

Abstract
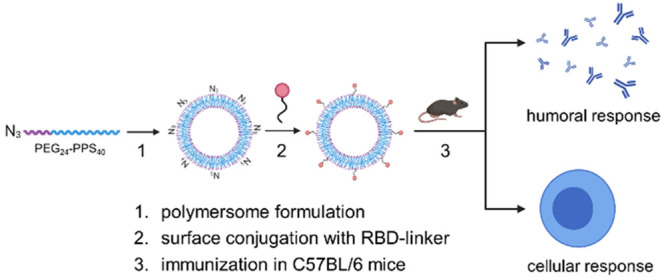
The COVID-19 pandemic underscores the need for rapid, safe, and effective vaccines. In contrast to some traditional vaccines, nanoparticle-based subunit vaccines are particularly efficient in trafficking antigens to lymph nodes, where they induce potent immune cell activation. Here, we developed a strategy to decorate the surface of oxidation-sensitive polymersomes with multiple copies of the SARS-CoV-2 spike protein receptor-binding domain (RBD) to mimic the physical form of a virus particle. We evaluated the vaccination efficacy of these surface-decorated polymersomes (RBDsurf) in mice compared to RBD-encapsulated polymersomes (RBDencap) and unformulated RBD (RBDfree), using monophosphoryl-lipid-A-encapsulated polymersomes (MPLA PS) as an adjuvant. While all three groups produced high titers of RBD-specific IgG, only RBDsurf elicited a neutralizing antibody response to SARS-CoV-2 comparable to that of human convalescent plasma. Moreover, RBDsurf was the only group to significantly increase the proportion of RBD-specific germinal center B cells in the vaccination-site draining lymph nodes. Both RBDsurf and RBDencap drove similarly robust CD4+ and CD8+ T cell responses that produced multiple Th1-type cytokines. We conclude that a multivalent surface display of spike RBD on polymersomes promotes a potent neutralizing antibody response to SARS-CoV-2, while both antigen formulations promote robust T cell immunity.
Short abstract
We developed nanoscale polymer vesicles (polymersomes) decorated with multiple copies of SARS-CoV-2 antigen as a COVID-19 vaccine that elicits neutralizing antibodies and cellular immunity.
Introduction
COVID-19, the disease caused by the novel coronavirus SARS-CoV-2, emerged in late 2019 and was declared a pandemic by the World Health Organization in March 2020. Since its emergence, researchers across the world have sought to rapidly develop vaccine candidates, some of which have received Emergency Use Authorization by the U.S. Food and Drug Administration.1,2 While the first vaccines that entered the clinic were based on nucleic acid technologies, subunit vaccines are gaining attention and have also shown promise in clinical trials.3,4 The primary antigens used in preclinical and clinical vaccine candidates are the spike protein and its constituent receptor-binding domain (RBD). The RBD of the spike protein binds to the ACE-2 receptor on host cell surfaces, enabling viral entry into host cells.5,6
Several highly potent neutralizing antibodies have been isolated that target RBD and prevent viral binding and uptake, making it an attractive vaccine target.7−10 Since RBD is smaller and more stable than the full homotrimeric spike fusion protein, it is also advantageous from a manufacturing and distribution perspective.11 However, RBD has been shown to have lower immunogenicity than the full spike protein or its RBD-containing S1 domain.12,13 Materials science and engineering approaches, particularly strategies involving nanotechnology, may improve RBD immunogenicity and thus aid in the development of next-generation vaccines.14−16 Indeed, several approaches of self-assembling RBD into virus-like particles have resulted in potent neutralizing antibody responses.17−20
To offer robust protection from infection, cellular in addition to humoral responses are needed.21−23 Almost all convalescent individuals show T cell immunity, and the majority have both CD4+ and CD8+ SARS-CoV-2-specific T cells.24−27 Conversely, severe disease is associated with lymphopenia and reduced T cell function.28−30 Furthermore, T cell immunity may be more durable than humoral responses, and T cells are expected to play an important role in immune memory.23,28,31 Therefore, the goals of this study were to improve both humoral and cellular immunogenicity of RBD and compare the efficacy of engineered nanoparticle formulations in order to inform the design of next-generation nanovaccines.
We have previously reported the development of polymersomes (PS) that self-assemble from the oxidation-responsive block copolymer poly(ethylene glycol)-bl-poly(propylene sulfide) (PEG–PPS)32 and shown their efficacy in delivering antigen and adjuvant to dendritic cell endosomes.33 In endolysosomal compartments, the PPS block becomes oxidized, which initiates the restructuring of the PS into micelles and concurrent release of encapsulated payload.33,34 These vaccine nanocarriers have been shown to activate dendritic cells, induce robust T cell immunity, and elicit high antibody titers with broad epitope coverage.33,35,36
In this study, we hypothesized that we could further improve the humoral responses elicited by PS while retaining their ability to induce T cell immunity by engineering them to mimic the physical form of a viral particle through multivalent surface display of antigen. We envisaged that multivalent surface display of RBD would result in enhanced cross-linking and clustering of B cell receptors (BCRs) and subsequent production of neutralizing antibodies. As such, we developed and evaluated PS displaying surface-bound RBD (RBDsurf) and PS encapsulating RBD (RBDencap) adjuvanted with monophosphoryl-lipid-A-encapsulated PS (MPLA PS). MPLA was chosen as an adjuvant due to its hydrophobicity and ability to be encapsulated within the PPS-rich shell of the polymersomes without affecting antigen loading in future single-particle vaccines. Furthermore, as MPLA has been combined with aluminum hydroxide to prolong its retention as a clinical adjuvant,37 we hypothesized that its incorporation into PS would similarly increase its efficacy. Here, we show that mice vaccinated with RBDsurf in combination with MPLA PS in a prime-boost schedule develop high titers of SARS-CoV-2-neutralizing antibodies with robust germinal center responses as well as CD4+ and CD8+ T cell immunity, thus meeting our design criteria.
Results
Formulated Polymersomes Exhibit Long-Term Stability and In Vitro Activity
Having previously encapsulated antigen into PS as nanovaccines,33 here we developed a conjugation strategy to attach antigens to their surface. To create a modular platform that could be generalized to virtually any antigen, we synthesized N3–PEG–PPS (Figure S1), which, when formulated into PS, yields particles displaying clickable surface moieties (Figure 1a). Upon the addition of RBD conjugated to a DBCO-containing linker, we generated PS decorated with RBD (RBDsurf, Figure S2). We also synthesized PEG–PPS (Figure S3) and formulated PS encapsulating RBD (RBDencap) or adjuvant (MPLA PS, Figure 1a). The loading capacities of RBDsurf and RBDencap were 1.57 and 1.75%, respectively, comparable to previous reports of encapsulated ovalbumin,33,35 while the loading capacity of MPLA PS was 6.46% (Table S1). We confirmed the vesicular structure of PS through cryo-electron microscopy (cryoEM) and demonstrated that the different formulations have similar sizes and morphologies (Figures 1b and S4). According to dynamic light scattering (DLS) measurements, the average PS diameter is around 150 nm (Figure 1c,d), which is comparable to the reported size of SARS-CoV-2 particles (60–140 nm).38 The polydispersity index (PDI) of each formulation was less than 0.2, indicative of a relatively homogeneous population of nanovesicles. As indicated by their consistent size and PDI, in addition to the absence of free RBD released into solution, PS remain stable at 4 °C for at least 180 days, which can be beneficial for distribution and shelf life considerations (Figures 1c and S5).
Figure 1.

RBD and MPLA are formulated into stable, biologically active polymersomes (PS). (a) Schematic of formulation of PS. RBD was conjugated to the surface (RBDsurf) or encapsulated inside (RBDencap) of PS, and MPLA was encapsulated in the vesicle membrane (MPLA PS) due to its hydrophobicity. (b) Representative cryo-electron microscopy images of PS, depicting vesicle structure. Scale = 50 nm. (c) Size and polydispersity index (PDI) from dynamic light scattering (DLS) measurements of PS upon formulation and after >180 days at 4 °C. (d) Representative DLS curves of PS. (e) Normalized mean fluorescence intensity (MFI) of AF647 conjugated to free RBD or RBDsurf by flow cytometry showing concentration-dependent binding to HEK-293 cells that express human ACE-2 (HEK-hACE2). Nonlinear regression was used to model data assuming specific binding to one site to determine equilibrium dissociation constants (KD). (f) Dose-dependent secretion of TNFα from cultured murine bone-marrow-derived dendritic cells (BMDCs) stimulated by free MPLA, MPLA PS, or empty PS. Data represent mean ± SD for n = 2 (e) or 3 (f) replicates.
We next characterized the biological activity of the PS formulations in vitro. To confirm that RBD structure is not substantially altered upon conjugation to the PS surface, we quantified its ability to bind to HEK-293 cells that express human ACE-2 (HEK-hACE2, Figure 1e). The normalized mean fluorescence intensity (MFI) versus RBD concentration curves were used to calculate the equilibrium dissociation constants (KD) for free RBD and RBDsurf conjugated to AF647 (AF647-RBDfree and AF647-RBDsurf, respectively). The curves and KD values are in excellent agreement, indicating that surface conjugation to PS does not impact ACE-2 binding of RBD. Empty PS conjugated to AF647 did not bind to HEK-hACE2, and neither PS formulation bound to HEK-293 cells lacking hACE-2 (Figure S6).
Next, we confirmed that MPLA retained its ability to serve as a TLR4 agonist upon formulation in PS with a HEK-Blue TLR4 reporter cell line (Figure S7). To further validate MPLA PS activity in a more physiologically relevant model, we stimulated murine bone-marrow-derived dendritic cells (BMDCs) with free MPLA, MPLA PS, or empty PS, and we measured the subsequent secretion of the proinflammatory cytokines TNFα, IL-6, IL-1α, and IL-1β (Figures 1f and S8). For each cytokine, there was a dose-dependent increase in secretion for free MPLA and MPLA PS with only background levels of secretion for empty PS, indicating that MPLA PS successfully activated antigen presenting cells (APCs) in vitro. Thus, we successfully synthesized two RBD formulations of PS in addition to MPLA PS and showed that they are homogeneous vesicular structures with long-term stability and in vitro biological activity.
All Adjuvanted Formulations Elicit RBD-Specific IgG Responses
Having confirmed that antigen- and adjuvant-loaded PS exhibit their expected bioactivity in vitro, we next evaluated their ability to enhance humoral and cellular immunity in 8-week-old, female C57BL/6 mice compared to RBDfree. We immunized mice via subcutaneous (s.c.) injection in the hocks in a prime-boost schedule 3 weeks apart and monitored antibody titers weekly (Figure 2a). The total RBD-specific IgG is represented by the area under the log-transformed ELISA absorbance curves (AUC), starting at a plasma dilution of 10–2 (see Methods, Figure S9). All adjuvanted groups had significant RBD-binding antibody responses within a week after their first dose, with RBDencap stimulating the highest responses (Figure 2b). The antibody responses in adjuvanted groups either increased gradually or remained constant until a week after the booster, when the mean AUC increased 1.3- to 1.6-fold.
Figure 2.

High levels of RBD-specific IgG antibodies are produced upon PS vaccination. (a) Vaccination schedule consisting of a priming dose followed by a booster 3 weeks later. (b) Total RBD-specific IgG antibodies over time reported as the area under the log-transformed curve (AUC) of absorbance vs dilution. (c) Comparison of RBD-specific IgG isotypes (IgG1, IgG2b, IgG2c, IgG3) on day 28. (d) Ratio of AUCs of IgG2b/IgG1 isotypes. (e) Quantification of RBD-specific IgG+ antibody secreting cells by ELISpot of splenocytes (Dunn’s post-test compared to unadjuvanted RBDfree). (f) Representative ELISpot wells from (e). Data plotted as mean ± SD and represent 1 of 2 experiments with n = 5 mice each. Symbols represent individual mice. (g) Vaccine and blood sampling schedule of long-term kinetics study. (h) Total RBD-specific IgG antibodies over time for the vaccination schedule in (g). Data represent mean ± SD for n = 5 mice. Comparisons were made using one-way ANOVA with Tukey’s post-test unless stated otherwise. #P < 0.0001 compared to unadjuvanted RBDfree.
In order to explore the humoral response in further detail, we then evaluated IgG subtypes of induced antibodies at the study end point (day 28, 1 week postboost). While plasma antibody levels of all adjuvanted groups were similar for IgG1 and IgG3, RBDsurf elicited significantly lower IgG2b and IgG2c antibody responses (Figure 2c). The ratio of IgG2b/IgG1 was then taken as an indication of a Th1/Th2-mediated response.39 While RBDencap and RBDfree + MPLA PS have a ratio of around 1, indicating a balanced Th1/Th2 response, RBDsurf shows a lower ratio of IgG2b to IgG1, indicating a slightly Th2-skewed response (Figure 2d). Since IgA is important for combating respiratory viruses at the mucosal sites, we also measured plasma levels of RBD-specific IgA antibodies.40,41 Although we did not expect high titers of circulating IgA from an s.c. administered vaccine, there were detectable anti-RBD IgA antibodies in all adjuvanted groups, which were significantly higher than the background-level responses elicited by unadjuvanted RBDfree (Figure S10).
We next aimed to determine whether the presence of RBD-specific antibodies from the priming dose affects the drainage of RBDsurf to the lymph nodes during the booster. To accomplish this goal, we compared the accumulation of fluorescently labeled RBDsurf in the lymph nodes of mice that had been previously immunized with RBDsurf or saline in a prime-boost schedule and had mean RBD-specific IgG AUCs of 9.9 and 0.23, respectively. AF647-RBDsurf and MPLA PS were injected s.c. in the hocks, and the brachial and popliteal draining lymph nodes were collected and fluorescently imaged 24 h later (Figure S11a). There was no significant difference in the total radiant efficiency of the lymph nodes between the preimmunized and saline control groups, indicating that the presence of antibodies does not substantially affect PS accumulation in the lymph nodes at this time point (Figure S11b).
Next, to determine if the higher antibody responses of adjuvanted groups stemmed from an expanded number of RBD-specific antibody secreting cells (ASCs), we performed an ex vivo RBD enzyme-linked immunosorbent spot (ELISpot) assay with splenocytes harvested 1 week postboost. All groups receiving adjuvanted RBD showed significantly higher RBD-specific IgG+ ASCs compared to unadjuvanted RBDfree, consistent with plasma antibody levels (Figure 2e,f).
Finally, we evaluated the kinetics and durability of the humoral response to demonstrate the persistence of elicited antibodies (Figure 2g). The RBD-specific IgG AUC for all adjuvanted groups increased until 1 week postboost (week 4) and then remained constant over the subsequent 12 weeks, indicating that the antibody responses stimulated by these vaccine formulations persist for at least 4 months in mice after the initial dose. The results of this later experiment correlate with the two experiments performed according to the timeline in Figure 2a, suggesting that not only are the PS stable upon storage at 4 °C but also maintain their activity. Specifically, our data suggest RBDsurf, RBDencap, and MPLA PS remain active for at least 4, 6, and 10 months, respectively. Taken together, MPLA PS-adjuvanted RBDsurf, RBDencap, and RBDfree all stimulated persistent anti-RBD antibodies with increased frequencies of splenic RBD-specific ASCs while maintaining their activity upon storage at 4 °C for extended durations.
RBD-Surface-Decorated Polymersomes, but Not RBD-Encapsulated Polymersomes, Induce Neutralizing Antibodies
After analyzing the quantity of RBD-specific antibodies produced by the vaccine candidates, we next sought to determine their neutralizing capacity and breadth of epitope recognition. Neutralizing antibodies were assessed against SARS-CoV-2 infection of Vero E6 cells in vitro. Although all adjuvanted groups elicited similarly high titers of RBD-binding IgG antibodies (105–107, Figure S12a), only RBDsurf neutralized the virus to a greater extent than unadjuvanted RBDfree at a plasma dilution of 10–2.11 (lower limit of quantification, Figure 3a). We then quantified the virus neutralization titer (VNT) as the dilution at which 50% of SARS-CoV-2-mediated cell death is neutralized. There was no significant difference between VNTs of human convalescent plasma and RBDsurf plasma, and both groups induced higher VNTs compared to unadjuvanted RBDfree (Figure 3b). Furthermore, the median VNT elicited by RBDsurf was 2.45, which falls within the FDA classification of “high titer” for convalescent plasma therapy (>2.40).42 To ensure reproducibility, the neutralization assay was repeated with 3 different cohorts of n = 5 mice each, and no significant differences were observed between the resulting VNTs (Figure S12b).
Figure 3.

Antibodies induced by vaccination with RBDsurf + MPLA PS are neutralizing and localized to the receptor-binding motif. (a) Plasma from mice 1 week postboost was tested for its ability to neutralize SARS-CoV-2 infection of Vero E6 cells in vitro. Percent neutralization for multiple plasma dilutions normalized to cells without virus (100%) or without plasma (0%). Data plotted as mean ± SEM for n = 5 human convalescent samples (human conv.) or 10–15 mice. Comparisons to unadjuvanted RBDfree were made for lowest dilution (10–2.11) using one-way ANOVA with Dunnett’s post-test. (b) Virus neutralization titers, representing the plasma dilution at which 50% of SARS-CoV-2-mediated cell death is neutralized. Dashed line: lower limit of quantification (LLOQ = 2.11). For values below the LLOQ, LLOQ/2 values were plotted. Solid line: FDA recommendation for “high titer” classification (=2.40). Comparisons were made using a Kruskal–Wallis nonparametric test with Dunn’s post-test or a Wilcoxon signed rank test (‡ ns, P > 0.05 compared to hypothetical value of 2.40). Symbols represent individual mice. (c) Pooled plasma was then tested for antibody binding to linear epitopes using overlapping 15-amino-acid peptides with 5-amino-acid offsets, spanning the entire RBD sequence. The x-axis represents sequential peptide number within the RBD amino acid sequence showing the position of the receptor-binding motif (RBM), and the y-axis represents average luminescence for each peptide epitope. (d) 3D structure and amino acid sequence of RBD with the RBM underlined and main peptide sequences recognized by mouse plasma highlighted in pink (Protein Data Bank Entry 7DDD).
We next aimed to determine whether differences in neutralizing ability resulted from the epitope recognition breadth elicited by each vaccine formulation. To test this, we mapped the epitopes recognized by vaccine-elicited antibodies using a linear peptide array from the full-length RBD sequence. While IgG antibodies elicited by RBDsurf primarily recognized linear epitopes concentrated within the receptor-binding motif of RBD (RBM; aa 438–508), RBDencap and RBDfree + MPLA PS exhibited broader linear epitope diversity (Figures 3c,d and S13). Within RBD, the RBM is particularly critical for interacting with ACE-2, so antibodies specific for this region may have potent neutralizing potential.43,44
Because RBDsurf appeared to offer the advantage of improved neutralizing activity while RBDencap offered epitope diversity, we asked if coadministration would synergize to further enhance protection. To test this, we mixed RBDsurf and RBDencap (with the total RBD dose remaining constant) with MPLA PS and treated mice using the same vaccination schedule. As an additional control, we also investigated the humoral response of RBDfree adjuvanted with free MPLA. While both groups produced high RBD-specific IgG AUCs, neither exhibited neutralizing potential against the SARS-CoV-2 virus above background levels (Figure S14a,b). Analysis of the peptide arrays for these groups shows the presence of high-intensity-binding antibodies outside of the RBM (Figure S14c).
In summary, while all adjuvanted groups elicit high titers of RBD-specific antibodies, only RBDsurf generated neutralizing antibodies against SARS-CoV-2 at titers comparable to human convalescent plasma. Additionally, these antibodies uniquely bound to linear epitopes localized within the RBM, while all other groups produced antibodies with greater epitope breadth.
All Adjuvanted Formulations Increase Tfh and B Cell Activation in the dLN
Given the differences in antibody responses and neutralizing activity elicited by RBDsurf versus RBDencap vaccination, we further investigated the phenotypes of the B and T cells involved in the initiation of the humoral immune response. All adjuvanted groups showed trends of higher frequencies of T follicular helper cells (Tfh; CD4+ CXCR5+ BCL6+) in the injection site draining lymph nodes (dLNs) 1 week postboost compared to unadjuvanted RBDfree (Figures 4a and S15), although differences were only statistically significant for RBDfree + MPLA PS. Interestingly, a greater percentage of Tfh cells in all adjuvanted groups highly upregulated expression of ICOS, a costimulatory receptor important in Tfh activation and maintenance (Figure 4b).45
Figure 4.

CD4+ T follicular helper cells (Tfh) and B cells are activated by PS vaccine 1 week postboost in the injection site draining lymph nodes (dLN). (a) Tfh cells (CXCR5+ BCL6+) of vaccinated mice quantified via flow cytometry as a percent of live CD4+ T cells. (b) Highly activated ICOS+ Tfh cells quantified as percent of Tfh cells. (c) Germinal center B cells (CD38– GL7+) quantified as a percentage of total B cells (B220+ CD19+). (d) Plasmablasts (B220+ CD138+) quantified as a percentage of total dLN cells. (e) Germinal center B cells quantified as a percentage of RBD-specific B cells. (f) Plasmablasts quantified as a percentage of RBD-specific dLN cells. (g,h) Concatenated flow cytometry contour plots for n = 5 mice/group showing RBD-specific GC B cells (g) or plasmablasts (h). Data plotted as mean ± SD and represent 1 of 2 experiments with n = 5 mice each. Symbols represent individual mice. Comparisons to unadjuvanted RBDfree were made using one-way ANOVA with Dunn’s post-test.
Following activation by Tfh cells, naïve B cells can either undergo a germinal center (GC)-dependent response, in which they become GC B cells and undergo cycles of class-switching and somatic hypermutation (SHM) before differentiation into long-lived plasma cells and class-switched memory B cells, or they can differentiate into short-lived plasmablasts or IgM memory cells in a GC-independent response.46 A stronger GC response is valuable in vaccination, because it results in higher affinity and longer-lived antibody production.47 Overall frequencies of B cells (CD19+ B220+) were unaffected across groups, but there were significantly lower frequencies of naïve IgD+ B cells in the adjuvanted groups compared to unadjuvanted RBDfree (Figures S16 and S17). All adjuvanted groups generated GC responses, characterized by increased frequencies of GC B cells (CD38– GL7+) in the dLN compared to unadjuvanted RBDfree (Figure 4c). Both RBDencap and RBDfree + MPLA PS formulations, but not RBDsurf, significantly increased the frequencies of plasmablasts (B220+ CD138+) in the dLN compared to unadjuvanted RBDfree (Figure 4d).
To determine the antigen specificity of the B cells, we developed a set of fluorescent RBD protein tetrameric probes. To ensure selectivity, B cells were considered RBD-specific if they were double-positive for both PE- and APC-conjugated RBD tetramers and negative for the nonspecific control protein tetramer (Figure S18). RBDfree + MPLA PS was the only formulation to significantly increase the frequency of RBD-specific B cells in the dLN (Figure S19). We next sought to further determine the phenotype of these RBD-specific B cells. RBDsurf, unlike the other adjuvanted formulations, led to a significantly higher fraction of RBD-specific B cells with GC B cell phenotype, suggesting a more robust GC response to RBD (Figure 4e). RBDsurf was also the only formulation with a significantly lower fraction of plasmablasts within the RBD-specific B cell subset compared to unadjuvanted RBDfree (Figure 4f). These differences are also visually apparent in pooled flow cytometry plots for RBD-specific GC B cells (Figure 4g) and plasmablasts (Figure 4h). In summary, all adjuvanted formulations of RBD increased activation of Tfh cells and GC B cells in the dLN, but within the RBD-specific B cell population, only RBDsurf generated a higher fraction of GC B cells and a lower fraction of plasmablasts.
Vaccination with Polymersome-Formulated RBD Generates RBD-Specific Th1 T Cell Responses
Having demonstrated that our PS vaccines can generate strong humoral responses, we next sought to determine their capacity to generate robust CD8+ and CD4+ T cell immunity. In order to assess the RBD-specific T cell response, we isolated cells from the dLNs of vaccinated mice 1 week postboost. Prior to intracellular staining, cells were restimulated with RBD peptide pools for 6 h. The RBD-specific response was quantified by subtracting the signal from cells incubated in media alone from those incubated with RBD peptide pools (Figure S20). Only PS-formulated RBD groups RBDsurf and RBDencap generated significantly higher frequencies of IFNγ+, bifunctional IFNγ+TNFα+, and IL-2+ CD8+ T cells compared to unadjuvanted RBDfree (Figure 5a). Similar trends were seen in the CD4+ T cell compartment. Treatment with RBDsurf and RBDencap but not RBDfree + MPLA PS led to significantly higher frequencies of IFNγ+ and IL-2+ CD4+ T cells, although the increase in bifunctional IFN γ+TNFα+ was not statistically significant (Figure 5b). The production of these cytokines implies a Th1 T cell response, which has been correlated with less severe cases of SARS-CoV infection.21
Figure 5.

Vaccination with polymersome-formulated RBD improves antigen-specific T cell responses in mice. Cells isolated from dLN of PS-vaccinated mice 1 week postboost were restimulated ex vivo for 6 h with RBD peptide pools or 3 days with full RBD protein and analyzed by flow cytometry or multiplexed ELISA, respectively. (a,b) Percentages of cytokine-positive CD8+ T cells (a) and CD4+ T cells (b) in response to RBD peptide pools, subtracting unstimulated controls. (c) Proinflammatory cytokine levels in the supernatant measured after restimulation with whole RBD protein. Data plotted as mean ± SD and represent 1 of 2 experiments with n = 5 mice each. Symbols represent individual mice. Comparisons to unadjuvanted RBDfree were made using one-way ANOVA with Dunn’s post-test.
For further validation of the cytokine response, cells from the dLN isolated from the same vaccinated mice were restimulated with full RBD protein ex vivo for 3 days, followed by quantification of cytokines in the supernatant. The RBD-specific response was quantified by subtracting unstimulated signal from stimulated signal as above. The levels of Th1-type cytokines detected were consistent with intracellular staining data. Levels of IFNγ and IL-2 were modestly but not significantly increased in the RBDsurf and RBDencap groups compared to the RBDfree group, while cells from RBDencap-treated mice secreted TNFα at significantly higher levels than unadjuvanted RBDfree (Figure 5c). RBDencap also led to increased production of IL-6 and IL-10, which are pleiotropic cytokines known to be secreted during Th1 responses (Figure 5c).48,49 Levels of secreted Th2-type cytokines were also measured. More IL-4 was produced in the supernatant of samples treated with RBDsurf and RBDencap compared to RBDfree, albeit at an overall low concentration. There was no significant elevation of IL-5 secretion across any of the treatment groups compared to the saline control and no detectable levels of IL-13 in any sample (Figure S21). In summary, vaccination with RBD delivered via polymersome formulations generated stronger RBD-specific Th1-type CD8+ and CD4+ T cell responses than unadjuvanted RBDfree, while RBDfree + MPLA PS did not.
Discussion
In this study, we developed antigen-decorated, oxidation-sensitive polymersomes that mimic virus particles as next-generation nanovaccines. While all adjuvanted formulations generated long-lived RBD-binding IgG responses, surface conjugation of antigen was necessary to generate neutralizing antibodies against SARS-CoV-2. More generally, this comparison of antigen formulation on the quality of immune response offers valuable insight into vaccine design, demonstrating the benefits of surface antigen display.
The differences in the immune responses elicited by the two PS antigen formulations, RBDsurf and RBDencap, suggest that surface display of antigen leads to stronger GC responses, while PS-encapsulated antigen elicits more predominantly an extrafollicular response. Though RBD-specific GC B cells are present in the dLN after treatment with both formulations, a much higher percentage of the RBD-specific B cells recovered after vaccination with RBDsurf exhibited a GC B cell phenotype. This higher percentage could be due to the induction of higher numbers of RBD-specific GC B cells after vaccination with RBDsurf or an increase in their affinity, leading to easier detection via RBD protein tetramer staining. Both possibilities suggest a more robust GC response, as GC responses are necessary for an efficient SHM leading to increased B cell affinity.50 Evidence for affinity maturation due to SHM also includes the relatively few epitopes on the RBD linear peptide array to which IgG from RBDsurf-treated groups were specific compared to the other adjuvanted groups. Clonal bursts in GC responses can lead to rapid expansion of high-affinity SHM variants and loss of overall clonal diversity.51 A difference in the affinity of RBD-specific IgG generated by these vaccines may also explain the neutralization ability of the plasma after vaccination with RBDsurf but not RBDencap. An interesting finding is that when RBDsurf and RBDencap were codelivered, the resulting antibodies recognized a greater diversity of epitopes compared to RBDsurf alone and were also non-neutralizing. These data suggest that in the context of PS vaccines, epitope breadth and neutralization capacity may be mutually exclusive. Therefore, the decision of encapsulating antigen versus displaying it on the PS surface should be determined based on vaccine design criteria.
Further data that suggest that RBDencap elicits a more extrafollicular response include the fast initial antibody response after priming by RBDencap, which resulted in RBD-specific IgG AUC 1 week after the priming dose that was significantly higher than that of RBDsurf. In an extrafollicular B cell response, B cells can differentiate immediately into plasmablasts and begin secreting antibodies after initial T cell help, whereas B cells that enter GCs delay the antibody response by several days.46 The preferential differentiation into plasmablasts rather than GC B cells was also evident in the increased fraction of plasmablasts within the RBD-specific B cell population generated by RBDencap. In vaccine development, the generation of robust GC reactions is typically preferable to extrafollicular responses, because GC formation usually results in higher affinity B cells, increased memory B cells, and increased long-lived plasma cells in the bone marrow.46
A potential rationale for these differences could be due to the multivalent display of RBD on RBDsurf. Multimeric structures have been associated with the induction of potent antibody responses due to BCR cross-linking in the presence of CD4+ T cell help.52 In our studies, RBDsurf most likely efficiently exposes for the BCR interaction the conformational epitopes of RBD reported to be targeted by neutralizing antibodies in plasma of convalescent or vaccinated individuals.7−10 Furthermore, increased antigen valency has been shown to enhance early activation and proliferation of antigen-specific B cells as well as B cell accumulation at the T-B border, leading to increased differentiation of antigen-specific B cells into GC B cells and plasma cells.53 Thus, the multimerization of RBD on the PS surface in addition to its increased availability to B cells led to an improved functional quality of humoral response compared to encapsulated or unformulated RBD.
The different formulations of RBD polymersomes likely access the follicular B cells via different mechanisms. In order for B cells to encounter their cognate antigen within the follicle, the antigen must first enter the subcapsular sinus of the lymph node and cross the subcapsular sinus barrier into the follicles. Particles 20–200 nm have been shown to gain ready access to lymphatics;54 thus, both RBDsurf and RBDencap particles likely enter into the draining lymph node with the afferent lymph into the subcapsular sinus after s.c. injection. Increased antigen exposure to B cells upon vaccination with RBDsurf may be due to protease activity in the sinus, which could cleave the surface-conjugated RBD from the larger particles, allowing the smaller proteins to diffuse into the follicular space, while the encapsulated RBD remains in the particle, which is too large to diffuse.55 Alternatively, the PS may be taken up by the subcapsular sinus macrophages. These macrophages are nondegradative phagocytes and have been shown to uptake full particulate antigens and subsequently present them to B cells on the follicular side of the subcapsular sinus.56 Presentation of antigen via subcapsular sinus macrophages is known to be increased for opsonized antigens via binding to complement receptor 3 and Fc receptor IIb.57 Immune-complex formation on RBDsurf upon a boost due to circulating RBD-specific antibodies could explain an increase in GC responses compared to RBDencap, which does not have exposed RBD-specific antibody epitopes. Cognate B cells in the follicle that encounter RBDsurf may become activated at the follicular border, avoiding the need to be trafficked into the B cell follicles.58 RBDencap particles, on the other hand, shield encapsulated RBD from B cell recognition and thus rely on uptake and degradation in APCs.
The type of immune response to SARS-CoV-2 may have important implications in how the infection is cleared and should be considered in vaccine design.21 Less severe cases of SARS-CoV were associated with increased Th1-type cell responses.59,60 In contrast, Th2-type responses were associated with increased pathology due to antibody-dependent enhancement, and several vaccine formulations against SARS-CoV showed signs of immunopathology due to Th2 cell-mediated eosinophil infiltration.61,62 For our vaccine formulations, RBDencap generated a IgG2b/IgG1 ratio around 1, suggesting a balance between type 1 and type 2 immunity, whereas RBDsurf generated a response with a slight IgG1 bias, suggestive of a more Th2-skewed response. The IgG2b/IgG1 ratio of RBDsurf is comparable to the IgG2a/c/IgG1 ratio reported for the mRNA-1273 SARS-CoV-2 vaccine although the mouse strain is different.63 Additionally, the BNT162b mRNA vaccine elicited large numbers of IgG1+ B cells in the spleen and LN of vaccinated mice.64 Both of these mRNA vaccines elicit strong Th1 cytokine responses in addition to the production of IgG1 antibodies. Similarly, both RBDsurf and RBDencap generated significant levels of the Th1-type cytokines IFNγ, TNFα, and IL-2, likely resulting from the use of MPLA as an adjuvant.65 RBDsurf did not result in an increase in Th2-type cytokines upon restimulation compared to RBDencap. Therefore, we conclude that RBDsurf promotes a balanced Th1/Th2 response, and the risk of adverse events related to Th2-type responses is low.
The PS-formulated RBD vaccines were able to generate stronger Th1-driven CD4+ and CD8+ T cell responses compared to RBDfree. We previously demonstrated that as an antigen delivery vehicle, PEG–PPS polymersomes improved cross-presentation to CD8+ T cells.33 This increased T cell response likely occurs as a result of both enhanced APC targeting and rapid endosomal antigen release.33 APCs uptake membrane-impermeable nanocarriers such as PS more efficiently than other cell types due to their constitutive macropinocytosis.66 Once endocytosed, PEG–PPS polymersomes require only a small amount of oxidation to release encapsulated antigen, and payload delivery to the cytosol is not restricted to endosomal compartments with reductive or acidic conditions.33 Unlike antigen-encapsulated PS, however, acidification may be important for proteolytic degradation of RBDsurf and translocation to the cytosol.67,68 Additionally, peptides derived from large antigen particles have been found to enter the cross-presentation pathways more efficiently than those derived from soluble antigens, which may provide rationale for the enhanced CD8+ response of RBDsurf.69 Therefore, antigen formulation using PS to improve T cell responses could be beneficial in the engineering of future vaccines against cancer or other infectious diseases for which T cell immune responses are thought to be necessary for protection, such as herpesviruses, human immunodeficiency virus, and hepatitis C virus.70
In summary, we have demonstrated that a polymersome-based antigen and adjuvant delivery system generates robust humoral immunity and neutralizing antibody titers as well as T cell responses against a key SARS-CoV-2 vaccine target, the RBD of the spike protein. This platform technology is amenable to a wide variety of antigens and formulated or soluble adjuvants. Once the type and dose of adjuvant has been optimized for a given application, a single particle could be used to deliver both antigen and adjuvant to APCs in the injection site draining lymph nodes. Additionally, multiple antigens, for example from different viruses or different strains or variants of the same virus, could be conjugated to the same particle as a strategy to induce cross-reactive neutralizing antibodies.20 Importantly, surface-decorated and antigen-encapsulated polymersomes remained stable and active at 4 °C for at least 6 and 4 months, respectively. Vaccines that exhibit long-term stability without requiring subzero temperatures will likely be important for widespread vaccine distribution, for example to rural populations or developing nations with poor cold chain networks. The evaluation of RBD-decorated polymersomes presented here could thus provide insight into the next generation of stable formulations of nanovaccines to combat the current COVID-19 pandemic as well as future viral outbreaks.
Acknowledgments
We are grateful for funding support through a pilot project grant from the Chicago Immunoengineering Innovation Center of the University of Chicago as well as the Chicago Biomedical Consortium COVID-19 Response Award (# CR-002) to M.A.S. In addition, a number of researchers were supported by fellowships via the NIH NHLBI (# T32-HL007605 to L.R.V.), Canadian Institutes of Health Research (#201910MFE-430736-73744 to N.M.), University of Chicago Comprehensive Cancer Center (Sigal Fellowship in Immuno-Oncology to N.M.), NIH NCI (# F30-CA221250 to M.R.S.), NIH NIGMS (# T32-GM007281 to M.R.S.), and NIH NIAID (# T32-AI007090 to T.M.M. and A.C.T.). We are grateful to the laboratory of Florian Krammer (Icahn School of Medicine at Mount Sinai, New York City, NY) for providing plasmids coding for the spike RBD, produced with support from the NIH NIAID (Contract # HHSN272201400008C). We are also grateful to the groups of Jesse Bloom (Fred Hutchinson Cancer Research Center, Seattle, WA) and Ali Ellebedy (Washington University School of Medicine, St. Louis, MO) for contributing reagents via the NIH NIAID BEI Resources repository. We acknowledge helpful discussions with Patrick C. Wilson, Jenna J. Guthmiller, Anne I. Sperling, and Aaron Esser-Kahn (University of Chicago, Chicago, IL) and with Robert Baker and David Boltz (Illinois Institute of Technology Research Institute, Chicago, IL) that were instrumental to experimental planning and model development. We acknowledge Suzana Gomes and Tera Lavoie for technical assistance. Parts of this work were carried out at the Cytometry and Antibody Technology Core Facility (Cancer Center Support Grant P30CA014599), the Soft Matter Characterization Facility, the Mass Spectrometry Facility (NSF instrumentation grant CHE-1048528), the Nuclear Magnetic Resonance Facility, the Advanced Electron Microscopy Facility (RRID:SCR_019198), and the Human Immunologic Monitoring Facility (RRID:SCR_017916) at the University of Chicago.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c00596.
Methods, Figures S1–S21, Tables S1–S4 (PDF)
Author Contributions
¶ L.R.V., R.P.W., S.C., M.M.R., and R.W. contributed equally to this work
Author Contributions
L.R.V., R.P.W., S.C., M.M.R., R.W., S.S.Y., M.K., M.A.S., and J.A.H. conceived the project and designed the research strategy. L.R.V., R.P.W., S.C., M.M.R., R.W., L.T.G., A.T.A., P.S.B., N.M., T.M.M., M.S.S., M.N., A.M., E.B., A.S., E.A.W., A.C.T., and J.W.R. performed experiments. L.R.V., R.P.W., S.C., M.M.R., A.T.A., P.S.B., T.M.M., and M.S.S. performed data analysis. P.S.B., V.N., K.F., S.D., and G.R. performed or advised on SARS-CoV-2 neutralization assays. M.R.S., J.L.L., and M.V.T. advised on surface modification of polymersomes. B.M. contributed the HEK-293-hACE2 cell line. L.R.V. and R.P.W. wrote the manuscript with contributions from S.C., S.S.Y., M.K., M.A.S., and J.A.H. All authors reviewed and approved the final version of the manuscript.
The authors declare the following competing financial interest(s): M.A.S. and J.A.H. have patents related to the polymersome technology and interests in LantaBio, which has licensed those patents.
Supplementary Material
References
- Le T. T.; Cramer J. P.; Chen R.; Mayhew S. Evolution of the COVID-19 vaccine development landscape. Nat. Rev. Drug Discovery 2020, 19, 667–668. 10.1038/d41573-020-00151-8. [DOI] [PubMed] [Google Scholar]
- Kyriakidis N. C.; López-Cortés A.; González E. V.; Grimaldos A. B.; Prado E. O. SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. npj Vaccines 2021, 6, 28. 10.1038/s41541-021-00292-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond P.; Hatchuel L.; Dong M.; Ma B.; Hu B.; Smolenov I.; Li P.; Liang P.; Han H. H.; Liang J.; Clemens R. Safety and immunogenicity of S-Trimer (SCB-2019), a protein subunit vaccine candidate for COVID-19 in healthy adults: a phase 1, randomised, double-blind, placebo-controlled trial. Lancet 2021, 397, 682–694. 10.1016/S0140-6736(21)00241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keech C.; Albert G.; Cho I.; Robertson A.; Reed P.; Neal S.; Plested J. S.; Zhu M.; Cloney-Clark S.; Zhou H.; et al. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N. Engl. J. Med. 2020, 383, 2320–2332. 10.1056/NEJMoa2026920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P.; Yang X.-L.; Wang X.-G.; Hu B.; Zhang L.; Zhang W.; Si H.-R.; Zhu Y.; Li B.; Huang C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Su B.; Guo X.; Sun W.; Deng Y.; Bao L.; Zhu Q.; Zhang X.; Zheng Y.; Geng C.; et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell 2020, 182, 73–84.e16. 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zost S. J.; Gilchuk P.; Case J. B.; Binshtein E.; Chen R. E.; Nkolola J. P.; Schäfer A.; Reidy J. X.; Trivette A.; Nargi R. S.; et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 2020, 584, 443–449. 10.1038/s41586-020-2548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R.; Shan C.; Duan X.; Chen Z.; Liu P.; Song J.; Song T.; Bi X.; Han C.; Wi L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- Liu L.; Wang P.; Nair M. S.; Yu J.; Rapp M.; Wang Q.; Luo Y.; Chan J. F-W.; Sahi V.; Figueroa A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- Sternberg A.; Naujokat C. Structural features of coronavirus SARS-CoV-2 spike protein: Targets for vaccination. Life Sci. 2020, 257, 118056. 10.1016/j.lfs.2020.118056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L.; Gao G. F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. 10.1038/s41577-020-00480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Wang L.; Cao H.; Liu C. SARS-CoV-2 S1 is superior to the RBD as a COVID-19 subunit vaccine antigen. J. Med. Virol. 2021, 93, 892–898. 10.1002/jmv.26320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine D. J.; Read B. J. Shaping humoral immunity to vaccines through antigen-displaying nanoparticles. Curr. Opin. Immunol. 2020, 65, 1–6. 10.1016/j.coi.2020.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M. D.; Shukla S.; Chung Y. H.; Beiss V.; Chan S. K.; Ortega-Rivera O. A.; Wirth D. M.; Chen A.; Sack M.; Pokorski J. K.; Steinmetz N. F. COVID-19 vaccine development and a potential nanomaterial path forward. Nat. Nanotechnol. 2020, 15, 646–655. 10.1038/s41565-020-0737-y. [DOI] [PubMed] [Google Scholar]
- Singh A. Eliciting B cell immunity against infectious diseases using nanovaccines. Nat. Nanotechnol. 2021, 16, 16–24. 10.1038/s41565-020-00790-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T. K.; Rijal P.; Rahikainen R.; Keeble A. H.; Schimanski L.; Hussain S.; Harvey R.; Hayes J. W. P.; Edwards J. C.; McLean R. K.; et al. A COVID-19 vaccine candidate using SpyCatcher multimerization of the SARS-CoV-2 spike protein receptor-binding domain induces potent neutralising antibody responses. Nat. Commun. 2021, 12, 542. 10.1038/s41467-020-20654-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.-F.; Sun C.; Zhuang Z.; Yuan R.-Y.; Zheng Q.; Li J.-P.; Zhou P.-P.; Chen X.-C.; Liu Z.; Zhang X.; et al. Rapid Development of SARS-CoV-2 Spike Protein Receptor-Binding Domain Self-Assembled Nanoparticle Vaccine Candidates. ACS Nano 2021, 15, 2738–2752. 10.1021/acsnano.0c08379. [DOI] [PubMed] [Google Scholar]
- Walls A. C.; Fiala B.; Schäfer A.; Wrenn S.; Pham M. N.; Murphy M.; Tse L. V.; Shehata L.; O’Connor M. A.; Chen C.; et al. Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for SARS-CoV-2. Cell 2020, 183, 1367–1382.e17. 10.1016/j.cell.2020.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A. A.; Gnanapragasam P. N. P.; Lee Y. E.; Hoffman P. R.; Ou S.; Kakutani L. M.; Keeffe J. R.; Wu H.-J.; Howarth M.; West A. P.; et al. Mosaic nanoparticles elicit cross-reactive immune responses to zoonotic coronaviruses in mice. Science 2021, 371, 735–741. 10.1126/science.abf6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyanathan M.; Afkhami S.; Smaill F.; Miller M. S.; Lichty B. D.; Xing Z. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 2020, 20, 615–632. 10.1038/s41577-020-00434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox R. J.; Brokstad K. A. Not just antibodies: B cells and T cells mediate immunity to COVID-19. Nat. Rev. Immunol. 2020, 20, 581–582. 10.1038/s41577-020-00436-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette A.; Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. 10.1016/j.cell.2021.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Wherry E. J. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020, 20, 529–536. 10.1038/s41577-020-0402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine T.; Perez-Potti A.; Rivera-Ballesteros O.; Strålin K.; Gorin J.-B.; Olsson A.; Llewellyn-Lacey S.; Kamal H.; Bogdanovic G.; Muschiol S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. 10.1016/j.cell.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A.; Weiskopf D.; Ramirez S. I.; Mateus J.; Dan J. M.; Moderbacher C. R.; Rawlings S. A.; Sutherland A.; Premkumar L.; Jadi R. S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Mentzer A. J.; Liu G.; Yao X.; Yin Z.; Dong D.; Dejnirattisai W.; Rostron T.; Supasa P.; Liu C.; et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacius A.; Tischer-Zimmermann S.; Dragon A. C.; Gussarow D.; Vogel A.; Krettek U.; Gödecke N.; Yilmaz M.; Kraft A. R. M.; Hoeper M. M.; et al. COVID-19 immune signatures reveal stable antiviral T cell function despite declining humoral responses. Immunity 2021, 54, 340–354. e6 10.1016/j.immuni.2021.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao B.; Wang C.; Tan Y.; Chen X.; Liu Y.; Ning L.; Chen L.; Li M.; Liu Y.; Wang G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.; Wu D.; Guo W.; Cao Y.; Huang D.; Wang H.; Wang T.; Zhang X.; Chen H.; Yu H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 2020, 130, 2620–2629. 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann D. M.; Boyton R. J. SARS-CoV-2 T cell immunity: Specificity, function, durability, and role in protection. Sci. Immunol. 2020, 5, eabd6160. 10.1126/sciimmunol.abd6160. [DOI] [PubMed] [Google Scholar]
- Napoli A.; Valentini M.; Tirelli N.; Müller M.; Hubbell J. A. Oxidation-responsive polymeric vesicles. Nat. Mater. 2004, 3, 183–189. 10.1038/nmat1081. [DOI] [PubMed] [Google Scholar]
- Scott E. A.; Stano A.; Gillard M.; Maio-Liu A. C.; Swartz M. A.; Hubbell J. A. Dendritic cell activation and T cell priming with adjuvant- and antigen-loaded oxidation-sensitive polymersomes. Biomaterials 2012, 33, 6211–6219. 10.1016/j.biomaterials.2012.04.060. [DOI] [PubMed] [Google Scholar]
- Cerritelli S.; O’Neil C. P.; Velluto D.; Fontana A.; Adrian M.; Dubochet J.; Hubbell J. A. Aggregation Behavior of Poly(ethylene glycol-bl-propylene sulfide) Di-and Triblock Copolymers in Aqueous Solution. Langmuir 2009, 25, 11328–11335. 10.1021/la900649m. [DOI] [PubMed] [Google Scholar]
- Stano A.; Scott E. A.; Dane K. Y.; Swartz M. A.; Hubbell J. A. Tunable T cell immunity towards a protein antigen using polymersomes vs. solid-core nanoparticles. Biomaterials 2013, 34, 4339–4346. 10.1016/j.biomaterials.2013.02.024. [DOI] [PubMed] [Google Scholar]
- Galan-Navarro C.; Rincon-Restrepo M.; Zimmer G.; Ollmann Saphire E.; Hubbell J. A.; Hirosue S.; Swartz M. A.; Kunz S. Oxidation-sensitive polymersomes as vaccine nanocarriers enhance humoral responses against Lassa virus envelope glycoprotein. Virology 2017, 512, 161–171. 10.1016/j.virol.2017.09.013. [DOI] [PubMed] [Google Scholar]
- Garçon N.; Morel S.; Didierlaurent A.; Descamps D.; Wettendorff M.; Van Mechelen M. Development of an AS04-Adjuvanted HPV Vaccine with the Adjuvant System Approach. BioDrugs 2011, 25, 217–226. 10.2165/11591760-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Zhu N.; Zhang D.; Wang W.; Li X.; Yang B.; Song J.; Zhao X.; Huang B.; Shi W.; Lu R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T. L.; Bossie A.; Sanders V. M.; Fernandez-Botran R.; Coffman R. L.; Mosmann T. R.; Vitetta E. S. Regulation of antibody isotype secretion by subsets of antigen-specific helper T cells. Nature 1988, 334, 255–258. 10.1038/334255a0. [DOI] [PubMed] [Google Scholar]
- Chao Y. X.; Rötzschke O.; Tan E.-K. The role of IgA in COVID-19. Brain, Behav., Immun. 2020, 87, 182–183. 10.1016/j.bbi.2020.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renegar K. B.; Small P. A.; Boykins L. G.; Wright P. F. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J. Immunol. 2004, 173, 1978–1986. 10.4049/jimmunol.173.3.1978. [DOI] [PubMed] [Google Scholar]
- Hinton D. M.Convalescent Plasma EUA Letter of Authorization, 2021.
- Yi C.; Sun X.; Ye J.; Ding L.; Liu M.; Yang Z.; Lu X.; Zhang Y.; Ma L.; Gu W.; et al. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. 10.1038/s41423-020-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Ye G.; Shi K.; Wan Y.; Luo C.; Aihara H.; Geng Q.; Auerbach A.; Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y. S.; Kageyama R.; Eto D.; Escobar T. C.; Johnston R. J.; Monticelli L.; Lao C.; Crotty S. ICOS Receptor Instructs T Follicular Helper Cell versus Effector Cell Differentiation via Induction of the Transcriptional Repressor Bcl6. Immunity 2011, 34, 932–946. 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkaya M.; Kwak K.; Pierce S. K. B cell memory: building two walls of protection against pathogens. Nat. Rev. Immunol. 2020, 20, 229–238. 10.1038/s41577-019-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama H.; Okada S.; Hatano M.; Takahashi Y.; Takeda N.; Ichii H.; Takemori T.; Kuroda Y.; Tokuhisa T. Memory B Cells without Somatic Hypermutation Are Generated from Bcl6-Deficient B Cells. Immunity 2002, 17, 329–339. 10.1016/S1074-7613(02)00387-4. [DOI] [PubMed] [Google Scholar]
- Mocellin S.; Marincola F. M.; Young H. A. Interleukin-10 and the immune response against cancer: a counterpoint. J. Leukocyte Biol. 2005, 78, 1043–1051. 10.1189/jlb.0705358. [DOI] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res. Ther. 2006, 8, S2. 10.1186/ar1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster J. G.; Allen C. D. C. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 2019, 177, 524–540. 10.1016/j.cell.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesin L.; Ersching J.; Victora G. D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. 10.1016/j.immuni.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohsen M. O.; Zha L.; Cabral-Miranda G.; Bachmann M. F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. 10.1016/j.smim.2017.08.014. [DOI] [PubMed] [Google Scholar]
- Kato Y.; Abbott R. K.; Freeman B. L.; Haupt S.; Groschel B.; Silva M.; Menis S.; Irvine D. J.; Schief W. R.; Crotty S. Multifaceted Effects of Antigen Valency on B Cell Response Composition and Differentiation In Vivo. Immunity 2020, 53, 548–563. e8 (2020) 10.1016/j.immuni.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster J. G. B cell follicles and antigen encounters of the third kind. Nat. Immunol. 2010, 11, 989–996. 10.1038/ni.1946. [DOI] [PubMed] [Google Scholar]
- Catron D. M.; Pape K. A.; Fife B. T.; van Rooijen N.; Jenkins M. K. A Protease-Dependent Mechanism for Initiating T-Dependent B Cell Responses to Large Particulate Antigens. J. Immunol. 2010, 184, 3609–3617. 10.4049/jimmunol.1000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T. G.; Grigorova I.; Okada T.; Cyster J. G. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat. Immunol. 2007, 8, 992–1000. 10.1038/ni1494. [DOI] [PubMed] [Google Scholar]
- Gonzalez S. F.; Lukacs-Kornek V.; Kuligowski M. P.; Pitcher L. A.; Degn S. E.; Turley S. J.; Carroll M. C. Complement-Dependent Transport of Antigen into B Cell Follicles. J. Immunol. 2010, 185, 2659–2664. 10.4049/jimmunol.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco Y. R.; Batista F. D. B Cells Acquire Particulate Antigen in a Macrophage-Rich Area at the Boundary between the Follicle and the Subcapsular Sinus of the Lymph Node. Immunity 2007, 27, 160–171. 10.1016/j.immuni.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Oh H. L. J.; Gan S. K. E.; Bertoletti A.; Tan Y. J. Understanding the T cell immune response in SARS coronavirus infection. Emerging Microbes Infect. 2012, 1, 1–6. 10.1038/emi.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H.-S.; Kim Y.; Kim G.; Lee J. Y.; Jeong I.; Joh J.-S.; Kim H.; Chang E.; Sim S. Y.; Park J.-S.; Lim D.-G. Immune Responses to Middle East Respiratory Syndrome Coronavirus During the Acute and Convalescent Phases of Human Infection. Clin. Infect. Dis. 2019, 68, 984–992. 10.1093/cid/ciy595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui F.; Kai C.; Kitabatake M.; Inoue S.; Yoneda M.; Yokochi S.; Kase R.; Sekiguchi S.; Morita K.; Hishima T.; et al. Prior Immunization with Severe Acute Respiratory Syndrome (SARS)-Associated Coronavirus (SARS-CoV) Nucleocapsid Protein Causes Severe Pneumonia in Mice Infected with SARS-CoV. J. Immunol. 2008, 181, 6337–6348. 10.4049/jimmunol.181.9.6337. [DOI] [PubMed] [Google Scholar]
- Liu L.; Wei Q.; Lin Q.; Fang J.; Wang H.; Kwok H.; Tang H.; Nishiura K.; Peng J.; Tan Z.; Wu T.; et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019, 4, e123158 10.1172/jci.insight.123158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett K. S.; Edwards D. K.; Leist S. R.; Abiona O. M.; Boyoglu-Barnum S.; Gillespie R. A.; Himansu S.; Schäfer A.; Ziwawo C. T.; DiPiazza A. T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. 10.1038/s41586-020-2622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel A. B.; Kanevsky I.; Che Y.; Swanson K. A.; Muik A.; Vormehr M.; Kranz L. M.; Walzer K. C.; Hein S.; Güler A.; et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 2021, 592, 283–289. 10.1038/s41586-021-03275-y. [DOI] [PubMed] [Google Scholar]
- Casella C. R.; Mitchell T. C. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell. Mol. Life Sci. 2008, 65, 3231–3240. 10.1007/s00018-008-8228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F.; Cella M.; Danieli C.; Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J. Exp. Med. 1995, 182, 389–400. 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morón V. G.; Rueda P.; Sedlik C.; Leclerc C. In Vivo, Dendritic Cells Can Cross-Present Virus-Like Particles Using an Endosome-to-Cytosol Pathway. J. Immunol. 2003, 171, 2242–2250. 10.4049/jimmunol.171.5.2242. [DOI] [PubMed] [Google Scholar]
- Bachmann M. F.; Jennings G. T. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- Gamvrellis A.; Leong D.; Hanley J. C.; Xiang S. D.; Mottram P.; Plebanski M. Vaccines that facilitate antigen entry into dendritic cells. Immunol. Cell Biol. 2004, 82, 506–516. 10.1111/j.0818-9641.2004.01271.x. [DOI] [PubMed] [Google Scholar]
- Panagioti E.; Klenerman P.; Lee L. N.; van der Burg S. H.; Arens R. Features of effective T Cell-inducing vaccines against chronic viral infections. Front. Immunol. 2018, 9, 276. 10.3389/fimmu.2018.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.