

Abstract
Genome-wide association studies (GWAS) have uncovered over a hundred genetic loci associated with atrial fibrillation (AF), the most common arrhythmia. Many of the top AF associated loci harbor key cardiac transcription factors, including PITX2, TBX5, PRRX1 and ZFHX3. Moreover, the vast majority of the AF-associated variants lie within non-coding regions of the genome where causal variants affect gene expression by altering the activity of transcription factors and the epigenetic state of chromatin. In this review, we discuss a transcriptional regulatory network model for AF defined by effector genes in GWAS loci. We describe the current state of the field regarding the identification and function of AF-relevant gene regulatory networks, including variant regulatory elements, dose-sensitive transcription factor functionality, target genes and epigenetic states. We illustrate how altered transcriptional networks may impact cardiomyocyte function and ionic currents that impact AF risk. Lastly, we identify the need for improved tools to identify and functionally test transcriptional components in order to define the links between genetic variation, epigenetic gene regulation and atrial function.
Keywords: Atrial fibrillation, Transcription Factor, Transcriptional regulation, epigenetics, Atrial Fibrillation, Epigenetics
Introduction
While historically considered an acquired disease, atrial fibrillation (AF) risk is now understood to include a major heritable component, with evidence including rare mutations in families and association with common variants in the population at large.1–3 Recent work by large consortia have identified over 100 genetic loci associated with AF by genome-wide association studies (GWAS).4,5 Together with epidemiological studies, the current model suggests that AF is a “complex trait”, influenced by a combination of multiple genetic and environmental risk factors that contribute cumulatively to disease predisposition.6,7,8. Genome-wide polygenic risk scores of common variants have recently been developed that identify individuals at greater than threefold increased risk of AF in the general population.9 In the near future, use of such approaches may identify high risk patients with a combination of inherited susceptibilities and potentially provide preventive treatment or lifestyle adjustments to decrease AF risk. However, the polygenic risk score currently does not help to determine a targeted treatment, as it does not give any specific information on the affected molecular pathways, such as ion flux deficiencies affecting conduction velocity or myocardial automaticity, or ancillary pathologies such as fibrosis.
Most AF-associated GWAS variants reside in the non-coding genome. The majority of disease-associated variants, as well as variants in high linkage disequilibrium, are enriched in predicted transcriptional regulatory elements, implying that the identified GWAS variants alter transcriptional regulation.10–12
A number of loci harboring TFs, including PITX2, ZFHX3, PRRX1, TBX5, NKX2–5 and HAND2, have been linked to AF susceptibility (Table 1). While information regarding the actual target genes of the noncoding variation (i.e. variant regulatory elements) in most of these loci is still lacking, experimental evidence from animal and cell models with modified transcription factor genes has implicated these transcription factors in normal atrial rhythm control and the pathology of AF (Table 1).13–25 These transcription factors target thousands of regulatory elements for genes involved in atrial function, including ion channels, gap junctions, structural/cytoskeletal components, and others.26–30 Thus, growing evidence supports a transcriptional model for the mechanisms underlying GWAS in AF: noncoding variation in regulatory sequences affects expression of genes encoding transcriptional regulators, and/or function of regulatory elements of given target genes, altering gene expression and conferring disease susceptibility (Fig. 1). Therefore, resolution of the individual components of the GWAS-associated transcriptional networks influenced by genetic variation is a fundamental goal of current research.
Table 1.
Overview of TFs near AF associated loci and their phenotypes
| Locus | (Cardiac) phenotype | eQTL (direction) | Refs. |
|---|---|---|---|
| PITX2 | At least 6 independent common genetic variants upstream of the PITX2 gene are strongly associated with AF in populations. PITX2 regulates left-right differentiation (including sinoatrial node restriction to the right atrium), and deficiency leads to structural and electrical remodeling of the heart, which may lead to AF through different mechanisms. | - | 4,45,59–62 |
| ZFHX3 | Gene involved in tumorigenesis. Heterozygous germline deletion results in preweaning mortality and increased cell proliferation. Also involved in circadian rhythm. | - | 63–66 |
| TBX5 | Heterozygous mutations cause limb and cardiac defects (Holt-Oram syndrome). TBX5 is essential for septal and atrioventricular conduction system development, and atrial ventricular bundle maintenance. TBX5 point mutations are known to cause AF. | TBX5 * (risk***) | 52,53,67 |
| PRRX1 | Involved in skeletogenesis, limb-, elastic artery-, ductus arteriosus and pulmonary vascular development. | PRRX1 * (protective***) | 68–71 |
| NKX2–5 | Essential for cardiogenesis and for the development of the conduction system. Mutations in this gene have been found to lead to congenital heart disease, conduction defects as well as atrial fibrillation. | - | 72,73–75 |
| BMP2 | Involved atrioventricular canal and in valve formation, cardiomyocyte differentiation, cushion formation and septation. | - | 76–80 |
| CREB5 | Together with ATF-2 responsible for adipocyte differentiation, and involved in the regulation of colorectal cancer metastasis. | - | 81,82 |
| HAND2 | KO of HAND2 leads to embryonic lethality between E9.5-E10.5, due to anterior heart tube hypoplasia and vascular development defects. OFT morphology is dependent on Hand2, and cardiac morphological development is sensitivity to Hand2 dosage. | HAND2-AS1** (risk***) | 83–86 |
| TBX3 | Heterozygous mutations cause defects of limbs, mammary glands, teeth, and additional tissues (ulnar-mammary syndrome). In heart required for specification of the sinoatrial node and pacemaker function, atrioventricular conduction system, and outflow tract development. | - | 87–89 |
MetaXcan heart,
eQTL GTEx heart.
Risk: higher expression associated with increased risk for AF. Protective: higher expression associated with decreased risk for AF
Figure 1.
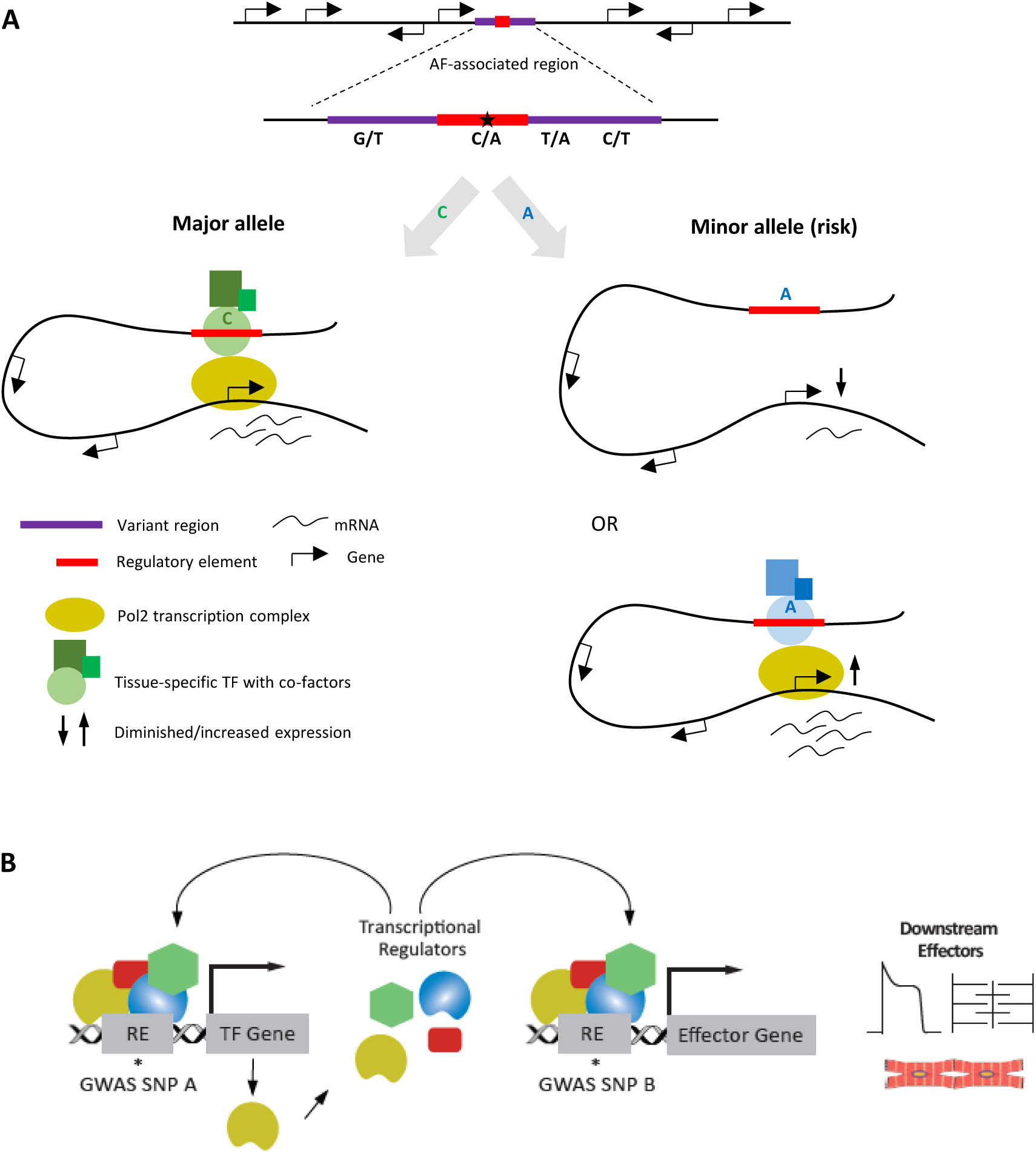
Simplified scheme depicting the relation between noncoding variants clustered in variant regions, variant regulatory elements, transcription factor (TF) dose and target genes. A) An AF-associated region is depicted with neighboring genes. The major allele of an AF-associated variant lies in a regulatory element, which interacts with transcription factors and cofactors leading to physiological levels of transcription of (a) target gene(s) through physical interaction. On the right, two situations are depicted that could result from the presence of a minor (risk) allele in the regulatory element. The risk allele could interfere with binding of the correct TF, leading to diminished expression of target genes. The risk allele could cause altered binding affinity favoring another TF, causing (tissue-specific) gain/loss of expression of target gene(s), that results in atrial fibrillation predisposition. B) Variation in a regulatory element (RE) can lead to the altered expression level of a gene for a TF (yellow) or target effector gene. The changed dose of the TF influences the expression of many effector genes, possibly including its own gene. (Illustration Credit: Ben Smith).
In this review, we summarize the progress of the field in linking transcription factor networks to AF, and discuss currently employed approaches, progress and difficulties in identifying the variants and target genes contributing to AF development. We describe the potential role and current knowledge of epigenetics (i.e. gene regulation by transcription factors, chromatin states, and conformation) in modulating the effect of variant regulatory elements. Moreover, we discuss the combinatorial effects of multiple common variants that could increase predisposition to AF. Finally, we discuss AF variants in the context of development and aging.
Transcription factors, atrial rhythm and AF risk
The identification of non-coding regulatory elements harboring AF risk implies the importance of transcriptional regulators in AF, borne out by the identification of AF risk signals at many transcription factor loci. There are hundreds of validated and suspected transcription factors, some of which have tissue- or condition- specific expression to regulate cell differentiation and developmental programs (reviewed in31). The binding of accessible regulatory elements is dependent on both availability and activity (including binding affinity) of transcription factors. Futhermore, transcription factor dose is crucial in the regulation of transcription. For example, mutations or haploinsufficiency of developmentally expressed transcription factors such as TBX5, NKX2–5, PRRX1, HAND2, and PITX2 result in genetic syndromes that have many symptoms including congenital defects of the heart and other tissues, and conduction abnormalities (Table 1).32–34 Many common variants associated with AF and cardiac conduction are near these and other transcription factor-encoding genes, suggesting that variation in their regulation may impact AF predisposition.
An example of the importance of transcription factor dosage in AF is found in the PITX2 locus. PITX2 expresses three isoforms, PITX2A, PITX2B, and PITX2C, the latter being expressed from an independent promoter and representing the major cardiac isoform.35–38 Mouse studies revealed that Pitx2c is required for asymmetric heart development, imposing left-sided identity on the left atrium and sinus venosus (including suppression of sinoatrial node development at the left side), and for proper development of the pulmonary vein myocardium.39–43 In addition, cardiac expression of PITX2 is almost exclusively restricted to the left atrium in the normal adult heart,36 and its expression increases after myocardial injury in the nenonatal left ventricle.44 Both the left atrium and the sleeves of the pulmonary vein are potential substrates for development of AF, and the pulmonary vein is often the origin of ectopic foci in AF patients.45 While homozygous Pitx2c mutant mice display embryonic lethality, reduced expression of Pitx2c during development results in AF inducibility upon stimulation, which implicates a developmental role of this gene in AF susceptibility.35–37 One of the key findings of the gene expression studies is that PITX2 is one of the most differentially expressed left atrial genes in healthy adult heart. In addition, genes associated with a right atrial phenotype such as BMP10 (bone morphogenic protein 10) appear to be repressed by PITX2, thus linking PITX2 to left-sided identity of the left atrium.17,36,44 Various possible mechanistic explanations of PITX2 deficiency resulting in AF have been proposed. It has been observed that reduction in Pitx2 resulted in potassium and calcium channel gene dysregulation, ultimately leading to a shortening of the atrial action potential, a depolarized resting membrane potential, and predisposition to AF (Fig. 2).21,45 Pitx2 may also regulate miR-17–92 and miR-106b-25; when deleted from the mouse genome, AF susceptibility occurred.46 Analysis of gene expression in an atrial-specific knockout of Pitx2 demonstrated similar transcriptional alterations in calcium homeostasis and AF-related microRNAs (Fig. 2).47 In addition, through the use of zebrafish and murine models of Pitx2 deficiency, changes in sarcomere length and metabolic dysfunction in cardiomyocytes were linked to AF.17,20,48,49 These findings suggest that, although Pitx2-deficient mice with a predisposition to AF apparently have morphologically normal left atria,36,44,45 there are critical Pitx2 targets beyond ion channels that have important implications for electrophysiology. The postnatal function of Pitx2 was investigated using conditional deletion techniques, resulting in altered expression of genes encoding ion channels, cell junction proteins, calcium handling genes and transcription factors, several of which were previously implicated by GWAS.17,21,36,44 Importantly, these mice show ECG abnormalities indicative of sinus node dysfunction, but surprisingly are not inducible for AF. These conditional deletion studies in mice appear to indicate that Pitx2 also has an important role in the postnatal left atria necessary for maintaining normal sinus rhythm, in addition to its more established developmental functions. Continued studies of the diverse roles of Pitx2 in both the developing and postnatal heart will deepen our understanding of this critical cardiac transcription factor. The multiple roles of Pitx2 are depicted in Fig. 2.
Figure 2.

Schematic of PITX2 functions within a left atrial cardiomyocyte. It has been established that TBX5 is a transcriptional activator of PITX2. PITX2 is then capable of regulating gene expression throughout the genome, having both activating and repressing capabilities. Direct PITX2 targets, validated largely by luciferase assays and ChIP-seq, are listed by cellular function. While direct ion channel and gap junction genes were some of the first PITX2 targets discovered, there is now evidence in multiple model organisms that PITX2 is also capable of regulating cardiomyocyte metabolism and the antioxidant response to stress. Furthermore, PITX2 has been shown to directly alter microRNA transcription, which in turn can globally regulate gene expression in the cardiomyocyte. It is important to note that there are additional putative PITX2 targets not listed here that have been discovered through RNA sequencing experiments of PITX2 deficient murine models; however, these transcriptional alterations need further validation to distinguish direct targets of PITX2 from secondary effects. Overall, these transcriptional targets of PITX2 are able to determine critical physiological outputs of the cardiomyocyte, such as regulating cardiac rhythm and sarcomere structure. (Illustration Credit: Ben Smith).
TBX5 is another well-studied example of a dose dependent transcription factor, which is at the center of the cardiac transcription factor network. TBX5 mutations resulting in haploinsufficiency cause Holt-Oram syndrome of congenital upper limb (hand) and heart defects.50–52 In contrast, a “gain-of-function” mutation in a large Dutch family causes atypical Holt-Oram syndrome, without obvious cardiac structural defects, but with over 65% of affected family members displaying early onset AF.53 Regions near TBX5 have been associated with prolongation of the PR-interval in addition to AF risk.4,5,54 Murine models of spontaneous AF are rare, however, conditional deletion of Tbx5 in the adult heart resulted in a model of primary, spontaneous AF.14,55 This model identified Tbx5 as direct activator of Pitx2, with Tbx5 and Pitx2 antagonistically regulating the expression of key ion channel protein-encoding genes such as Scn5a, Gja1, Ryr2 and Atp2a2 (Fig. 2, 3). AF in Tbx5 deficient mice was mechanistically connected to calcium transport mechanisms and expression of Atp2a2, which encodes the main cardiac sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2).16,18 Genetic deletion of the SERCA2 inhibitor phospholamban (Pln) normalized SERCA function and rescued Tbx5 deficiency-associated AF. Interestingly, variants in the vicinity of PLN have also been associated with AF.4 Moreover, conditional haploinsufficiency of Tbx5 in adult mice caused reduced expression of predicted target genes and a predisposition to AF; additional Pitx2 haploinsufficiency rescued these phenotypes, supporting the notion of antagonistic gene regulation in vivo. These data resulted in a model describing an incoherent feed forward loop driven by TBX5 and modulated by PITX2 (Fig. 2). This Tbx5 driven regulatory network was further interrogated using the sequencing of non-coding RNAs (ncRNAs) to identify regulatory elements crucial for cardiac rhythm homeostasis. This differential deep sequencing approach identified lncRNA-associated regulatory elements which controlled the expression of calcium handling genes, including Ryr2 and Atp2a2.56 These data elucidated important aspects of the regulatory network driving calcium handling physiology, and also identified a lncRNA required for Ryr2 expression.56 This lncRNA was also tightly associated with chromatin and required for stabilization of RNA PolII at the Ryr2 promoter, all of which suggested it had a functional role in gene expression.56 While non-coding RNA transcripts in AF will not be generally discussed in this review, the investigation of functional lncRNAs in future efforts could provide instrumental insight into the maintenance of gene expression for cardiac rhythm.
Figure 3.

Transcription factor networks control the expression of effector genes defining properties relevant for atrial structure, function and rhythm. Tbx5 and Pitx2 interact antagonistically as transcriptional activator and repressor, respectively, to co-regulate a gene regulatory network that governs effector genes involved in calcium cycling, sodium currents, potassium currents, and cell-cell conduction. Other transcription factors such as Gata4 and Nkx2–5 are also known to interact with Tbx5 and Pitx2 to co-regulate genes involved in atrial structural development and electrophysiological properties. The interactions of other transcription factors (such as Prrx1) in AF physiology and their role in atrial rhythm remain to be determined.
The cooperative interactions between the transcription factors TBX5, GATA4 and NKX2–5 is key for proper binding at their respective binding sites, and cooperative regulation of the expression of the downstream target genes and of cardiac development.57,58 In contrast, Tbx5 and Gata4 were demonstrated to interact antagonistically for regulation of atrial rhythm control (Fig. 3).18 The adult-specific Tbx5 haploinsufficiency phenotype of arrhythmia susceptibility and prolonged action potential duration14 was rescued by Gata4 haploinsufficiency, but not by Nkx2–5 haploinsufficiency.18 Because Gata4 haploinsufficiency normalized the reduced expression of calcium channel genes Ryr2 and Atp2a2 in the Tbx5 haploinsufficient mice, it was hypothesized that the beneficial effect was mediated by rescue of calcium homeostasis (Fig. 3). In the healthy cardiomyocyte, the inhibiting effect of Gata4, coupled with the stimulating effect of Tbx5 results in balanced Atp2a2 expression. However, in Tbx5 haploinsufficient mice, Gata4 overtakes the balance causing over-repression of Atp2a2 expression. Decreased Atp2a2 expression causes Ca2+ cycling dysfunction, resulting in reduced Ca2+-influx into the SR. Consequently, there is excess cytosolic Ca2+, leading to prolonged action potential duration and arrhythmia susceptibility. Gata4 haploinsufficiency in Tbx5 haploinsufficient mice restored the transcription factor dosage balance and sinus rhythm. Pln haploinsufficiency also had a similar rescuing effect on Tbx5 haploinsufficiency, validating the involvement of SERCA. This study illustrates the potential collaboration of transcription factors in a network, contributing to AF (Fig. 3).
For a number of AF-associated genes, including TBX5, eQTL analysis predicts increased, not decreased expression will result in increased AF susceptibility.4 In line with this, the familial “gain-of-function” mutation in TBX5 caused early onset AF development in humans with atypical Holt-Oram syndrome.53 Consistently, when the mouse orthologs of PR interval or AF associated regions were removed from the Tbx3 and Tbx5 loci, respectively, moderate (<2 fold) increases in expression of the respective genes were observed, resulting in target gene deregulation and conduction disorders (VMC, unpublished data). These data indicate expression levels of these transcription factors are tightly balanced in sinus rhythm, and that similar to haploinsufficiency, mildly increased transcription factor expression can have physiologically relevant consequences. To date, loss of function models have been used to explore the role of particular genes in AF, however the above findings suggest that physiologic overexpression models need to be considered to appropriately recapitulate AF risk-associated changes in gene expression and accurately model AF molecular and clinical features.
Transcription is controlled by a combination of active and repressive transcription factors in concert with epigenetics, i.e. “molecules and mechanisms that can perpetuate alternative gene activity states in the context of the same DNA sequence”.90 This definition includes DNA methylation, histone modifications, chromatin conformation and (nuclear/noncoding) RNAs, all of which are involved in regulation of gene transcription. For summaries of the literature on the role of noncoding RNAs, mostly microRNAs, in AF we refer to others.91–96
Causative variant regions: identification of regulatory elements and single nucleotide polymorphisms that alter their activity
The vast majority of trait-associated variants are located in noncoding regions of the genome. These regions are posited to contain elements acting as regulatory chromatin. Therefore, identification of those elements harboring functional phenotype-associated genetic variation is required to understand how transcriptional regulation contributes to AF. Genetic variation may alter the function of regulatory elements by disrupting the binding of transcription factors to the associated binding motif in the regulatory element (Fig. 1) even if the variant does not directly disrupt the motif itself (reviewed in12).
Several factors complicate the search for transcription-modulating variants and their target genes. Multiple regulatory elements could be present in a single disease-associated linkage disequilibrium block, where the function could be either redundant, additive, synergistic or modulating.97,98 These properties are of critical physiological importance but are not captured in current functional regulatory element assays (Table 2). Furthermore, while the mouse epigenome is more thoroughly characterized than that of human, conservation of mouse-human orthologous regulatory elements (based on H3K27ac ChIP-seq) conservation is limited,99 and species-specific acquisition of new TF-binding sites within enhancers occurs frequently.100 Therefore, mouse (or other non-human) data sets used to predict regulatory elements are very useful but should be interpreted with care. Currently, our ability to predict the function of regulatory elements from epigenetic data is poor. Additionally, regulatory element in vivo function (e.g. repression, modulating chromatin conformation) is poorly captured by most assays. Limited anecdotal examples of in vivo characterization of the physiological role of regulatory elements in atrial biology and AF has revealed the vast complexity of the gene regulatory mechanisms which control and establish normal cardiac rhythms.15,18,19,101
Table 2:
Techniques and characteristics
| Technique | Output | Advantages | Disadvantages | Refs |
|---|---|---|---|---|
| STARR-seq, Cap-STARR-seq, ChIP-STARR | Quantifies enhancer strength in complex candidate libraries | High throughput and size of test element | Elements tested episomally, in combination with a single selected gene promoter. Difficult to perform in physiologically relevant cell type. | 123–125 |
| SuRE, Variant MPRA | Survey of regulatory elements, Quantifies allele-specific enhancer strength | Allele-specific, high sensitivity and throughput | Elements tested episomally. Difficult to perform in physiologically relevant cell type. | 117,126 |
| MPRA, CRE-seq | Multiplexed measurement of transcriptional regulatory activity via synthetic reporter gene | High throughput | Restricted size of test element. Elements tested episomally. Difficult to perform in physiologically relevant cell type. |
127–129 |
| Cellular genetic screens (CRISPR/Cas9-based) | Cellular CRISPRi perturbations followed by single cell RNA-seq | Enhancer-target gene pairing | Perturbation not equivalent to natural situation | 121,122 |
| ATAC-seq | Assay for transposase accessible chromatin | Low cell number input, fast protocol | No functional information | 130 |
| ChIP-seq | Sequence reads obtained by antibody-captured protein-DNA interactions | Genome-wide | Dependent on crosslinking (PFA) and specificity of antibody | 131,132 |
| ncRNA-seq | Sequencing of RNA to identify active regulatory elements | Endogenous, in vivo, requires small input, genome-wide | Dependent on accessibility data, on level of expressed RNA and its stability. | 56,103,105,133 |
| CUT&RUN | Antibody-targeted controlled cleavage by nuclease | Low background signal, suitable for single cell analysis | Likelihood of over-digestion, dependence on antibody quality | 134 |
| 3C | Chromatin architecture analysis “one-versus-one” | High resolution | Low throughput, use of primers, questionable quantification | 112,135 |
| 4C | High-throughput chromatin architecture analysis “one-versus-all” | High-throughput | Dependence on restriction enzyme, missed close-range interactions, semi-quantitative (UMI-4C is quantitative) | 136–138 |
| 5C | Medium-throughput chromatin architecture analysis “many-versus-many” | Medium-throughput | Limited in terms of size of genomic region assayed, dependence on restriction enzyme, missed close-range interactions | 135,139–141 |
| Hi-C, SPRITE | High-throughput chromatin architecture analysis “all-versus-all” | High-throughput, genome-wide | Dependence on restriction enzyme, missed close-range interactions | 142,143 |
| PC Hi-C | High-throughput chromatin architecture analysis “one-versus-all” | Enrichment for promoter interactions | Dependence on restriction enzyme, missed close-range interactions | 144–146 |
Several epigenomic features can define regulatory elements: occupancy by specific transcription factors, association with activating histone post-translational modifications (such as H3K27ac), localization within DNase-hypersensitivity or ATAC-seq identified region of open chromatin, or the production of non-coding RNAs. For cell type or organ-specific occupation by a transcription factor or histones with an activating post-translational modification, various assays have been developed over the decades (Table 2). These epigenetic signatures have provided semi-comprehensive lists of candidate cell type specific regulatory elements and insight into their behavior.102 A significant body of literature indicates that active regulatory elements are transcribed, producing non-coding RNAs (ncRNAs).56,103–106 The production of ncRNA from regulatory elements has been used to quantitatively define regulatory element activity,104,107 including from native regulatory elements from the mouse atria in vivo.56 The identification of regulatory elements based on ncRNA transcription has been applied to identify functionally active context dependent enhancers and has allowed identification of functional transcription factor-dependent regulatory elements.56 In addition to their limited conservation across species, regulatory elements are often specific to cell-type and condition (developmental stage, stress).108,109 Therefore, epigenetic datasets derived from human atrial tissues, stages of development and (disease) conditions are required to accurately identify AF-relevant regulatory elements.
While regulatory elements have been identified over one megabase (Mb) away from the target promoter(s), they are thought to act in close proximity, by the three-dimensional conformation of the DNA.110,111 Because regulatory elements need to be in close physical proximity to their target promoters to directly regulate transcriptional activity,112–114 knowledge of the conformation of the DNA is necessary in the search for target genes of variant regulatory elements.
More than a million candidate regulatory elements have been identified across the human genome, many of which are cell type- or condition-specific and temporally restricted.115,116 Nevertheless, the majority of regulatory elements lack functional validation and their target genes are largely unknown. Therefore, the invention and application of new high throughput methods for functional evaluation of regulatory elements is a current focus of functional genomics research. Currently, high-throughput enhancer assays such as STARR-seq and MPRA are utilized for the large scale discovery of regulatory elements (Table 2), and have identified disease-relevant variant regulatory elements and expression modulating variants.97,117–120 While these approaches enable the functional screening of large regions of the genome, a major drawback is that they are extra/chromosomal and are thus not subject to chromatin packaging (Table 2). Moreover, for practical reasons, most studies thus far have identified regulatory elements with an enhancing effect on transcription, while active repression of transcription is also physiologically relevant. New methods of identification and characterization of repressive regulatory elements is therefore essential for a comprehensive understanding of the relevant regulatory networks. Recently developed approaches consider regulatory element activity, and regulatory element-promoter contacts within native genomic context in model cell lines.120–122 Application of such assays in AF-relevant cell types, such as atrial cardiomyocytes or fibroblasts derived from human with or without AF or from animals modeling AF, will be technically challenging, but may uncover functional variant regulatory elements at scale.
Several efforts have been made to identify human cardiac enhancers.147,148 One study used >35 epigenomic data sets from mouse and human pre- and postnatal hearts to generate a reference of >80,000 putative human heart enhancers.149 Recently, the enhancer prediction tool EMERGE150 was trained using 70 human cardiac epigenetic data sets and a set of confirmed cardiac enhancers151 to predict human cardiac enhancers, 1750 of which are located in over 100 AF loci.15 The accessible chromatin in non-diseased human left atrial cardiomyocytes was identified using ATAC-seq.15,19 These regions represent >83,000 putative regulatory elements in human atrial cardiomyocytes. An example of the output of such assays of the locus PRRX1 is shown in Fig. 4. Cross-referencing both this data set and the EMERGE predicted regulatory element set with AF-associated SNPs4 revealed 876 putative variant regulatory elements genome-wide.15 Several candidates were validated by luciferase assay, showing the potential of such a strategy for identification of disease-specific variant regulatory elements. Future efforts should provide epigenetic data from AF-prone heart tissue including pulmonary veins and from other cell types in the heart, such as fibroblasts, to identify tissue-specific AF-relevant variant regulatory elements.
Figure 4.

UCSC track showing the locus containing PRRX1 associated with AF and different datasets used for regulatory element and target gene identification. USCS browser view with lead variants, topologically associated domains (Juicebox TADs)(Durand et al., 2016;Robinson et al., 2018), identified enhancers (Tucker et al., 2017), SNPs associated with AF (p<10–4) (Roselli et al., 2018), annotated genes, promoter capture Hi-C (PCHi-C) data (Montefiori et al., 2018), ATAC-seq representing accessible chromatin in cardiomyocytes of left atria (Hill et al., 2019; van Ouwerkerk et al., 2019), EMERGE enhancer prediction (van Duijvenboden et al., 2015;van Ouwerkerk et al., 2019) and expression of left atria whole tissue (van Ouwerkerk et al., 2019).
Identification of the relationship between variant SNPs and biological effect
A second challenge is to link the variant regulatory elements to target genes (Fig. 1). Regulatory element-promoter targeting is selective and regulated.97,114,152 The three principal approaches to define specific target genes of variant regulatory elements in any complex disease are 1) identification of the variant regulatory elements as described in the previous section; 2) identification of which genes at a locus are affected by associated variants (Fig. 5); and 3) the variant regulatory element as well as the directional effect on transcription should be validated in vitro or in vivo, by e.g. genome editing of cultured human cells or mice, to enable evaluation of the biological effect of different components on transcriptional networks and (electro)physiological function (Fig. 5).
Figure 5.
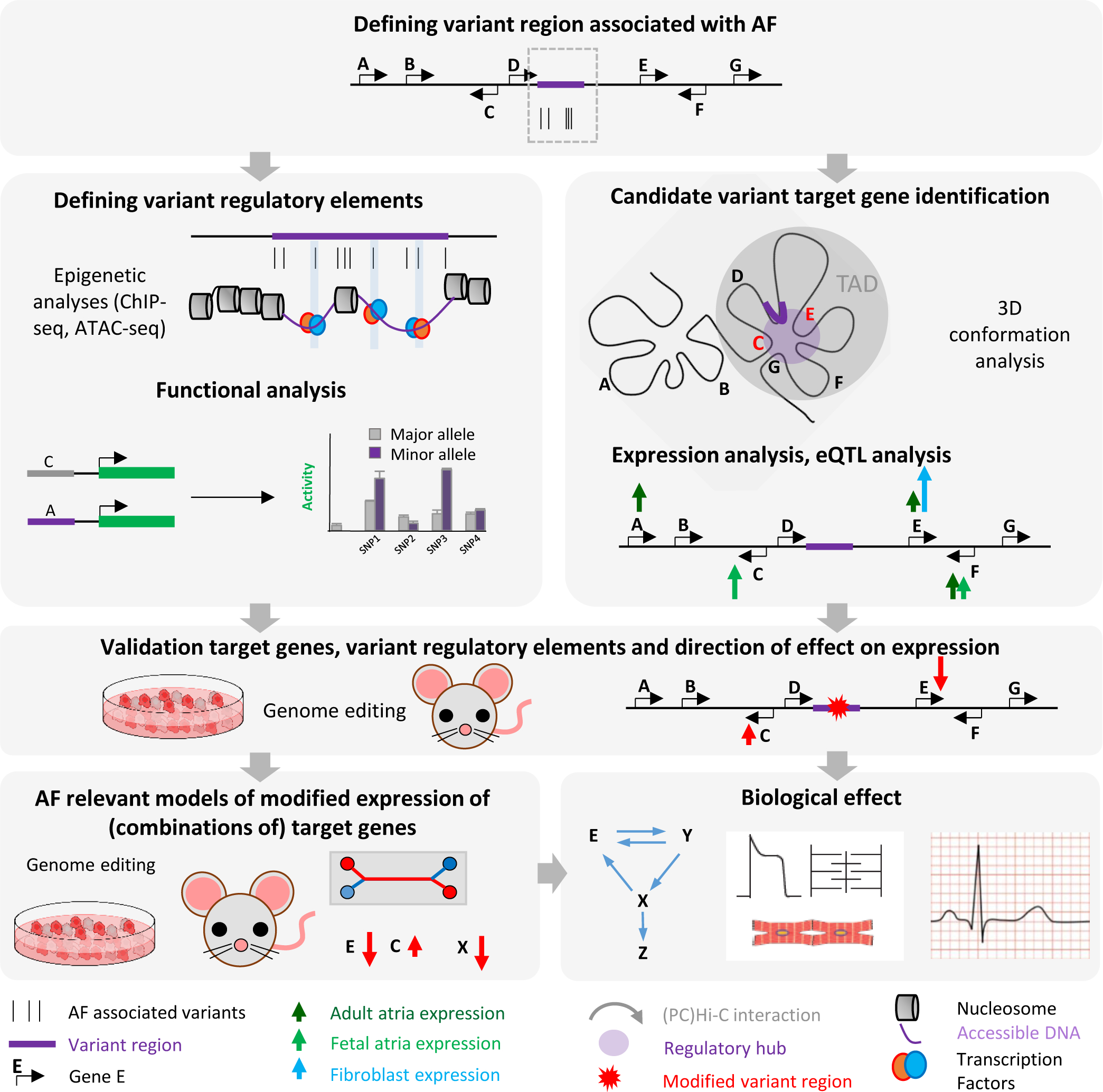
Commonly employed approaches and challenges to study the mechanistic link between genetic variation and AF predisposition. Technical challenges and unmet needs include: 1) Relevant cell type- and condition-specific (hypertension, aging etc.) conformation and transcriptome data are required to define candidate AF associated genes. 2) Relevant cell type- and condition-specific epigenetic data are required to identify physiologically relevant variant regulatory elements. 3) Assays to define physiologically relevant functions of regulatory elements (activation, repression, conformation, combinatorial) are required. 4) Sensitive models to recapitulate the effect of variants are needed.
A common and productive approach to resolve which genes within a locus are targeted by the identified GWAS variants is to link the signals to levels of gene expression. Such expression quantitative trait locus (eQTL) analysis involves the correlation of risk variants with gene expression levels in a specific tissue relevant to the disease studied.4,5 However, only a fraction of the AF-associated sentinel SNPs could be linked to one or multiple candidate target genes. Due to the limited availability of relevant genotyped tissue or cell type (e.g. left atrial cardiomyocytes, pulmonary vein wall, atrial fibroblasts) for expression analysis, lack of knowledge of relevant tissue for particular genes, and the inability to detect small but biological relevant differences in expression, we currently lack sufficient power to identify target genes for all AF-associated loci.153
Regulatory elements and target genes usually share the same topologically associated domain (TAD), a self-interacting genomic region in which DNA sequences physically interact with each other more frequently than with sequences outside the TAD.154,155 TAD structures and loops within TADs are mediated by cohesin and CCCTC-binding factor (CTCF).152,156–159 Once regulatory elements have been identified, the potential target genes can be prioritized by interrogating their three-dimensional chromatin contacts in the relevant tissue. Recent Hi-C analyses of human atrial and ventricular tissue combined with improved statistical analyses of interactions have provided a highly relevant contact map facilitating the identification of target genes.160 While regulatory element-promoter interactions are required for transcriptional regulation, close proximity observed by 3C-derived technologies does not necessarily imply a functional interaction.111,126 Moreover, Hi-C and promoter-capture (PC)-Hi-C technologies do not identify all possible interactions, as contacts within 30kb are technically challenging to capture. Therefore, interactions between regulatory element and promoters, or a lack thereof, should be interpreted with care and require functional validation. In an effort to map cardiovascular disease risk loci interactions, two studies used PCHi-C in human iPSC-derived cardiomyocytes to map the promoter interactome, identifying interactions (physical proximity) between promoters and disease-associated regions with regulatory elements.161,162 Indeed, both studies found that interactions involving cardiovascular disease and heart rhythm GWAS SNPs were enriched in promoter interactions with genes relevant for heart development and disease. Similarly, Hi-C of primary cardiac tissue combined with a new more sensitive and quantitative loop calling has been used to link the heart regulatory interactome and disease variants.160 These genome-wide interaction maps of genome organization have also been useful to identify candidate target genes of AF variant regulatory elements, as PCHi-C datasets identify interactions between potential regulatory elements and target gene promoters (Table 2, Fig. 4).15
While the variant regulatory elements and their target genes have been predicted based on features such as epigenetic signatures, expression, contact, and eQTL,15 a few functional variant regulatory elements and their target genes have been experimentally identified (Table 3). The strongest AF risk locus -with associations orders of magnitude more significant than the subsequent loci- is found 150 kb upstream of the homeobox transcription factor PITX2 located on chromosome 4q25.4,5 One study found a risk variant in high LD with the sentinel SNP that confers differential expression mediated by a transcription factor in an intron of PITX2a/b, upstream of the promoter of the cardiac specific isoform PITX2c.163 The region containing the variant of interest (rs2595104) showed enhancer activity in zebrafish, and the risk allele, which is known to predispose to AF, demonstrated reduced enhancer activity in vitro. Subsequent CRISPR/Cas9-mediated deletion of the rs2595104 region in human stem cell-derived cardiomyocytes showed reduced PITX2c expression compared to the unedited allele.163 Moreover, rs2595104 lies in a TFAP2a binding site, a ubiquitously expressed transcription factor that was previously shown to be important for PITX2 expression.164,165 The authors verified that TFAP2a binds preferentially to the non-risk allele, and that TFAP2a knock down mediated the reduction of PITX2c expression.
Table 3.
Currently identified AF-associated (variant) regulatory elements and target genes
| Locus | Regulatory element/variant | Models used | Target gene identified | Refs |
|---|---|---|---|---|
| PITX2 | Intron of PITX2a/b / rs2595104 | Enhancer reporter assays in zebrafish | PITX2c expression down after deletion of the rs2595104 region in human stem cell derived cardiomyocytes | 163 |
| PITX2 | 20 kb region upstream of Pitx2 | Orthologues region deleted from mouse genome | Pitx2c expression down (males) Enpep expression up (females) | 19 |
| PRRX1 | Upstream of Prrx1 / rs577676 | Enhancer reporter assays in embryonic zebrafish / HL-1 cells | - | 13 |
| GJA1 | 0.7 Mbp downstream of GJA1 | Orthologues region deleted from mouse genome | Gja1 expression decreased in atrial tissue | 15 |
| KCNN3 | First exon and promoter of Kcnn3 | Orthologues region deleted from mouse genome | Reduced expression of several nearby genes | 15 |
| SCN5a | Downstream of Scn5a | Orthologues region deleted from mouse genome | Scn5a expression decreased in atrial tissue | 166 |
| SCN10a | Intronic of Scn10a/ rs6801957 | Orthologues region deleted from mouse genome | Scn5a expression decreased in cardiac tissue | 167,168 |
Many more (876) putative regulatory elements have been identified in AF associated regions using EMERGE and ATAC-seq, and although a handful of these showed allele-specific activity in luciferase assays, the vast majority require in vivo validation as well as target gene identification.15
It was hypothesized that the highest risk-associated region near 4q25 was regulated by TBX5.14 Using a combination of ATAC-seq, 4C, and ChIP-qPCR (Table 2), TBX5 was confirmed to occupy the regulatory element that is in contact with the PITX2 promoter in human LA. Furthermore, this RE regulatory element harbors an AF risk variant rs1906595, the major allele of which abolishes regulatory element activity.
In the mouse orthologue of the AF-associated variant region 170 kb upstream of Pitx2, a non-tissue-specific regulatory element specifically contacts the promoters of Pitx2c as well as Enpep, a gene adjacent to Pitx2.22 Further analyses are required to establish whether both Pitx2c and Enpep are targets of variant regulatory elements in the upstream gene desert, and what could be mechanisms underlying the change in target gene expression.
In a recent study the mouse orthologues of two variant noncoding regions in the gene desert upstream of Pitx2 were deleted from the mouse genome using CRISPR/Cas9 genome editing.19 One of these regions, a 20 kb region upstream of Pitx2 with regulatory activity, is in contact with the promoter of Pitx2 as shown by 4C-seq (Table 2). Deletion of the regulatory element reduced expression of Pitx2c specifically in male mice, and led to inducible AF susceptibility. Of note, regulatory element deletion caused increased Enpep expression only in females. Moreover, the regulatory element region was essential for and specific to maintaining the active chromatin state of the Pitx2c promoter specifically. Disruption of the Pitx2 intronic CTCF binding site caused reduced Pitx2 expression, AF predisposition and reduced active chromatin marks on Pitx2 indicating that long-range looping was mediated by CTCF. However, the separate deletion of the two enhancer regions did not lead to major phenotypic abnormalities without stress, and the authors speculate that a pair-wise deletion could uncover that these two enhancers confer phenotypic robustness.19 Further research should uncover whether these two identified enhancers are both needed in collaboration to reduce Pitx2 expression (or other downstream mechanisms) to a degree sufficient for an arrhythmogenic phenotype. Despite these efforts, AF relevant target genes of the highest risk-associated region near 4q25 are still uncertain. Although there is no correlation between PITX2 expression changes and AF, most models suggest that regulatory elements present in AF risk variant loci are linked to PITX2 regulation.
A region near PRRX1, a transcriptional co-activator important for development of the heart,68 associated with Agnathia-Otocephaly and Dysgnathia Complex,169 also harbors a variant regulatory element. Putative enhancers were selected in the AF-associated region on 1q24 upstream of PRRX1 using conservation and genomic markers of transcriptional enhancers.13 Two of these putative enhancers showed strong eGFP expression in cardiac and skeletal muscle tissue when tested in embryonic zebrafish (Fig. 4). Twenty-one common SNPs within these regulatory elements were tested for enhancer activity of the risk versus non-risk allele in HL1 cells, demonstrating that only SNP rs577676 led to decreased regulatory element activity. Possible target genes in the variant region were identified using Hi-C and 3C, suggesting physical contact between the identified regulatory element and the promoter of PRRX1 in cardiac fibroblasts compared to human embryonic stem cells. This supports the idea that the risk variant negatively regulates the expression of PRRX1, providing a direct mechanism linking the risk variant and target gene for this locus. Additionally, optical mapping of cardiomyocytes differentiated from PRRX1 knockout human embryonic stem cells and Prrx1a morpholino-treated zebrafish embryos revealed the action potential duration was significantly shortened in PRRX1-knockout cardiomyocytes, a phenotype often seen in AF patients.
The mouse orthologue of another AF-associated regulatory element was found regulating expression of the AF-relevant gene Gja1 in the heart.15 Gja1 encodes the major gap junctional protein with a role in impulse propagation and cell-cell coupling.170,171 Using CRISPR/Cas9 genome editing a region containing the mouse homologue of a 28 kb AF associated locus 0.7 Mb downstream of GJA1 was deleted. Mice homozygous for the deletion showed atrial-specific decreased expression of Gja1. The same study found that deletion of the mouse orthologue of the variant region close to the potassium-calcium activated channel KCNN3 caused the reduced expression of several nearby genes, whereas deletion of the 33 kb orthologue of the variant region in the first intron of the zinc-finger transcription factor, ZFHX3 did not alter cardiac expression of any gene within the TAD in vivo.
Multiple regulatory elements were found around the SCN5A-SCN10A locus, which has frequently been associated with cardiac rhythm and conduction as identified through GWAS and with ECG parameters being linked to AF4,5,168,172–176 and Brugada syndrome.177 One of the regulatory elements of SCN5A is positioned in an intron of SCN10A,168 and contains a common variant that is associated with PR interval.54 The variant disrupts a T-box binding site in the regulatory element, and is associated with decreased SCN5A expression.167,168 Two other regulatory elements are located in introns and downstream of SCN5A, the latter of which also contains variants associated with PR interval and QRS duration.54,178–180 The regulatory element cluster (“super enhancer”) downstream of SCN5A is evolutionary conserved, and drives gene expression in the heart in a pattern resembling that of Scn5a. When deleted from the mouse genome, the cluster, which harbors conduction parameter-associated SNPs, was found to be essential for Scn5a expression and for bringing the other regulatory elements and promoters in the locus physically together to regulate Scn5a.101
Epigenetic states link environment and gene regulation
There are several risk factors for cardiovascular disease, the most important of which is age. Interestingly, epigenetic alterations have been proposed to underlie age-related transcriptional alterations.181 Indeed, changes in epigenetic state induced by aging (or other risk factors such as hypertension and metabolic disease) could provide the basis for the “uncovering” of genetic risk factors at AF-associated loci. Moreover, combined GWAS loci only account for a portion of heritability, suggesting that a part of the so-called missing heritability might be found in non-sequence related epigenetics.182 An example of the importance of changes in epigenetic state is the epigenetic clock. Chronological age is highly correlated with epigenetic alterations of the methylome,183 and deviation of this methylation state could be used to predict healthspan184 and is correlated with cardiovascular disease incidence.185 Altered DNA methylation was found near or on the promoters of poised genes, which contributed to transcriptional heterogeneity of these genes.186 A methylome-wide association study of an AF cohort revealed 14 AF-associated SNPs to be associated with a CpG site in blood-derived genomic DNA.187 In a smaller study, a CpG islet proximal to PITX2 was found to be hypermethylated in AF patients compared to controls, along with decreased PITX2 expression in AF patients.188 Furthermore, hypermethylation in the AF susceptible loci containing PITX2 and CCDC141, as well as hypomethylation in the locus containing CACNA1C locus, were identified in all 7 patients with permanent AF.189 These data suggest that changes in DNA methylation could be involved in AF.
Not only methylation, but also the alteration of other epigenetic mechanisms such as histone modification and chromatin remodeling could contribute to disease risk.190 Transcription factors regulate or maintain cell fate by activating certain and repressing other target genes in cooperativity with chromatin remodelers and histone modifying enzymes. Chromatin remodelers respond to modifications to histone post-translational modifications including methylation, phosphorylation, acetylation, ubiquitylation, and sumoylation.191,192 Recognizing these modifications, chromatin remodelers make distant regulatory elements as well as promoters accessible (with tissue-specificity) by regulating nucleosome dynamics.193 There is extensive cross-talk between histone modifications and chromatin remodelers, the balance of which determines the recruitment of transcription factors.194 Evidence of a role for both histone modifications and chromatin remodelers in “complex trait” diseases such as AF is emerging. For example, the interplay between transcription factors and chromatin remodeling complex dosage of Brg1 were shown to be important during development,195 which suggests a mechanism of interdependence between transcription factors and remodeling proteins in the heart. Additionally, histone deacetylases, were found to modulate pathogenic gene expression in many diseases including AF progression.196 Moreover, there is evidence that increased expression of histone modifier enhancer of zeste homolog 2 (EZH2), which catalyzes the deposition of methylation to histone 3 at lysine 27 (H3K27me3) is associated with fibrosis in AF patients.197
Another possible mechanism linking epigenetics and AF are reactive oxygen species (ROS) levels. Increased levels of ROS are known to accelerate aging, and they are associated with AF pathogenesis.198,199 It is known that ROS levels have an impact on the mechanisms which govern the epigenome, such as DNA methylation, histone modification and ncRNAs.200 Increased ROS levels can disrupt the epigenetic balance and thus the epigenetic state of a cell, causing potentially altered mechanisms (such as “uncovering” of the effect of risk variants) leading to increased AF predisposition. Moreover, one study found that oxidative stress promotes AF via increased intracellular Ca2+ release by oxidized RyR2.201 In mice, ROS is increased in response to injury, but also during the natural transition from glycolytic to oxidative metabolism shortly after birth as well as during aging. This increase in ROS inhibits cardiomyocyte regeneration.202 Indeed, Pitx2 is known to regulate the transcriptional response to oxidative stress (cooperatively with Yap).49 In response to injury, Pitx2 expression is induced, which in turn activates ROS scavengers, protecting cells from injury. Future models should provide insight into the interplay between ROS, PITX2, TBX5 and AF, but as of yet the interaction between these factors and the effect on effectors has not been studied.
In addition to the contribution of aging via ROS, fetal and developmental origins of disease may also be mediated by epigenetic responses to environmental factors.203 In a generalized view, heritable epigenetic states of chromatin are subject to developmental changes, and are sensitive to internal variation (e.g. variable transcription factor activity) and external signals (e.g. metabolic state) during development. This leads to interindividual variability in regulatory element deployment, gene expression and cellular and tissue phenotype. Indeed, the same could hold for AF, as there is a high density of AF-risk variants near cardiac developmental transcription factor genes such as NKX2–5, PITX2, TBX5, and TBX3 as well as their target genes such as ion channels (HCN4, KCNN3) and gap junctions (GJA1). Two of the genes, PITX2 and NKX2–5, well-known players in AF, are responsible for the correct development of the left atrial myocardium as well as the myocardial sleeves of the pulmonary veins.39 The myocardial sleeves of the pulmonary veins are composed of cardiomyocytes resembling working atrial myocardium, and it are the sites where ectopic foci that initiate AF are often found.204,205 It has been speculated that a developmental change in expression of these genes combined with other structural or epigenetic remodeling could ultimately lead to the development of ectopic foci in the pulmonary vein myocardial sleeves.206–208 Likewise, HCN4 encodes a potassium channel responsible for the generation of pacemaker current in the sinoatrial node during early development, as well as atrial rhythm.209,210 However, the strict requirement of HCN4 function is confined to embryonic development209–211 as it seems to be dispensable in adult heart rhythm.212 Indeed, AF is characterized by irregularly irregular heart rates, suggesting that developmental changes in expression of this gene could contribute to AF in later life.
Conclusion
AF is a collective name for an expressed disease phenotype with many distinct pathological mechanisms. GWAS and genetically modified animal models have indicated that AF predisposition likely involves many additive small expression changes induced by variant regulatory elements. While several parameters have been clearly associated with AF, including impulse conduction, action potential duration, intracellular calcium handling, and fibrosis, the origins of AF and the cell-types involved are still not fully known. An additional confounding factor is that although many genetic components have been associated with AF, some heritable components remain unexplained. This missing heritability could be caused by epigenetics not identified by sequence-based GWAS. Indeed, there are developmental and aging related epigenetic changes that could contribute to interindividual variability. This could result in differential regulatory element deployment and gene expression in a tissue- or developmental stage-specific manner.
Here we conclude that there is growing evidence that a transcription factor network could modulate mechanisms underlying AF. Essential cardiac developmental transcription factors that are situated close to AF associated variants have been linked to AF or factors that predispose to AF. Here we propose a threefold model that can link genetic risk variants and transcription factor networks to AF predisposition. First, AF-associated genetic variation in binding sites of transcription factors can lead to altered expression of target effector genes (e.g. ion channels). Second, genetic variants could interfere with transcriptional regulation of these AF relevant transcription factors themselves (Fig. 1B). Third, transcription factors are now also known to interact as cofactors of each other in the regulation of target gene expression (Fig. 3). Therefore, it is likely that a slight alteration in expression levels of any of these transcription factors –as a result of variant regulatory element activity- can alter the delicate balance in the transcriptional network, affecting its output, which results in altered expression of target effector genes and AF predisposition (Fig. 3).
To uncover the contribution of epigenetic state to AF risk in relation to genetic variation there is a pressing need for availability of datasets from relevant cell types and developmental stages. Additionally, we require knowledge of the effect of disease states such as high blood pressure, stress and aging on these epigenetic parameters. This would allow the identification of variants with a biologically relevant impact on the system in the context of complex disease. Specifically, assays are required that subsequently test variant regulatory element function and their effect on target gene expression. Moreover, we lack the means to test the impact of combinations of different variant regulatory elements on target gene expression and the transcriptional network. Most importantly, there is an unmet need for the possibility to perform such techniques in relevant cell or tissue types. With the likely diverse causes of AF in different patients, a valuable addition to AF treatment would be to make patient-specific classifications of the disease based on genetic and epigenetic parameters.
Funding:
The work was supported by Fondation Leducq 14CVD01 to Dr. Ellinor, Dr. Kirchhof, Dr. Martin, Dr. Moskowitz, Dr. de Laat and Dr. Christoffels. Dr. Ellinor was also supported by grants from the American Heart Association (18SFRN34110082), and the National Institutes of Health (1RO1HL092577, R01HL128914, K24HL105780). Dr. Moskowitz was supported by R01 HL147571, R01 HL148719, R01 HL126509, AHA 17CSA33610126. Dr. Nadadur was supported by NIH F30HL131298 and T32-HL007381, Dr. Lazarevic by NIH F31HL144023 and T32-HL007381, T32-HD055164. Dr. Hall was supported by an AHA Strategically Focused Research Networks (SFRN) postdoctoral fellowship (18SFRN34110082). Dr. Tucker was supported by a grant from the National Institutes of Health (5K01HL140187). Z. Kadow was supported by NHLBI F30HL145908; Baylor Research Advocates for Student Scientists (BRASS). Dr. Martin was supported by grants from the National Institutes of Health (HL127717, HL130804, HL118761); Vivian L. Smith Foundation, State of Texas funding. This work was partially supported by European Union (grant agreement No 633196 [CATCH ME], European Union BigData@Heart (grant agreement EU IMI 116074), British Heart Foundation (FS/13/43/30324 to Dr. Kirchhof and Dr. Fabritz; PG/17/30/32961 to Dr. Kirchhof; AA/18/2/34218 to PK and LF), and German Centre for Cardiovascular Research supported by the German Ministry of Education and Research (DZHK, via a grant to AFNET to Dr. Kirchhof).
Disclosures: Dr. Ellinor is supported by a grant from Bayer AG to the Broad Institute focused on the genetics and therapeutics of cardiovascular diseases. Dr. Ellinor has also served on advisory boards or consulted for Bayer AG, Quest Diagnostics, and Novartis. Dr. Kirchhof receives research support for basic, translational, and clinical research projects from European Union, British Heart Foundation, Leducq Foundation, Medical Research Council (UK), and German Centre for Cardiovascular Research, from several drug and device companies active in atrial fibrillation, and has received honoraria from several such companies in the past. Dr. Kirchhof and Dr. Fabritz are listed as inventors on two patents held by University of Birmingham (Atrial Fibrillation Therapy WO 2015140571, Markers for Atrial Fibrillation WO 2016012783). Dr. Fabritz receives research support for basic, translational, and clinical research projects from European Union, British Heart Foundation, Medical Research Council (UK), and drug and device companies involved in atrial fibrillation. Dr. Martin is a founder and owns shares in Yap Therapeutics. The other authors declare no competing interests.
Non-standard abbreviations:
- AF
Atrial fibrillation
- GWAS
Genome-wide association studies
- eQTL
expression quantitative trait locus
- TAD
topologically associated domain
- CTCF
CCCTC-binding factor
- PCHi-C
Promoter capture Hi-C
- iPSC
induced pluripotent stem cell
- SNP
single nucleotide polymorphism
- ChIP-seq
chromatin immunoprecipitation sequencing
- ATAC-seq
assay for transposase accessible chromatin
References
- 1.Fox CS. Parental Atrial Fibrillation as a Risk Factor for Atrial Fibrillation in Offspring. Jama [Internet]. 2004;291:2851. Available from: http://jama.jamanetwork.com/article.aspx?doi=10.1001/jama.291.23.2851 [DOI] [PubMed] [Google Scholar]
- 2.Lubitz SA, Sinner MF, Lunetta KL, Makino S, Pfeufer A, Rahman R, Veltman CE, Barnard J, Bis JC, Danik SP, et al. Independent Susceptibility Markers for Atrial Fibrillation on Chromosome 4q25. Circulation. 2010;122:976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nattel S, Dobrev D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat Rev Cardiol [Internet]. 2016;13:575–590. Available from: 10.1038/nrcardio.2016.118 [DOI] [PubMed] [Google Scholar]
- 4.Roselli C, Chaffin MD, Weng L-C, Aeschbacher S, Ahlberg G, Albert CM, Almgren P, Alonso A, Anderson CD, Aragam KG, , et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet [Internet]. 2018;1. Available from: http://www.nature.com/articles/s41588-018-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, Herron TJ, McCarthy S, Schmidt EM, Sveinbjornsson G, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet 2018;50:1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutierrez A, Chung MK. Genomics of Atrial Fibrillation. Curr. Cardiol. Rep 2016;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalstø SM, Siland JE, Rienstra M, Christophersen IE. Atrial Fibrillation Genetics Update: Toward Clinical Implementation. Front. Cardiovasc. Med 2019;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fabritz L, Guasch E, Antoniades C, Bardinet I, Benninger G, Betts TR, Brand E, Breithardt G, Bucklar-Suchankova G, Camm AJ, et al. Expert consensus document: Defining the major health modifiers causing atrial fibrillation: A roadmap to underpin personalized prevention and treatment. Nat. Rev. Cardiol 2016;13:230–237. [DOI] [PubMed] [Google Scholar]
- 9.Khera AV., Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome TL - 22. Genome Res [Internet]. 2012;22 VN-r:1748–1759. Available from: 10.1101/gr.136127.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic Localization of Common Disease-Associate Variation in Regulatorty DNA. Science (80-). 2012;337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deplancke B, Alpern D, Gardeux V. The Genetics of Transcription Factor DNA Binding Variation. Cell. 2016;166:538–554. [DOI] [PubMed] [Google Scholar]
- 13.Tucker NR, Dolmatova EV., Lin H, Cooper RR, Ye J, Hucker WJ, Jameson HS, Parsons VA, Weng LC, Mills RW, et al. Diminished PRRX1 Expression Is Associated with Increased Risk of Atrial Fibrillation and Shortening of the Cardiac Action Potential. Circ Cardiovasc Genet. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadadur RD, Broman MT, Boukens B, Mazurek SR, Yang X, Van Den Boogaard M, Bekeny J, Gadek M, Ward T, et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci Transl Med. 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Ouwerkerk AF, Bosada FM, van Duijvenboden K, Hill MC, Montefiori LE, Scholman KT, Liu J, de Vries AAF, Boukens BJ, Ellinor PT, et al. Identification of atrial fibrillation associated genes and functional non-coding variants. Nat Commun. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dai W, Laforest B, Tyan L, Shen KM, Nadadur RD, Alvarado FJ, Mazurek SR, Lazarevic S, Gadek M, Wang Y, et al. A calcium transport mechanism for atrial fibrillation in Tbx5-mutant mice. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao Y, Zhang M, Li L, Bai Y, Zhou Y, Moon AM, Kaminski HJ, Martin JF. Pitx2, an atrial fibrillation predisposition gene, directly regulates ion transport and intercalated disc genes. Circ Cardiovasc Genet. 2014;7:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laforest B, Dai W, Tyan L, Lazarevic S, Shen KM, Gadek M, Broman MT, Weber CR, Moskowitz IP. Atrial fibrillation risk loci interact to modulate Ca2+-dependent atrial rhythm homeostasis. J Clin Invest. 2019;129:4937–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang M, Hill MC, Kadow ZA, Suh JH, Tucker NR, Hall AW, Tran TT, Swinton PS, Leach JP, Margulies KB, et al. Long-range Pitx2c enhancer–promoter interactions prevent predisposition to atrial fibrillation. Proc Natl Acad Sci U S A. 2019;116:22692–22698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collins MM, Ahlberg G, Hansen CV, Guenther S, Marín-Juez R, Sokol AM, El-Sammak H, Piesker J, Hellsten Y, Olesen MS, et al. Early sarcomere and metabolic defects in a zebrafish pitx2c cardiac arrhythmia model. Proc Natl Acad Sci U S A. 2019;116:24115–24121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Syeda F, Holmes AP, Yu TY, Tull S, Kuhlmann SM, Pavlovic D, Betney D, Riley G, Kucera JP, Jousset F, et al. , Kirchhof P. PITX2 Modulates Atrial Membrane Potential and the Antiarrhythmic Effects of Sodium-Channel Blockers. J Am Coll Cardiol. 2016;68:1881–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aguirre L a, Alonso ME, Badía-Careaga C, Rollán I, Arias C, Fernández-Miñán A, López-Jiménez E, Aránega A, Gómez-Skarmeta JL, Franco D, et al. Long-range regulatory interactions at the 4q25 Atrial Fibrillation risk locus involve PITX2c and ENPEP. BMC Biol [Internet]. 2015;13:1–13. Available from: http://www.biomedcentral.com/1741-7007/13/26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franco D, Christoffels VM, Campione M. Homeobox transcription factor Pitx2: The rise of an asymmetry gene in cardiogenesis and arrhythmogenesis. Trends Cardiovasc Med [Internet]. 2014;24:23–31. Available from: 10.1016/j.tcm.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 24.Furtado MB, Wilmanns JC, Chandran A, Tonta M, Biben C, Eichenlaub M, Coleman HA, Berger S, Bouveret R, Singh R, et al. A novel conditional mouse model for NKX2–5 reveals transcriptional regulation of cardiac ion channels. Differentiation. 2015;91:29–41. [DOI] [PubMed] [Google Scholar]
- 25.Posch MG, Boldt LH, Polotzki M, Richter S, Rolf S, Perrot A, Dietz R, Özcelik C, Haverkamp W. Mutations in the cardiac transcription factor GATA4 in patients with lone atrial fibrillation. Eur J Med Genet. 2010;53:201–203. [DOI] [PubMed] [Google Scholar]
- 26.Bapat A, Anderson CD, Ellinor PT, Lubitz SA. Genomic basis of atrial fibrillation. Heart. 2018;104:201–206. [DOI] [PubMed] [Google Scholar]
- 27.Fatkin D, Santiago CF, Huttner IG, Lubitz SA, Ellinor PT. Genetics of Atrial Fibrillation: State of the Art in 2017. Hear. Lung Circ 2017;26:894–901. [DOI] [PubMed] [Google Scholar]
- 28.Christophersen IE, Ellinor PT. Genetics of atrial fibrillation: from families to genomes. J Hum Genet [Internet]. 2015;1–10. Available from: http://www.nature.com/doifinder/10.1038/jhg.2015.44 [DOI] [PubMed] [Google Scholar]
- 29.Tucker NR, Ellinor PT. Emerging directions in the genetics of atrial fibrillation. Circ Res. 2014;114:1469–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tucker NR, Clauss S, Ellinor PT. Common variation in atrial fibrillation: Navigating the path from genetic association to mechanism. Cardiovasc Res. 2016;109:493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT. The Human Transcription Factors. Cell. 2018;172:650–665. [DOI] [PubMed] [Google Scholar]
- 32.Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–948. [DOI] [PubMed] [Google Scholar]
- 33.Seifi M, Walter MA. Axenfeld-Rieger syndrome. Clin. Genet 2018; [DOI] [PubMed] [Google Scholar]
- 34.Dasouki M, Andrews B, Parimi P, Kamnasaran D. Recurrent agnathia-otocephaly caused by DNA replication slippage in PRRX1. Am J Med Genet Part A. 2013;161:803–808. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Klysik E, Sood S, Johnson RL, Wehrens XHT, Martin JF. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc Natl Acad Sci U S A. 2010;107:9753–9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirchhof P, Kahr PC, Kaese S, Piccini I, Vokshi I, Scheld HH, Rotering H, Fortmueller L, Laakmann S, Verheule S, et al. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ Cardiovasc Genet. 2011;4:123–133. [DOI] [PubMed] [Google Scholar]
- 37.Chinchilla A, Daimi H, Lozano-Velasco E, Dominguez JN, Caballero R, Delpo E, Tamargo J, Cinca J, Hove LM, Aranega AE, et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ Cardiovasc Genet. 2011;4:269–279. [DOI] [PubMed] [Google Scholar]
- 38.Liu C, Liu W, Lu MF, Brown NA, Martin JF. Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development. 2001;128:2039–2048. [DOI] [PubMed] [Google Scholar]
- 39.Mommersteeg MTM, Brown NA, Prall OWJ, De Gier-De Vries C, Harvey RP, Moorman AFM, Christoffels VM. Pitx2c and Nkx2–5 are required for the formation and identity of the pulmonary myocardium. Circ Res. 2007;101:902–909. [DOI] [PubMed] [Google Scholar]
- 40.Campione M, Steinbeisser H, Schweickert A, Deissler K, Van Bebber F, Lowe LA, Nowotschin S, Viebahn C, Haffter P, Kuehn MR, Blum M. The homeobox gene Pitx2: Mediator of asymmetric left-right signaling in vertebrate heart and gut looping. Development. 1999;126:1225–1234. [DOI] [PubMed] [Google Scholar]
- 41.Liu C, Liu W, Palie J, Lu MF, Brown NA, Martin JF. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development. 2002;129:5081–5091. [DOI] [PubMed] [Google Scholar]
- 42.Hill MC, Kadow ZA, Li L, Tran TT, Wythe JD, Martin JF. A cellular atlas of Pitx2-dependent cardiac development. Dev. 2019;146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu MF, Pressman C, Dyer R, Johnson RL, Martin JF. Function of rieger syndrome gene in left-right asymmetry and craniofacial development. Nature. 1999;401:276–278. [DOI] [PubMed] [Google Scholar]
- 44.Kahr PC, Piccini I, Fabritz L, Greber B, Schöler H, Scheld HH, Hoffmeier A, Brown NA, Kirchhof P. Systematic analysis of gene expression differences between left and right atria in different mouse strains and in human atrial tissue. PLoS One. 2011;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Syeda F, Kirchhof P, Fabritz L. PITX2-dependent gene regulation in atrial fibrillation and rhythm control. J. Physiol 2017;595:4019–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Bai Y, Li N, Ye W, Zhang M, Greene SB, Tao Y, Chen Y, Wehrens XHT, Martin JF. Pitx2-microRNA pathway that delimits sinoatrial node development and inhibits predisposition to atrial fibrillation. Proc Natl Acad Sci U S A. 2014;111:9181–9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lozano-Velasco E, Hernández-Torres F, Daimi H, Serra SA, Herraiz A, Hove-Madsen L, Aránega A, Franco D. Pitx2 impairs calcium handling in a dose-dependent manner by modulating Wnt signalling. Cardiovasc Res. 2016;109:55–66. [DOI] [PubMed] [Google Scholar]
- 48.Li L, Tao G, Hill MC, Zhang M, Morikawa Y, Martin JF. Pitx2 maintains mitochondrial function during regeneration to prevent myocardial fat deposition. Dev. 2018;145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tao G, Kahr PC, Morikawa Y, Zhang M, Rahmani M, Heallen TR, Li L, Sun Z, Olson EN, Amendt BA, Martin JF. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis ARJ, Yi CH, Gebühr T, Bullen PJ, Robson SC, Strachan T, et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet 1997;15:21–29. [DOI] [PubMed] [Google Scholar]
- 51.Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, et al. Mutations in human cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet 1997;15:30–35. [DOI] [PubMed] [Google Scholar]
- 52.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, et al. A murine model of Holt-Oram syndrome defines roles of the T-Box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–721. [DOI] [PubMed] [Google Scholar]
- 53.Postma AV., Van De Meerakker JBA, Mathijssen IB, Barnett P, Christoffels VM, Ilgun A, Lam J, Wilde AAM, Deprez RHL, Moorman AFM. A gain-of-function TBX5 mutation is associated with atypical Holt-Oram syndrome and paroxysmal atrial fibrillation. Circ Res. 2008;102:1433–1442. [DOI] [PubMed] [Google Scholar]
- 54.van Setten J, Brody JA, Jamshidi Y, Swenson BR, Butler AM, Campbell H, Del Greco FM, Evans DS, Gibson Q, Gudbjartsson DF,, et al. PR interval genome-wide association meta-analysis identifies 50 loci associated with atrial and atrioventricular electrical activity. Nat Commun. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riley G, Syeda F, Kirchhof P, Fabritz L. An introduction to murine models of atrial fibrillation. Front. Physiol 2012;3 August. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang XH, Nadadur RD, Hilvering CR, Bianchi V, Werner M, Mazurek SR, Gadek M, Shen KM, Goldman JA, Tyan L, Bekeny J, Hall JM, Lee N, Perez-Cervantes C, Burnicka-Turek O, Poss KD, Weber CR, et al. Transcription-factor-dependent enhancer transcription defines a gene regulatory network for cardiac rhythm. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luna-zurita L, Stirnimann CU, Glatt S, Pollard KS, Mu CW, Bruneau BG, Luna-zurita L, Stirnimann CU, Glatt S, Kaynak BL, et al. Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell. 2016;1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ang YS, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TMA, Fu JD, Spencer CI, et al. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell. 2016;167:1734–1749.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yashiro K, Shiratori H, Hamada H. Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature. 2007;450:285–288. [DOI] [PubMed] [Google Scholar]
- 60.Kääb S, Darbar D, Van Noord C, Dupuis J, Pfeufer A, Newton-Cheh C, Schnabel R, Makino S, Sinner MF, Kannankeril PJ, et al. Large scale replication and meta-analysis of variants on chromosome 4q25 associated with atrial fibrillation. Eur Heart J. 2009;30:813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sigurdsson A, Jonasdottir A, Baker A, Thorleifsson G, Kristjansson K, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357. [DOI] [PubMed] [Google Scholar]
- 62.Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, Arking DE, Müller-Nurasyid M, Krijthe BP, Lubitz S a, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet [Internet]. 2012;44:670–675. Available from: 10.1038/ng.2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parsons MJ, Brancaccio M, Sethi S, Maywood ES, Satija R, Edwards JK, Jagannath A, Couch Y, Finelli MJ, et al. The Regulatory Factor ZFHX3 Modifies Circadian Function in SCN via an at Motif-Driven Axis. Cell. 2015;162:607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berry FB, Miura Y, Mihara K, Kaspar P, Sakata N, Hashimoto-Tamaoki T, Tamaoki T. Positive and negative regulation of myogenic differentiation of C2C12 cells by isoforms of the multiple homeodomain zinc finger transcription factor ATBF1. J Biol Chem. 2001;276:25057–65. [DOI] [PubMed] [Google Scholar]
- 65.Sun X, Fu X, Li J, Xing C, Martin DW, Zhang HH, Chen Z, Dong J-T. Heterozygous deletion of Atbf1 by the Cre-loxP system in mice causes preweaning mortality. Genesis. 2012;50:819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wilcox AG, Vizor L, Parsons MJ, Banks G, Nolan PM. Inducible Knockout of Mouse Zfhx3 Emphasizes Its Key Role in Setting the Pace and Amplitude of the Adult Circadian Clock. J Biol Rhythms. 2017;32:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moskowitz IPG, Pizard A, Patel VV., Bruneau BG, Kim JB, Kupershmidt S, Roden D, Berul CI, Seidman CE, Seidman JG. The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development. 2004;131:4107–4116. [DOI] [PubMed] [Google Scholar]
- 68.Martin JF, Bradley A, Olson EN. The paired-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes Dev. 1995; [DOI] [PubMed] [Google Scholar]
- 69.Berge D Ten, Brouwer A, Korving J, Martin JF, Meijlink F. Prx1 and Prx2 in skeletogenesis: Roles in the craniofacial region, inner ear and limbs. Development. 1998;125:3831–3842. [DOI] [PubMed] [Google Scholar]
- 70.Bergwerff M, Gittenberger-de Groot AC, Wisse LJ, DeRuiter MC, Wessels A, Martin JF, Olson EN, Kern MJ. Loss of function of the Prx1 and Prx2 hameobox genes alters architecture of the great elastic arteries and ductus arteriosus. Virchows Arch. 2000;436:12–19. [DOI] [PubMed] [Google Scholar]
- 71.Ihida-Stansbury K, McKean DM, Gebb SA, Martin JF, Stevens T, Nemenoff R, Akeson A, Vaughn J, Jones PL. Paired-related homeobox gene Prx1 is required for pulmonary vascular development. Circ Res. 2004;94:1507–1514. [DOI] [PubMed] [Google Scholar]
- 72.Yu H, Xu JH, Song HM, Zhao L, Xu WJ, Wang J, Li RG, Xu L, Jiang WF, Qiu XB, Jiang JQ, Qu XK, Liu X, Fang WY, Jiang JF, Yang YQ. Mutational spectrum of the NKX2–5 gene in patients with lone atrial fibrillation. Int J Med Sci. 2014;11:554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakashima Y, Yanez DA, Touma M, Nakano H, Jaroszewicz A, Jordan MC, Pellegrini M, Roos KP, Nakano A. Nkx2–5 suppresses the proliferation of atrial myocytes and conduction system. Circ Res. 2014;114:1103–1113. [DOI] [PubMed] [Google Scholar]
- 74.Abou Hassan OK, Fahed AC, Batrawi M, Arabi M, Refaat MM, Depalma SR, Seidman JG, Seidman CE, Bitar FF, Nemer GM. NKX2–5 mutations in an inbred consanguineous population: Genetic and phenotypic diversity. Sci Rep. 2015;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Biben C, Weber R, Kesteven S, Stanley E, McDonald L, Elliott DA, Barnett L, Köentgen F, Robb L, Feneley M, Harvey RP. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2–5. Circ Res. 2000;87:888–895. [DOI] [PubMed] [Google Scholar]
- 76.Saxon JG, Baer DR, Barton JA, Hawkins T, Wu B, Trusk TC, Harris SE, Zhou B, Mishina Y, Sugi Y. BMP2 expression in the endocardial lineage is required for AV endocardial cushion maturation and remodeling. Dev Biol. 2017;430:113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang J, Greene SB, Bonilla-Claudio M, Tao Y, Zhang J, Bai Y, Huang Z, Black BL, Wang F, Martin JF. Bmp Signaling Regulates Myocardial Differentiation from Cardiac Progenitors Through a MicroRNA-Mediated Mechanism. Dev Cell. 2010;19:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luna-Zurita L, Prados B, Grego-Bessa J, Luxán G, Del Monte G, Benguría A, Adams RH, Pérez-Pomares JM, De La Pompa JL. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest. 2010;120:3493–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goldman DC, Donley N, Christian JL. Genetic interaction between Bmp2 and Bmp4 reveals shared functions during multiple aspects of mouse organogenesis. Mech Dev. 2009;126:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601–5611. [DOI] [PubMed] [Google Scholar]
- 81.Maekawa T, Jin W, Ishii S. The Role of ATF-2 Family Transcription Factors in Adipocyte Differentiation: Antiobesity Effects of p38 Inhibitors. Mol Cell Biol. 2010;30:613–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Qi L, Ding Y. Involvement of the CREB5 regulatory network in colorectal cancer metastasis. Yi Chuan. 2014;36:679–684. [DOI] [PubMed] [Google Scholar]
- 83.Srivastava D, Thomas T, Kirby ML, Brown D, Olson EN. Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nat Genet. 1997;16:154–160. [DOI] [PubMed] [Google Scholar]
- 84.Liu N, Barbosa AC, Chapman SL, Bezprozvannaya S, Qi X, Richardson JA, Yanagisawa H, Olson EN. DNA binding-dependent and -independent functions of the Hand2 transcription factor during mouse embryogenesis. Development. 2009;136:933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xia M, Luo W, Jin H, Yang Z. HAND2-mediated epithelial maintenance and integrity in cardiac outflow tract morphogenesis. Dev. 2019;146. [DOI] [PubMed] [Google Scholar]
- 86.Yamagishi H, Olson EN, Srivastava D. The basic helix-loop-helix transcription factor, dHAND, is required for vascular development. J Clin Invest. 2000;105:261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bakker ML, Boukens BJ, Mommersteeg MTM, Brons JF, Wakker V, Moorman AFM, Christoffels VM. Transcription factor Tbx3 is required for the specification of the atrioventricular conduction system. Circ Res. 2008;102:1340–1349. [DOI] [PubMed] [Google Scholar]
- 88.Mesbah K, Harrelson Z, Théveniau-Ruissy M, Papaioannou VE, Kelly RG. Tbx3 is required for outflow tract development. Circ Res. 2008;103:743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoogaars WMH, Engel A, Brons JF, Verkerk AO, De Lange FJ, Wong LYE, Bakker ML, Clout DE, Wakker V, Barnett P, Ravesloot JH, Moorman AFM, Verheijck EE, Christoffels VM. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 2007;21:1098–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571:489–499. [DOI] [PubMed] [Google Scholar]
- 91.Duygu B, Poels EM, Costa Martins PA da. Genetics and epigenetics of arrhythmia and heart failure. Front Genet. 2013;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tao H, Shi KH, Yang JJ, Li J. Epigenetic mechanisms in atrial fibrillation: New insights and future directions. Trends Cardiovasc. Med 2016;26:306–318. [DOI] [PubMed] [Google Scholar]
- 93.Komal S, Yin JJ, Wang SH, Huang CZ, Tao HL, Dong JZ, Han SN, Zhang LR. MicroRNAs: Emerging biomarkers for atrial fibrillation. J. Cardiol 2019;74:475–482. [DOI] [PubMed] [Google Scholar]
- 94.Zhang H, Liu L, Hu J, Song L. MicroRNA regulatory network revealing the mechanism of inflammation in atrial fibrillation. Med Sci Monit. 2015;21:3505–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang S, Guo C, Zhang W, Che W, Zhang J, Zhuang S, Wang Y, Zhang Y, Liu B. The integrative regulatory network of circRNA, microRNA, and mRNA in atrial fibrillation. Front Genet. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Briasoulis A, Inampudi C, Akintoye E, Alvarez P, Panaich S, Vaughan-Sarrazin M. Safety and efficacy of novel oral anticoagulants versus warfarin in medicare beneficiaries with atrial fibrillation and valvular heart disease. J Am Heart Assoc. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Long HK, Prescott SL, Wysocka J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell. 2016;167:1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Osterwalder M, Barozzi I, Tissiéres V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Lee EA, Zhu Y, Plajzer-Frick I, Pickle CS, Kato M, Garvin TH, Pham QT, Harrington AN, Akiyama JA, Afzal V, Lopez-Rios J, Dickel DE, Visel A, Pennacchio LA. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature [Internet]. 2018;554:239–243. Available from: 10.1038/nature25461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ, Turner JMA, Bertelsen MF, Murchison EP, Flicek P, Odom DT. Enhancer evolution across 20 mammalian species. Cell. 2015;160:554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Flores MA, Ovcharenko I. Enhancer reprogramming in mammalian genomes. BMC Bioinformatics. 2018;19:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Man JCK, Mohan RA, Boogaard M van den, Hilvering CRE, Jenkins C, Wakker V, Bianchi V, Laat W de, Barnett P, Boukens BJ, Christoffels VM. An enhancer cluster controls gene activity and topology of the SCN5A-SCN10A locus in vivo. Nat Commun. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yen A, Kheradpour P, Zhang Z, Heravi-moussavi A, Liu Y, Amin V, Ziller MJ, Whitaker JW, Schultz MD, Sandstrom RS, Eaton ML, Wu Y, Wang J, Ward LD, Sarkar A, Quon G, Pfenning A, Wang X, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Andrew J, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh K, Feizi S, Abdennur N, Adli M, Akerman M, Barrera L, Antosiewicz-bourget J, Barnes MJ, Bates D, Bell RJA, Bennett DA, Bianco K, Bock C, Boyle P, Brinchmann J, Caballero-campo P, Camahort R, Carrasco-alfonso MJ, Chen H, Chen Z, Cheng JB, Cho S, Chu A, Chung W, Cowan C, Deng A, Deshpande V, Diegel M, Ding B, Durham T, Echipare L, Edsall L, Genbacev-krtolica O, Gifford C, Gillespie S, Giste E, Glass IA, Gnirke A, Gu H, Gu J, Hafler DA, Hangauer MJ, Hariharan M, Hatan M. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, Glass CK, Rosenfeld MG, Fu XD. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu H, Nord AS, Akiyama JA, Shoukry M, Afzal V, Rubin EM, Pennacchio LA, Visel A. Tissue-Specific RNA Expression Marks Distant-Acting Developmental Enhancers. PLoS Genet. 2014;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaikkonen MU, Adelman K. Emerging Roles of Non-Coding RNA Transcription. Trends Biochem. Sci 2018;43:654–667. [DOI] [PubMed] [Google Scholar]
- 107.Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, Adelman K. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 2018;32:26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ, Turner JMA, Bertelsen MF, Murchison EP, Flicek P, Odom DT. Enhancer evolution across 20 mammalian species. Cell. 2015;160:554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Berthelot C, Villar D, Horvath JE, Odom DT, Flicek P. Complexity and conservation of regulatory landscapes underlie evolutionary resilience of mammalian gene expression. Nat Ecol Evol. 2018;2:152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, Herman B, Happe S, Higgs A, Leproust E, Follows GA, Fraser P, Luscombe NM, Osborne CS. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet. 2015;47:598–606. [DOI] [PubMed] [Google Scholar]
- 111.de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature [Internet]. 2013;502:499–506. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24153303 [DOI] [PubMed] [Google Scholar]
- 112.Tolhuis B, Palstra RJ, Splinter E, Grosveld F, De Laat W. Looping and interaction between hypersensitive sites in the active β-globin locus. Mol Cell. 2002; [DOI] [PubMed] [Google Scholar]
- 113.Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zabidi MA, Stark A. Regulatory Enhancer–Core-Promoter Communication via Transcription Factors and Cofactors. Trends Genet. 2016;32:801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV., Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer-Frick I, Shoukry M, Afzal V, Rubenstein JLR, Rubin EM, Pennacchio LA, Visel A. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell. 2013;155:1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van Arensbergen J, Pagie L, FitzPatrick VD, de Haas M, Baltissen MP, Comoglio F, van der Weide RH, Teunissen H, Võsa U, Franke L, de Wit E, Vermeulen M, Bussemaker HJ, van Steensel B. High-throughput identification of human SNPs affecting regulatory element activity. Nat Genet. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, Sabeti PC. Erratum: Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay (Cell (2016) 165(6) (1519–1529)(S0092867416304214)(10.1016/j.cell.2016.04.027)). Cell. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Engel KL, Mackiewicz M, Hardigan AA, Myers RM, Savic D. Decoding transcriptional enhancers: Evolving from annotation to functional interpretation. Semin. Cell Dev. Biol 2016;57:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Patwardhan TA, Nguyen TH, Kane M, Doughty B, Perez EM, Durand NC, Stamenova EK, Aiden EL, Lander ES, Engreitz JM. Activity-by-Contact model of enhancer specificity from thousands of CRISPR perturbations (CRISPRi FlowFISH). bioRxiv [Internet]. 2019;51:529990. Available from: https://www.biorxiv.org/content/10.1101/529990v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, Jackson D, Leith A, Schreiber J, Noble WS, Trapnell C, Ahituv N, Shendure J. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell. 2019;176:377–390.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fei T, Li W, Peng J, Xiao T, Chen CH, Wu A, Huang J, Zang C, Liu XS, Brown M. Deciphering essential cistromes using genome-wide CRISPR screens. Proc Natl Acad Sci U S A. 2019;116:25186–25195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science [Internet]. 2013;339:1074–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23328393 [DOI] [PubMed] [Google Scholar]
- 124.Vanhille L, Griffon A, Maqbool MA, Zacarias-Cabeza J, Dao LTM, Fernandez N, Ballester B, Andrau JC, Spicuglia S. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat Commun [Internet]. 2015;6:6905. Available from: http://www.nature.com/doifinder/10.1038/ncomms7905 [DOI] [PubMed] [Google Scholar]
- 125.Vockley CM, D’Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, Crawford GE, Reddy TE. Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell. 2016;166:1269–1281.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, Sabeti PC. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell. 2016;165:1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, Kellis M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013;23:800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kinney JB, Murugan A, Callan CG, Cox EC. Using deep sequencing to characterize the biophysical mechanism of a transcriptional regulatory sequence. Proc Natl Acad Sci U S A. 2010;107:9158–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Kinney JB, Kellis M, Lander ES, Mikkelsen TS. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol [Internet]. 2012;30:271–277. Available from: 10.1038/nbt.2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods [Internet]. 2013;10:1213–8. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3959825&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. [DOI] [PubMed] [Google Scholar]
- 132.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science (80-). 2007;316:1497–1502. [DOI] [PubMed] [Google Scholar]
- 133.Han J, Zhang J, Chen L, Shen B, Zhou J, Hu B, Du Y, Tate PH, Huang X, Zhang W. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 2014;11:829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Skene PJ, Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science (80-). 2002;295:1306–1311. [DOI] [PubMed] [Google Scholar]
- 136.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, De Wit E, Van Steensel B, De Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet. 2006;38:1348–1354. [DOI] [PubMed] [Google Scholar]
- 137.Van De Werken HJG, Landan G, Holwerda SJB, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Öz Y, Bouwman BAM, Verstegen MJAM, De Wit E, Tanay A, De Laat W. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat Methods. 2012;9:969–972. [DOI] [PubMed] [Google Scholar]
- 138.Schwartzman O, Mukamel Z, Oded-Elkayam N, Olivares-Chauvet P, Lubling Y, Landan G, Izraeli S, Tanay A. UMI-4C for quantitative and targeted chromosomal contact profiling. Nat Methods. 2016;13:685–691. [DOI] [PubMed] [Google Scholar]
- 139.Dostie J, Dekker J. Mapping networks of physical interactions between genomic elements using 5C technology. Nat Protoc. 2007;2:988–1002. [DOI] [PubMed] [Google Scholar]
- 140.Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schoenfelder S, Javierre BM, Furlan-Magaril M, Wingett SW, Fraser P. Promoter capture Hi-C: High-resolution, genome-wide profiling of promoter interactions. J Vis Exp. 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lieberman-Aiden E, Van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (80-). 2009;326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, Trinh V, Aznauryan E, Russell P, Cheng C, Jovanovic M, Chow A, Cai L, McDonel P, Garber M, Guttman M. Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell. 2018;174:744–757.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Davies JOJ, Telenius JM, McGowan SJ, Roberts NA, Taylor S, Higgs DR, Hughes JR. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat Methods. 2015;13:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hughes JR, Roberts N, Mcgowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, Higgs DR. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat Genet. 2014;46:205–212. [DOI] [PubMed] [Google Scholar]
- 146.Schoenfelder S, Sugar R, Dimond A, Javierre B-M, Armstrong H, Mifsud B, Dimitrova E, Matheson L, Tavares-Cadete F, Furlan-Magaril M, Segonds-Pichon A, Jurkowski W, Wingett SW, Tabbada K, Andrews S, Herman B, LeProust E, Osborne CS, Koseki H, Fraser P, Luscombe NM, Elderkin S. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet [Internet]. 2015;1–11. Available from: http://www.nature.com/doifinder/10.1038/ng.3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama J a, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Afzal V, Simpson PC, Rubin EM, Black BL, Bristow J, Pennacchio L a, Visel A. Large-scale discovery of enhancers from human heart tissue. Nat Genet. 2011;44:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, Ovcharenko I. Genome-wide discovery of human heart enhancers. Genome Res. 2010;20:381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dickel DE, Barozzi I, Zhu Y, Fukuda-Yuzawa Y, Osterwalder M, Mannion BJ, May D, Spurrell CH, Plajzer-Frick I, Pickle CS, Lee E, Garvin TH, Kato M, Akiyama JA, Afzal V, Lee AY, Gorkin DU, Ren B, Rubin EM, Visel A, Pennacchio LA. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat Commun [Internet]. 2016;7:1–13. Available from: 10.1038/ncomms12923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Van Duijvenboden K, De Boer BA, Capon N, Ruijter JM, Christoffels VM. EMERGE: A flexible modelling framework to predict genomic regulatory elements from genomic signatures. Nucleic Acids Res. 2015;44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser - A database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Krijger PHL, De Laat W. Regulation of disease-associated gene expression in the 3D genome. Nat Rev Mol Cell Biol [Internet]. 2016;17:771–782. Available from: 10.1038/nrm.2016.138 [DOI] [PubMed] [Google Scholar]
- 153.Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16:197–212. [DOI] [PubMed] [Google Scholar]
- 154.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, Van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell. 2017;169:930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, Huang X, Shamim MS, Shin J, Turner D, Ye Z, Omer AD, Robinson JT, Schlick T, Bernstein BE, Casellas R, Lander ES, Aiden EL. Cohesin Loss Eliminates All Loop Domains. Cell. 2017;171:305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zuin J, Dixon JR, Van Der Reijden MIJA, Ye Z, Kolovos P, Brouwer RWW, Van De Corput MPC, Van De Werken HJG, Knoch TA, Van Ijcken WFJ, Grosveld FG, Ren B, Wendt KS. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A. 2014;111:996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell [Internet]. 2015;1012–1025. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867415003773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bianchi V, Geeven G, Tucker N, Hilvering CRE, Hall AW, Roselli C, Hill M, Martin JF, Margulies KB, Ellinor PT, de Laat W. Detailed Regulatory Interaction Map of the Human Heart Facilitates Gene Discovery for Cardiovascular Disease. SSRN Electron J. 2019; [Google Scholar]
- 161.Montefiori LE, Sobreira DR, Sakabe NJ, Aneas I, Joslin AC, Hansen GT, Bozek G, Moskowitz IP, McNally EM, Nóbrega MA. A promoter interaction map for cardiovascular disease genetics. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Choy MK, Javierre BM, Williams SG, Baross SL, Liu Y, Wingett SW, Akbarov A, Wallace C, Freire-Pritchett P, Rugg-Gunn PJ, Spivakov M, Fraser P, Keavney BD. Promoter interactome of human embryonic stem cell-derived cardiomyocytes connects GWAS regions to cardiac gene networks. Nat Commun [Internet]. 2018;9. Available from: 10.1038/s41467-018-04931-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ye J, Tucker NR, Weng LC, Clauss S, Lubitz SA, Ellinor PT. A Functional Variant Associated with Atrial Fibrillation Regulates PITX2c Expression through TFAP2a. Am J Hum Genet. 2016;99:1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shiratori H, Sakuma R, Watanabe M, Hashiguchi H, Mochida K, Sakai Y, Nishino J, Saijoh Y, Whitman M, Hamada H. Two-step regulation of left-right asymmetric expression of Pitx2: Initiation by nodal signaling and maintenance by Nkx2. Mol Cell. 2001;7:137–149. [DOI] [PubMed] [Google Scholar]
- 165.Bamforth SD, Bragança J, Farthing CR, Schneider JE, Broadbent C, Michell AC, Clarke K, Neubauer S, Norris D, Brown NA, Anderson RH, Bhattacharya S. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat Genet. 2004;36:1189–1196. [DOI] [PubMed] [Google Scholar]
- 166.Man JCK, Mohan RA, Boogaard M van den, Hilvering CRE, Jenkins C, Wakker V, Bianchi V, Laat W de, Barnett P, Boukens BJ, Christoffels VM. An enhancer cluster controls gene activity and topology of the SCN5A-SCN10A locus in vivo. Nat Commun. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Van Den Boogaard M, Wong LYE, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, Bezzina CR, ‘T Hoen P a C, Bakkers J, Barnett P, Christoffels VM. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. 2012;122:2519–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Boogaard M Van Den. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J … [Internet]. 2014;124. Available from: http://www.jci.org/articles/view/73140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Leussink B, Brouwer A, El Khattabi M, Poelmann RE, Gittenberger-de Groot AC, Meijlink F. Expression patterns of the paired-related homeobox genes MHox Prx1 and S8 Prx2 suggest roles in development of the heart and the forebrain. Mech Dev. 1995;52:51–64. [DOI] [PubMed] [Google Scholar]
- 170.Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast Network in Rabbit Sinoatrial Node: Structural and Functional Identification of Homogeneous and Heterogeneous Cell Coupling. Circ Res. 2004;94:828–835. [DOI] [PubMed] [Google Scholar]
- 171.Rohr S Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res. 2004;62:309–322. [DOI] [PubMed] [Google Scholar]
- 172.Sotoodehnia N, Isaacs A, De Bakker PIW, DÖrr M, Newton-Cheh C, Nolte IM, Van Der Harst P, Müller M, Eijgelsheim M, Alonso A, Hicks AA, Padmanabhan S, Hayward C, Smith AV, Polasek O, Giovannone S, Fu J, Magnani JW, Marciante KD, Pfeufer A, Gharib SA, Teumer A, Li M, Bis JC, Rivadeneira F, Aspelund T, KÖttgen A, Johnson T, Rice K, Sie MPS, Wang YA, Klopp N, Fuchsberger C, Wild SH, Leach IM, Estrada K, VÖlker U, Wright AF, Asselbergs FW, Qu J, Chakravarti A, Sinner MF, Kors JA, Petersmann A, Harris TB, Soliman EZ, Munroe PB, Psaty BM, Oostra BA, Cupples LA, Perz S, De Boer RA, Uitterlinden AG, VÖlzke H, Spector TD, Liu FY, Boerwinkle E, Dominiczak AF, Rotter JI, Van Herpen G, Levy D, Wichmann HE, Van Gilst WH, Witteman JCM, Kroemer HK, Kao WHL, Heckbert SR, Meitinger T, Hofman A, Campbell H, Folsom AR, Van Veldhuisen DJ, Schwienbacher C, O’Donnell CJ, Volpato CB, Caulfield MJ, Connell JM, Launer L, Lu X, Franke L, Fehrmann RSN, Te Meerman G, Groen HJM, Weersma RK, Van Den Berg LH, Wijmenga C, Ophoff RA, Navis G, Rudan I, Snieder H, Wilson JF, Pramstaller PP, Siscovick DS, Wang TJ, Gudnason V, Van Duijn CM, Felix SB, Fishman GI, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Smith JG, Magnani JW, Palmer C, Meng YA, Soliman EZ, Musani SK, Kerr KF, Schnabel RB, Lubitz SA, Sotoodehnia N, Redline S, Pfeufer A, Müller M, Evans DS, Nalls MA, Liu Y, Newman AB, Zonderman AB, Evans MK, Deo R, Ellinor PT, Paltoo DN, Newton-Cheh C, Benjamin EJ, Mehra R, Alonso A, Heckbert SR, Fox ER. Genome-wide association studies of the PR interval in African Americans. PLoS Genet. 2011;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chambers JC, Zhao J, Terracciano CMN, Bezzina CR, Zhang W, Kaba R, Navaratnarajah M, Lotlikar A, Sehmi JS, Kooner MK, Deng G, Siedlecka U, Parasramka S, El-Hamamsy I, Wass MN, Dekker LRC, De Jong JSSG, Sternberg MJE, McKenna W, Severs NJ, De Silva R, Wilde AAM, Anand P, Yacoub M, Scott J, Elliott P, Wood JN, Kooner JS. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–152. [DOI] [PubMed] [Google Scholar]
- 175.Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, Gudjonsson SA, Jonasdottir A, Mathiesen EB, Njølstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Løchen ML, Kong A, Thorsteinsdottir U, Stefansson K. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–122. [DOI] [PubMed] [Google Scholar]
- 176.Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Müller M, Sotoodehnia N, Sinner MF, Verwoert GC, Li M, Kao WHL, Köttgen A, Coresh J, Bis JC, Psaty BM, Rice K, Rotter JI, Rivadeneira F, Hofman A, Kors J a, Stricker BHC, Uitterlinden AG, van Duijn CM, Beckmann BM, Sauter W, Gieger C, Lubitz S a, Newton-Cheh C, Wang TJ, Magnani JW, Schnabel RB, Chung MK, Barnard J, Smith JD, Van Wagoner DR, Vasan RS, Aspelund T, Eiriksdottir G, Harris TB, Launer LJ, Najjar SS, Lakatta E, Schlessinger D, Uda M, Abecasis GR, Müller-Myhsok B, Ehret GB, Boerwinkle E, Chakravarti A, Soliman EZ, Lunetta KL, Perz S, Wichmann H-E, Meitinger T, Levy D, Gudnason V, Ellinor PT, Sanna S, Kääb S, Witteman JCM, Alonso A, Benjamin EJ, Heckbert SR. Genome-wide association study of PR interval. Nat Genet [Internet]. 2010;42:153–159. Available from: 10.1038/ng.517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Schulze-Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt-Hansen J, Olesen MS, Kääb S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bézieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S, Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Le Marec H, Wilde AA, Probst V, Schott JJ, Dina C, Redon R. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Verweij N, Leach IM, Van Den Boogaard M, Van Veldhuisen DJ, Christoffels VM, Hillege HL, Van Gilst WH, Barnett P, De Boer RA, Van Der Harst P. Genetic Determinants of P Wave Duration and PR Segment. Circ Cardiovasc Genet. 2014;7:475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hemerich D, Pei J, Harakalova M, Van Setten J, Boymans S, Boukens BJ, Efimov IR, Michels M, Van Der Velden J, Vink A, Cheng C, Van Der Harst P, Moore JH, Mokry M, Tragante V, Asselbergs FW. Integrative Functional Annotation of 52 Genetic Loci Influencing Myocardial Mass Identifies Candidate Regulatory Variants and Target Genes. Circ Genomic Precis Med. 2019;12:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.van der Harst P, van Setten J, Verweij N, Vogler G, Franke L, Maurano MT, Wang X, Mateo Leach I, Eijgelsheim M, Sotoodehnia N, Hayward C, Sorice R, Meirelles O, Lyytikäinen LP, Polašek O, Tanaka T, Arking DE, Ulivi S, Trompet S, Müller-Nurasyid M, Smith AV., Dörr M, Kerr KF, Magnani JW, Del Greco MF, Zhang W, Nolte IM, Silva CT, Padmanabhan S, Tragante V, Esko T, Abecasis GR, Adriaens ME, Andersen K, Barnett P, Bis JC, Bodmer R, Buckley BM, Campbell H, Cannon MV., Chakravarti A, Chen LY, Delitala A, Devereux RB, Doevendans PA, Dominiczak AF, Ferrucci L, Ford I, Gieger C, Harris TB, Haugen E, Heinig M, Hernandez DG, Hillege HL, Hirschhorn JN, Hofman A, Hubner N, Hwang SJ, Iorio A, Kähönen M, Kellis M, Kolcic I, Kooner IK, Kooner JS, Kors JA, Lakatta EG, Lage K, Launer LJ, Levy D, Lundby A, Macfarlane PW, May D, Meitinger T, Metspalu A, Nappo S, Naitza S, Neph S, Nord AS, Nutile T, Okin PM, Olsen JV., Oostra BA, Penninger JM, Pennacchio LA, Pers TH, Perz S, Peters A, Pinto YM, Pfeufer A, Pilia MG, Pramstaller PP, Prins BP, Raitakari OT, Raychaudhuri S, Rice KM, Rossin EJ, Rotter JI, Schafer S, et al. 52 Genetic Loci Influencing Myocardial Mass. J Am Coll Cardiol. 2016;68:1435–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol 2015;16:593–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Koch L Epigenetics: An epigenetic twist on the missing heritability of complex traits. Nat. Rev. Genet 2014;15:218. [DOI] [PubMed] [Google Scholar]
- 183.Horvath S DNA methylation age of human tissues and cell types. Genome Biol. 2013;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, Hou L, Baccarelli AA, Stewart JD, Li Y, Whitsel EA, Wilson JG, Reiner1 AP, Aviv1 A, Lohman K, Liu Y, Ferrucci L, Horvath S. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10:573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lind L, Ingelsson E, Sundström J, Siegbahn A, Lampa E. Methylation-based estimated biological age and cardiovascular disease. Eur J Clin Invest. 2018;48. [DOI] [PubMed] [Google Scholar]
- 186.Hernando-Herraez I, Evano B, Stubbs T, Commere PH, Jan Bonder M, Clark S, Andrews S, Tajbakhsh S, Reik W. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat Commun. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lin H, Yin X, Xie Z, Lunetta KL, Lubitz SA, Larson MG, Ko D, Magnani JW, Mendelson MM, Liu C, McManus DD, Levy D, Ellinor PT, Benjamin EJ. Methylome-wide Association Study of Atrial Fibrillation in Framingham Heart Study. Sci Rep. 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Doñate Puertas R, Meugnier E, Romestaing C, Rey C, Morel E, Lachuer J, Gadot N, Scridon A, Julien C, Tronc F, Chapuis B, Valla C, Janin A, Pirola L, Méjat A, Rome S, Chevalier P. Atrial fibrillation is associated with hypermethylation in human left atrium, and treatment with decitabine reduces atrial tachyarrhythmias in spontaneously hypertensive rats. Transl Res. 2017;184:57–67.e5. [DOI] [PubMed] [Google Scholar]
- 189.Zhao G, Zhou J, Gao J, Liu Y, Gu S, Zhang X, Su P. Genome-wide DNA methylation analysis in permanent Atrial fibrillation. Mol Med Rep. 2017;16:5505–5514. [DOI] [PubMed] [Google Scholar]
- 190.Zhang W, Song M, Qu J, Liu GH. Epigenetic modifications in cardiovascular aging and diseases. Circ. Res 2018;123:773–786. [DOI] [PubMed] [Google Scholar]
- 191.Berger SL. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev 2002;12:142–148. [DOI] [PubMed] [Google Scholar]
- 192.Lawrence M, Daujat S, Schneider R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016;32:42–56. [DOI] [PubMed] [Google Scholar]
- 193.Zaret KS, Mango SE. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr. Opin. Genet. Dev 2016;37:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Li B, Carey M, Workman JL. The Role of Chromatin during Transcription. Cell. 2007;128:707–719. [DOI] [PubMed] [Google Scholar]
- 195.Takeuchi JK, Lou X, Alexander JM, Sugizaki H, Delgado-OlguÃ- N P, Holloway AK, Mori AD, Wylie JN, Munson C, Zhu Y, Zhou YQ, Yeh RF, Henkelman RM, Harvey RP, Metzger D, Chambon P, Stainier DYR, Pollard KS, Scott IC, Bruneau BG. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat Commun. 2011;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhang D, Hu X, Henning RH, Brundel BJJM. Keeping up the balance: Role of HDACs in cardiac proteostasis and therapeutic implications for atrial fibrillation. Cardiovasc. Res 2016;109:519–526. [DOI] [PubMed] [Google Scholar]
- 197.Song S, Zhang R, Mo B, Chen L, Liu L, Yu Y, Cao W, Fang G, Wan Y, Gu Y, Wang Y, Li Y, Yu Y, Wang Q. EZH2 as a novel therapeutic target for atrial fibrosis and atrial fibrillation. J Mol Cell Cardiol. 2019;135:119–133. [DOI] [PubMed] [Google Scholar]
- 198.Korantzopoulos P, Kolettis TM, Galaris D, Goudevenos JA. The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation. Int. J. Cardiol 2007;115:135–143. [DOI] [PubMed] [Google Scholar]
- 199.Dai DF, Rabinovitch PS. Cardiac Aging in Mice and Humans: The Role of Mitochondrial Oxidative Stress. Trends Cardiovasc. Med 2009;19:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev 2016;2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX, Marks AR. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Calkins K, Devaskar SU. Fetal origins of adult disease. Curr Probl Pediatr Adolesc Health Care. 2011;41:158–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Haïssaguerre M, Jaïs P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Métayer P, Clémenty J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666. [DOI] [PubMed] [Google Scholar]
- 205.Ho SY, Cabrera JA, Tran VH, Farré J, Anderson RH, Sánchez-Quintana D. Architecture of the pulmonary veins: Relevance to radiofrequency ablation. Heart. 2001;86:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Postma AV., Dekker LRC, Soufan AT, Moorman AFM. Developmental and Genetic Aspects of Atrial Fibrillation. Trends Cardiovasc. Med 2009;19:123–130. [DOI] [PubMed] [Google Scholar]
- 207.Mommersteeg MTM, Christoffels VM, Anderson RH, Moorman AFM. Atrial fibrillation: A developmental point of view. Hear Rhythm. 2009;6:1818–1824. [DOI] [PubMed] [Google Scholar]
- 208.Kapur S, MacRae CA. The developmental basis of adult arrhythmia: Atrial fibrillation as a paradigm. Front. Physiol 2013;4 September. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Garcia-Frigola C, Shi Y, Evans SM. Expression of the hyperpolarization-activated cyclic nucleotide-gated cation channel HCN4 during mouse heart development. Gene Expr Patterns. 2003;3:777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Stieber J, Herrmann S, Feil S, Löster J, Feil R, Biel M, Hofmann F, Ludwig A. The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc Natl Acad Sci U S A. 2003;100:15235–15240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Harzheim D, Pfeiffer KH, Fabritz L, Kremmer E, Buch T, Waisman A, Kirchhof P, Kaupp UB, Seifert R. Cardiac pacemaker function of HCN4 channels in mice is confined to embryonic development and requires cyclic AMP. EMBO J. 2008;27:692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hoesl E, Stieber J, Herrmann S, Feil S, Tybl E, Hofmann F, Feil R, Ludwig A. Tamoxifen-inducible gene deletion in the cardiac conduction system. J Mol Cell Cardiol. 2008;45:62–69. [DOI] [PubMed] [Google Scholar]