

ABSTRACT
We previously reported that the normally essential step of integration of the HIV-1 proviral DNA intermediate into the host cell genome becomes dispensable in T cells that express the human T cell leukemia virus 1 (HTLV-1) Tax protein, a known activator of cellular NF-κB. The rescue of integrase (IN)-deficient HIV-1 replication by Tax results from the strong activation of transcription from the long terminal repeat (LTR) promoter on episomal HIV-1 DNA, an effect that is closely correlated with the recruitment of activating epigenetic marks, such as H3Ac, and depletion of repressive epigenetic marks, such as H3K9me3, from chromatinized unintegrated proviruses. In addition, activation of transcription from unintegrated HIV-1 DNA coincides with the recruitment of NF-κB to the two NF-κB binding sites found in the HIV-1 LTR enhancer. Here, we report that the recruitment of NF-κB to unintegrated viral DNA precedes, and is a prerequisite for, Tax-induced changes in epigenetic marks, so that an IN− HIV-1 mutant lacking both LTR NF-κB sites is entirely nonresponsive to Tax and fails to undergo the epigenetic changes listed above. Interestingly, we found that induction of Tax expression at 24 h postinfection, when unintegrated HIV-1 DNA is already fully repressed by inhibitory chromatin modifications, is able to effectively reverse the epigenetic silencing of that DNA and rescue viral gene expression. Finally, we report that heterologous promoters introduced into IN-deficient HIV-1-based vectors are transcriptionally active even in the absence of Tax and do not increase their activity when the HIV-1 promoter and enhancer, located in the LTR U3 region, are deleted, as has been recently proposed.
IMPORTANCE Integrase-deficient expression vectors based on HIV-1 are becoming increasingly popular as tools for gene therapy in vivo due to their inability to cause insertional mutagenesis. However, many IN− lentiviral vectors are able to achieve only low levels of gene expression, and methods to increase this low level have not been extensively explored. Here, we analyzed how the HTLV-1 Tax protein is able to rescue the replication of IN− HIV-1 in T cells, and we describe IN− lentiviral vectors, lacking any inserted origin of replication, that are able to express a heterologous gene effectively.
KEYWORDS: integrase, NF-κB, Tax, chromatin, epigenetics
INTRODUCTION
Integration of the linear DNA proviral intermediate into host cell DNA by the viral integrase (IN) enzyme is not only a key step in the life cycle of all retroviral species examined so far but also essential for effective retroviral gene expression (1, 2). As a result, the HIV-1 IN protein has emerged as a major target for antiviral drugs, such as raltegravir and dolutegravir, that function as IN strand transfer inhibitors and thereby effectively prevent integration (3–5). Importantly, these drugs block HIV-1 replication and gene expression not only in dividing cells, such as T cells, but also in nondividing cells, such as macrophages, thereby demonstrating that loss of unintegrated proviral DNA during cell division, though this clearly does occur, is at least not the only inhibitory process that proviral integration has evolved to avoid (6–9). In fact, it is now clear that the inability of unintegrated proviral DNA to support effective retroviral gene expression and replication is due to the rapid addition of repressive epigenetic marks to chromatinized episomal retroviral DNA and a concomitant lack of addition of activating epigenetic modifications (10–14). This involves the rapid recruitment of repressive epigenetic marks, such as H3K9me3, to chromatinized retroviral episomes shortly after nuclear entry and also correlates with the inability of unintegrated retroviral DNA to recruit the activating epigenetic marks, such as H3Ac, that are abundant on actively transcribed, integrated proviruses.
While proviral integration is therefore normally an essential step in the HIV-1 life cycle, we recently demonstrated that HIV-1 IN function is dispensable in T cells that express an inducible form of the human T cell leukemia virus 1 (HTLV-1) Tax protein (13, 15). Specifically, induction of Tax expression in the CD4+ T cell line CEM-SS led to a high level of transcription from the HIV-1 long terminal repeat (LTR) promoter present on unintegrated viral DNA that was sufficient to support the robust replication and spread of HIV-1 in culture. The Tax-mediated activation of the HIV-1 LTR not only coincided with the recruitment of activating epigenetic marks, such as H3Ac, to unintegrated HIV-1 DNA at levels that actually exceeded the level of H3Ac detected on integrated proviruses but also prevented the acquisition of repressive epigenetic marks, such as H3K9me3.
Because integration of retroviral proviruses into the host cell genome has the potential to cause insertional mutagenesis, which could potentially lead to oncogenic transformation (16, 17), there has been considerable interest in the question of whether one could design lentiviral vectors based on HIV-1 or other lentiviruses that remain able to express significant levels of a heterologous gene of interest in the absence of IN function (18, 19). Initially, several groups reported that nonintegrating vectors based on HIV-1, feline immunodeficiency virus (FIV), or equine infectious anemia virus (EIAV) produced negligible levels of an encoded indicator gene, >100-fold lower than that seen with the matched IN+ control vector (20–23). However, more recently, reasonable levels of gene expression by IN− vectors have been reported by several groups, though no clear explanation of why some designs appear more effective than others has been presented (24–27). While it has been reported that deletion of almost the entire U3 region in the 3′ LTR, to generate a so-called self-inactivating (SIN) lentivector, increased expression of the encoded green fluorescent protein (GFP) by ∼3-fold, this level remained ∼7-fold lower than that seen with the matched IN+ vector (28).
In this study, we sought to further clarify why an HIV-1 mutant lacking a functional IN protein is replication defective in normal T cells but able to replicate in the presence of the HTLV-1 Tax protein. We now report that this effect is due to the activation of NF-κB by Tax, resulting in the effective recruitment of the NF-κB subunits Rel A and Rel B to the two NF-κB binding sites located in the HIV-1 LTR U3 region on unintegrated proviral DNA. This in turn modifies the chromatin marks recruited to these retroviral episomes, including 2-LTR circular DNA, leading to a highly significant increase in the activating H3Ac epigenetic mark and a marked reduction in the repressive H3K9me3 epigenetic mark. Importantly, even after repressive chromatin modifications have accumulated on unintegrated HIV-1 DNA at 24 h postinfection (hpi), induction of Tax expression can still fully reverse this epigenetic repression and rescue viral gene expression. Finally, we report the efficient expression of an indicator gene product from several HIV-1-based IN− vectors even in the absence of Tax and find that, in our hands, the SIN vector design is not more active.
RESULTS
We previously reported that expression of the HTLV-1 Tax protein in human T cells, in particular in the CD4+ human T cell lines C8166 and CEM-SS, rescues gene expression from unintegrated DNA episomes derived from an IN− HIV-1 mutant and allows their robust replication despite their lack of integrase function (13). We further reported that this effect correlated both with the recruitment of the NF-κB subunits Rel A and Rel B to the two NF-κB sites located in the HIV-1 LTR enhancer and with the addition of the activating epigenetic marks H3Ac and H3K4me3 and the loss of the repressive epigenetic mark H3K9me3 in chromatin bound to unintegrated HIV-1 episomes. However, our previous work only looked at the epigenetic status of unintegrated viral DNA late after infection, and therefore, we could not determine whether the observed rescue of HIV-1 gene expression was due to the observed epigenetic changes leading to the effective recruitment of NF-κB to unintegrated HIV-1 DNA, or vice versa. We reasoned that it might be possible to resolve this question by addressing the temporal order underlying the recruitment of the NF-κB subunits, relative to the addition of epigenetic marks, to unintegrated HIV-1 DNA. For this purpose, we infected Tax-inducible CEM-SS cells with IN+ or IN− forms of the previously described replication-competent indicator virus NL-NLuc (29) in the presence or absence of doxycycline (Dox) and then harvested the infected cells at 16, 19, 21, or 24 hpi. We then performed chromatin immunoprecipitation linked to quantitative PCR (ChIP-qPCR) to quantify the level of Rel A, Rel B, H3Ac, and H3K9me3 present on proviral DNA.
As shown in Fig. 1A and B, analysis of the recruitment of Rel A and Rel B to the LTR on unintegrated HIV-1 DNA revealed a highly significant (P < 0.01) boost in the recruitment of both proteins to the HIV-1 LTR in Tax-expressing CEM-SS cells, relative to uninduced CEM-SS cells, as early as 16 hpi, the earliest time point examined, and this effect was even clearer at subsequent times. In contrast, as shown in Fig. 1C and D, we did not detect any increase in the level of H3Ac on HIV-1 DNA-bound chromatin or loss of H3K9me3 marks from viral chromatin at 16 hpi, though this effect did become significant (P < 0.01) at 19 hpi and was even more pronounced at later times, as previously reported (13). Analysis by ChIP-qPCR of H3Ac and H3K9me3 addition to other sites on unintegrated HIV-1 DNA, specifically to the HIV-1 gag gene (Fig. 1E and F) or the NanoLuc indicator gene (NLuc) inserted in place of nef (Fig. 1G and H), revealed the same pattern seen when the HIV-1 LTR was analyzed. Again, there was no significant difference at 16 hpi in H3Ac and H3K9me3 modification levels between the unintegrated IN− episomal DNA with or without Tax, but significantly more H3Ac histone modifications and fewer H3K9me3 modifications on the IN− episomal viral chromatin in Tax-expressing cells were seen at 19 hpi.
FIG 1.

NF-κB subunits RelA and RelB are recruited to the HIV-1 LTR prior to activating or repressive epigenetic histone modifications. ChIP-qPCR detection of RelA/p65 (A) and Rel B (B) as well as H3Ac and H3K9me3 on the HIV-1 LTR promoter (C and D), the viral gag gene (E and F), and the NLuc gene inserted in place of nef (G and H). Assays were determined at the indicated times in CEM-SS cells expressing or not expressing HTLV-1 Tax. Levels were normalized to input DNA and are expressed as percentage of input. n = 3 biological replicates. Error bars show standard deviations (SD). **, P < 0.01, and ***, P < 0.001, calculated by two-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test.
The Tax-induced recruitment of NF-κB to unintegrated HIV-1 DNA is essential for rescue of viral gene expression.
The data presented in Fig. 1 reveal that Tax-induced Rel A and Rel B are recruited to unintegrated HIV-1 proviruses before any Tax-induced changes in activating (H3Ac) and repressive (H3K9me3) chromatin marks can be detected. However, these data do not address whether the Tax-induced activation and recruitment of NF-κB to viral DNA are necessary for the rescue of IN− HIV-1 replication. To address this question, we generated derivatives of the previously described IN− form of the indicator virus NL-NLuc (13) in which either one LTR NF-κB site (1Δ) or both NF-κB sites (2Δ) were mutationally inactivated. We then asked whether loss of NF-κB sites affected the rescue of this IN− virus by Tax. In this experiment, we infected CEM-SS cells bearing the Tet-inducible tax gene with IN+ NL-NLuc, with IN− NL-NLuc, or with the new 1Δ or 2Δ NF-κB mutants of IN− NL-NLuc. We then measured the level of viable cells in each culture over time (Fig. 2A), the level of expression of the virally encoded NLuc indicator (Fig. 2B), the level of viral DNA (Fig. 2C), and the level of total viral RNA (Fig. 2D) at up to 7 days postinfection (dpi).
FIG 2.

At least one HIV-1 LTR NF-κB binding site is required for the rescue of IN− HIV-1 replication by Tax. Clonal CEM-SS T cell lines with Tet-inducible Tax were infected with WT or IN− NL-NLuc with either neither, one, or both of the NF-κB sites mutated. Live cell counts (A), virally encoded NLuc expression (B), total HIV-1 DNA measured by qPCR (C), and HIV-1 RNA expression measured by qRT-PCR (D) were quantified at 1, 2, 3, 5, and 7 days postinfection (dpi). The WT culture was devoid of live cells at 7 dpi. DNA and RNA levels were normalized to those in WT HIV-1-infected cells in the absence of Tax at 1 dpi, which was set to 1. The epigenetic state of the viral LTR promoter was characterized by quantifying the bound NF-κB subunits Rel A and Rel B (E) and the activating H3Ac and repressive H3K9me3 histone modification (F) at 2 dpi using ChIP-qPCR. n = 3 biological replicates. Error bars show SD. ***, P < 0.001, calculated by two-way ANOVA with Tukey’s multiple-comparison test.
As shown in Fig. 2A, and as previously reported (13), we observed that infection of CEM-SS cells with the IN− NL-NLuc derivative bearing an intact LTR did not reduce cellular viability, relative to the uninfected culture, in the absence of Tax but induced the death of almost the entire CEM-SS culture by 7 days postinfection (dpi) if Tax expression was induced by Dox addition. Interestingly, while the 1Δ mutant of the IN− NL-NLuc virus also induced a strong cytopathic effect, the 2Δ mutant phenocopied the lack of cell killing seen with the parental IN- NL-Nluc virus in the absence of Tax (Fig. 2A). The lack of cytopathic effect seen with the 2Δ mutant of NL-NLuc correlated with an almost complete lack of viral gene expression, as measured by NLuc activity (Fig. 2B). In contrast, the 1Δ mutant, which retains a single NF-κB site, gave rise to a lower level of NLuc activity than the parental IN− NL-NLuc virus, which retains both NF-κB sites, that was nevertheless readily detectable (Fig. 2B). Similarly, the 2Δ mutant failed to generate a spreading infection in Tax-expressing CEM-SS cells, as measured by our inability to measure any increase in either viral DNA or viral RNA expression over time (Fig. 2C and D). The 1Δ mutant was again intermediate in phenotype between the 2Δ mutant and the parental NL-NLuc vector in that it remained able to spread through the Tax-expressing CEM-SS culture, albeit more slowly than NL-NLuc, as revealed by increasing levels of both HIV-1 DNA and HIV-1 RNA expression (Fig. 2C and D).
The data presented in Fig. 2A to D demonstrate that the loss of both NF-κB sites present in the HIV-1 LTR prevents the rescue of viral gene expression by Tax seen when both or even only one LTR NF-κB site is retained in the LTR of an IN− HIV-1 mutant, a result that is consistent with the known ability of Tax to activate NF-κB in expressing cells by inducing the phosphorylation and degradation of IκB (15). These data could suggest that recruitment of NF-κB is required for the subsequent epigenetic changes (i.e., increased H3Ac marks and reduced H3K9me3 marks) seen on unintegrated HIV-1 proviruses after rescue by Tax induction (Fig. 1). Alternatively, it could be argued that these epigenetic changes are still induced by Tax but, in the absence of NF-κB recruitment, are insufficient to rescue transcription of unintegrated HIV-1 DNA. To resolve this question, we performed ChIP-qPCR on IN− NL-NLuc either bearing a wild-type (WT) LTR promoter or the 2Δ LTR lacking both NF-κB sites. As expected, in the absence of Tax, we observed almost no detectable recruitment of either the Rel A or Rel B NF-κB subunit to the intact viral LTR on unintegrated viral DNA (Fig. 2E), and similarly, in the absence of Tax, the chromatin added to unintegrated HIV-1 episomes was modified by addition of high levels of the repressive H3K9me3 mark, while the activating H3Ac mark was essentially absent (Fig. 2F).
In contrast, in the presence of Tax, both Rel A and Rel B were effectively recruited to the intact LTR promoter present on unintegrated HIV-1 DNA (Fig. 2E), and we now saw efficient recruitment of the activating H3Ac mark and very little addition of the repressive H3K9me3 mark (Fig. 2F), as expected. Importantly, however, when the 2Δ mutant of IN− NL-NLuc was examined in the presence of Tax it phenocopied the pattern observed for IN− NL-NLuc bearing the WT LTR in the absence of Tax. That is, while the 2Δ indicator virus mutant failed to recruit either the Rel A or Rel B NF-κB subunit, as expected (Fig. 2E), it was heavily modified by addition of the repressive H3K9me3 mark to chromatinized viral DNA while remaining devoid of the activating H3Ac marks seen on the unintegrated WT LTR in the presence of Tax (Fig. 2F). Thus, these data argue that recruitment of the NF-κB subunits Rel A and/or Rel B to the viral LTR present on unintegrated HIV-1 DNA is a prerequisite for the subsequent recruitment of the epigenetic marks that correlate with active transcription of a DNA template.
Tax induction reverses the epigenetic repression of unintegrated HIV-1 DNA.
The data presented in Fig. 1 and 2 demonstrate that the activation of cellular NF-κB by Tax expression induces NF-κB binding to the HIV-1 LTR and then prevents the epigenetic repression of unintegrated viral DNA. We were curious to see if Tax could also reverse the preexisting epigenetic repression of viral unintegrated DNA, which, as shown in Fig. 1, is complete by 24 hpi. For this purpose, we induced Tax expression at 24 hpi and monitored viral gene expression, in this case by measuring production of the encoded NLuc indicator protein (Fig. 3A), as well as the epigenetic state of the unintegrated HIV-1 DNA, by ChIP-qPCR (Fig. 3B and C). As shown in Fig. 3A, Tax induction at 24 hpi was able to rescue gene expression from the otherwise silenced unintegrated IN− HIV-1 DNA by 48 hpi, and this increase in viral gene expression continued at 72 hpi. Similarly, induction of Tax expression at 24 hpi fully rescued recruitment of activating H3Ac marks, and effectively removed inhibitory H3K9me3 marks, by 48 hpi and maintained this active epigenetic state at 72 hpi (Fig. 3B and C). Therefore, the induction of Tax expression not only can prevent the epigenetic repression of unintegrated HIV-1 DNA but also can fully reverse this repression after inhibitory chromatin marks have been allowed to assemble on the viral DNA.
FIG 3.
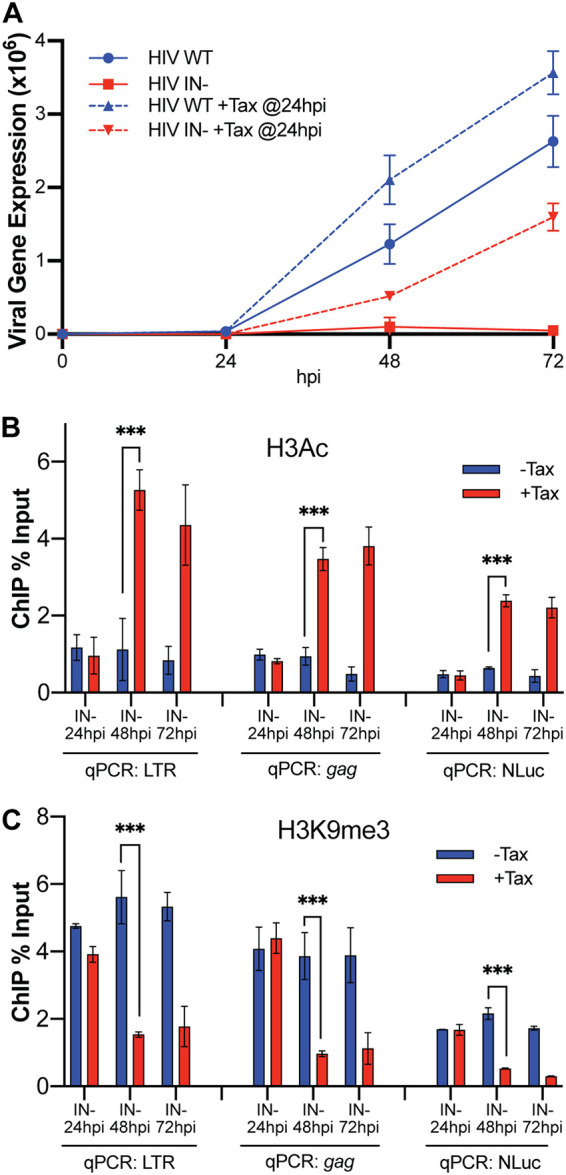
Induction of Tax expression after unintegrated HIV-1 DNA has been epigenetically silenced reverses silencing and rescues viral gene expression. (A) Tax-inducible CEM-SS cells were infected with WT or IN− HIV-1 and treated with nevirapine at 16 hpi to prevent viral spread; then, treatment with or without Dox was added at 24 hpi to induce Tax expression. Cells were harvested to assay NLuc expression at the indicated time points. n = 3 biological replicates. Error bars show SD. Cells were also harvested for ChIP-qPCR to determine the levels of H3Ac (B) and H3K9me3 (C) on the HIV-1 LTR promoter, the viral gag gene, and the NLuc indicator gene inserted in place of nef in NL-NLuc. ChIP levels are expressed as percentage of input. n = 3 biological replicates. Error bars show SD. ***, P < 0.001, calculated by two-way ANOVA with Tukey’s multiple-comparison test.
Analysis of the effect of Tax on gene expression from IN− lentiviral vectors.
While there is considerable evidence indicating that integration of the retroviral proviral intermediate into the host cell genome is an essential step in the retroviral life cycle (1, 2) and demonstrating that this is largely due to the efficient epigenetic silencing of unintegrated retroviral DNA (10–14), there are also a number of reports documenting the effective expression of cDNAs with a gene of interest from some, but not all, integrase-deficient lentiviral vectors bearing inserted heterologous enhancer/promoter combinations (24–27). To our knowledge, the issue of why some, but not all, lentiviral vectors are able to escape epigenetic silencing has not been resolved, though it has been suggested that deletion of the retroviral promoter/enhancer located in the LTR U3 region, in so-called self-inactivating or SIN lentiviral vectors, can promote the function of inserted heterologous promoters (28).
To address this question, we generated a set of vectors from the WT and IN− NL-NLuc vectors in which we first deleted almost the entire U3 region, from position −18 to −418, in the 3′ LTR, while leaving cis-acting sequences required for reverse transcription and integration intact, to generate SIN vectors (30). We then inserted either the LTR enhancer/promoter from Rous sarcoma virus (RSV) (31) or the highly active cytomegalovirus (CMV) immediate early enhancer/promoter (32), 5′ to the NLuc indicator gene (Fig. 4A). These vectors were then packaged and used to infect Tax-inducible CEM-SS cells in the presence and absence of Dox. At 48 hpi, the cells were lysed and the level of virus-encoded NLuc determined. As may be seen in Fig. 4B, the level of NLuc expression induced upon infection with lentiviral vectors containing internal heterologous promoters was comparable to that seen with the parental NL-NLuc vector and, unlike NL-NLuc, was only modestly inhibited, by <3-fold, when the integrase gene was mutationally inactivated. While expression of Tax did not affect the level of NLuc expression from either the IN+ or IN− form of NL-SIN-RSV-NLuc, Tax did exert a significant positive effect on expression from the CMV promoter, which contains NF-κB binding sites (33), in both the IN+ and IN− versions of NL-SIN-CMV-NLuc (Fig. 4B).
FIG 4.

Unlike the HIV-1 LTR, heterologous internal Pol II and Pol III promoters are not silenced on unintegrated HIV-1 proviruses. (A) Schematic of pNL-NLuc and its derivatives. (B) CEM-SS cells with and without Tax were infected with IN+ or IN− HIV-1 variants expressing NLuc from the viral LTR or with self-inactivating (SIN) derivatives lacking the LTR U3 region from −18 to −418, including the HIV-1 promoter and enhancer. Instead, these viruses express NLuc from an internal RSV LTR promoter (that has no NF-κB binding sites) or an internal CMV promoter (that has NF-κB binding sites). Nevirapine was added at 16 hpi to prevent virus spread, and cells were harvested for NLuc assays at 48 hpi. NLuc activity was normalized to WT NL-NLuc activity in the absence of Tax, which was set to 1. (C) Similar to panel B but using a non-SIN vector design. (D) CEM-SS cells with and without Tax were infected with WT or IN− virus expressing KSHV miR-K2 from the Pol III-dependent H1 promoter. Nevirapine was added to the cells at 16 hpi, and the cells were harvested for quantification of miR-K2 by qRT-PCR at 48 hpi. Endogenous U6 RNA served as an internal control, and data were normalized to WT minus Tax, which was set to 1. n = 3 biological replicates. Error bars show SD. (E) HIV-1 Gag expression levels in CEM-SS cells with and without Tax infected with the indicated WT or IN− viruses. Endogenous GAPDH was used as a loading control. *, P < 0.05, **, P < 0.01, and ***, P < 0.001, calculated using an unpaired Holm-Šidák multiple-comparison t test.
As noted above, the SIN design has been reported to enhance the level of gene expression from heterologous promoters inserted into IN− retroviral vectors (28), possibly due to the loss of the potential for transcriptional interference inherent in this design (34). To test whether this is indeed the case, we generated a set of four IN+ or IN− vectors, bearing either the RSV or CMV enhancer/promoter driving NLuc expression, that differed from the vectors analyzed in Fig. 4B only in that the HIV-1 LTR was now left intact. As shown in Fig. 4C, these vectors in fact expressed up to 10-fold-higher levels of NLuc than the similar SIN vectors. This higher level of NLuc expression was slightly more subject to inhibition upon loss of IN function than seen with the SIN design and was also more responsive to the HTLV-1 Tax protein. The latter effect was again particularly prominent with vectors bearing the CMV enhancer/promoter, which, unlike the RSV LTR, contains NF-κB binding sites (33). Nevertheless, overall it was clear that the tested lentiviral vectors, bearing internal heterologous promoters, were all transcriptionally active regardless of IN activity and that the SIN design did not increase expression from unintegrated lentiviral vector DNA and in fact seemed to modestly inhibit NLuc expression.
Finally, while the data presented in Fig. 4A and B reveal that a heterologous polymerase II (Pol II)-dependent promoter inserted into a lentiviral vector lacking integrase function is still essentially fully active, they do not address whether other types of promoters, for example, promoters that utilize Pol III, would also be functional on a nonintegrated proviral DNA template. This could be important if, for example, one wished to express a microRNA (miRNA) from a nonintegrating retroviral vector. To test this, we therefore inserted a previously described expression cassette containing the H1 Pol III promoter linked to an RNA hairpin encoding the miR-K2 miRNA expressed by Kaposi’s sarcoma herpesvirus (KSHV) (35). As may be seen in Fig. 4C, this vector expressed high levels of mature miR-K2, as measured by quantitative reverse transcription-PCR (qRT-PCR), regardless of whether an IN+ or IN− vector design was used. Unexpectedly, the expression of miR-K2 appeared to be enhanced, especially in the IN+ vector, by induction of Tax expression.
The CMV immediate early promoter contains an exceptionally active enhancer (32), and, given that both the CMV and RSV promoters were active in the context of the NL-NLuc vectors bearing intact LTRs, we wondered if the CMV or RSV promoter/enhancer might be able to rescue expression from the 5′ HIV-1 LTR in cis when integrase was mutationally inactivated. If so, this should result in the expression of the HIV-1 Gag protein. To test this, we infected CEM-SS cells with NL-NLuc, with NL-RSV-NLuc and with NL-CMV-NLuc in the presence and absence of an intact integrase gene and in the presence or absence of Dox, to induce Tax expression, As expected, and as previously reported (13), the IN− form of NL-NLuc failed to express Gag unless Tax expression had been induced (Fig. 4E, compare lanes 3 and 5). Similarly, for both NL-RSV-NLuc and NL-CMV-NLuc, we failed to detect Gag expression from the IN− form of this vector unless Tax expression was induced (Fig. 4E, lanes 7, 9, 11, and 13). We therefore conclude that, even though both the RSV LTR and the CMV promoter/enhancer remain essentially fully active when inserted into a lentiviral vector lacking integrase function, they are nevertheless both unable to prevent the epigenetic silencing of the immediately adjacent HIV-1 LTR promoter.
DISCUSSION
Previously, we reported that expression of the HTLV-1 Tax protein in human CD4+ T cells is able to rescue the transcription and robust replication of an integrase-deficient HIV-1 mutant, bearing the D64V IN mutation, in the absence of any detectable proviral DNA integration (13). This rescue correlated both with the recruitment of activated NF-κB to the HIV-1 LTR on unintegrated DNA and with the acquisition of high levels of the activating epigenetic mark H3Ac and loss of the repressive mark H3K9me3. While our previous research did not address whether the rescue of HIV-1 gene expression by Tax was primarily due to NF-κB activation and recruitment or, instead, due to changes in the epigenetic status of the HIV-1 LTR promoter, we did observe that a Tax mutant called M22, previously shown to have selectively lost the ability to activate NF-κB (36), also failed to rescue IN− HIV-1 replication (13). Moreover, we reported that direct activation of NF-κB function using TNF-α also rescues gene expression from unintegrated HIV-1 proviral DNA, albeit far less effectively than seen upon Tax expression (13).
Here, we extend the earlier work by demonstrating that recruitment of the NF-κB subunits Rel A and Rel B to the HIV-1 LTR on unintegrated viral DNA precedes the acquisition of activating H3Ac marks, and loss of repressive H3K9me3 marks, by the same viral DNA. Moreover, we observed that mutational inactivation of both NF-κB sites on the HIV-1 LTR not only blocks the recruitment of NF-κB subunits to unintegrated viral DNA but also entirely blocks the epigenetic changes, noted above, that mark DNA as being transcriptionally active. We have therefore demonstrated that activation of NF-κB by Tax (37), leading to its recruitment to unintegrated HIV-1 DNA, is the key step that results in the epigenetic changes that correlate with the active transcription and rescue of IN− HIV-1 mutants. Moreover, we now also report that Tax expression can not only prevent the epigenetic silencing of unintegrated HIV-1 DNA but can also reverse the epigenetic silencing of that DNA by inducing the removal of preexisting inhibitory epigenetic marks and promoting the recruitment of activating marks (Fig. 3).
In addition to addressing the effect of Tax on IN− full-length HIV-1, we also looked at how loss of IN function, and activation of Tax expression, affects gene expression from a heterologous reporter cassette inserted into a SIN configuration of the HIV-1 genome. As shown in Fig. 4B, we observed a high level of expression from inserted RSV- and CMV-derived enhancer/promoter combinations driving expression of the NLuc indicator gene, and this high level of expression was, unexpectedly, only minimally impacted by loss of IN function. This high level of NLuc expression, in both the presence and absence of IN function, was also observed when an HIV-1-based vector retaining a fully intact LTR promoter was analyzed, thus arguing that, at least in this vector design, the HIV-1 LTR does not exert any inhibitory effect in cis. To confirm that the intact HIV-1 LTR present in these vectors was indeed active, we were able to demonstrate high levels of HIV-1 Gag protein expression from the IN+ version of the NL-RSV-NLuc and NL-CMV-NLuc vectors as well as from the IN− version, but only in the presence of Tax in the latter case (Fig. 4E). Therefore, while both the RSV LTR and the CMV immediate early promoter-enhancer are highly active in the context of unintegrated HIV-1 DNA, they are both unable to activate the HIV-1 LTR promoter in cis.
MATERIALS AND METHODS
Cells and culture conditions.
The cell lines used in this study are HEK293T (293T), a human kidney epithelial cell line of female origin, and CEM-SS, a human CD4+ T cell line of female origin. CEM-SS cells transduced with a lentiviral vector bearing a Tet-inducible promoter that controls the expression of the HTLV-1 Tax protein were described previously (13). All cells were cultured at 37°C with 5% CO2. 293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotic-antimycotic. CEM-SS cells were cultured in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% FBS and antibiotic-antimycotic.
HIV-1 production.
In experiments that measure the level of viral NLuc expression in cells infected with replication competent HIV-1, we used a previously described replication-competent NLuc reporter virus (pNL-NLuc) where the viral nef gene in NL4-3 was replaced with the NLuc gene (29). The 1Δ and 2Δ NF-κB mutant viruses were created by mutating either one or both of the NF-κB binding sites in the HIV-1 LTR U3 region, respectively, by using overlap extension PCR to change the NF-κB binding site from the functional 5′-GGGACTTTCC-3′ to a nonfunctional 5′-GGGACTGGAT-3′ sequence.
HIV-1 expressing miR-K2 (pNL-miR-K2) was created by PCR amplification of an H1 promoter-driven miR-K2 expression cassette from pSUPER-miR-K2 (35), followed by cloning into pNL4-3 using NotI and XhoI. HIV-1 SIN viruses expressing NLuc from an RSV- or CMV-derived promoter were created by first cloning the promoters into the NotI site 5′ of the NLuc gene in pNL-NLuc (Fig. 3A), then replacing the 3’LTR with the corresponding SIN LTR from the pNL-SIN-CMV-RLuc plasmid described previously (38), which bears a deletion extending from −18 to −418 in U3.
All viruses express either WT integrase (IN) or IN bearing a point mutation, D64V, that blocks IN function (39). Plasmids expressing the NL-NLuc provirus, or the relevant mutant constructs, were transfected into 293T cells using polyethylenimine (PEI). At 24 h posttransfection, the spent media were replaced with fresh media. At 72 h posttransfection, supernatant media were harvested and passed through a 0.45-μm filter to remove cell debris. HIV-1-containing supernatants were normalized using a p24 enzyme-linked immunosorbent assay (ELISA) before being used to infect target cells.
ChIP-qPCR.
Tet-inducible HTLV-1 Tax CEM-SS cells were cultured in 10 ml of RPMI medium (at 106 cells/ml) and infected with WT or IN− HIV-1 virus in the presence or absence of 0.5 μg/ml Dox to induce Tax production. Prior to infection, viral supernatants were incubated with 5 U/ml of DNase I at 37°C for 1 h to remove residual plasmid contamination.
Cells were harvested at the indicated times postinfection, rinsed twice with 1× phosphate-buffered saline (PBS), and cross-linked with 1% formaldehyde for 20 min at 25°C, before being quenched in 0.125 M glycine for 5 min. The rinsed cells were then lysed and subjected to chromatin immunoprecipitation (ChIP) as previously described (13) to pull down the DNA associated with the indicated NF-κB subunits or modified histones. DNA was then purified using DNA cleanup columns (Zymo Research), digested with DpnI (New England Biolabs [NEB]) to remove any residual plasmid contamination, and then used for qPCR analysis using primers that amplify either U5-R on the HIV-1 promoter (forward primer, 5′-CTCTCTGGTTAGACCAGATC-3′; reverse primer, 5′-GCTAGAGATTTTCCACACTG-3′), gag (forward primer, 5′-GCGAGAGCGTCGGTATTAAGCG-3′; reverse primer, 5′-AATCGTTCTAGCTCCCTGCTTGC-3′), or the NLuc gene (forward primer, 5′-TTCAGAATCTCGGGGTGTCCGTA-3′; reverse primer, 5′-TTCGATCTGGCCCATTTGGTCG-3′) in a SYBR green master mix (Thermo Fisher). ChIP data are expressed as a percentage of input DNA, which is calculated by dividing the ChIP copy number for each qPCR target by the copy number from input DNA and multiplying by 100.
Luciferase assays.
Cells were washed three times in PBS, lysed in passive lysis buffer (Promega), and assayed for NLuc activity using the Nano-Glo luciferase assay on a Lumat LB9507 luminometer (Berthold Technologies).
Quantifying changes in HIV-1 replication and viral gene expression.
A total of 107 Tet-inducible Tax CEM-SS cells were suspended in 10 ml of RPMI medium and infected with NLuc reporter viruses in the presence or absence of 0.5 μg/ml Dox to induce Tax expression. Viral supernatants were normalized by p24 and pretreated with 5 U/ml DNase I for 1 h at 37°C to remove residual plasmid DNA. All IN− HIV-1 infections were supplemented with raltegravir (20 μM) to prevent any revertant mutations. Live cells were quantified, and 106 live cells were harvested on days 1, 2, 3, 5, and 7 and equally split into 3 aliquots to assay NLuc or to extract and analyze viral DNA and RNA.
For DNA analysis, cells were spun down, washed three times in ice-cold PBS, and incubated with DpnI (NEB) to remove any residual plasmid contamination. DNA was then extracted using Quick-DNA Miniprep Plus columns (Zymo Research).
For RNA analysis, cells were lysed in TRIzol (Thermo Fisher), and RNA was harvested according to the manufacturer's instructions and treated with DNase I for 2 h. The DNase I was then heat inactivated, and the RNA was converted to cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems).
qPCR assays to quantify total HIV-1 DNA or RNA were performed in triplicate in a QuantStudio 6 Pro real-time PCR system according to the manufacturer's instructions. HIV-1 DNA/cDNA was amplified with a custom total HIV-1 TaqMan probe/primer set that amplifies the U5-gag region of HIV-1 (40). β-Actin DNA was PCR amplified using a premade TaqMan probe/primer set (Thermo Fisher; catalog no. 431182), while β-actin cDNA in RNA analyses was quantified using a separate TaqMan probe/primer set that amplifies across a splice junction (forward primer, 5′-CAATGAAGATCAAGATCATTGC-3′; reverse primer, 5′-AAGCATTTGCGGTGGAC-3′; probe, 5′-FAM [6-carboxyfluorescein]-CCACCTTCCAGCAGATGTGGATCAGCAAG-TAMRA [6-carboxytetramethylrhodamine]-3′). Relative quantification of HIV-1 DNA/RNA levels was performed using the ΔΔCT method by normalizing total HIV-1 DNA/RNA to the β-actin internal control (41).
Quantification of miR-K2.
Tet-inducible HTLV-1 Tax CEM-SS cells were infected with IN+ or IN− pNL-miR-K2 in the presence or absence of 0.5 μg/ml Dox. Nevirapine (Sigma) was added to the cells at 16 hpi, and the cells were harvested for RNA analysis using TRIzol (Thermo Fisher). RNA was diluted to 2 ng/μl and then reverse transcribed with either a specific miR-K2 primer or a U6 RNA internal control primer (Applied Biosystems) using a microRNA reverse transcription kit (Applied Biosystems). The converted cDNA was then quantified by TaqMan qPCR small RNA assays that target either miR-K2 or U6. All qPCR assays were performed in a QuantStudio 6 Pro real-time PCR machine. Quantification was performed by correcting the observed miR-K2 level to the U6 internal control. Corrected miR-K2 expression levels were then normalized to the level of miR-K2 detected in cells infected with IN+ pNL-miR-K2 in the absence of Tax, which was set to 1.
Western blot for HIV-1 Gag.
Cells were lysed in Laemmli buffer, sonicated, and denatured at 95°C for 10 min. Lysates were run on a 4-to-20% SDS-polyacrylamide gel (Bio-Rad), transferred to a nitrocellulose membrane, and then blocked in 5% milk in PBS plus 0.1% Tween (PBS-T). Membranes were incubated with a p24-specific monoclonal antibody (no. 24-3) (AIDS Reagent Program [ARP] no. 6458) or a glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (Proteintech; 60004-1), followed by a horseradish peroxidase (HRP)-conjugated secondary anti-mouse immunoglobulin antibody (Sigma; A9044) in 5% milk in PBS-T for 1 h each, and then washed in PBS-T. The membranes were incubated with a luminol-based enhanced chemiluminescent (ECL) substrate (Advansta), and signals were visualized using GeneSnap (Syngene).
Reactivation of epigenetically repressed IN− HIV-1 by Tax.
Tax-inducible CEM-SS T cells were infected with NL-NLuc reporter virus, and nevirapine (Sigma) was added 16 hpi to prevent secondary viral spread. Then, either 0.5 μg/ml doxycycline in dimethyl sulfoxide (DMSO) or DMSO alone was added at 24 hpi to induce a “with or without Tax” condition. Cells were harvested at 24, 48, and 72 hpi for measurement of NLuc expression and ChIP-qPCR analysis.
ACKNOWLEDGMENTS
This research was supported in part by NIH grant R21-AI157616 to B.R.C., who is also supported through the Center for HIV RNA Studies (CRNA, U54-AI150470).
This research received infrastructure support from the Duke University CFAR (P30-AI064518).
The p24 Gag monoclonal antibody (no. 24-3) was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from Michael Malim.
Contributor Information
Bryan R. Cullen, Email: bryan.cullen@duke.edu.
Frank Kirchhoff, Ulm University Medical Center.
REFERENCES
- 1.Schwartzberg P, Colicelli J, Goff SP. 1984. Construction and analysis of deletion mutations in the pol gene of Moloney murine leukemia virus: a new viral function required for productive infection. Cell 37:1043–1052. 10.1016/0092-8674(84)90439-2. [DOI] [PubMed] [Google Scholar]
- 2.Sakai H, Kawamura M, Sakuragi J, Sakuragi S, Shibata R, Ishimoto A, Ono N, Ueda S, Adachi A. 1993. Integration is essential for efficient gene expression of human immunodeficiency virus type 1. J Virol 67:1169–1174. 10.1128/JVI.67.3.1169-1174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sayana S, Khanlou H. 2008. Raltegravir: the first in a new class of integrase inhibitors for the treatment of HIV. Expert Rev Anti Infect Ther 6:419–426. 10.1586/14787210.6.4.419. [DOI] [PubMed] [Google Scholar]
- 4.Hare S, Smith SJ, Métifiot M, Jaxa-Chamiec A, Pommier Y, Hughes SH, Cherepanov P. 2011. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol Pharmacol 80:565–572. 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. 2010. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Natl Acad Sci U S A 107:20057–20062. 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scopelliti F, Pollicita M, Ceccherini-Silberstein F, Di Santo F, Surdo M, Aquaro S, Perno C-F. 2011. Comparative antiviral activity of integrase inhibitors in human monocyte-derived macrophages and lymphocytes. Antiviral Res 92:255–261. 10.1016/j.antiviral.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Pollicita M, Surdo M, Di Santo F, Cortese MF, Fabeni L, Fedele V, Malet I, Marcelin A-G, Calvez V, Ceccherini-Silberstein F, Perno CF, Svicher V. 2014. Comparative replication capacity of raltegravir-resistant strains and antiviral activity of the new-generation integrase inhibitor dolutegravir in human primary macrophages and lymphocytes. J Antimicrob Chemother 69:2412–2419. 10.1093/jac/dku144. [DOI] [PubMed] [Google Scholar]
- 8.Wiskerchen M, Muesing MA. 1995. Human immunodeficiency virus type 1 integrase: effects of mutations on viral ability to integrate, direct viral gene expression from unintegrated viral DNA templates, and sustain viral propagation in primary cells. J Virol 69:376–386. 10.1128/JVI.69.1.376-386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Englund G, Theodore TS, Freed EO, Engelman A, Martin MA. 1995. Integration is required for productive infection of monocyte-derived macrophages by human immunodeficiency virus type 1. J Virol 69:3216–3219. 10.1128/JVI.69.5.3216-3219.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geis FK, Goff SP. 2019. Unintegrated HIV-1 DNAs are loaded with core and linker histones and transcriptionally silenced. Proc Natl Acad Sci U S A 116:23735–23742. 10.1073/pnas.1912638116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang GZ, Wang Y, Goff SP. 2016. Histones are rapidly loaded onto unintegrated retroviral DNAs soon after nuclear entry. Cell Host Microbe 20:798–809. 10.1016/j.chom.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsai K, Cullen BR. 2020. Epigenetic and epitranscriptomic regulation of viral replication. Nat Rev Microbiol 18:559–570. 10.1038/s41579-020-0382-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irwan ID, Karnowski HL, Bogerd HP, Tsai K, Cullen BR. 2020. Reversal of epigenetic silencing allows robust HIV-1 replication in the absence of integrase function. mBio 11:e01038-20. 10.1128/mBio.01038-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Y, Wang GZ, Cingöz O, Goff SP. 2018. NP220 mediates silencing of unintegrated retroviral DNA. Nature 564:278–282. 10.1038/s41586-018-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Currer R, Van Duyne R, Jaworski E, Guendel I, Sampey G, Das R, Narayanan A, Kashanchi F. 2012. HTLV Tax: a fascinating multifunctional co-regulator of viral and cellular pathways. Front Microbiol 3:406. 10.3389/fmicb.2012.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seggewiss R, Pittaluga S, Adler RL, Guenaga FJ, Ferguson C, Pilz IH, Ryu B, Sorrentino BP, Young WS, Donahue RE, von Kalle C, Nienhuis AW, Dunbar CE. 2006. Acute myeloid leukemia is associated with retroviral gene transfer to hematopoietic progenitor cells in a rhesus macaque. Blood 107:3865–3867. 10.1182/blood-2005-10-4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. 2003. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302:415–419. 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 18.Wanisch K, Yáñez-Muñoz RJ. 2009. Integration-deficient lentiviral vectors: a slow coming of age. Mol Ther 17:1316–1332. 10.1038/mt.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw A, Cornetta K. 2014. Design and potential of non-integrating lentiviral vectors. Biomedicines 2:14–35. 10.3390/biomedicines2010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loewen N, Leske DA, Chen Y, Teo W-L, Saenz DT, Peretz M, Holmes JM, Poeschla EM. 2003. Comparison of wild-type and class I integrase mutant-FIV vectors in retina demonstrates sustained expression of integrated transgenes in retinal pigment epithelium. J Gene Med 5:1009–1017. 10.1002/jgm.447. [DOI] [PubMed] [Google Scholar]
- 21.Naldini L, Blömer U, Gage FH, Trono D, Verma IM. 1996. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci U S A 93:11382–11388. 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park F, Ohashi K, Kay MA. 2000. Therapeutic levels of human factor VIII and IX using HIV-1-based lentiviral vectors in mouse liver. Blood 96:1173–1176. 10.1182/blood.V96.3.1173.015k34_1173_1176. [DOI] [PubMed] [Google Scholar]
- 23.Mitrophanous K, Yoon S, Rohll J, Patil D, Wilkes F, Kim V, Kingsman S, Kingsman A, Mazarakis N. 1999. Stable gene transfer to the nervous system using a non-primate lentiviral vector. Gene Ther 6:1808–1818. 10.1038/sj.gt.3301023. [DOI] [PubMed] [Google Scholar]
- 24.Yáñez-Muñoz RJ, Balaggan KS, MacNeil A, Howe SJ, Schmidt M, Smith AJ, Buch P, MacLaren RE, Anderson PN, Barker SE, Duran Y, Bartholomae C, von Kalle C, Heckenlively JR, Kinnon C, Ali RR, Thrasher AJ. 2006. Effective gene therapy with nonintegrating lentiviral vectors. Nat Med 12:348–353. 10.1038/nm1365. [DOI] [PubMed] [Google Scholar]
- 25.Philippe S, Sarkis C, Barkats M, Mammeri H, Ladroue C, Petit C, Mallet J, Serguera C. 2006. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc Natl Acad Sci U S A 103:17684–17689. 10.1073/pnas.0606197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Apolonia L, Waddington SN, Fernandes C, Ward NJ, Bouma G, Blundell MP, Thrasher AJ, Collins MK, Philpott NJ. 2007. Stable gene transfer to muscle using non-integrating lentiviral vectors. Mol Ther 15:1947–1954. 10.1038/sj.mt.6300281. [DOI] [PubMed] [Google Scholar]
- 27.Philpott NJ, Thrasher AJ. 2007. Use of nonintegrating lentiviral vectors for gene therapy. Hum Gene Ther 18:483–489. 10.1089/hum.2007.013. [DOI] [PubMed] [Google Scholar]
- 28.Bayer M, Kantor B, Cockrell A, Ma H, Zeithaml B, Li X, McCown T, Kafri T. 2008. A large U3 deletion causes increased in vivo expression from a nonintegrating lentiviral vector. Mol Ther 16:1968–1976. 10.1038/mt.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mefferd AL, Bogerd HP, Irwan ID, Cullen BR. 2018. Insights into the mechanisms underlying the inactivation of HIV-1 proviruses by CRISPR/Cas. Virology 520:116–126. 10.1016/j.virol.2018.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee M-TM, Coburn GA, McClure MO, Cullen BR. 2003. Inhibition of human immunodeficiency virus type 1 replication in primary macrophages by using Tat- or CCR5-specific small interfering RNAs expressed from a lentivirus vector. J Virol 77:11964–11972. 10.1128/jvi.77.22.11964-11972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cullen BR, Raymond K, Ju G. 1985. Functional analysis of the transcription control region located within the avian retroviral long terminal repeat. Mol Cell Biol 5:438–447. 10.1128/mcb.5.3.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boshart M, Weber F, Jahn G, Dorsch-Häsler K, Fleckenstein B, Schaffner W. 1985. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 41:521–530. 10.1016/s0092-8674(85)80025-8. [DOI] [PubMed] [Google Scholar]
- 33.Assogba BD, Choi BH, Rho HM. 2002. Transcriptional activation of the promoter of human cytomegalovirus immediate early gene (CMV-IE) by the hepatitis B viral X protein (HBx) through the NF-kappaB site. Virus Res 84:171–179. 10.1016/s0168-1702(01)00445-2. [DOI] [PubMed] [Google Scholar]
- 34.Cullen BR, Lomedico PT, Ju G. 1984. Transcriptional interference in avian retroviruses–implications for the promoter insertion model of leukaemogenesis. Nature 307:241–245. 10.1038/307241a0. [DOI] [PubMed] [Google Scholar]
- 35.Gottwein E, Cai X, Cullen BR. 2006. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J Virol 80:5321–5326. 10.1128/JVI.02734-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith MR, Greene WC. 1990. Identification of HTLV-I tax trans-activator mutants exhibiting novel transcriptional phenotypes. Genes Dev 4:1875–1885. 10.1101/gad.4.11.1875. [DOI] [PubMed] [Google Scholar]
- 37.Böhnlein E, Siekevitz M, Ballard DW, Lowenthal JW, Rimsky L, Bogérd H, Hoffman J, Wano Y, Franza BR, Greene WC. 1989. Stimulation of the human immunodeficiency virus type 1 enhancer by the human T-cell leukemia virus type I tax gene product involves the action of inducible cellular proteins. J Virol 63:1578–1586. 10.1128/JVI.63.4.1578-1586.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gottwein E, Cullen BR. 2007. Protocols for expression and functional analysis of viral microRNAs. Methods Enzymol 427:229–243. 10.1016/S0076-6879(07)27013-2. [DOI] [PubMed] [Google Scholar]
- 39.Leavitt AD, Robles G, Alesandro N, Varmus HE. 1996. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J Virol 70:721–728. 10.1128/JVI.70.2.721-728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler SL, Hansen MST, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7:631–634. 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 41.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]