

Summary
Hepatocellular carcinoma (HCC) is an aggressive malignancy with its global incidence and mortality rate continuing to rise, although early detection and surveillance are suboptimal. We performed serological profiling of the viral infection history in 899 individuals from an NCI-UMD case-control study using a synthetic human virome, VirScan. We developed a viral exposure signature and validated the results in a longitudinal cohort with 173 at-risk patients who had long-term follow-up for HCC development. Our viral exposure signature significantly associated with HCC status among at-risk individuals in the validation cohort [Area Under the Curve: 0.91 (95% CI 0.87–0.96) at baseline and 0.98 (95% CI 0.97–1) at diagnosis]. The signature identified cancer patients prior to a clinical diagnosis and was superior to alpha-fetoprotein. In summary, we established a viral exposure signature that can predict HCC among at-risk patients prior to a clinical diagnosis, which may be useful in HCC surveillance.
In brief
Lui et al. demonstrate how viral infection history, obtained using human blood samples and VirScan analysis of antiviral antibodies, can be used to detect hepatocellular carcinoma in at-risk patients prior to clinical cancer diagnoses.
Graphical Abstract
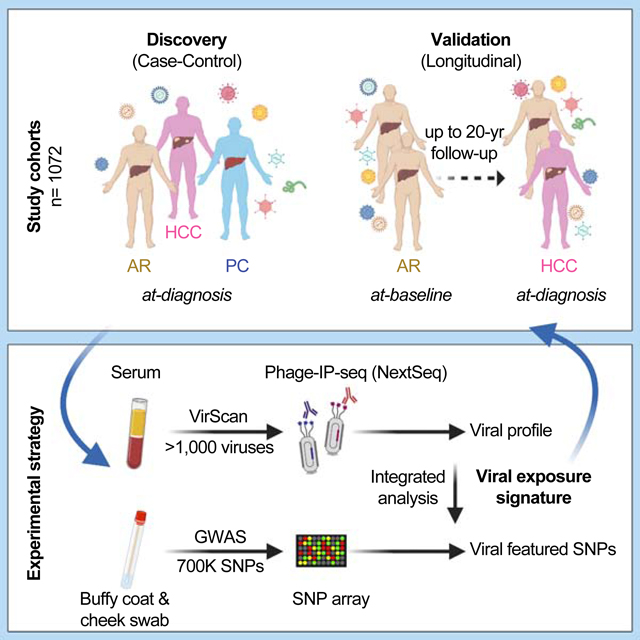
Introduction
Hepatocellular carcinoma (HCC), a main histological type of primary liver cancer, is considered a virus-related malignancy in which hepatitis B and C viruses (HBV and HCV) are major etiological factors (Farazi and DePinho, 2006). Viral hepatitis causes inflammation and chronic liver diseases, which may lead to fibrosis, cirrhosis and eventually, HCC. While HBV or HCV chronic carriers have an increased risk of developing HCC, the risk varies among individuals and not all patients with liver disease develop liver cancer (Arzumanyan et al., 2013). An effective strategy to prevent HCC is to eliminate causative factors. However, while direct-acting antiviral treatment is remarkably effective in eliminating HCV infection, it reduces but cannot eliminate HCC risk (Carrat et al., 2019; Janjua et al., 2017). Similarly, HBV vaccination, when introduced in the early 1980s, has been successful in significantly reducing the number of HBV carriers. Still, it only modestly reduces HCC burden in HBV-prevalent areas (Chang et al., 2016). It is puzzling that the control of HBV infection in HBV-prevalent areas as well as HCV infection has been remarkably successful for decades, while the global HCC incidence and mortality rates continues to increase since the 1990s (Liu et al., 2019b). Changing trends of etiological factors such as alcohol and non-alcohol/non-viral related liver diseases likely contribute to the observed increase. Thus, in addition to cancer prevention, early detection remains a key approach to prevent HCC-inflicted mortality. Currently, medical society guidelines recommend biannual surveillance using ultrasound with or without alpha-fetoprotein (AFP) for certain individuals with chronic liver disease and those with cirrhosis (Sherman et al., 2012). However, these practices have yielded mixed results related to the effectiveness in detecting HCC at an early stage or to provide survival benefit (Moon et al., 2018; Sherman et al., 1995; Tzartzeva et al., 2018). Notably, a majority of HCC patients are still diagnosed at an advanced stage, which precludes their chance to receive potentially curative therapies, leading to poor survival. Thus, there is an unmet need to identify an effective biomarker-guided surveillance program for early liver cancer detection.
Viruses are known to affect human health by altering host immunity, which makes the interplay between the virome and the host crucial in the pathogenesis of human chronic diseases, including cancer (Cadwell, 2015; Foxman and Iwasaki, 2011). Diverse pathogenic and non-pathogenic viruses may interact with one another as well as their host to shape host immunity, which may alter its response to new infections and cancer risk. Consequently, viruses that persist or are cleared in the host may leave unique molecular footprints that can affect host susceptibility to cancer and may serve as an excellent window of early onset of disease (Cadwell, 2015). We hypothesize that unique viral exposure signatures resulting from virus-host interactions could reflect a cascade of events that may alter the risk of developing HCC. Such signatures may serve as early detection biomarkers and offer knowledge about potentially modifiable factors for early onset of HCC. In this study, we profiled serological samples from 899 individuals currently enrolled in an NCI-UMD case-control study of liver cancer (NCT00913757; clinicaltrials.gov). We used a synthetic virome technology, VirScan, based on a high-throughput sequencing method, to detect the exposure history to all known human viruses (Xu et al., 2015). Using the high-throughput method, we developed a unique viral exposure signature (VES) that can discriminate HCC cases from at-risk or healthy volunteers, and then validated this signature in a prospective NIDDK at-risk cohort for HCC (NCT0001971; clinicaltrials.gov).
Results
The landscape of viral exposure profiles.
VirScan applies a phage display library that covers 93,904 viral epitopes, representing 206 human viral species and over 1,000 viral strains, to screen for previous exposure history (Xu et al., 2015). A phage particle with an epitope that was recognized by a participant’s antibody was immunoprecipitated (Phage-IP) and the encoding DNA barcode was then sequenced (Fig. 1A). We used a case-control design of the Maryland (NCI-UMD) cohort for the discovery of viral exposure profiles. The inclusion and enrollment of the study subjects are outlined in Suppl. Fig. S1A, following the CONSORT guideline (Schulz et al., 2010) (Suppl. Table S1A). For the NCI-UMD cohort, VirScan phage-IP products yielded 0.5–5 million single-end reads per serum sample, with mean mapped read matching accuracy of 0.93 (Fig. 1B). A total of 30,033 viral epitopes were significantly enriched with a -log10(p-value) greater than the reproducibility threshold of 2.358, which was determined based on both replicates (Fig. S1B–C). It was noted that the composition of the viral types at the viral taxonomic level showed small yet noticeable differences between the obtained Phage-IP products and the library input (Fig. S1D–E), indicating a measurable difference between patients-derived data and the original input. When assessing viral richness among healthy volunteers as a population control (PC), chronic liver disease (CLD) patients as at-risk (AR), and HCC, we found that the number of detected viral infections increases initially with the sample size but reaches saturation at a sample size of 200 or more (Fig. 1C). We detected a median of 7 species of virus per sample, with four individuals showing cross-reactivity to more than 20 virus species (Fig. 1D). Overall, the number of viral species was similarly distributed among PC, AR, and HCC (Fig. 1D), indicating no bias in the landscape of overall viral exposure profiles between these groups. The abundance of the most prevalent viral species, including human herpesvirus 4 (EBV) and human herpesvirus 5 (CMV), was similar in our study populations to what was reported in other populations (Fig. 1E, Suppl. Table S1B) (Xu et al., 2015), and is consistent with previous epidemiology reports (Ho, 1990; Straus et al., 1993). However, the HCV infection rate (26%) in our study was relatively high, which is explained by the high rates in AR (48%) and HCC (39%) (Suppl. Table S1B, Suppl. Fig. S2). Intriguingly, we detected a wide range of unique viral epitopes for each viral species that were recognized among different participants, indicating that B-cell antigenicity to the same viral species is rather diverse among the participants (right panel in Fig. 1E, Suppl. Fig. S2).
Figure 1. Assessment of Viral Infection History in a Case-control and a Prospective Cohort Using VirScan.

(A) Schema of screening of a NCI-UMD cohort consisting of 899 serum samples (analyzed by VirScan) and 849 matching buffy coat or cheek swab samples (analyzed by GWAS) with integrated analysis across population groups, namely population controls (PC, n=412), at-risk chronic liver disease cases (AR, n=337) and hepatocellular carcinoma cases (HCC, n=150); the resulting viral exposure signatures (VES) were validated in a prospective NIDDK cohort with cancer-free (n=129) and HCC (n=44) patients. (B) Sequencing read statistics of VirScan with mean matching accuracy of 0.93. (C) Rarefaction plot showing the viral species richness detected in PC, AR and HCC groups. (D) Raincloud plot showing the number of viral species in each individual across populations. (E) Left panel: viral infection prevalence across all samples. Right panel: number of unique epitopes per sample; vertical bars represent mean values.
To further assess the quality of VirScan, we compared VirScan results with available medical chart entries for HCV, HBV, and HIV testing results and found that VirScan had 45%, 47%, and 70% specificity in detecting HCV, HBV, and HIV, respectively, when compared to these medical record data (Fig. 2A). In contrast, its sensitivity was 84% for HCV, 48% for HBV, and 73% for HIV. It should be noted that a majority of viral status data from medical charts are unknown or missing (Suppl. Table S2), which makes this comparison suboptimal. We also examined epitope enrichment of HCV1b, a major viral subtype associated with HCC (Bruno et al., 2007). An increase in peptide enrichment, corresponding mainly to the core, NS4 and NS5A of HCV1b, was consistently observed among AR and HCC when compared to PC, which could be due to a high B-cell antigenicity of the epitopes as indicated by a prediction score (Fig. 2B). We also examined the presence of HIV and other viruses known to have co-infection with HIV (Chang et al., 2013; Echavarria, 2008; Stover et al., 2003; Xu et al., 2015). Consistently, we found an increased co-infection prevalence for HIV with human herpesvirus 5, human adenovirus C, human adenovirus D, influenza B virus, human herpesvirus B and HBV at false discovery rate FDR < 0.05 (Fig. 2C). Taken together, the above results revealed that VirScan is a reliable method to capture a broad spectrum of viral exposures with a simple serological assay.
Figure 2. Comparison of VirScan with Medical Charts, Antigenicity of HCV1b and HIV Coinfection Viruses.

(A) Contingency matrices comparing HCV, HBV and HIV detection with VirScan against viral detection laboratory tests reported in the patient medical charts. For the purpose of computing binary classification test statistics, clinical results were considered true values and VirScan results were considered predicted values. (B) Left panel: Heatmap showing HCV proteomic enrichment among PC, AR and HCC groups. Each row represents the significant peptide tiling. Each column is a sample. The proteomic location of the tiling peptides is annotated with a vertical bar on the left. The top-most horizontal annotation bar indicates patients grouped as PC, AR and HCC, as indicated. Second from the top, the annotation bar indicates positive HCV species based on VirScan data. The color intensity of each cell corresponds to the scaled - log10(p-value) measure of significance of enrichment for a peptide in a sample (where greater values indicate stronger antibody response). Right panel: B-cell epitope prediction score for each peptide. (C) Coinfection viral status in HIV positive versus HIV negative cases. Asterisks denote false discovery rate FDR < 0.05.
HCC-associated viral exposure signature (VES)
We applied a gradient boosting approach to search for the best-fit virus composition that can discriminate HCC from PC (Fig. 3A). Using 10-fold cross validation and 1,000 random permutations first where 90% of samples were used for training and remaining 10% of samples were used as an independent validation, we found that a viral exposure signature (referred to as VES) can significantly discriminate HCC from PC with Area under the Curve (AUC) values above 0.9 and 0.7 for training and cross validation, respectively (Fig. 3B). This signature consisted of unique epitopes corresponding to 61 viral strains (Fig. 3C). Among them, 18 viral strains were positively associated, while the remaining viruses were negatively associated, with HCC. A total of 11 HCV strains, including unique variants such as 3b or Taiwan 1b, were among the main contributing viruses in the signature. This observation was not surprising since 39% of HCC cases from this cohort were HCV positive. We also found that herpesvirus 5, HDV and influenza virus strains H1N1 and H3N2 were enriched in the HCC group. In contrast, 43 viruses, such as human respiratory syncytial virus and human rhinovirus 23, were preferentially depleted in the HCC group (Suppl. Table S3A, Fig. 3C). Weighed VES scores of the 61 viral strains differed significantly between HCC and PC (p < 0.0001), as well as between HCC and AR (p < 0.0001), which was not included for initial signature discovery, and also between AR and PC (p < 0.0001) (Fig. 3D). There was a significant increase of the VES score among PC, AR and HCC (p-trend < 0.0001), suggesting that VES was positively linked to hepatocarcinogenesis. We performed phylogenetic analysis of the reactive epitopes of the 61 viral strains to determine similarity among these HCC-related viruses (Fig. 3E). To search common reactive viral epitopes either enriched or depleted in HCC, we restricted viral epitopes that rank the top for its association with HCC. These viruses can be divided into eight main branches where different HCV epitopes are clustered together with other viral epitopes, with an exception of cluster #6 that contains 6 HCV variants (out of 12 viruses) (Fig. 3E, Suppl. Table S3B). In general, there was no clear enrichment within each branch for increased or decreased viruses, suggesting varying viral epitopes involved in immunoreactivity are commonly shared among HCC.
Figure 3. Extraction of Predictive VES Signatures Associated with HCC.

(A) VES are identified using the Xgboost machine learning algorithm. The flow chart shows the procedure to compare HCC versus PC viral profiles in training and cross validation sets. The resulting model provides scored results leading to a predictive VES score for each individual in the PC, AR and HCC groups. (B) Gradient boosting plot showing the area under the curve (AUC) value of training and cross validation sets. Gradient boosting stops at round 108 (vertical line) to avoid overfitting. (C) Bar plot showing the VES identified by comparing HCC with PC using Xgboost in the NCI-UMD cohort. Red bars denote an increase of viral infections in HCC versus PC, whereas blue bars represent a decrease. (D) Predictive VES score among PC, AR and HCC groups (**** two-tailed Mann-Whitney test p-value < 0.0001). (E) Phylogenetic analysis of the 61 viral strains, which results in 8 well-defined branches.
Since a majority of HCC patients has an evidence of chronic liver diseases, to avoid this confounding variable, we also compared AR to HCC using the same gradient boosting approach. We found that an AR vs HCC VES can significantly discriminate HCC from AR or PC with AUC values similar to VES for training and cross validation (Suppl. Fig. S3A–B). A majority of these VES-related viral strains overlap (Suppl. Fig. S3C). To further test the robustness of VES, we performed 60/40 split where 60% of cases were used for VES discovery while the remaining 40% of cases were used for an independent prediction. We performed 1000 permutations of the split to establish the confidence interval. Again, we found similar VES with a mean of AUC 0.7 for prediction (Suppl. Fig. S3D–G). Collectively, these results indicate that a VES consisting of a certain set of viral epitopes is relatively stable and robust in discriminating HCC from AR or PC. Given a limited number of HCC cases in the NCI-UMD cohort, we decided to keep the original VES for further analysis.
Phenotype-genotype association with VES
To determine if the host genetic background may be linked to VES, we performed a genome-wide association study (GWAS) in the NCI-UMD cohort, as this approach may help to identify genetic variants susceptible to viral infection and cancer (Fumagalli et al., 2010; McKay et al., 2017; Pharoah et al., 2013). After quality assessment of genomic data using genetic quality control measures, 849 participants (PC=402; AR=323; HCC=124) were included in the analysis. Following the removal of mono-allelic SNPs and the ones that deviate away from Hardy-Weinberg equilibrium, we performed an association test for all the remaining SNPs. To further assess the quality of our GWAS data, we determined whether there was an association between a SNP, rs12979860 in IL28B and HCV infection as its favorable genotype CC has been shown to be associated with better HCV treatment response or natural clearance. We found that rs12979860-CC was significantly associated with HCV Genotype 3 with Odds Ratio 2.74 (95% CI 1.14–7.97) in a dominant model manner (Suppl. Table S4A). Furthermore, we looked into the SNP associated with 375 epitopes abundances of HCV Genotype 2 and 3. We found that the CC allele is associated with a decreased abundance of core epitopes but an increased abundance of NS5B epitopes in the HCV genome (Suppl. Fig. 4A and Suppl. Table S4B), consistent with a recent study (Ansari et al., 2017). To assess VES-associated SNPs, HCC and PC groups were combined and then divided into two groups based on dichotomization of VES scores. In the associated quantile-quantile plots (Suppl. Fig. S5B), a wider spread with small differences in allele frequencies was evident with increased slope of the line. PCA analysis based on genotyping revealed differences in ethnicity (Suppl. Fig. S5C). Manhattan plot revealed significant SNPs between the high- and low-VES score groups (Fig. 4A). Three SNPs, namely rs34725101, rs4483229 and rs16960234, located in three different genomic regions corresponding to the RHOA, EPB41L4B and CDH13 loci, respectively, showed associations with VES score groups at the p-value < 10−7 significance level (Fig. 4A and Suppl. Table S4C). Among them, rs16960234 was further analyzed because both major and minor alleles of this variant could be detected in this cohort. We also found 127 SNPs with a high linkage disequilibrium (LD) (r2>0.6) for rs16960234, but none for rs34725101 and rs4483229 (Fig. 4B–C and Suppl. Table S4D). Seven of the high LD SNPs of rs16960234 showed the expression profile of CDH13 as expression quantitative trait loci (eQTL) in the genotype-tissue expression (GTEx) database (McKay et al., 2017). The CDH13 expression levels in the artery tibial tissues from the carriers with risk/protective G/G genotype of rs16960234 were significantly higher than the carriers with protective/risk genotype A/A (Fig. 4D). To obtain the genotypic effects of rs16960234 in HCC or AR, we generated logistic regression models to calculate the genotypic odds ratio of this SNP in AR or HCC compared to PC (Fig. 4E). The G/G genotype of rs16960234 showed an increase in risk of HCC in AR compared to PC, OR=1.89 (95% CI 0.30–11.4), and the risk was higher and statistically significant for HCC, OR=7.22 (95% CI 1.30–40.0) (Fig.4E and Suppl. Table S4E). Consistent with the genotypic effect in HCC, the VES score also increased gradually from heterozygous A/G to G/G when compared with A/A (Fig. 4F). Thus, rs16960234 and its linked gene CDH13 may be associated with VES and contribute to disease risk.
Figure 4. Genome-wide Scan Identifies Specific Genetic Variants Linked to VES. Related to Figure 3, Figure 5 and Genotyping and GWAS analysis in STAR Method section.

(A) Manhattan plot showing the detected genetic variants from GWAS associated with the viral featural phenotype of the NCI-UMD cohort. Annotated names of gene loci with P-value less than 10−7 (B) Locus Zoom plot showing the LD structure one of the lead SNPs, rs16960234, around the region of CDH13 and RP11-543N12.1. (C) Heatmap showing the high linkage disequilibrium (LD) SNPs of rs16960234 from 1000 Genomes database (R2>0.6). The density of the heatmap indicates the r2 value of the correlation. The labeled SNPs are the ones with eQTL available. (D) The eQTL of CDH13 in tissue artery tibial across genotypes of SNP rs1690234 from GTEx database. (E) The genotypic Odds Ratios (OR) of rs1690234 among AR (black) and HCC (red) relative to PC. (F) VES score fold changes (FD) in genotypes AA, AG and GG of rs1690234 based on VES among HCC relative to PC.
Validation of the VES in a prospective HCC cohort
To further validate the VES identified above for its clinical utility, we analyzed VirScan profiles in the at-risk NIDDK cohort for HCC. This cohort consisted of 173 CLD patients who were enrolled for a natural history study of liver disease with a follow-up of up to 20 years (Table 1, Suppl. Fig. S5A). Among them, 44 individuals developed HCC. The median number of viral species in the NIDDK cohort was 6, i.e. similar to the NCI-UMD cohort. This cohort contained serum samples collected at enrollment (baseline) and at various follow-up timepoints until a diagnosis of HCC by imaging was made (diagnosis). We then performed logistic regression analysis using VES. In order to capture their performance in either the NCI-UMD cohort or the NIDDK cohort, we generated receiver-operating characteristic (ROC) curves. Area under the curve (AUC) performance measures thus obtained were 0.89 (95% CI 0.86–0.92) in the NCI-UMD cohort (Fig. 5A). We observed that VES scores varied substantially among HCC cases in the NCI-UMD cohort with some having scores below the detection limit and others having quite high scores (Fig. 5B). Interestingly, patients with a high score had a significantly worse survival compared to patients with a low score or a score below the detection limit (logrank p=0.026 and p-trend = 0.024) (Fig. 5C). Suppl. Table S5A shows the results from univariable and multivariable Cox model survival analysis on several clinicopathologic variables to clarify the independent and additional prognostic value of VES. Among patients from the NIDDK cohort, VirScan data were available for 40 HCC cases at baseline, 129 controls at baseline, 44 HCC cases at diagnosis and 106 controls at diagnosis. We found that the AUC values were 0.91 (95% CI 0.87–0.96) at baseline (Fig. 5D) and 0.98 (95% CI 0.97–1) at diagnosis (Fig. 5E). The performance of VES was superior to alpha-fetoprotein (AFP), a known HCC diagnostic marker used in clinical practice. The DeLong test showed a significant improvement between VES and AFP (p-values 4×10−12 and 8×10−10 at baseline and diagnosis, respectively) (Fig. 5D–E). We also found similar trends (p-trend=0.19) between the levels of VES and overall survival among 44 patients in the NIDDK cohort (Suppl. Fig. S5B). In order to assess the time-dependent performance of VES to predict the onset of HCC, we analyzed 104 cancer-free controls and 40 HCC cases (from the NIDDK validation cohort) for which at least two timepoints were available.
Table 1.
Clinical Characteristics of the Patients.*
| Variable | Without HCC (N=129) | With HCC (N=44) | P Value |
|---|---|---|---|
| Age – year | 0.12 | ||
| Median (Range) | 51 (23–79) | 54 (23–79) | |
| Missing data | 1 | 0 | |
| Sex — no. (%) | 1.00 | ||
| Female | 40 (31.0) | 14 (31.8) | |
| Male | 89 (69.0) | 30 (68.2) | |
| Missing data | 0 | 0 | |
| Race — no. (%) | 0.69 | ||
| European American | 63 (48.8) | 22 (50.0) | |
| African American | 29 (22.5) | 12 (27.3) | |
| Asian American | 26 (20.2) | 8 (18.2) | |
| Other | 2 (1.6) | 0 | |
| Missing data | 9 (7.0) | 2 (4.6) | |
| HCV only — no. (%) | 98 (76.0) | 27 (61.4) | 0.61 |
| HBV only — no. (%) | 18 (14.0) | 7 (15.9) | |
| HBV + HCV — no. | 2 (1.6) | 1 (2.3) | |
| HBV + HDV — no. | 4 (3.1) | 3 (6.8) | |
| Others not hepatitis | 7 (5.4) | 6 (13.6) | |
| Cirrhosis — no. (%) | 15 (11.6) | 28 (63.6) | <0.001 |
| Missing data | 2 (1.6) | 4 (9.1) | |
| Alanine aminotransferase — no. (%) | < 0.01 | ||
| Elevated (>50 U/L) | 84 (65.1) | 28 (63.6) | |
| Normal (≤50 U/L) | 45 (34.9) | 16 (36.4) | |
| Alpha-fetoprotein — no. (%) | <0.001 | ||
| >20 ng/mL | 9 (7.0) | 21 (47.7) | |
| ≤20 ng/mL | 120 (93.0) | 23 (52.3) | |
| Missing data | 0 | 0 | |
| Survival (months) | |||
| Median | NA | 15.2 | |
| Range | NA | 0.07–131.8 | |
| Missing data (%) | NA | 1 (2.3) | |
The clinical characteristics of the 173 at-risk patients in the prospective NIDDK cohort.
Figure 5. Assessment of VES Signatures: Predictive Performance and Association with Clinical Outcomes.

(A) Receiver operating characteristic (ROC) curves for the NCI-UMD cohort at HCC diagnosis. Area under the curve (AUC) values are shown for PC vs HCC prediction, as well as for AR vs HCC prediction, as indicated. (B) Feature score distributions of NCI-UMD subcohorts grouped by VES level, which were classified as below (threshold=0.50, dashed line), low and high. Low and high VES levels are separated by the median among patients with VES above the threshold. (C) Kaplan-Meier survival plots for NCI-UMD subcohorts grouped by VES level. (D, E) ROC curves for the NIDDK validation cohort at HCC baseline and diagnosis, respectively. AUC values are shown for alpha-fetoprotein (AFP), VES, and the combination of both. (F) Time-dependent AUC showing the landmark timepoints performance of VES from 1 to 10 years relative to baseline. (G) Baseline and follow-up viral feature scores of the NIDDK cancer-free group compared with those of high-risk patients that ultimately developed HCC. (H) AUC values corresponding to predictions based on clinical indicators from patient charts compared with those based on VES, as well as those based on the combination clinical and VES for the NIDDK cohort at baseline.
In the context of survival modeling, an event was defined as the occurrence of an HCC diagnosis. Under this interpretation, censoring time was defined as the time difference between baseline and follow-up within the cancer-free control group, whereas event time was defined as the time difference between baseline and HCC diagnosis within the HCC group. Suppl. Table S5B shows results from a multivariable Cox regression model generated to predict the occurrence of HCC diagnosis based on VES scores at baseline, adjusted for clinical prognostic variables. Moreover, we performed time-dependent ROC curve analysis (Bansal and Heagerty, 2019; Blanche et al., 2013) to assess the performance of VES over a range of landmark timepoints from 1 to 10 years relative to baseline (Fig. 5F, Suppl. Fig. S5C), which appears very robust and stable across this range. Interestingly, we found that patients who developed HCC had on average much higher VES scores at baseline and at different times of follow-up until HCC diagnosis, when compared to cancer-free at-risk patients who were followed up at a similar time interval without developing HCC (Fig. 5G). A statistically significant increase in viral exposures (p < 0.05) was observed only for patients who developed HCC over time during the surveillance period in the NIDDK cohort. It appears that HCC cases with a high viral exposure had a more aggressive disease than those with a low viral exposure, and that VES was a robust indicator of early onset of HCC in this prospective cohort. Furthermore, the prediction performance of AR vs HCC based on VES was superior to other clinical indicators from the patient charts, such as AFP, ALT, cirrhosis and platelet counts, as well as the combination of all key clinical variables, as shown by analyses of the NIDDK cohort at baseline (Fig. 5H), which agree qualitatively with those of NIDDK at diagnosis (Suppl. Fig. S5D) and the NCI-UMD cohort (Suppl. Fig. S5E). An association of VES and HCC was similarly found in both HCV-positive and HCV-negative patients (Suppl. Table S5C).
DISCUSSION
Detecting cancer at an early stage, preferably before it is symptomatic, may provide an opportunity in achieving a cure and improving cancer-related mortality. Evidence suggests that earlier detection of cancer may potentially improve survival for some cancer types such as cervical and colon cancers. A conventional approach is to develop biomarkers specific for cancer cells in order to determine cancer early diagnosis. CancerSEEK is an emerging platform with a good sensitivity and specificity to clinically-detected multiple cancer types by profiling circulating cell-free DNA (ctDNA) for driver mutations presumably shed from tumor cells (Cohen et al., 2018). However, a recent study offers a cautionary note using cancer gene panels in ctDNA because of its high false positive rate among healthy individuals (Liu et al., 2019a). Molecular and biological heterogeneity of cancer cells contributed by complex etiological landscape creates a dilemma as how best to design cancer-specific diagnostic panels effective for early cancer detection. As such, a continuous debate has been carried out in recent decades for many malignant diseases including HCC as to whether available methods are adequate in achieving this goal (Sherman et al., 2012; Shieh et al., 2016).
HCC is a unique malignancy in which we know most of the major causative etiologies (Wang and Thorrgeirsson, 2014). However, defining biomarkers specific for HCC cells has been challenging because of its complex genomic landscape with extensive intratumor and intertumor heterogeneities. Are there common features shared among HCC patients to be used as a surrogate for early detection? An emerging concept is that an interplay between viral infection and host genetic background is crucial for maintaining virome homeostasis or causing human disease (Virgin, 2014). In this study, we have attempted to assess how a history of viral exposures in an individual is associated with its risk to develop HCC. Using a synthetic viral scan technology (VirScan) with a simple blood test (Xu et al., 2015), we found a VES that could discriminate HCC with a high confidence from at risk individuals or from healthy volunteers. Remarkably, this signature was able to identify individuals at a median follow-up year of 8.8 prior to a clinical diagnosis of HCC. Thus, our results may offer a sensitive tool applicable to the HCC surveillance program to improve early diagnosis.
The current study took the advantage of a simple tool to profile serological samples to link an individual’s history of viral infection and corresponding response to early onset of HCC. Our strategy was first to search VES using a case control design that include HCC cases as well as at-risk individuals with chronic liver diseases and healthy volunteers matched by age, sex and race. A VES that can discriminate HCC from at-risk and healthy individuals was then validated using a prospective cohort of sequentially enrolled at-risk patients who were followed up for the development of HCC. Interestingly, the VES consists of known HCC etiologies such as HCV, HBV and HDV, but also includes other viruses such as EBV (HHV-4), CMV (HHV-5), Crimean-Congo hemorrhagic fever virus, cytomegalovirus, influenza A virus, among others. A few features are noted. First, HCV appears a major etiological factor driving VES but an extended heterogeneity in various HCV subtypes are noted in both NCI-UMD and NIDDK cohorts. Second, a set of viruses are enriched while many others including HBV are depleted in HCC patients. The nature of a potential impact of detected viruses in VES other than HBV, HCV and HDV on HCC pathogenesis is unknown at the present time. One plausible explanation is that some of the detected viruses and the levels of viral burden may reflect how individuals react to a history of viral exposure and how an individual’s intrinsic immune surveillance unique to linked genetic background may react to viral infection. Encouragingly, GWAS analysis revealed that several SNPs have a strong association with a history of viral infection. Among them, rs16960234 in CDH13 has the strongest link to viral infection, a gene previously linked to cytomegalovirus infection (Borglum et al., 2014). Although the molecular mechanisms of CDH13 in human cancer are unknown, some studies suggest that its expression may be associated with tumorigenesis (Takeuchi et al., 2000). Whether this variant is linked to viral infection in general remains to be further validated.
It should be noted that VES discovery was based on a case-control study design with 150 HCC cases. Given the nature of both molecular and clinical heterogeneity in HCC, we were unable to test all possible clinical covariables that may confound VES classification. To overcome this limitation, we used both 10-fold cross validation and 60/40 split approaches with random permutations to test the robustness of VES. Remarkably, various approaches yielded VES with similar viral compositions and HCC classification when compared to healthy volunteers or at-risk individuals. Moreover, the VES could independently classify HCC in a prospective at-risk population, confirming the robustness of VES in HCC diagnosis. These results suggest a clinical utility of VES as a surveillance tool to screen early onset of HCC, thereby increasing the opportunity for HCC patients receiving curative therapies. While an initial discovery of VES was validated using a prospective cohort with a longitudinal follow up, these cohorts were used retrospectively. It is important to develop a CLIA-certified diagnostic assay that consists of VES and then test its ability to reduce HCC mortality in a randomized trial, similar to the study of lung cancer (de Koning et al., 2020).
The current method of VirScan is based on the phage immunoprecipitation sequencing (PhIP-Seq) technology that provides a powerful approach for analyzing antibody-repertoire binding specificities with high throughput and at low cost to all known human viruses (Mohan et al., 2018). How accurate and sensitive is the VirScan method? Comparing VirScan results (as predicted values of HCV and HBV status) against those from medical charts of the NCI-UMD cohort (as true values), we found that VirScan shows better accuracy (0.73, 95% CI 0.67–0.79), sensitivity (0.84, 95% CI: 0.77–0.89) and positive predictive value (0.80, 95% CI 0.76–0.84) for HCV than the accuracy (0.48, 95% CI 0.41–0.54), sensitivity (0.48, 95% CI 0.40–0.57) and positive predictive value (0.55, 95% CI 0.49–0.61) for HBV. HCV encodes a large polyprotein consisting of ~3,000 amino acids, which is cleaved co- and post-translationally into ten different proteins associated with intracellular membranes (Bartenschlager et al., 2013). Consistently, we found that the HCV antigen reactivity is largely overlapped with the predicted antigenicity score by the B-cell epitope prediction method coinciding with the epitopes to be presented at the surface of the cellular membrane. Consistent with early reports for the likelihood of coinfection of HIV and other viruses associated with AIDS and non-AIDS diseases (Lichtner et al., 2015; Slyker et al., 2013; Xu et al., 2015), we found evidence of coinfection between HIV and viruses such as HBV, herpesvirus 8 and adenovirus D, influenza B virus, adenovirus C, and herpesvirus 5 in patients enrolled in the NCI-UMD cohort. Interestingly, we found that history of HCV infection is prevalent among at-risk (48%), HCC patients (39%) and healthy volunteers (4%) who reside in Maryland. This is in contrast to an estimate prevalence of about 4.6 million persons (~1.5%) infected with HCV in the U.S. (Edlin et al., 2015). It should be noted that 7.5%−44% of incarcerated individuals and 4%−38% of hospitalized patients tested positive for HCV (Edlin et al., 2015), suggesting that the current surveys underestimate the prevalence of HCV infection. In contrast, while we observed 2.6% of healthy individuals in the NCI-UMD study showing evidence of HBV infection, more than 800,000 chronic HBV carriers were detected during 2011–2012 in the noninstitutional U.S. population (Roberts et al., 2016). It seems that the current survey methods may underestimate the prevalence of HBV and HCV. This is important as both HBV and HCV are major causative factors for HCC. It should also be noted that the sensitivity and specificity of HBV detection by VirScan are relatively low. Due to a significant fraction of missing data for medical test results for HBV, we were not able to perform in-depth correlation analysis of HBV genotypes and VES-based viral loads. This is a limitation of this study. It is worth to speculate that cancer-prone individuals may perceive viral epitopes differently compared to healthy individuals. An in-depth correlative analysis between individual viral epitopes, a host’s TCR/BCR profile and genetic background may help to understand mechanisms of anti-tumor immunity. VirScan is a reliable method for profiling viral exposure collectively and is both scalable (regarding to sample throughput) and non-invasive, which makes it amenable for surveillance and early detection of HCC.
In conclusion, we performed the largest study to date analyzing serological samples using a high throughput virome technology in patients with HCC and the first one, to our knowledge, utilizing VES as cancer biomarkers for early onset of HCC. The VES was validated in at-risk patients with a longitudinal design for HCC diagnosis. A prospective randomized study with a large population is warranted to evaluate its utility in HCC surveillance.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xin Wei Wang, xw3@nih.gov
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The viral exposure profiles supporting the current study have not been deposited in a public repository because of the requirement for reporting only the aggregate data by the study protocol but are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study Cohorts
NCI-UMD cohort.
To measure virome-host interplay, 899 participants were recruited; all were from the greater Baltimore area between 2003 and 2016. All participants voluntarily signed informed consent for collection and analysis of blood and cheek swab samples under Institutional Review Board (IRB)-approved protocols at the National Institutes of Health. Participants were classified into three groups, as follows: healthy individuals without any diagnosis of liver disease were classified as population controls (PC, n=412); patients diagnosed with chronic liver diseases due to hepatitis B virus (HBV), hepatitis C virus (HCV), hepatitis delta virus (HDV), aflatoxins from fungal contamination, alcohol, nonalcoholic fatty-liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) were classified as at-risk (AR, n=337); and patients diagnosed with hepatocellular carcinoma were classified in the third group (HCC, n=150). All clinic measurements were covered by NCT0091375 (clinicaltrials.gov) with liver disease status as the enrollment criteria. Serum and matching buffy coat or cheek swab were collected at the time of interview.
NIDDK cohort.
This cohort consisted of 173 patients with chronic liver disease that included 44 HCC cases with 129 controls matched by liver disease etiology, age and sex. Patients were at-risk individuals for the development of HCC who were enrolled in a natural history protocol (clinicaltrials.gov number; NCT0001971) between 1991 and 2017 with longitudinal follow-up, at least annually with serologic testing and imaging, for up to 20 years. Only cases with complete clinical and laboratory data and available longitudinal serologic samples were selected for analysis. The 44 HCC cases were sequentially identified out of 3,067 patients followed in this natural history study on chronic liver disease, and the controls were matched on a 2:1 basis as described above. HCC was diagnosed by radiologic imaging and/or liver biopsy as described by the American Association for the Study of Liver Disease (AASLD) practice guidelines (Marrero et al., 2018). For the purposes of this analysis, stored serum samples (−80°C) were analyzed at study entry (baseline) and at recurrent time points until the time of HCC diagnosis.
All participants were volunteers with informed consent for collection and analysis of blood samples under the IRB-approved protocols at NCI and NIDDK.
Sample collection.
Sera were prepared from blood samples and then stored at −80°C for research (protocols previously listed) (n=899 from NCI-UMD, n=488 from NIDDK). Buffy coat and cheek swab samples were collected and stored at −80°C for research (protocols previously listed) (n=849 from NCI-UMD).
Study Oversight.
The study was approved by the NCI and NIDDK institutional review boards. All participants provided written informed consent.
METHOD DETAILS
VirScan T7 phage library proliferation.
The T7 phage library displays the virome peptide library. It was created to cover 56-amino acid (aa) peptide titling from all viral proteins with 28-aa overlap, which has been published previously2. The T7 library was proliferated using BLT5403 E.coli. Briefly, to keep the diversity of phage-displayed libraries during T7 phage proliferation, the agar plates were used as isolated compartments of the T7 phage library with M9LB/Ampicillin with for 3 to 4 hr. The plates were checked at every 20 minutes after 2.5 hours until the plates were just cleared, so that the final phage titer was high and also maintain the heterogeneity of the phage library. To elute the phage, each plate was covered with 10 ml of Phage Extraction Buffer and placed on a rocking platform at 4°C overnight. The phage library was harvested by tipping the plate slightly. Combine the extraction buffer from all the plates in several Falcon tubes. 0.5 ml chloroform was added to each 50 ml Falcon tube and gently mixed. The tubes were centrifuged at 4500 rpm for 15 min to clarify the lysate and transfer the supernatant to a sterile bottle. DMSO was added to final volume of 10% to the supernatant. The expanded library was aliquot and snapped freeze in liquid nitrogen. Then the expanded library was stored in −80°C freezer. The titer of the amplified library was determined by plaque assay.
Library Quality Control.
The library was sampled and lysed at 95C for 10min. After two rounds of PCR were performed to amplify and index the lysed bacteriophage DNA product. After gel extraction, the size and quality of libraries were assessed on a Bioanalyzer instrument from Agilent. The DNA samples were sent for sequencing. Then, the sequencing was done at Sequencing Facility - Illumina (CCR) using 50bp single round sequenced the DNA base read cycle on Illumina HiSeq 4000 platform (1X50 bp) obtained ~100 million to 200 million reads per lane (around 1,000,000 reads per sample). Total coverage rate is more than 99.99% of designed peptides displayed in T7 phage library.
VirScan PhIP-seq.
We performed phage immunoprecipitation and sequencing by using a slightly modified version of previously published PhIP-Seq protocols (Mohan et al., 2018; Xu et al., 2015). First, 96-deep-well plates were blocked with bovine serum albumin in TBST overnight on a rotator at 4°C. The diluted 1ml bacteriophage library were added in each blocked well. Serum samples, containing 2mg IgG, were mixed with the bacteriophage library. Two technical replicates for each sample were set up. After overnight rotation as the incubation, protein A and protein G Dynabeads were added to each well. With another 4-hours incubation on a rotator at 4°C, with a 96-well magnetic stand, the beads were washed for three times with 400 ml of PhIP-Seq wash buffer. Next, the beads were resuspended in water and adhered phages were lysed at 95°C for 10 min. The blank PBS samples instead of serum were also set up as negative controls on each plate. Two rounds of PCR were performed to amplify and multiplex on the lysed bacteriophage DNA product. After the second round of PCR, PCR products were pooled equimolar amounts of all 192 samples for gel extraction. After gel extraction, the size and quality of libraries were assessed on a Bioanalyzer instrument from Agilent. The DNA samples were aliquot and stored at −80°C until sequencing. Then, the sequencing was done at Sequencing Facility - Illumina (CCR) using 50bp single round sequenced the DNA base read protocol on Illumina HiSeq 4000 platform (1X50 bp) obtained ~100 million to 200 million reads per lane (around 1 million reads per sample). Raw data from Illumina HiSeq 4000 platform was processed by BCL2FASTQ2 for demultiplexing and converting binary base calls and qualities to fastq format. The fastq files were mapped to original virome peptide reference sequences by using Bowtie program. Two sequencing samples were cut off from next-step analysis.as their reads were less than 30,000. The initial informatics and statistical analysis were performed by using a slightly modified version of the previously published technique (8, 10). Briefly, the scatter plots of the –log10(p-values) and a sliding window of width 0.005 from 0 to 2 across the axis of one replicate were used. We found that the distribution of the threshold –log10 (p-value) was centered around a mode of ~2.358 (Suppl. Fig. S1B). We eliminated the 593 hits that came up in at least 3 of the 22 immunoprecipitations with PBS beads alone blank sample. We also filtered out any peptides that were not enriched in at least two of the samples. A threshold number of hits per virus was set based on the size of the virus. If the hit shared a subsequence of at least 7 aa with any hit previously observed in any of the viruses from that sample, that hit was considered to be from a cross-reactive antibody and would be ignored for that virus. The peptide hits, which do not share any linear epitopes, were summed to be strain and species score data. The final score was compared for each virus to the threshold for that virus to determine whether the sample is positive for exposure to that virus. The raw count data were calculated based on – log10(p-value)=2.358 cutoff.
Predicted antigenicity score.
Peptide sequence of HCV1b from PhIP is used to generate the prediction score based on bepipred linear epitope prediction for B-cell.
DNA sample extraction.
DNA extraction from buffy coat or lymphocyte samples was performed following the manufacturer’s instruction (DNeasy Blood & Tissue Kit, Qiagen). The eluted DNA was stored at −20°C for further analysis.
Genotyping and GWAS analysis.
Illumina OmniExpress was applied for the SNP array. Genotyping was performed on 200 ng of genomic DNA using Illumina Infinium HTS Global Screening Arrays on an Illumina iScan system at the Genomic Share Resource of University of Hawaii Cancer Center. The raw genotyping data were processed by Illumina GenomeStudio software 2.0. Quality control was performed using PLINK version 2.0 (http://www.cog-genomics.org/plink/2.0). Samples with a genotyping call rate<95% were removed and 849 individuals remained. SNPs with mono-allelic and MAF (Minor Allele Frequency) <0.05, HWE (Hardy-Weinberg equilibrium) <10–4, and call rate<95%, were excluded. After further considering the ones in high linkage disequilibrium, we obtained roughly 500,000 SNPs for the association study. Therefore, the suggestive genome-wide significant p-value threshold in current study was set at 10−7 as 0.05/5×105 using Bonferroni correction. We identified three and 849 individuals remained. SNPs with mono-allelic and MAF (Minor Allele Frequency) <0.05, HWE (Hardy-Weinberg equilibrium) <10–4, and call rate<95%, were excluded. After further considering the ones in high linkage disequilibrium, we obtained roughly 500,000 SNPs for the association study. Therefore, the suggestive genome-wide significant p-value threshold in current study was set at 10−7 as 0.05/5×105 using Bonferroni correction. We identified three independent loci in three regions were associated with virus feature phenotype at P < 10−7 using PLINK. LocusZoom was used to plot regional signals associated with phenotype with LD and recombination rate calculated from 1000 Genomes. LD structure of signals were further investigated with Haploview. A linear regression with additive model was applied to estimate the genotypic effect of SNP contributed to the disease or phenotype.
ELISA assay.
IgG, IgA and IgG4 levels in serum were measured using human ELISA kits (Bethyl and Thermo Fisher) according to the manufacturers’ instructions. The ELISA results were obtained using a microplate absorbance reader from (Bio-Rad).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Methods.
To identify differences between populations, Xgboost was used to calculate the significance of association of virus exposure traits with HCC versus PC.
XGBoost.
XGBoost is a recently developed software, which implements a machine learning approach of regression and classification using ensemble learning with gradient tree boosting (https://xgboost.readthedocs.io/en/latest/). It is designed to increase the scalability and acceleration of optimized computation for practical use. XGBoost includes three types of parameters: general, booster and task. Each of these types has several hyperparameters, such as maximum depth of the regression trees, number of weak learners, learning rate, regularization, etc, that need to be tuned. We tuned these parameters using a grid search to maximize the mean AUC value computed from 10-fold cross validation on the training data. After finding the optimal values of the hyperparameters, we constructed the model using the following main parameter setting: max_depth =3, eta =0.1, subsample=1, colsample_bytree =0.5, and min_child_weight=1. Then Xgboost was applied to the entire data set with 200 boosting iterations.
To avoid over-fitting, we applied several approaches according to the document of XGBoost. First, we controlled model complexity, and adjust parameters max_depth, min_child_weight and gamma. Second, we added randomness to make training robust to noise and adjust parameters, subsample and colsample. Third, we introduce early stopping to training models to avoid overfitting. It stops the training procedure once the performance on the test dataset has not improved after a fixed number of training iterations. It avoids overfitting by attempting to automatically select the inflection point where performance on the test dataset starts to decrease while performance on the training dataset continues to improve as the model starts to overfit. We set to stop model training at least 20 rounds when no improvement was observed in AUC value was set (early_stopping_rounds=20). The best iteration model was then used as the final model. XGBoost automatically conducts feature selection and calculates the importance for each feature. We tested multiple subsets of the features to achieve the highest AUC and decided to take all the output features for further analysis. For each training and testing sample, we also generated a virus feature score based on the features we selected implemented in the XGBoost classification prediction.
Phylogenetic tree.
The full protein library sequences of 61 feature virus species were obtained from VirScan reference library and stored in fasta format. A multiple sequence alignment tool, Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) was used to generate alignments with default parameters. The phylogenetic trees were then imported to ITOL (https://itol.embl.de/itol.cgi) for further visualization and labelling in normal display mode.
Additional Statistical Methods.
All analyses were conducted in R and GraphPad Prism 7 (La Jolla, CA) used for statistical analyses. Data are presented either as means +/− s.e.m. or medians of continuous values and were analyzed by a two-sided Student’s t-test or Mann-Whitney test used for comparison of two groups, respectively. Fisher’s exact X2 t-test was used to calculate statistical significance of categorical values between groups. Two-tail P values with no more than 0.05 were considered significant. Linear regression was used to determine the correlation between two different variables.
Viral feature level, clinical outcome and ROC curve.
All HCC patients were classified into high, low or below viral feature score groups based on viral feature levels. Kaplan-Meier estimates of overall survival were estimated for each group and compared using the log rank test. Hazard ratios and 95% confidence intervals were calculated using univariate and multivariate Cox proportional hazards models to assess associations between different viral feature level along with several clinical factors. The ability of clinical and viral features in predicting HCC was assessed by computing receiver operating characteristic (ROC) curves using the logistic regression in R. Area under the curve (AUC) values were calculated for these variables. Comparison of ROC curves obtained from different feature sets was assessed by the DeLong test.
Survival modeling and time-dependent ROC assessment.
Cox regression was performed using the R package survival (version 3.1–8). In order to analyze time-dependent ROC curves and corresponding AUC values, the R package timeROC (version 0.4)(Blanche et al., 2013) was used.
Participants and VirScan analysis.
The NCI-UMD (Maryland) cohort consisted of 899 sequentially enrolled participants (clinicaltrials.gov number: NCT0091375) and included 150 hepatocellular carcinoma (HCC) cases, 337 at-risk (AR) individuals and 412 population control (PC) healthy volunteers matched by age and sex (Suppl. Table S1A). The NIDDK cohort (clinicaltrials.gov number; NCT0001971) consisted of 129 AR individuals without HCC and 44 AR individuals with HCC (Table 1).
Supplementary Material
(A) The diagram includes detailed information on the excluded participants from initial enrollment, sample allocation with indicated criteria, QC and final data analysis. (B) Distribution of reproducibility threshold -log10(p-value). The mode of the distribution is 2.358. (C) Examples of the experimental repeats in VirScan showing background signals (blue) of the blank PBS samples (bottom panel) and the hits (green) with significant -log10(p-value) more than 2.358 of serum samples (top panel). (D) DNA and RNA viral compositions before and after immunoprecipitation in VirScan, as library input and Phage-IP, respectively. (E) Phylogenetic composition of common viral taxa (>0.1% abundance) at the viral family level across PC, HR and HCC groups.
For PC, AR and HCC respectively, the viral infection prevalence across all samples shown on bar plot; the count of unique epitopes per sample shown on dot plot and the vertical lines represent the mean values of the count of unique epitopes.
(A) XGBoost performance evaluated by AUC on HCC vs AR with 10x cross-validation. (B) ROC curves for PC vs HCC prediction, as well as for AR vs HCC prediction, using features from HCC vs AR predication. (C) Features selected by HCC vs AR predication was highly overlapped with VES signature. (D) XGBoost performance evaluated by AUC on HCC vs PC with 60/40 train-test split. (E) ROC curves showed the train and test datasets performance. (F) 1000 permutation with the 60/40 train-test split. (G) The selected features and feature importance after 1000 permutation test.
(A) SNP rs12979860 was significantly associated with epitopes in Core and NS5B regions of HCV. Related to Genotyping and GWAS analysis in STAR Method section. Left panel: Heatmap showed the significance of SNP associated with 375 epitopes abundances of HCV Genotype 2 and 3. Core and NS5B regions were highly associated with the genotypes. Right panel: Boxplots represented the difference of the epitope abundance between the genotypes in Core region and NS5B region respectively. (B) Quantile-quantile plot for all tested SNPs represented in the GWAS. (C) Principal component analysis of all samples after quality control in different racial groups.
(A) The diagram includes detailed information on the excluded participants from initial enrollment, sample allocation with indicated criteria, follow-up, QC and final data analysis. (B) Kaplan-Meier survival curves for NIDDK cohorts grouped by VES level. (C) Time-dependent ROC curve analysis of VES performance for landmark timepoints 1–10 years relative to baseline. (D) AUC prediction performance based on univariate and multivariate clinical indicators compared to VES (purple band) for the NIDDK cohort at diagnosis. (E) AUC prediction performance based on univariate and multivariate clinical indicators compared to VES (purple band) for the NCI-UMD cohort.
Supplemental Table S1. Clinical characteristics of NCI-UMD cohort and its viral frequency. Related to Figure 1.
Supplemental Table S1A. Clinical characteristics of 899 patients and volunteers from NCI-UMD cohort. Related to Figure 1.
Supplemental Table S1B. Viral Frequency in 899 patients and volunteers from NCI-UMD cohort. Related to Figure 1.
Supplemental Table S2. Comparison of VirScan with HBV, HCV and HIV from medical charts. Related to Figure 2.
Supplemental Table S3. VES signature and its epitopes. Related to Figure 3.
Supplemental Table S3A. List of viruses in the VES signature. Related to Figure 3.
Supplemental Table S3B. Most frequent epitopes from the VES. Related to Figure 3.
Supplemental Table S4. Association between GWAS SNPs and HCV genotypes and epitopes. Related to Figure 4.
Supplemental Table S4A. The association between SNP in IL28B gene (rs12979860) and HCV genotype 2 & genotype 3. Related to Figure 4.
Supplemental Table S4B. SNP in IL28B gene (rs12979860) association with the epitopes of HCV genotype 2 & 3. Related to Figure 4.
Supplemental Table S4C. The top significant SNPs associated with VES with p-value < 10−7. Related to Figure 4.
Supplemental Table S4D. High Linkage Disequilibrium (LD) SNPs. Related to Figure4.
Supplemental Table S4E. VES linked SNPs were associated with disease risk. Related to Figure 4.
Supplemental Table S5. Assessment of VES Signatures: Predictive Performance and Association with Clinical Outcomes. Related to Figure 5.
Supplemental Table S5A. Univariable and multivariable analyses of factors associated with survival of NCI-UMD cohort. Related to Figure 5.
Supplemental Table S5B. Multivariable Cox regression of HCC diagnosis on the NIDDK cohort predicted with the VES signature at baseline (adjusted for clinical prognostic variables). Related to Figure 5.
Supplemental Table S5C. Prediction performance within HCV+ and HCV- subcohorts. Related to Figure 5.
Highlights.
A host-virome infection map at epitope resolution of 1,072 people in the U.S.
A history of viral exposure predicts liver cancer among at-risk populations
A viral exposure signature identifies liver cancer prior to a clinical diagnosis
GWAS uncovers genetic variants that link viral exposure signature to liver cancer
ACKNOWLEDGEMENTS
We are grateful to all study participants as well as clinicians, nurses and study coordinators who helped with patient enrollment. We also thank Sankar Adhya for advice on phage production. This work was supported in part by grants (ZIA-BC010313, ZIA-BC010876, ZIA BC 010877, ZIA BC 011870) from the Intramural Research Program of the Center for Cancer Research of the National Cancer Institute and from the NIH DDIR Innovation Award. YZ, BT and XW were supported by federal funds from the National Cancer Institute under the contract No. HHSN261200800001E.
Footnotes
DECLARATION OF INTERESTS
JL, WT and XWW are inventors of a U.S. patent application (No. 62/914,138) for the viral exposure signature for detection of early stage hepatocellular carcinoma. SJE is an inventor of the VirScan technology. All other authors declare no Conflicts of Interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ansari MA, Pedergnana V, C LCI, Magri A, Von Delft A, Bonsall D, Chaturvedi N, Bartha I, Smith D, Nicholson G, et al. (2017). Genome-to-genome analysis highlights the effect of the human innate and adaptive immune systems on the hepatitis C virus. Nat Genet 49, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzumanyan A, Reis HM, and Feitelson MA (2013). Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer 13, 123–135. [DOI] [PubMed] [Google Scholar]
- Bansal A, and Heagerty PJ (2019). A comparison of landmark methods and time-dependent ROC methods to evaluate the time-varying performance of prognostic markers for survival outcomes. Diagn Progn Res 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R, Lohmann V, and Penin F. (2013). The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol 11, 482–496. [DOI] [PubMed] [Google Scholar]
- Blanche P, Dartigues JF, and Jacqmin-Gadda H. (2013). Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med 32, 5381–5397. [DOI] [PubMed] [Google Scholar]
- Borglum AD, Demontis D, Grove J, Pallesen J, Hollegaard MV, Pedersen CB, Hedemand A, Mattheisen M, investigators G, Uitterlinden A, et al. (2014). Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol Psychiatry 19, 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno S, Crosignani A, Maisonneuve P, Rossi S, Silini E, and Mondelli MU (2007). Hepatitis c virus genotype 1b as a major risk factor associated with hepatocellular carcinoma in patients with cirrhosis: A seventeen-year prospective cohort study. Hepatology 46, 1350–1356. [DOI] [PubMed] [Google Scholar]
- Cadwell K. (2015). The virome in host health and disease. Immunity 42, 805–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrat F, Fontaine H, Dorival C, Simony M, Diallo A, Hezode C, De Ledinghen V, Larrey D, Haour G, Bronowicki JP, et al. (2019). Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: a prospective cohort study. Lancet. [DOI] [PubMed] [Google Scholar]
- Chang CC, Crane M, Zhou J, Mina M, Post JJ, Cameron BA, Lloyd AR, Jaworowski A, French MA, and Lewin SR (2013). HIV and co-infections. Immunol Rev 254, 114–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MH, You SL, Chen CJ, Liu CJ, Lai MW, Wu TC, Wu SF, Lee CM, Yang SS, Chu HC, et al. (2016). Long-term Effects of Hepatitis B Immunization of Infants in Preventing Liver Cancer. Gastroenterology 151, 472–480 e471. [DOI] [PubMed] [Google Scholar]
- Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox A, et al. (2018). Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science (New York, NY) 359, 926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning HJ, van der Aalst CM, de Jong PA, Scholten ET, Nackaerts K, Heuvelmans MA, Lammers JJ, Weenink C, Yousaf-Khan U, Horeweg N, et al. (2020). Reduced Lung-Cancer Mortality with Volume CT Screening in a Randomized Trial. N Engl J Med 382, 503–513. [DOI] [PubMed] [Google Scholar]
- Echavarria M. (2008). Adenoviruses in immunocompromised hosts. Clin Microbiol Rev 21, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlin BR, Eckhardt BJ, Shu MA, Holmberg SD, and Swan T. (2015). Toward a more accurate estimate of the prevalence of hepatitis C in the United States. Hepatology 62, 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farazi PA, and DePinho RA (2006). Hepatocellular carcinoma pathogenesis: from genes to environment. NatRevCancer 6, 674–687. [DOI] [PubMed] [Google Scholar]
- Foxman EF, and Iwasaki A. (2011). Genome-virome interactions: examining the role of common viral infections in complex disease. Nat Rev Microbiol 9, 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Pozzoli U, Cagliani R, Comi GP, Bresolin N, Clerici M, and Sironi M. (2010). Genome-wide identification of susceptibility alleles for viral infections through a population genetics approach. PLoS Genet 6, e1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M. (1990). Epidemiology of cytomegalovirus infections. Rev Infect Dis 12 Suppl 7, S701–710. [DOI] [PubMed] [Google Scholar]
- Janjua NZ, Chong M, Kuo M, Woods R, Wong J, Yoshida EM, Sherman M, Butt ZA, Samji H, Cook D, et al. (2017). Long-term effect of sustained virological response on hepatocellular carcinoma in patients with hepatitis C in Canada. J Hepatol 66, 504–513. [DOI] [PubMed] [Google Scholar]
- Lichtner M, Cicconi P, Vita S, Cozzi-Lepri A, Galli M, Lo Caputo S, Saracino A, De Luca A, Moioli M, Maggiolo F, et al. (2015). Cytomegalovirus Coinfection Is Associated With an Increased Risk of Severe Non-AIDS-Defining Events in a Large Cohort of HIV-Infected Patients. Journal of Infectious Diseases 211, 178–186. [DOI] [PubMed] [Google Scholar]
- Liu J, Chen X, Wang J, Zhou S, Wang CL, Ye MZ, Wang XY, Song Y, Wang YQ, Zhang LT, et al. (2019a). Biological background of the genomic variations of cf-DNA in healthy individuals. Ann Oncol 30, 464–470. [DOI] [PubMed] [Google Scholar]
- Liu Z, Jiang Y, Yuan H, Fang Q, Cai N, Suo C, Jin L, Zhang T, and Chen X. (2019b). The trends in incidence of primary liver cancer caused by specific etiologies: Results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J Hepatol 70, 674–683. [DOI] [PubMed] [Google Scholar]
- Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM, Roberts LR, and Heimbach JK (2018). Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 68, 723–750. [DOI] [PubMed] [Google Scholar]
- McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, Caporaso NE, Johansson M, Xiao X, Li Y, et al. (2017). Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet 49, 1126–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan D, Wansley DL, Sie BM, Noon MS, Baer AN, Laserson U, and Larman HB (2018). PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat Protoc 13, 1958–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AM, Weiss NS, Beste LA, Su F, Ho SB, Jin GY, Lowy E, Berry K, and Ioannou GN (2018). No Association Between Screening for Hepatocellular Carcinoma and Reduced Cancer-Related Mortality in Patients With Cirrhosis. Gastroenterology 155, 1128–1139 e1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharoah PD, Tsai YY, Ramus SJ, Phelan CM, Goode EL, Lawrenson K, Buckley M, Fridley BL, Tyrer JP, Shen H, et al. (2013). GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet 45, 362–370, 370e361–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts H, Kruszon-Moran D, Ly KN, Hughes E, Iqbal K, Jiles RB, and Holmberg SD (2016). Prevalence of chronic hepatitis B virus (HBV) infection in U.S. households: National Health and Nutrition Examination Survey (NHANES), 1988–2012. Hepatology 63, 388–397. [DOI] [PubMed] [Google Scholar]
- Schulz KF, Altman DG, Moher D, and Group C. (2010). CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 340, c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman M, Bruix J, Porayko M, Tran T, and Committee APG (2012). Screening for hepatocellular carcinoma: the rationale for the American Association for the Study of Liver Diseases recommendations. Hepatology 56, 793–796. [DOI] [PubMed] [Google Scholar]
- Sherman M, Peltekian KM, and Lee C. (1995). Screening for hepatocellular carcinoma in chronic carriers of hepatitis B virus: incidence and prevalence of hepatocellular carcinoma in a North American urban population. Hepatology 22, 432–438. [PubMed] [Google Scholar]
- Shieh Y, Eklund M, Sawaya GF, Black WC, Kramer BS, and Esserman LJ (2016). Population-based screening for cancer: hope and hype. Nat Rev Clin Oncol 13, 550–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slyker JA, Casper C, Tapia K, Richardson B, Bunts L, Huang ML, Maleche-Obimbo E, Nduati R, and John-Stewart G. (2013). Clinical and virologic manifestations of primary Epstein-Barr virus (EBV) infection in Kenyan infants born to HIV-infected women. J Infect Dis 207, 1798–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CT, Smith DK, Schmid DS, Pellett PE, Stewart JA, Klein RS, Mayer K, Vlahov D, Schuman P, Cannon MJ, et al. (2003). Prevalence of and risk factors for viral infections among human immunodeficiency virus (HIV) - Infected and high-risk HIV-uninfected women. Journal of Infectious Diseases 187, 1388–1396. [DOI] [PubMed] [Google Scholar]
- Straus SE, Cohen JI, Tosato G, and Meier J. (1993). NIH conference. Epstein-Barr virus infections: biology, pathogenesis, and management. Ann Intern Med 118, 45–58. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Misaki A, Liang SB, Tachibana A, Hayashi N, Sonobe H, and Ohtsuki Y. (2000). Expression of T-cadherin (CDH13, H-Cadherin) in human brain and its characteristics as a negative growth regulator of epidermal growth factor in neuroblastoma cells. J Neurochem 74, 1489–1497. [DOI] [PubMed] [Google Scholar]
- Tzartzeva K, Obi J, Rich NE, Parikh ND, Marrero JA, Yopp A, Waljee AK, and Singal AG (2018). Surveillance Imaging and Alpha Fetoprotein for Early Detection of Hepatocellular Carcinoma in Patients With Cirrhosis: A Meta-analysis. Gastroenterology 154, 1706–1718 e1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW (2014). The virome in mammalian physiology and disease. Cell 157, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XW, and Thorrgeirsson SS (2014). The Biological and clinical challenge of liver cancer heterogeneity. Hepatic Oncology 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GJ, Kula T, Xu Q, Li MZ, Vernon SD, Ndung’u T, Ruxrungtham K, Sanchez J, Brander C, Chung RT, et al. (2015). Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science (New York, NY) 348, aaa0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) The diagram includes detailed information on the excluded participants from initial enrollment, sample allocation with indicated criteria, QC and final data analysis. (B) Distribution of reproducibility threshold -log10(p-value). The mode of the distribution is 2.358. (C) Examples of the experimental repeats in VirScan showing background signals (blue) of the blank PBS samples (bottom panel) and the hits (green) with significant -log10(p-value) more than 2.358 of serum samples (top panel). (D) DNA and RNA viral compositions before and after immunoprecipitation in VirScan, as library input and Phage-IP, respectively. (E) Phylogenetic composition of common viral taxa (>0.1% abundance) at the viral family level across PC, HR and HCC groups.
For PC, AR and HCC respectively, the viral infection prevalence across all samples shown on bar plot; the count of unique epitopes per sample shown on dot plot and the vertical lines represent the mean values of the count of unique epitopes.
(A) XGBoost performance evaluated by AUC on HCC vs AR with 10x cross-validation. (B) ROC curves for PC vs HCC prediction, as well as for AR vs HCC prediction, using features from HCC vs AR predication. (C) Features selected by HCC vs AR predication was highly overlapped with VES signature. (D) XGBoost performance evaluated by AUC on HCC vs PC with 60/40 train-test split. (E) ROC curves showed the train and test datasets performance. (F) 1000 permutation with the 60/40 train-test split. (G) The selected features and feature importance after 1000 permutation test.
(A) SNP rs12979860 was significantly associated with epitopes in Core and NS5B regions of HCV. Related to Genotyping and GWAS analysis in STAR Method section. Left panel: Heatmap showed the significance of SNP associated with 375 epitopes abundances of HCV Genotype 2 and 3. Core and NS5B regions were highly associated with the genotypes. Right panel: Boxplots represented the difference of the epitope abundance between the genotypes in Core region and NS5B region respectively. (B) Quantile-quantile plot for all tested SNPs represented in the GWAS. (C) Principal component analysis of all samples after quality control in different racial groups.
(A) The diagram includes detailed information on the excluded participants from initial enrollment, sample allocation with indicated criteria, follow-up, QC and final data analysis. (B) Kaplan-Meier survival curves for NIDDK cohorts grouped by VES level. (C) Time-dependent ROC curve analysis of VES performance for landmark timepoints 1–10 years relative to baseline. (D) AUC prediction performance based on univariate and multivariate clinical indicators compared to VES (purple band) for the NIDDK cohort at diagnosis. (E) AUC prediction performance based on univariate and multivariate clinical indicators compared to VES (purple band) for the NCI-UMD cohort.
Supplemental Table S1. Clinical characteristics of NCI-UMD cohort and its viral frequency. Related to Figure 1.
Supplemental Table S1A. Clinical characteristics of 899 patients and volunteers from NCI-UMD cohort. Related to Figure 1.
Supplemental Table S1B. Viral Frequency in 899 patients and volunteers from NCI-UMD cohort. Related to Figure 1.
Supplemental Table S2. Comparison of VirScan with HBV, HCV and HIV from medical charts. Related to Figure 2.
Supplemental Table S3. VES signature and its epitopes. Related to Figure 3.
Supplemental Table S3A. List of viruses in the VES signature. Related to Figure 3.
Supplemental Table S3B. Most frequent epitopes from the VES. Related to Figure 3.
Supplemental Table S4. Association between GWAS SNPs and HCV genotypes and epitopes. Related to Figure 4.
Supplemental Table S4A. The association between SNP in IL28B gene (rs12979860) and HCV genotype 2 & genotype 3. Related to Figure 4.
Supplemental Table S4B. SNP in IL28B gene (rs12979860) association with the epitopes of HCV genotype 2 & 3. Related to Figure 4.
Supplemental Table S4C. The top significant SNPs associated with VES with p-value < 10−7. Related to Figure 4.
Supplemental Table S4D. High Linkage Disequilibrium (LD) SNPs. Related to Figure4.
Supplemental Table S4E. VES linked SNPs were associated with disease risk. Related to Figure 4.
Supplemental Table S5. Assessment of VES Signatures: Predictive Performance and Association with Clinical Outcomes. Related to Figure 5.
Supplemental Table S5A. Univariable and multivariable analyses of factors associated with survival of NCI-UMD cohort. Related to Figure 5.
Supplemental Table S5B. Multivariable Cox regression of HCC diagnosis on the NIDDK cohort predicted with the VES signature at baseline (adjusted for clinical prognostic variables). Related to Figure 5.
Supplemental Table S5C. Prediction performance within HCV+ and HCV- subcohorts. Related to Figure 5.
Data Availability Statement
The viral exposure profiles supporting the current study have not been deposited in a public repository because of the requirement for reporting only the aggregate data by the study protocol but are available from the corresponding author on request.