

Abstract
Chronic, unresolved tissue inflammation is a well described feature of obesity, type 2 diabetes mellitus (T2DM), and other insulin resistant states and adipose tissue and liver inflammation have been particularly well studied. However, this proinflammatory state is not restricted to liver and adipose tissue, and abundant evidence demonstrates that inflammatory processes are also activated in islets from obese animals, humans, and subjects with T2DM. The nature of the inflammatory response in each of these tissues is quite different, and in this review we focus on the characteristics of immune cell-mediated inflammation in islets and the consequences of this with respect to beta cell function. In contrast to type 1 diabetes mellitus (T1DM), the dominant immune cell type causing inflammation in obese and T2DM islets is the macrophage. This emphasizes the importance of the innate immune system, as opposed to adaptive immunity, in this pathophysiologic process. The increased macrophage accumulation in these islets primarily arises through local proliferation of resident macrophages. These macrophages then provide signals, such as PDGF, which drive beta cell hyperplasia which is a classic feature of obesity. In addition, through a number of mechanisms, islet macrophages also impair beta cell insulin secretory capacity. Through these mechanisms, resident islet macrophages underly the inflammatory response in obesity and mechanistically participate in the beta-cell hyperplasia and dysfunction which characterizes this insulin resistant state. These findings point to the possibility that methods to interrupt islet inflammation could have beneficial effects on beta cell function and glycemia.
Introduction
The global prevalence of type 2 diabetes mellitus (T2DM) continues to increase at an alarming rate, with a corresponding rise in morbidity and mortality, representing an enormous burden on healthcare costs1. Insulin resistance is a key antecedent pathophysiologic feature in T2DM. The majority of patients with T2DM are obese, and obesity is far and away the major cause of insulin resistance in humans. Thus, obesity and T2DM are closely linked, and the parallel worldwide increase in rates of obesity is the driver of the T2DM epidemic. However, not all obese patients advance to T2DM. In those subjects that do progress, a second metabolic defect occurs involving beta cell dysfunction 2,3. Thus, when beta cells can no longer secrete excessive amounts of insulin to compensate for the insulin resistance, hyperglycemia ensues. There is an enormous literature on beta cell dysfunction in T2DM. Glucotoxicity, lipotoxicity, genetic defects, oxidative responses, ER stress, inflammation, and other mechanisms have all been implicated as causally related to decreased insulin secretion. With respect to the insulin resistance side of this metabolic disease, it is well known that obesity confers a state of chronic low-grade inflammation, particularly in adipose tissue and liver 4–6. Many studies point to this inflammatory state as an underlying mechanism for the development of insulin resistance in obesity and T2DM. A number of papers have now demonstrated that this chronic tissue inflammatory state also includes pancreatic islets 7–9. Islet inflammation has been described in a variety of mouse models of obesity and T2DM as well as in human islets from obese and/or type 2 diabetic patients. The mechanisms leading to islet inflammation are incompletely understood, but it is likely that islet inflammation contributes to the beta cell dysfunction which characterizes Type 2 diabetes. In this review, we will focus on the concept of islet inflammation with particular attention on the innate immune system, i.e. macrophages. We will discuss the distribution and accumulation of macrophages in pancreatic islets in normal and obese conditions and how macrophages influence beta cell abnormalities.
Macrophages in pancreatic islets.
1. Pancreas-resident macrophages during embryonic development and at steady state.
Studies in mice show that during embryonic development, macrophages in the pancreas are derived from yolk sac-derived primitive hematopoiesis10. These pancreatic macrophages are found in close proximity to insulin+ beta cells 10 and additional studies suggested that these macrophages are involved in islet morphogenesis 11. Consistent with this idea, the deficit of macrophage development in osteopetrotic op/op mice, lacking colony stimulating factor 1 (CSF-1), dampened pancreatic islet development and beta cell expansion 11,12.
Pancreatic macrophages display heterogenous phenotypes, depending on developmental stage 11,13, anatomical location 9,13, or metabolic setting 9. With respect to the latter, using immunostaining and flow cytometry analyses, Ying and colleagues found that pancreatic islets harbored two phenotypically distinct macrophage subsets at steady state. The F4/80lo CD11c+ population was enriched within the islets, whereas F4/80hi CD11c- macrophages largely resided in the peripheral islet area 9. Knowledge about the role of these pancreas- or islet- resident cells at steady state remains incomplete. In addition to promoting islet organogenesis 12,13, how these macrophages participate in tissue homeostasis and function of beta cells remains to be fully defined.
2. Inflammation in T2DM Islets
Numerous studies have revealed that macrophage infiltration is increased in T2DM islets, and islet macrophage content generally correlates with the degree of beta cell dysfunction 14–19. While it is now clear that insulitis is a common feature of T1DM and T2DM islets, there is a clear distinction between these two conditions. For example, in T2DM islet inflammation, macrophages dominate, whereas T lymphocytes dominates in T1DM islets 8. Thus, Ehses et al. reported that the number of CD68+ macrophages were increased in T2DM islets without changes in CD3+ T lymphocytes 14. They reported that the majority of CD68+ macrophages were positive for resident macrophage makers such as CD163 and HLA-2. In another study, Kamata et al. showed that, the number of CD68 and iNOS double-positive macrophages increased in amyloid-positive T2DM islets, without changes in CD163 and CD204-positive macrophages 15, suggesting that T2DM increases M1-like macrophage accumulation in the islets.
3. Accumulation of macrophages in obesity-associated islet inflammation.
Tissue inflammation is characterized by the accumulation and differentiation of various types of immune cells in local pathological lesions20–23. Macrophages are key cell types that orchestrate the initiation, specification and resolution of tissue-specific inflammation 6,24,25. During the course of obesity, islet inflammation has been reported, characterized by the accumulation of immune cells 7–9,19 along with elevated production of inflammatory cytokines and chemokines 19,26,27. Studies have reported increased numbers of myeloid-lineage cells (primarily monocytes and macrophages) in the islets of obese animal models 7,14,17,28. The first comprehensive analysis of obesity-associated islet macrophages was performed by Ehses and colleagues 14. Using CD68 and CD11b as markers, they found an increased number of macrophages in the pancreas of HFD-fed C57BL/6J mice and GK rats. Another key observation from this study was that the islets from T2D subjects released substantial amounts of inflammatory cytokines and chemokines such as IL-6, IL-8, CXCL1, G-CSF and MIP1a. While these data showed a correlation between the abundance of macrophages and increased inflammatory cytokines, the cellular origins of these cytokines and their roles in beta cell function remained undetermined. Using leptin receptor deficient db/db mice, Cucak and colleagues found significantly increased accumulation of pancreatic macrophages in diabetic mice. Their further characterization showed that both CD68+ F4/80- and CD68+ F4/80+ subsets exhibited a proinflammatory M1-like phenotype 28. Eguchi et al reported that saturated fatty acids induced beta cells to produce chemokines that attracted CD11b+ Ly-6C+ M1-type proinflammatory monocytes/macrophages to the islets 7. Using both diet- and genetically- induced rodent models of obesity, Ying et al provided an in-depth assessment of obesity-associated islet metaflammation in mice 9. They found that obesity-associated islet inflammation is dominated by macrophages, with negligible involvement of adaptive immune cells 9 (Figure 1).
Figure 1. Macrophages dominate obesity-associated islet inflammation.
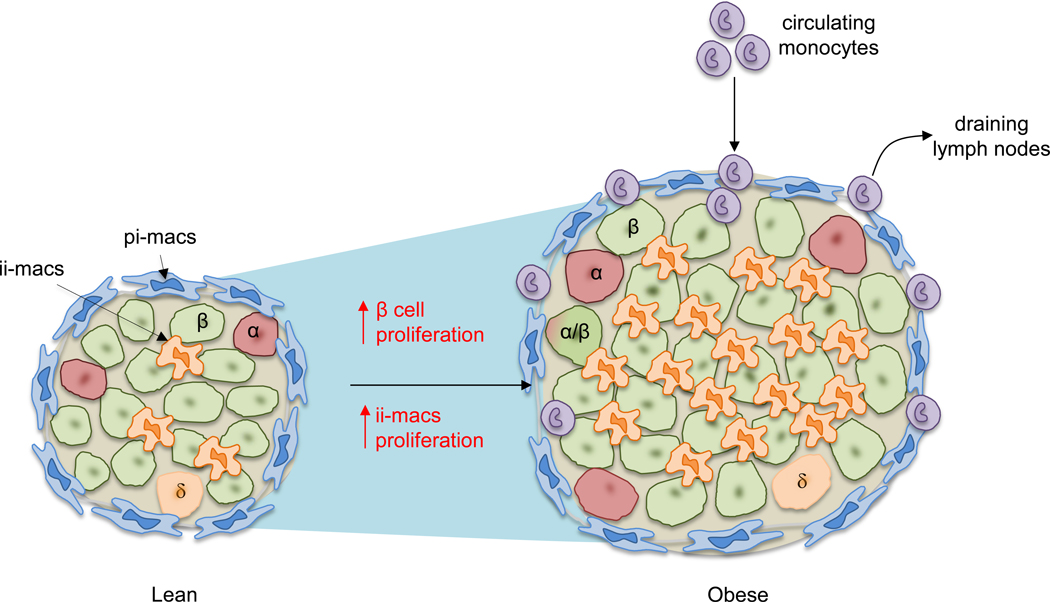
Illustrated here is a comparison of mouse islets between lean and obese conditions. In lean mice, two major populations of macrophages can be detected based on their anatomical distributions: peri-islet macrophages (pi-macs) (F4/80hi CD11c-) and intra-islet macrophages (ii-macs) (F4/80lo CD11chi). Both are islet-resident cells. In contrast, in obesity, the size of islet is increased due to increased beta cell replication and cell size. Multiple studies have demonstrated the increase of islet macrophages. However, different mechanisms have been proposed to explain obesity-associated macrophage accumulation in the islet. In one model, stressed beta cells recruit circulating monocytes which differentiate into pro-inflammatory macrophages after infiltrating into the islets. This model has been challenged by other studies showing that even though monocytes can be detected in the pancreas, they do not infiltrate into the islets. Instead, the accumulation of intra-islet macrophages is caused by local proliferation of resident macrophages.
Thus, macrophages in islets and in the exocrine stroma are distinct in origin and phenotypic properties. While it is not certain whether this is due to location or different lineages, the reconstitution after irradiation experiments performed by Calderon et al and Ying et al suggest that location plays a key role in programming macrophages to gain specific phenotypes and functions 9.
4. What causes the accumulation of islet macrophages?
4.1. Obesity induces local proliferation of islet macrophages.
Ying et al revealed a new mechanism of intra-islet macrophage accumulation in obese mice 9. They found that in lean mice, both intra-islet and peri-islet macrophages were maintained at a very low turnover rate. This is consistent with a previous report showing that, at steady state, islet macrophages are only minimally derived from blood cells and replicate locally at a low rate 13. However, under obese conditions, local proliferation of islet resident macrophages was significantly enhanced 9. Local proliferation has also been demonstrated for adipose tissue macrophages (ATMs) under obese condition 29–32. Reports have shown that IL-6 29, CCL2 31, CSF-1 33 or osteopontin 32 might promote ATM replication.
Macrophages can adapt to local environmental cues 34,35 and it is possible that intra-islet resident macrophage proliferation is an adaptive response to pathophysiological stimuli. However, the factors that mediate this adaptation remain to be defined. An interesting question is whether elevated glucose and/or fatty acids (FAs) in obesity can trigger the release of stimulating factors that promote the local proliferation of islet macrophages.
4.2. Circulating monocytes and islet macrophages.
During chronic inflammation, circulating monocytes infiltrate and accumulate in inflamed tissue sites. Whether this also explains obesity-associated islet inflammation is an important question. In a saturated FA treatment model, Eguchi et al found that ethyl palmitate infusion significantly increased the number of CD11b+ Ly6C+ cells in pancreatic islets 7. However, the exact distribution of these monocytes (intra- or peri- islet) was not determined. Using adoptive transfer, Ying et al found that transferred fluorescently labeled Ly6C+ monocytes reached the boundary between the exocrine and endocrine pancreas but did not penetrate into the islets in obese mice. Furthermore, these transferred cells retained their monocyte phenotype and did not differentiate into macrophages in the pancreas. Interestingly, time-course analysis showed that these peri-islet monocytes eventually migrate to pancreas-draining lymph nodes 9 (Figure 1). However, it remains possible that the lack of penetration of transferred monocytes into the HFD islets was due to the lack of an available tissue niche. Indeed, a previous study reported that monocytes can replace islet macrophages in mice after lethal irradiation 13.
4.3. The initiating factors for islet inflammation.
An important and unanswered question is what triggers islet inflammation in obese animals. Since obesity is associated with systemic low-grade inflammation, it is possible that systemic metabolic or inflammatory factors could impinge on the islet microenvironment. For example, in vitro high glucose stimulation can induce IL-1β secretion from islets through activation of the NLRP3 inflammasome 36,37. Circulating SFAs could also promote islet inflammation, particularly in the presence of hyperglycemia 7,38,39. While circulating cytokine levels are often increased in obesity/diabetes, these serve primarily as biomarkers of systemic inflammation, since the concentration of circulating cytokines is much lower than the biologically active levels within the local tissue microenvironment. Thus, the blood concentration of cytokines such as TNFα or IL-1β, are 10–100 fold lower than the levels required to induce pro-inflammatory biologic effects and, thus, are unlikely to trigger obesity-induced islet inflammation. Alternatively, islet-derived local signals could also play an important role in initiating the inflammatory cascade40. Islet-resident macrophages could act as first-line responders to sense systemic metabolic changes. As a consequence, these macrophages could alter their function and/or expand locally. On the other hand, as mentioned above, Eguchi et al suggested that beta cells were early responders, as these cells can sense excessive fatty acids and produce chemokines to recruit Ly6C+ monocytes/macrophages to the islets 7. Weitz et al reported that ATP released from stressed beta cells led to macrophage activation 41. In T2DM patients, aggregated islet amyloid polypeptide (IAPP), the major component forming amyloid deposits, can induce proinflammatory responses in islet macrophages. Using synthetic human IAPP (hIAPP) treatment or the hIAPP transgenic mouse model, Westwell-Roper et al demonstrated that hIAPP can enhance the synthesis of IL-1β in islet resident macrophages42. While it remains unclear whether islet macrophages, beta cells, or both are the first responders to metabolic insults, local interactions between these two cell types seem to accelerate the inflammatory process.
5. Transcriptional profile of islet macrophages.
Obesity affects islet macrophage transcriptomes, resulting in altered functions. Earlier studies have reported that macrophages within the islets of obese mice exhibited a pro-inflammatory M1-like phenotype 7,28. However, Calderon et al found that in lean mice, islet-resident macrophages constitutively expressed M1 cytokines, including IL-1β and TNF-α 13. In a recent study 9, RNA-seq analysis of isolated macrophages from lean and obese mice showed no clear shift between M1 and M2 profiles in either intra- or peri- islet macrophage subsets. Interestingly, Cucak et al proposed that islet macrophages in mice exhibited a shift from an early stage pro-inflammatory phenotype toward late stage profibrotic characteristics 28. Thus, during the development of obesity and T2D, the nature of the inflammation in the islets is not simply polarization from an M2 to M1 state, but, rather, a mixed continuum. Consistent with this notion, in adipose tissue of obese mice, adipose tissue macrophages exhibit diverse phenotypes across a broad spectrum of M1/M2 polarization states 43–45.
6. Other immune and non-immune cells
A few studies reported the presence of T and B lymphocytes 19,46 in pancreatic islets. In contrast, Ying et al did not detect the presence of T cells or B cells in the islets of HFD mice, arguing against the involvement of the adaptive immune system in obesity-associated islet pathology 9. Instead, T cells can be detected in the exocrine pancreas at very low abundancy and HFD did not alter their number 9. Another study reported that type 2 innate lymphoid cells (ILC2) were present in the islets and promoted insulin secretion through retinoic acid produced by myeloid cells in the islets 47. They also found that an IL-33-ILC2 axis was activated after acute beta cell stress but was defective during chronic obesity. Thus, it is possible that other types of immune cells may be involved in obesity-associated islet inflammation in a model, or stage-dependent manner. In addition, islet endothelial cells and neurons also play important roles in modulating the islet microenvironment, influencing beta cell function 41,48.
Effects of islet macrophages on beta cell function.
The deficit of beta cell function in T2DM is characterized by decreased mass and impaired GSIS 49. Incubation of primary islets or beta cell lines with pro-inflammatory cytokines such as IL-1β or TNF-α increases beta cell apoptosis and decreases glucose-stimulated insulin secretion (GSIS) 50. On the other hand, clodronate liposome-mediated depletion of macrophages in the islets of HFD mice improves GSIS 9. Consistent with this, depletion of tissue macrophages by clodronate liposomes improves glucose tolerance in obese mice 7. While the full mechanisms by which islet macrophages impair GSIS are not clearly understood, it likely involves both macrophage-derived soluble factors, and direct cell-cell contact between islet macrophages and beta cells (Figure 2).
Figure 2. Interactions of islet macrophages and beta cells in obesity.
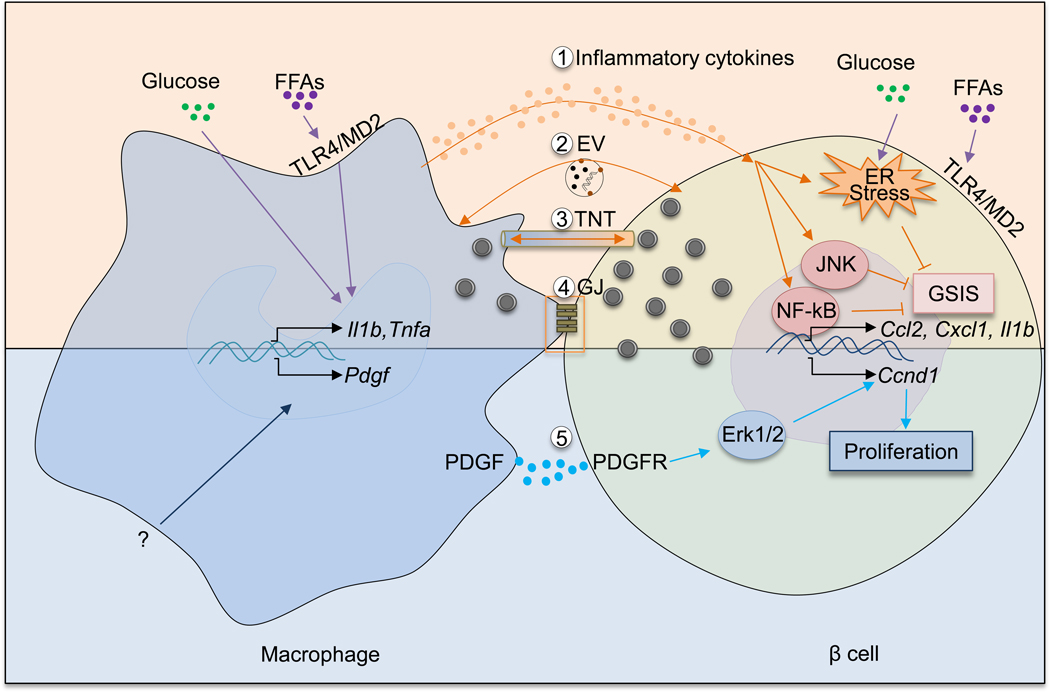
An increasing body of evidence supports the idea that islet macrophages influence beta cells in multiple ways. In obesity, elevated levels of glucose and free fatty acids can induce a pro-inflammatory phenotype of islet macrophages. As a result, macrophages produce increased amounts of proinflammatory cytokines such as IL-1β and TNF-α. These cytokines activate NF-κB and JNK pathways in beta cells and also exacerbate ER stress. Synergistically, these responses dampen beta cell GSIS. In addition to inflammatory cytokines, other mechanisms involving macrophage-mediated beta cell dysfunction exist. There mechanisms include: extracellular vesicles (EV) containing insulin released by beta cells and phagocytosed by islet macrophages; the formation of tunneling nanotubes (TNT) or gap junctions (GJ) between macrophages and beta cells allowing for bidirectional exchange of cellular contents. Obesity increases PDGF expression in islet macrophages via unclear mechanisms. Through PDGFR expressed in beta cells, PDGF promotes beta cell proliferation by activating downstream Erk signaling and inducing cell cycle gene (e.g., Ccnd1) expression.
1. Soluble factors.
Several cytokines such as TNF-α and IL-1β are increased in obese and T2DM islets and suppress beta cell GSIS 50,51. The most well-studied cytokine produced by islet macrophages is IL-1β. Beta cells express the IL-1 receptor, and obesity increases IL-1β expression in islet macrophages 7,52. In vitro incubation of macrophages in media containing high concentrations of glucose or free fatty acids (FAA) induce increased IL-1β production, suggesting that metabolic stress in obesity and/or T2DM can stimulate islet macrophages to produce more IL-1β. Indeed, while both beta cells and macrophages can produce IL-1β, macrophages appear to be the major source of islet IL-1β production in obesity 7,52. Incubation of primary human or mouse islets, or beta cell lines, with IL-1β decreases GSIS and increases beta cell apoptosis. Moreover, secretion of inflammatory cytokines from islet macrophages amplifies lipotoxic effects of chronic palmitate treatment, including decreased GSIS and decreased expression of the genes involved in beta cell differentiation and function 7. Thus, the enhanced lipotoxic effects of FFAs by co-culture with macrophages is ameliorated by addition of neutralizing antibodies against the pro-inflammatory cytokines TNF-α and IL-1β to the culture media.
The intracellular events through which inflammation impairs beta cell function involve the JNK and NF-κB signaling pathways. For example, TNF-α-induced NF-κB activation in beta cells decreases expression of genes involved in beta cell differentiation and insulin secretion. Beta cell NF-κB activation can reduce expression of an ER calcium ion (Ca2+) pump (sarcoplasmic reticulum Ca2+ ATPase type 2b), leading to decreased GSIS by promoting ER stress 53. Unlike TNF-α and IFN-γ, IL-1β also stimulates JNK in beta cells 54–57. IL-1β-induced JNK activation decreases GSIS by suppressing the IRS/PI3K/Akt signaling pathway. This results in reduced FoxO1 phosphorylation with increased nuclear localization of active FoxO1 and decreased DNA binding to PDX-1 58–60. It has also been suggested that normal FoxO1 function is necessary for maintenance of the full beta cell differentiation state 61. Additionally, Grunnet et al have shown that combination IL-1β/TNF-α/IFN-γ treatment can result in beta cell apoptosis by inducing mitochondrial stress and activation of proapoptotic Bcl-2 family proteins 62.
However, IL-1β treatment does not fully recapitulate the glucolipotoxicity-induced gene expression changes in islets, suggesting some divergence of mechanisms may also exist 63,64. Welsh et al reported data that questioned the importance of IL-1β in human islets showing high glucose in vitro, or the diabetic milieu in vivo, did not induce IL-1β production or NF-κB activation 65. Together, these results indicate the existence of other factors and mechanisms involved in high glucose or FFA-induced beta cell dysfunction.
2. Cell-cell contact and macrophage-mediated impaired beta cell insulin secretion.
Another mechanism by which macrophages can decrease beta cell insulin secretion is through direct cell-cell contact. Ying et al found that co-incubation of the Min6 beta cell line with intra-islet macrophages isolated from HFD mice decreases GSIS. Interestingly, this effect was seen only when the macrophages were directly added to the Min6 cells. Preventing cell-cell direct contact by culturing the two cell types in Transwell plate chambers, which allows interactions through soluble factors, blocks the effect to impair GSIS 9. These results suggest that the inhibitory effect of intra-islet macrophages on beta cell GSIS includes a cell-cell contact process. The concept of beta cell-macrophage interaction through direct cell-cell contact is not new, and was previously suggested by Vomund et al 66.They found that, in the context of mouse T1D, islet macrophages can engulf insulin secretory vesicles 66. Similar to their findings, Ying et al observed that intra-islet macrophages contain intact insulin vesicles, and this was greatly increased in obesity 9. While it remains unclear how islet macrophages incorporate insulin secretory vesicles from beta cells and how obesity promotes this process, it has been demonstrated that macrophages can generate open-ended channels called tunneling nanotubes (TNT) to transport cytoplasmic materials between connected cells 67. Therefore, it is theoretically possible that tunneling nanotubes mediate macrophage uptake of beta cell secretory granules, and that obesity and islet inflammation promote this process (Figure 2). It is also possible that transport of macrophage-derived factors to beta cells through these nanotubes impacts beta cell insulin secretion.
Effect of macrophages on beta cell proliferation.
In adult humans, the turnover rate of beta cells is extremely low. However, beta cells are not permanently quiescent. Under certain conditions, such as pregnancy and obesity, these cells are able to reenter the cell cycle. In addition, adaptive expansion of beta cell mass has been observed in the pre-diabetic stage in rodent models of obesity 68–72. Multiple factors, including glucose 73–75, insulin 76, and hepatocyte growth factor 77–79 can stimulate beta cell replication under obese conditions. Recently, a number of studies suggest that macrophages can play an important role in beta cell replication.
1. Macrophages are involved in recovery of beta cell mass after injury or acute beta cell depletion.
An increasing body of evidence has supported a role for macrophages in beta cell expansion. Using a pancreatic injury model induced by partial duct ligation, Xiao et al observed that islet macrophages can promote beta cell proliferation through TGF and downstream SMAD7 signaling 80. Conversely, depletion of islet macrophages using in vivo or ex vivo clodronate liposome treatment results in a decreased number of the BrdU+ beta cells 80. Using a mouse model with 50% beta cell depletion, followed by beta cell specific overexpression of connective tissue growth factor (CTGF), Riley et al found that beta cells released CTGF as a chemoattractant to recruit macrophages which then promote the recovery of beta cell mass 81. In another study, transiently increased expression of VEGF-A in beta cells led to beta cell loss and withdrawal of VEGF-A stimulated a robust, but transient, burst in proliferation of pre-existing beta cells 48. They also found that bone marrow-derived macrophages were recruited to the site of beta cell injury and are important for beta cell proliferation. Taken together, these studies indicate that macrophages play a critical role in maintaining steady state beta cell mass and promote increased beta cell proliferation and mass in pathophysiologic insulin resistant states.
2. Islet Macrophages from obese mice promote beta cell proliferation.
Obesity-induced insulin resistance provokes a compensatory expansion of pancreatic beta cells. Ying et al showed that islet macrophages from obese mice, regardless of their anatomical location, exhibited an enhanced capacity to promote beta cell proliferation and both intra-islet and peri-islet macrophages showed similar effects. This effect was mediated by PDGF-PDGFR signaling 9. Thus, the PDGF/PDGFR pathway plays an important role in mouse and human beta-cell proliferation and Chen et al have suggested that the reduction of PDGFR signaling in beta cells accounts for the decline of beta cell replication in aged mice and humans 82. Macrophages are a major source of PDGF 83–86 and the expression of Pdgfa was increased in both CD11c+ intra-islet macrophages and CD11c- peri-islet macrophages 9. Furthermore, inhibition of PDGFR signaling blocked the effect of islet macrophages to promote beta cell replication 9. Therefore, in addition to the expression of PDGFR on beta cells, the production of PDGF by islet macrophages forms a signaling system promoting beta cell proliferation (Figure 2).
3. Proinflammatory cytokines and macrophage-mediated beta cell proliferation.
Proinflammatory cytokines, such as TNF-α and IL-1β, are key components of the islet inflammatory microenvironment 27, and the role of IL-1b has been extensively studied 87. Knockout of beta cell IL1R causes beta cell dedifferentiation 88. In addition, low concentrations of IL-1β can promote beta cell proliferation by activating the FAS-FLIP pathway 89. Lean mice lacking IL-1β production exhibited down-regulation of FAS-FLIP activation, as well as decreased beta cell proliferation. IL-1β can also suppress the expression of genes associated with a fully differentiated beta cell phenotype. This can facilitate beta cell apoptosis and can also induce beta cell dedifferentiation into islet endocrine cells which express both insulin and glucagon. These latter dual positive endocrine cells generally demonstrate impaired GSIS, providing another mechanism whereby IL-1β adversely effects beta cell GSIS. Given all of these findings, the IL-1β “tone” within the islet provides an important control point for regulation of beta cell mass. IL-1 receptor antagonist (IL-1Ra) expression is increased in the blood during obesity and T2DM but decreased in islets from T2DM patients 90,91. Beta-cell specific IL-1Ra ablation resulted in a reduction in beta cell proliferation and impaired insulin secretion. Decreased levels of IL-1Ra promote IL-1β action and studies have shown that treatment of GK rats with IL-1Ra led to a reduction in islet inflammation and improved beta cell GSIS. Taken together, these results provide compelling evidence for the role of this inflammatory cytokine in islet inflammation and beta cell dysfunction.
Beneficial effects of macrophages on beta cells.
Although depletion of islet macrophages improves GSIS in HFD/obese mice, macrophage depletion dampens beta cell GSIS in lean/normal islets 9. This suggests that in the normal lean state, islet macrophages may have a beneficial role on beta cell insulin secretion. It remains unclear why islet macrophages from lean and obese mice apparently play opposite roles in beta cell function. It is possible that in obesity, islet macrophages are reprogrammed to impair beta cell GSIS. This could be due to effects of FFAs, glucose, or cytokines, such as IL-1β. One possible explanation is the level of IL-1β. As reported by Calderon et al, islet macrophages in non-obese mice exhibit a proinflammatory activation state and produce IL-1β 13. Dror et al demonstrated that acute post-prandial rise in IL-1β production by myeloid cells contributes to meal-induced insulin secretion in a fasting-refeeding setting and is necessary for normal glycemic control 92. In line with this, IL-1 receptor 1 (IL-1R1) deficient mice exhibited an impaired adaptive increase of plasma insulin after HFD feeding 93. However, chronic accumulation or elevated production of IL-1β (mainly by islet macrophages) is detrimental to beta cell GSIS 87.
In addition to IL-1β, other factors and mechanisms could also play a role in shifting the effect of islet macrophages on beta cells. For instance, Ying et al found that pathways related to synapse-formation were more activated in islet macrophages in obese mice compared to lean mice 9, suggesting that obesity alters cell-cell interaction between macrophages and beta cells.
The above discussion raises several questions: what induces islet macrophages to change their effects on beta cell GSIS going from the lean to obese state? At which stage of obesity does this functional switch of islet macrophages happen? What modifications can reverse the macrophage-mediated “detrimental” effects on beta cells? To answer these questions, it will be necessary to perform longitudinal studies that can parse out the functional properties of islet macrophages and assess the effects of these cells on beta cell function in a stage-dependent manner.
Lipids as mediators of crosstalk between islet macrophages and beta cells.
Several studies have indicated that saturated fatty acids (SFAs), and perhaps lipoproteins, are circulating factors that might contribute to islet inflammation, beta cell dysfunction, and beta cell hyperplasia 7,38,39. Thus, experimental elevation of palmitate levels in mice promotes islet inflammation 7. In vivo treatment with SFAs increased the accumulation of islet macrophages and other markers of islet inflammation, as well as decreased GSIS. SFAs activate intracellular proinflammatory pathways by binding to TLR4 and studies show that TLR4 is required for the SFA-induced effect on islet inflammation and further show that islet macrophage TLR4 was essential for this process. TLR4 is also expressed on beta cells, and SFAs induce beta cells to secrete chemokines and other factors which promote islet macrophage accumulation. The SFAs act on macrophage TLR4 to stimulate the NF-κB pathway, resulting in cytokine secretion, particularly IL1β and TNFα, which can negatively influence beta cell GSIS. In addition to these effects of SFAs on beta cell secretory function, elevated SFA levels can also affect beta cell proliferation. This is particularly true when elevated SFA levels are combined with hyperglycemia, since this combination appears to cause additive effects on beta cell proliferation. Apoptosis is the other side of the equation regulating overall beta cell mass and, within the islet, SFAs can be converted to ceramides which promote apoptosis 94,95. Cunha et al reported that exposure to palmitate can trigger human beta cell apoptosis by promoting death protein 5-mediated ER stress 96. Thus, by enhancing apoptosis and promoting proliferation, SFAs may have important effects on increasing overall beta cell mass in the context of obesity and T2DM.
Another interesting connection between dyslipidemia and beta cell function has been reported. LDL receptor related protein 1 (LRP1) can interact with extracellular lipoproteins. Ye et al generated beta cell specific LRP1 KO mice and showed that these animals were partially protected from the adverse effects of HFD on beta cell insulin secretion, as well as HFD-induced beta cell hyperplasia 39. They also found that this could be related to the effect of LRP1 deletion to upregulate beta cell PPARγ2 in HFD mice since transgenic overexpression of PPARγ2 in beta cells partially recapitulated the LRP1 phenotype.
Senescence.
Cellular senescence is characterized by stable cell-cycle arrest and a pro-inflammatory secretory phenotype 97. The current theory is that the senescent signal can spread from one senescent cell to other surrounding cells. How this occurs remain to be resolved, but some indications suggest that T1D humans and the non-obese diabetic (NOD) mouse express a set of senescent beta cells with increased levels of the pro-survival mediator Bcl-2 98. Interestingly, treatment with Bcl-2 inhibitor can efficiently deplete senescent beta cells and prevent T1D occurrence in the NOD mouse model 98. It is possible that obesity might promote a beta cell senescence response which drives the pathogenesis of type 2 diabetes. In an early study, Sone and Kagawa observed that the number of acidic beta-galactosidase-positive senescent beta cells was increased in HFD-induced mice 99. Recently, Aguayo-Mazzucato et al. found that insulin resistance can promote senescence state of beta cells, leading to decreased insulin secretion 100. Consistent with this idea, the number of senescent beta cells is increased in T2DM human islets 100. However, it remains unknown whether islet macrophages also develop senescent phenotypes in the context of obesity or if senescent beta cells can regulate islet macrophage activation.
Clinical studies.
A number of clinical trials targeting inflammatory pathways have been tested in patients with T2DM, including the salicylic acid derivative, salsalate and anti-inflammatory TNFα inhibitors 101,102. In some clinical trials, the effects on beta cells have been assessed. In one study, the administration of the recombinant a IL-1 receptor antagonist, Anakinra (recombinant IL-1Ra) confers a moderate but significant decrease in fasting blood glucose and glycated hemoglobin (−0.3–0.4%) levels 103. In this study, the authors reported that Anakinra treatment increases insulin secretion without affecting insulin sensitivity. Moreover, the larger scale CANTOS trial using the anti-IL-1β antibody Canakinumab demonstrated that IL-1β neutralization lowers glycated hemoglobin levels in the plasma and delays new onset of T2DM by 2–3 years 104. Since Canakinumab treatment significantly reduces recurrence of cardiovascular events, these results suggest that anti-inflammatory therapy can provide beneficial effects for T2DM. A recent meta-analysis of 2921 individuals from eight studies found a significant overall HbA1c-lowering effect of IL-1 antagonism, through either anti-IL-1 antibodies or IL-1R antagonists105. In this meta-analysis, a significant correlation between baseline C-reactive protein and C-peptide, and HbA1c outcomes was also revealed. However, the authors also cautioned that identification of further biomarkers are needed to define the potential of anti-IL-1 therapies in T2DM.
Concluding remarks and future perspectives.
Islet inflammation has emerged as a key feature of obesity and T2DM, and islet macrophages are the defining immune cell-type in these conditions. While much has been learned about macrophage-beta cell interactions, many important questions remain unresolved. What are the triggering events with respect to islet macrophage proliferation and expansion? Are initiating signals derived from the beta-cell or are they extrinsic to the islet? Islet macrophage proliferation is not significantly increased until 12–16 weeks HFD. If beta-cell insulin secretory function is affected prior to this time, then what is the cause? Have macrophages already started to reprogram before proliferation ensues, or is there a macrophage-independent mechanism causing the initial defect in GSIS? Why do circulating monocytes migrate to the peri-islet area but do not enter the islets? Finally, more information is needed on the role of islet inflammation and macrophages in the insulin secretory defects which characterize human T2DM?
Table 1.
The roles of islet macrophages in non-inflammatory state versus obese/T2DM conditions
| Non-Inflammatory State | Obesity/T2DM |
|---|---|
| Intra-islet macrophages and peri-islet macrophages9,13 | Increased number of intra-islet macrophages7,9,14,17,28 |
| Supports islet (β cell) development and regeneration11–13 | Promotes compensatory β cell proliferation9 |
| Promotes GSIS9 | Suppresses GSIS with decreased expression of β cell-specific genes7,9,49,50 |
| Low concentration of IL-1β 7,13,88,91 | Elevated production of IL-1β7,40,49 |
Box 1: main points of this review.
Obesity is associated with low-grade tissue inflammation, which is characterized by the accumulation of immune cells, metabolic and inflammatory mediators at multiple tissue sites, including pancreatic islets. In both animal models and human subjects, it is evident that increased number of immune cells (predominantly macrophages) is seen in pancreatic islets. The origin, phenotype and function of these macrophages are subjects of active investigations. Different mechanisms (local proliferation versus replenishment by blood monocytes) have been proposed to explain the increase of islet macrophages in obesity. The interactions between macrophages and beta cells are multifaceted. In rodent models of obesity, islet macrophages can dampen beta cell insulin secretion and beta cell proliferation through distinct mechanisms, suggesting that macrophages may exert both beneficial and detrimental roles in beta cells. Metabolic factors (glucose and free fatty acids), inflammatory cytokines or cell-cell contact may each play a critical role in the crosstalk between macrophages and beta cells. In addition to macrophages, other types of immune and non-immune cells are also found to participate in composing obesity-associated islet inflammation and influencing beta cell activities.
Box 2: Protective and harmful inflammation.
Inflammation is an adaptive response exploited by the host to deal with harmful conditions 24. Acute inflammation is best exemplified as a result of infection whereas, whereas chronic inflammation is widely associated with more complex disorders, such as diabetes and cardiovascular diseases20,21,23. Evolutionarily, inflammation is a beneficial protective mechanism to eliminate infectious pathogens, elicit tissue repair and restore tissue homeostasis24. In contrast, it is generally appreciated that chronic, low-grade inflammation results in pathological consequences in the targeted tissue or cell, such as functional impairment, cell death and tissue damage 6.
Macrophages are key players in orchestrating the initiation, specification and resolution of tissue inflammation6,24,25. Macrophages are heterogenous in terms of their origin, tissue phenotype and function106. By adapting to tissue environmental cues, macrophages can acquire different functionalities and are programmed into pro-inflammatory, anti-inflammatory, or reparative states.
Dissecting the phenotypic and functional heterogeneity of islet macrophages and the interactions between macrophages and beta cells will improve our understanding of obesity and diabetes. This could lead to the development of new immunomodulatory regimens to prevent or reverse these disorders.
Key points.
Macrophages are the primary immune-type cells involved in obesity-associated islet inflammation in both mice and humans.
Obesity reconstructs the islet immune microenvironment by promoting local replication of islet-resident macrophages or recruiting circulating monocytes.
Islet macrophages in obese mice display multiple functions, including dampening beta cell insulin secretion and promoting beta cell proliferation.
Islet macrophages are potential therapeutic targets to modulate beta cell function.
ACKNOWLEDGEMENTS
This study was funded by the US National Institute of Diabetes and Digestive and Kidney Diseases (DK063491 and DK101395 to J.M.O, and DK114427 to W.F.), the US National Institute of Diabetes and Digestive and Kidney Diseases K99/R00 award (1K99DK115998 to W.Y.), the UCSD/UCLA Diabetes Research Center Pilot and Feasibility grants (W.Y., Y.S.L, and W.F.), UCSD CTRI UL1 TR000100 (W.F.).
Footnotes
DECLARATION OF INTERESTS
The authors have no conflicts of interests to declare.
References
- 1.da Rocha Fernandes J et al. IDF Diabetes Atlas estimates of 2014 global health expenditures on diabetes. Diabetes Res Clin Pract 117, 48–54, doi: 10.1016/j.diabres.2016.04.016 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Saisho Y Importance of Beta Cell Function for the Treatment of Type 2 Diabetes. J Clin Med 3, 923–943, doi: 10.3390/jcm3030923 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitamura T The role of FOXO1 in beta-cell failure and type 2 diabetes mellitus. Nat Rev Endocrinol 9, 615–623, doi: 10.1038/nrendo.2013.157 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Donath MY & Shoelson SE Type 2 diabetes as an inflammatory disease. Nat Rev. Immunol 11, 98–107, doi:nri2925 [pii]; 10.1038/nri2925 [doi] (2011). [DOI] [PubMed] [Google Scholar]
- 5.Lackey DE & Olefsky JM Regulation of metabolism by the innate immune system. Nat Rev Endocrinol 12, 15–28, doi: 10.1038/nrendo.2015.189 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Lee YS, Wollam J & Olefsky JM An Integrated View of Immunometabolism. Cell 172, 22–40, doi: 10.1016/j.cell.2017.12.025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eguchi K et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab 15, 518–533, doi:S1550–4131(12)00064–2 [pii]; 10.1016/j.cmet.2012.01.023 [doi] (2012). [DOI] [PubMed] [Google Scholar]
- 8.Boni-Schnetzler M & Meier DT Islet inflammation in type 2 diabetes. Semin Immunopathol, doi: 10.1007/s00281-019-00745-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ying W et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting beta Cell Proliferation and Function in Obesity. Cell Metab 29, 457–474 e455, doi: 10.1016/j.cmet.2018.12.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulz C et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90, doi:science.1219179 [pii]; 10.1126/science.1219179 [doi] (2012). [DOI] [PubMed] [Google Scholar]
- 11.Geutskens SB, Otonkoski T, Pulkkinen MA, Drexhage HA & Leenen PJ Macrophages in the murine pancreas and their involvement in fetal endocrine development in vitro. J. Leukoc. Biol 78, 845–852, doi:jlb.1004624 [pii]; 10.1189/jlb.1004624 [doi] (2005). [DOI] [PubMed] [Google Scholar]
- 12.Banaei-Bouchareb L et al. Insulin cell mass is altered in Csf1op/Csf1op macrophage-deficient mice. J. Leukoc. Biol 76, 359–367, doi: 10.1189/jlb.1103591 [doi];jlb.1103591 [pii] (2004). [DOI] [PubMed] [Google Scholar]
- 13.Calderon B et al. The pancreas anatomy conditions the origin and properties of resident macrophages. J Exp Med 212, 1497–1512, doi: 10.1084/jem.20150496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehses JA et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56, 2356–2370, doi: 10.2337/db06-1650 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Kamata K et al. Islet amyloid with macrophage migration correlates with augmented beta-cell deficits in type 2 diabetic patients. Amyloid 21, 191–201, doi: 10.3109/13506129.2014.937857 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao HL et al. Prevalence and clinicopathological characteristics of islet amyloid in chinese patients with type 2 diabetes. Diabetes 52, 2759–2766, doi: 10.2337/diabetes.52.11.2759 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Richardson SJ, Willcox A, Bone AJ, Foulis AK & Morgan NG Islet-associated macrophages in type 2 diabetes. Diabetologia 52, 1686–1688, doi: 10.1007/s00125-009-1410-z (2009). [DOI] [PubMed] [Google Scholar]
- 18.Marselli L et al. beta-Cell inflammation in human type 2 diabetes and the role of autophagy. Diabetes Obes Metab 15 Suppl 3, 130–136, doi: 10.1111/dom.12152 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Butcher MJ et al. Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 57, 491–501, doi: 10.1007/s00125-013-3116-5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathis D Immunological goings-on in visceral adipose tissue. Cell Metab 17, 851–859, doi:S1550–4131(13)00198–8 [pii]; 10.1016/j.cmet.2013.05.008 [doi] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLaughlin T, Ackerman SE, Shen L & Engleman E Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest 127, 5–13, doi: 10.1172/JCI88876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sell H, Habich C & Eckel J Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol 8, 709–716, doi: 10.1038/nrendo.2012.114 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Shalapour S & Karin M Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest 125, 3347–3355, doi: 10.1172/JCI80007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medzhitov R Origin and physiological roles of inflammation. Nature 454, 428–435, doi:nature07201 [pii]; 10.1038/nature07201 [doi] (2008). [DOI] [PubMed] [Google Scholar]
- 25.Murray PJ & Wynn TA Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol 11, 723–737, doi:nri3073 [pii]; 10.1038/nri3073 [doi] (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donath MY, Storling J, Berchtold LA, Billestrup N & Mandrup-Poulsen T Cytokines and beta-cell biology: from concept to clinical translation. Endocr Rev 29, 334–350, doi: 10.1210/er.2007-0033 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Eguchi K & Nagai R Islet inflammation in type 2 diabetes and physiology. J Clin Invest 127, 14–23, doi: 10.1172/JCI88877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cucak H, Grunnet LG & Rosendahl A Accumulation of M1-like macrophages in type 2 diabetic islets is followed by a systemic shift in macrophage polarization. J Leukoc Biol 95, 149–160, doi: 10.1189/jlb.0213075 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Braune J et al. IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J Immunol 198, 2927–2934, doi: 10.4049/jimmunol.1600476 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Haase J et al. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 57, 562–571, doi: 10.1007/s00125-013-3139-y (2014). [DOI] [PubMed] [Google Scholar]
- 31.Amano SU et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab 19, 162–171, doi: 10.1016/j.cmet.2013.11.017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tardelli M et al. Osteopontin is a key player for local adipose tissue macrophage proliferation in obesity. Mol Metab 5, 1131–1137, doi: 10.1016/j.molmet.2016.09.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamilton JA Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 8, 533–544, doi: 10.1038/nri2356 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Gosselin D et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340, doi: 10.1016/j.cell.2014.11.023 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavin Y et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326, doi: 10.1016/j.cell.2014.11.018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou R, Tardivel A, Thorens B, Choi I & Tschopp J Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11, 136–140, doi: 10.1038/ni.1831 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Maedler K et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110, 851–860, doi: 10.1172/JCI15318 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boni-Schnetzler M et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 150, 5218–5229, doi: 10.1210/en.2009-0543 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Ye R et al. Intracellular lipid metabolism impairs beta cell compensation during diet-induced obesity. J Clin Invest 128, 1178–1189, doi: 10.1172/JCI97702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aamodt KI & Powers AC Signals in the pancreatic islet microenvironment influence beta-cell proliferation. Diabetes Obes Metab 19 Suppl 1, 124–136, doi: 10.1111/dom.13031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weitz JR et al. Mouse pancreatic islet macrophages use locally released ATP to monitor beta cell activity. Diabetologia 61, 182–192, doi: 10.1007/s00125-017-4416-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Westwell-Roper CY, Ehses JA & Verchere CB Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1beta production and beta-cell dysfunction. Diabetes 63, 1698–1711, doi: 10.2337/db13-0863 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Kratz M et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20, 614–625, doi: 10.1016/j.cmet.2014.08.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lumeng CN, DelProposto JB, Westcott DJ & Saltiel AR Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57, 3239–3246, doi: 10.2337/db08-0872 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li P et al. Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J Biol Chem 285, 15333–15345, doi: 10.1074/jbc.M110.100263 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J & von Herrath MG Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes 63, 3880–3890, doi: 10.2337/db14-0549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalmas E et al. Interleukin-33-Activated Islet-Resident Innate Lymphoid Cells Promote Insulin Secretion through Myeloid Cell Retinoic Acid Production. Immunity 47, 928–942 e927, doi: 10.1016/j.immuni.2017.10.015 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Brissova M et al. Islet microenvironment, modulated by vascular endothelial growth factor-A signaling, promotes beta cell regeneration. Cell Metab 19, 498–511, doi: 10.1016/j.cmet.2014.02.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Halban PA et al. beta-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care 37, 1751–1758, doi: 10.2337/dc14-0396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donath MY, Boni-Schnetzler M, Ellingsgaard H, Halban PA & Ehses JA Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab 21, 261–267, doi: 10.1016/j.tem.2009.12.010 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Burns SM et al. High-throughput luminescent reporter of insulin secretion for discovering regulators of pancreatic Beta-cell function. Cell Metab 21, 126–137, doi: 10.1016/j.cmet.2014.12.010 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Westwell-Roper C, Denroche HC, Ehses JA & Verchere CB Differential Activation of Innate Immune Pathways by Distinct Islet Amyloid Polypeptide (IAPP) Aggregates. J Biol Chem 291, 8908–8917, doi: 10.1074/jbc.M115.712455 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cardozo AK et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 54, 452–461, doi: 10.2337/diabetes.54.2.452 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Major CD & Wolf BA Interleukin-1beta stimulation of c-Jun NH(2)-terminal kinase activity in insulin-secreting cells: evidence for cytoplasmic restriction. Diabetes 50, 2721–2728, doi: 10.2337/diabetes.50.12.2721 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Ammendrup A et al. The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 49, 1468–1476, doi: 10.2337/diabetes.49.9.1468 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Bonny C, Oberson A, Negri S, Sauser C & Schorderet DF Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50, 77–82, doi: 10.2337/diabetes.50.1.77 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Welsh N Interleukin-1 beta-induced ceramide and diacylglycerol generation may lead to activation of the c-Jun NH2-terminal kinase and the transcription factor ATF2 in the insulin-producing cell line RINm5F. J Biol Chem 271, 8307–8312, doi: 10.1074/jbc.271.14.8307 (1996). [DOI] [PubMed] [Google Scholar]
- 58.Bouzakri K et al. Rab GTPase-activating protein AS160 is a major downstream effector of protein kinase B/Akt signaling in pancreatic beta-cells. Diabetes 57, 1195–1204, doi: 10.2337/db07-1469 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Kawamori D et al. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J Biol Chem 281, 1091–1098, doi: 10.1074/jbc.M508510200 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Kaneto H et al. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 277, 30010–30018, doi: 10.1074/jbc.M202066200 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Kim-Muller JY et al. Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab 20, 593–602, doi: 10.1016/j.cmet.2014.08.012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grunnet LG et al. Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells. Diabetes 58, 1807–1815, doi: 10.2337/db08-0178 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cardozo AK, Kruhoffer M, Leeman R, Orntoft T & Eizirik DL Identification of novel cytokine-induced genes in pancreatic beta-cells by high-density oligonucleotide arrays. Diabetes 50, 909–920, doi: 10.2337/diabetes.50.5.909 (2001). [DOI] [PubMed] [Google Scholar]
- 64.Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S & Sharma A Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 50 Suppl 1, S154–159, doi: 10.2337/diabetes.50.2007.s154 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Welsh N et al. Is there a role for locally produced interleukin-1 in the deleterious effects of high glucose or the type 2 diabetes milieu to human pancreatic islets? Diabetes 54, 3238–3244, doi: 10.2337/diabetes.54.11.3238 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Vomund AN et al. Beta cells transfer vesicles containing insulin to phagocytes for presentation to T cells. Proc Natl Acad Sci U S A 112, E5496–5502, doi: 10.1073/pnas.1515954112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamashita YM, Inaba M & Buszczak M Specialized Intercellular Communications via Cytonemes and Nanotubes. Annu Rev Cell Dev Biol 34, 59–84, doi: 10.1146/annurev-cellbio-100617-062932 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hull RL et al. Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia 48, 1350–1358, doi: 10.1007/s00125-005-1772-9 (2005). [DOI] [PubMed] [Google Scholar]
- 69.Peyot ML et al. Beta-cell failure in diet-induced obese mice stratified according to body weight gain: secretory dysfunction and altered islet lipid metabolism without steatosis or reduced beta-cell mass. Diabetes 59, 2178–2187, doi: 10.2337/db09-1452 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ebato C et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8, 325–332, doi: 10.1016/j.cmet.2008.08.009 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Stamateris RE, Sharma RB, Hollern DA & Alonso LC Adaptive beta-cell proliferation increases early in high-fat feeding in mice, concurrent with metabolic changes, with induction of islet cyclin D2 expression. Am J Physiol Endocrinol Metab 305, E149–159, doi: 10.1152/ajpendo.00040.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mosser RE et al. High-fat diet-induced beta-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am J Physiol Endocrinol Metab 308, E573–582, doi: 10.1152/ajpendo.00460.2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alonso LC et al. Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes 56, 1792–1801, doi: 10.2337/db06-1513 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Levitt HE et al. Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia 54, 572–582, doi: 10.1007/s00125-010-1919-1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Porat S et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab 13, 440–449, doi: 10.1016/j.cmet.2011.02.012 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Assmann A, Ueki K, Winnay JN, Kadowaki T & Kulkarni RN Glucose effects on beta-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol Cell Biol 29, 3219–3228, doi: 10.1128/MCB.01489-08 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garcia-Ocana A et al. Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J Biol Chem 275, 1226–1232 (2000). [DOI] [PubMed] [Google Scholar]
- 78.Araujo TG et al. Hepatocyte growth factor plays a key role in insulin resistance-associated compensatory mechanisms. Endocrinology 153, 5760–5769, doi: 10.1210/en.2012-1496 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Demirci C et al. Loss of HGF/c-Met signaling in pancreatic beta-cells leads to incomplete maternal beta-cell adaptation and gestational diabetes mellitus. Diabetes 61, 1143–1152, doi: 10.2337/db11-1154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xiao X et al. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc. Natl. Acad. Sci. U. S. A 111, E1211–E1220, doi:1321347111 [pii]; 10.1073/pnas.1321347111 [doi] (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Riley KG et al. Macrophages are essential for CTGF-mediated adult beta-cell proliferation after injury. Mol Metab 4, 584–591, doi: 10.1016/j.molmet.2015.05.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen H et al. PDGF signalling controls age-dependent proliferation in pancreatic [bgr]-cells. Nature 478, 349–355 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jaguin M, Fardel O & Lecureur V AhR-dependent secretion of PDGF-BB by human classically activated macrophages exposed to DEP extracts stimulates lung fibroblast proliferation. Toxicol Appl Pharmacol 285, 170–178, doi: 10.1016/j.taap.2015.04.007 (2015). [DOI] [PubMed] [Google Scholar]
- 84.Onogi Y et al. PDGFRbeta Regulates Adipose Tissue Expansion and Glucose Metabolism via Vascular Remodeling in Diet-Induced Obesity. Diabetes 66, 1008–1021, doi: 10.2337/db16-0881 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Shimokado K et al. A significant part of macrophage-derived growth factor consists of at least two forms of PDGF. Cell 43, 277–286 (1985). [DOI] [PubMed] [Google Scholar]
- 86.Zhou X et al. Circuit Design Features of a Stable Two-Cell System. Cell 172, 744–757 e717, doi: 10.1016/j.cell.2018.01.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Donath MY, Dalmas E, Sauter NS & Boni-Schnetzler M Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab 17, 860–872, doi: 10.1016/j.cmet.2013.05.001 (2013). [DOI] [PubMed] [Google Scholar]
- 88.Burke SJ et al. Pancreatic deletion of the interleukin-1 receptor disrupts whole body glucose homeostasis and promotes islet beta-cell de-differentiation. Mol Metab 14, 95–107, doi: 10.1016/j.molmet.2018.06.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maedler K et al. Low concentration of interleukin-1beta induces FLICE-inhibitory protein-mediated beta-cell proliferation in human pancreatic islets. Diabetes 55, 2713–2722, doi: 10.2337/db05-1430 (2006). [DOI] [PubMed] [Google Scholar]
- 90.Boni-Schnetzler M et al. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab 93, 4065–4074, doi: 10.1210/jc.2008-0396 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boni-Schnetzler M et al. beta Cell-Specific Deletion of the IL-1 Receptor Antagonist Impairs beta Cell Proliferation and Insulin Secretion. Cell Rep 22, 1774–1786, doi: 10.1016/j.celrep.2018.01.063 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Dror E et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol 18, 283–292, doi: 10.1038/ni.3659 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Hajmrle C et al. Interleukin-1 signaling contributes to acute islet compensation. JCI Insight 1, e86055, doi: 10.1172/jci.insight.86055 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kelpe CL et al. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem 278, 30015–30021, doi: 10.1074/jbc.M302548200 (2003). [DOI] [PubMed] [Google Scholar]
- 95.Lang F, Ullrich S & Gulbins E Ceramide formation as a target in beta-cell survival and function. Expert Opin Ther Targets 15, 1061–1071, doi: 10.1517/14728222.2011.588209 (2011). [DOI] [PubMed] [Google Scholar]
- 96.Cunha DA et al. Death protein 5 and p53-upregulated modulator of apoptosis mediate the endoplasmic reticulum stress-mitochondrial dialog triggering lipotoxic rodent and human beta-cell apoptosis. Diabetes 61, 2763–2775, doi: 10.2337/db12-0123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tchkonia T, Zhu Y, van Deursen J, Campisi J & Kirkland JL Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123, 966–972, doi: 10.1172/JCI64098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thompson PJ et al. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab 29, 1045–1060 e1010, doi: 10.1016/j.cmet.2019.01.021 (2019). [DOI] [PubMed] [Google Scholar]
- 99.Sone H & Kagawa Y Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 48, 58–67, doi: 10.1007/s00125-004-1605-2 (2005). [DOI] [PubMed] [Google Scholar]
- 100.Aguayo-Mazzucato C et al. beta Cell Aging Markers Have Heterogeneous Distribution and Are Induced by Insulin Resistance. Cell Metab 25, 898–910 e895, doi: 10.1016/j.cmet.2017.03.015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Donath MY Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov 13, 465–476, doi: 10.1038/nrd4275 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Goldfine AB et al. Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann Intern Med 159, 1–12, doi: 10.7326/0003-4819-159-1-201307020-00003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Larsen CM et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356, 1517–1526, doi: 10.1056/NEJMoa065213 (2007). [DOI] [PubMed] [Google Scholar]
- 104.Everett BM et al. Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. J Am Coll Cardiol 71, 2392–2401, doi: 10.1016/j.jacc.2018.03.002 (2018). [DOI] [PubMed] [Google Scholar]
- 105.Kataria Y, Ellervik C & Mandrup-Poulsen T Treatment of type 2 diabetes by targeting interleukin-1: a meta-analysis of 2921 patients. Semin Immunopathol 41, 413–425, doi: 10.1007/s00281-019-00743-6 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Gordon S & Pluddemann A Tissue macrophages: heterogeneity and functions. BMC Biol 15, 53, doi: 10.1186/s12915-017-0392-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]