

Abstract
There has been an increased interest in the use of protein therapeutics, especially antibodies, for the treatment of a variety of diseases due to their high specificity to tissues and biological pathways of interest. However, the use of antibodies can be hindered by physical aggregation, degradation, and diffusion when injected in vivo leading to the need for antibody-releasing depots for the controlled and localized delivery within tissues of interest. Here, we investigated photolabile hydrogel chemistries for creating on-demand and tunable antibody release profiles. Innovative, scalable synthetic procedures were established and applied for fabricating hydrogels with nitrobenzyl (NB) and coumarin (CMR) photolabile crosslinks that responded to clinically relevant doses of long-wavelength UV and short-wavelength visible light. This synthetic procedure includes a route to make a CMR linker possessing two functional handles at the same ring position with water-stable bonds. The photocleavage properties of NB and CMR crosslinked hydrogels were characterized, as well as their potential for translational studies by degradation through pig skin, a good human skin mimic. The mechanism of hydrogel degradation, bulk versus surface eroding, was determined to be dependent on the wavelength of light utilized and the molar absorptivity of the different photolabile linkers, providing a facile means for altering protein release upon hydrogel degradation. Further, the encapsulation and on-demand release of a model monoclonal antibody was demonstrated, highlighting the ability to control antibody release from these hydrogels through the application of light while retaining its bioactivity. In particular, the newly designed CMR hydrogels undergo surface erosion-based protein release using visible light, which is more commonly used clinically. Overall, this work establishes scalable syntheses and relevant pairings of formulation-irradiation conditions for designing on-demand and light-responsive material systems that provide controlled, tunable release of bioactive proteins toward addressing barriers to preclinical translation of light-based materials and ultimately improving therapeutic regimens.
Keywords: biomaterials, drug delivery, photolabile materials, light chemistry, protein delivery, synthetic hydrogels, photolabile linker synthesis
Graphical Abstract
Introduction
Protein therapeutics, including antibodies and cytokines, have become an increasingly popular option for the treatment and prevention of various diseases from cancer to rheumatoid arthritis.1 In the case of antibodies, there is significant potential for them to be utilized in novel treatments for cancers, as well as for communicable and autoimmune diseases.2–4 However, like other large protein therapeutics, antibodies can suffer from physical aggregation, limited diffusion, and degradation, all of which negatively affect the bioactivity and effectiveness of the antibody when administered in vivo.5, 6 Additionally, while antibody-based therapies have been engineered for longer circulations times (e.g., PEGylation),7 localization of antibodies to specific sites where they are most needed (e.g., wounds, tumors) remains a challenge due to the need for intravenous injections for effective delivery. For example, antibodies injected intravenously are known to have issues penetrating into dense tumor tissues, impacting their efficacy for stimulating immune cells within the tumor microenvironment.8, 9 To address this, antibodies can be locally injected but only persist in the site for a few days owing to diffusion,10 where the uneven distribution of the antibody within tumors can lead to resistant mutants.11 Issues related to keeping antibodies and other therapeutic proteins at relevant concentrations when and where needed exist for other indications, such as chronic wounds for which antibody-based therapies are used to modulate inflammation and scar tissue formation,12 as well as for proteins with vaccination potential, which require multiple doses over time.13 Consequently, most antibodies and proteins require repetitive dosing to achieve therapeutic efficacy, leading to decreased patient compliance caused by discomfort and inconvenience.5 Hydrogels can be designed as delivery vehicles to create either sustained or on-demand delivery of antibodies or other proteins toward achieving the desired therapeutic or vaccination strategy. By encapsulating proteins within hydrogel networks, they can be protected from aggregation and degradation in vitro and in vivo preserving their bioactivity.14–16 However, strategies are still needed to maintain high local concentrations over relevant timescales to improve treatment efficiency and reduce off target side effects.
The release of encapsulated antibodies can be achieved through the incorporation of cleavable moieties or crosslinks into the hydrogel, with different levels of temporal control imparted through the selection of the cleavable bond. Materials systems, made from natural or synthetic polymers, have been designed to respond to cues within the body for the programmed and sustained delivery of proteins by taking advantage of the distinct rates of degradation provided by different cleavable motifs.17–21 For example, the use of enzymatically or hydrolytically degradable crosslinks can provide delivery of large proteins over days to weeks.17, 18, 22 In addition to providing a means for antibody release, hydrogel degradation also provides a mechanism for clearance of these materials, mitigating the need for surgical removal after the treatment. These controlled delivery hydrogel formulations have the potential to decrease the administration frequency and the antibody dose required for therapeutic efficacy to be realized, with the possibility for less adverse side effects and improved patient compliance. Although these systems have proven valuable for the controlled release of antibodies, they seldom offer tunability over the rate or timing of protein release after injection, a desirable property for certain therapeutic modalities (e.g., cancer therapies, vaccination regimens). To achieve this type of on-demand control over protein delivery, hydrogels that respond to external stimuli, such as heat, ultrasound, electrical stimulation, or light, have been developed, where the user controls the application of the desired stimulus.15, 23–25 The control over hydrogel degradation afforded by these systems not only allows on-demand delivery of proteins, but also the ability to ask questions about what release profiles are ‘best’ for achieving effectiveness, an opportunity not easily provided by preprogrammed hydrogel platforms. In particular, light-responsive chemistries can be designed such that the light wavelength and intensity used for degradation produces altered protein release profiles without changes to the hydrogel composition.
To fully take advantage of the spatiotemporal control provided by light-responsive hydrogels in creating tunable therapeutic release profiles of proteins, well-characterized photolabile chemistries are needed with a focus toward properties necessary for clinical translation. For example, these properties include scalable syntheses, responsiveness to clinically relevant light doses, modularity for use with a range of therapeutic proteins without significant protein modification, and, depending on the application, orthogonality to other stimuli. Within the biomaterials community, nitrobenzyl (NB) and coumarin (CMR) based moieties, amongst others, have been incorporated into hydrogels to create on-demand therapeutic release profiles. Their extensive use has been facilitated by their biocompatibility, as well as that of their cleavage products, and their ability to respond to cytocompatible doses of both long-wavelength UV and short-wavelength visible light.26–29 For example, in one light-responsive hydrogel platform, NB moieties with complementary absorbances of different light wavelengths were utilized to achieve the biased release of hydrogel-tethered model therapeutics via irradiation wavelength selection.30 The combination of NB and CMR moieties, which respond differently to long-wavelength UV and blue light, also has been utilized to selectively control the release of hydrogel-tethered growth factors by using different wavelengths of light.27 To integrate NB or CMR moieties within hydrogels, chemical modifications to the ring structures are needed to create reactive handles; unfortunately, these modifications can lead to large and often undesirable shifts in the photo-absorption properties of these compounds.28, 30–32 This is a particular challenge when these photolabile moieties are used as crosslinks within hydrogel networks due to the need for integration of two or more functional handles for crosslink formation, where multiple ring substitutions can result in low photocleavage efficiency.31 Consequently, many prior strategies have focused on utilization of different photolabile moieties as cages or tethers to achieve controlled release of molecules covalently linked to the network;28, 33 however, such approaches may face hurdles for clinical translation as the modified molecule would be considered a new therapeutic and would have to be evaluated as such by various international regulatory agencies.34, 35
Toward addressing underlying materials design challenges, an innovative strategy was recently developed for creating bi-functional photolabile crosslinks through a single ring substitution utilizing the Passerini reaction.36 A variety of different photolabile moieties that respond to a range of wavelengths of light (300 – 600 nm) could be incorporated as crosslinks within hydrogels using this pioneering approach, and the resulting materials were shown effective for the selective release of live cells in vitro.36 While useful, the long-term stability of the resulting hydrogels in aqueous microenvironments (e.g., ester linkages) and the scalability of the synthetic protocol (e.g, instability of reactants) may present challenges for the creation of bulk materials that enable triggered release of therapeutics for translational studies. More broadly, no studies to date with NB or CMR based hydrogel protein depots have addressed how degradation of the photolabile linkages is affected by wavelength-specific attenuation and scattering of light when passing through skin, which may lead to varied degradation profiles and presents another hurdle for translation in vivo. To facilitate future translation into different applications, photolabile hydrogel designs are needed that can be degraded with light doses that are used clinically (e.g, applied through skin, 10 – 130 J cm−2 visible (400 – 500 nm) or long wavelength UV light (UVA1, 340 – 400 nm))37–40 and result in degradation and release profiles that can be achieved over different timescales (e.g, minutes, hours, days).
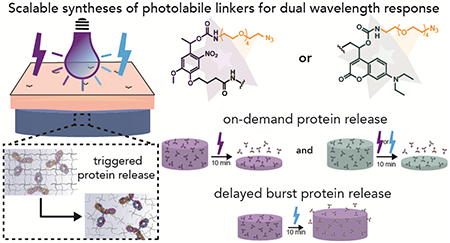
In this work, we have designed photolabile polyethylene glycol) (PEG)-based hydrogels for the controlled release of an encapsulated bioactive antibody to address barriers to translation of light responsive materials and toward realizing single-injection treatments or vaccination strategies (Figure 1). First, a method was developed for synthesizing CMR-azide, in addition to NB-azide, photolabile linkers in a scalable manner while achieving desirable visible light absorbance and integrating photolabile carbamate bonds that are resistant to hydrolysis.41 Next, the degradation and protein release properties of NB and CMR crosslinked hydrogels in response to blue and long-wavelength UV light were compared. To investigate the utility of photodegradable hydrogels as therapeutic delivery vehicles, hydrogel degradation timescales were determined using in situ rheology and by monitoring volume for equilibrium swollen hydrogels irradiated through excised pig skin, which is a good model for human skin owing in part to its similar light penetration properties.42 These studies were designed to provide insights into how both photolabile moiety and light selection influence the mode and rate of hydrogel degradation and how light-responsiveness is altered through the skin layer toward bridging between traditional in vitro and in vivo studies. After the light-responsive properties of the different hydrogel formulations were characterized, we demonstrated the encapsulation of the bioactive antibody PANG and its controlled release in response to light-driven hydrogel degradation. PANG, a non-glycosylated (NG) monoclonal antibody (mAb) against the anthrax protective antigen (PA), was developed by Fraunhofer USA Center for Molecular Biotechnology (FhCMB) and has been shown to provide full protection against an anthrax spore challenge in non-human primates.43 PANG is currently being examined for the treatment of anthrax intoxication and is used here as a model antibody for which assays related to detection and bioactivity are well established.18, 43 The light-responsive systems described herein are readily extendable for the delivery of many large proteins of interest for the treatment of or vaccination against cancers, autoimmune diseases, or communicable illnesses.
Figure 1. Overview of approach.
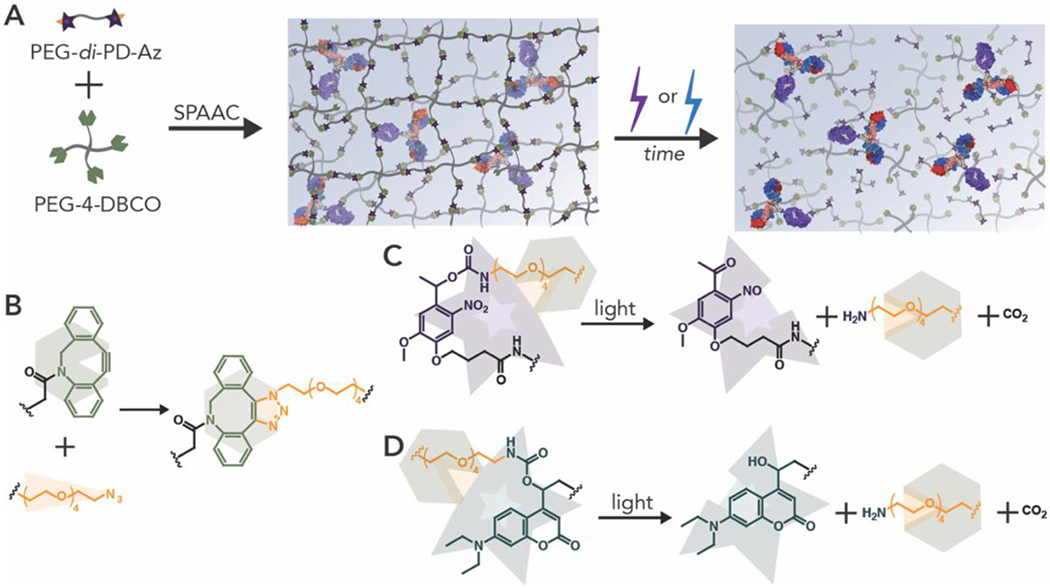
(A) Hydrogels were formed in the presence of PANG, a model monoclonal antibody, and subsequently degraded by irradiation with cytocompatible doses of long-wavelength UV (365 nm) or blue light (400-500 nm) to release an encapsulated bioactive antibody. (B) Hydrogels were fonned using biologically orthogonal strain promoted azide-alkyne cycloaddition (SPAAC) click reactions. (C) Nitrobenzyl or (D) coumarin groups synthesized using scalable synthetic approaches (Scheme 1 and Scheme 2) were incorporated as crosslinks within the hydrogel network to impart light-responsive properties.
Materials and Methods
Materials and General Characterization.
General organic reagents were purchased from commercial sources and used as received unless otherwise stated. Four-arm PEG amine (Mn = 10 kDa) and linear PEG-bis-amine (Mn = 3.4 kE)a) were purchased from JenKem Technology USA Inc. (Allen, TX). Heterofunctional azide-PEG4-amine was purchased from BroadPharm (San Diego, CA). Deionized (DI) water (18 MΩ-cm) was purified onsite using a Milli-DI water purification system (Millipore Sigma, Burlington, MA). All other specialty reagents used are specified below.
All flash chromatography was done on a CombiFlash Rf flash column using standard RediSep Rf silica columns (Teledyne ISCO, Lincoln, NE). 1H and 13C NMR spectrum were collected with a Bruker NMR spectrometer (Bruker Daltonics, Billerica, MA) under standard quantitative conditions (small molecules at 600 MHz and 16 scans for 1H and 101 MHz for 13C, polymers at 600 MHz and 128 scans for 1H). Chemical shifts for protons are reported in parts per million and data are represented as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, dd = doublet of doublets, h = heptet), coupling constants in Hertz, integration. Mass spectral data for small molecules at a concentration of 0.1 mg/mL was obtained using a ThermoFisher Q-Extractive Orbitrap (ThermoFisher Scientific, Waltham, MA). NMRs for all synthesized molecules can be found in the SI.
Synthesis of nitrobenzyl azide.
NB-azide was synthesized by a two-step synthetic route (Scheme 1). First, compound 1 was synthesized using previously published methods.41 To a nitrogen purged oven dried 25 mL round bottom flask was added compound 1 (133 mg, 0.27 mmol, 1 eq) followed by anhydrous dichloromethane (DCM) (2 mL). To the solution was then added azido-tetraethyleneglycol-amine (129 mg, 0.5 mmol, 1.8 eq). The reaction was left to stir overnight. The following day, the solution was concentrated by rotary evaporation and directly purified via flash chromatography using a gradient of 0% to 75% ethyl acetate in hexanes. The product containing fractions were combined and dried via rotary evaporation to produce compound 2 as a yellow oil (124 mg, 74%). 1H NMR (600 MHz, deuterated dimethyl sulfoxide (DMSO-d6)) δ 7.58 (s, 1H), 7.40 (t, J = 5.8 Hz, 1H), 7.14 (s, 1H), 6.12 (q, J = 6.5 Hz, 1H), 4.14 – 4.03 (m, 4H), 3.92 (s, 3H), 3.65 – 3.35 (m, 18H), 3.09 (dd, J = 6.0, 2.3 Hz, 2H), 2.46 (t, J = 7.3 Hz, 2H), 2.05 – 1.93 (m, 2H), 1.53 (d, J = 6.5 Hz, 3H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 172.85, 155.67, 154.11, 147.14, 139.66, 133.95, 126.68, 116.38, 109.01, 108.98, 70.28, 70.26, 70.24, 70.18, 70.16, 69.97, 69.71, 69.56, 68.37, 67.63, 60.37, 56.68, 55.36, 50.47, 30.51, 24.48, 22.35, 14.54. Mass: C26H41N5O12 [M+H]+ Calculated: 616.64, Found: 616.28.
Scheme 1. Synthetic route for small molecule nitrohemyl-carhamate-PEG4-azide (NB-azide).
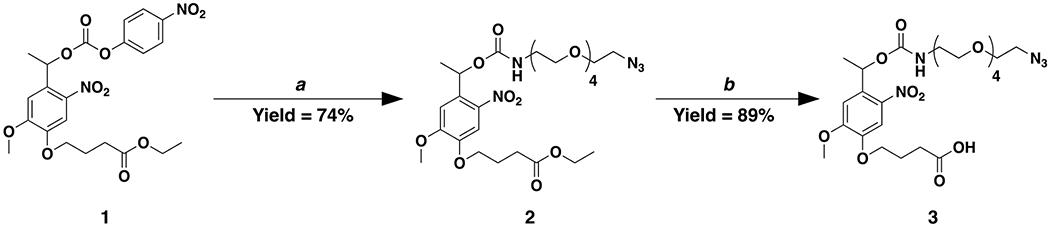
Reagents and conditions as follows: a) azido-tetraethyleneglycol-amine in anhydrous DCM at room temperature and b) LiOH in 2:1 THF to DI water at room temperature.
To a 50 mL round bottom flask was added compound 2 (1.37 g, 2.22 mmol, 1 eq) followed by a 2:1 mixture of tetrahydrofuran (THF) (16.7 mL) and DI water (8.3 mL). To the stirred solution was added a 1 M solution of lithium hydroxide (LiOH) (64 mg, 2.67 mmol, 1.2 eq), and the solution was left to react overnight. The next day, 1 M HCl (50 mL) was added to the solution. Then the aqueous solution was transferred to an extraction vessel followed by the addition of DCM (100 mL). The biphasic solution was mixed thoroughly, and upon reforming two phases, the organic phase (bottom phase) was extracted and set aside. The extraction process was repeated two additional times with DCM (100 mL) and the organic phases were collected and set aside both times. The collected organic phases were combined, dried over sodium sulfate (Na2SO4), filtered, and concentrated by rotary evaporation. The residue was subjected to column chromatography with a gradient of 0% to 5% methanol in DCM. The product containing fractions were combined and dried via rotary evaporation to produce compound 3 as a yellow oil (1.15 g, 89%). 1H NMR (600 MHz, DMSO-d6) δ 12.16 (s, 1H), 7.59 (s, 1H), 7.40 (t, J = 5.8 Hz, 1H), 7.14 (s, 1H), 6.12 (q, J = 6.4 Hz, 1H), 4.08 (t, J = 6.5 Hz, 2H), 3.92 (s, 3H), 3.65 – 3.36 (m, 19H), 3.09 (qd, J = 5.9, 2.9 Hz, 2H), 2.39 (t, J = 7.3 Hz, 2H), 1.97 (h, J = 6.9 Hz, 2H), 1.53 (d, J = 6.5 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 174.42, 155.68, 154.12, 147.19, 139.68, 133.90, 126.63, 116.25, 109.04, 109.00, 70.27, 70.26, 70.24, 70.18, 70.16, 69.97, 69.70, 69.57, 68.44, 67.63, 56.69, 55.36, 50.47, 30.41, 24.49, 22.36. Mass: C24H37N5O12 [M+H]+ Calculated: 588.58, Found: 588.63.
Synthesis of coumarin azide.
CMR-azide was synthesized by a multi-step synthetic route (Scheme 2). First, compound 4 was synthesized based on previously published methods.44 To an oven dried nitrogen purged 250 mL round bottom flask was added coumarin 7-diethylamino-4-methylcoumarin (11.6 g, 50 mmol, 1 eq) followed by dimethylformamide (DMF) (20 mL). To the solution was added dimethylformamide dimethyl acetal (10 mL, 75 mmol, 1.5 eq) followed by heating at reflux for 24 h. The following day, the reaction was cooled to room temperature and diluted with DCM (300 mL). The organic solution was transferred to an extraction vessel with an aqueous solution of saturated sodium bicarbonate (150 mL), forming a biphasic solution. The biphasic solution was mixed thoroughly, and upon reforming two phases, the organic phase (bottom phase) was collected while the aqueous phase was discarded. The organic phase was again added to the extraction vessel and the extraction process was repeated with DI water (150 mL) three additional times. Finally, the organic phase was collected, dried over Na2SO4, and filtered. Upon concentrating the organic solution by rotary evaporation, a yellow solid formed. The solid was triturated with minimal acetone and filtered, producing 10.8 g of the dimethylamino coumarin intermediate. The solid was collected and used directly in the next step without further purification.
Scheme 2. Synthetic route for small molecule coumarin-carbamate-PEG4-azide (CMR-azide).
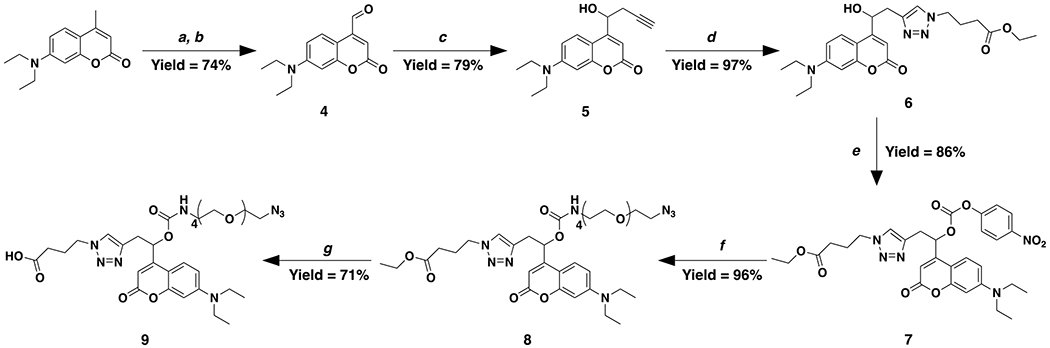
Reagents and conditions as follows: a) dimethylformamide dimethyl acetal in DMF under reflux; b) sodium periodate in 1:1 THF to DI water at room temperature; c) propargyl zinc bromide in THF at 0 °C; d) ethyl 4-azidobutanoate, CuSO4, and sodium ascorbate in 9:1 methanol to DI water at room temperature; e) 4-nitrophenyl chloroformate, anhydrous pyridine, and DMAP in anhydrous DCM at 0 °C; f) azido-tetraethyleneglycol-amine, anhydrous pyridine, and DMAP in anhydrous DCM at 0 °C to room temperature; and g) LiOH in 2:1 THF to DI water at room temperature.
To a 1 L round bottom flask was added the intermediate coumarin (10.8 g, 36.8 mmol, 1 eq) followed by the addition of a 1:1 mixture of THF to water (500 mL). Next, sodium periodate (NaIO4) (24.2 g, 113 mmol, 3 eq) was added slowly and the reaction was allowed to proceed for 2 h. Once finished, ethyl acetate (300 mL) was added to the reaction and the solution was then subsequently transferred to an extraction vessel. To the organic solution was added an aqueous solution of saturated sodium bicarbonate (200 mL), forming a biphasic solution. The biphasic solution was mixed thoroughly, and upon reforming two phases, the aqueous phase (bottom phase) was discarded while retaining the organic phase in the extraction vessel. The extraction process was repeated an additional time with saturated sodium bicarbonate (200 mL) followed by brine (200 mL). The aqueous phases were all discarded. The organic phase was then collected, dried over Na2SO4, filtered, and concentrated via rotary evaporation. Once concentrated to an oil, the residue was dissolved in DCM (25 mL) and concentrated via rotary evaporation to coevaporate off residual ethyl acetate producing compound 4 (9 g, 74% over two steps) as a red oil that solidified upon standing. 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 8.19 (d, J = 9.2 Hz, 1H), 6.84 – 6.44 (m, 3H), 3.44 (q, J = 7.0 Hz, 4H), 1.12 (t, J = 7.0 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 194.68, 161.55, 157.31, 150.98, 144.22, 127.07, 116.45, 109.89, 103.70, 97.45, 68.94, 66.02, 62.30, 44.47, 12.77. Mass: C14H15NO3 [M+H]+ Calculated: 246.28, Found: 246.13.
To an oven dried nitrogen purged 100 mL round bottom flask was added zinc dust (4 g, 61.2 mmol, 5 eq) followed by anhydrous THF (20 mL). To the solution, a catalytic amount of trimethyl chlorosilane (388 μL, 3.1 mmol, 0.25 eq) was added and the reaction was stirred at room temperature for 30 min. After 30 min the round bottom flask was placed in an ice bath to cool. In a separate 50 mL oven dried and nitrogen purged round bottom flask was added 80% propargyl bromide in toluene (3.32 mL, 30.6 mmol, 2.5 eq) followed by anhydrous THF (20 mL). The bromide solution was then added dropwise to the cooled zinc solution over the course of 30 min. The reaction was allowed to proceed over an ice bath for an hour upon full addition of the bromide. Once complete, compound 4 (3 g, 12.2 mmol, 1 eq) dissolved in anhydrous THF (10 mL) was added dropwise to the zinc solution. The reaction proceeded for 1 h over an ice bath until the reaction was finished as determined by thin-layer chromatography (TLC) (1:1 hexanes to ethyl acetate, Rf = 0.32). To quench the reaction, 5 mL of concentrated ammonium chloride was added, and the solution was further diluted with 1 M HCl (150 mL). The aqueous solution was transferred to an extraction vessel to which ethyl acetate (100 mL) was added. The biphasic solution was mixed thoroughly; upon reforming two phases, the aqueous phase (bottom phase) and the organic phase (top phase) were collected separately and set aside for further use. The aqueous phase was again added to the extraction vessel and the extraction process was performed two additional times with ethyl acetate (100 mL). All the collected organic phases were then combined, dried over Na2SO4, filtered, and concentrated via rotary evaporation. The residue was purified by column chromatography with a gradient from 0% to 40% ethyl acetate in hexanes. The product containing fractions were collected and dried via rotary evaporation to give compound 5 as a red oil that solidified upon standing (2.75 g, 79%). 1H NMR (600 MHz, DMSO-d6) δ 7.50 (d, J = 9.1 Hz, 1H), 6.59 (dd, J = 9.1, 2.6 Hz, 1H), 6.44 (d, J = 2.6 Hz, 1H), 6.01 (d, J = 0.9 Hz, 1H), 5.76 (d, J = 4.6 Hz, 1H), 4.96 (q, J = 5.0 Hz, 1H), 3.34 (q, J = 7.1 Hz, 4H), 2.74 (t, J = 2.6 Hz, 1H), 2.54 (dddd, J = 70.6, 17.0, 5.7, 2.7 Hz, 2H), 1.04 (t, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 161.55, 157.98, 156.48, 150.58, 126.19, 109.06, 106.06, 105.66, 97.51, 81.22, 73.84, 67.10, 44.40, 27.52, 12.81. Mass: C17H19NO3 [M+H]+ Calculated: 286.34, Found: 286.45.
To a 100 mL nitrogen purged round bottom flask was added compound 5 (1.9 g, 6.7 mmol, 1 eq) followed by a 9:1 mixture of methanol (47.5 mL) and deionized water (2.5 mL). To the stirring solution was added ethyl 4-azidobutanoate (1.3 g, 8.3 mmol, 1.25 eq) synthesized using a previously reported procedure.45 Next was added copper sulfate (CuSO4) (482 mg, 3.0 mmol, 0.45 eq) and the reaction was stirred for 10 min at room temperature. Finally, sodium ascorbate (1.65 g, 8.33 mmol, 1.25 eq) was added and the reaction was allowed to mix overnight. Upon completion of the reaction as determined by TLC (100% ethyl acetate, Rf = 0.44), the reaction was diluted with DCM (150 mL) and water (100 mL). The biphasic solution was then transferred to an extraction vessel, mixed thoroughly, and upon reforming two phases, the organic phase (bottom phase) was extracted and set aside. The aqueous phase (top phase) remained in the extraction vessel. The aqueous phase was washed two additional times with DCM (100 mL) and each time the organic phase was collected and set aside. Finally, all of the collected organic phases were combined and washed with a 0.1 N aqueous solution of 1:2 ethylenediaminetetraacetic acid (EDTA) and NaOH (100 mL) to remove the residual copper. The organic phase was then collected, dried over Na2SO4, and concentrated via rotary evaporation followed by purification by column chromatography with a gradient of 0% to 100% ethyl acetate in hexanes. The fractions containing the product were collected and dried via rotary evaporation to produce compound 6 as a red solid (2.87 g, 97%). 1H NMR (600 MHz, DMSO-d6) δ 7.91 (s, 1H), 7.60 (d, J = 9.1 Hz, 1H), 6.71 (dd, J = 9.1, 2.6 Hz, 1H), 6.53 (d, J = 2.6 Hz, 1H), 6.02 (d, J = 0.9 Hz, 1H), 5.70 (d, J = 4.8 Hz, 1H), 5.14 (dt, J = 8.5, 4.8 Hz, 1H), 4.35 (t, J = 7.0 Hz, 2H), 4.07 (q, J = 7.1 Hz, 2H), 3.44 (q, J = 7.1 Hz, 4H), 3.13 – 2.88 (m, 2H), 2.27 (t, J = 7.4 Hz, 2H), 2.12 – 1.95 (m, 2H), 1.16 (dt, J = 32.8, 7.1 Hz, 9H). 13C NMR (151 MHz, DMSO-d6) δ 172.50, 161.59, 159.39, 156.46, 150.54, 143.67, 125.99, 123.72, 109.10, 106.11, 104.98, 97.47, 68.33, 60.43, 48.79, 44.41, 34.27, 30.76, 25.74, 14.54, 12.82. Mass: C23H30N4O5 [M+2H]+ Calculated: 444.52, Found: 444.08.
To an oven dried nitrogen purged 250 mL round bottom flask was added compound 6 (3 g, 6.8 mmol, 1 eq) followed by anhydrous DCM (75 mL), anhydrous pyridine (1.2 mL, 13.6 mmol, 2 eq), and a catalytic amount of 4-dimethylaminopyridine (DMAP) (82.7 mg, 0.68 mmol, 0.1 eq). The solution was cooled in an ice bath for 15 min. Next, 4-nitrophenyl chloroformate (1.37 g, 6.8 mmol, 1.2 eq) was added and the reaction was left to react in an ice bath for 4 h. Upon completion of the reaction as determined by TLC (100% ethyl acetate, Rf = 0.5), the reaction was quenched with saturated sodium bicarbonate (100 mL). The biphasic solution was then transferred to an extraction vessel, mixed thoroughly, and upon reforming two phases, the organic phase (bottom phase) was extracted and set aside. The aqueous phase (top phase) remained in the extraction vessel. The aqueous phase was washed two additional times with DCM (150 mL) and each time the organic phase was collected and set aside. Finally, all the organic phases were combined, dried over Na2SO4, filtered, concentrated via rotary evaporation, and purified by flash chromatography with a gradient of 0% to 100% ethyl acetate in hexanes. The fractions containing the product were collected and dried via rotary evaporation producing compound 7 as an orange oil (2.92 g, 86%). 1H NMR (600 MHz, DMSO-d6) δ 8.40 – 7.45 (m, 5H), 6.76 – 5.90 (m, 4H), 4.36 (td, J = 7.0, 2.9 Hz, 2H), 4.12 – 3.92 (m, 2H), 3.63 – 3.35 (m, 6H), 2.28 – 1.95 (m, 4H), 1.15 (dt, J = 20.1, 7.1 Hz, 9H). 13C NMR (151 MHz, DMSO-d6) δ 172.44, 161.22, 160.91, 156.55, 155.45, 153.85, 152.76, 151.52, 150.95, 150.91, 145.78, 142.67, 141.36, 126.20, 125.90, 125.78, 124.15, 123.93, 122.97, 109.41, 109.26, 106.87, 105.70, 105.39, 105.26, 97.60, 97.56, 75.71, 60.42, 60.40, 48.91, 44.47, 33.42, 31.08, 30.70, 30.66, 25.71, 25.69, 14.53, 14.51, 12.78. Mass: C30H33N5O9 [M+2H]+ Calculated: 609.62, Found: 609.23.
To an oven dried nitrogen purged 250 mL round bottom flask was added compound 7 (3.3 g, mmol, 1 eq) followed by anhydrous DCM (40 mL). The solution was cooled in an ice bath followed by slow addition of azido-tetraethyleneglycol-amine (1.43 g, 5.4 mmol, 1 eq), anhydrous pyridine (962 μL, 10.9 mmol, 2 eq), and a catalytic amount of DMAP (66.4 mg, 0.54 mmol, 0.1 eq). The reaction was allowed to warm to room temperature and was left to proceed overnight. Upon completion of the reaction, as determined by TLC, the solution was diluted with DCM (150 mL) and transferred to an extraction vessel. To the extraction vessel was added 1 M HCl (100 mL), the contents were thoroughly mixed, and upon reforming a biphasic solution, the organic phase (bottom phase) was extracted, dried over Na2SO4, filtered, and concentrated via rotary evaporation. The red residue was column purified using a gradient of 0% to 5% methanol in DCM. The fractions containing the product were collected and dried via rotary evaporation producing compound 8 as an orange oil (3.82 g, 96%). 1H NMR (600 MHz, DMSO-d6) δ 8.26 – 6.85 (m, 4H), 6.71 (dd, J = 9.1, 2.6 Hz, 1H), 6.54 (d, J = 2.5 Hz, 1H), 6.08 (dd, J = 7.2, 4.9 Hz, 1H), 5.85 (s, 1H), 4.34 (t, J = 7.0 Hz, 2H), 4.06 (q, J = 7.1 Hz, 2H), 3.71 – 3.35 (m, 22H), 3.27 – 3.06 (m, 4H), 2.25 (t, J = 7.4 Hz, 2H), 2.01 (p, J = 7.2 Hz, 2H), 1.16 (dt, J = 30.6, 7.1 Hz, 9H). 13C NMR (151 MHz, DMSO-d6) δ 172.46, 164.46, 161.17, 156.47, 155.44, 150.81, 142.21, 126.64, 125.81, 123.83, 116.28, 109.31, 105.62, 104.60, 97.52, 70.27, 70.25, 70.23, 70.18, 70.16, 70.05, 69.70, 69.49, 60.42, 50.47, 48.89, 44.46, 31.53, 30.68, 25.70, 14.52, 12.78. Mass: C34H50N8O10 [M+2H]+ Calculated: 732.82, Found: 732.53.
To a nitrogen purged 50 mL round bottom flask was added compound 8 (3.26 mmol, 1 eq) followed by a 2:1 mixture of THF (30 mL) to water (15 mL). Next, LiOH (3.92 mmol, 1.2 eq) was added and the reaction was left to react overnight. The next day cold 1 M HCl (50 mL) was added to the reaction vessel. The solution was extracted three times with DCM (100 mL) with the organic phase (bottom phase) collected and set aside each time. The organic phases were combined, dried over Na2SO4, filtered, and concentrated via rotary evaporation. The residue was purified by column chromatography with a gradient of 0 to 5% methanol in DCM. The fractions containing the product were collected and dried via rotary evaporation producing compound 9 as an orange oil (1.61 g, 71%). 1H NMR (600 MHz, DMSO-d6) δ 12.21 (s, 1H), 7.98 (s, 1H), 7.65 – 7.41 (m, 2H), 6.71 (dd, J = 9.1, 2.5 Hz, 1H), 6.54 (d, J = 2.6 Hz, 1H), 6.14 – 5.99 (m, 1H), 5.85 (s, 1H), 4.34 (t, J = 7.0 Hz, 2H), 3.70 – 3.35 (m, 23H), 3.27 – 3.04 (m, 4H), 2.18 (t, J = 7.3 Hz, 2H), 1.98 (h, J = 7.3, 6.6 Hz, 2H), 1.14 (t, J = 7.0 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 174.04, 161.20, 156.46, 155.44, 150.80, 142.20, 125.80, 123.79, 109.31, 105.61, 104.54, 97.51, 70.25, 55.37, 50.47, 49.01, 44.47, 30.75, 25.81, 12.78. Mass: C32H46N8O10 [M+2H]+ Calculated: 702.77, Found: 702.65.
Synthesis of PEG-di-nitrobenzyl-azide and PEG-di-coumarin-azide.
PEG-di-nitrobenzylazide (PEG-di-NB-Az) and PEG-di-coumarin-azide (PEG-di-CMR-Az) were synthesized following the same protocol. The photolabile molecule to be coupled (0.132 mmol NB-azide (compound 2) or CMR-azide (compound 9), 2.2 eq) was dissolved in anhydrous DMF followed by the addition of 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (0.12 mmol, 2.1 eq) and diisopropylethylamine (DIPEA) (0.27 mmol, 5 eq). To this mixture PEG-di-amine (3.4 kDa, 0.03 mmol, 1 eq) dissolved in anhydrous DMF was added. The reaction was stirred overnight at room temperature followed by precipitation in cold ethyl ether. The product was recovered by filtration and washed with cold ethyl ether twice more. The polymer was dried overnight at room temperature, dissolved in DI water (2 mL), and dialyzed (Spectra/Por 7 dialysis tubing, 1000 Da MWCO, Repligen) against DI water (1000 mL, 3 changes over 20 h) followed by lyophilization. The functionality of the resulting polymer was confirmed using 1H NMR. PEG-di-NB-Az (functionality = 89%) 1H NMR (600 MHz, DMSO-d6) δ 1H NMR (600 MHz, DMSO-d6) δ 7.92 (t, J = 5.7 Hz, 1H), 7.56 (s, 1H), 7.40 (t, J = 5.8 Hz, 1H), 7.13 (s, 1H), 6.11 (q, J = 6.4 Hz, 1H), 4.04 (t, J = 6.5 Hz, 2H), 3.91 (s, 3H), 3.68 – 3.37 (m, 176H), 3.20 (q, J = 5.8 Hz, 3H), 3.15 – 3.00 (m, 2H), 2.24 (t, J = 7.4 Hz, 2H), 1.94 (p, J = 6.8 Hz, 2H), 1.52 (d, J = 6.4 Hz, 3H). PEG-di-CMR-Az (functionality = 91%) 1H NMR (600 MHz, DMSO-d6) δ 4.30 (q, J = 7.0, 5.6 Hz, 1H), 3.64 – 3.56 (m, 1H), 3.50 (s, 10H), 3.51 – 3.47 (m, 3H), 3.46 – 3.35 (m, 4H), 3.33 (s, 17H), 3.25 – 3.17 (m, 1H), 3.17 – 3.08 (m, 1H), 2.06 (t, J = 6.9 Hz, 1H), 1.97 (p, J = 6.9 Hz, 1H), 1.13 (t, J = 7.0 Hz, 2H).
Synthesis of dibenzocyclooctyne end-functionalized four-arm PEG.
Four-arm dibenzocyclooctyne (DBCO) end-functionalized PEG (PEG-4-DBCO) was synthesized following a similar protocol as outlined above. Briefly, to a solution of dibenzocyclooctyne-acid (DBCO-C6-Acid, 0.088 mmol, 2.2 eq), HATU (0.08 mmol, 2.1 eq), and DIPEA (0.18 mmol, 5 eq) dissolved in anhydrous DMF was added amine end-functionalized 4-arm PEG (0.01 mmol, 1 eq) dissolved in anhydrous DMF. The reaction was stirred overnight at room temperature, precipitated in cold ethyl ether, recovered by filtration, and washed three more times with cold ethyl ether. The resulting polymer precipitate was dried overnight at room temperature and the functionality of the resulting polymer was confirmed using 1H NMR. PEG-4-DBCO (functionality = 86%) 1H NMR (600 MHz, DMSO-d6) δ 7.52 – 7.43 (m, 3H), 7.36 (dtd, J = 24.0, 7.4, 1.4 Hz, 2H), 3.62 (dd, J = 8.3, 2.8 Hz, 2H), 3.49 (d, J = 5.2 Hz, 34H), 3.33 (s, 57H), 3.31 (s, 2H), 3.10 (q, J = 5.9 Hz, 2H), 1.84 (t, J = 7.0 Hz, 2H), 1.31 – 1.23 (m, 2H), 1.24 (s, 2H), 1.16 (tdd, J = 11.9, 8.5, 6.2 Hz, 2H).
Formation and characterization of photodegradable SPAAC hydrogels.
Hydrogels were formed through a SPAAC reaction between the azide on the linear photolabile PEG and the DBCO on the four arm PEG monomers. For characterization of photodegradation, hydrogels were formed to contain 10 wt% of polymer with functional groups on stoichiometry (20 mM final DBCO concentration and 20 mM final NB-Az or CMR-Az concentration). The mechanical properties of hydrogels formed with either PEG-di-NB-Az or PEG-di-CMR-Az were determined on a shear rheometer (ARG2, TA Instruments) as described in detail below. Minor adjustments to the formulations were made to account for any differences that arose due to differences in the precursor functionality: final formulations of hydrogels with NB-Az were formed at 11.0 wt% and with CMR-Az were formed at 11.3 wt%.
For rheological characterization, the rheometer was fitted with an 8 mm flat plate geometry and measurements were conducted at a gap height of 120 μm at an applied strain of 1% and a frequency of 2 rad s-1, which is within the linear viscoelastic (LVE) region for these hydrogels. Hydrogel precursor solutions were prepared at a total volume of 14 μL, 6.2 μL of which was pipetted onto the quartz plate of a UV-vis curing accessory on the rheometer. The geometry was lowered to a 120 μm gap height and mineral oil was placed around the outside of the sample to ensure the hydrogel remained hydrated during the experiment. Dynamic time sweep measurements were conducted over a 30 min polymerization time for all hydrogels, over which time complete gelation of all formulations was achieved. After 30 min the hydrogels were irradiated with light, supplied through an Exfo Omnicure Series 2000 light source for 15 min (Excelitas Technologies, Waltham, MA). The Omnicure is a high pressure mercury arc lamp with bands of light emitted between 300 – 600 nm, where certain bands of interest can be selected for using bandpass filters. Hydrogels were degraded with either a bandpass filter centered at 365 nm (I0 = 4 mW cm−2) or a 400 - 500 nm bandpass filter (I0 = 6.7 mW cm−2). The reported intensity is the integrated intensity from 400 to 500 nm, which primarily includes the 405 and 435 nm high pressure mercury emission bands; toward accurate assessment, the integrated intensity between 400-500 nm was measured with calibration settings on the radiometer at 405 nm and 435 nm, and those measurements of the intensity were averaged.
Determination of photodegradation effective rate constants.
The photodegradation rates of hydrogels containing NB or CMR crosslinks were determined using measurements of the storage modulus over time upon light irradiation of hydrogels that had been formed in situ on the rheometer. Using rubber elasticity theory, the storage modulus of hydrogels can be expressed as a function of the crosslinking density of equilibrium swollen hydrogels (ρx) and the volumetric swelling ratio (Q),
where, R is the universal gas constant and T is the temperature.19, 46 The cleavage of photolabile linkages (PLs) contained within the hydrogel crosslinks can be directly related to the rate of degradation of the hydrogel due to crosslink cleavage. This allows for the kinetics of photodegradation to be modeled as first order process.
Here, the concentration of PL is defined as [PL] and the effective first order degradation rate constant for the hydrogel is defined as keff. This equation can be separated and integrated over time, t = 0 to t = t, and from the initial concentration of PLs to the concentration of PLs to yield,
Looking at rubber elasticity theory and based on the network structure, a correlation between the hydrogel modulus, crosslink density, and first order degradation rate constant can be obtained as [PL] is directly correlated to the crosslink density for these hydrogels.
Using this generalization, the first order degradation rate for the NB or CMR moiety can be determined by utilizing the change in storage modulus over time upon irradiation.
The factor of two arises due two photolabile linkages being present per crosslink, where cleavage of either of these moieties leads to a decrease in the storage modulus and thus the crosslink density of the network.47 To ensure data remained within the linear region of the photodegradation process, the first two minutes of degradation were fit for all samples. The first order rate constant is a function of the light intensity used for degradation, the quantum yield (φ), and molar absorptivity (ε), and, to allow for ease of comparison, the rate constants were normalized by the incident light intensity (I0) applied for photodegradation.
Where, Na is Avogadro’s number, h is Plank’s constant, and v is the frequency of light.
Molar absorptivity of NB and CMR moieties.
The UV-Vis absorbance profile for small molecule NB-azide and CMR-azide were collected at 0.1 mM using a NanoDrop™ 2000/2000c Spectrophotometer (ThermoFisher, Waltham, MA). NB-azide and CMR-azide small molecules were first solubilized in DMSO at 20 mM then diluted to 0.1 mM in lx PBS. Molar absorptivity was determined at different wavelengths by transforming the data using the Beer-Lambert law,
where A is the absorbance, ε is the molar absorptivity, l is the path length, and c is the concentration. Given the molar absorptivity the amount of light transmitted through a layer of photolabile hydrogel was calculated at different wavelengths using the following relationship,
where I0 is the incident light intensity applied to the top of sample, It is the transmitted light intensity at the bottom of the sample, and T is the transmittance.
Photodegradation of thick bulk hydrogels.
Hydrogels (21 μL volume) were prepared in cylindrical molds (1 mL BD plastic syringe with tip cut off, inner diameter (ID) = 4.78 mm) and allowed to polymerize for 2 hours at room temperature, which was sufficient time for hydrogel polymerization to have occurred based on rheological characterization. Polymerized hydrogels were transferred to a 48-well plate and incubated in PBS (500 μL) overnight. The volume of the equilibrium swollen hydrogels was determined using three diameter measurements made with Vernier calipers and a height measurement made using the gap size of the rheometer once a 0.01 N normal force was applied to the hydrogel surface. The measured hydrogels were transferred to a black 96-well plate, covered with PBS (200 μL), and irradiated with light in 2.5-min bursts, followed by replacement of buffer after each burst as described below, for a total of 20 min irradiation. The time between light bursts was 15 min providing enough time to measure the height and volume of the hydrogels and to collect and replace the buffer surrounding the hydrogels. This irradiation pattern was selected as 2.5 min was similar to the half-life of the fastest degrading hydrogel condition based on the kinetics determined using rheological measurements. Between each 2.5 min burst, the hydrogel volume was determined using the methodology described above and the buffer was replaced to minimize any light attenuation caused by degraded components. Hydrogels were irradiated with 365 nm (I0 = 10 mW cm−2) or 400 - 500 nm (I0 = 17.1 mW cm−2). The light intensity was determined using a radiometer as described above.
A separate set of equilibrium swollen hydrogels were irradiated with light through a thin layer of pig skin (Herman’s Deli, Newark, DE) using a slightly modified protocol than that stated above. First, frozen pig skin obtained from a local butcher was defrosted and biopsy punched into 8 mm rounds. The fat was removed from these rounds using a flat razor blade. The average thickness of the defatted pig skin samples was 0.73 ± 0.05 mm. After the initial volumes of the hydrogels were determined, the hydrogels were transferred to a black 96-well plate followed by the addition of 200 μL of PBS and then covered with a layer of defatted pig skin. The hydrogels were irradiated using the same conditions as above. Buffer exchanges were conducted every 2.5 min and hydrogel volume was determined every 5 min for a total of 60 min. The time between light bursts was 15 min as stated above.
Encapsulation and release of PANG.
Engineering, expression, and purification of monoclonal antibody PANG followed methods described previously.43 PANG was encapsulated within the photolabile hydrogel networks. Hydrogels (21 μL volume) were prepared in cylindrical molds as described above with the addition of PANG to the aqueous precursor solution for a final concentration of 2.5 mg/mL PANG (52.5 μg PANG loaded per hydrogel). Hydrogels were allowed to polymerize for 2 h at room temperature. Polymerized hydrogels were transferred to a polypropylene 24-well plate blocked with 1 wt% bovine serum albumin (BSA) and swollen overnight in PBS (1 mL). The entirety of the buffer was collected and replaced with 500 μL of fresh buffer and the hydrogels were incubated for another 24 h. A set of NB hydrogels and CMR hydrogels were left at room temperature with daily buffer exchanges throughout the experimental timeframe. After 48 h, another set of NB and CMR hydrogels were irradiated with either 365 nm light (I0 = 10 mW cm−2) or 400 - 500 nm light (I0 = 17.1 mW cm−2). The entirety of the buffer was exchanged and replaced with fresh buffer after every 2.5 min of irradiation for a total of 10 min of irradiation. The time between each light burst was 5 min, providing enough time to collect and replace the buffer surrounding the hydrogels. Any hydrogel that remained after the degradation process was incubated in buffer overnight followed by buffer collection. Collected samples were stored at −80 °C until further analysis.
Characterization of released PANG.
The concentration of released PANG was determined by characterization of 30 μL of release sample on reverse phase ultra-performance liquid chromatography (RPC) using water and acetonitrile mobile phases containing 0.1% TFA and a C4 column (P/N 186004496, Waters Corporation, Milford, MA). PANG was resolved during a linear portion of the gradient from 20 to 80% acetonitrile. Prior to loading, the release samples were thawed at room temperature and then centrifuged for 10 min at 4 °C to remove any large PEG oligomers or antibody aggregates. The integrated peak areas for a set of PANG standards were used to generate a standard curve. This standard curve was fit using a four-parameter regression model. The test sample integrated peak areas were interpolated onto the four-parameter regression model in order to determine PANG sample concentration loaded.
The toxin binding activity of the released PANG was characterized using an anthrax lethal toxin neutralization assay following a previously reported protocol.18 Briefly, J774A.1 cells (ATCC TIB-67, Manassa, VA) were plated in 96-well tissue culture plates at 250,000 cells per well and incubated overnight at 37 °C with 5% CO2. Release samples were diluted to 1 ng/mL based on the concentration determined using RPC and titrated onto the plated cells in the presence or absence of lethal toxin. The plates were incubated at 37 °C with 5% CO2 for 4 h followed by assessment of cell viability using WST-1 (Roche Applied Sciences, Indianapolis, IN), a proliferation measuring reagent. After a 2 h incubation with the WST-1 reagent a spectrophotomertric measurement was conducted on a plate reader following manufacture instructions (Molecular Devices, San Jose, CA). The data were plotted as optical density (OD) versus the concentration of each sample dilution and a four-parameter logistic-log regression was used to fit the data. The inflection point on the established curve reports the concentration of the sample that provides 50% inhibition of lethal toxin termed as EC50.
Statistics.
All reported values are represented as the mean and standard error (n = 3 unless otherwise noted). A one-way analysis of variance (ANOVA) with post-hoc Tukey’s Honest Significant Difference (HSD) test was performed to compare the hydrogel mechanical properties, hydrogel photodegradation rates, and concentration of PANG released from the four different hydrogel degradation conditions. To compare the EC50 value of the released PANG to pristine PANG two-sided equal variance Student’s t-tests were performed.
Results and Discussion
Synthesis of nitrobenzyl-azide and coumarin-azide.
Both NB-azide and CMR-azide were successfully synthesized using the synthetic strategies shown in Scheme 1 and Scheme 2. The NB-azide moiety containing a water-stable carbamate bond was synthesized with slight modifications to a previously established protocol with functional handles incorporated at two ring positions (Scheme 1), as detailed further below.41 For the CMR molecule, a more sophisticated synthetic scheme was developed to create a visible-light responsive moiety that could be integrated within crosslinks. Indeed, as noted in the Introduction, substitutions to the base structure of coumarins are known to drastically change their molar absorptivity and degradation properties.28 For example, incorporating a CMR that acts as a crosslink within a hydrogel through substitutions to two ring positions (e.g., the first and seventh ring positions) has been observed to shift the absorbance spectrum of the coumarin from the visible to the UV resulting in slowed photodegradation kinetics upon irradiation with visible light.31 Recent work highlighted how the Passerini reaction can be used to modify a CMR with two functional handles at the first ring position, allowing its incorporation into the crosslinks while also maintaining its absorbance properties within the visible light range.36 Taking inspiration from this approach, we developed a synthetic scheme to synthesize a CMR moiety with two functional handles incorporated through water-resistance bonds at the first ring position (Scheme 2). Further, the synthetic route for CMR-azide presented here was designed to addressed obstacles faced with current synthetic strategies, including conjugations that resulted in depreciation of the max light absorbance, required numerous steps that resulted in lower yields, or produced bonds that degrade under physiological conditions (i.e., esters).31, 32, 36, 48 This new approach resulted in a CMR-azide with strong absorbance in both UV and visible light regions and that is facilely integrated within crosslinks of hydrogel networks.
NB-azide and CMR-azide groups have been previously synthesized using 3-azidopropan-1-amine41 or 1-azido-3-isocyanopropane,36 respectively. To improve the safety of the synthetic route and reaction scalability, a four repeat PEG spacer was added between the amine and azide moieties (Schemes 1 and 2). Through increasing the length of the alkyl carbon chain, the carbon and oxygen to nitrogen ratio was increased from 0.75 to 3.5, where values above 3 are generally considered safe for synthesis and storage of purified azides.49, 50 This modification to the synthesis of azide-containing NB and CMR moieties improved the safety of the synthetic workflow, the accessibility of these protocols to laboratories without expertise in organic synthesis of azide-containing compounds, and the scalability of the procedure by use of a shelf-stable commercially available PEG-azide spacer, supporting translation of these materials.
Excitingly, these synthetic strategies produced a NB moiety with an absorbance profile similar to those previously reported41 and a CMR molecule with good absorbance properties within the visible light region (Figure 2, Table S1). Further, the functional handle installation onto the NB and CMR moieties did not negatively affect their absorbance properties in the regions of interest when compared to the unmodified versions (Figure S1). The NB and CMR linkers were subsequently attached to PEG-di-amine via amide coupling to produce linear end-functionalized NB-azide PEG (PEG-di-NB-azide) or linear end-functionalized CMR-azide PEG (PEG-di-CMR-azide) polymers for use as photolabile crosslinks within hydrogel networks (Figure 1C–D). Photolabile NB and CMR functionalized PEG-based monomers and hydrogels, as well as their degradation products, have previously been shown to have good biocompatibility in both two-dimensional and three-dimensional cell culture applications, where monomers were designed such that the photolabile molecules were attached to PEG both at hydrogel formation and upon degradation to minimize any potential toxicity.36, 51, 52 Additionally, more broadly, PEG-based hydrogels have been shown to have good biocompatibility, including in skin wound healing applications,53 and PEGs of the size used here can be cleared by the renal system within animal models and humans, providing a mechanism for clearance of degraded hydrogel components.54, 55 Taken together, the materials investigated here were designed for good biocompatibility based off prior reports that support such PEG-based hydrogels having minimal toxicity. While utilized as PEG-based hydrogel crosslinks here, the presentation of two orthogonal functional handles, an azide and a carboxylic acid, provides opportunities for facile conjugation of these NB and CMR linkers to other amine, hydroxyl, alkyne, or cyclooctyne presenting molecules or surfaces.
Figure 2. Molar absorptivity of NB-azide and CMR-azide.
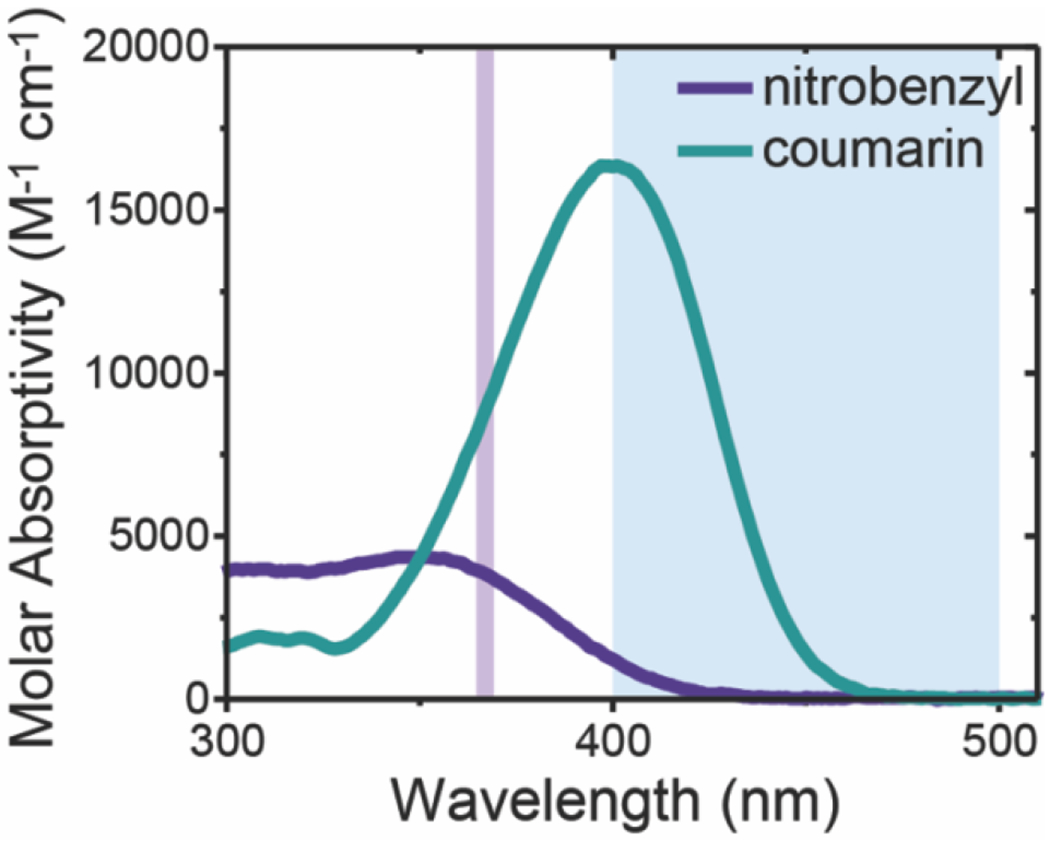
The molar absorptivity of the small molecule NB-azide and CMR-azide moieties was determined using UV-Vis spectrophotometry. The light absorbance of NB-azide and CMR-azide moieties in the wavelengths of interest are shown in the shaded regions: long-wavelength UV (365 nm, purple line) and visible blue light (400 – 500 nm, blue region).
Formation and characterization of photodegradable SPAAC hydrogels.
Photodegradable PEG-based hydrogels were formed using a SPAAC reaction between a dibenzocyclooctyne end-functionalized four-arm PEG (PEG-4-DBCO) and either PEG-di-NB-azide or PEG-di-CMR-azide (Figure 1). For all experiments, hydrogels were formed at approximately 10 wt% of polymer. As the storage modulus is directly correlated to bond formation and cleavage, it can be used to monitor hydrogel polymerization and degradation using in situ dynamic time sweep measurements (Figure S2). To ensure there were no confounding effects of the starting moduli of the different formulations on their degradation behavior, the hydrogel formulations were slightly altered (11.0 or 11.3 wt% polymer for NB-azide or CMR-azide formulations, respectively) to ensure the final storage moduli were statistically the same for both formulations (Figure S3).
The degradation behavior of the resulting photolabile hydrogels upon exposure to either UV or visible light was determined by irradiating thin hydrogels (120 μm) formed in situ on a photorheometer with either 365 nm (I0 = 4 mW cm−2) or 400 – 500 nm (I0 = 6.7 mW cm−2) light and monitoring the storage modulus over time (Figure S2). As described above, the storage modulus is directly correlated with the network crosslink density and thus bond cleavage, which can be modeled at early times using first-order reaction kinetics.19, 46, 56 The relationship between the storage modulus and bond cleavage allows for linearized and normalized rheological data to be used to obtain the first-order effective degradation constant for each hydrogel formulation (Figure 3A). All first-order effective rate constants were normalized to the incident intensity of light being applied to the sample to ensure fair comparisons between the different conditions.
Figure 3. Photodegradation of NB and CMR hydrogels.
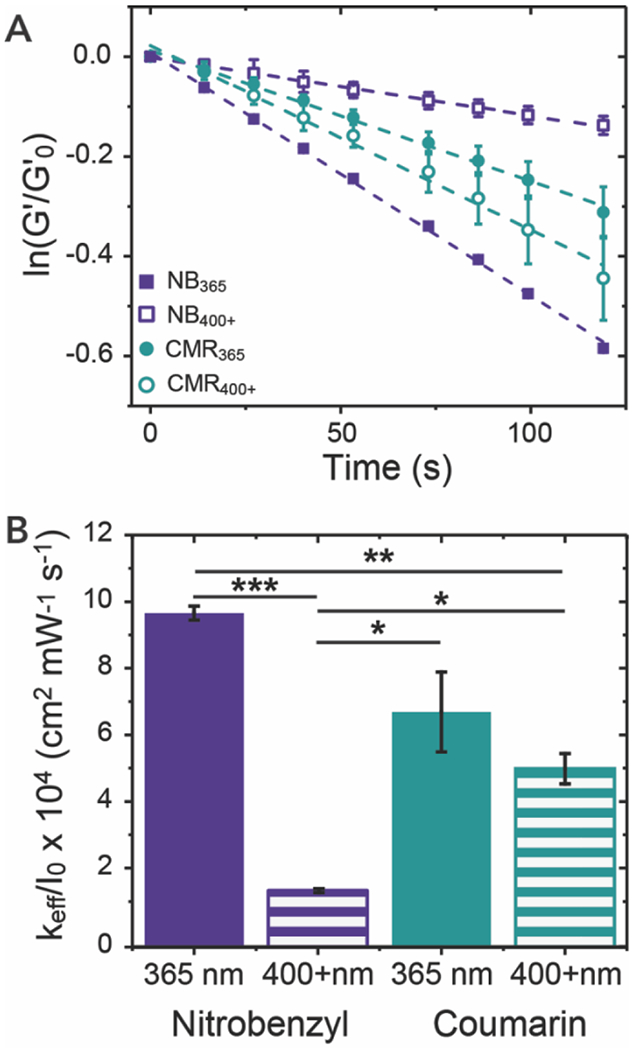
(A) Photolabile NB or CMR hydrogels were irradiated with either 365 nm (I0 = 4 mW cm−2) or 400 – 500 nm (I0 = 6.7 mW cm−2) light and their photodegradation rate was determined using linear regression of normalized storage modulus data. (B) The effective first-order rate constants were determined by fits of these data for each hydrogel formulation-light pairing, here normalizing each keff by the intensity of the applied light for ease of comparison. The data shown illustrate the mean (n = 3) with error bars representing standard error. * signifies p < 0.05, ** signifies p < 0.01, and *** signifies p < 0.001 as determined using an ANOVA with a Tukey’s post-hoc means comparison test.
NB hydrogels degraded using 365 nm light (NB365) had similar degradation rates to those previously observed41 and showed significantly slower degradation rates when irradiated using 400 – 500 nm irradiation (NB400+) (Figure 3B). Notably, CMR hydrogels exhibited fast degradation upon irradiation with both 365 nm (CMR365) and 400 – 500 nm light (CMR400+) (Figure 3B). Further, thin NB365 and CMR400+ hydrogels were fully degraded over the 15 min of irradiation, and NB400+ and CMR365 achieved approximately 70% and 80% degradation, respectively. Importantly, the photolabile linkers investigated here achieved varied degradation properties between the NB and CMR hydrogels using light exposure doses that are on the lower end of the ranges typically used in clinical applications. For example, UVA1 (λ = 340 – 400 nm) and visible light (BLU-U lamp centered at 417 nm) based therapies have been applied in the clinic for the treatment of dermatologic conditions and phototherapy of cancers at I0 = 10 mW cm−2 or 17 mW cm−2 in 10 to 15 min intervals, respectively.37, 57 To mimic the difference in light intensities utilized within the biomaterials community and the clinic for UVA1 versus visible light, higher light intensities were used for the degradation of hydrogels irradiated with blue light versus those irradiated with 365 nm light. 27, 30, 37, 40 A lower light intensity was utilized in these rheological experiments to capture effectively the degradation kinetics.
The first-order effective rate constant is a function of the intensity of light, quantum yield, and molar absorptivity, as described in the methods, which provides additional insight into the photodegradation of the NB and CMR hydrogels.45, 56 The normalization of the first-order effective rate constants by the light intensity provides a more direct comparison of how differences in molar absorptivity and quantum yield of the NB and CMR photocleavable moieties affect photodegradation. As expected, based on their respective molar absorptivity, faster degradation was observed for NB hydrogels with 365 nm, whereas faster degradation was achieved for CMR hydrogels with 400 – 500 nm light. The ability to rapidly degrade CMR crosslinked hydrogels with visible light highlights the power of using a single ring substitution over multiple ring substitutions when adding functional handles for integration within hydrogel crosslinks. Interestingly, despite higher light absorbance at 365 nm, the CMR hydrogels degraded slower than the NB365 hydrogels (Table S1); this likely owes to the differences in the quantum yield of the distinct photocleavable groups at the respective irradiation wavelengths. Indeed, it has been reported that the NB-azide small molecule has a quantum yield of approximately 0.053,41 whereas a small molecule CMR, of similar structure to the one used here, had a reported quantum yield of 0.03.58 This highlights the balance between molar absorptivity and quantum yield that dominates the degradation rate of these moieties and accordingly the importance of the wavelength of light selected for irradiation. These parameters are essential for controlling the photodegradation properties of these systems, whether for use as therapeutic delivery depots or cell culture scaffolds.
Photodegradation of optically thick hydrogels.
Optically thick equilibrium swollen hydrogels can be used as depots for therapeutic release via light-driven erosion. Hydrogels with high light absorbances have limited to no light transmission through the entirety of the hydrogel sample geometry, resulting in degradation through a surface erosion mechanism (i.e., linear decreases in hydrogel volume over time). This mechanism arises due to the incident light being significantly absorbed as it traverses through the sample, leading to degradation of the top layers before sufficient light has reached lower layers and thereby erosion of the hydrogel surface occurs in a layer-by-layer fashion.26, 56 Alternatively, if a hydrogel is considered optically thin, the light penetrates the full sample thickness leading to degradation throughout the bulk of the hydrogel {i.e., decrease in bulk crosslink density and with a concomitant increase in hydrogel volume over time until reverse gelation). In practice, the photodegradation of thick samples, such as the mm-thick cylindrical disks studied here (average hydrogel height of 1.25 mm and diameter of 5.5 mm after equilibrium swelling), will be a complex mixture of bulk degradation and surface erosion depending on the selected photolabile moiety and irradiation wavelength.56 To investigate these effects, NB and CMR hydrogels were degraded in black 96-well plates by light applied to the hydrogel surface with buffer exchanges and volume measurements every 2.5 minutes.
Here, a linear decrease in the normalized volume was observed for NB365, CMR365, and CMR400+ hydrogels, which is indicative of a surface erosion dominated degradation profile (Figure 4A). In contrast, NB400+ hydrogels were observed to undergo a more bulk degradation profile with increases in the normalized volume observed over time. Based on the Beer-Lambert law, over 40% of the light applied to the NB400+ bulk hydrogels was estimated to be transmitted through its thickness, whereas no light was transmitted through the NB365, CMR365, or CMR400+ conditions (Figure S4). The final time point for all conditions was compared to further assess the differences in degradation mechanism. The final volume for the NB365, CMR365, and CMR400+ hydrogels were statistically different than that of the NB400+ hydrogels. These data further highlight how the trends in the observed light absorbance and the photodegradation rates of NB and CMR at the respective irradiation wavelengths enables the design of hydrogels with a specific degradation type (full statistical analysis Table S2).
Figure 4. Degradation of bulk NB and CMR hydrogels.
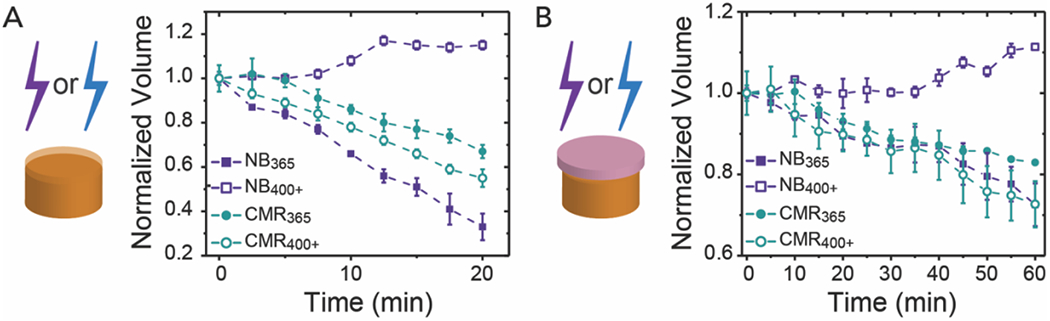
(A) Bulk cylindrical hydrogels degrade through varied mechanisms upon irradiation with either 365 nm (I0 = 10 mW cm−2) or 400 – 500 nm (I0 = 17.1 mW cm−2) light over 20 min as monitored by the normalized volume. (B) Bulk hydrogels irradiated through a layer of pig skin over 60 min exhibit similar trends in degradation profiles, albeit with slower overall rates as expected relative to those irradiated in buffer alone. Note, the expanded y-axis used to show differences in degradation profile when hydrogels were irradiated through pig skin. The data shown illustrate the mean (n = 3) with error bars representing standard error. Full statistical analysis comparing the final time points in each condition can be found in the SI (Table S2 and Table S3).
Photodegradation of optically thick hydrogels through skin.
Skin consists of an epidermis, dermis, and hypodermis. The epidermis and dermis combined are usually between 2 – 3 mm in thickness and the hypodermis contains a subcutaneous fat layer.59, 60 Owing to wavelength-dependent scattering and absorbance by tissues, long wavelength UV, visible light, near infrared, and infrared light can penetrate through these layers of skin with varying levels of attenuation.60 Light within the visible region is known to penetrate skin deeper than long-wavelength UV light (e.g., ~2 mm for blue light versus ~1 mm for long-wavelength UV),60 leading to an increased interest in the development of longer wavelength-responsive photolabile systems. As skin attenuates light approximately 10-fold or greater between 365 – 500 nm, depending on the skin thickness,42 we sought to better understand how this attenuation of light through skin will affect hydrogel photodegradation properties. To investigate the potential for future translation of photolabile hydrogels to preclinical models (e.g, in vivo or ex vivo systems), optically thick photoresponsive hydrogels were covered with a thin layer of pig skin (height = 0.73 ± 0.05 mm), which is a good mimic for human skin due to similarities in light attenuation and porosity,42, 61 and photodegradation was monitored as described above (Figure 4B). Here, the fat layer below the epidermis and dermis was removed from pig skin to best mimic the light conditions that a hydrogel would be exposed to if injected intradermally, where the hydrogel would sit above the subcutaneous fat layer of the hypodermis. This approach is consistent with previous studies aimed at examining materials for intradermal applications42 and provides an opportunity to bridge between traditional in vitro and in vivo assays on the road to translation. Hydrogel samples irradiated through a layer of pig skin exhibited similar trends in degradation profiles, albeit at slower rates as compared with hydrogel not irradiated through pig skin. For example, ~50% to ~70% of the volume of CMR365, CMR400, or NB365 hydrogel samples were degraded after 20 min of irradiation in buffer alone, with relative differences commensurate with observations of respective keff, whereas ~10% of the same hydrogel formulations were degraded after 20 min of irradiation through pig skin. Given the slower degradation rates in the presence of pig skin, differences in volume changes over time between hydrogel formulation-light pairs became less significant for the surface eroding formulations.
Overall, these data indicate that light attenuation due to the pig skin layer mostly serves to decrease the incident light intensity (i.e., leading to a decreased degradation rate) and has minimal effect on the degradation mechanism (i.e., surface erosion vs. bulk degradation). Thus, the light attenuation properties of the hydrogels continue to be the dominant factor driving the observed degradation mechanism, making the precise control over degradation and release predictable. If desired for applications of interest, light attenuation within photolabile hydrogel systems can be tuned by controlling the wavelength of light used for photodegradation (e.g., using band pass filters at specific wavelengths associated with desired absorbance) or changing the concentration of photocleavable moieties within the formulation. Alternatively, one could investigate the incorporation of upconverting nanoparticles, a promising technology that upconverts near infrared (NIR) light into higher energy wavelengths of light, into the hydrogels to provide opportunities for degradation in response to NIR light, which would not be attenuated by the NB or CMR moieties.62 Importantly, the degradation of hydrogel samples through pig skin was significant and achieved using clinically relevant doses of long-wavelength UV37 or visible blue light57 as used in dermatologic treatments. Specifically, samples irradiated with long-wavelength UV light were exposed to a light dose of 12 J cm−2, which is well within the low dosing region of 10 – 20 J cm−2 used for UVA1 phototherapy.37, 38 Hydrogels irradiated with blue light were exposed to 21 J cm−2 of light, which is within FDA approved dosing regimens for the treatment of dermatological conditions (7.5 – 135 J cm−2).39, 63 Taken together, these results highlight the potential for photolabile hydrogels to be degraded intradermally and serve as responsive delivery vehicles within a translational setting.
Encapsulation and release of antibody from photolabile hydrogels.
For release studies, PANG was selected as a model bioactive antibody due to its well-established detection and activity assays and interest for use in therapeutic intervention. PANG was encapsulated within thick PEG-based NB or CMR hydrogels, as described above, and subsequently released through irradiation with long-wavelength UV or blue light. The concentration of PANG released from the hydrogels into buffer was determined using reversed-phase chromatography (RPC), where a four-parameter fit was used to generate a calibration curve to correlate the amount of PANG loaded onto the column with area under the elution curve. Hydrogels were loaded with approximately 53 μg of PANG, 35 μg of which was lost in the initial burst release as the hydrogels reached equilibrium swelling. The burst release was designed to be high in the formulations tested, as we envisioned it could be utilized to provide the first dose of the therapeutic upon injection. Following equilibrium swelling of the hydrogels, the on-demand release of PANG from photolabile hydrogels was achieved by cyclic irradiation bursts of 2.5 min, separated by 5 min intervals of no light for a total irradiation time of 10 min. Between each 2.5 min irradiation burst, the buffer surrounding the hydrogel was rapidly collected and replaced with fresh buffer to help i) mitigate optical attenuation caused by released polymer components and ii) monitor the release of PANG from hydrogels upon irradiation.
With this cyclic irradiation approach, we observed that PANG was released at a similar rate from NB365, CMR365, and CMR400+ hydrogels (Figure 5), which was anticipated given similarities in their relative rates of degradation and mechanism dominated by surface erosion. For NB400+ hydrogels that are dominated by bulk degradation, minimal PANG was released until the final timepoint, indicating that the degradation-induced increases in mesh size alone at these early times are insufficient for significant release of entrapped protein. Over the first 7.5 min, the amount of PANG released from NB365, CMR365, and CMR400+ hydrogels was found to be significantly different than that released from NB400+ hydrogels, demonstrating that bulk degrading versus surface eroding hydrogels can be utilized to achieve diverse control over protein release (Table S4). Moreover, this difference in photodegradation mechanism and thereby protein release profile was achieved for the NB hydrogels simply by changing the wavelength of light. Of particular interest for therapeutic applications is the controlled protein dosing that is achieved by surface eroding photolabile hydrogels. The coumarin-based hydrogels therefore may be more broadly advantageous for translation as the surface erosion mechanism is maintained upon irradiation with visible light wavelengths, which are used clinically in a wider range of indications due to decreased potential for damage to cells as compared to UV light.
Figure 5. Release of PANG from photodegradable NB and CMR hydrogels.
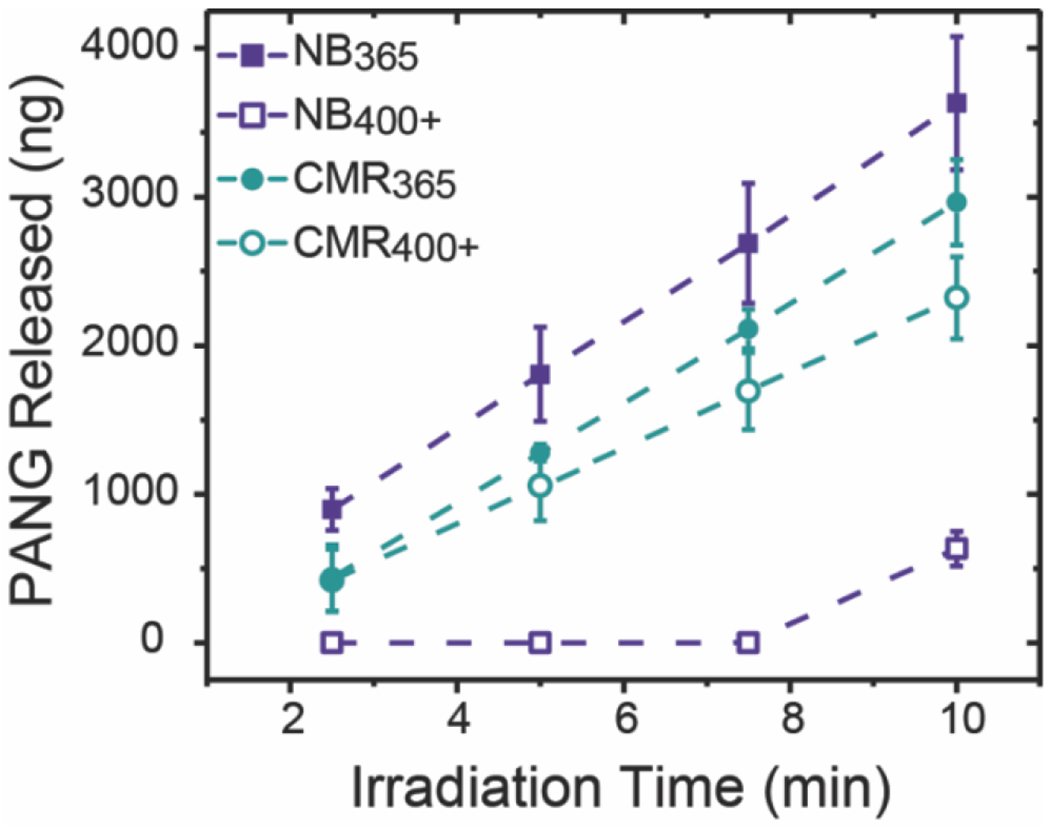
PANG encapsulated within either NB or CMR hydrogels was released upon hydrogel degradation in response to either 365 nm (I0 = 10 mW cm−2) or 400 – 500 nm (I0 = 17.1 mW cm−2) light. Increases in the cumulative amount of PANG released over the irradiation time course were observed for all formulations. The data shown illustrate the mean (n = 3) with error bars representing standard error. Full statistical analysis comparing the formulations at each time point can be found in the SI (Table S4).
Hydrogels irradiated with light were subsequently incubated in buffer overnight to determine the amount of PANG released after the irradiation was terminated. Additionally, the total amount of PANG released from hydrogels irradiated with light was compared to a set of non-irradiated control NB and CMR hydrogels that were in buffer and protected from light over the experimental timeframe. As the hydrodynamic radius of PANG, an IgG-1 monoclonal antibody, is similar in size to the average mesh size of these hydrogel formulations (on the order of 10 nm) there is potential for the encapsulated PANG to escape from the network over time by a hindered Fickian diffusion mechanism.64
All hydrogels irradiated with 10 min of light and swelled overnight released statistically the same amount of PANG (Figure 6). Importantly, more PANG was released from formulations irradiated with light than those incubated solely in buffer, illustrating the ability of protein release to be temporally controlled and enhanced upon application of light. Upon comparison of the different formulations, we observed that additional PANG was released from NB365, CMR365, and CMR400+ hydrogels upon overnight swelling and that the percent of PANG released overnight, as depicted by the solid part of the bars in Figure 6, was similar to release from non-irradiated controls (i.e., hydrogels solely incubated in buffer with release by hindered diffusion alone). These data may suggest that the average mesh size of the remaining hydrogel after irradiation of these formulations was similar to control hydrogels yielding similar rates of hindered antibody diffusion out of the network. Interestingly, NB400+ hydrogels were observed to have a significant burst of antibody after light irradiation followed by overnight swelling, with about ~10x more antibody released upon swelling than directly after irradiation and with ~3x more antibody released compared with hindered diffusion. The observation that changes in antibody release after overnight swelling for NB400+ hydrogels further supports that degradation occurs through the bulk degradation mechanism and illustrates its relevance for controlling release of entrapped proteins. In all cases, hydrogels remained after light irradiation and overnight swelling indicating that more PANG likely remained within the hydrogels based on a rough mass balance and could be released with further light irradiation.
Figure 6. Cumulative release of PANG from NB and CMR hydrogels after light irradiation and overnight swelling.
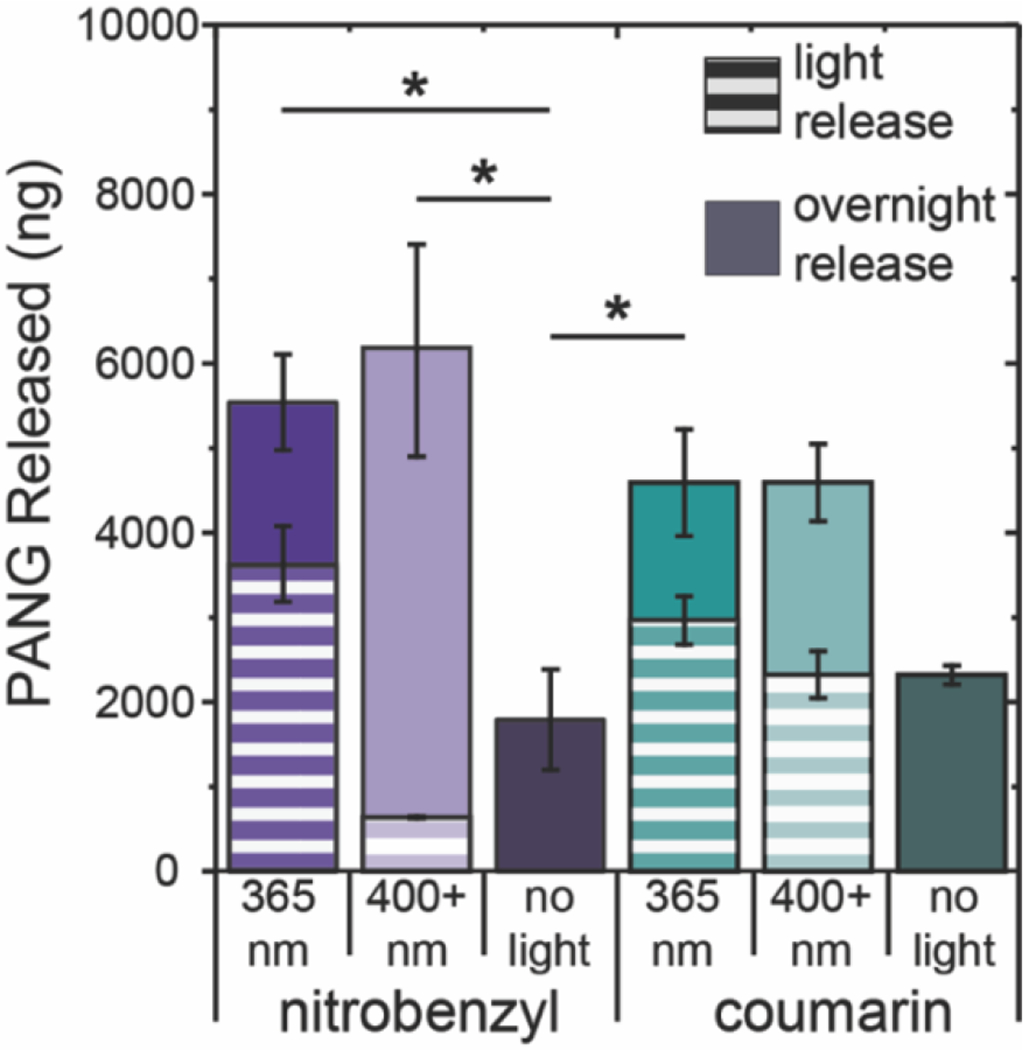
The cumulative release of PANG from NB and CMR hydrogels irradiated with long-wavelength UV or blue light compared to those not exposed to light after overnight swelling. The striped portion of the bar indicates the amount of PANG released after 10 min of light irradiation and the solid bars represent PANG released from hydrogels after incubation in PBS overnight. The data shown illustrate the mean (n = 3) with error bars representing standard error. * signifies p < 0.05 as determined using an ANOVA with a Tukey’s post-hoc means comparison test on total protein release.
Taken together, more PANG was released from formulations irradiated with light than those incubated solely in buffer and the rate of protein release can be controlled by the mechanism of degradation. For example, surface eroding hydrogels can be used to ‘dial-in’ a specific amount of protein release, whereas bulk degrading hydrogels provide a delayed burst release of protein. Importantly, these results highlight how rationale selection of both light wavelength and photolabile moiety can be used to control hydrogel degradation and protein release over minutes to days. The timescale of antibody release from the formulations shown here, which afford controlled release on the order of minutes to days, could be beneficial in a range of applications, including mitigation of maladaptive wound healing responses. For example, in the healing of chronic wounds, persistence of high levels of TNF-α, amongst other cytokines, leads to immune cell overactivation and a prolonged inflammatory response that is detrimental to healing.65 In one potential future application, the materials investigated here could be used to control the delivery of a TNF-α neutralizing antibody over several days with the application of light providing a methodology for increasing the rate of antibody release dependent on the patients response or for probing what antibody release profiles are most efficacious in promoting healing more broadly. Depending on the application of interest, additional control over protein release can be achieved by changing the composition of the hydrogel network to decrease the mesh size (e.g., increasing the polymer concentration)66 or through the encapsulation of larger proteins (e.g., an IgM or fibrinogen).67 For example, to achieve controlled release over longer timescales, the hydrogel formulation could be adjusted to limit any Fickian diffusion of the cargo prior to light-triggered degradation, which may be useful for controlled dosing of immunotherapies, such as anti-PD1 or anti-CTLA4 over several weeks, toward improving the immune response while minimizing dosing outside of the therapeutic region.68
Finally, the bioactivity of the released PANG was determined using a lethal toxin (LeTx) neutralization assay that measures the dose of PANG required to protect cells from exposure to anthrax lethal factor and protective antigen (PA). The binding of PANG to PA prevents the formation of LeTx in the presence of lethal factor, protecting the cells against anthrax intoxication.18 By exposing the cells to dilutions of PANG, the half-maximal effective concentration (EC50) was determined. To assess the effect of encapsulation and release on PANG bioactivity, the EC50 was determined for PANG released after 10 min of irradiation followed by overnight swelling (Figure 7). By comparing the PANG released from both NB and CMR hydrogels, we observed EC50 values that were statistically the same as pristine PANG (represented as a solid pink line) at all time points (Table S5). In addition, the EC50 values were found to be much lower than that of degraded or heat inactivated PANG (in which the disulfides of the antibody were oxidized with 1x SDS-buffer and elevated temperature; represented by the solid red line), where a higher EC50 value indicates less bioactivity. Further, the PANG control solutions (i.e., pristine vs. degraded/heat-inactivated) prepared at the same concentration, based on the calculated solution concentration obtained with RPC (Figure S5), exhibited significantly different bioactivities. Taken together these assays confirm that the protein encapsulation and hydrogel degradation processes lead to the release of a bioactive protein.
Figure 7. Bioactivity of PANG released from photolabile NB and CMR hydrogels.
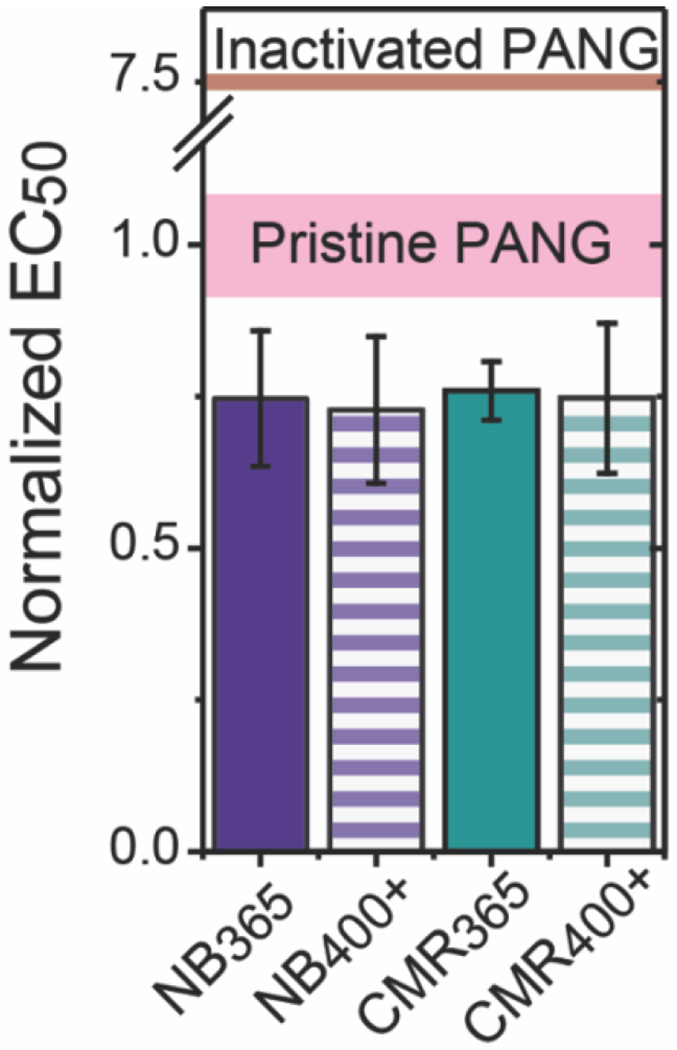
Normalized EC50 values for PANG released from photolabile hydrogels after 10 min of light irradiation and incubation in PBS overnight as determined using a lethal toxin (LeTx) neutralization assay. All samples were normalized against the EC50 value of pristine PANG. The pink line represents the EC50 values of pristine PANG and the red line represents that of degraded/heat-inactivated PANG (note the broken axis). The data shown illustrate the mean (n = 3) with error bars representing the standard error. All release formulations were determined to be statistically the same as pristine PANG using a p of 0.05 (Table S5).
Conclusions
Photolabile hydrogels formed with NB or CMR moieties were investigated for encapsulation and on-demand antibody release for the design of controlled protein delivery depots. A synthetic methodology was introduced to safely synthesize a CMR linker, in addition to a NB linker, with azide functional handles through a water-stable carbamate bond while achieving the desired photoactivity and allowing facile integration within hydrogel network crosslinks. The resulting NB and CMR linkers exhibited differential peak molar absorptivity within long-wavelength UV and visible light regions, respectively, and the light-responsive properties of their individual hydrogels were found to be commensurately dictated by the wavelength of light used for degradation. Selection of both photolabile linker and irradiation wavelength regulated the mechanism of photodegradation for thick bulk hydrogels, where high light attenuation led to a surface erosion dominated mechanism and low light attenuation led to a bulk degradation dominated mechanism. Importantly, all formulations could be degraded upon irradiation with clinically relevant doses of light through a thin layer of pig skin providing a model to help bridge the gap between in vitro and in vivo models. Additionally, the surface erosion mechanism obtained upon degradation of CMR hydrogels with 400 – 500 nm light is appealing for translational applications as it can provide controlled protein dosing using visible light wavelengths used more widely in the clinic. Further, the successful encapsulation and release of a bioactive antibody, PANG, was demonstrated from both NB and CMR hydrogels, with triggered increases in release controlled by simply tuning the wavelength of light used for hydrogel degradation. Taken together these data suggest that irradiation conditions clinically approved for dermatologic applications could be sufficient to degrade photolabile hydrogels intradermally, providing an opportunity for future preclinical studies of on-demand delivery of bioactive proteins with injected or implanted depots of these materials. Overall, this work establishes innovative synthetic protocols and important design parameters for creating photodegradable hydrogels relevant for overcoming current translational barriers facing light responsive material systems and demonstrates the utility of these materials for the encapsulation and on-demand release of a bioactive antibody toward improved therapeutic or vaccination regimens.
Supplementary Material
Acknowledgments
The authors would like to thank Fraunhofer Center for Molecular Biotechnology (CMB) for providing the PANG used in these studies. We additionally would like to thank Dr. Konstantin Musiychuk, Dr. Yoko Shoji, and Mr. Kevin Stecca for helpful conversations regarding PANG release and bioactivity characterization. PJL would like to thank the University of Delaware (UD) for financial support through the UD Dissertation Fellowship. The research presented here was supported by a grant from the Delaware Bioscience Center for Advanced Technology (DelCAT, 12A00448) and, in part by, a NIH Director’s New Innovator Award (DP2 HL152424-01) for work related to photolabile materials design. Core instrument support was provided in part by the Delaware COBRE programs with grants from the NIGMS (P20GM104316 and P30GM110758-02). We additionally would like to thank the University of Delaware NMR and Mass Spectrometry Laboratories for core instrument access and support.
Footnotes
Representative rheological data; NB and CMR molar absorptivity spectra; Hydrogel mechanical properties; Attenuation properties of thick bulk hydrogels; Statistical analysis; Representative RPC traces; NMRs spectra
The authors declare no conflicts of interest.
References
- 1.Chames P; Van Regenmortel M; Weiss E; Baty D, Therapeutic antibodies: successes, limitations and hopes for the future. British Journal of Pharmacology 2009,157 (2), 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gotwals P; Cameron S; Cipolletta D; Cremasco V; Crystal A; Hewes B; Mueller B; Quaratino S; Sabatos-Peyton C; Petruzzelli L; Engelman JA; Dranoff G, Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nature Reviews Cancer 2017, 47(5), 286–301. [DOI] [PubMed] [Google Scholar]
- 3.Dyck L; Mills KHG, Immune checkpoints and their inhibition in cancer and infectious diseases. European Journal of Immunology 2017, 47 (5), 765–779. [DOI] [PubMed] [Google Scholar]
- 4.Randall KL, Rituximab in autoimmune diseases. Australian Prescriber 2016, 39 (4), 131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitragotri S; Burke PA; Langer R, Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nature Reviews Drug Discovery 2014,13 (9), 655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pisal DS; Kosloski MP; Balu-Iyer SV, Delivery of Therapeutic Proteins. Journal of Pharmaceutical Sciences 2010, 99 (6), 2557–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turecek PL; Bossard MJ; Schoetens F; Ivens IA, PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. Journal of Pharmaceutical Sciences 2016,105 (2), 460–475. [DOI] [PubMed] [Google Scholar]
- 8.Singh SK; Luisi DL; Pak RH, Antibody-Drug Conjugates: Design, Formulation and Physicochemical Stability. Pharmaceutical Research 2015, 32 (11), 3541–3571. [DOI] [PubMed] [Google Scholar]
- 9.Tabrizi M; Bomstein GG; Suria H, Biodistribution Mechanisms of Therapeutic Monoclonal Antibodies in Health and Disease. Aaps Journal 2010,12 (1), 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C; Ye YQ; Hochu GM; Sadeghifar H; Gu Z, Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Letters 2016, 16(4), 2334–2340. [DOI] [PubMed] [Google Scholar]
- 11.Thurber GM; Schmidt MM; Wittrup KD, Factors determining antibody distribution in tumors. Trends in Pharmacological Sciences 2008, 29 (2), 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Streit M; Beleznay Z; Braathen LR, Topical application of the tumour necrosis factor-α antibody infliximab improves healing of chronic wounds. International Wound Journal 2006, 3 (3), 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korupalli C; Pan WY; Yeh CY; Chen PM; Mi FL; Tsai HW; Chang Y; Wei HJ; Sung HW, Single-injecting, bioinspired nanocomposite hydrogel that can recruit host immune cells in situ to elicit potent and long-lasting humoral immune responses. Biomaterials 2019, 216, 119268. [DOI] [PubMed] [Google Scholar]
- 14.Li JY; Mooney DJ, Designing hydrogels for controlled drug delivery. Nature Reviews Materials 2016,1 (12), 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kharkar PM; Scott RA; Olney LP; LeValley PJ; Maverakis E; Kiick KL; Kloxin AM, Controlling the Release of Small, Bioactive Proteins via Dual Mechanisms with Therapeutic Potential. Advanced Healthcare Materials 2017, 6 (24), 1700713–1700713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sridhar BV; Janczy JR; Hatlevik Ø; Wolfson G; Anseth KS; Tibbitt MW, Thermal Stabilization of Biologies with Photoresponsive Hydrogels. Biomacromolecules 2018,19 (3), 740–747. [DOI] [PubMed] [Google Scholar]
- 17.Holloway JL; Ma H; Rai R; Burdick JA, Modulating hydrogel crosslink density and degradation to control bone morphogenetic protein delivery and in vivo bone formation. Journal of Controlled Release 2014,191, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang YK; Coffin MV; Manceva SD; Chichester JA; Jones RM; Kiick KL, Controlled release of an anthrax toxin-neutralizing antibody from hydrolytically degradable polyethylene glycol hydrogels. Journal of Biomedical Materials Research Part A 2016, 104 (1), 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kharkar PM; Kiick KL; Kloxin AM, Design of thiol- and light-sensitive degradable hydrogels using Michael-type addition reactions. Polymer Chemistry 2015, 6 (31), 5565–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gregoritza M; Abstiens K; Graf M; Goepferich AM, Fabrication of antibody-loaded microgels using microfluidics and thiol-ene photoclick chemistry. European Journal of Pharmaceutics and Biopharmaceutics 2018,127, 194–203. [DOI] [PubMed] [Google Scholar]
- 21.D’Amato AR; Puhl DL; Ellman SAT; Balouch B; Gilbert RJ; Palermo EF, Vastly extended drug release from poly(pro-17 beta-estradiol) materials facilitates in vitro neurotrophism and neuroprotection. Nature Communications 2019, 10, 4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashley GW; Henise J; Reid R; Santi DV, Hydrogel drug delivery system with predictable and tunable drug release and degradation rates. Proceedings of the National Academy of Sciences of the United States of America 2013, 110 (6), 2318–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bae KH; Kurisawa M, Emerging hydrogel designs for controlled protein delivery. Biomaterials Science 2016, 4 (8), 1184–1192. [DOI] [PubMed] [Google Scholar]
- 24.Brudno Y; Mooney DJ, On-demand drug delivery from local depots. Journal of Controlled Release 2015, 219, 8–17. [DOI] [PubMed] [Google Scholar]
- 25.Said SS; Campbell S; Hoare T, Externally Addressable Smart Drug Delivery Vehicles: Current Technologies and Future Directions. Chemistry of Materials 2019, 31 (14), 4971–4989. [Google Scholar]
- 26.Kloxin AM; Kasko AM; Salinas CN; Anseth KS, Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science 2009, 324 (5923), 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azagarsamy MA; Anseth KS, Wavelength-Controlled Photocleavage for the Orthogonal and Sequential Release of Multiple Proteins. Angewandte Chemie-International Edition 2013, 52 (51), 13803–13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruskowitz ER; DeForest CA, Photoresponsive biomaterials for targeted drug delivery and 4D cell culture. Nature Reviews Materials 2018, 3 (2), 17087–17087. [Google Scholar]
- 29.Foster AA; Greco CT; Green MD; Epps TH; Sullivan MO, Light-Mediated Activation of siRNA Release in Diblock Copolymer Assemblies for Controlled Gene Silencing. Advanced Healthcare Materials 2015, 4 (5), 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffin DR; Kasko AM, Photoselective Delivery of Model Therapeutics from Hydrogels. Acs Macro Letters 2012, 1 (11), 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azagarsamy MA; McKinnon DD; Age DL; Anseth KS, Coumarin-Based Photodegradable Hydrogel: Design, Synthesis, Gelation, and Degradation Kinetics. Acs Macro Letters 2014, 3 (6), 515–519. [DOI] [PubMed] [Google Scholar]
- 32.Gandioso A; Contreras S; Melnyk I; Oliva J; Nonell S; Velasco D; Garcia-Amoros J; Marchan V, Development of Green/Red-Absorbing Chromophores Based on a Coumarin Scaffold That Are Useful as Caging Groups. Journal of Organic Chemistry 2017, 82 (10), 5398–5408. [DOI] [PubMed] [Google Scholar]
- 33.Shadish JA; Strange AC; DeForest CA, Genetically Encoded Photocleavable Linkers for Patterned Protein Release from Biomaterials. Journal of the American Chemical Society 2019, 141 (39), 15619–15625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serban MA, Translational biomaterials - the journey from the bench to the market - think ‘product’. Current Opinion in Biotechnology 2016, 40, 31–34. [DOI] [PubMed] [Google Scholar]
- 35.FDA, Chemistry, Manufacturing, and Controls Changes to an Approved Application: Certain Biological Products. Administration, F. a. D., Ed. 2017; pp 5–10. [Google Scholar]
- 36.Truong VX; Li FY; Forsythe JS, Photolabile Hydrogels Responsive to Broad Spectrum Visible Light for Selective Cell Release. Acs Applied Materials & Interfaces 2017, 9 (38), 32441–32445. [DOI] [PubMed] [Google Scholar]
- 37.Gambichler T; Terras S; Kreuter A, Treatment regimens, protocols, dosage, and indications for UVA1 phototherapy: Facts and controversies. Clinics in Dermatology 2013, 31 (4), 438–454. [DOI] [PubMed] [Google Scholar]
- 38.Arndt S; Lissner C; Unger P; Baeumler W; Berneburg M; Karrer S, Biological effects of a new ultraviolet A(1)prototype based on light-emitting diodes on the treatment of localized scleroderma. Experimental Dermatology 2020, 1–10. [DOI] [PubMed] [Google Scholar]
- 39.Jagdeo J; Austin E; Mamalis A; Wong C; Ho D; Siegel DM, Light-emitting diodes in dermatology: A systematic review of randomized controlled trials. Lasers in Surgery and Medicine 2018, 50 (6), 613–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maytin EV; Kaw U; Ilyas M; Mack JA; Hu B, Blue light versus red light for photodynamic therapy of basal cell carcinoma in patients with Gorlin syndrome: A bilaterally controlled comparison study. Photodiagnosis and Photodynamic Therapy 2018, 22, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.LeValley PJ; Neelarapu R; Sutherland BP; Dasgupta S; Kloxin CJ; Kloxin AM, Photolabile Linkers: Exploiting Labile Bond Chemistry to Control Mode and Rate of Hydrogel Degradation and Protein Release. Journal of the American Chemical Society 2020, 142 (10), 4671–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elisseeff J; Anseth K; Sims D; McIntosh W; Randolph M; Langer R, Transdermal photopolymerization for minimally invasive implantation. Proceedings of the National Academy of Sciences of the United States of America 1999, 96 (6), 3104–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mett V; Chichester JA; Stewart ML; Musiychuk K; Bi H; Reifsnyder CJ; Hull AK; Albrecht MT; Goldman S; Baillie LWJ; Yusibov V, A non-glycosylated, plant-produced human monoclonal antibody against anthrax protective antigen protects mice and non-human primates from B. anthracis spore challenge. Human Vaccines 2011, 7, 183–190. [DOI] [PubMed] [Google Scholar]
- 44.Laguerre A; Haukei S; Qiu J; Kelly MJ; Schultz C, Photorelease of 2-Arachidonoylglycerol in Live Cells. Journal of the American Chemical Society 2019, 141 (42), 16544–16547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeForest CA; Anseth KS, Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nature Chemistry 2011, 3 (12), 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tibbitt MW; Kloxin AM; Sawicki LA; Anseth KS, Mechanical Properties and Degradation of Chain and Step-Polymerized Photodegradable Hydrogels. Macromolecules 2013, 46 (7), 2785–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong MJ; Wang R; Kawamoto K; Olsen BD; Johnson JA, Quantifying the impact of molecular defects on polymer network elasticity. Science 2016, 353 (6305), 1264–1268. [DOI] [PubMed] [Google Scholar]
- 48.Pahattuge TN; Jackson JM; Digamber R; Wijerathne H; Brown V; Witek MA; Perera C; Givens RS; Peterson BR; Soper SA, Visible photorelease of liquid biopsy markers following microfluidic affinity-enrichment. Chemical Communications 2020, 56 (29), 4098–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keicher T. L. Stefan, Lab-scale Synthesis of Azido Compounds: Safety Measures and Analysis. John Wiley and Sons: Hoboken, 2010. [Google Scholar]
- 50.Brase S; Gil C; Knepper K; Zimmermann V, Organic azides: An exploding diversity of a unique class of compounds. Angewandte Chemie-International Edition 2005, 44 (33), 5188–5240. [DOI] [PubMed] [Google Scholar]
- 51.Kloxin AM; Benton JA; Anseth KS, In situ elasticity modulation with dynamic substrates to direct cell phenotype. Biomaterials 2010, 31 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kloxin AM; Tibbitt MW; Anseth KS, Synthesis of photodegradable hydrogels as dynamically tunable cell culture platforms. Nature Protocols 2010, 5 (12), 1867–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen SL; Fu RH; Liao SF; Liu SP; Lin SZ; Wang YC, A PEG-Based Hydrogel for Effective Wound Care Management. Cell Transplantation 2018, 27 (2), 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.PEGylated Protein Drugs: Basic Science and Clinical Applications. Pegylated Protein Drugs: Basic Science and Clinical Applications 2009, 1–287. [Google Scholar]
- 55.Baumann A; Tuerck D; Prabhu S; Dickmann L; Sims J, Pharmacokinetics, metabolism and distribution of PEGs and PEGylated proteins: quo vadis? Drug Discovery Today 2014, 19 (10), 1623–1631. [DOI] [PubMed] [Google Scholar]
- 56.Tibbitt MW; Kloxin AM; Anseth KS, Modeling controlled photodegradation in optically thick hydrogels. Journal of Polymer Science Part a-Polymer Chemistry 2013, 51 (9), 1899–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lane JE; Allen JH; Lane TN; Lesher JL, Unilateral basal cell carcinomas: An unusual entity treated with photodynamic therapy. Journal of Cutaneous Medicine and Surgery 2005, 9 (6), 336–340. [DOI] [PubMed] [Google Scholar]
- 58.Grimm JB; English BP; Chen JJ; Slaughter JP; Zhang ZJ; Revyakin A; Patel R; Macklin JJ; Normanno D; Singer RH; Lionnet T; Lavis LD, A general method to improve fluorophores for live-cell and single-molecule microscopy. Nature Methods 2015, 12 (3), 244-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim EJ; Kim YK; Kim JE; Kim S; Kim MK; Park CH; Chung JH, UV Modulation of Subcutaneous Fat Metabolism. Journal of Investigative Dermatology 2011, 131 (8), 1720–1726. [DOI] [PubMed] [Google Scholar]
- 60.Ash C; Dubec M; Donne K; Bashford T, Effect of wavelength and beam width on penetration in light-tissue interaction using computational methods. Lasers in Medical Science 2017, 32 (8), 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Godin B; Touitou E, Transdermal skin delivery: Predictions for humans from in vivo, ex vivo and animal models. Advanced Drug Delivery Reviews 2007, 59 (11), 1152–1161. [DOI] [PubMed] [Google Scholar]
- 62.Ruggiero E; Alonso-de Castro S; Habtemariam A; Salassa L, Upconverting nanoparticles for the near infrared photoactivation of transition metal complexes: new opportunities and challenges in medicinal inorganic photochemistry. Dalton Transactions 2016, 45 (33), 13012–13020. [DOI] [PubMed] [Google Scholar]
- 63.Barolet D, Light-Emitting Diodes (LEDs) in Dermatology. Seminars in Cutaneous Medicine and Surgery 2008, 27 (4), 227–238. [DOI] [PubMed] [Google Scholar]
- 64.Bertz A; Wohl-Bruhn S; Miethe S; Tiersch B; Koetz J; Hust M; Bunjes H; Menzel H, Encapsulation of proteins in hydrogel carrier systems for controlled drug delivery: Influence of network structure and drug size on release rate. Journal of Biotechnology 2013, 163 (2), 243–249. [DOI] [PubMed] [Google Scholar]
- 65.Ferrari G; Bignami F; Giacomini C; Franchini S; Rama P, Safety and Efficacy of Topical Infliximab in a Mouse Model of Ocular Surface Scarring. Investigative Ophthalmology & Visual Science 2013, 54 (3), 1680–1688. [DOI] [PubMed] [Google Scholar]
- 66.Rehmann MS; Skeens KM; Kharkar PM; Ford EM; Maverakis E; Lee KH; Kloxin AM, Tuning and Predicting Mesh Size and Protein Release from Step Growth Hydrogels. Biomacromolecules 2017, 18 (10), 3131–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Armstrong JK; Wenby RB; Meiselman HJ; Fisher TC, The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophysical Journal 2004, 87 (6), 4259–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maverakis E; Cornelius LA; Bowen GM; Phan T; Patel FB; Fitzmaurice S; He Y; Burrall B; Duong C; Kloxin AM; Sultani H; Wilken R; Martinez SR; Patel F, Metastatic Melanoma - A Review of Current and Future Treatment Options. Acta Dermato-Venereologica 2015, 95 (5), 516–524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.