

Abstract
The striatin-interacting phosphatase and kinase (STRIPAK) complex is a large multisubunit protein phosphatase 2A (PP2A) assembly that integrates diverse cellular signals in the Hippo pathway to regulate cell proliferation and survival. The architecture and assembly mechanism of this critical complex are poorly understood. Using cryo-EM, we determine the structure of the human STRIPAK core comprising PP2AA, PP2AC, STRN3, STRIP1, and MOB4 at 3.2 Å resolution. Unlike the canonical trimeric PP2A holoenzyme, STRIPAK contains four copies of STRN3 and one copy of each the PP2AA–C heterodimer, STRIP1, and MOB4. The STRN3 coiled-coil domains form an elongated homotetrameric scaffold that links the complex together. An inositol hexakisphosphate (IP6) is identified as structural cofactor of STRIP1. Mutations of key residues at subunit interfaces disrupt the integrity of STRIPAK, causing aberrant Hippo pathway activation. Thus, STRIPAK is established as a noncanonical PP2A complex with four copies of regulatory STRN3 for enhanced signal integration.
Introduction
Protein phosphatase 2A (PP2A) is a major serine/threonine phosphatase with myriad functions and forms many distinct complexes with different regulatory subunits 1-10. The PP2A core enzyme consists of the scaffold subunit A (PP2AA) and the catalytic subunit C (PP2AC). This heterodimeric core associates with a variable regulatory B subunit to form a trimeric holoenzyme, with the B subunit mediating substrate recruitment and subcellular localization. There are four families of regulatory B subunits: B (B55 or PR55), B′ (B56 or PR61), B″ (PR72), and B‴ (striatin).
The striatin-interacting phosphatase and kinase (STRIPAK) complex is a large multisubunit PP2A complex with striatin being the regulatory subunit 2,11-17. In addition to the PP2AA–C heterodimer and striatins (STRN1, STRN3 and STRN4), human STRIPAK contains several other core subunits, including STRN-interacting protein 1 or 2 (STRIP1 or STRIP2), and the MOB family member 4 protein (MOB4). This invariable STRIPAK core further associates with the cerebral cavernous malformation 3 protein (CCM3), which recruits one GCKIII family kinase (MST3, MST4, or STK25) to form distinct intact STRIPAK complexes 18. Thus, STRIPAK is a non-canonical phosphatase complex that contains both kinase and phosphatase subunits for enhanced regulation. In response to extracellular stimuli, STRIPAK can assemble into different complexes with additional variable subunits to regulate diverse cellular processes, including cell proliferation and survival, cell polarity, cell migration, and stem cell differentiation.
STRIPAK is a key negative regulator of the Hippo pathway that controls tissue homeostasis and suppresses tumorigenesis 19-27. Aside from the invariable catalytic core, the STRIPAK complex requires the addition of another two specific regulatory modules – SLMAP–SIKE1 and STK25–CCM3 – to regulate the Hippo pathway 2,13,28. The Hippo kinases MST1 and MST2 (MST1/2) are activated through trans-autophosphorylation at their activation loop (T-loop) 21,29-31. Once activated, active MST1/2 can further autophosphorylate multiple threonines in the linker between the N-terminal kinase domain and the C-terminal dimerization domain 32. These phospho-threonines bind the adaptor protein SLMAP, which in turn recruits STRIPAK to reverse T-loop phosphorylation of MST1/2 26,27. Thus, STRIPAK restrains MST1/2 activation through feedback inhibition. Remarkably, STRIPAK also serves as a signal integrator to mediate diverse upstream regulatory inputs into the Hippo pathway 26,33-35. Dysregulation of STRIPAK has been linked to tumorigenesis 11,12,36-39.
To better understand the molecular mechanism underlying the assembly of STRIPAK, we have determined the cryo-EM structure of the human STRIPAK core at 3.2 Å resolution. Our structure provides the first high-resolution depiction of STRIPAK, the most complicated PP2A holoenzyme, and reveals an unexpected subunit stoichiometry that has implications for the mechanisms and functions of STRIPAK in the Hippo pathway and beyond.
Results
Overall structure of human STRIPAK core complex.
The human STRIPAK core complex was produced in insect cells using the biGBac expression system 40 (Fig. 1a). The protein complexes were purified using a combination of affinity and size exclusion chromatography (Fig. 1b and Extended Data Fig. 1a). In the presence of recombinant SLMAP–SIKE1, the STRIPAK core complex dephosphorylated MST2 pT180 much more efficiently than the PP2AA–C heterodimer did (Fig. 1c), indicating that the recombinant STRIPAK core complex was functional.
Fig. 1 ∣. Cryo-EM structure of human STRIPAK core.

a, Domain organization of human STRIPAK core components. Residue numbers for domain boundaries are indicated. CC, coiled-coil; NTD, N-terminal domain; CTD, C-terminal domain; CTT, C-terminal tail. b, The elution profiles of the STRIPAK core (blue line) and STRIPAK (orange line) complexes from size exclusion chromatography (SEC) purification using a Superose 6 column. Fraction numbers are indicated. c, In vitro phosphatase assay with PP2AA–C core enzyme or STRIPAK core. The relative anti-pT180 intensities were normalized against MST2 treated with PP2A inhibitor microcystin-LR (MCLR, 100%). Data are mean ± SEM of three independent experiments. d, Overall structure of STRIPAK core in surface (left) and cartoon (right) representations in front and back views. Source data for panel c are available online.
The STRIPAK core complex was subjected to single-particle cryo-EM analysis, resulting in a 3D reconstruction of the STRIPAK core with an overall resolution of 3.9 Å (Extended Data Fig. 1b-d). The cryo-EM map at this resolution did not, however, allow de novo building of the complete model for STRIP1 and the coiled-coil domain (CC) of STRN3. To improve the resolution, we performed cryo-EM analysis of a larger version of the STRIPAK complex that included both the core and STK25–CCM3 (Fig. 1b and Extended Data Figs. 1a and 2). A total of 87,779 selected particles yielded a 3D reconstruction of the STRIPAK complex with an overall resolution of 3.3 Å (Extended Data Figs. 3 and 4). A focused refinement approach was used to further improve the resolution to 3.2 Å, allowing us to build a reliable model for the STRIPAK core, including STRIP1 and STRN3 CC (Fig. 1d and Table 1). There was no definitive cryo-EM density for STK25–CCM3, presumably because of structural flexibility. However, we did observe additional weak density in the map of the STRIPAK complex, which could be attributed to STK25–CCM3. Therefore, STK25–CCM3 may stabilize the STRIPAK core through the weak binding to the core subunits.
Table 1 ∣.
Cryo-EM data collection, refinement and validation statistics
| STRIPAK (EMD- 22650, PDB 7K36) |
|
|---|---|
| Data collection and processing | |
| Magnification | 60,241 |
| Voltage (kV) | 300 |
| Electron exposure (e−/Å2) | 60 |
| Defocus range (μm) | 1.5 - 2.5 |
| Pixel size (Å) | 0.83 |
| Symmetry imposed | C1 |
| Initial particle images (no.) | 2,451,582 |
| Final particle images (no.) | 87,779 |
| Map resolution (Å) | 3.3 |
| FSC threshold | 0.143 |
| Map resolution range (Å) | 3.3 – 7.0 |
| Refinement | |
| Initial model used | PDB 2NPP, PDB 4N6J, PDB 2YMU, PDB 5YF4 |
| Model resolution (Å) | 3.4 / 3.4 / 3.5 |
| FSC threshold | 0 / 0.143 / 0.5 |
| Model resolution range (Å) | 3.5 |
| Map sharpening B factor (Å2) | −100 |
| Model composition | |
| Nonhydrogen atoms | 16,614 |
| Protein residues | 2,171 |
| Ligands | 5 (2 MN, 2 ZN, 1 IHP) |
| B factors (Å2) | |
| Protein | 131.34 |
| Ligand | 109.49 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 0.665 |
| Validation | |
| MolProbity score | 2.72 |
| Clashscore | 14.89 |
| Poor rotamers (%) | 7.2 |
| Ramachandran plot | |
| Favored (%) | 94.17 |
| Allowed (%) | 5.83 |
| Disallowed (%) | 0 |
The STRIPAK core adopts an elongated architecture with a size of 200 x 100 x 80 Å (Fig. 1d). At the bottom part of the STRIPAK complex, four STRN3 CCs form two parallel dimers, which interact anti-parallelly through their N-terminal end to further assemble into a rod-like homotetramer. The STRN3 CC tetramer extends to the entire length of STRIPAK and acts as a central platform to organize the assembly of STRIPAK. Located in the center of the complex, STRIP1 directly interacts with PP2AC, STRN3, and MOB4, and has a pivotal role in stabilizing the entire complex. The PP2AA–C heterodimer interacts with one end of the STRN3 CC tetramer through the first HEAT-repeat of PP2AA. At the opposite end of the tetramer, MOB4 connects STRIP1 to the WD40 domain of STRN3. Notably, although there are four STRN3 molecules in the STRIPAK complex, only one WD40 domain of STRN3 is visible in the cryo-EM map. The other three WD40 domains and all four linkers between the CC and the WD40 domain (residues 139-384) that contain the CCM3-binding region were completely unresolved in the cryo-EM map. Because the STRN3 CC tetramer only binds to one copy each of the other core components, the overall architecture of STRIPAK is asymmetric.
Tetramerization of STRN3 CC.
STRN3 comprises the N-terminal CC, the CCM3-binding middle region, and the C-terminal WD40 domain (Fig. 1a). Previous biochemical studies have suggested that STRN3 CC forms a homodimer that directly binds to PP2AA 41,42. The crystal structure of a shorter version of STRN3 CC (residues 86-131) reveals a parallel dimer 41. Based on clear side-chain densities in our cryo-EM map of the STRIPAK complex, we could unambiguously build a complete structural model of STRN3 CC, which reveals an unanticipated homotetrameric configuration (Fig. 2a, b). The four copies of STRN3 CC enable multiple interactors to engage a short sequence of STRNs. Therefore, STRN CC tetramer provides a central platform to organize STRIPAK assembly.
Fig. 2 ∣. Interactions between PP2AA and the STRN3 CC tetramer.

a, Cryo-EM density map and atomic model of human STRIPAK core at a resolution of 3.5 Å. b, Cartoon representation of the STRN3 CC tetramer. Protomers A-D are colored light blue, slate, cyan and teal, respectively. (Below) Detailed views of the boxed tetramer region: (I) Overall view of tetramer interface. The black oval indicates the 2-fold symmetry axis. (II) Interface between protomer A and C. (III) Interface between protomer B and C. c, Zoomed-in view of the interface between PP2AA and STRN3 CC expanded from the boxed region in a. d, Quantification of the relative binding intensity between the indicated STRIPAK core components and FLAG-tagged PP2AA wild type (WT) or its mutants in 293A cells, derived from co-immunoprecipitation (co-IP) experiments. e, Quantification of the relative binding intensity between the indicated STRIPAK core components and FLAG-tagged STRN3 WT or its mutants in STRN1/3/4 KO 293A cells, derived from co-IP experiments. The relative binding intensities of the mutants (d,e) were normalized against the WT (100%). f, Mock vector or indicated plasmids were transfected in control or STRN1/3/4 KO 293A cells. The pMST and MST levels were quantified based on immunoblots of transfected cell lysates. The quantified values were used to calculate the ratios. g, Quantification of YAP localization based on immunofluorescence staining of YAP. Approximately 100 cells were counted for quantification. N<C (blue), N=C (grey), and N>C (red) categories indicate YAP localization in cytoplasm, both cytoplasm and nucleus, and nucleus, respectively. h, Relative CTGF gene expression based on qRT-PCR analysis. Data in d–h are mean ± SEM of three independent experiments. Results were evaluated by Two-tailed unpaired t tests (*, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; ns, non-significant). Source data for graphs are available online.
Two STRN3 CC dimers, formed by protomers A-B and C-D, interact with each other in a head-to-head manner through their N-terminal end to form the anti-parallel tetramer (Fig. 2b). STRN3 CC can be divided into the N-terminal tetramer region (62-76) and the C-terminal dimer region (77-135). Each dimer region closely resembles the previous crystal structure of the STRN3 CC fragment with RMSDs of 1.05 and 1.62 Å, respectively (Supplementary Fig. 1a). The STRN3 CC tetramer region appears to have a pseudo-D2 symmetry, with each protomer associating with the other three protomers in a similar manner through exclusive hydrophobic interactions (Fig. 2b and Supplementary Fig. 1b).
Previous studies have reported that three striatin family members can form homo- or hetero-oligomers through the CC domain 43. Deletion of STRN1 residues 53-66 (corresponding to STRN3 residues 69-79) abolishes striatin oligomerization 42, which is consistent with our structural model that the tetramer region is critical for the formation of striatin oligomers. Interestingly, the tetramer region contains a caveolin-binding motif (residues 71-79) (Supplementary Fig. 1b) 44. It is, therefore, tempting to speculate that caveolin may cooperate with STRNs to organize membrane-associated signaling complexes.
Interaction between PP2AA–C and STRN3.
PP2AA consists of 15 huntingtin-elongation-A subunit-TOR (HEAT) repeats, which are arranged in a horseshoe-shape 1. Each HEAT repeat comprises two anti-parallel α helices. The catalytic subunit PP2AC binds to the interhelical loops of HEAT repeats 11-15. Compared to the crystal structure of the PP2AA–C heterodimer, the N-terminal HEAT repeats 1-10 of PP2AA in STRIPAK twist and swing by as much as 42 Å (Supplementary Fig. 1c). Similar drastic conformational differences of PP2AA are observed in trimeric PP2A holoenzymes involving other B subunits (Supplementary Fig. 1d) 7,10. Unlike the B subunits in trimeric PP2A holoenzymes, STRN3 does not make direct contacts with PP2AC, and its interaction with PP2AA is also less extensive. Only protomer A of the STRN3 tetramer makes extensive contacts with PP2AA HEAT repeat 1 (Fig. 2a, c). All STRN3 residues that contact PP2AA are conserved among STRNs (Supplementary Fig. 1b), indicating that all STRNs likely interact with PP2AA in a similar manner.
To validate the interface between PP2AA and STRN3 observed in our structure, we mutated the interface residues in PP2AA and STRN3, and tested the effects of these FLAG-tagged mutants in 293 cells with three STRNs deleted (STRN1/3/4 KO). Anti-FLAG immunoprecipitation was used to evaluate in vivo binding activity. Mutations of PP2AA that target STRN3-binding residues abolished its binding to STRN3, STRIP1, and MOB4, supporting our STRIPAK model (Fig. 2d). Additionally, these mutations also slightly reduced the binding of PP2AA to PP2AC. Thus, although PP2AA and PP2AC can interact with each other in the absence of regulatory subunits, incorporation into STRIPAK might further strengthen their interaction. Likewise, STRN3 mutations targeting PP2AA-binding residues greatly reduced the binding of STRN3 to PP2AA, PP2AC, and STRIP1 (Fig. 2e and Extended Data Fig. 5a). These mutations did not affect MOB4 binding, indicating that the STRN3-MOB4 interaction does not depend on the formation of intact STRIPAK complex. Consistent with the role of STRIPAK in suppressing the Hippo pathway, unstimulated STRN1/3/4 KO cells at low cell density exhibited aberrant Hippo pathway activation, as evidenced by elevated levels of phospho-MST1/2, phospho-MOB1, phospho-LATS1/2 (LATS1 and LATS2) and phospho-YAP levels, as well as decreased nuclear YAP and Hippo-target gene expression (Fig. 2f-h and Extended Data Fig. 5b-d). Expression of STRN3 wild-type (WT) in these cells restored STRIPAK formation and suppressed Hippo signaling, whereas expression of PP2AA-binding-deficient STRN3 mutants failed to do so. However, due to transient expression, STRN3 WT cannot fully rescue YAP nuclear localization in STRN1/3/4 KO cells (Fig. 2g). These results collectively demonstrate that the PP2AA–STRN3 interface is critical for the assembly and function of STRIPAK.
Methylation of the carboxyl terminus of PP2AC plays an important role in the formation of certain PP2A holoenzymes (Supplementary Fig. 1d) 7. The C-terminal tail of PP2AC does not contact STRN3, suggesting that methylation does not promote the association of striatin with the PP2AA–C heterodimer. Consistent with this structural observation, deletion of nine C-terminal residues of PP2AC does not prevent the striatin-PP2A interaction 17.
Structure and inositol hexakisphosphate (IP6) binding of STRIP1.
STRIP1 consists of two domains: the N-terminal N1221 domain (NTD) and the C-terminal DUF3402 domain (CTD) (Fig. 1a). The structures of both domains are previously unknown. With the 3.2 Å cryo-EM map, we were able to build a complete model of STRIP1 de novo (Fig. 3a). The N-terminal 68 residues, residues 335-420 in the NTD-CTD linker, residues 825-837 at the C-terminus, and several short loops are disordered, thus cannot be modelled. The well-folded part of STRIP1 contains 12 α helices (α1-α12) in the NTD and 19 α helices (α13-α31) in the CTD (Supplementary Fig. 2).
Fig. 3 ∣. IP6 is a structural cofactor of STRIP1.

a, Cartoon representation of human STRIP1. Binding interface for PP2AC, STRN3 or MOB4 was indicated as green, blue or yellow curve, respectively. ARM: armadillo repeat. b, Surface electrostatic potential representation (blue, positive; red, negative) of the STRIP1 ARM repeats in front and side views. The flanking helices of NTD (wheat) and CTD (orange) are shown as ribbons. The linker (residues 335-420) between NTD and CTD is as dashed line, and IP6 is as stick. c, Zoomed-in view of the IP6-binding site of STRIP1. IP6 is shown as stick, along with its cryo-EM density map (black mesh). IP6-binding residues are shown as sticks and labeled. d, Effects of mutations at the IP6-binding site of STRIP1 on STRIPAK formation. The mock vector, FLAG-STRIP1 WT or mutants were transfected into control or STRIP1 KO 293A cells. The total cell lysates (input) and anti-FLAG IP were blotted with the indicated antibodies. e, Quantification of the relative binding intensity between the indicated STRIPAK components and STRIP1 or its mutants in d. The relative binding intensities of the mutants were normalized against the WT (100%). f, Effects of mutations in STRIP1 on the ratios of pMST/MST, pMOB1/MOB1, pLATS/LATS, and pYAP/YAP in control or STRIP1 KO 293A cells transfected with mock vector or indicated plasmids. The total and phosphorylated protein levels were individually quantified based on immunoblots of transfected cell lysates. The quantified values were used to calculate the ratios. Data in e and f are mean ± SEM of three independent experiments. Results were evaluated by Two-tailed unpaired t tests (*, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; ns, non-significant). Source data for graphs are available online.
Unexpectedly, helices α2-α10 from the NTD and α16-α28 from the CTD fold into one contiguous structural entity that contains seven armadillo repeats (ARM) 45. Consistent with this observation, a Dali search identified several ARM repeat proteins, such as HspBP1 (PDB 1XQR), importin-α (PDB 5GXW), and β-catenin (PDB 4HM9), as the closest structural homologs of STRIP1 46. Each canonical ARM repeat consists of three α helices (H1-3), in which H2 and H3 pack against each other in an antiparallel fashion, and H1 lies perpendicular to H2 and H3. STRIP1 contains irregular ARM repeats (Fig. 3a and Supplementary Fig. 2). In particular, ARM4 and ARM5 have an extra short helix between H2 and H3 and ARM7 lacks H3, while ARM4 H3 is an extra-long helix that scaffolds the flanking helices α1 and α12 of the NTD.
The tandem ARM repeats in STRIP1 has a right-handed superhelical curvature with a long groove extending along the superhelix axis (Fig. 3b). This groove is formed by a ladder of parallel H3s and is positively charged. The flanking helices α1, α11, and α12 of the NTD bind to the groove. A similar array of parallel H2s, flanked by H1s, forms the convex surface of STRIP1. The flanking helix α29 of the CTD interacts extensively with the ridge formed by H2s from ARM4-6, whereas the flanking helices α13-15 of CTD loosely associate with the ARM repeats. Therefore, the ARM repeats of STRIP1 interact with the flanking regions to form an overall compact fold.
During the model building of STRIP1, we noticed an extra density in a positively charged pocket formed by H3s from ARM4-6. Intriguingly, inositol hexakisphosphate (IP6) can be unequivocally fit into this density (Fig. 3c). The identity of this cofactor as IP6 was further confirmed by mass spectrometry (Supplementary Fig. 3). IP6 is the most abundant lipid-derived metabolite in eukaryotes with diverse signaling properties. It is a structural cofactor frequently involved in protein-protein and protein-ligand interactions. STRIP1 binds to IP6 through nine basic residues and one hydrophobic residue (Fig. 3c and Supplementary Fig. 2), all of which are highly conserved among STRIP1 proteins of various species, suggesting that IP6 binding is a conserved feature of STRIP1. The flanking helices α11 and α12 of the NTD form a ‘lid’ to cover the IP6-binding pocket. IP6 orients the NTD lid to make extensive contacts with the α14-α15 loop of the CTD. These contacts in turn stabilize the tongs-like structure formed by helices α13-α15. Thus, IP6 is a structural cofactor of STRIP1 and connects the peripheral structural elements to the ARM repeats.
To test the functional importance of IP6 binding, we mutated IP6-binding basic residues K427 and R744 of STRIP1 to glutamate and tested whether IP6 binding was required for STRIPAK formation in human cells.6 binding was required for STRIPAK formation in human cells. When expressed at similar levels in STRIP1 KO cells, the STRIP1 K427E and R744E mutants had decreased binding to STRN3, PP2AA, PP2AC, and MOB4, as compared to the WT (Fig. 3d,e). Furthermore, STRIP1 K427E and R744E were deficient in suppressing aberrant activation of the Hippo pathway in STRIP1 KO cells (Fig. 3f). These results indicate that IP6 binding by STRIP1 is required for the assembly and function of STRIPAK.
STRIP1 as a hub for STRIPAK assembly.
STRIP1 is the central hub in the STRIPAK structure (Fig. 4a). It directly interacts with PP2AC, STRN3, and MOB4 through six discrete interfaces (denoted as interfaces 1-6). STRIP1 and PP2AA bind at two opposite sides of PP2AC. Specifically, STRIP1 binds to the structural elements of PP2AC that are close to its active site. It interacts with PP2AC through interfaces 1 and 2. Both interfaces are mainly mediated by hydrophobic interactions that together bury about 1,300 Å2 of exposed surface area. Mutations of PP2AC and STRIP1 that target these two interfaces disrupted STRIPAK formation to various degrees (Fig. 4b,c). We noticed that the PP2AC W209A mutant almost completely lost its binding to PP2AA, indicating that it might be misfolded. In addition, some single point mutations with drastic effects presumably targeted “hot spots” of these interfaces. We also tested some of these STRIP1 mutants in functional assays and showed that they were indeed deficient in suppressing the aberrant activation of the Hippo pathway in STRIP1 KO cells (Fig. 4d,e and Extended Data Fig. 6). Thus, the STRIP1–PP2AC interfaces revealed by our structure are functionally relevant.
Fig. 4 ∣. STRIP1 scaffolds the STRIPAK core.

a, Overlaid cartoon and surface representations of the STRIPAK core. The six interfaces between STRIP1 and other core components are highlighted by dashed boxes. Closeup views of interfaces 1-5 are shown and labeled. The potential hydrogen bonds are indicated by dashed red lines. b, Quantification of the relative binding intensity between the indicated STRIPAK core components and FLAG-tagged PP2AC WT or its mutants in 293A cells, derived from co-immunoprecipitation (co-IP) experiments. Each interacting protein level was quantified based on immunoblot and normalized against corresponding FLAG-tagged protein level. Normalized values were used to calculate the ratios. c, Quantification of the relative binding intensity between the indicated STRIPAK core components and FLAG-tagged STRIP1 WT or its mutants in STRIP1 KO 293A cells, derived from co-IP experiments. d, Quantification of the ratio of pMST/MST derived from immunoblot analysis of control or STRIP1 KO 293A cell lysates transfected with mock vector or indicated plasmids. e, YAP localization in control or STRIP1 KO 293A cells transfected with mock vector or indicated plasmids based on immunofluorescence staining analysis. Approximately 100 cells were counted for quantification. f, Quantification of the relative binding intensity between the indicated STRIPAK core components and FLAG-tagged STRIP1 WT or ΔCTT in STRIP1 KO 293A cells, derived from co-IP experiments. The relative binding intensities of the mutants in b,c,f were normalized against the WT (100%). g, Effect of STRIP1 ΔCTT mutant on YAP localization in control or STRIP1 KO 293A cells transfected with mock vector or indicated plasmids, derived from immunofluorescence staining analysis of YAP. Approximately 100 cells were counted for quantification. Data in b–g are mean ± SEM of three independent experiments. Results were evaluated by Two-tailed unpaired t tests (*, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; ns, non-significant). Source data for graphs are available online.
STRIP1 CTD associates with STRN3 through interfaces 3 to 5. The flanking structural elements of STRIP1 CTD span almost the entire STRN3 C-D dimer through these spatially separated interfaces. In particular, two structural elements—α13-loop-α14 and α15-turn-loop—form a tongs-like structure, which grabs the middle part of STRN3 CC. The extended α29-α30 loop interacts with the mid-section of the C-D CC dimer. Helices α30 and α31 bind at the C-terminal end of the C-D CC dimer. α30 also contacts the WD40 domain of STRN3. The α31-binding site on the C-D CC dimer overlaps with the PP2AA-binding site of protomer D, preventing a second PP2AA molecule from binding to this site on the C-D CC dimer. Thus, each STRN3 tetramer can only bind to one PP2AA molecule, contributing to the overall asymmetry of STRIPAK.
Mutations targeting these STRN3-binding interfaces of STRIP1, including R491A, L494A, L799A, V802A (Data not shown), and deletion of the C-terminal tail (ΔCTT; with residues 796-837 deleted), decreased STRIPAK formation (Fig. 4f and Extended Data Fig. 7a). Furthermore, expression of STRIP1 ΔCTT in STRIP1 KO cells did not suppress the aberrant activation of the Hippo pathway (Extended Data Fig. 7b-d). Finally, mutations of STRIP1 CTT have been linked to human diseases 47. These results together demonstrate that the observed STRIP1-STRN3 interactions are also critical for the assembly and function of STRIPAK.
MOB4 as a molecular glue connecting STRIP1 and STRN3.
MOB4 belongs to the Mps one binder (MOB) family of adaptor proteins that can bind phospho-peptides and regulate kinase activities 48. For example, MOB1 binds to LATS1/2 and promotes its activation by MST1/2 32. MOB4 adopts a canonical MOB fold that consists of a four-helix bundle at its core (Extended Data Fig. 8a). The N-terminal extension (NTE, residues 1-61) forms additional three short α helices: Nα1 (residues 20-23), Nα2 (residues 39-49), and Nα3 (residues 54-57). Although the sequences of MOB NTE are divergent (Extended Data Fig. 8b), Nα2 and Nα3 of MOB4 share structure homology with MOB1 NTE (Extended Data Fig. 8c). It has been shown that apo-MOB1 adopts an autoinhibited conformation with Nα2 blocking its LATS1/2 binding surface 32,49. As a result, binding of LATS1/2 requires the conformational change of MOB1 triggered by the phosphorylation of MOB1 NTE (Extended Data Fig. 8d). In the STRIPAK complex, MOB4 is sandwiched between STRIP1 and the WD40 domain of STRN3 and acts as a molecular glue to tether STRN3 WD40 to the main body of STRIPAK (Fig. 5a). Strikingly, MOB4 in STRIPAK structure adopts an autoinhibited conformation similar to that of apo-MOB1 (Extended Data Fig. 8c), indicating that phosphorylation-dependent conformational change is not required for its binding to the WD40 domain of STRN3 (Extended Data Fig. 8d).
Fig. 5 ∣. MOB4 tethers the WD40 domain of STRN3 to STRIP1.

a, Front and back views of MOB4 bound to STRIP1 NTD and STRN3 WD40. MOB4 and WD40 are in cartoon. STRIP1 and STRN3 CC are in both cartoon and surface representations. b, Superposition of MOB4–pMST4 (colored in salmon and blue, respectively; PDB code 5YF4) and MOB4–STRIP1 (colored in yellow and wheat, respectively). Potential hydrogen bonds are shown as dashed lines. c, Quantification of the relative binding intensity between the indicated STRIPAK core components and FLAG-tagged STRIP1 WT or STRIP1 D131K,E134K. d, Quantification of the relative binding intensity between the indicated STRIPAK core components and FLAG-tagged MOB4 WT or MOB4 3RE in MOB4 KO 293A cells, derived from co-immunoprecipitation (co-IP) experiments. e, Effect of MOB4 3RE on YAP localization in control or MOB4 KO 293A cells transfected with mock vector or indicated plasmids, derived from immunofluorescence staining analysis of YAP. Approximately 100 cells were counted for quantification. f, Quantification of the relative binding intensity between the indicated STRIPAK core components and FLAG-tagged MOB4 WT or MOB4 ΔN40, derived from co-IP experiments. g, Quantification of the relative binding intensity between the indicated STRIPAK core components and FLAG-tagged STRN3 WT or its mutants in STRN1/3/4 KO 293A cells, derived from co-IP experiments. The relative binding intensities of the mutants in c,d,f,g were normalized against the WT (100%). Data in c–g are mean ± SEM of three independent experiments. Results were evaluated by Two-tailed unpaired t tests (*, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; ns, non-significant). Source data for graphs are available online. h, A proposed model for STRIPAK in cell signaling.
On one side, MOB4 interacts with STRIP1NTD through interface 6 (Figs. 4a and 5a). Residues from helix H3 and the C-terminal tail of MOB4 interact with residues from ARM1 H2 and ARM2 H2 of STRIP1 through extensive electrostatic interactions (Extended Data Fig. 8e and Supplementary Fig. 2). In particular, STRIP1 E134 binds to the phospho-threonine (pT)-binding site formed by R161, R162 and R165 of MOB4 (Fig. 5b) 50. Mutations targeting this site, including double mutant STRIP1 D131K,E134K and triple mutant MOB4 R161E,R162E,R165E (3RE), decreased the formation of the STRIPAK core complex and led to deregulated Hippo signaling (Fig. 5c-e and Supplementary Fig. 4). Because pT328 of MST4 has been reported to bind to MOB4 at the same site (Fig. 5b) 50, MST4 binding to STRIPAK might lead to the reorganization of the STRIPAK core.
On the other side, MOB4 interacts with the WD40 domain of STRN3 through two discrete sites (Extended Data Fig. 8f,g). First, the WD40 domain of STRN3 folds into a canonical seven-bladed β propeller, which has a deep ligand-binding pocket at its top face (Extended Data Fig. 9a,b). Residues 12-25 of MOB4, including Nα1, bind to this pocket (Extended Data Figs. 4b and 8g). Second, H1 and H2 of MOB4 forms numerous contacts with a highly conserved insert (residues 484-491) in the β2C-β2D loop of STRN3 WD40 through both sidechain and backbone interactions (Extended Data Fig. 8f). Deletion of the N-terminal 40 residues (ΔN40) of MOB4 abolished the formation of STRIPAK (Fig. 5f and Extended Data Fig. 9c). Likewise, deletion of the WD40 domain (ΔWD40) of STRN3 or point mutations of key residues (such as R446E) also decreased STRIPAK formation (Fig. 5g and Extended Data Fig. 10a). Furthermore, disrupting the interacting sites between MOB4 and STRN3 caused aberrant regulation of Hippo signaling (Extended Data Figs. 9d and 10b-d). Thus, the MOB4-STRN3 interactions play important roles in STRIPAK assembly and function.
Discussion
Dynamic protein phosphorylation regulates virtually all biological processes. Selective dephosphorylation of specific substrates or specific sites on a given substrate is critical for the successful execution of key cellular events. How the relatively few phosphatases regulate the phospho-proteome in a substrate- and site-specific manner is poorly understood. One emerging theme is that a given phosphatase can be assembled into multiple complexes with different scaffolding and substrate-recruiting subunits, which confer specificity. As an unusually large PP2A assembly, STRIPAK is an epitome of phospho-regulatory enzymes. Through multiple scaffolding subunits and adaptors, STRIPAK integrates upstream signals to control Hippo pathway activation. The cryo-EM structure of human STRIPAK core complex presented in this study reveals key features of this non-canonical PP2A phosphatase complex, providing a foundation for understanding how STRIPAK acts as a signaling hub to recruit myriad binding partners for catalysis and regulation (Fig. 5h).
As a single polypeptide chain, the B/B′/B″ regulatory subunit of the canonical trimeric PP2A holoenzyme contacts both PP2AA and PP2AC and recruits substrates. In STRIPAK, STRN3 contacts PP2AA whereas STRIP1 contacts PP2AC. STRIP1 has also been reported to directly interact with phosphorylated MST2 (pMST2), a key STRIPAK substrate 15,34. MOB4 further stabilizes the STRIP1-STRN3 interaction. Thus, multiple subunits of STRIPAK, including STRN3, STRIP1, and MOB4, together form one structural core to fulfill the roles of the regulatory subunit of PP2A. This division-of-duty enables dynamic, functionality-specific regulation of STRIPAK. For example, STRIP1 binding to STRN3 has been shown to be regulated by upstream stimuli 15,34. Without STRIP1, the PP2AA–C and STRN3–MOB4 heterodimers do not interact efficiently, leading to inefficient substrate recruit and catalysis. Future structural studies of STRIPAK bound to substrates are needed to reveal how STRIP1 orients pMST2 for dephosphorylation by PP2AC.
The canonical trimeric PP2A holoenzyme contains one copy each of the PP2AA–C heterodimer and the B/B′/B″ regulatory subunit. By contrast, our structure reveals that STRIPAK has four copies of STRN3 (B‴ subunit) for each PP2AA–C heterodimer. The CC domain of STRN3 forms an elongated head-to-head tetramer. Only one out of four WD40 domains of the STRN3 tetramer is visible in the cryo-EM map, suggesting that the other three WD40 domains are tethered to the STRIPAK core through long, flexible linkers. These WD40 domains can sample large distances and fetch potential regulators and substrates to the catalytic core. For example, STRN3 WD40 has been reported to interact with adenomatous polyposis coli protein (APC) to regulate WNT signaling 51,52. None of the linkers that connect the CC and WD40 domains of STRN3 are visible in the cryo-EM map, consistent with their flexibility. The linker contains both binding motifs for additional STRIPAK components, such as CCM3 and SIKE1–SLMAP. Thus, STRIPAK has a structured core with multiple flexible tentacles that reach out in space to grab regulators and substrates. These tentacles can act independently to engage distinct interactors. This unexpected multivalency of STRN3 underlies the ability of STRIPAK to interact with myriad interacting proteins and integrate upstream signals.
About 10% STRIPAK particles form a dimer with C2 symmetry (Extended Data Figs. 2b and 3). Although we have no evidence to support the formation of the STRIPAK dimer in solution, its existence on the cryo-EM grid suggests that STRIPAK might further oligomerize, particularly when associated with membranes. The phosphorylated MST1/2 linker contains multiple pTM sites that can simultaneously recruit multiple copies of SLMAP. Each STRIPAK complex can potentially bind to multiple copies of SIKE1–SLMAP. In the future, it will be interesting to test whether these multivalent interactions involving flexible linkers lead to the formation of biomolecular condensates through liquid-liquid phase separation in vitro and in human cells.
STRIPAK represents the pinnacle of protein phosphatases with unusually intricate protein network that is readily tunable to regulate diverse cellular signaling. Our structural analysis reveals that STRIPAK has a rigid core with multiple, flexible regulatory tentacles. This unique architecture allows many regulators to engage the same catalytic scaffold, leading to the assembly of different mature complexes that can integrate upstream signals.
Methods
Construct design and cloning of human STRIPAK.
biGBac, baculoviral co-expression system was used to generate the recombinant human STRIPAK core complex. The coding sequences of human PP2AA, PP2AC with an N-terminal His10-TEV-HA-tag, STRN3 with an N-terminal His6-TEV-MBP-3C-tag and a C-terminal His6-tag, STRIP1 with an N-terminal Strep-tag, MOB4 with an N-terminal His6-TEV-2XStrep-3C-tag, SIKE1 with a C-terminal His6-tag, and SLMAP (residues 1-799) with an N-terminal His6-TEV-MBP-3C-tag were cloned into a baculoviral library vector pLIB, respectively (TEV, tobacco etch virus protease; 3C, PreScission protease from human rhinovirus (HRV 3C)). To assemble the cDNAs of five STRIPAK core components into a single expression vector, the gene expression cassettes (GECs) consisting of polyhedrin promoter, cDNA, and SV40-terminator were amplified by PCR from pLIBs of PP2AA, PP2AC, STRN3, STRIP1, and MOB4, respectively. Linearized pBIG1a vector and 5 PCR products were combined in a Gibson assembly reaction, leading to the assembly of a polygene cassette in a circular pBIG1a construct. For PP2AA–C core enzyme, PCR-amplified GECs of PP2AA and PP2AC were combined in a Gibson assembly reaction with the pBIG1a vector. For the SLMAP–SIKE1 complex, PCR-amplified GECs of SLMAP and SIKE1 are combined in a Gibson assembly reaction with the pBIG1a vector. The assembled pBIG1a constructs were analyzed by digestion with SwaI to release individual GECs.
The coding regions of human STK25 and CCM3 were cloned into a modified pET-28 vector (Novagen) that included a TEV cleavage site at the N-terminus, respectively. Human MST2D146N catalytic inactive mutant was generated using QuikChange mutagenesis kit (Agilent Technologies) from a modified pGEX-6p-MST2 vector as previously described 32. Mutant constructs were verified by DNA sequencing.
Protein expression and purification.
Protein expression and purification for the STRIPAK complexes are described in Supplementary Note 1. To generate the phosphorylated MST2D146N (pMST2 D146N), GST-MST2D146N was purified using similar procedure for GST-MST2 as previously described 32. The kinase domain (MST2KD, residues 16-313) of MST2 31 was used to phosphorylate MST2D146N. GST-MST2D146N was mixed with MST2KD at 100:1 ratio supplemented with 1 mM ATP and 5 mM MgCl2, and the reaction was incubated at room temperature for 30 min. Next, the phosphorylated GST-MST2D146N was bound to Glutathione Sepharose 4B beads (GE Healthcare). The beads were washed and cleaved with TEV to remove the GST moiety. pMST2D146N was further purified by SEC with a 10/300 Superdex 200 column equilibrated with buffer E, and fractions containing pMST2D146N were pooled and concentrated to 100 μM using a 30 kDa cut-off filter and stored at −80°C.
Mass spectrometry analysis.
The coding region of human STRIP1 (residues 41-800) was cloned into a modified pFastBac-MBP vector with MBP-tag fused at the N-terminus. MBP-STRIP1 was purified from High Five cells (Sigma). MBP-tag was removed by amylose beads after TEV cleavage overnight at 4°C. STRIP1 was further purified by SEC with a Superdex 200 Increase column (GE Healthcare) to over 90% purity based on SDS-PAGE. Next, STRIP1 was dialyzed against a buffer containing 20 mM ammonium formate and then concentrated to 0.5 mg/ml. Samples were directly injected onto a Sciex X500B Q-ToF mass spectrometer running Sciex OS v.1.3. Data was acquired from m/z = 100-1000 Da in negative ion mode. The presence of IP6 was confirmed by the presence of a number of peaks, including those at m/z = 658.8577 (M-H)−, 578.8908 (M-HPO3)−, 498.9241 (M-2HPO3)−, and 328.9250 (M-2H)2− (Supplementary Fig. 3). The two most abundant peaks are shown in Supplementary Fig. 3b.
In vitro phosphatase assays.
50 nM PP2AA–C or STRIPAK core was incubated with pMST2D146N (2 μM) and SLMAP–SIKE1 (2 μM) in phosphatase assay buffer (20 mM Tris-HCl, pH 8.0, 85 mM NaCl, 10 mM MgCl2, and 0.5 mM TCEP) at 30°C for 30 min, respectively. The PP2A inhibitor Microcystin-LR (Enzo Life Science) was used to block PP2A catalytic activity as control. Reaction mixtures (5 μL) were separated by SDS-PAGE and blotted with an antibody against pT180 of MST2 (Cell Signaling).
Cryo-EM data collection.
The STRIPAK sample at 2 mg/ml was applied to a glow-discharged Quantifoil R1.2/1.3 300-mesh gold holey carbon grid (Quantifoil, Micro Tools GmbH, Germany), blotted under 100% humidity at 4°C and plunged into liquid ethane using a Mark IV Vitrobot (FEI). Micrographs were acquired on a Titan Krios microscope (FEI) with a K3 Summit direct electron detector (Gatan) in the super-resolution counting mode, operated at 300 kV using the SerialEM software 53. The slit width of the GIF-Quantum energy filter was set to 20 eV. Micrographs were dose-fractioned into 32 frames at the dose rate of ~2 e−/Å2/frame.
Image processing and 3D reconstruction.
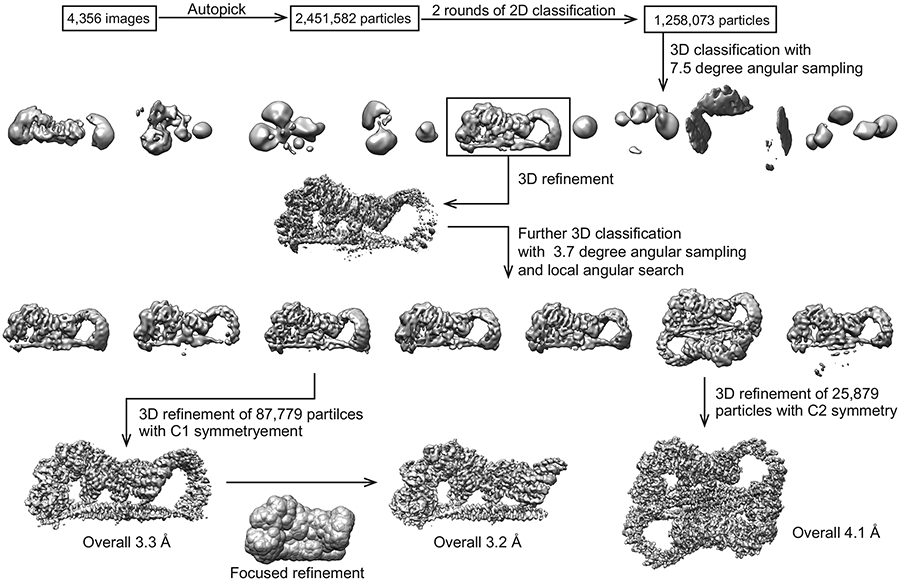
Movie frames of the STRIPAK complex were motion-corrected and binned 2-fold, resulting in the pixel size of 0.83 Å, and dose-weighted using the Motioncorr2 program 54. CTF correction were performed using the GCTF programs 55. Most of the subsequent processing steps were carried out using RELION 3 56,57. A few micrographs from the dataset were used for manual picking of ~1000 particles. These particles were subjected to 2D classification. Class averages representing projections of the complex in different orientations were used as templates for automated particle picking from the full datasets. A total of 2,451,582 particles were picked from 4,356 micrographs. Particles were extracted and binned by 4 times (leading to 3.32 Å/pixel) and subjected to two rounds of 2D classification. Particles from those 2D classes showing sharp structural features were chosen (1,258,073 in total) for 3D classification. The initial model for 3D classification was generated by the angular reconstruction method in EMAN2 58. Specifically, the angle relationships between selected class averages were calculated by using “common-line” method, and the initial map was reconstructed and optimized by the standard EMAN2 refinement procedure. After the first round of 3D classification with RELION, particles from one 3D class showing good secondary structural features was selected and re-extracted into the original pixel size of 0.83 Å. 3D refinements of this particle subset with C1 symmetry imposed resulted in a 3D reconstruction with relatively blurred density for many regions of the complex, indicating the remaining structural heterogeneities in the structure. To further improve the map, we performed another round of 3D classification by using local search in combination with finer angular sampling, resulting two major classes. One of the two classes is similar to the map from previous 3D refinement with C1 symmetry, but showed improved density. The final reconstruction of this class with C1 symmetry applied was resolved at overall 3.3 Å resolution. Another major class was determined as twice larger than the C1 map. Close inspection of this class revealed that this 3D reconstruction comprises two identical C1 complexes, related by a C2 symmetry. The final reconstruction of this class with C2 symmetry applied was resolved at overall 4.1 Å resolution.
7,375 micrographs were collected for the STRIPAK core complex. 377,072 particles were selected by 2D classification. The same image processing procedure was used to obtain the 3D reconstruction of the STRIPAK core complex, yielding a map at 4.3 Å resolution. Local resolution for both STRIPAK and STRIPAK core maps were calculated in RELION. Resolution was estimated by applying a soft mask around the protein density with the Fourier Shell Correlation (FSC) 0.143 criterion 59.
Model building, refinement and validation.
The model building was carried out by fitting the crystal structures of PP2AA–C (PDB 2NPP) 60, STRN3 coiled-coil (PDB 4N6J) 41, WD40 domain (a structure model by SWISS-MODEL from PDB 2YMU), MOB4 (PDB 5YF4) 50 in the electron microscopy density maps of STRIPAK core using Chimera 61, and subsequently manual building in Coot 62. Model building for the WD40 domain of STRN3 was initiated by docking the SWISS-MODEL structure into the density. The top face of the β-propeller that interacts with MOB4 is substantially smaller than the opposite face, which allowed the determination of the overall orientation of the domain. The 7 β-sheets in the WD40 domain are similar in structure and therefore impose a certain degree of ambiguity in their assignment into the density. The assignment was largely guided by bulky residues, such as W386, Y390, F396, F693, Y699, and F712. In addition, the assignment took into consideration that residues in the WD40 domain that are conserved are involved in interacting with MOB4, including R446 and the conserved long loop spanning residues 484-491, and must be placed close to MOB4. STRIP1 did not have a previous structural model, thus it was de novo built based on the predicted secondary structures. The local resolution of the cryo-EM map for STRIP1 is ~ 3Å, which is substantially better than typical X-ray crystallographic maps at similar nominal resolution. The sidechains for the majority of the residues in STRIP1 and the bound IP6 molecule are unambiguously resolved. The fact that most of the residues in the protein form α helices also greatly reduces the level of difficulty in model building. The assignment of residues into the density took into consideration of bulky sidechains, charge complementarity of interacting charged residues, hydrophobicity of residues and secondary structure prediction of the sequence. Real-space refinement was carried out with secondary structure restraints using Phenix 63. Model geometries were assessed with MolProbity 64. Structures and maps in the figures were rendered with PyMOL (The PyMOL Molecular Graphics System, v.2.0, Schrödinger) or Chimera. The sequence-alignment figures were generated with ESPript 65.
Mammalian cell culture and transfection.
293A and 293FT cells were purchased from Thermo Fisher Scientific, and were authenticated by STR (short tandem repeat) DNA profiling. All cells were incubated at 37°C in a humidified 5% CO2 atmosphere. All cells were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 1% penicillin/streptomycin. Cells were routinely tested and confirmed free of mycoplasma contamination. Lipojet (Signagen) or Lipofectamine 2000 (Invitrogen) used for transient transfection into 293A or 293FT cells according to the manufacturer’s instructions.
For Hippo signaling analysis, mock vector or appropriate plasmids were transfected into control or KO 293A cell lines with Lipojet. 24 hours after transfection, 5 x 105 cells were re-plated into 60 mm dishes for immunoblotting and qRT-PCR, and 1 x 105 cells were seeded on Lab-Tek II 4 well chamber slides for immunofluorescence. 24 hours later, cells were either collected or fixed for further experiments.
Generation of knockout (KO) cell lines.
The sgRNAs of STRIP1, STRN3, and MOB4 were cloned into psCas9 (Addgene). The individual psCas9-sgRNA plasmids were transfected into 293A cells with the Lipojet reagent. One day after transfection, cells were selected with 1 μg/ml puromycin. After two days of selection, single cells were re-plated into individual wells of 96-well plates without treatment of puromycin. Single clones were tested by immunoblotting and DNA sequencing. For generating the STRN1/3/4 KO cell lines, the sgRNAs of STRN and STRN4 were cloned into plentiCRISPR v2 (Addgene). The plentiCRISPR v2-sgRNA, pMD2.G and psPax2 plasmids were co-transfected into 293FT cells with the Lipofectamine 2000 reagent. Two and three days after transfection, the lentiviral supernatants were harvested and concentrated with Lenti X-concentrator (Clontech). STRN3 KO cells were infected with the lentiviruses and 4 μg/ml polybrene. Two days after infection, cells were selected with 1 μg/ml puromycin. After three days of selection, single cells were sorted into individual wells of 96-well plates. But none of any triple or double KO cells were found. Instead, pools of STRN1/3/4 KO cells were used for the experiments. The following sgRNAs were used in this study: STRIP1, GGTGGGCACGTTCAATGCTT; STRN, AACAACCACCCGGGCGCCGG; STRN4, GCACTTTATCCAGCACGAGT; STRN3, TGCCTTGCTTCCACGTTAAC; MOB4, TACTTCTAGGGTTAGCATAA.
Antibodies, immunoblotting and immunoprecipitation.
The following antibodies were purchased from the indicated sources: anti-pMST1/2 (T183/T180; GTX133948, GeneTex); anti-STRN (A304-537A), anti-STRIP1 (A304-644A, Bethyl Laboratories Inc); anti-MYC (Roche); anti-FLAG (F1804 and F7425 , Sigma); anti-YAP (sc-101199), anti-STRN3 (sc-13562), anti-PP2AA (sc-6112, Santa Cruz Biotechnology); anti-MST1 (3682), anti-MOB1 (13730), anti-pMOB1 (T35; 8699), anti-LATS1 (3477), anti-pLATS1/2 (HM; 8654), anti-pYAP (4911), anti-GAPDH (2118), anti-PP2AC (2259), anti-rabbit immunoglobulin G (IgG) (H+L) (Dylight 800 or 680 conjugates), and anti-mouse IgG (H+L) (Dylight 800 or 680 conjugates, Cell Signaling); anti-MOB4 (A4590, ABclonal); anti-Tubulin (ab4074, Abcam); anti-STRN4 (PA5-63343, Thermo Fisher Scientific). Anti-FLAG M2 resin (A2220) was purchased from Sigma-Aldrich.
To check the effects of designed mutations on Hippo pathway activity or formation of STRIPAK complex, FLAG-tagged mutants were transfected into control or appropriate KO 293A cells. 24 hours later, cells were harvested and lysed with lysis buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.2% Triton X-100, protease inhibitors (Roche), and PhosSTOP (Roche) on ice for 20 min. Cell lysates were separated by centrifugation at 20,000 g for 20 min at 4°C. Cleared cell lysates were collected and subjected to further immunoblotting or immunoprecipitation. For immunoblotting, cell lysates and immunoprecipitates were analyzed by standard immunoblotting protocol. The membranes were scanned and band intensities were quantified by an Odyssey Infrared Imaging System (LI-COR). For immunoprecipitation, collected cell lysates were incubated with anti-FLAG M2 resin for 2 hr at 4°C. After incubation, resins were washed three times by wash buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1 mM EDTA, and 1% Triton X-100, and eluted by SDS sampling buffer. Eluates were separated by SDS-PAGE and blotted with appropriate antibodies.
Immunofluorescence.
Cells were washed once with PBS and fixed with 4% paraformaldehyde for 20 min. Cells were then permeabilized with PBS containing 0.2% Triton X-100 (PBS-T) for 20 min and blocked with PBS-T containing 3% BSA for 30 min at room temperature. Cells were incubated with the primary antibody in PBS-T containing 3% BSA for 1 hr. Cells were then washed with PBS-T three times and incubated with secondary antibody for 1 hr. Cells were washed again with PBS-T three times and mounted in ProLong Gold Antifade reagent with DAPI (Invitrogen). Cells were visualized with a DeltaVision microscope system (Applied Precision). Approximately 100 cells were counted in randomly chosen fields for each sample. Experiments were repeated three times for statistical analysis.
RNA isolation and real-time qRT-PCR.
Total RNA was isolated from cells using the Trizol reagent (Invitrogen). cDNA was obtained by reverse transcription reactions using the Reverse transcription kit (Applied Biosystems). Real-time PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad) and the 7900HT Fast Real-Time PCR System (Applied Biosystems). Relative abundance of mRNA was normalized to GAPDH.
Statistical analysis.
For quantification of phospho- and total protein, the phospho- and total band intensities were individually normalized to the intensity of GAPDH from the same gel. The ratios of normalized phospho-proteins versus normalized total proteins were calculated and plotted. For quantification of relative interaction intensity, every interacting protein level was normalized by FLAG-tagged mutant protein level. Normalized values were used for calculating the ratios. Data were plotted as mean ± SEM of three independent experiments, unless otherwise noted. Results were evaluated by Two-tailed unpaired t tests (*, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; ns, non-significant). The graphs and statistical calculations were performed using Prism (GraphPad).
Reporting Summary.
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 ∣. Purification of human STRIPAK complexes and single-particle cryo-EM analysis of the STRIPAK core.

a, Coomassie blue staining of fractions from size exclusion chromatography (SEC) purification of the STRIPAK core (left) and STRIPAK (right) complexes. Highlighted fraction (red) was used for cryo-EM analysis. b, Cryo-EM data processing for initial model generation. c, Cryo-EM density map and atomic model of the STRIPAK core at a resolution of ~3.9 Å. d, Final reconstruction with C1 symmetry colored based on local resolution.
Extended Data Fig. 2 ∣. Cryo-EM analyses of the STRIPAK complex.

a, Representative micrograph and 2D classes. b, Final reconstructions with C1 or C2 symmetry colored based on local resolution. c, Gold-standard Fourier shell correlation (FSC) curves of the final 3D reconstruction (C1 in orange and C2 in blue). d, Euler angle distribution of the particles used in the final C1 3D reconstruction.
Extended Data Fig. 3 ∣. Flow chart of cryo-EM image processing for the STRIPAK complex.
Extended Data Fig. 4 ∣. Cryo-EM map of the STRIPAK complex.

a, Overall cryo-EM map of human STRIPAK at a resolution of 3.5 Å in front and back views. b, Representative cryo-EM densities of the STRIPAK complex. The densities from the cryo-EM maps and the corresponding protein fragments are shown in surface and sticks, respectively. Residues numbers of each sample fragment are labeled. Representative residues from STRN3 WD40 or at the PP2AC–STRIP1 interface are labeled.
Extended Data Fig. 5 ∣. Mutational studies of STRN3 at the STRN3-PP2AA interface.

a, Effects of mutations in STRN3 on STRIPAK formation. Mock vector or FLAG-STRN3 mutants were transfected into control or STRN1/3/4 KO 293A cells as indicated. Lysates and FLAG IPs were subjected to immunoblotting. b, Effects of mutations in STRN3 on the ratios of pMST/MST, pMOB1/MOB1, pLATS/LATS, and pYAP/YAP in control or STRN1/3/4 KO 293A cells transfected with mock vector or indicated plasmids. c, Immunofluorescence staining of YAP localization in 293A control or STRN1/3/4 KO cells transfected with mock vector or indicated plasmids. Scale bar, 10 μm. d, Relative expression of YAP target genes CYR61 in 293A control or STRN1/3/4 KO cells transfected with mock vector or indicated plasmids. Data in b,d are plotted as mean ± SEM of three biologically independent experiments. Results were evaluated by Two-tailed unpaired t tests (*, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; ns, non-significant). Source data for graphs are available online.
Extended Data Fig. 6 ∣. Functional validation of PP2AC-interacting residues of STRIP1.

a, Effects of mutations in STRIP1 on the ratios of pMOB1/MOB1, pLATS/LATS, and pYAP/YAP in control or STRIP1 KO 293A cells transfected with mock vector or indicated plasmids. b, Immunofluorescence staining of YAP localization in 293A control or STRIP1 KO cells transfected with mock vector or indicated plasmids. Scale bar, 10 μm.
Extended Data Fig. 7 ∣. Functional validation of the C-terminal tail (CTT) of STRIP1.

a, Effects of mutation in STRIP1 on STRIPAK formation. Control or STRIP1 KO 293A cells were transfected with mock vector, FLAG-STRIP1 WT or ΔCTT mutant plasmid as indicated. Lysates and FLAG IPs were subjected to immunoblotting. b, Immunoblot of lysates of control or STRIP1 KO 293A cells transfected with mock vector, STRIP1 WT or ΔCTT mutant plasmid as indicated. HM indicates hydrophobic motif in LATS1/2. c, Quantification of the ratios of phospho- and total proteins in c. d, Immunofluorescence staining of YAP localization in 293A control or STRIP1 KO cells transfected with mock vector, STRIP1 WT or ΔCTT mutant plasmids. Scale bar, 10 μm.
Extended Data Fig. 8 ∣. MOB4 interacts with STRIP1 and the WD40 domain of STRN3.

a, Cartoon representation of MOB4. The N-terminal extension (NTE) and core of MOB4 are colored in slate and yellow, respectively. b, Sequence alignment of human MOB NTEs. The N-terminal α helices (Nαs) and residue numbers of MOB1 and MOB4 are shown above and below the sequences, respectively. c, Superposition of MOB4 with the crystal structure of unphosphorylated MOB1 (colored in grey; PDB 5B5V). The NTE and core of MOB1 are colored in teal and grey, respectively. d, Superposition of MOB4–STRN3 WD40 (this study) and pMOB1–LATS1 (colored in grey and salmon, respectively; PDB 5BRK). The NTE of pMOB1 is colored in red. e, Interface between MOB4 and STRIP1 NTD (colored in yellow and wheat, respectively). Potential hydrogen bonds are indicated with red dashed lines. f, Interface between MOB4 core and STRN3 WD40 (colored in yellow and blue, respectively). g, Interface between MOB4 Nα1 and STRN3 WD40.
Extended Data Fig. 9 ∣. Interactions between MOB4 and STRN3 WD40.

a,b, Overall structure of the STRN3 WD40 domain with cartoon (a) and surface representation (b) in the same view. Residues at the MOB4–STRIP1 interface are colored salmon. The seven blades of the WD40 domain are numbered from 1 to 7, and the strands of each blade are numbered A-D from the innermost strand to the outermost strand. The N- and C-termini are indicated. c, Immunoblot of lysates of control or MOB4 KO 293A cells transfected with mock vector, MOB4 WT or ΔN40 mutant plasmids. d, Quantification of the ratios of phospho- and total proteins in c.
Extended Data Fig. 10 ∣. Interactions between STRN3 WD40 and MOB4.

a, Immunoblot of lysates of control or STRN1/3/4 KO 293A cells transfected with mock vector or indicated plasmids. b, Quantification of the ratios of phospho- and total proteins in a. c, Quantification of YAP localization in control or STRN1/3/4 KO 293A cells transfected with mock vector or indicated plasmids. d, Relative expression of YAP target genes CTGF and CYR61 in control or STRN1/3/4 KO 293A cells transfected with mock vector or indicated plasmids. Data in b–d are plotted as mean ± SEM of three biologically independent experiments. Results were evaluated by Two-tailed unpaired t tests (*, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; ns, non-significant). Source data for graphs are available online.
Supplementary Material
Acknowledgements
We thank Hongtao Yu for helpful discussion. We thank Shun Liu for assistant in mutant analysis. Single particle cryo-EM data were collected at University of Texas Southwestern Medical Center (UTSW) Cryo-Electron Microscopy Facility, which is funded by a Cancer Prevention and Research Institute of Texas (CPRIT) Core Facility Support Award (Grant no. RP170644). We thank D. Nicastro for facility access and data acquisition. This study is supported in part by grants from the National Institutes of Health (CA220283 to X.Z.; GM132275 to X.L.), and grants from the Welch Foundation (I-1702 to X.Z.; I-1944 to X.-c.B.; I-1932 to X.L.). X.Z. and X.-c.B. are Virginia Murchison Linthicum Scholars in Medical Research at UTSW.
Footnotes
Competing interests
The authors declare no competing interests.
Data Availability
Cryo-EM density maps and atomic coordinates for human STRIPAK have been deposited in the Electron Microscopy Data Bank and wwPDB, respectively, under accession codes EMD-22650 and PDB 7K36. Source data are provided with this paper.
References
- 1.Shi Y Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484, doi: 10.1016/j.cell.2009.10.006 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Glatter T, Wepf A, Aebersold R & Gstaiger M An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Molecular systems biology 5, 237, doi: 10.1038/msb.2008.75 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janssens V, Goris J & Van Hoof C PP2A: the expected tumor suppressor. Current opinion in genetics & development 15, 34–41, doi: 10.1016/j.gde.2004.12.004 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Janssens V & Goris J Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353, 417–439 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Virshup DM & Shenolikar S From promiscuity to precision: protein phosphatases get a makeover. Mol Cell 33, 537–545, doi: 10.1016/j.molcel.2009.02.015 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Kamibayashi C et al. Comparison of heterotrimeric protein phosphatase 2A containing different B subunits. J Biol Chem 269, 20139–20148 (1994). [PubMed] [Google Scholar]
- 7.Cho US & Xu W Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 445, 53–57, doi: 10.1038/nature05351 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Lambrecht C, Haesen D, Sents W, Ivanova E & Janssens V Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol Biol 1053, 283–305, doi: 10.1007/978-1-62703-562-0_17 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Wlodarchak N et al. Structure of the Ca2+-dependent PP2A heterotrimer and insights into Cdc6 dephosphorylation. Cell Res 23, 931–946, doi: 10.1038/cr.2013.77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, Chen Y, Zhang P, Jeffrey PD & Shi Y Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol Cell 31, 873–885, doi: 10.1016/j.molcel.2008.08.006 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Z, Jiao S & Zhou Z STRIPAK complexes in cell signaling and cancer. Oncogene, doi: 10.1038/onc.2016.9 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Hwang J & Pallas DC STRIPAK complexes: structure, biological function, and involvement in human diseases. The international journal of biochemistry & cell biology 47, 118–148, doi: 10.1016/j.biocel.2013.11.021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goudreault M et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Molecular & cellular proteomics : MCP 8, 157–171, doi: 10.1074/mcp.M800266-MCP200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuck U, Radchenko D & Teichert I STRIPAK, a highly conserved signaling complex, controls multiple eukaryotic cellular and developmental processes and is linked with human diseases. Biol Chem, doi: 10.1515/hsz-2019-0173 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Tang Y et al. Architecture, substructures, and dynamic assembly of STRIPAK complexes in Hippo signaling. Cell discovery 5, 3, doi: 10.1038/s41421-018-0077-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herzog F et al. Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352, doi: 10.1126/science.1221483 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Moreno CS et al. WD40 repeat proteins striatin and S/G(2) nuclear autoantigen are members of a novel family of calmodulin-binding proteins that associate with protein phosphatase 2A. J Biol Chem 275, 5257–5263 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kean MJ et al. Structure-function analysis of core STRIPAK Proteins: a signaling complex implicated in Golgi polarization. J Biol Chem 286, 25065–25075, doi: 10.1074/jbc.M110.214486 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng Y & Pan D The Hippo Signaling Pathway in Development and Disease. Dev Cell 50, 264–282, doi: 10.1016/j.devcel.2019.06.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu V, Plouffe SW & Guan KL The Hippo pathway in organ development, homeostasis, and regeneration. Current opinion in cell biology 49, 99–107, doi: 10.1016/j.ceb.2017.12.012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misra JR & Irvine KD The Hippo Signaling Network and its Biological Functions. Annu Rev Genet, doi: 10.1146/annurev-genet-120417-031621 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu FX, Zhao B & Guan KL Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163, 811–828, doi: 10.1016/j.cell.2015.10.044 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis JR & Tapon N Hippo signalling during development. Development 146, doi: 10.1242/dev.167106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ribeiro PS et al. Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Mol Cell 39, 521–534, doi: 10.1016/j.molcel.2010.08.002 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Couzens AL et al. Protein interaction network of the Mammalian hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal 6, rs15, doi: 10.1126/scisignal.2004712 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Bae SJ et al. SAV1 promotes Hippo kinase activation through antagonizing the PP2A phosphatase STRIPAK. eLife 6, doi: 10.7554/eLife.30278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng Y et al. Homeostatic Control of Hpo/MST Kinase Activity through Autophosphorylation-Dependent Recruitment of the STRIPAK PP2A Phosphatase Complex. Cell reports 21, 3612–3623, doi: 10.1016/j.celrep.2017.11.076 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bae SJ, Ni L & Luo X STK25 suppresses Hippo signaling by regulating SAV1-STRIPAK antagonism. eLife 9, doi: 10.7554/eLife.54863 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bae SJ & Luo X Activation mechanisms of the Hippo kinase signaling cascade. Biosci Rep 38, doi: 10.1042/BSR20171469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng Z, Moroishi T & Guan KL Mechanisms of Hippo pathway regulation. Genes Dev 30, 1–17, doi: 10.1101/gad.274027.115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ni L et al. Structural basis for autoactivation of human Mst2 kinase and its regulation by RASSF5. Structure 21, 1757–1768, doi: 10.1016/j.str.2013.07.008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ni L, Zheng Y, Hara M, Pan D & Luo X Structural basis for Mob1-dependent activation of the core Mst-Lats kinase cascade in Hippo signaling. Genes Dev 29, 1416–1431, doi: 10.1101/gad.264929.115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu B et al. Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell 164, 406–419, doi: 10.1016/j.cell.2015.12.029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen R, Xie R, Meng Z, Ma S & Guan KL STRIPAK integrates upstream signals to initiate the Hippo kinase cascade. Nat Cell Biol 21, 1565–1577, doi: 10.1038/s41556-019-0426-y (2019). [DOI] [PubMed] [Google Scholar]
- 35.Gil-Ranedo J et al. STRIPAK Members Orchestrate Hippo and Insulin Receptor Signaling to Promote Neural Stem Cell Reactivation. Cell reports 27, 2921–2933 e2925, doi: 10.1016/j.celrep.2019.05.023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang Y et al. Selective Inhibition of STRN3-Containing PP2A Phosphatase Restores Hippo Tumor-Suppressor Activity in Gastric Cancer. Cancer Cell 38, 115–128 e119, doi: 10.1016/j.ccell.2020.05.019 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Kim JW et al. STRIPAK directs PP2A activity toward MAP4K4 to promote oncogenic transformation of human cells. eLife 9, doi: 10.7554/eLife.53003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanti GK, Pandey S & Goswami SK SG2NA enhances cancer cell survival by stabilizing DJ-1 and thus activating Akt. Biochem Biophys Res Commun 463, 524–531, doi: 10.1016/j.bbrc.2015.05.069 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Wong M et al. Silencing of STRN4 suppresses the malignant characteristics of cancer cells. Cancer Sci 105, 1526–1532, doi: 10.1111/cas.12541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weissmann F et al. biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc Natl Acad Sci U S A 113, E2564–2569, doi: 10.1073/pnas.1604935113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen C et al. Striatins contain a noncanonical coiled coil that binds protein phosphatase 2A A subunit to form a 2:2 heterotetrameric core of striatin-interacting phosphatase and kinase (STRIPAK) complex. J Biol Chem 289, 9651–9661, doi: 10.1074/jbc.M113.529297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gordon J et al. Protein phosphatase 2a (PP2A) binds within the oligomerization domain of striatin and regulates the phosphorylation and activation of the mammalian Ste20-Like kinase Mst3. BMC biochemistry 12, 54, doi: 10.1186/1471-2091-12-54 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moreno CS, Lane WS & Pallas DC A mammalian homolog of yeast MOB1 is both a member and a putative substrate of striatin family-protein phosphatase 2A complexes. J Biol Chem 276, 24253–24260, doi: 10.1074/jbc.M102398200 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castets F et al. Zinedin, SG2NA, and striatin are calmodulin-binding, WD repeat proteins principally expressed in the brain. J Biol Chem 275, 19970–19977, doi: 10.1074/jbc.M909782199 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Groves MR & Barford D Topological characteristics of helical repeat proteins. Current opinion in structural biology 9, 383–389, doi: 10.1016/s0959-440x(99)80052-9 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Holm L DALI and the persistence of protein shape. Protein Sci 29, 128–140, doi: 10.1002/pro.3749 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madsen CD et al. STRIPAK components determine mode of cancer cell migration and metastasis. Nat Cell Biol 17, 68–80, doi: 10.1038/ncb3083 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duhart JC & Raftery LA Mob Family Proteins: Regulatory Partners in Hippo and Hippo-Like Intracellular Signaling Pathways. Front Cell Dev Biol 8, 161, doi: 10.3389/fcell.2020.00161 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim SY, Tachioka Y, Mori T & Hakoshima T Structural basis for autoinhibition and its relief of MOB1 in the Hippo pathway. Scientific reports 6, 28488, doi: 10.1038/srep28488 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen M et al. The MST4-MOB4 complex disrupts the MST1-MOB1 complex in the Hippo-YAP pathway and plays a pro-oncogenic role in pancreatic cancer. J Biol Chem 293, 14455–14469, doi: 10.1074/jbc.RA118.003279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Breitman M, Zilberberg A, Caspi M & Rosin-Arbesfeld R The armadillo repeat domain of the APC tumor suppressor protein interacts with Striatin family members. Biochim Biophys Acta 1783, 1792–1802, doi: 10.1016/j.bbamcr.2008.04.017 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Buda A & Pignatelli M E-cadherin and the cytoskeletal network in colorectal cancer development and metastasis. Cell Commun Adhes 18, 133–143, doi: 10.3109/15419061.2011.636465 (2011). [DOI] [PubMed] [Google Scholar]
Methods-only References
- 53.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51, doi: 10.1016/j.jsb.2005.07.007 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature methods 14, 331–332, doi: 10.1038/nmeth.4193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang K Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12, doi: 10.1016/j.jsb.2015.11.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, doi: 10.7554/eLife.42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530, doi: 10.1016/j.jsb.2012.09.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang G et al. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46, doi: 10.1016/j.jsb.2006.05.009 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Scheres SH & Chen S Prevention of overfitting in cryo-EM structure determination. Nature methods 9, 853–854, doi: 10.1038/nmeth.2115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu Y et al. Structure of the protein phosphatase 2A holoenzyme. Cell 127, 1239–1251, doi: 10.1016/j.cell.2006.11.033 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612, doi: 10.1002/jcc.20084 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501, doi: 10.1107/S0907444910007493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21, doi: 10.1107/S0907444909042073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gouet P, Courcelle E, Stuart DI & Metoz F ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308, doi: 10.1093/bioinformatics/15.4.305 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM density maps and atomic coordinates for human STRIPAK have been deposited in the Electron Microscopy Data Bank and wwPDB, respectively, under accession codes EMD-22650 and PDB 7K36. Source data are provided with this paper.