

ABSTRACT
The antiapoptotic protein BCL2 inhibits death of HIV-infected cells. Previously, we showed that the BCL2 inhibitor venetoclax selectively kills acutely HIV-infected cells and reduces HIV DNA in latently infected CD4 T cells ex vivo after reactivation with anti-CD3/anti-CD28. However, there is a need to identify a combination therapy with venetoclax and a clinically relevant latency reversal agent. Ixazomib is an oral proteasome inhibitor which we have shown reactivates latent HIV and predisposes reactivated cells to cell death. Here, we determined that the combination of venetoclax and ixazomib kills more latently HIV-infected cells and leads to greater reduction in HIV replication than either treatment alone in vitro in a T cell model. However, combination treatment of ex vivo CD4 T cells from antiretroviral therapy (ART)-suppressed, HIV-positive participants resulted in unanticipated and unacceptable nonspecific toxicity in primary cells. Therefore, while we show proof of concept that multiple agents can enhance selective killing of HIV-infected cells, the combination of venetoclax and ixazomib has unacceptable toxicity in primary cells, and so further investigation is needed to identify a clinically relevant latency reversal agent to combine with venetoclax as a novel strategy to reduce the size of the HIV reservoir.
IMPORTANCE A cure for HIV would require eliminating cells that contain the virus in a latent form from the body. Current antiretroviral medications are unable to rid the body of latently infected cells. Here, we show that a combination of investigational agents—ixazomib plus venetoclax—which reactivate latent virus and predispose infected cells to apoptosis may reduce latent virus in a T cell model, but at the expense of nonspecific toxicity in primary cells.
KEYWORDS: HIV cure, HIV latency reversal, BCL2, venetoclax, proteasome inhibitor, ixazomib, human immunodeficiency virus, latency reversal, proteasome
INTRODUCTION
A major obstacle for an HIV cure is the presence of latent HIV reservoirs, defined as infected cells that harbor proviral DNA but do not actively replicate virus, and therefore cannot be eliminated by neither current antiretroviral therapy (ART) or host immune responses. The “kick-and-kill” strategy for eliminating these latent reservoirs posits that proviral reactivation with latency-reversing agents (LRAs) exposes “reactivated” cells to cell death by immunologic clearance or viral cytopathic effects, such as apoptosis (1, 2). However, LRAs alone have been ineffective at inducing HIV-infected-cell death after reactivation and can independently suppress CD8 T cell function, and therefore, the shock-and-kill strategy has been ineffective to date (1–3). This suggests a need for combination therapy to eradicate HIV.
One mechanism by which HIV-infected CD4 T cells can persist after viral reactivation is by resisting the Casp8p41 pathway of apoptosis (reviewed in reference 4). When HIV protease cleaves procaspase 8, it generates an HIV-specific protein Casp8p41. Casp8p41 can bind and activate BAK (BCL2 antagonist/killer 1) to induce mitochondrion-dependent apoptosis of HIV-infected cells (4). Alternatively, Casp8p41 can bind to the antiapoptotic protein BCL2, following which the Casp8p41/BCL2 complex is degraded by the proteasome. BCL2 can thereby counteract the proapoptotic effects of Casp8p41 to promote HIV-infected-cell survival (5, 6).
Venetoclax, a BCL2-selective inhibitor, has been shown to antagonize Casp8p41 binding to BCL2, causing preferential killing of HIV-infected CD4 T cells and a reduction in latent reservoir size ex vivo after reactivation with anti-CD3/anti-CD28 (5, 7). Other studies have begun investigating how other LRAs, such as protein kinase C (PKC) agonists and histone deacetylase inhibitors (HDACi), affect the latent reservoir when administered in combination with venetoclax (3, 8, 9). However, the extent by which venetoclax synergizes with other clinically relevant LRAs remains unknown. In this study, we evaluated the combination of venetoclax with a panel of LRAs to determine if any selectively target HIV-infected CD4 T cells and reduce the HIV latent reservoir in combination with venetoclax.
RESULTS
BCL2 inhibition affects reactivation with specific latency reversal agents.
Our previous study reported the ability of the BCL2 inhibitor venetoclax for preferential killing of acutely HIV-infected cells in vitro and primary CD4 T cells acutely HIV infected ex vivo (7). In unstimulated J-Lat 10.6 cells, a chronically HIV-infected T-cell lymphoma cell line that expresses green fluorescent protein (GFP) upon HIV production, venetoclax causes reactivated cells to die (7). Since LRAs reactivate latent HIV transcription by various pathways (10), we compared HIV production between combination treatments of venetoclax and a panel of LRAs. To model a mix of infected and uninfected cells, J-Lat 10.6 cells were cocultured 1:1 with uninfected parental Jurkat T cells. Cells were treated with venetoclax and one of several LRAs, including interleukin-15 (IL-15), disulfiram, the HDACi vorinostat, panobinostat, romidepsin, and resminostat, the PKC agonists bryostatin and ingenol, the BET (bromodomain and extraterminal motif) bromodomain inhibitor JQ1, and the proteasome inhibitor ixazomib, at concentrations previously observed to induce HIV production (6, 10–13).
GFP expression was measured in the J-Lat 10.6 cells over time by live-cell imaging to quantify HIV production, as the parental Jurkat T cells would not express GFP upon treatment. The number of GFP+ cells after LRA treatment was compared to that after phorbol myristate acetate (PMA)/ionomycin treatment, considered maximum reactivation (Fig. 1A and B). While there was a range of GFP expression, including in the positive-control samples, bryostatin, JQ1, and ixazomib had the highest number of GFP+ cells and the highest percentage of maximum GFP expression compared to PMA/ionomycin (mean, 109%, 94%, and 62%, respectively) (Fig. 1C). Venetoclax significantly decreased GFP expression after 48 h with JQ1 and ixazomib (P = 0.0004 and P < 0.0001, respectively) but did not change GFP expression with bryostatin treatment, despite effective reactivation (Fig. 1D). We previously showed that the loss of GFP expression in J-Lat 10.6 cells occurs with apoptotic cell death (5, 6), which suggests that venetoclax may kill the subset of latent cells that have become actively productive with these LRAs. Therefore, JQ1 and ixazomib were selected for downstream analyses with venetoclax.
FIG 1.

Venetoclax reduces J-Lat 10.6 cell GFP expression stimulated by JQ1 or ixazomib treatment. Jurkat and J-Lat 10.6 cells were cocultured and treated with or without 1 μM venetoclax and a latency reversal agent for 48 h in triplicates of 15,000 cells/well. The number of GFP+ cells was measured over time by live-cell imaging. (A) Maximum reactivation with 50 ng/ml PMA and 1 μg/ml ionomycin compared to DMSO control or venetoclax treatment alone. The graph is representative of 20 experiments. (B) GFP expression was compared between PMA/ionomycin and 50 ng/ml interleukin-15, 1 μM disulfiram, 500 nM vorinostat, 30 nM panobinostat, 100 nM romidepsin, 10 μM resminostat, 10 ng/ml bryostatin-1, 1 μM ingenol, 10 μM JQ1, and 100 nM ixazomib for one representative experiment. (C) Mean percentage of maximum reactivation from PMA/ionomycin of each LRA from 2 or 3 independent experiments. (D) GFP expression of each LRA with or without venetoclax. Error bars indicate SD. P values were calculated from linear regression. ***, P < 0.0001.
Treatment with ixazomib in the presence of venetoclax increases the proportion of HIV-producing cells that die by apoptosis and reduces HIV replication.
We next examined whether the combination of venetoclax and JQ1 or venetoclax and ixazomib preferentially induces death of latently HIV-infected cells compared to treatment with either LRA alone. Jurkat and J-Lat 10.6 cells were cocultured and treated with venetoclax or dimethyl sulfoxide (DMSO) vehicle, and either JQ1, ixazomib, or DMSO (Fig. 2A). In order to determine whether the decrease in GFP expression in Fig. 1 after JQ1 or ixazomib treatment with venetoclax was due to the apoptosis of HIV-producing cells, we measured active caspase 3 and GFP expression by flow cytometry (Fig. 2B).
FIG 2.
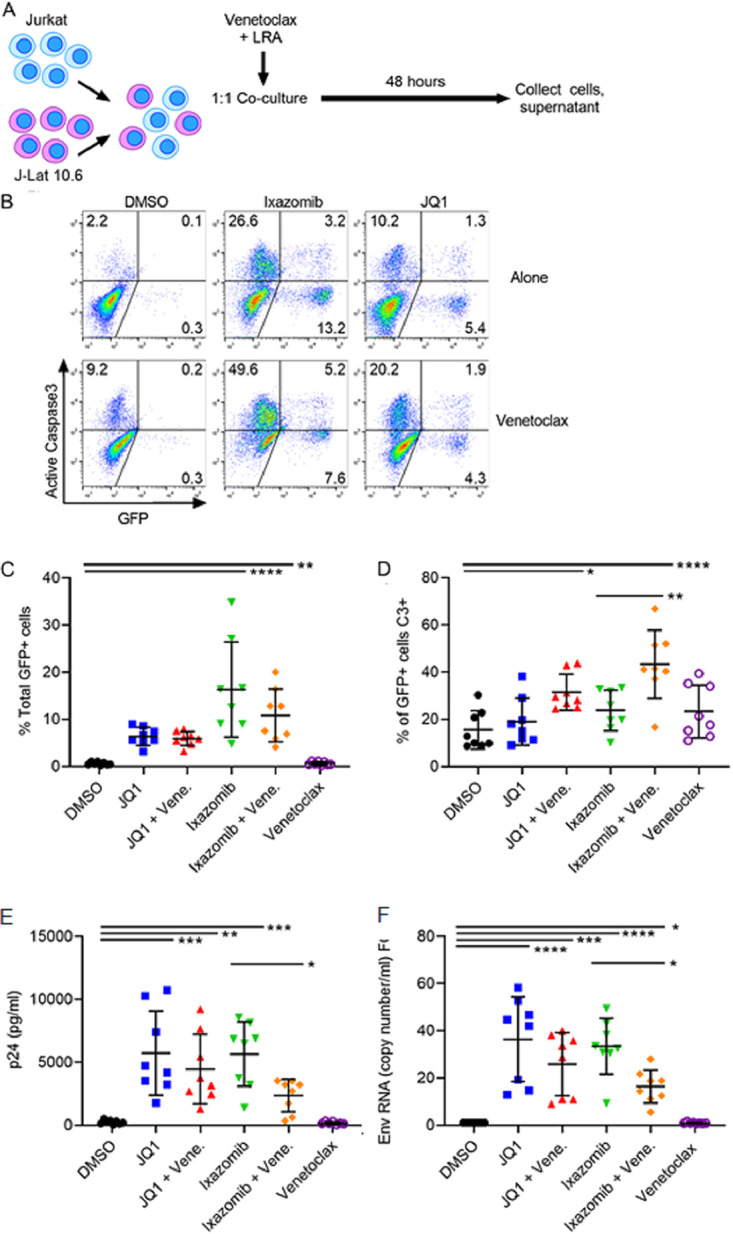
Venetoclax sensitizes infected cell death and reduces HIV replication after treatment of chronically infected T cells with ixazomib. (A) A 1:1 coculture of Jurkat and J-Lat 10.6 cells was treated with 1 μM venetoclax and either 1 μM JQ1, 100 nM ixazomib, or DMSO vehicle. (B) GFP and caspase 3 expression was measured by flow cytometry. Results from one representative experiment of eight are shown. (C) Total percentage of GFP+ cells after 48 h, assessed by flow cytometry. (D) Total percentage of GFP+ cells that were caspase 3 positive. (E) HIV supernatant p24 levels were measured by p24 ELISA. (F) HIV env RNA copies/ml were determined by RT-qPCR, and relative fold change (FC) was calculated for each treatment compared to the DMSO control. Error bars indicate SD. P values were calculated from one-way ANOVA after Dunnett’s multiple-comparison test. *, P < 0;05; **, P < 0;01; ***, P < 0;001; ****, P < 0.0001.
Both ixazomib and JQ1 increased the proportion of GFP+ cells, consistent with stimulating HIV production (with means of 16.3% and 10.81%, respectively [P < 0.0001 and P = 0.0016 compared to DMSO, respectively]) (Fig. 2C). Furthermore, when venetoclax was added, the proportion of reactivated (GFP+) cells that were positive for active caspase 3 was increased for both JQ1 and ixazomib compared to each LRA plus DMSO (P = 0.037 and P < 0.0001, respectively) (Fig. 2D). However, these numbers may be underrepresentative of the true increase in infected-cell death, because intracellular GFP degradation can occur during apoptosis (5, 6).
To confirm that the increased cell death in HIV-infected cells was due to killing of productively infected cells, we measured supernatant p24 from the same experiment (Fig. 2E). Both JQ1 and ixazomib significantly increased supernatant p24 levels compared to DMSO, again confirming their induction of HIV production. However, the combination of venetoclax and ixazomib had significantly lower supernatant p24 levels than ixazomib alone (P = 0.041), suggesting a greater magnitude of killing in the infected-cell population. This phenomenon was not seen with the addition of venetoclax to JQ1 compared to JQ1 alone. HIV env supernatant RNA levels were measured by reverse transcription-quantitative PCR (RT-qPCR), and the fold change over the DMSO control was calculated (Fig. 2F). While all treatments with JQ1 or ixazomib had significantly higher fold changes than DMSO, there was a significant decrease in env RNA when treated with both venetoclax and ixazomib compared to ixazomib alone (P = 0.03).
Treatment with ixazomib in the presence of venetoclax increases apoptosis in HIV-producing cells in association with the unfolded-protein response.
Ixazomib can potentiate venetoclax-induced killing of productively HIV-infected cells by one of two nonexclusive mechanisms: (i) increased reactivation of latent virus, resulting in more HIV protease expression, leading to more Casp8p41 expression, which is then disinhibited by venetoclax, resulting in mitochondrion-dependent apoptosis, or (ii) increased accumulation of the Casp8p41/BCL2 complex through inhibition of proteasomal degradation, followed by disinhibition by venetoclax, resulting in apoptosis. To investigate the first potential mechanism, we confirmed that ixazomib increases GFP expression in J-Lat 10.6 cells in a time- and dose-dependent manner (Fig. 3A). Second, we demonstrated that venetoclax increased apoptosis in HIV protein-expressing cells after reactivation with ixazomib, by assessing active caspase 3 expression in GFP and HIV p24 double-positive J-Lat 10.6 cells (Fig. 3B and C). Finally, since Casp8p41 induces mitochondrion-dependent caspase activation by binding and activating BAK (9), we assessed BAK activation in GFP+ cells after reactivation (Fig. 3D). The addition of venetoclax to ixazomib increased BAK activation in GFP+ but not GFP− J-Lat 10.6 cells compared to ixazomib treatment alone (Fig. 3D). Together, these data indicate that venetoclax increases apoptosis in HIV-expressing cells reactivated by ixazomib in a manner associated with BAK and caspase 3 activation.
FIG 3.

Ixazomib plus venetoclax induces intrinsic apoptosis associated with the unfolded protein response in chronically infected T cells. (A) J-Lat 10.6 cells were treated with increasing doses of ixazomib and assessed for GFP expression over time by flow cytometry. PMA was used as a positive control. (B) J-Lat 10.6 cells were treated with PMA, ixazomib (50 nM), or ixazomib plus venetoclax (100 nM) and assessed for active caspase 3 expression in GFP+ and HIV p24+ cells by flow cytometry. (C) Representative data as for panel B. (D) J-Lat 10.6 cells were treated with ixazomib with or without venetoclax and assessed for active BAK expression in GFP+ versus GFP− cells by flow cytometry. Vincristine was used as a positive control for BAK activation. (E) Primary CD4 T cells were transfected with HA-Casp8p41, treated with venetoclax, ixazomib, or the combination, and assessed for PARP cleavage, caspase 3 cleavage, and HA expression by Western blotting. (F) J-Lat 10.6 cells were treated with ixazomib, venetoclax, or the combination, in the presence or absence of ISRIB (an eIF-2α inhibitor), and assessed for PARP cleavage by Western blotting. (G) J-Lat 10.6 cells treated as described for panel F were assessed for active caspase 3 expression in GFP+ and HIV p24+ cells by flow cytometry. Error bars indicate SD from at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To investigate the second potential mechanism in isolation without HIV reactivation, uninfected CD4 T cells were transfected with HA-Casp8p41 and were either untreated or treated with venetoclax, ixazomib, or the combination. As we showed previously (6), ixazomib treatment alone increased HA-Casp8p41 expression in transfected cells through inhibition of proteasomal degradation (Fig. 3E). However, venetoclax increased apoptosis, as measured by PARP [poly(ADP-ribose) polymerase 1] cleavage and caspase 3 activation, in Casp8p41-transfected cells compared to control treated, transfected cells, regardless of cotreatment with ixazomib (Fig. 3E). Therefore, these data suggest that the increased killing with venetoclax in combination with ixazomib may occur upstream of inhibition of Casp8p41 degradation by the proteasome. In this setting of combination treatment, then, the potential latency reversal activity of ixazomib may be more influential than the specific inhibition of degradation of Casp8p41 by the proteasome.
Next, proteasome inhibitors have also been shown to activate the unfolded-protein response (UPR) in association with PERK, the endoplasmic reticulum (ER) stress–specific eIF-2α kinase, and ATF4, an ER stress–induced transcription factor. Therefore, we questioned whether ER stress and the UPR were involved in the synergistic killing of J-Lat 10.6 cells by ixazomib and venetoclax. J-Lat 10.6 cells were treated with the combination of ixazomib and venetoclax in the presence or absence of ISRIB (integrated stress response inhibitor) and eIF-2a inhibitor. ISRIB reduced apoptosis as measured by PARP cleavage in J-Lat 10.6 cells treated with ixazomib, venetoclax, or the combination of both (Fig. 3F). Likewise, ISRIB significantly inhibited caspase 3 activation in GFP and p24 double-positive cells treated with ixazomib and venetoclax (Fig. 3G). Together, these data indicate that the increased killing of HIV-expressing cells by the combination of ixazomib and venetoclax is at least partially due to ER stress and the UPR.
Treatment with ixazomib in the presence of venetoclax is toxic in primary CD4 T cells.
We next questioned whether the combination of ixazomib plus apoptosis sensitization with venetoclax would reduce HIV provirus in ex vivo CD4 T cells from HIV-positive persons. First, we tested combinations of doses of venetoclax and ixazomib on loss of viability or activation of caspase 3 in uninfected primary CD4 T cells (Fig. 4A and B). These experiments suggested a potentially narrow therapeutic window of toxicity with the combined agents, even at low doses, in primary uninfected CD4 T cells. Next, primary CD4 T cells were then isolated from eight ART-suppressed, HIV positive participants, and cultured ex vivo in the presence of venetoclax (100 nM) or ixazomib (25 nM) alone, or in combination, in the presence of ART to prevent spreading infection. The combination of venetoclax and ixazomib significantly reduced both ATP content (Fig. 4C) and proteasome activity (Fig. 4D) compared to untreated cells, consistent with expected pharmacodynamic effects on the mitochondria and proteasome, respectively, but at levels that would be unacceptable clinically. The combination of venetoclax and ixazomib did not reduce total HIV proviral DNA content (Fig. 4E), nor did it consistently reduce the percentage of provirus with intact sequences (Fig. 4F). In 3 of 6 experiments in which intactness of HIV proviral DNA was able to be assessed (there was technical failure of the assay for patients 2 and 3; see Materials and Methods), the combination of venetoclax plus ixazomib reduced intact HIV proviral DNA (Fig. 4F and G); however, there were no phenotypic associations to indicate that results for this subset of samples had more than stochastic probability or were due to nonspecific toxicity.
FIG 4.

Ixazomib plus venetoclax reduces intact HIV provirus in ex vivo CD4 T cells in a subset of participants. (A and B) CD4 T cells from 3 uninfected donors were treated with increasing concentrations of venetoclax (venet.) and/or ixazomib and assessed for viability by trypan blue exclusion (A) or apoptosis by active caspase 3 expression (B). CD4 T cells from 8 ART-suppressed, HIV-positive participants were treated ex vivo with venetoclax (100 nM), ixazomib (25 nM), or the combination for 72 h in the presence of ART to prevent spreading infection. (C) Treated CD4 T cells were assessed for viability by ATP content at 72 h. (D) Treated CD4 T cells were assessed for proteasome activity at 72 h. (E and F) Total and intact cell associated HIV provirus was measured at 72 h by IPDA. (F) The percentage of the total HIV provirus that was intact provirus is depicted for 6 of 8 participants. Two samples were excluded due to technical failure. (G) Proportions of intact (blue), 3′ defective (red), and 5′ defective (green) provirus from participants 1, 4, and 8.
DISCUSSION
The shock-and-kill strategy necessitates measures beyond ART and LRAs alone to increase death of reactivated latent cells. BCL2 has emerged as a promising therapeutic target to shift infected cells from a survival to an apoptotic phenotype (reviewed in reference 14), but few studies have investigated the combination of venetoclax with clinically relevant LRAs on the latent HIV reservoir. We evaluated reactivation of latently infected J-Lat 10.6 cells with venetoclax and a panel of established LRAs and further examined the BET inhibitor JQ1 and proteasome inhibitor ixazomib based on nontoxic concentrations and the indication that the combination treatment reduced GFP expression compared to the LRA alone (Fig. 1). While JQ1 did not have a synergistic effect with venetoclax, ixazomib augmented productive cell killing and the reduction in productive HIV infection (Fig. 2) in the J-Lat 10.6 cell model.
From a mechanistic standpoint, our data are consistent with three potential ways in which venetoclax and ixazomib could be synergistic in the context of reactivated HIV. First, ixazomib may reactivate latent HIV, which produces Casp8p41 via HIV protease activity, and venetoclax inhibits the interaction of Casp8p41 with BAK, causing cell death (consistent with Fig. 3A to D). Second, ixazomib may inhibit proteasomal degradation of existing Casp8p41, resulting in increased intracellular concentrations of Casp8p41, which then binds to BAK, which is released from BCL2 sequestration by venetoclax (consistent with Fig. 3E). Finally, the combination of venetoclax and ixazomib may induce ER stress and the unfolded-protein response in HIV-infected, but not uninfected, cells (consistent with Fig. 3F and G). It is likely that these mechanisms are not mutually exclusive but may variously combine in different T cell lines, subsets, or stages of cellular activation or stress.
Others have also observed the ability of venetoclax to sensitize latent cells reactivated by clinically relevant LRAs to cell death. Kim et al. similarly observed synergy between venetoclax and the HDACi romidepsin and saw a significant decrease of integrated HIV DNA in total CD4 T cells from ART-suppressed participants ex vivo (9). Huang et al. recorded significant increases in CTL-mediated cell killing of participant CD4 T cells with both venetoclax and either romidepsin or bryostatin compared to each LRA alone (3). However, this cytotoxic activity was not reproducible after treatment with venetoclax alone or with bryostatin or anti-CD3/CD28 antibodies without the addition of HIV-1-specific CD8 T cells (8). We did not find romidepsin to be effective at inducing HIV expression in our J-Lat 10.6 in vitro latency model. Although romidepsin has been shown to increase HIV transcriptional initiation and elongation, a decrease in viral RNA maturation has also been documented in ART-suppressed HIV participants due to obstructions of splicing (15). This potential drawback may explain why treatment with HDACi has shown impaired ex vivo autologous CD8 T cell killing if CD4 T cells cannot properly process and present viral antigens (16).
In this study, we determined that the combination of venetoclax and ixazomib increased death of productively infected cells and significantly reduced productive infection in a stable T cell line. However, when ex vivo primary CD4 T cells were treated with the combination of venetoclax and ixazomib, unacceptable toxicity was noted, even at low concentrations, suggesting that this combination would not be suitable for in vivo studies of HIV infection. During the course of our studies, the results of the BELLINI study were published, which was a randomized, double-blind, placebo-controlled trial of venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (17). The combination of venetoclax with the proteasome inhibitor bortezomib was associated with more cytopenias and treatment-emergent fatal infections than placebo, consistent with the combinatorial nonspecific toxicity of venetoclax and ixazomib evident in Fig. 4. Therefore, further investigations are needed to define optimal combination agents with either venetoclax or ixazomib that more selectively target the HIV reservoir with less toxicity.
MATERIALS AND METHODS
Cells.
Jurkat cells were obtained from the American Type Culture Collection (Manassas, VA). J-Lat 10.6 T-cells were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from Eric Verdin. All cells were cultured in RPMI 1640 medium (Corning) enriched with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals), 2 mM l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin (Gibco) at 37°C and 5% CO2.
IncuCyte analysis.
Jurkat and J-Lat 10.6 coculture was treated simultaneously with either 1 μM venetoclax or DMSO and 50 ng/ml PMA, 1 μg/ml ionomycin, 50 ng/ml interleukin-15, 1 μM disulfiram, 500 nM vorinostat, 30 nM panobinostat, 100 nM romidepsin, 10 μM resminostat, 10 ng/ml bryostatin-1, 1 μM ingenol, 10 μM JQ1, 100 nM ixazomib, or DMSO. Treatments were plated in triplicates of 15,000 total cells/well in a 96-well plate. J-Lat 10.6 GFP expression was measured with a real-time imaging IncuCyte system (Essen BioScience) and analyzed using IncuCyte Zoom software (v2018A). The absolute number of GFP+ cells per well is reported, comparing an LRA treatment to the positive control (PMA/ionomycin) for that plate and run. Data are representative of at least 2 independent experimental repeats.
Coculture experiments.
Parental Jurkat and J-Lat 10.6 T cells were cultured 1:1 and treated with 1 μM venetoclax or DMSO diluent control and simultaneously with 1 μM JQ1, 100 nM ixazomib, or DMSO for 48 h. At 48 h, cells were fixed for flow cytometry, and supernatant was collected for HIV p24 measurement by Retrotek enzyme-linked immunosorbent assay (ELISA) kits (Zeptometrix Corporation) according to the manufacturer’s protocol.
Flow cytometry.
Cells were stained intracellularly for active caspase 3 as described previously (18) with phycoerythrin (PE)-conjugated rabbit IgG (sc-3871; Santa Cruz) and PE–rabbit anti-active caspase 3 antibodies (550821; BD Pharmingen). J-Lat 10.6 cell reactivation was measured by GFP fluorescence. Flow cytometry was performed on a Fortessa flow cytometer (BD Biosciences) and analyzed with FlowJo software (version 10.6.2; Tree Star, Inc.), using unstained controls to determine gating.
Western blotting and transfection.
Activated CD4 cells were transfected with a Casp8p41-HA expression vector using the Lanza Nucleofector IIb system. After 1 h of electroporation, cells were treated with or without venetoclax (100 nM) and/or ixazomib (100 nM) for 6 h. Cells were collected and washed and lysed with cell lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1.0% Tween 20, protease inhibitors). For protein expression analysis, 20 μg of cell lysate was run on 10% polyacrylamide gels and then transferred onto polyvinylidene difluoride (PVDF) membranes for 2 h at 1,200 mA in transfer buffer (24 mM Tris, 192 mM glycine). The membranes were then blocked in Tris-buffered saline–Tween (TBST) (20 mM Tris, 150 mM NaCl, 0.05% Tween 20) with 3% bovine serum albumin (BSA) (Sigma, St. Louis, MO) for 1 h at room temperature or overnight at 4°C. Membranes were blotted with primary antibodies to PARP (clone 4C10-5; BD Pharmingen, San Jose, CA), hemagglutinin (HA) peroxidase 3F10 (Roche, St. Louis, MO), cleaved caspase 3 (D175; Cell Signaling, Danvers, MA), or actin (Sigma, St. Louis, MO). Membranes were then washed three times with TBST, and a horseradish peroxidase-linked secondary antibody was used when necessary. All blots were developed using a SuperSignal West Pico chemiluminescent substrate kit from Thermo Fisher Scientific (Waltham, MA).
Quantitative real-time reverse transcription-PCR.
Jurkat–J-Lat 10.6 coculture HIV-1 env RNA was measured by RT-qPCR (forward primer, 5′-TGCTGTTAGCTTGCTCAATGCCAC-3′; reverse primer, 5′-TGGCGAATAGCTCTACAAGCTCCT-3′). RNA was extracted from 48-h supernatant (Qiagen QIAamp viral RNA minikit) according to the manufacturer’s protocol. cDNA was generated using a high-capacity cDNA reverse transcription kit and RNase inhibitor (Applied Biosystems) with 5 μM reverse HIV env primer, in a final volume of 20 μl. The RT-PCR was run according to the manufacturer’s protocol using a Mastercycler gradient (Eppendorf) cycler. After dilution with 80 μl nuclease-free water, 5 μl cDNA templates was added to PowerUp SYBR green master mix (Applied Biosystems) and 1 μM HIV env primers, in a final volume of 15 μl. The RT-qPCR was run according to the manufacturer’s protocol using the QuantStudio 7 Flex real-time system (Applied Biosystems). Average gene expression from three sample replicates was determined by a standard curve.
Ex vivo primary cell experiments.
Primary human peripheral blood mononuclear cells (PBMCs) were previously obtained following informed consent according to Mayo Clinic Institutional Review Board (IRB)-approved protocols, and cryopreserved. CD4 T cells were isolated from frozen PBMC samples from eight HIV+ participants using an EasySep human CD4+ T cell isolation kit (STEMCELL Technologies, Vancouver, BC, Canada) according to the manufacturer’s protocol. Cells were allowed to rest for 3 to 4 h at 37°C before being plated in 12-well plates at a concentration of 5 × 106/ml with IL-2 (10 U/ml) and with enfuvirtide (NIH AIDS reagent) and raltegravir (NIH AIDS reagent) at doses of 1 μl/ml to prevent HIV replication. Treatment conditions of DMSO, 100 nM venetoclax, 25 nM ixazomib, or both 100 nM venetoclax and 25 nM ixazomib were added. Cells were incubated at 37°C for 72 h before harvesting. Cells were collected for a CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI) and Proteasome-Glo assays (Promega), which were performed according to the manufacturer’s protocols. Intact proviral DNA analysis (IPDA) was performed on the frozen cell pellets by AccelevirDx (Baltimore, MD). IPDA on samples from two participants (patients 2 and 3) was excluded from the analysis due to technical failure of the assay. In these two instances, the digital droplet PCR (ddPCR) droplet spread was inconsistent across the experimental samples from each participant, and specimen mislabeling could not be definitively proven. Because there was no residual DNA to retest from the original experiments, the data from the IPDA on these two participants were excluded from statistical analysis.
Study approval.
PBMCs were previously obtained following written informed consent according to Mayo Clinic IRB-approved protocols (IRB 13–005646) in accordance with all institutional and national guidelines.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism v8.0.0. (GraphPad Software). Results are presented as means and standard deviations (SD) and were compared using linear regression or analysis of variance (ANOVA), as appropriate. Dunnett’s multiple-comparison was conducted for ANOVA. P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health, including AI110173 and AI120698 (A.D.B.) and AI145407 (N.W.C.), and by grant no. 109593–62–RGRL (A.D.B.) from amfAR, the Foundation for AIDS Research.
A.D.B. and N.W.C. have filed a patent for use of Bcl-2 inhibitors in HIV infection.
Study design, A.A., S.N., N.W.C., and A.D.B.; conducting experiments, A.A., S.N., A.P.C., A.K., A.M., F.S., C.V., and N.W.C.; analyzing data, A.A., S.N., J.D.Y., and N.W.C.; providing reagents, J.D.Y. and A.D.B.; writing the manuscript, A.A., S.N., N.W.C., and A.D.B.; critical review and editing the manuscript, all authors; approval of final manuscript, all authors.
Contributor Information
Andrew D. Badley, Email: Badley.Andrew@mayo.edu.
Guido Silvestri, Emory University.
REFERENCES
- 1.Deeks SG. 2012. HIV: shock and kill. Nature 487:439–440. 10.1038/487439a. [DOI] [PubMed] [Google Scholar]
- 2.Lewin SR, Rasmussen TA. 2020. Kick and kill for HIV latency. Lancet 395:844–846. 10.1016/S0140-6736(20)30264-6. [DOI] [PubMed] [Google Scholar]
- 3.Huang S-H, Ren Y, Thomas AS, Chan D, Mueller S, Ward AR, Patel S, Bollard CM, Cruz CR, Karandish S, Truong R, Macedo AB, Bosque A, Kovacs C, Benko E, Piechocka-Trocha A, Wong H, Jeng E, Nixon DF, Ho Y-C, Siliciano RF, Walker BD, Jones RB. 2018. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Invest 128:876–889. 10.1172/JCI97555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sampath R, Cummins NW, Badley AD. 2016. Casp8p41: the protean mediator of death in CD4 T-cells that replicate HIV. J Cell Death 9:9–17. 10.4137/JCD.S39872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cummins NW, Sainski AM, Dai H, Natesampillai S, Pang YP, Bren GD, de Araujo Correia MCM, Sampath R, Rizza SA, O'Brien D, Yao JD, Kaufmann SH, Badley AD. 2016. Prime, shock, and kill: priming CD4 T cells from HIV patients with a BCL-2 antagonist before HIV reactivation reduces HIV reservoir size. J Virol 90:4032–4048. 10.1128/JVI.03179-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Natesampillai S, Cummins NW, Nie Z, Sampath R, Baker JV, Henry K, Pinzone M, O'Doherty U, Polley EC, Bren GD, Katzmann DJ, Badley AD. 2018. HIV protease-generated Casp8p41, when bound and inactivated by Bcl2, is degraded by the proteasome. J Virol 92:e00037-18. 10.1128/JVI.00037-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cummins NW, Sainski-Nguyen AM, Natesampillai S, Aboulnasr F, Kaufmann S, Badley AD. 2017. Maintenance of the HIV reservoir is antagonized by selective BCL2 inhibition. J Virol 91:e00012-17. 10.1128/JVI.00012-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren Y, Huang S-H, Patel S, Conce Alberto WD, Magat D, Ahimovic DJ, Macedo AB, Durga R, Chan D, Zale E, Mota TM, Truong R, Rohwetter T, McCann CD, Kovacs CM, Benko E, Wimpelberg A, Cannon CM, Hardy WD, Bosque A, Bollard CM, Jones RB. 2020. BCL-2 antagonism sensitizes cytotoxic t cell-resistant hiv reservoirs to elimination ex vivo. J Clin Invest 130:2542–2559. 10.1172/JCI132374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim YR, Solomon A, Zerbato JM, Cameron PU, McMahon JH, Anderson JL, Lewin SR. 2020. Romidepsin combined with pro-apoptotic drugs reduce integrated HIV DNA, abstr 354. In Conference on Retroviruses and Opportunistic Infections. International Antiviral Society–USA, San Francisco, CA. [Google Scholar]
- 10.Pardons M, Fromentin R, Pagliuzza A, Routy J-P, Chomont N. 2019. Latency-reversing agents induce differential responses in distinct memory CD4 T cell subsets in individuals on antiretroviral therapy. Cell Rep 29:2783–2795.e5. 10.1016/j.celrep.2019.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palermo E, Acchioni C, Di Carlo D, Zevini A, Muscolini M, Ferrari M, Castiello L, Virtuoso S, Borsetti A, Antonelli G, Turriziani O, Sgarbanti M, Hiscott J. 2019. Activation of latent HIV-1 T cell reservoirs with a combination of innate immune and epigenetic regulators. J Virol 93:e01194-19. 10.1128/JVI.01194-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das B, Dobrowolski C, Luttge B, Valadkhan S, Chomont N, Johnston R, Bacchetti P, Hoh R, Gandhi M, Deeks SG, Scully E, Karn J. 2018. Estrogen receptor-1 is a key regulator of HIV-1 latency that imparts gender-specificrestrictions on the latent reservoir. Proc Natl Acad Sci U S A 115:E7795–E7804. 10.1073/pnas.1803468115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xing S, Bullen CK, Shroff NS, Shan L, Yang H-C, Manucci JL, Bhat S, Zhang H, Margolick JB, Quinn TC, Margolis DM, Siliciano JD, Siliciano RF. 2011. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ Tcell model without inducing global T cell activation. J Virol 85:6060–6064. 10.1128/JVI.02033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandrasekar AP, Cummins NW, Badley AD. 2019. The role of the BCL-2 family of proteins in HIV-1 pathogenesis and persistence. Clin Microbiol Rev 33:e00107-19. 10.1128/CMR.00107-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moron-Lopez S, Kim P, Søgaard OS, Tolstrup M, Wong JK, Yukl SA. 2019. Characterization of the HIV-1 transcription profile after romidepsin administration in ART-suppressed individuals. AIDS 33:425–431. 10.1097/QAD.0000000000002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mota TM, McCann CD, Danesh A, Huang S-H, Magat DB, Ren Y, Leyre L, Bui TD, Rohwetter TM, Kovacs CM, Benko E, MacLaren L, Wimpelberg A, Cannon CC, Hardy WD, Safrit JT, Jones RB. 2020. Integrated assessment of viral transcription, antigen presentation, and CD8+ T cell function reveal multiple limitations of class I selective HDACi during HIV-1 latency reversal. J Virol 94:e01845-19. 10.1128/JVI.01845-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar SK, Harrison SJ, Cavo M, de la Rubia J, Popat R, Gasparetto C, Hungria V, Salwender H, Suzuki K, Kim I, Punnoose EA, Hong WJ, Freise KJ, Yang X, Sood A, Jalaluddin M, Ross JA, Ward JE, Maciag PC, Moreau P. 2020. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol 21:1630–1642. 10.1016/S1470-2045(20)30525-8. [DOI] [PubMed] [Google Scholar]
- 18.Sainski AM, Dai H, Natesampillai S, Pang YP, Bren GD, Cummins NW, Correia C, Meng XW, Tarara JE, Ramirez-Alvarado M, Katzmann DJ, Ochsenbauer C, Kappes JC, Kaufmann SH, Badley AD. 2014. Casp8p41 generated by HIV protease kills CD4 T cells through direct Bak activation. J Cell Biol 206:867–876. 10.1083/jcb.201405051. [DOI] [PMC free article] [PubMed] [Google Scholar]