

Abstract
This work describes a modification of the pRH2521 vector of the pRH2502/pRH2521 system for CRISPR-dCas9-mediated RNA interference. The modification enabled an increase in the cloning efficiency of guide RNA spacers. The ability of the modified pRH2502/pRH2521 system to suppress the transcription of certain genes was evaluated with the use of genes of Mycobacterium tuberculosis adenylate cyclases. The results revealed the limitations of the pRH2502/pRH2521 system for CRISPR interference associated with the probability of the detection of a protospacer adjacent motif (PAM) in the gene promoter region.
Keywords: CRISPR, CRISPRi, tuberculosis, mycobacteria, adenylate cyclase
INTRODUCTION
Despite the efforts of the medical and scientific community, tuberculosis (TB) remains the leading cause of death among infectious diseases. According to the WHO, it kills approximately 4000 people every day [1], which is comparable to the number of daily deaths from COVID-19 at the peak of the coronavirus infection pandemic in 2020. The functions of almost a quarter of the 4000 Mycobacterium tuberculosis (Mtb) genes remain unknown, although 5% of them are presumably significant for growth [2] and may be potential targets in the development of antituberculosis drugs and vaccines. The technology Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) is a multifunctional genetic engineering tool [3] created from the bacterial system of acquired resistance to phages and plasmids. CRISPR-associated nuclease forms a complex with guide RNA (gRNA) designed to recognize the target sequence of interest (protospacer) and the adjacent region (motif) (protospacer adjacent motif, PAM), which consists of two to six DNA base pairs. The recognition of the gRNA-CRISPR-associated nuclease complex and its binding to the target sequence results in a double-stranded break. Directional editing of the DNA sequence is possible during break repair. Later, the method was adapted for targeted suppression of gene expression without changes to the DNA sequence. The specific suppression of transcription with CRISPR has been termed “CRISPR interference” (CRISPRi) [4–6].
For CRISPR-Cas9 interference, the Cas9 nuclease was deprived of endonuclease activity via the replacement of two amino acids (D10A and H841A) in the RuvC and HNH nickase domains of the original protein [5]. Since DNA-binding activity of dCas9 was not affected, a complex of this nuclease with the gRNA can reversibly bind the target DNA, blocking the transcription of the target genes. The presence of an inducible promoter makes the regulation of the CRISPRi system more subtle and precise [7]. As a result, CRISPRi-mediated gene repression can reach 99% and lead to phenotypic changes similar to gene knockout [8]. This approach makes it possible to study the functions of unknown genes without resorting to lengthy and complex production of deletions.
The CRISPR-dCas9-mediated RNA interference system optimized for mycobacteria [9] includes a vector for the expression of dCas9 pRH2502 and a vector pRH2521 to clone spacers that encode the gRNA in order to target a gene of interest. The authors of [9] demonstrated the suppression of the expression of several Mtb genes and the potential to modulate the degree of transcription inhibition. The key factor affecting the efficiency of expression suppression was the proximity of the protospacer to the promoter region.
As a method, CRISPR interference with the pRH2502/pRH2521 system includes the following steps. The first is the cloning of the selected gRNAs into the pRH2521 vector and then sequential transformation of the pRH2502 vector with dCas9 into Mtb (obtaining Mtb-pRH2502 cells), followed by transformation of the vector pRH2521-gRNA into Mtb-pRH2502 (obtaining Mtb-pRH2502/pRH2521-gRNA cells). This is followed by the induction of CRISPR interference and analysis of the results via real-time polymerase chain reaction (PCR). It turned out that the efficiency of the cloning of gRNA spacers into the pRH2521 vector is rather low. In this regard, we set the task of improving the efficiency of cloning by modifying the pRH2521 vector. The modified system was used to assess the possibility of suppressing the transcription of adenylate cyclase genes (AC) mycobacteria, which are of significant scientific interest from the standpoint of their influence on the Mtb physiology.
Cyclic 3', 5'-AMP (cAMP) is one of the most widespread secondary messengers of bacteria, archaea, and eukaryotes [10].
In mycobacteria, AC, which synthesize cAMP, are encoded by an unusually large number of different genes. At the same time, cAMP signaling in Mtb is not associated with the classical system of catabolic repression characteristic of many bacteria [11]. The cAMP level in tubercle bacillus practically does not depend on the amount of glucose in the cultivation medium and regulates other metabolic pathways, as well as the interaction of the pathogen with the host [12].
We previously showed [13] that overexpression of the AC Rv2212 gene prevents the transition of Mtb to a dormant state. Cells overexpressing ACRv2212 demonstrated a significantly higher growth rate in the lungs and spleen of infected mice as compared to the control strain and, in contrast to the control strain, led to the death of TB-resistant mice [13]. The contribution of the remaining AC genes to the virulence of Mtb is currently unknown.
The goal of this work is to modify the pRH2502/pRH2521 system used for the CRISPR-dCas9-mediated RNA interference of mycobacteria and to assess its potential for CRISPR interference of the AC genes Rv2212, Rv1320c, Rv1319c, Rv1318c.
MATERIALS AND METHODS
Vectors for the expression of dCas9 and sgRNA. In order to create a system for CRISPR/Cas knockdown the following vectors (Addgene, Great Britain), optimized for work with mycobacteria [9], were acquired: plasmid pRH2502 carrying dCas9Spy from Streptococcus pyogenes and plasmid for Cas9 pRH2521 gRNA.
Modification of the pRH2521 vector to optimize the cloning of gRNA spacers. The α peptide β-galactosidase gene lacZα was amplified to obtain vector pRH2521-α amplified from the pCRCT plasmid. The PCR product was hydrolyzed at the BbsI sites and cloned into the pRH2521 vector, which was previously hydrolyzed by BbsI. As a result, the created plasmid allowed the selection of bacterial colonies containing an insert of spacers via blue–white selection in the presence of the substrate β-galactosidase X-Gal. Colonies of cells carrying the empty vector pRH2521-α were stained blue, while bacterial colonies containing an insert of spacers lost the α peptide and did not show blue staining in the presence of the chromogenic substrate β-galactosidase X-Gal. Table 1 shows the names and sequences of primers; the BbsI site is in bold.
Table 1. .
Oligonucleotides and primers used in the study
| Name | Sequence | Amplified area |
|---|---|---|
| UpBbsLacZα |
ATTTCTACGGGAATGTCTTCTCAG CCGCTACAGGGCGCGTCCCAT |
Β-galactosidase α-peptide gene |
| LowBbsLacZα |
AGTAGAAAAACGAGTCTTCTGGAA AGCGGGCAGTGAGCGCAA |
|
| gRNA Rv2212 | CTTGGACTTCGACGCCCTCG | Guide RNA for the Rv2212 gene |
| gRNA Rv1320c | GCCACCACTCGACACTTGCC | Guide RNA for the Rv1320c gene |
| UpRT2212 | GCGGGCCGGCTTGCTCACCTACCT | Gene Rv2212 |
| LowRT2212 | CGGCGTTCGGCTTGCACCATCTCT | |
| UpRT1320 | CCGCATTGGGTGTCGTTCG | Gene Rv1320c |
| LowRT1320 | TGAGCCAGTAGGTGCCCAGTATGA | |
| UpRT1319 | CCGACGGGCGCAACACCTT | Gene Rv1319с |
| LowRT1319 | ACGCCCCGCCGATATCCCACAGAA | |
| UpRT1318 | TCCCGAAGACAAGGCACTGG | Gene Rv1318c |
| LowRT1318 | CCTGGCACTCGGGCATTTCGT T |
Selection of gRNA spacers for Mtb adenylate cyclase genes. At the request of the authors of this work, the Mtb genome was added to the resource http://grna. ctegd.uga.edu/, which was used for the selection of gRNA spacers. gRNAs were selected for the knockdown of selected AC genes with Cas9 endonuclease. Table 1 shows the sequences of the guide RNA spacers.
The gene expression of operons Rv1320c, Rv1319c, and Rv1318c was suppressed with one gRNA guide.
Cloning of gRNA spacers for Mtb adenylate cyclase genes. Guide RNA spacers were selected according to the principle of maximum proximity to the start of transcription, as far as the presence of a PAM motif in the target region allowed (Fig. 1).
Fig. 1.
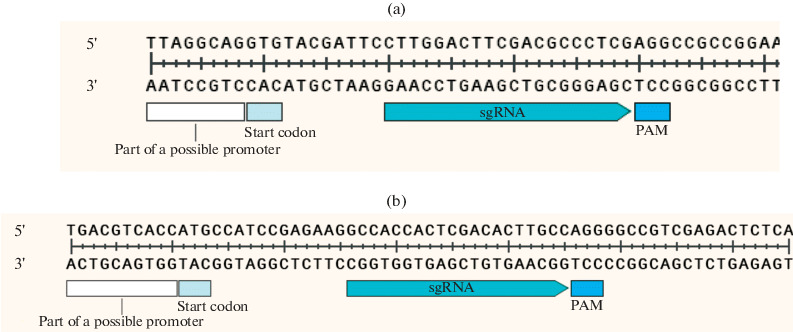
Mutual arrangement of gene interference elements Rv2212 (a) and Rv1320с (b).
Oligonucleotides were designed in order to clone gRNA spacers. After annealing to each other, the oligonucleotides formed sticky ends compatible with the sticky ends of the pRH2521 α vector, which formed after its hydrolysis by restriction endonuclease BbsI (Fig. 2).
Fig. 2.
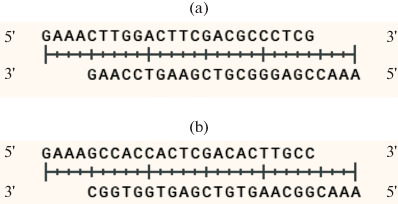
Guide RNA spacer for suppression the gene expression of Rv2212 (a) and the operon genes Rv1320c-Rv1319c-Rv1318c (b).
Bacterial strains, media, and cultivation conditions. The study used the bacterial strains Escherichia coli DH5α and M. tuberculosis H37Rv.
E. coli DH5α cells were grown in Luria-Bertani medium (LB) with the addition of antibiotics when necessary (50 μg/mL kanamycin (Km) and/or 50 μg/mL hygromycin (Hg). Mtb was cultured in Middlebrook 7H9/7H10 media supplemented with the growth additive OADC (Himedia, India), 0.025% tyloxapol, and, if necessary, antibiotics (20 μg/mL Km and/or 25 μg/mL Hg). The mycobacteria were cultivated in accordance with the recommendations of T. Parish [14].
Vectors of the CRISPRi system were transformed into Mtb sequentially (pRH2502, then pRH2521-α-gRNA) via electroporation on a Gene Pulser Xcell device (Bio-Rad, United States) according to the protocol [14].
After electroporation Mtb with vector pRH2502, single colonies were obtained on medium with Km after 4 weeks. Cells with pRH2502 plasmid were grown in liquid medium and transformed with pRH2521-α-gRNA plasmids. Colonies containing a pair of vectors pRH2502/pRH2521-α-gRNA were obtained after 4 weeks on a medium with Km and Hg. The transformants were then grown in liquid medium for 2–3 weeks, after which gRNA synthesis and dCas9 expression were induced via the addition of tetracycline at a concentration of 20 ng/mL.
Sample preparation for real-time PCR. After 24 h of induction, the cell biomass was collected and the total RNA was isolated with a commercial RNeasy Mini Kit (Qiagen, Germany). It was then purified of DNA impurities via treatment with DNase I Ambion DNase I RNase free (Thermo Scientific, United States), which does not contain RNase. The removal of DNA contaminants was confirmed via PCR. The prepared RNA samples served as a template for cDNA synthesis with a random primer. The resulting cDNA was used as a template for real-time PCR.
Real-time PCR. IQ SYBR Green Supermix Sample kits (Bio-Rad, United States) were used for real-time PCR. Reactions were carried out with a Mini Opticon Real-time PCR System (Applied Biosystems, United States) according to the manufacturer’s protocol. The gene was used as a 16S rRNA reference gene. The experiments were carried out in three technical repetitions. Table 1 presents the specific primers used in the reaction.
The qRT-PCR results were processed with the LinRegPCR v. 2017.0 program (Heart Failure Research Center, Netherlands), and the obtained data were analyzed with the 2–∆∆Ct method.
RESULTS AND DISCUSSION
Modification of the pRH2521 vector to optimize the cloning of gRNA spacers. The pRH2521/pRH2502 two-plasmid system was originally developed for CRISPR interference (CRISPR knockdown) of mycobacterial genes [9]; the corresponding modifications and properties of the vectors are described above. However, it turned out that the efficiency of the cloning of gRNA spacers into the BbsI restriction sites of the pRH2521 vector was low, less than 10%, which made it difficult to select clones carrying the plasmid with spacers. To facilitate the cloning of gRNA spacers into the vector pRH2521, we cloned the lacZα gene of the β-galactosidase α peptide using the BbsI sites. As a result, cell colonies carrying the empty vector pRH2521-α were stained blue, and bacterial colonies containing the vector with inserted spacers lost the α peptide and did not have a blue color in the presence of X-Gal, a chromogenic substrate of β-galactosidase. This modification of the pRH2521 vector made it easy to select target clones via blue–white selection.
Evaluation of the efficiency of the suppression of the transcription of genes Rv2212, Rv1318, Rv1319, and Rv1320 with the pRH2502 /pRH2521-α CRISPRi system. The expression of the Rv2212 gene was suppressed by 43% as a result of CRISPRi with the system pRH2502/pRH2521-α (Fig. 3). The genes Rv1320c, Rv1319c, and Rv1318c are included in a united operon and are transcribed in the direction from Rv1320c to Rv1318c. Transcription of genes of the Rv1320-Rv1319-Rv1318 operon with the selected gRNA was not suppressed. This result is apparently associated with the low expression level of the studied operon under conditions in vitro. It cannot be ruled out that the operon expression under the conditions of infected macrophages will be higher and, accordingly, it will be possible to suppress the expression of selected genes and to observe the phenotypic effects of such suppression.
Fig. 3.
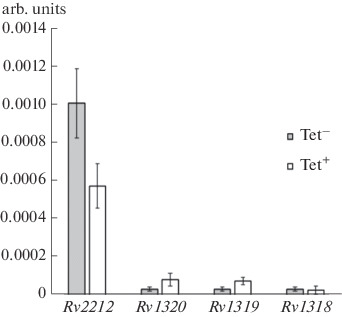
Relative expression (rel. units) of the adenylate cyclase genes of Rv2212 and operon Rv1320c-Rv1319c-Rv1318c under the activation of the pRH2502/pRH2521-α system with tetracycline (Tet+). The expression level in the absence of tetracycline induction (Tet–). The mean values of the ± standard deviation are presented.
The authors of the pRH2502/pRH2521 system reported suppression of the expression level from 10–50 to 80–90% for genes inhA, dfrA, wag31, and ftsZ. The first two are targets for isoniazid and methotrexate, respectively [15, 16], while the other two are involved in the formation of cell morphology [17, 18]. The specificity of the choice of interference targets made it possible to detect the suppression of expression and to observe phenotypic manifestations of CRISPRi work.
A more effective reduction in gene expression could possibly be achieved via the selection of a DNA region complementary to the promoter region of the analyzed genes as a protospacer; however, the location of the promoter requires clarification for inhibited adenylate cyclase genes. Another limitation of the method was the need for a specific PAM (NGG) to be present in the immediate vicinity of the protospacer. Singh et al. [9] studied different target variants (capturing element 35 of the promoter, a part of the 5'‑untranslated region between the transcription start site and the translation initiation codon, and the sequence at the 5'-end of the protein-coding region of DNA). Based on their results, the authors concluded that it is important to choose a protospacer near the promoter region. Unfortunately, it is not possible to find suitable gRNAs in functionally different regions of the template for all genes. Likewise, the promoter region is not known for all genes. Moreover, it is far from always possible to detect PAM in the narrow region of the desired annealing site of the gRNA. This is also a limitation of the method and, apparently, one of the reasons for the unsuccessful suppression of the Rv1320-Rv1319-Rv1318 operon.
Our results indicate that the performed modification of the pRH2521 vector for optimization the cloning of gRNA spacers makes it easy to select target clones. The PRH2502/pRH2521-α system can be effectively used for CRISPR interference, but it has limitations that are determined by the possibility of the detection of PAM in the region of the mapped promoter.
FUNDING
The study was partly supported by the Russian Foundation for Basic Research (project no. 1901500149A).
COMPLIANCE WITH ETHICAL STANDARDS
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
REFERENCES
- 1.Global Tuberculosis Report, Geneva: World Health Organization, 2020.
- 2.Zhang Y., Ioerger T., Huttenhower C., Long J., Sassetti C., Sacchettini J. PLoS Pathog. 2012;8:e1002946. doi: 10.1371/journal.ppat.1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang, F., Wen, Y., and Guo, X., Hum. Mol. Genet., 2014, vol. 23, no. R 1, pp. R40–R46. [DOI] [PubMed]
- 4.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qi L., Larson M., Gilbert L., Doudna J., Weissman J., Arkin A., Lim W. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larson M., Gilbert L., Wang X., Lim A., Weissman J., Qi L. Nat. Protoc. 2013;8:2180–2196. doi: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehrt S., Schnappinger D. Future Microbiol. 2006;1:177–184. doi: 10.2217/17460913.1.2.177. [DOI] [PubMed] [Google Scholar]
- 8.Cleto S., Jensen J., Wendisch V., Lu T. ACS Synth. Biol. 2016;5:375–385. doi: 10.1021/acssynbio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh A., Carette X. L. Nucleic Acids Res. 2016;44:e143. doi: 10.1093/nar/gkw625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Botsford J., Harman J. Microbiol. Rev. 1992;56:100–122. doi: 10.1128/mr.56.1.100-122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Carvalho L., Fischer S., Marrero J., Nathan C., Ehrt S., Rhee K. Chem. Biol. 2010;17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Bai G., Schaak D., McDonough K. FEMS Immunol. Med. Microbiol. 2009;55:68–73. doi: 10.1111/j.1574-695X.2008.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shleeva, M., Kondratieva, T., Demina, G., Rubakova, E., Goncharenko, A., Apt, A., and Kaprelyants, A., Front. Cell. Infect. Microbiol., 2017, vol. 7, no. 370. [DOI] [PMC free article] [PubMed]
- 14.Mycobacteria Protocols, Parish, T. and Roberts, D., Eds., New York: Media Springer Science + Business, 2015.
- 15.Larsen M., Vilchèze C. Mol. Microbiol. 2002;46:453–466. doi: 10.1046/j.1365-2958.2002.03162.x. [DOI] [PubMed] [Google Scholar]
- 16.White E., Ross L., Cunningham A., Escuyer V. FEMS Microbiol. Lett. 2004;232:101–105. doi: 10.1016/S0378-1097(04)00038-2. [DOI] [PubMed] [Google Scholar]
- 17.Kang C., Nyayapathy S., Lee J., Suh J., Husson R. Microbiology. 2008;154:725–735. doi: 10.1099/mic.0.2007/014076-0. [DOI] [PubMed] [Google Scholar]
- 18.Dziadek J., Rutherford S., Madiraju M., Atkinson M., Rajagopalan M. Microbiology. 2003;149:1593–1603. doi: 10.1099/mic.0.26023-0. [DOI] [PubMed] [Google Scholar]