase to ~ 30 Å relative to its position in the closed state (Figures 6 and S7, best-fit to the S2 core CH helices). The attached CTD1 domain also shifts, but remains much closer to the position in the closed RBD state, deviating less than 5 Å over the pathway. These values largely reflect rigid-body shifts in the individual domains, with much smaller RMSD values of ~ 1–2 Å when these domains are self-fit (Figure S8), likely due to the presence of multiple disulfide bonds proximal to the hinge pocket (Table S2). Thus, the most significant change in RBD opening is in the relative position of the RBD and CTD1 domains, with smaller outward shifts of NTD and CTD1 that are consistent with experiments26–27.
The hinge connecting the RBD to the CTD1 is composed of two antiparallel strands, one connecting the C-terminal end of the first β-strand in CTD1 to the N-terminal end of the RBD (“N-connector”), and the other connecting the C-terminal of the RBD back to form the remainder of CTD1 (“C-connector”). Both connectors are located on one side of the domain interface, on the exterior of the spike, allowing it to serve as a hinge around which the RBD can rotate up and outward (Figure 6A). As hinges, both connectors undergo local conformational changes as the spike RBD opens, with backbone dihedral changes present in I332, T333 and P527 (Figure S9).
Specific interactions in the hinge region may stabilize the closed RBD
In the all-closed cryo-EM structure 6VXX27, a salt bridge is apparent between K528 on the C-connector and D389 on the RBD α−3 helix. This salt bridge is also present at the beginning of the RBD opening pathway, but the interaction is broken as the RBD moves upward and away from CTD1 (Figure 6, with distance vs. pathway shown in Figure S10). The absence of this salt bridge with an open RBD supports the hypothesis that the hinge region can modulate RBD opening. Cryo-EM structures also suggest increased hinge flexibility when the RBD is open; many experimental structures are missing density in the hinge region, including K528 and the entire N-connector in the initial model for our 1-up spike system (6VSB).26
The hinge region is strongly conserved both within identified SARS-CoV-2 isolates, as well as other betacoronaviruses (Figure S11, Table S6), supporting a mechanistic function for this region. In particular, the salt-bridge pair D389 and K528 are both well conserved, while the immediately adjacent K529 is poorly conserved; this difference is consistent with our suggested role for K528 in stabilizing the RBD:CTD1 interface when the RBD is closed and the lack of specific interactions observed in MD for K529. N331 on the C-connector and nearby N343 on the RBD are both glycosylated, with the glycans extending into solution during MD and shielding the surface of the conserved hinge region from antibody recognition (Figure S12).
RBD opening via the hinge opens a pocket between RBD and CTD1
In the RBD-closed state, both hinge strands typically are resolved in cryo-EM structures, and the sulfur atoms of RBD disulfide C391-C525 dock into the hydrophobic space between the connector strands. The two domains are tightly packed, with no gaps in the solvent-accessible surface. A520 on the RBD and Q564 on CTD1 are in close contact in the interior of the hinge pocket when the RBD is closed, but move apart to ~ 27 Å as the RBD opens (Figure S13). The disulfide on the RBD base moves upward, leaving a short tunnel between the connector strands and creating a pocket in the expanding space at the former interface between the RBD and CTD1 (Figures 4, 6). SASA and volume analysis indicates that the pocket becomes solvent accessible halfway along the opening pathway (Figure S14).
Free energy landscapes for RBD opening in wild-type and substituted spike systems
The pathways provide insight into structural changes along the transition, but do not provide information about locations of local minima, their relative free energies, or how the energies are influenced by the observed interactions. We calculated free energy landscapes in the form of potentials of mean force (PMFs) for the RBD closed/open transition in the complete, glycosylated spike. Uncertainties in our structure (such as model quality for flexible loops) likely prevent us from obtaining accurate quantitative free energy values for the overall open-to-closed RBD transition, but the changes resulting from amino acid substitutions are likely to be qualitatively informative due to cancellation of systematic errors. The free energy landscapes were obtained using umbrella sampling using the CVs described above, sampling a broad area surrounding the pathway obtained from NEB (see Methods for details).
The PMFs are shown in Figure 7. Local minima suggest that RBD opening involves three states: the closed state, open state, and a more diffuse, weakly populated intermediate state. The intermediate state (CoM angle 65–70°; CoM dihedral 18–26°) is stabilized in part by a transient salt bridge between K386 on the RBD and D985 at the top of the central helix (CH) in the S2 subunit. An on-path intermediate RBD state also has been proposed based on smFRET experiments, where the fluorophores were attached via insertion of an additional six and eight amino acids to the RBD and CTD1, respectively.83 The length and potential flexibility of these linkers, and size of the fluorophores, preclude a quantitative comparison to the data for the wild-type system presented here.
Figure 7:
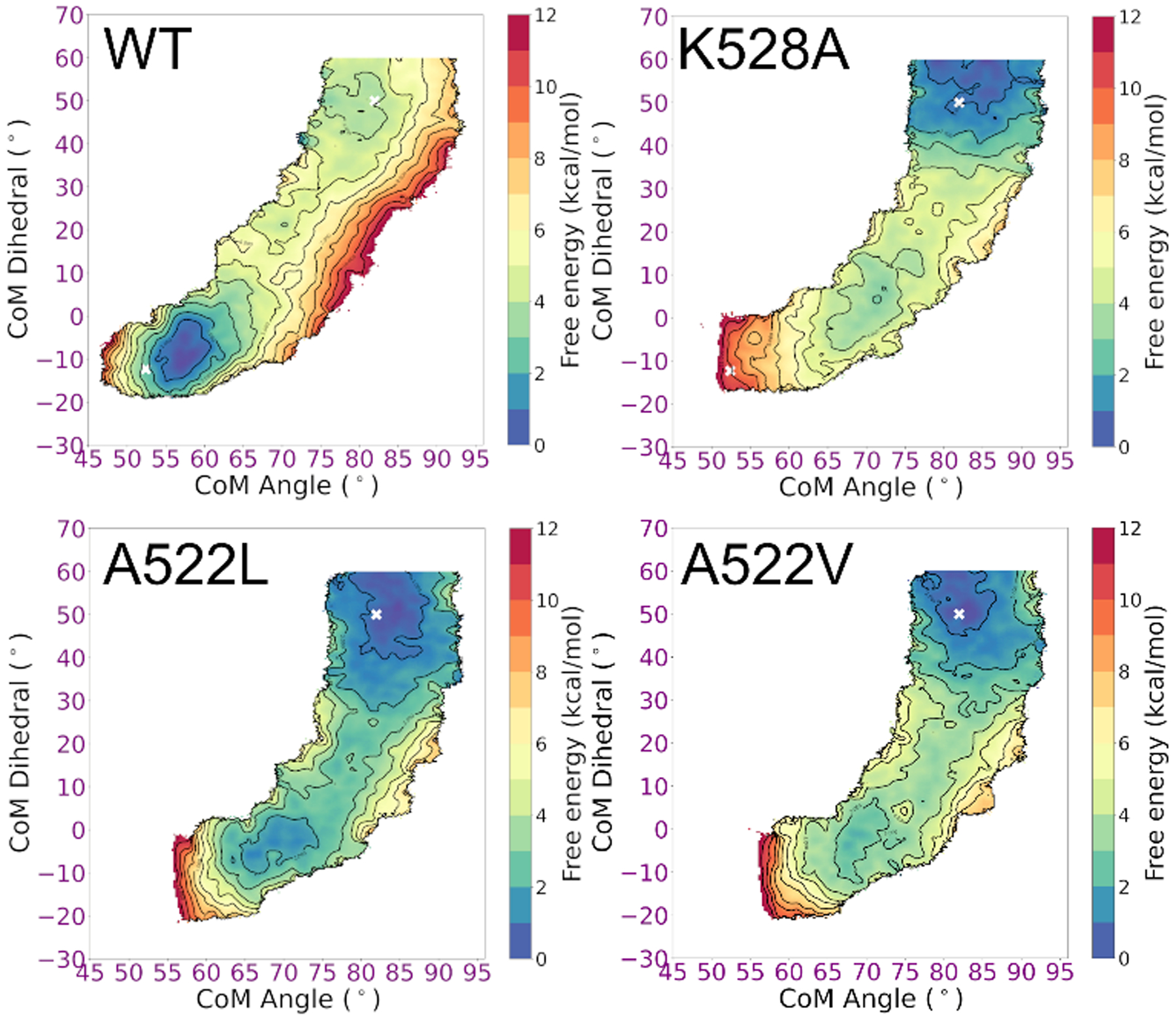
Free energy landscapes for RBD opening in spike variants. Contour lines indicate 1 kcal/mol intervals. WT has a global minimum with closed RBD, K528A prefers an open RBD, and the system with branched side chains in the hinge pocket (A522L, A522V) show significant flattening of the free energy landscape. Values obtained from the experimental open and 1-up spike structures are indicated with a white x.
In the closed and intermediate state, K528 on the C-connector helps stabilize the RBD by forming a network of electrostatic interactions involving D389-K386-S383 to D985 at the top of CH. Further opening results in separation of the K528-D389 salt bridge. In the open state, a cluster of salt bridges forms between D427/D428 on the RBD and R403/R408 on the closed clockwise RBD (See Figure 1 for clockwise/counter-clockwise definition).
The WT spike shows a strong preference for the closed RBD state (Figure 7), consistent with cryo-EM experiments6, 23 on the wild-type system that refined only a closed RBD form, in contrast to more frequent observation of open RBD in experiments using the common “2P” stabilizing substitutions82, 84. Our results are further supported by the suggestion (based on experimental structures) that the 2P substitution may weaken23, 36 the interaction between the closed RBD and the S2 subunit.
Focusing next on the hinge pocket, we estimated the impact of deleting the salt bridge involving conserved K528 and D389 via simulations of the K528A spike. Consistent with our structure-based hypothesis, the K528A substitution significantly destabilizes the closed RBD, and the free energy surface is shifted toward easier RBD opening (Figure 7). The intermediate state is flatter, likely due to increased flexibility of D389-K386-S383 in the absence of K528.
A shift in the open RBD position is seen in a cryo-EM structure of the spike with amino acid substitutions at the CTD2:S2 interface.39 We hypothesized that substitutions inside the hinge pocket interface may also affect the RBD. The small, nonpolar A522 is tightly packed into the hinge pocket when the RBD is closed, but becomes exposed when the RBD opens (Figure 6B). Replacement of A522 with a bulkier side chain such as Leu could be readily accommodated with an open RBD, but may introduce a steric clash in the tightly packed RBD:CTD1 interface in the closed-RBD structure. The simulated free energy landscape for the A522L substitution supports our hypothesis, and is strongly shifted to favoring the intermediate and open RBD (Figure 7). The minimum for the closed RBD is shifted to higher angles, consistent with a steric clash preventing full closing, and the poorly packed interface destabilizes the closed RBD. These A522L results suggest that introducing new chemical matter into the hinge pocket interface could disrupt the ability of the spike to adopt a closed-RBD state.
The A522V substitution is present in a small subset of circulating clinical strains47, despite the report48 of stronger antibody binding to the spike with an A522V substitution. It is possible that this substitution plays a role in increasing RBD opening via looser hinge packing, perhaps to counter the effect of other substitutions that reduce RBD opening. An increased population of open RBD could lead to stronger binding affinity due to an increased effective concentration17 of binding-competent open RBD. We calculated the free energy landscape for A522V and observed flattening of the landscape similar to that for A522L (Figure 7), providing a possible rationale for these experimental observations.
Although these proof-of-principle sequence substitutions support the proposed allosteric nature of the hinge region, it may be preferable to alter RBD dynamics without modification of the viral genome. Spike variants with higher flexibility are typically more difficult to express and purify;37 in particular, the A522V variant was reported to be “especially deleterious” for yield, preventing detailed analysis of the spike47. The location of the hinge pocket at the base of the RBD, facing the interior of the spike, makes it unlikely that it could be accessible for antibody or nanobody binding. In particular, the pocket is ~ 20 Å farther down the RBD than a cryptic epitope observed in the crystal structure85 of a complex between the CR3022 antibody and isolated RBD (Figure S15). However, the pocket appears more readily accessible to small molecules diffusing from the outside of the protein, through a 20+ Å gap between the NTD of the clockwise protomer and CH of the counterclockwise protomer; this space was previously occupied by the closed RBD (Figure 4). Small molecules also are more likely to bypass the glycan shield. A useful strategy to explore the effects of increased RBD opening, and possible coupling to S1 shedding, could be to purify the wild-type spike, then add a small molecule probe that binds to the hinge pocket and shifts the free energy landscape. Virtual screening against our simulation models may aid the development of such probes.
Conclusions
Experimental structures have provided a wealth of data on most of the spike structure, but less is known about flexible regions, and especially the detailed mechanisms by which the spike transitions between the observed states of the RBD. We performed computational analysis of the fully atomic detail SARS-CoV-2 spike glycoprotein dynamics in explicit solvent, using standard MD simulations, steered MD, RBD opening pathway mapping, and umbrella sampling calculations of the associated free energy landscape. We identified a conserved allosteric pocket adjacent to a hinge region that is critical for the RBD opening and closing motion. This pocket exists when the RBD is in the open state, and collapses when the RBD closes and contacts the CTD1 domain.
In order to understand the effect of amino acid substitutions on the RBD opening equilibrium, we calculated free energy landscapes as a function of two collective variables that quantify RBD motion along our pathway. The free energy landscapes were consistent with expected changes based on analysis of changes in side-chain interactions along the pathway. Furthermore, they confirmed the allosteric nature of the pocket in controlling dynamics of the RBD and exposure of the ACE-binding region. The results suggest that the pocket may be an interesting target for screening of small molecules with the goal of altering the energy profile for RBD closing.
Modulation of RBD dynamics in SARS-CoV-2 could have several consequences. As with the clinically-observed A522V substitution, small-molecule binding in the hinge pocket could increase antibody binding affinity. Furthermore, the molecular events that trigger irreversible S1 shedding and spike inactivation, experimentally observed in the absence of ACE2, remain unclear. A molecular tool that can reduce RBD closing may help stabilize intermediate states for structural analysis on spike constructs that are easily accessible to experiments. Such probes also may facilitate investigation of the coupling between RBD opening and S1 shedding, thought to be the crucial link between S1-mediated ACE2 binding and S2-mediated membrane fusion. Any insight could be valuable in the development of therapeutics that promote premature shedding and, thereby, irreversibly disable the ability of the virus to infect host cells.
Supplementary Material
Acknowledgements
We are grateful to Dan Raleigh, Ronit Freeman, and Arvind Ramanathan for helpful discussions, and to Sarina Bromberg for assistance with Figure 2. We thank Rommie Amaro and her group for helpful discussions and for early access to the spike models.
This work was funded in part by the SUNY Research Seed Grant Program, the Stony Brook University Office of the Vice President for Research, the Research Corporation for Science Advancement (COVID Initiative grant #27350), NIH grants R01GM107104 (C.S.), R35GM126906 (R.C.R) and the DOE Office of Science through the National Virtual Biotechnology Laboratory, a consortium of DOE national laboratories focused on response to COVID-19, with funding provided by the Coronavirus CARES Act.
This work used resources services, and support provided via the COVID-19 HPC Consortium (https://covid19-hpc-consortium.org/), which is a unique private-public effort to bring together government, industry, and academic leaders who are volunteering free compute time and resources in support of COVID-19 research. Additional computer time was provided by Brookhaven National Laboratory and AiMOS at Rensselaer Polytechnic Institute.
Footnotes
Supplementary Information
Additional figures, tables, methods and PDB coordinates for snapshots of the spike along the RBD opening pathway.
Competing Interests
The authors declare no competing interests.
References
- 1.World Health Organization Coronavirus Disease (COVID-19) Dashboard. https://covid19.who.int/ (accessed 2020-12-27).
- 2.Belouzard S; Chu VC; Whittaker GR, Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proceedings of the National Academy of Sciences of the United States of America 2009, 106 (14), 5871–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li F, Structure, Function, and Evolution of Coronavirus Spike Proteins. Annual Rev Virol 2016, 3 (1), 237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosch BJ; van der Zee R; de Haan CA; Rottier PJ, The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 2003, 77 (16), 8801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang T; Bidon M; Jaimes JA; Whittaker GR; Daniel S, Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Research 2020, 178, 104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ke Z; Oton J; Qu K; Cortese M; Zila V; McKeane L; Nakane T; Zivanov J; Neufeldt CJ; Cerikan B; Lu JM; Peukes J; Xiong X; Kräusslich H-G; Scheres SHW; Bartenschlager R; Briggs JAG, Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588 (7838), 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turoňová B; Sikora M; Schürmann C; Hagen WJH; Welsch S; Blanc FEC; von Bülow S; Gecht M; Bagola K; Hörner C; van Zandbergen G; Landry J; de Azevedo NTD; Mosalaganti S; Schwarz A; Covino R; Mühlebach MD; Hummer G; Krijnse Locker J; Beck M, In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370 (6513), 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Premkumar L; Segovia-Chumbez B; Jadi R; Martinez DR; Raut R; Markmann AJ; Cornaby C; Bartelt L; Weiss S; Park Y; Edwards CE; Weimer E; Scherer EM; Rouphael N; Edupuganti S; Weiskopf D; Tse LV; Hou YJ; Margolis D; Sette A; Collins MH; Schmitz J; Baric RS; de Silva AM, The receptor-binding domain of the viral spike protein is an immunodominant and highly specific target of antibodies in SARS-CoV-2 patients. Science Immunology 2020, 5 (48), eabc8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corey L; Mascola JR; Fauci AS; Collins FS, A strategic approach to COVID-19 vaccine R&D. Science 2020, 368 (6494), 948–950. [DOI] [PubMed] [Google Scholar]
- 10.Callaway E, The race for coronavirus vaccines: a graphical guide. Nature 2020, 580 (7805), 576–577. [DOI] [PubMed] [Google Scholar]
- 11.Coutard B; Valle C; de Lamballerie X; Canard B; Seidah NG; Decroly E, The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Research 2020, 176 (February), 104742–104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffmann M; Kleine-Weber H; Schroeder S; Krüger N; Herrler T; Erichsen S; Schiergens TS; Herrler G; Wu N-H; Nitsche A; Müller MA; Drosten C; Pöhlmann S, SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181 (2), 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaimes JA; André NM; Chappie JS; Millet JK; Whittaker GR, Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J Mol Biol 2020, 432 (10), 3309–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shang J; Wan Y; Luo C; Ye G; Geng Q; Auerbach A; Li F, Cell entry mechanisms of SARS-CoV-2. Proceedings of the National Academy of Sciences 2020, 117 (21), 11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe Y; Allen JD; Wrapp D; McLellan JS; Crispin M, Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369 (6501), 330–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H; Penninger JM; Li Y; Zhong N; Slutsky AS, Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Medicine 2020, 46 (4), 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shang J; Ye G; Shi K; Wan Y; Luo C; Aihara H; Geng Q; Auerbach A; Li F, Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581 (7807), 221–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benton DJ; Wrobel AG; Xu P; Roustan C; Martin SR; Rosenthal PB; Skehel JJ; Gamblin SJ, Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588 (7837), 327–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q; Zhang Y; Wu L; Niu S; Song C; Zhang Z; Lu G; Qiao C; Hu Y; Yuen KY; Wang Q; Zhou H; Yan J; Qi J, Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181 (4), 894–904.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan R; Zhang Y; Li Y; Xia L; Guo Y; Zhou Q, Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science (New York, N.Y.) 2020, 2 (March), 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lan J; Ge J; Yu J; Shan S; Zhou H; Fan S; Zhang Q; Shi X; Wang Q; Zhang L; Wang X, Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581 (7807), 215–220. [DOI] [PubMed] [Google Scholar]
- 22.Walls AC; Tortorici MA; Snijder J; Xiong X; Bosch BJ; Rey FA; Veesler D, Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci U S A 2017, 114 (42), 11157–11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Y; Zhang J; Xiao T; Peng H; Sterling SM; Walsh RM; Rawson S; Rits-Volloch S; Chen B, Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369 (6511), 1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C; Mendonça L; Yang Y; Gao Y; Shen C; Liu J; Ni T; Ju B; Liu C; Tang X; Wei J; Ma X; Zhu Y; Liu W; Xu S; Liu Y; Yuan J; Wu J; Liu Z; Zhang Z; Liu L; Wang P; Zhang P, The Architecture of Inactivated SARS-CoV-2 with Postfusion Spikes Revealed by Cryo-EM and Cryo-ET. Structure 2020, 28 (11), 1218–1224.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y; Yang C; Xu X.-f.; Xu W; Liu S.-w., Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacologica Sinica 2020, 41 (9), 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wrapp D; Wang N; Corbett KS; Goldsmith JA; Hsieh C-L; Abiona O; Graham BS; McLellan JS, Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (New York, N.Y.) 2020, 1263 (March), 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walls AC; Park YJ; Tortorici MA; Wall A; McGuire AT; Veesler D, Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181 (2), 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tai W; He L; Zhang X; Pu J; Voronin D; Jiang S; Zhou Y; Du L, Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cellular & Molecular Immunology 2020, 17 (6), 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gui M; Song W; Zhou H; Xu J; Chen S; Xiang Y; Wang X, Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Research 2017, 27 (1), 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song W; Gui M; Wang X; Xiang Y, Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog 2018, 14 (8), e1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barros EP; Casalino L; Gaieb Z; Dommer AC; Wang Y; Fallon L; Raguette L; Belfon K; Simmerling C; Amaro RE, The flexibility of ACE2 in the context of SARS-CoV-2 infection. Biophys J 2021, 120 (6), 1072–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casalino L; Gaieb Z; Goldsmith JA; Hjorth CK; Dommer AC; Harbison AM; Fogarty CA; Barros EP; Taylor BC; McLellan JS; Fadda E; Amaro RE, Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Central Science 2020, 6 (10), 1722–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grant OC; Montgomery D; Ito K; Woods RJ, Analysis of the SARS-CoV-2 spike protein glycan shield reveals implications for immune recognition. Scientific Reports 2020, 10 (1), 14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walls AC; Xiong X; Park YJ; Tortorici MA; Snijder J; Quispe J; Cameroni E; Gopal R; Dai M; Lanzavecchia A; Zambon M; Rey FA; Corti D; Veesler D, Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019, 176 (5), 1026–1039 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zou J; Yin J; Fang L; Yang M; Wang T; Wu W; Bellucci MA; Zhang P, Computational Prediction of Mutational Effects on SARS-CoV-2 Binding by Relative Free Energy Calculations. J Chem Inf Model 2020, 60 (12), 5794–5802. [DOI] [PubMed] [Google Scholar]
- 36.Xiong X; Qu K; Ciazynska KA; Hosmillo M; Carter AP; Ebrahimi S; Ke Z; Scheres SHW; Bergamaschi L; Grice GL; Zhang Y; Bradley J; Lyons PA; Smith KGC; Toshner M; Elmer A; Ribeiro C; Kourampa J; Jose S; Kennet J; Rowlands J; Meadows A; O’Brien C; Rastall R; Crucusio C; Hewitt S; Price J; Calder J; Canna L; Bucke A; Tordesillas H; Harris J; Ruffolo V; Domingo J; Graves B; Butcher H; Caputo D; Le Gresley E; Dunmore BJ; Martin J; Legchenko E; Treacy C; Huang C; Wood J; Sutcliffe R; Hodgson J; Shih J; Graf S; Tong Z; Mescia F; Tilly T; O’Donnell C; Hunter K; Pointon L; Pond N; Wylot M; Jones E; Fawke S; Bullman B; Bergamaschi L; Turner L; Jarvis I; Omarjee O; De Sa A; Marsden J; Betancourt A; Perera M; Epping M; Richoz N; Bower G; Sharma R; Nice F; Huhn O; Stark H; Walker N; Stirrups K; Ovington N; Dewhust E; Li E; Papadia S; Nathan JA; Baker S; James LC; Baxendale HE; Goodfellow I; Doffinger R; Briggs JAG; The C-NC-BC, A thermostable, closed SARS-CoV-2 spike protein trimer. Nature Structural & Molecular Biology 2020, 27 (10), 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsieh C-L; Goldsmith JA; Schaub JM; DiVenere AM; Kuo H-C; Javanmardi K; Le KC; Wrapp D; Lee AG; Liu Y; Chou C-W; Byrne PO; Hjorth CK; Johnson NV; Ludes-Meyers J; Nguyen AW; Park J; Wang N; Amengor D; Lavinder JJ; Ippolito GC; Maynard JA; Finkelstein IJ; McLellan JS, Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369 (6510), 1501–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCallum M; Walls AC; Bowen JE; Corti D; Veesler D, Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nature Structural & Molecular Biology 2020, 27 (10), 942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henderson R; Edwards RJ; Mansouri K; Janowska K; Stalls V; Gobeil SMC; Kopp M; Li D; Parks R; Hsu AL; Borgnia MJ; Haynes BF; Acharya P, Controlling the SARS-CoV-2 spike glycoprotein conformation. Nature Structural & Molecular Biology 2020, 27 (10), 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toelzer C; Gupta K; Yadav SKN; Borucu U; Davidson AD; Kavanagh Williamson M; Shoemark DK; Garzoni F; Staufer O; Milligan R; Capin J; Mulholland AJ; Spatz J; Fitzgerald D; Berger I; Schaffitzel C, Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370 (6517), 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bangaru S; Ozorowski G; Turner HL; Antanasijevic A; Huang D; Wang X; Torres JL; Diedrich JK; Tian J-H; Portnoff AD; Patel N; Massare MJ; Yates JR; Nemazee D; Paulson JC; Glenn G; Smith G; Ward AB, Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science 2020, 370 (6520), 1089–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J; Wang W; Chen Z; Lu S; Yang F; Bi Z; Bao L; Mo F; Li X; Huang Y; Hong W; Yang Y; Zhao Y; Ye F; Lin S; Deng W; Chen H; Lei H; Zhang Z; Luo M; Gao H; Zheng Y; Gong Y; Jiang X; Xu Y; Lv Q; Li D; Wang M; Li F; Wang S; Wang G; Yu P; Qu Y; Yang L; Deng H; Tong A; Li J; Wang Z; Yang J; Shen G; Zhao Z; Li Y; Luo J; Liu H; Yu W; Yang M; Xu J; Wang J; Li H; Wang H; Kuang D; Lin P; Hu Z; Guo W; Cheng W; He Y; Song X; Chen C; Xue Z; Yao S; Chen L; Ma X; Chen S; Gou M; Huang W; Wang Y; Fan C; Tian Z; Shi M; Wang F-S; Dai L; Wu M; Li G; Wang G; Peng Y; Qian Z; Huang C; Lau JY-N; Yang Z; Wei Y; Cen X; Peng X; Qin C; Zhang K; Lu G; Wei X, A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity. Nature 2020, 586 (7830), 572–577. [DOI] [PubMed] [Google Scholar]
- 43.Hansen J; Baum A; Pascal KE; Russo V; Giordano S; Wloga E; Fulton BO; Yan Y; Koon K; Patel K; Chung KM; Hermann A; Ullman E; Cruz J; Rafique A; Huang T; Fairhurst J; Libertiny C; Malbec M; Lee W.-y.; Welsh R; Farr G; Pennington S; Deshpande D; Cheng J; Watty A; Bouffard P; Babb R; Levenkova N; Chen C; Zhang B; Romero Hernandez A; Saotome K; Zhou Y; Franklin M; Sivapalasingam S; Lye DC; Weston S; Logue J; Haupt R; Frieman M; Chen G; Olson W; Murphy AJ; Stahl N; Yancopoulos GD; Kyratsous CA, Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369 (6506), 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walls A; Tortorici MA; Bosch BJ; Frenz B; Rottier PJ; DiMaio F; Rey FA; Veesler D, Crucial steps in the structure determination of a coronavirus spike glycoprotein using cryo-electron microscopy. Protein Sci 2017, 26 (1), 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan X; Cao D; Kong L; Zhang X, Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nature Communications 2020, 11 (1), 3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huo J; Zhao Y; Ren J; Zhou D; Duyvesteyn HME; Ginn HM; Carrique L; Malinauskas T; Ruza RR; Shah PNM; Tan TK; Rijal P; Coombes N; Bewley KR; Tree JA; Radecke J; Paterson NG; Supasa P; Mongkolsapaya J; Screaton GR; Carroll M; Townsend A; Fry EE; Owens RJ; Stuart DI, Neutralization of SARS-CoV-2 by Destruction of the Prefusion Spike. Cell Host & Microbe 2020, 28 (3), 445–454.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Long SW; Olsen RJ; Christensen PA; Bernard DW; Davis JJ; Shukla M; Nguyen M; Saavedra MO; Yerramilli P; Pruitt L; Subedi S; Kuo H-C; Hendrickson H; Eskandari G; Nguyen HAT; Long JH; Kumaraswami M; Goike J; Boutz D; Gollihar J; McLellan JS; Chou C-W; Javanmardi K; Finkelstein IJ; Musser JM, Molecular Architecture of Early Dissemination and Massive Second Wave of the SARS-CoV-2 Virus in a Major Metropolitan Area. mBio 2020, 11 (6), e02707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q; Wu J; Nie J; Zhang L; Hao H; Liu S; Zhao C; Zhang Q; Liu H; Nie L; Qin H; Wang M; Lu Q; Li X; Sun Q; Liu J; Zhang L; Li X; Huang W; Wang Y, The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182 (5), 1284–1294.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation 2015, 11 (8), 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirschner KN; Yongye AB; Tschampel SM; González-Outeiriño J; Daniels CR; Foley BL; Woods RJ, GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem 2008, 29 (4), 622–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Izadi S; Onufriev AV, Accuracy limit of rigid 3-point water models. The Journal of Chemical Physics 2016, 145 (7), 074501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joung IS; Cheatham TE, Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. The Journal of Physical Chemistry B 2009, 113 (40), 13279–13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joung IS; Cheatham TE, Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. The Journal of Physical Chemistry B 2008, 112 (30), 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Case DA; Belfon KAA; Ben-Shalom I; Brozell SR; Cerutti D; Cheatham T; Cruzeiro VWDD,T; Duke RE; Giambasu G; Gilson M; Gohlke H; Götz A; Harris R; Izadi S; Измайлов СА; Kasavajhala K; Kovalenko A; Krasny R; Kurtzman T; Lee T; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Man V; Merz KM; Miao Y; Monard G; Nguyen H; Onufriev A; Pan F; Pantano S; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling C; Скрынников HP; Smith J; Swails J; Walker R; Wang J; Wilson L; Wolf RM; Wu X; Xiong Y; Xue Y; York D; Kollman PA, AMBER 2020, University of California, San Francisco. 2020. [Google Scholar]
- 55.Hopkins CW; Le Grand S; Walker RC; Roitberg AE, Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. Journal of Chemical Theory and Computation 2015, 11 (4), 1864–1874. [DOI] [PubMed] [Google Scholar]
- 56.Darden T; York D; Pedersen L, Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics 1993, 98 (12), 10089–10092. [Google Scholar]
- 57.Humphrey W; Dalke A; Schulten K, VMD: visual molecular dynamics. J Mol Graph 1996, 14 (1), 33–8, 27–8. [DOI] [PubMed] [Google Scholar]
- 58.Roe DR; Cheatham TE, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. Journal of Chemical Theory and Computation 2013, 9 (7), 3084–3095. [DOI] [PubMed] [Google Scholar]
- 59.Hunter JD, Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering 2007, 9 (3), 90–95. [Google Scholar]
- 60.Shajahan A; Supekar NT; Gleinich AS; Azadi P, Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30 (12), 981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ghoreishi D; Cerutti DS; Fallon Z; Simmerling C; Roitberg AE, Fast Implementation of the Nudged Elastic Band Method in AMBER. Journal of Chemical Theory and Computation 2019, 15 (8), 4699–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergonzo C; Campbell AJ; Walker RC; Simmerling C, A partial nudged elastic band implementation for use with large or explicitly solvated systems. International Journal of Quantum Chemistry 2009, 109 (15), 3781–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.JÓNSSON H; MILLS G; JACOBSEN KW, Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations, pp 385–404. [Google Scholar]
- 64.Bergonzo C; Campbell AJ; de los Santos C; Grollman AP; Simmerling C, Energetic Preference of 8-oxoG Eversion Pathways in a DNA Glycosylase. Journal of the American Chemical Society 2011, 133 (37), 14504–14506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuznetsov NA; Bergonzo C; Campbell AJ; Li HQ; Mechetin GV; de los Santos C; Grollman AP; Fedorova OS; Zharkov DO; Simmerling C, Active destabilization of base pairs by a DNA glycosylase wedge initiates damage recognition. Nucleic Acids Research 2015, 43 (1), 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lai CT; Li HJ; Yu WX; Shah S; Bommineni GR; Perrone V; Garcia-Diaz M; Tonge PJ; Simmerling C, Rational Modulation of the Induced-Fit Conformational Change for Slow-Onset Inhibition in Mycobacterium tuberculosis InhA. Biochemistry 2015, 54 (30), 4683–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li HJ; Lai CT; Pan P; Yu WX; Liu NN; Bommineni GR; Garcia-Diaz M; Simmerling C; Tonge PJ, A Structural and Energetic Model for the Slow-Onset Inhibition of the Mycobacterium tuberculosis Enoyl-ACP Reductase InhA. Acs Chem Biol 2014, 9 (4), 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li HQ; Endutkin AV; Bergonzo C; Campbell AJ; de los Santos C; Grollman A; Zharkov DO; Simmerling C, A dynamic checkpoint in oxidative lesion discrimination by formamidopyrimidine-DNA glycosylase. Nucleic Acids Research 2016, 44 (2), 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li HQ; Endutkin AV; Bergonzo C; Fu L; Grollman A; Zharkov DO; Simmerling C, DNA Deformation-Coupled Recognition of 8-Oxoguanine: Conformational Kinetic Gating in Human DNA Glycosylase. Journal of the American Chemical Society 2017, 139 (7), 2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Izrailev S; Stepaniants S; Isralewitz B; Kosztin D; Lu H; Molnar F; Wriggers W; Schulten K In Steered Molecular Dynamics, Berlin, Heidelberg, Springer Berlin Heidelberg: Berlin, Heidelberg, 1999; pp 39–65. [Google Scholar]
- 71.Kumar S; Rosenberg JM; Bouzida D; Swendsen RH; Kollman PA, Multidimensional free-energy calculations using the weighted histogram analysis method. 1995, 16 (11), 1339–1350. [Google Scholar]
- 72.Grossfield A WHAM: an implementation of the weighted histogram analysis method. Version 2.0.10. http://membrane.urmc.rochester.edu/content/wham/ (accessed 2020-07-25).
- 73.Shimazaki H; Shinomoto S, A method for selecting the bin size of a time histogram. Neural computation 2007, 19 (6), 1503–27. [DOI] [PubMed] [Google Scholar]
- 74.Le Guilloux V; Schmidtke P; Tuffery P, Fpocket: An open source platform for ligand pocket detection. BMC Bioinformatics 2009, 10 (1), 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmidtke P; Bidon-Chanal A; Luque FJ; Barril X, MDpocket: open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics (Oxford, England) 2011, 27 (23), 3276–85. [DOI] [PubMed] [Google Scholar]
- 76.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004, 25 (13), 1605–12. [DOI] [PubMed] [Google Scholar]
- 77.Cagliani R; Forni D; Clerici M; Sironi M, Computational Inference of Selection Underlying the Evolution of the Novel Coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2. Journal of Virology 2020, 94 (12), e00411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Benson DA; Cavanaugh M; Clark K; Karsch-Mizrachi I; Lipman DJ; Ostell J; Sayers EW, GenBank. Nucleic Acids Res 2013, 41 (Database issue), D36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elbe S; Buckland-Merrett G, Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges 2017, 1 (1), 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Notredame C; Higgins DG; Heringa J, T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 2000, 302 (1), 205–17. [DOI] [PubMed] [Google Scholar]
- 81.Crooks GE; Hon G; Chandonia JM; Brenner SE, WebLogo: a sequence logo generator. Genome Res 2004, 14 (6), 1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kirchdoerfer RN; Wang N; Pallesen J; Wrapp D; Turner HL; Cottrell CA; Corbett KS; Graham BS; McLellan JS; Ward AB, Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Scientific Reports 2018, 8 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lu M; Uchil PD; Li W; Zheng D; Terry DS; Gorman J; Shi W; Zhang B; Zhou T; Ding S; Gasser R; Prévost J; Beaudoin-Bussières G; Anand SP; Laumaea A; Grover JR; Liu L; Ho DD; Mascola JR; Finzi A; Kwong PD; Blanchard SC; Mothes W, Real-Time Conformational Dynamics of SARS-CoV-2 Spikes on Virus Particles. Cell Host & Microbe 2020, 28 (6), 880–891.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pallesen J; Wang N; Corbett KS; Wrapp D; Kirchdoerfer RN; Turner HL; Cottrell CA; Becker MM; Wang L; Shi W; Kong W-P; Andres EL; Kettenbach AN; Denison MR; Chappell JD; Graham BS; Ward AB; McLellan JS, Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proceedings of the National Academy of Sciences 2017, 114 (35), E7348–E7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu H; Wu NC; Yuan M; Bangaru S; Torres JL; Caniels TG; van Schooten J; Zhu X; Lee C-CD; Brouwer PJM; van Gils MJ; Sanders RW; Ward AB; Wilson IA, Cross-Neutralization of a SARS-CoV-2 Antibody to a Functionally Conserved Site Is Mediated by Avidity. Immunity 2020, 53 (6), 1272–1280.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.