

Abstract
Hepatocellular carcinoma (HCC), the most common primary liver cancer, of which ~800,000 new cases will be diagnosed worldwide this year, portends a five-year survival rate of merely 17% in patients with unresectable disease. This dismal prognosis is due, at least in part, from the late stage of diagnosis and the limited efficacy of systemic therapies. As a result, there is an urgent need to identify risk factors that contribute to HCC initiation and provide targetable vulnerabilities to improve patient survival. While myriad risk factors are known, elevated copper (Cu) levels in HCC patients and the incidence of hepatobiliary malignancies in Wilson disease patients, which exhibit hereditary liver Cu overload, suggests the possibility that metal accumulation promotes malignant transformation. Here we found that expression of the Cu transporter genes ATP7A, ATP7B, SLC31A1, and SLC31A2 were significantly altered in liver cancer samples and were associated with elevated Cu levels in liver cancer tissue and cells. Further analysis of genomic copy number data revealed that alterations in Cu transporter gene loci correlates with poorer survival in HCC patients. Genetic loss of the Cu importer SLC31A1 (CTR1) or pharmacologic suppression of Cu decreased the viability, clonogenic survival, and anchorage-independent growth of human HCC cell lines. Mechanistically, CTR1 knockdown or Cu chelation decreased glycolytic gene expression and downstream metabolite utilization and as a result forestall tumor cell survival after exposure to hypoxia, which mimics oxygen deprivation elicited by transarterial embolization, a standard-of-care therapy used for patients with unresectable HCC. Taken together, these findings established an association between altered Cu homeostasis and HCC and suggest that limiting Cu bioavailability may provide a new treatment strategy for HCC by restricting the metabolic reprogramming necessary for cancer cell survival.
Graphical Abstract
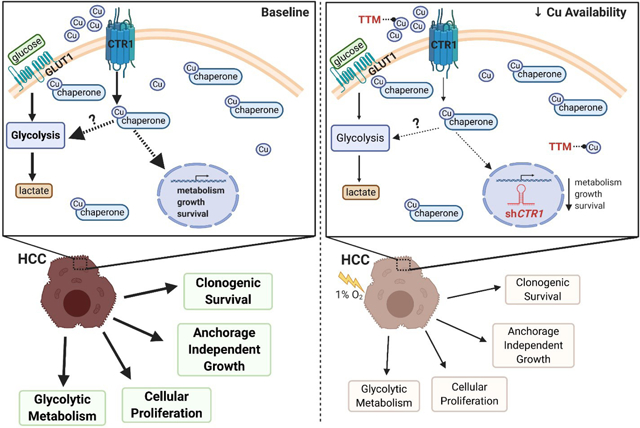
In glycolytic-addicted HCC cells, bioavailable Cu fuels oncogenic pathways that drive tumorigenesis. Intriguingly, genetic manipulation or pharmacologic inhibition of intracellular Cudiminishes hypoxia-induced glycolytic metabolism and attenuates HCC tumorigenic properties.
Introduction
Liver cancer represents the second and the sixth leading cause of cancer-related death in men and women, respectively.1 HCC accounts for 80% of liver cancer cases in the world,2 and pre-existing conditions associated with cirrhosis, such as hepatitis B viral infection, chronic hepatitis C virus infection, non-alcoholic fatty liver disease, hereditary hemochromatosis, and Wilson disease contribute to HCC onset.3 Genomic alterations within HCC, irrespective of etiologic risk factor, emphasize the heterogeneous nature of the disease and thus, advances in molecular medicine targeting the genetic mutations underlying HCC have been unsuccessful.4 Like most cancers, surgical therapies (resection and liver transplantation) and locoregional procedures (radiofrequency ablation) are an effective first line of treatment for localized HCC, with five-year survival rates of up to 60 to 70%.5 Unfortunately, the majority of HCC cases are diagnosed at late stage with only 20% of patients meeting criteria for curative intervention, and the treatment options for intermediate and advanced disease are limited.6 Specifically for intermediate or advanced stage HCC, locoregional therapies including, transarterial embolization (TAE) and transarterial chemoembolization (TACE),5 which take advantage of obstructing tumor blood supply with or without local chemotherapy, or systemic therapy with sorafenib,7,8 a small molecule multikinase inhibitor, provide limited survival benefits. Although obstruction of tumor blood flow limits the delivery of oxygen and nutrients to promote cancer cell death, surviving cells contribute to recurrent HCC following TAE or TACE.6,9–11 The local hypoxic environment induced by TAE and TACE stabilizes the oxygen sensitive master transcription factor, hypoxia inducible factor 1 (HIF-1), which upregulates numerous genes in pro-survival pathways, including glycolysis, essential for cancer cell adaptation.12 Thus, there is a great need to discover unique cellular and molecular features of HCC that can be exploited as novel approaches to treat advanced disease and limit resistance.
Case studies of patients with Wilson Disease, which harbor germline mutations in the gene encoding the P-type ATPase ATP7B,13,14 demonstrate that persistently elevated levels of intracellular Cu impair liver function such that HCC results as a complication.15–18 Beyond the extensive research from both Wilson disease patients and animals that supports a connection between excessive Cu accumulation and hepatobiliary malignancies,19–21 multiple clinical investigations observed elevated serum22–25 and intratumoral26–28 Cu levels in liver cancer patients. Notably, Cu lies at a unique intersection between chemistry and biology, as Cu-driven redox chemistry is required for a multitude of biological programs. To minimize oxidative damage, Cu homeostasis is a highly regulated process, beginning when Cu enters the cell through the essential copper transporter 1, CTR1.29,30 CTR1 is the predominant high-affinity Cu transporter at the plasma membrane, while its low-affinity homolog, CTR2, localizes and facilitates transport from vesicular compartments into the cytoplasm.31,32 To complement Cu influx via CTR1, cells are also equipped with Cu efflux machinery. Specifically, the P-type ATPases ATP7A and ATP7B are membrane-bound transport pumps that function to export cytosolic Cu in an ATP-dependent fashion.33,34 The importance of maintaining a balance between Cu influx and efflux is clear given that deletions in CTR1 lead to embryonic lethality, while mutations in ATP7A and ATP7B manifest in Menkes or Wilson disease, respectively. Interestingly, emerging evidence indicates that regulation of Cu homeostasis coincides with other cellular processes, from lipid metabolism35 to cellular proliferation.36 Our lab has discovered that Cu can enhance oncogenic signaling in the MAPK pathway via a direct interaction with MEK1 and MEK2, resulting in a Cu dependency in mutant BRAFV600E positive melanoma.37 Similarly, Cu is required for the activity of the autophagic kinases ULK1 and ULK2 and autophagosome formation downstream of ULK1 and ULK2 is sensitive to fluctuations in Cu availability.38 Collectively, these findings create a molecular foundation that directly links Cu homeostasis to deregulated signal transduction events that drive pathological conditions. However, the contribution of Cu to intrinsic and extrinsic cellular mechanisms necessary for liver tumorigenesis and resistance driven by treatment-mediated metabolic reprogramming in HCC patients remains unclear.
In the current study, we sought to bridge this gap by taking a bioinformatic approach utilizing publicly available cancer genome datasets. We evaluated the expression of Cu homeostasis genes, namely those facilitating Cu transport such as ATP7A, ATP7B, SLC31A1, which encodes CTR1, and SLC31A2, which encodes CTR2, across HCC and normal tissue samples. From this analysis, we identified significant alterations in the aforementioned Cu transporter genes, and further, found that copy number variations in the Cu transporters correlated with poorer survival and disease-free progression, and was associated with increased Cu levels. Given the importance of Cu modulation in disease management, the manipulation of Cu homeostasis has been previously exploited as an alternative cancer therapy through the usage of various Cu chelating agents.37,39–41 We explored the relevance of perturbed Cu availability, through genetic ablation of CTR1 or pharmacologic inhibition via tetrathiomolybdate (TTM), in HCC. Here we demonstrate that targeting Cu homeostasis exposes a unique vulnerability in HCC, as we discovered Cu-dependent contributions to both the glycolytic metabolism and the tumorigenic properties of HCC.
Methods
Data mining
ATP7A, ATP7B, SLC31A1, or SLC31A2 mRNA expression in normal or tumor liver tissue samples was obtained from the Gene Expression across Normal and Tumor tissue (GENT2) web-based genome database (http://gent2.appex.kr/gent2/, Korean Research Institute and Biotechnology). A total of 1095 patients and 1089 samples across six hepatocellular adenoma or carcinoma data sets [Memorial Sloan Kettering (MSK), Clin Cancer Res 2018; INSERM, Nat Genet 2015; MSK, PLOS One 2018; AMC, Hepatology 2014; RIKEN, Nat Genet 2012; The Cancer Genome Atlas (TCGA), Firehorse Legacy] were selected for query from cBioportal for ATP7A, ATP7B, SLC31A1, or SLC31A2 copy number alterations and patient survival data. For overall survival plot, n = 13 patients with altered copy number of Cu transporters and n = 594 for patients with unaltered copy number Cu transporters. For disease-free progression, n = 11 patients with altered copy number of Cu transporters and n = 545 for patients with unaltered copy number Cu transporters.
ICP-MS sample preparation of human HCC cell lines and rat liver tissue
Human HCC cell lines were seeded at 2.0 × 106 cells in 100mm dishes. After incubation for 48 hours in either 21% O2 or 1% O2, cells were washed twice and harvested with 1X Phosphate Buffered Saline. Cell pellets were collected by centrifugation at 2,000 xg for 5 minutes, and were flash-frozen in a dry ice-ethanol bath prior to storing at −80°C. HCC tumors and adjacent liver tissues were harvested from the livers of Wistar rats subjected to the Dietylnitrosamine-induced (DEN) diet according to an established protocol42. Tissue samples were harvested, weighed, flash-frozen in a dry ice-ethanol bath, and immediately stored at −80°C. All samples were processed by the PADLS New Bolton Center Toxicology Laboratory in the School of Veterinary Medicine at the University of Pennsylvania.
X-Ray Fluorescence Microscopy
For XFM experiments 10 μm sections were transferred to Ultralene®, a XFM compatible window material, mounted on in-house developed lucite sample holders and air-dried. XFM data were collected on beamline 2-ID-E at the Advanced Photon Source (APS), Argonne National Laboratory, Argonne, IL. The incident X-ray energy was tuned to 10 keV using a Simonochromator and focused with a Fresnel zone plate. X-ray fluorescence was collected using an energy dispersive 4-element detector (Vortex ME-4, SII Nanotechnology, Northridge, CA). Raster scans were collected in fly-scan mode, using 1 μm × 1 μm step size with 200 msec dwell time per point. 2-dimensional elemental maps were created by extracting, background subtracting, and fitting the fluorescence photon counts at each point using the program MAPS43. The fluorescent counts were transformed into μg/cm2 using calibrated X-ray standards (AXO products, Dresden, Germany). Quantitative analysis was performed by extracting the fluorescent spectra and fitting and quantifying them against the calibration standards as mentioned above. Area concentrations were converted into volume concentrations using the tissue thickness of 10 μm under the assumption that the X-ray beam fully penetrated the sample.
Cell lines & cell culture
SNU387, SNU398, and SNU449 HCC cell lines and human plateable hepatocytes, 5-Donor were obtained from the American Type Culture Collection (ATCC) and ThermoFisher Scientific, respectively. Parental cell lines were cultured in Roswell Park Memorial Institute (RPMI 1640, Gibco) Media and supplemented with 10% v/v fetal bovine serum (FBS, GE Lifesciences), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). SNU398 and SNU449 cell lines stably expressing the pLKO.1puro constructs were maintained as above supplemented with 5μg/mL puromycin (Invitrogen). SNU398 and SNU449 were stably infected with lentiviruses derived from the pLKO.1 plasmid (see plasmids below) using established protocols. Unless specified, all cell lines were maintained in a humidified Heracell (ThermoFischer Scientific) incubator set to 37°C and 5% CO2. For hypoxic cell culture, cells were maintained at 37°C and 1% O2 in Whitley H35 Hypoxystation (Don Whitley Scientific). MycoAlert® mycoplasma test detection kit (Lonza, LT07–418) was used to test for mycoplasma contamination.
Immunoblot analysis
The indicated HCC cell lines were washed with cold 1x Phosphate-buffered Saline (PBS), and lysed in cold RIPA buffer supplemented with 1x EDTA-free Halt™ protease and phosphatase inhibitor cocktail (ThermoFisher Scientific, #78441). Total protein was quantified using the BCA assay (Pierce, # PI23228), where sample concentrations were interpolated to a BSA standard curve. Equivalent amounts of lysate were resolved by SDS-PAGE using lab established protocols, and protein was detected using the following antibodies (dilution, catalog#, manufacturer): rabbit anti-CCS (1:2000, 20141, Santa Cruz) or mouse anti-β-actin (1:5000, 3700, Cell Signaling Technologies (CST)), followed by detection with one of the following horseradish-peroxidase-conjugated secondary antibodies: goat anti-mouse IgG (1:4000, 7076, CST) or goat anti-rabbit IgG (1:4000, 7074, CST) using SignalFire ECL (CST, # 6883S) detection reagents.
Plasmids
pLKO.1puro lentiviral shRNA plasmids were obtained from High-Throughput Screening Core at the University of Pennsylvania to express: nontargeted control (shSCR), human CTR1 target sequence #1 5’-GATGCCTATGACCTTCTACTT-3’ (shCTR1#1), or human CTR1 target sequence #2 5’-CGGTACAGGATACTTCCTCTT-3’ (shCTR1#2).
RT-qPCR
To examine the expression of Cu transporter and glycolytic genes, 3.0 × 105 cells of the indicated HCC cells were seeded into 60mm dishes. Sixteen hours post-seeding, cells were treated with the indicated concentrations of TTM and/or were moved to hypoxic conditions for 48 hours. To isolate RNA, cells were harvested in TRIzol™ reagent (Invitrogen, #15596018) and RNA was extracted following manufacturer’s protocol. Purified RNA was reverse transcribed (RT) into cDNA using the Applied Biosystems™ Taqman™ Reverse Transcription Reagents (Applied Biosystems, # N8080234) and corresponding protocol. Subsequent cDNA was loaded onto a clear 384-well plate (Genesee, #24–305) and quantified on a ViiA 7 Real-Time PCR System with standard protocols using the following Taqman™ probes: Hs00163707_m1 to detect human ATPase copper transporter A (ATP7A), Hs01075310_m1 to detect human ATPase copper transporter B (ATP7B), Hs00977266_g1 to detect human SLC31A1 (CTR1), Hs00156984_m1 to detect human copper transporter 2 SLC31A2 (CTR2), Hs00761782_s1 to detect human pyruvate kinase muscle isoform (PKM), Hs01378790_g1 to detect human lactate dehydrogenase A (LDHA), Hs00892681_m1 to detect human glucose transporter 1 (GLUT1), and Hs00427620_m1 to detect human TATA-binding proteins (TBP). The Δ2−Ct or comparative ΔΔCt method was used as described previously44 to analyze mRNA after transcript levels were normalized to TBP. For validation of CTR1 knockdown, cells transduced with shRNA targeting CTR1 were harvested and processed as above upon selection with puromycin, and then assayed for relative CTR1 expression following the procedure above.
Reagents
The Cu chelator TTM (#323446) and the crystal violet (# C0775–100G) used for colony staining were purchased from Sigma-Aldrich.
Clonogenic assay
SNU398 and SNU449 cell lines stably expressing indicated constructs were seeded at 3.0 × 103 cells per well in six-well plates. After incubation for seven days, cells were washed once with 1X Phosphate Buffered Saline (PBS) and stained with 1mL of a crystal violet staining solution (0.5% w/v crystal violet (CV), 20% v/v methanol, distilled water) for 15 minutes. After 15 minutes, all wells were washed three times with distilled water to minimize background staining. CV stained colonies were imaged using a ChemiDoc Touch Imagining System (Bio-Rad). To quantify colony abundance, stained cell colonies were dissolved in a 10% acetic acid solution for 30 minutes at room temperature, and extracted CV was measured at an absorbance of 590nm in a plate reader (Synergy, BioTek). For TTM treatments, cells were treated 24 hours after seeding with either a vehicle or a final indicated concentration of TTM for seven days and then processed as mentioned above. For hypoxia (1% O2) exposures, cells were seeded in normoxic (21% O2) conditions for 24 hours, and then moved to the hypoxic condition where pre-equilibrated hypoxic media was applied for the remainder of the seven-day incubation time.
Measurement of cell proliferation with trypan blue
SNU398 and SNU449 cell lines stably expressing the indicated constructs were seeded at 1.5 × 104 cells per well in a six-well plate on Day 0. Proliferation curves using cell lines stably expressing indicated constructs in the presence or absence of TTM were performed at identical times. On Day 1, DMSO or TTM treatment was added to a final concentration of 25 μM. Cell counts were performed every other day by washing cells with 1x PBS, detaching cells with 0.05% Trypsin (Gibco, #25300054). Cells were then resuspended in an equal volume of complete DMEM, and centrifuged at 1000xg for 5 mins. Following aspiration of media, cell pellets were then resuspended in identical volumes of complete DMEM. Cell counting was performed using an automated cell counter (Invitrogen Cell Countess II) by taking an aliquot of cell culture and diluting 1:1 with 0.4% Trypan Blue Solution (Life Technologies/Invitrogen, #15250061) before plating on and reading with a hemocytometer.
Anchorage-independent growth on ultra-low attachment plates
SNU398 and SNU449 cell lines stably expressing the indicated constructs were seeded at 2.0 × 103 cells per well in a 96-well clear flat bottom ultra-low attachment plates (Corning, #3474) on Day 0. For the TTM-treated groups, indicated concentrations of TTM were added 16 hours post-seeding. On Day 5, cells were imaged using an EVOS XL Core Imaging System brightfield microscope with a x10 dry objective. The mean number of spheroids (cells) per field was quantified by a researcher blinded to the genetic manipulations or TTM treatment groups using Fiji software (https://imagej.net/Fiji). Data was normalized to the respective control (shSCR) or vehicle-treated (DMSO) control group.
Measurement of ATP using CellTiter-Glo ® viability assay
SNU398 and SNU449 cell lines stably expressing indicated constructs were seeded in triplicate at 5.0 × 102 cells per well in 96-well white walled flat bottom plates (Fisher Scientific, #655098) or 96-well clear flat bottom ultra-low attachment plates for IC50 determinations in anchorage-dependent (2D) or anchorage-independent (3D) conditions, respectively. Sixteen hours post-seeding, indicated concentrations of TTM were added to appropriate wells and cells were incubated for 72 hours. Cell viability was assessed using the CellTiter-Glo® Luminescent (Promega, #G7572) or 3D (Promega, #9682) Cell Viability Assay for 2D or 3D conditions, respectively, following the manufacturer’s protocol. Normalized %ATP values were calculated by normalizing the raw luminescent values of wells containing the vehicle (DMSO) to wells containing each dose of TTM. To determine the IC50 values, the data was transformed using the nonlinear fit of Log(TTM) versus normalized response (%ATP Normalized to DMSO) with a variable slope function in GraphPad Prism8 software.
Measurement of glucose consumption and lactate production
To determine relative glucose consumption and lactate production, 3.0 × 105 cells of the indicated HCC cells were seeded into 60mm dishes (GenClon, #25–260). Approximately 16 hours post-seeding, cells were treated with indicated concentrations of TTM for 48 hours. For hypoxic conditions, cells were moved to the hypoxic condition where pre-equilibrated hypoxic media was applied for the 48-hour incubation time. Both spent media, collected upon completion of the incubation time, and fresh media, collected at the start of the experiment, were harvested from cell cultures, and following a brief centrifugation (500 × g for five minutes), supernatant was transferred to a fresh Eppendorf tube, flash frozen on an ethanol-dry ice bath, and stored at −80°C until further processing. Glucose consumption and lactate production were measured using a YSI2950 immobilized enzyme analyzer from YSI Life Sciences. Prior to sample analysis, the linearity of the analyzer was calibrated using standards from the manufacturer. Metabolite consumption or production was calculated following the cell number area under the curve normalization as previously described.45 Briefly, metabolite consumption is defined as v = V(xfresh medium − xspent medium)/A, where v is glucose consumption or lactate production, V is the culture volume used, x is the metabolite concentration, and A is the cell number area under the curve. A is obtained by integrating the final cell count and doubling time across the duration (time) of the experiment. Initial and final cell counts used to determine A were obtained using an automated cell counter.
Statistical analysis
Data are reported as mean ±s.e.m. Each sample size (n) represents biologically independent experiments or fields of view. For biologically independent experiments, data was collected from three independent experiments unless otherwise specified within the figure legend. For fields of view, data presented are from 9 fields of view taken from three biologically independent experiments. Statistical significance was determined using an unpaired one- or two-tailed Student’s t-test, a Mantel-Cox test, a one-way or two-way ANOVA followed by Dunnett’s or Tukey’s multiple comparisons test, where significance was defined as P ≤0.05. All statistical analysis was performed in GraphPad Prism 8 software.
Results
Expression of Cu transporters is dysregulated in hepatocellular carcinoma
Clinical measurements of transition metals demonstrated that HCC tumors exhibit elevated Cu levels when compared to normal liver tissue27 and higher serum Cu levels correlate with the presence of cirrhosis or HCC.24 In agreement, Wilson disease patients accumulate Cu in the liver and exhibit cirrhosis and thus, have a higher propensity to develop HCC. To further investigate these clinically relevant observations that connect Cu to HCC development15–18,27,24, we examined the expression of Cu influx and efflux transporters across human liver cancer and normal liver tissue utilizing the Gene Expression database of Normal and Tumor tissues 2 (GENT2). We found that mRNA expression of both Cu transporters SLC31A1 and SLC31A2, along with the Cu exporter ATP7B was significantly lower, while the mRNA expression of the Cu exporter ATP7A was significantly higher in liver cancer (Fig. 1a,b). Intrigued by this finding, we investigated the relationship between patient survival and copy number alterations of Cu transporter loci in six HCC patient data sets in cBioPortal. Although a small cohort, HCC patients with copy number alterations of Cu influx or efflux transporters had a significantly worse outcome with respect to overall survival and likelihood of recurrence (Fig. 1c,d). Taken together, these data suggest that Cu transport into or out of the cell is dysfunctional in a subset of HCC patients. Consistent with clinical findings, we observed elevated Cu levels in HCC tumors compared to adjacent liver tissue collected from a DEN-induced rat model of HCC using inductively coupled plasma mass spectrometry (ICP-MS) (Fig. 1e). Closer examination of the Cu concentration in the rat liver tumor tissue with X-Ray Fluorescence Microscopy (XFM), revealed heterogeneous Cu concentration. In an example shown in Figure 1f from HCC rat tumor tissue, Cu was localized to highly concentrated focal areas of approximately 10 to 15μm diameter that exceeded 5mg/g (~80mM), while the average concentration across the scan was 40μg/g (~60μM). To establish a model that is more amenable to genetic and pharmacologic perturbations, we measured total intracellular Cu levels using ICP-MS from a panel of human HCC cell lines (Fig. 1g). In agreement with the rodent model data, HCC cell lines exhibited significant elevation in Cu levels when compared to a primary hepatocyte line, HMCPP. Recognizing that Cu transporters mediate cellular Cu influx and efflux, we investigated whether there was evidence of differential Cu transporter expression as measured by quantitative PCR (qPCR) (Fig. 1h). Interestingly, mRNA expression of ATP7B, SLC31A1, and SLC31A2 was significantly lower in HCC cells, which parallels the observations made from patient genomic data sets (Fig 1a,b). Further, elevated Cu levels were confirmed by lower protein expression of CCS, which is degraded in a Cu-dependent fashion,46 in all of the HCC cell lines when compared to normal hepatocytes (Fig. 1i), suggesting that HCC cells accumulate excess Cu levels that may contribute to oncogenic properties.
Fig. 1. Aberrant Cu homeostasis is observed in liver cancer and specifically in HCC.
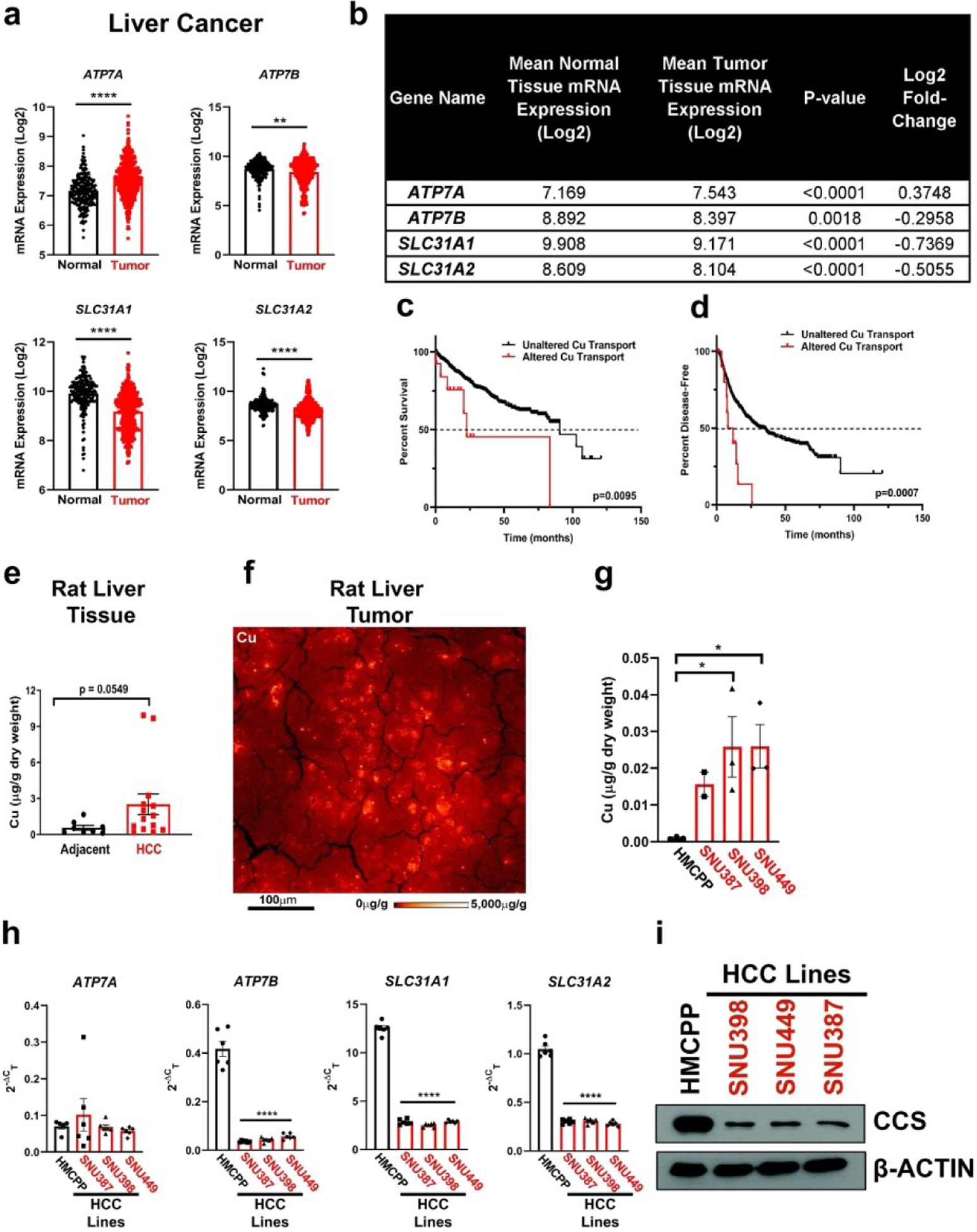
(a) Scatter dot plot with bar at mean ±s.e.m. of mRNA expression of ATP7A, ATP7B, SLC31A1 (CTR1), and SLC31A2 (CTR2) from normal and tumor liver tissue samples from the online, open-access database GENT2. Statistical analysis was performed using an unpaired, two-tailed Student’s t-test. *P < 0.0332,**P < 0.0021, ***P < 0.0002, ****P < 0.0001. (b) Summary table of mRNA expression data shown in (a). (c and d) Kaplan-Meier analysis of overall survival (c) and disease-free progression (d) with median (dashed black lines) from HCC patients with either altered (solid red lines) or unaltered (solid black lines) copy number of Cu transporter genes mentioned in (a) and (b). For overall survival plot, n = 25 patients with altered and n = 582 for patients with unaltered copy number of Cu transporter genes. For disease-free progression, n = 20 patients with altered expression and n = 536 for patients with unaltered copy number of Cu transporter genes. Results were compared using a Mantel-Cox test. *P < 0.0332,**P < 0.0021. (e) Scatter dot plot of inductively coupled plasma mass spectrometry (ICP-MS) detection with bar at mean Cu (μg/g dry weight) from HCC tumors or adjacent liver tissue from rats per sample dry weight ± s.e.m. n = 8 adjacent liver tissue or n = 14 HCC tumors. Results were compared using an unpaired, one-tailed t-test. (f) XFM elemental distribution for Cu of a representative tumor section. The sections was scanned with 1 μm spot size in x and y. Elemental concentrations are shown using false coloring (red temperature, logarithmic scale). (g) Scatter dot plot of ICP-MS detection with bar at mean Cu (μg/g dry weight) from human liver cell lines per sample dry weight ± s.e.m. n = 3 biologically independent samples. Results were compared using a one-way ANOVA followed by a Tukey’s multiple-comparisons test where *P < 0.0332. (h) Scatter dot plot with bar at mean ±s.e.m. of Δ2−Ct normalized quantitative PCR (qPCR) expression of ATP7A, ATP7B, SLC31A1, and SLC31A2 mRNA from normal liver cells (HMCPP) or HCC cell lines (SNU387, SNU398, SNU449) (i) Immunoblot detection of CCS or β-Actin from normal liver cells (primary hepatocytes) or HCC cell lines (SNU398, SNU449, or SNU387). n = 1 biologically independent experiment.
Genetic depletion of CTR1 diminished HCC tumorigenic properties
To interrogate whether these increased Cu levels are essential for HCC tumorigenic properties, we generated stable genetic knockdown of CTR1 with two independent short-hairpin RNAs (shRNA) targeting the SLC31A1 gene in both SNU398 and SNU 449 cells, as measured by qPCR (Fig. 2a,b). Disruption of CTR1 significantly reduced the proliferation of SNU398 and SNU449 cells (Fig. 2c,d). In addition to cellular proliferation, colony formation of SNU398 and SNU449 cells when plated at low density, a property that distinguishes tumorigenic cells from healthy cells, was dependent on CTR1 expression (Fig. 2e,f). Furthermore, patients with HCC may develop metastases which requires detachment from the extracellular matrix and invasion into nearby organs47. Thus, to complement our findings of reduced HCC proliferation and colony formation in the context of CTR1 deficiency, ultra-low attachment (ULA) polystyrene plates were used to evaluate the effects of CTR1 depletion on anchorage-independent growth in SNU398 and SNU449 cells. Knockdown of CTR1 significantly diminished the anchorage-independent growth of SNU398 and SNU449 cells (Fig. 2g,h). Taken together, these findings demonstrate that HCC cell lines depend on Cu transport via CTR1 for proliferation, colony formation, and anchorage-independent growth, suggesting that altered Cu availability contributes to hepatocarcinogenesis.
Fig. 2. Loss of the major Cu transporter CTR1 reduces tumorigenic properties of HCC.
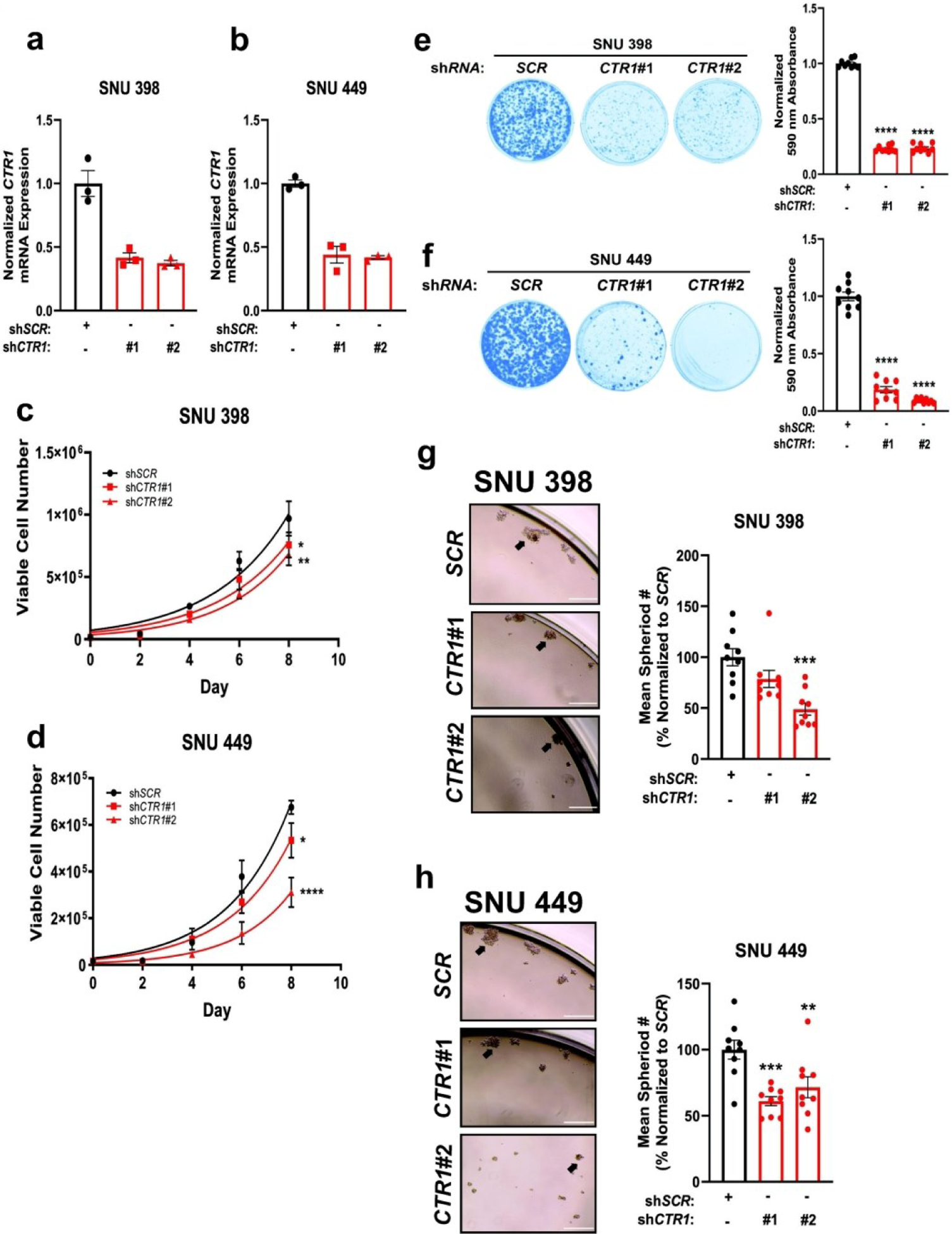
(a and b) Scatter dot plot with bar at mean ±s.e.m. of normalized quantitative PCR (qPCR) expression of CTR1 mRNA from SNU398 (a) and SNU449 (b) HCC cell lines stably expressing shRNA against CTR1 or a non-targeting scramble sequence (SCR). n = 1 biologically independent experiment performed in technical triplicate. (c and d) Non-linear fit to the exponential growth equation of cellular proliferation from SNU398 (c) and SNU449 (d) HCC cell lines expressing shRNA against CTR1 or SCR. n = 3 independent biological experiments, with each experiment plated in technical triplicate. Statistical analysis was performed using a two-way ANOVA followed by Dunnett’s multiple comparison test. (e and f) Representative images (left) of crystal violet stained colonies from SNU398 (e) and SNU449 (f) HCC cells expressing shRNA against CTR1 or SCR, and scatter dot plot (bottom) of mean absorbance of extracted crystal violet at 590 nm ±s.e.m. of crystal violet staining from three independent experiments plated in technical triplicate. The results were compared using a one-way ANOVA followed by Dunnett’s multiple comparison test. *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001. (g and h) Representative images of anchorage-independent growth in ultra-low attachment plates in SNU398 (g, left) or SNU449 (h, left) cell lines stably expressing shRNA against CTR1 or SCR, with the normalized mean number of spheroids per field of view represented as a scatter dot plot for SNU398 (g, right) and SNU449 (h, right) from n = 9 fields of view per condition from three independent biological experiments. Data was analyzed using a one-way ANOVA followed by Dunnett’s multiple comparison test *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001. 10x magnification, scale bar = 400 μm.
TTM treatment attenuated HCC proliferation and anchorage-independent growth
The repurposing of TTM, a Cu-specific chelator used in the treatment of Wilson disease, has been explored as a cancer therapy in several contexts.39,41,48,49 Mechanistically, preclinical studies and phase I/II clinical trials suggest that Cu-dependent components of the tumor microenvironment41, oncogenic kinase signaling pathways38,49,50, and metabolic pathways51 mediate sensitivity to TTM.40,41 Considering that HCC tumors are well-vascularized52 and demonstrate amplified receptor tyrosine kinase signaling4 as well as a dependency on glycolytic metabolism53,54, we hypothesized that disruption of Cu accessibility through pharmacological interventions would diminish HCC tumorigenic properties. To begin evaluating this hypothesis, SNU398 and SNU449 cells were treated with increasing concentrations of TTM and then cell viability was measured using a high-throughput luminescent-based assay that detects ATP. TTM treatment decreased ATP, and thus viability of SNU398 and SNU449 cells, in a dose-dependent manner (Fig. 3a–c). To determine whether TTM would remain as efficacious in 3D cultures, the IC50 of TTM-treated SNU398 and SNU449 cells seeded in ULA plates was determined (Fig. 3d–f). Interestingly, there was nearly a two-fold and three and a half-fold increase in the IC50 concentration of TTM from 2D to 3D cultures in SNU398 cells and SNU449 cells, respectively (Fig. 3c,f). This finding is in agreement with other observations that HCC cells grown in 3D have enhanced drug-resistance and aggressiveness as compared to their 2D counterparts.55 In accordance, when treated with TTM at a concentration at or above the IC50, SNU398 and SNU449 cells still exhibited reduced proliferation as measured by trypan blue exclusion staining (Fig. 3g,h). Moreover, when cultured with increasing concentrations of TTM in anchorage-independent conditions, spheroid formation was significantly reduced in SNU449 cells and trended downwards in SNU398 cells (Fig. 3i,j). Collectively, these results are early indications that Cu chelation through TTM may be effective in reducing the properties that comprise HCC tumorigenesis.
Fig. 3. TTM, a Cu specific chelator, hinders anchorage-dependent and anchorage-independent growth.
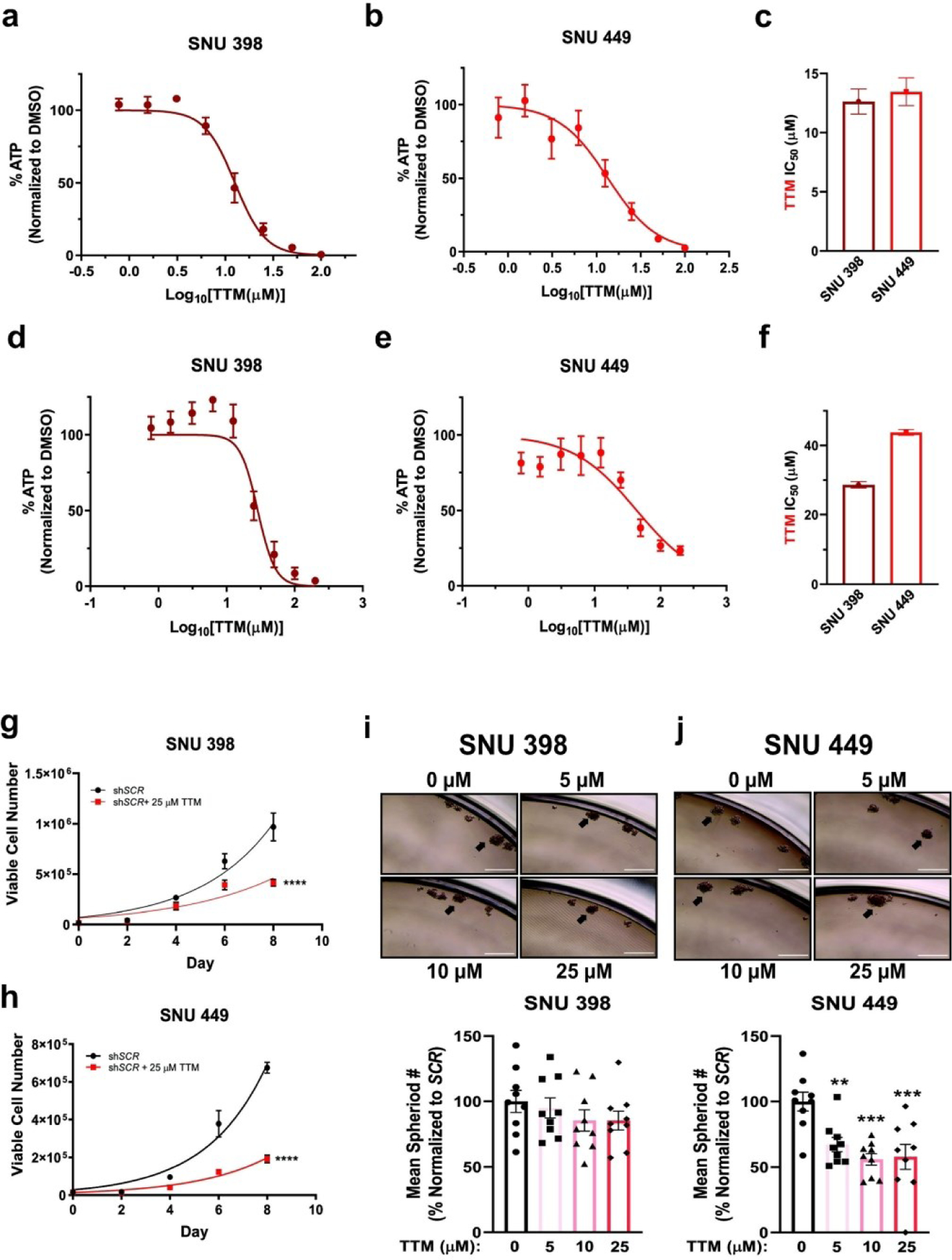
(a and b) Relative CellTiter-Glo® cell viability ± s.e.m. of SNU398 (a) or SNU449 (b) HCC cells treated with the indicated concentrations of TTM upon plating in anchorage-dependent (2D) conditions. n = 2 independent biological experiments, with each experiment plated in technical triplicate. (c) Bar graph of TTM IC50 values from (a) and (b) ± s.e.m. (d and e) Relative CellTiter-Glo 3D® cell viability ± s.e.m. of SNU398 (d) or SNU449 (e) HCC cells treated with the indicated concentrations of TTM upon seeding into ultra-low attachment (3D) plates. n = 3 independent biological experiments, with each experiment plated in technical triplicate. (f) Bar graph of TTM IC50 values from (d) and (e) + s.e.m. (g and h) Non-linear fit to the exponential growth equation of cellular proliferation from SNU398 (g) and SNU449 (h) cell lines, seeded side-by-side with cells in Fig. 2c and 2d, except treated with 25 μM TTM. n = 3 independent biological experiments, with each experiment plated in technical triplicate. Statistical analysis was performed using a two-way ANOVA followed by Dunnett’s multiple comparison test. ****P < 0.0001. (i and j) Representative images of anchorage-independent growth in ultra-low attachment plates in SNU398 (i, top) or SNU449 (j, top) cell lines treated with the indicated concentration of TTM, with the normalized mean number of spheroids per field of view represented as a scatter dot plot for SNU398 (i, bottom) and SNU449 (j, bottom) from n = 9 fields of view per condition from three independent biological experiments. Data was analyzed using a one-way ANOVA followed by Dunnett’s multiple comparison test *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001. 10x magnification, scale bar = 400 μm.
Genetic loss of CTR1 under hypoxic conditions hinders HCC metabolism
Intriguingly, HCC cells adapt to accommodate the larger energy demands required for rapid growth and proliferation. HCC cells reprogram their glucose metabolism56 to fulfill these requirements at baseline and upon stress induced by the hypoxic environment driven by the metabolic zonation of the liver57 and treatment-induced ischemia through TAE/TACE.10,11,58,59 In the latter, oxygen-depleted conditions stabilize HIF-1, which induces the transcription of genes in multiple hypoxia response pathways. In particular, HIF-1 elevates the mRNA expression of genes that encode critical glycolytic proteins, such as glucose importer 1 (GLUT1), pyruvate kinase muscle isoform (PKM), and lactate dehydrogenase A (LDHA). It has been well-established that cells under hypoxia survive by relying on glycolytic metabolism to produce ATP. However, questions remain regarding the effects of Cu depletion in a hypoxic environment, which more closely recapitulates the glycolytic state of HCC cells under standard-of-care TAE and TACE treatment. Given the requirement for elevated intracellular Cu levels in HCC cells,27 we investigated whether Cu depletion would alter the metabolic flexibility of HCC cells under hypoxic conditions. To address this question, SNU398 or SNU449 cells harboring stable genetic knockdown of CTR1 were exposed to hypoxic (1% O2) or normoxic (21% O2) conditions and evaluated for expression of key glycolytic genes (Fig 4a,b). As predicted, exposure to hypoxia for 48 hours significantly induced expression of the glycolytic genes PKM, GLUT1, and LDHA. However, mRNA knockdown of CTR1 significantly blocked the hypoxia-mediated increase in GLUT1 transcripts, while also reducing PKM or LDHA transcripts in SNU398 or SNU449 cells, respectively. These findings that reduced CTR1 expression decreases the hypoxia-dependent transcription of several glycolytic genes suggest that this response is partly Cu-dependent. In parallel to monitoring glycolytic gene expression upon hypoxic exposure, we investigated whether hypoxia would alter Cu transporter expression in a similar fashion to that previously observed in human pulmonary arterial smooth muscle cells,60 murine macrophages,61 or rodent intestinal epithelial cells.62 Similar to previous work, hypoxia exposure significantly elevated transcription of ATP7A, but only illustrated modest changes in SLC31A1, SLC31A2, and ATP7B across both cell lines (Fig. 4a,b). Notably, this response was further enhanced in the presence of reduced CTR1 upon hypoxic exposure. Following the trends in the transcriptional data, glucose consumption and lactate production increased upon exposure to hypoxia but significantly decreased upon stable knockdown of CTR1 in SNU398 and SNU449 cells (Fig. 4c,d). To examine the functional consequences of a Cu-dependent alteration in metabolic rewiring, we assessed whether SNU398 or SNU449 cells deficient in CTR1 could survive in a hypoxic environment (Fig. 4e,f). While CTR1 knockdown alone was sufficient to significantly reduce clonogenic survival, this effect was further accentuated under hypoxic conditions after seven days. Taken as a whole, a sustained reduction in CTR1 is sufficient to alter the glycolytic metabolism required to propel the tumorigenic properties of HCC cells undergoing hypoxic stress.
Fig. 4. Genetic loss of CTR1 under hypoxic conditions hinders HCC metabolism.
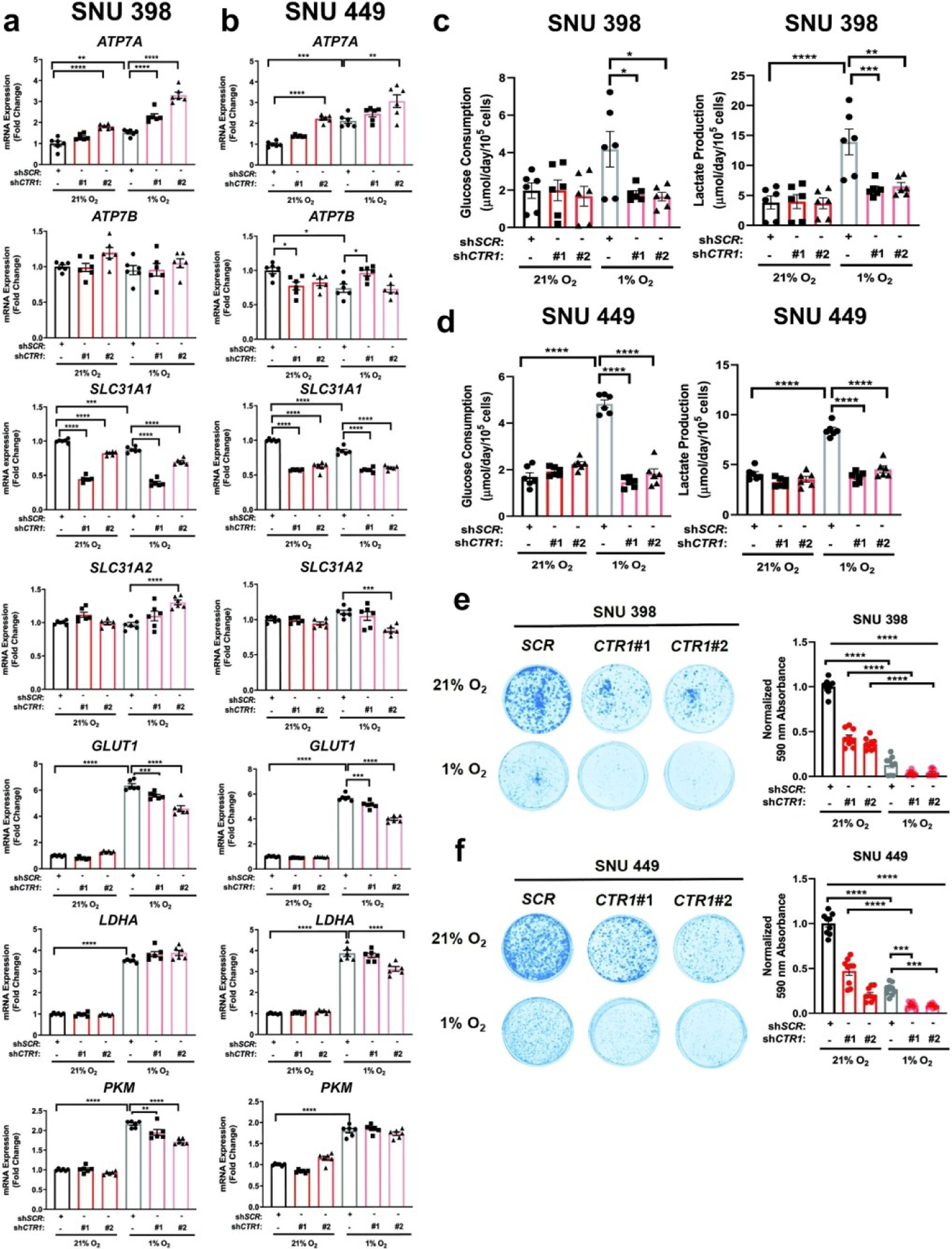
(a and b) Scatter dot plot with bar at mean ±s.e.m. of normalized quantitative PCR (qPCR) mRNA expression of indicated genes from SNU398 (a) and SNU449 (b) cells stably expressing shRNA against CTR1 or a non-targeting scramble sequence (SCR) with exposure to hypoxic (1% O2) for conditions for 48 hours. n = 3 biologically independent experiments performed in technical duplicate. (c and d) Scatter dot plot with bar at mean ±s.e.m. of glucose consumption (left) or lactate production (right) rates from SNU398 (c) and SNU449 (d) cells stably expressing shRNA against CTR1 or a non-targeting scramble sequence (SCR) with exposure to hypoxic conditions for 48 hours. n = 3 biologically independent experiments performed in technical duplicate. (e and f) Representative images (left) of crystal violet stained colonies from SNU398 (e) and SNU449 (f) cells stably expressing shRNA against CTR1 or a non-targeting scramble sequence (SCR) in normoxic (21% O2) or hypoxic conditions for seven days and scatter dot plot (right) of mean absorbance of extracted crystal violet at 590 nm ±s.e.m. of crystal violet staining from three independent experiments plated in technical triplicate. All statistical analysis was performed using a one-way ANOVA followed by Tukey’s multiple comparison test. *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001.
Hypoxia in combination with TTM treatment curtailed HCC metabolism
To determine whether Cu chelation would alter metabolic flexibility in HCC, SNU 398 and SNU449 cells treated with TTM were exposed to hypoxic or normoxic conditions and expression of key glycolytic genes was evaluated (Fig. 5a,b). As expected, exposure to hypoxia induced the robust and significant transcription of PKM, GLUT1, and LDHA genes (Fig. 5a,b). However, treatment with TTM revealed a significant Cu-dependent reduction in PKM, GLUT1, and LDHA when compared to hypoxia treatment alone in SNU398 cells, while trending similarly in SNU449 cells (Fig. 5a,b). These results agree with a previous study from Feng and collegues63 that demonstrated that the Cu and Zn chelator tetraethylenepentamine (TEPA) could suppress HIF-1 transcriptional activity and the subsequent expression of hypoxia-responsive genes. Next, to assess whether differential oxygen tensions foster differential Cu uptake, we measured intracellular Cu levels using ICP-MS analysis from SNU398 and SNU449 cells after exposure to hypoxia (1% O2) for 48 hours. Accordingly, intracellular Cu levels were significantly elevated upon hypoxic conditions in SNU398 cells and trended upwards in SNU449 cells (Fig 5g). These data support the idea that hypoxia induces differential Cu utilization within HCC cells.
Fig. 5. Hypoxic conditions in combination with TTM alter HCC metabolism.
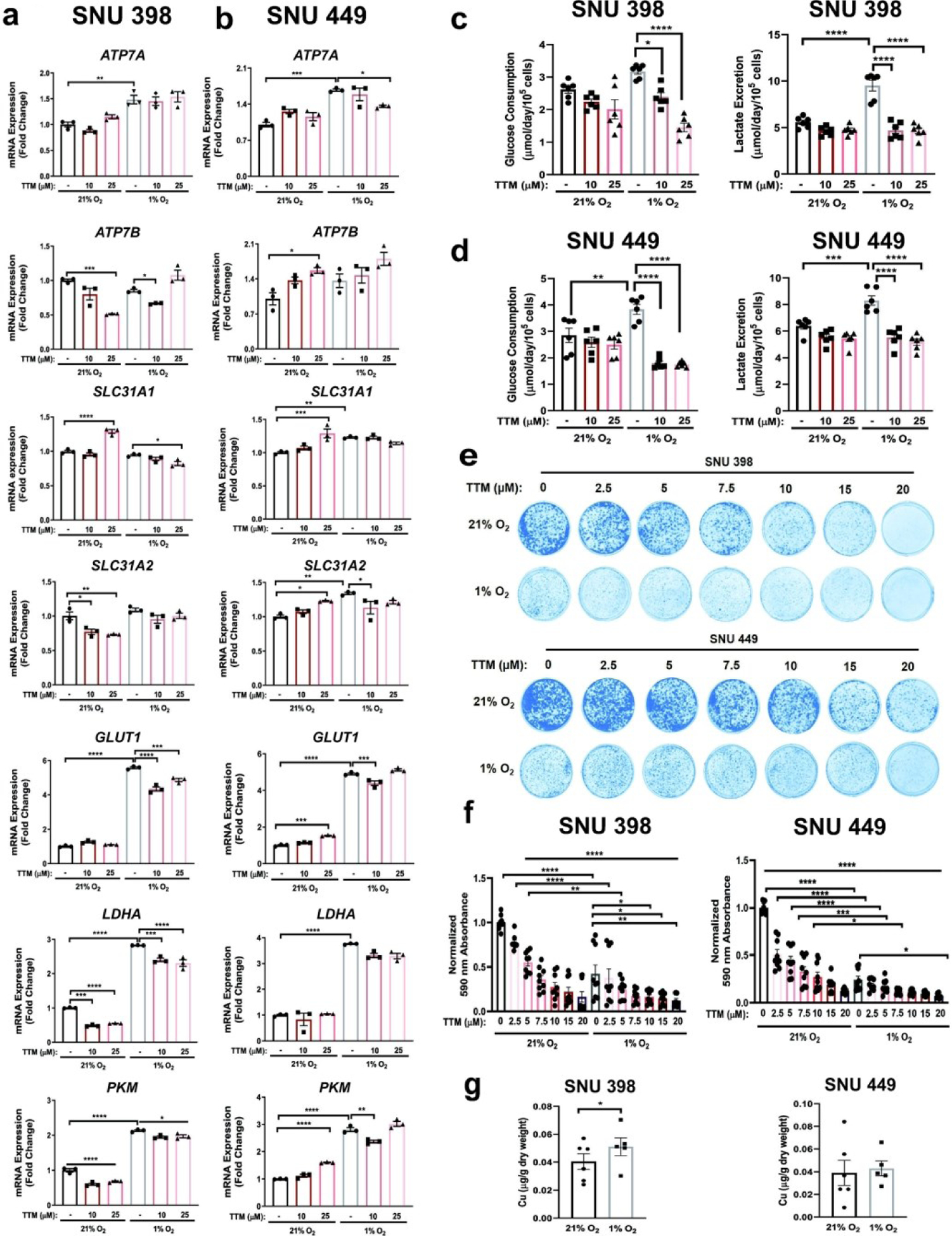
(a and b) Scatter dot plot with bar at mean ±s.e.m. of normalized quantitative PCR (qPCR) mRNA expression of indicated genes from SNU398 (a) and SNU449 (b) cells upon treatment with 10 μM or 25 μM TTM or exposure to hypoxic (1% O2) conditions for 48 hours. Representative n of three biologically independent experiments performed in technical triplicate. (c and d) Scatter dot plot with bar at mean +s.e.m. of glucose consumption (left) or lactate excretion (right) rates from SNU398 (c) and SNU449 (d) cells upon treatment with 10 μM or 25 μM TTM or exposure to hypoxic conditions for 48 hours. n = 3 biologically independent experiments performed in technical duplicate. (e) Representative images of crystal violet stained colonies from SNU398 (top) and SNU449 (bottom) HCC cells treated with vehicle (DMSO) or increasing concentrations of TTM in normoxic (21% O2) or hypoxic conditions for seven days of three independent experiments plated in technical triplicate. (f) Scatter dot plot of mean absorbance of extracted crystal violet at 590 nm ±s.e.m. of crystal violet staining experiments in (e). (g) Scatter dot plot of ICP-MS detection with bar at mean Cu (μg/g dry weight) from indicated HCC cell lines exposed to normoxic or hypoxic conditions. n = 6 biologically independent experiments performed and statistical analysis was performed with a paired, two-tailed student t-test *P = 0.0257. For panels a–f, statistical analysis was performed using a one-way ANOVA followed by Tukey’s multiple comparison test. *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001.
Importantly, both glucose consumption and lactate production aligned with the transcriptional findings, as these measurements significantly diminished upon the TTM-hypoxia combination compared to hypoxia exposure alone (Fig. 5c,d). Hence, these observations indicate that pharmacologic reductions in Cu availability hinder the reprogramming of glucose metabolism in HCC. Finally, to evaluate whether targeting intracellular Cu levels within a hypoxic environment will change HCC tumorigenic properties, we assessed the ability for TTM-treated SNU398 and SNU449 cells to survive under hypoxia upon seeding at low density. TTM treatment alone significantly blunted clonogenic growth in a dose-dependent manner and when combined with hypoxic conditions (Fig. 5e,f). Taken together, these results support the notion that limiting Cu availability within a uniform hypoxic microenvironment works to restrict both glucose metabolism and survival ability of HCC cells.
Discussion
In the present study, we highlighted Cu homeostasis as a targetable vulnerability within HCC and provided early evidence to suggest Cu chelation as a supplemental treatment option for patients with this disease. The mining of publicly available, cancer genomic data sets uncovered several features that may explain the consistent clinical findings of elevated Cu levels across HCC patients. We demonstrated that dysregulation of Cu homeostasis, through altered expression of Cu transporter genes, correlated with unfavorable outcomes in HCC patients. In support of this data, Cu transporter expression in a panel of human liver cell lines reflected these observations. While the SLC31A1 (CTR1) levels were decreased in both human liver tumor tissues and cell lines, which suggests HCC tumors could be Cu deficient, Cu levels were significantly elevated in rat liver tumors and several human HCC cell lines that aligned with reduced CCS protein abundance. Thus, we have demonstrated utilizing both molecular and genetic approaches that Cu levels are indeed elevated in the context of liver cancer and this marked increase is associated with reduced mRNA expression of SLC31A1, SLC31A2, and ATP7B, suggesting that increased Cu content of HCC tumors may be driven by the loss of ATP7B instead of the downregulation of SLC31A1 (CTR1) and SLC31A2 (CTR2). Alternatively, cancer cell lines increase nutrient scavenging in the form of macropinocytosis,64 which was recently proven to be a mechanism for cancer cells to acquire Cu and could be responsible for the increased Cu levels in HCC cell lines and tumor tissue. However, future studies must be conducted to further investigate this concept. To the best of our knowledge, this study is the first to identify a relationship between the expression of Cu transporter genes and prognosis in HCC patients. These results support previous studies that described Cu levels as significantly higher in both patient serum22–25 and resected tumors.26–28 Considering the trend of poorer disease-free survival for HCC patients with altered Cu transporter copy number (Fig. 1d), and a previous report where small tumors (<35 mm) yielded high Cu levels that correlated positively with differentiation status of the tumor,27 it is likely that Cu homeostasis contributes more to hepatocarcinogenesis than to HCC progression. Even though our findings reveal dysfunctional Cu transport and provide essential insights toward the development of alternative therapeutics in the treatment of HCC, further molecular studies are needed define the role of cuproproteins in the pathogenesis of HCC.
In addition, we demonstrated that CTR1 is necessary for cellular proliferation, clonogenic survival, and anchorage-independent growth of HCC cell lines. These results complement experiments performed by Porcu et al., where transient knockdown of CTR1 reduced cell viability, cell cycle progression, and cell migration in human immortalized hepatic progenitor or hepatoma cells, while treatment with CuSO4 slightly enhanced these properties.24 Although the authors suggested that MYC is responsive to Cu levels, whether other transcriptional regulators mediate these Cu-dependent responses remains to be determined. However, elementary proof may lie in the correspondence between MYC target genes and microarray data that revealed an upregulation in genes associated with cell growth, cell migration, angiogenesis, and small GTPase mediated signal transduction upon exposure to high concentrations of CuSO4 in HepG2 cells.65 Taken together, protein factors that regulate the above processes, such as the frequently mutated TERT, β-Catenin, or p16, may influence Cu-dependent responses and represent important targets for future studies. Furthermore, we demonstrated that hypoxia in combination with CTR1 deficiency attenuated HCC glycolytic metabolism and tumorigenic properties. Importantly, the liver is one of few organs that maintains an oxygen gradient, however, liver cancers seem to nurture a remarkedly hypoxic environment.66,67 Hypoxia is a known driver of oncogenesis and regulates processes including epithelial-to-mesenchymal transition,68 angiogenesis,69 and invasion.70 Considered in conjunction, manipulating Cu availability may therefore represent a useful approach for modulating one or more of these hypoxic-dependent responses which shape tumorigenesis in HCC.
In parallel, we demonstrated that treatment with the Cu specific chelator TTM inhibits HCC tumorigenic properties. Although animal studies in HCC have not yet been performed using TTM, an early study of HCC tumor xenografts showed that Cu chelation with trientine, another Cu chelator, can induce apoptosis and reduce tumor volume.71 Despite this preclinical study that explored Cu chelation as a treatment option for HCC,71 the efficacy of Cu chelation in combination with the standard-of-care therapies, TAE and TACE, has yet to be investigated. Here we provide evidence that TTM reduces the hypoxia-induced expression of glycolytic genes, which in turn, reduces both glucose utilization and lactate excretion. Based on previous work, several molecular phenomena may further support the contribution of our findings. Firstly, HCC cells exhibit “metabolic flexibility”, where glucose metabolism is reprogrammed in response to increased demand for biological building blocks required of rapidly proliferating cells or stress associated with the nutrient deprivation induced by TAE or TACE.11 Key elements of liver rewiring include upregulation of GLUT1, LDHA, and PKM, which drive flux through glycolysis and towards lactate production.56 Additionally, the consistent upregulation of ATP7A upon hypoxia suggests that liver rewiring may even extend to the reprogramming of trace metal metabolism. Moreover, metabolic flexibility can influence the angiogenic properties of tumors under hypoxia.72 In linking angiogenesis to metabolism, Végran and colleagues provided evidence to assert that lactate is sufficient to promote tumor angiogenesis through the lactate/NF-kB/IL-8 axis.73 In accordance with these studies, our data suggest that TTM may reduce tumorigenic properties in the context of hypoxia by significantly blocking glucose consumption and lactate production, which constrains vascularization. Secondly, findings from Martin et al. and Feng et al. demonstrate that Cu contributes to the regulation of HIF-1α. Specifically, excessive Cu in the presence of hypoxia resulted in a Cu-dependent enhancement of both HRE-reporter activity and mRNA expression of hypoxia-responsive genes, while Cu chelation with TEPA suppressed these outputs by blocking HIF-1α-mediated interactions.63,74 Thus, another explanation may be that Cu merely functions to inhibit the upstream negative regulators of HIF-1α, perhaps by competing with iron for binding to prolyl-4-hydroxylases (PHD) enzymes. Finally, while the concentration of TTM required to reduce the tumorigenic properties of HCC cell lines was in the micromolar range, Cu chelation could improve outcomes with locoregional therapies like TACE in which chemotherapy and embolic are applied directly to the tumor vasculature. In conclusion, this work provides genetic evidence of disrupted Cu homeostasis in the context of HCC. Moreover, we have provided a foundation for further exploration of TTM treatment in HCC. Specifically, future studies will focus on the combination of TTM with standard-of-care locoregional therapy, as this pairing may prove a suitable strategy to combat the recurrence observed in a majority of HCC patients.
Significance to metallomics.
HCC represents an alarming global healthcare problem with an increasing incidence. Current treatment strategies for HCC demonstrate limited efficacy because each is agnostic to molecular and genetic features of the disease. Although significant elevation of the transition metal Cu has been associated with HCC, the contribution of Cu to hepatocarcinogenesis is not well understood. Here, we present an analysis of altered Cu transporter expression in HCC that elucidated a unique Cu-dependent vulnerability within glycolytic metabolism necessary for tumorigenesis. These findings uncover a clinically tractable alternative treatment to combat HCC by repurposing Cu chelators.
Acknowledgements
We thank O. Antipova for support and assistance with X-Ray Fluorescence Microscopy data collection and analysis at the Advanced Photon Source part of the Argonne National laboratory supported by the Department of Energy, Office of Basic Energy Sciences, under contract no. DEAC02-06CH11357, A. Mancuso for materials, reagents, technical assistance, and discussions supporting this manuscript, and D. Sneddon for administrative support. This research was supported by W.W. Smith Charitable Trust #C1604 (D.C.B.), the Pilot & Feasibility Program Award (D.C.B) from National Institutes of Health (NIH) grant P30DK050306-21S1 for the University of Pennsylvania Center for Molecular Studies in Digestive and Liver Diseases, and Oregon Health and Science University Foundation (M.R.). R.M. Kiefer was supported by Howard Hughes Medical Insitute (HHMI) Medical Research Fellows Program.
Footnotes
Conflicts of Interest
D.C.B. holds ownership in Merlon Inc. D.C.B. is an inventor on the patent application 20150017261 entitled “Methods of treating and preventing cancer by disrupting the binding of copper in the MAP kinase pathway”. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Siegel RL, Miller KD and Jemal A, CA. Cancer J. Clin, 2019, 7–34. [DOI] [PubMed] [Google Scholar]
- 2.Altekruse SF, Henley SJ, Cucinelli JE and McGlynn KA, Am. J. Gastroenterol, 2014, 109, 542–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Serag HB, Hepatocellular Carcinoma, 2011, vol. 365. [DOI] [PubMed] [Google Scholar]
- 4.Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, Calatayud A-L, Pinyol R, Pelletier L, Balabaud C, Laurent A, Blanc J-F, Mazzaferro V, Calvo F, Villanueva A, Nault J-C, Bioulac-Sage P, Stratton MR, Llovet JM and Zucman-Rossi J, Nat Genet, 2015, 47, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruix J and Sherman M, Hepatology, 2011, 53, 1020–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terzi E, Piscaglia F, Forlani L, Mosconi C, Renzulli M, Bolondi L and Golfieri R, BMC Cancer, 2014, 14, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, de Oliveira AC, Santoro A, Raoul J-L, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz J-F, Borbath I, Häussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D and Bruix J, N. Engl. J. Med, 2008, 359, 378–390. [DOI] [PubMed] [Google Scholar]
- 8.Sanoff HK, Chang Y, Lund JL, O’Neil BH and Dusetzina SB, Oncologist, 2016, 21, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paul SB, Gamanagatti S, Sreenivas V, Chandrashekhara SH, Mukund A, Gulati MS, Gupta AK and Acharya SK, Indian J. Radiol. Imaging, 2011, 21, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gade TPF, Tucker E, Nakazawa MS, Hunt SJ, Wong W, Krock B, Weber CN, Nadolski GJ, Clark TWI, Soulen MC, Furth EE, Winkler JD, Amaravadi RK and Simon MC, Radiology, 2017, 000, 160728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perkons N, Kiefer R, Noji M, Pourfathi M, Ackerman D, Siddiqui S, Tischfield D, Profka E, Johnson O, Pickup S, Mancuso A, Pantel A, Denburg G, MR Nadolski S, Hunt, Furth E, Kadlecek S and Gade T, Hepatology, 2019, Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, Kwon JH, Moon YH, Kim YB, Yu YS, Lee N, Choi KY, Kim YS, Park YK, Kim BW and Wang HJ, J. Cancer Res. Clin. Oncol, 2014, 140, 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bull PC, Thomas GR, Rommens JM, Forbes JR and Cox DW, Nat. Genet, 1993, 5, 327–337. [DOI] [PubMed] [Google Scholar]
- 14.Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L and Brzustowicz LM, Nat. Genet, 1993, 5, 344–350. [DOI] [PubMed] [Google Scholar]
- 15.Kumagi T, Horiike N, Michitaka K, Hasebe A, Kawai K, Tokumoto Y, Nakanishi S, Furukawa S, Hiasa Y, Matsui H, Kurose K, Matsuura B and Onji M, J. Gastroenterol, 2004, 39, 1165–1169. [DOI] [PubMed] [Google Scholar]
- 16.Kumagi T, Horiike N, Abe M, Kurose K, Iuchi H, Masumoto T, Joko K, Akbar SMF, Michitaka K and Onji M, Intern. Med, 2005, 44, 439–443. [DOI] [PubMed] [Google Scholar]
- 17.Iwadate H, Ohira H, Suzuki T, Abe K, Yokokawa J, Takiguchi J, Rai T, Orikasa H, Irisawa A, Obara K, Kasukawa R and Sato Y, Intern. Med, 2004, 43, 1042–1045. [DOI] [PubMed] [Google Scholar]
- 18.Thattil R and Dufour JF, World J. Gastroenterol, 2013, 19, 2110–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfeiffenberger J, Mogler C, Gotthardt DN, Schulze-Bergkamen H, Litwin T, Reuner U, Hefter H, Huster D, Schemmer P, Członkowska A, Schirmacher P, Stremmel W, Cassiman D and Weiss KH, Liver Int, 2015, 35, 1615–1622. [DOI] [PubMed] [Google Scholar]
- 20.Huster D, Purnat TD, Burkhead JL, Ralle M, Fiehn O, Stuckert F, Olson NE, Teupser D and Lutsenko S, J Biol Chem, 2007, 282, 8343–8355. [DOI] [PubMed] [Google Scholar]
- 21.Huster D, Ann. N. Y. Acad. Sci, 2014, 1315, 37–44. [DOI] [PubMed] [Google Scholar]
- 22.Poo JL, Rosas-Romero R, Montemayor AC, Isoard F and Uribe M, J. Gastroenterol, 2003, 38, 45–51. [DOI] [PubMed] [Google Scholar]
- 23.El Fotouh OA, El Aziz HA, Galal M and El Nakeeb N, Egypt. Liver J, 2012, 2, 7–11. [Google Scholar]
- 24.Porcu C, Antonucci L, Barbaro B, Illi B, Nasi S, Martini M, Licata A, Miele L, Grieco A and Balsano C, Oncotarget, 2018, 9, 9325–9343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Attia AM, Attalla SM, Barakat EAME, Zaki MES and Elkhouly NY, Indian J. Forensic Med. Toxicol, 2019, 13, 398–404. [Google Scholar]
- 26.Ebara M, Fukuda H, Hatano R, Saisho H, Nagato Y, Suzuki K, Nakajima K, Yukawa M, Kondo F, Nakayama A and Sakurai H, J. Hepatol, 2000, 33, 415–422. [DOI] [PubMed] [Google Scholar]
- 27.Ebara M, Fukuda H, Hatano R, Yoshikawa M, Sugiura N, Saisho H, Kondo F and Yukawa M, Oncology, 2003, 65, 323–330. [DOI] [PubMed] [Google Scholar]
- 28.Jie J, Hao S, Hongxiu Y, Huiying Y, Jun M, Chenji W, Mingjie Y and Yong M, J. Trace Elem. Med. Biol, 2007, 21, 255–260. [DOI] [PubMed] [Google Scholar]
- 29.Lee J, Prohaska JR and Thiele DJ, Proc. Natl. Acad. Sci. U. S. A, 2001, 98, 6842–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuo YM, Zhou B, Cosco D and Gitschier J, Proc. Natl. Acad. Sci. U. S. A, 2001, 98, 6836–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Den Berghe PVE, Folmer DE, Malingré HEM, Van Beurden E, Klomp AEM, Van De Sluis B, Merkx M, Berger R and Klomp LWJ, Biochem. J, 2007, 407, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertinato J, Swist E, Plouffe LJ, Brooks SPJ and L’Abbé MR, Biochem. J, 2008, 409, 731–740. [DOI] [PubMed] [Google Scholar]
- 33.Nyasae L, Bustos R, Braiterman L, Eipper B and Hubbard A, Am. J. Physiol. Liver Physiol, 2007, 292, G1181–G1194. [DOI] [PubMed] [Google Scholar]
- 34.Pase L, Voskoboinik I, Greenough M and Camakaris J, Biochem. J, 2004, 378, 1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnamoorthy L, Cotruvo JA, Chan J, Kaluarachchi H, Muchenditsi A, Pendyala VS, Jia S, Aron AT, Ackerman CM, Wal MNV, Guan T, Smaga LP, Farhi SL, New EJ, Lutsenko S and Chang CJ, Nat. Chem. Biol, 2016, 12, 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turski ML and Thiele DJ, J Biol Chem, 2009, 284, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ and Counter CM, Nature, 2014, 509, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsang T, Posimo JM, Gudiel AA, Cicchini M, Feldser DM and Brady DC, Nat. Cell Biol,, DOI: 10.1038/s41556-020-0481-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brewer GJ, Dick RD, Grover DK, LeClaire V, Tseng M, Wicha M, Pienta K, Redman BG, Jahan T, Sondak VK, Strawderman M, LeCarpentier G and Merajver SD, Clin. Cancer Res, 2000, 6, 1–10. [PubMed] [Google Scholar]
- 40.Pan Q, Kleer CG, Van Golen KL, Irani J, Bottema KM, Bias C, De Carvalho M, Mesri EA, Robins DM, Dick RD, Brewer GJ and Merajver SD, Cancer Res, 2002, 62, 4854–4859. [PubMed] [Google Scholar]
- 41.Chan N, Willis A, Kornhauser N, Mward M, Lee SB, Nackos E, Seo BR, Chuang E, Cigler T, Moore A, Donovan D, Cobham MV, Fitzpatrick V, Schneider S, Wiener A, Guillaume-Abraham J, Aljom E, Zelkowitz R, Warren JD, Lane ME, Fischbach C, Mittal V and Vahdat L, Clin. Cancer Res, 2017, 23, 666–76. [DOI] [PubMed] [Google Scholar]
- 42.Kiefer RM, Hunt SJ, Pulido S, Pickup S, Furth EE, Soulen MC, Nadolski GJ and Gade TP, J. Vasc. Interv. Radiol, 2017, 28, 1043–1050.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogt S, Maser J and Jacobsen C, in Journal De Physique. IV : JP, 2003. [Google Scholar]
- 44.Schmittgen TD and Livak KJ, Nat. Protoc, 2008, 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- 45.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB and Mootha VK, Science (80-.), 2012, 336, 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brady GF, Galbán S, Liu X, Basrur V, Gitlin JD, Elenitoba-Johnson KSJ, Wilson TE and Duckett CS, Mol Cell Biol, 2010, 30, 1923–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zimmermann A, in Tumors and Tumor-Like Lesions of the Hepatobiliary Tract, Springer International Publishing, 2016, pp. 1–29. [Google Scholar]
- 48.Cox C, Teknos TN, Barrios M, Brewer GJ, Dick RD and Merajver SD, Laryngoscope, 2001, 111, 696–701. [DOI] [PubMed] [Google Scholar]
- 49.Brady DC, Crowe MS, Greenberg DN and Counter CM, Cancer Res, 2017, 77, 6240–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ and Counter CM, Nature, 2014, 509, 492–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishida S, Andreux P, Poitry-Yamate C, Auwerx J and Hanahan D, Proc Natl Acad Sci U S A, 2013, 110, 19507–19512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fodor D, Jung I, Turdean S, Satala C and Gurzu S, World J. Hepatol, 2019, 11, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu H, Pan L, Gao C, Xu H, Li Y, Zhang L, Ma L, Meng L, Sun X and Qin H, Molecules,, DOI: 10.3390/molecules24101993. [DOI] [Google Scholar]
- 54.Amann T, Maegdefrau U, Hartmann A, Agaimy A, Marienhagen J, Weiss TS, Stoeltzing O, Warnecke C, Schölmerich J, Oefner PJ, Kreutz M, Bosserhoff AK and Hellerbrand C, Am. J. Pathol, 2009, 174, 1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jung HR, Kang HM, Ryu JW, Kim DS, Noh KH, Kim ES, Lee HJ, Chung KS, Cho HS, Kim NS, Im DS, Lim JH and Jung CR, Sci. Rep, 2017, 7, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hay N, Nat. Rev. Cancer, 2016, 16, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang YBA, Eo J, Mert S, Yarmush ML and Usta OB, Sci. Rep, 2018, 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wong CCL, Kai AKL and Ng IOL, Front. Med. China, 2014, 8, 33–41. [Google Scholar]
- 59.Kung-Chun Chiu D, Pui-Wah Tse A, Law CT, Ming-Jing Xu I, Lee D, Chen M, Kit-Ho Lai R, Wai-Hin Yuen V, Wing-Sum Cheu J, Wai-Hung Ho D, Wong CM, Zhang H, Oi-Lin Ng I and Chak-Lui Wong C, Cell Death Dis, 2019, 10, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zimnicka AM, Tang H, Guo Q, Kuhr FK, Oh M-J, Wan J, Chen J, Smith KA, Fraidenburg DR, Choudhury MSR, Levitan I, Machado RF, Kaplan JH and Yuan JXJ, PLoS One, 2014, 9, e90544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.White C, Kambe T, Fulcher YG, Sachdev SW, Bush AI, Fritsche K, Lee J, Quinn TP and Petris MJ, J Cell Sci, 2009, 122, 1315–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie L and Collins JF, Am. J. Physiol. - Cell Physiol,, DOI: 10.1152/ajpcell.00023.2011. [DOI] [Google Scholar]
- 63.Feng W, Ye F, Xue W, Zhou Z and Kang YJ, Mol. Pharmacol, 2009, 75, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aubert L, Nandagopal N, Steinhart Z, Lavoie G, Nourreddine S, Berman J, Saba-El-Leil MK, Papadopoli D, Lin S, Hart T, Macleod G, Topisirovic I, Gaboury L, Fahrni CJ, Schramek D, Meloche S, Angers S and Roux PP, Nat. Commun, 2020, 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Min OS, Li J and Freedman JH, Physiol. Genomics, 2009, 38, 386–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brooks AJ, Eastwood J, Beckingham IJ and Girling KJ, Br. J. Anaesth, 2004, 92, 735–737. [DOI] [PubMed] [Google Scholar]
- 67.Leary TS, Klinck JR, Hayman G, Friend P, Jamieson NV and Gupta AK, Anaesthesia, 2002, 57, 1128–1133. [DOI] [PubMed] [Google Scholar]
- 68.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M and Haase VH, J. Clin. Invest, 2007, 117, 3810–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E, Song H, VandenBerg S, Johnson RS, Werb Z and Bergers G, Cancer Cell, 2008, 13, 206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krishnamachary B, Berg-Dixon S, Kelly B, Agani F, Feldser D, Ferreira G, Iyer N, LaRusch J, Pak B, Taghavi P and Semenza GL, Cancer Res, 2003, 63, 1138–1143. [PubMed] [Google Scholar]
- 71.Yoshii J, Yoshiji H, Kuriyama S, Ikenaka Y, Noguchi R, Okuda H, Tsujinoue H, Nakatani T, Kishida H, Nakae D, Gomez DE, De Lorenzo MS, Tejera AM and Fukui H, Int. J. Cancer, 2001, 94, 768–773. [DOI] [PubMed] [Google Scholar]
- 72.Sonveaux P, Copetti T, De Saedeleer CJ, Végran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frérart F, Gallez B, Ribeiro A, Michiels C, Dewhirst MW and Feron O, PLoS One, 2012, 7, e33418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Végran F, Boidot R, Michiels C, Sonveaux P and Feron O, Cancer Res, 2011, 71, 2550–2560. [DOI] [PubMed] [Google Scholar]
- 74.Martin F, Linden T, Katschinski DM, Oehme F, Flamme I, Mukhopadhyay CK, Eckhardt K, Tröger J, Barth S, Camenisch G and Wenger RH, Blood, 2005, 105, 4613–4619. [DOI] [PubMed] [Google Scholar]