

ABSTRACT
The endospore-forming pathogen Clostridioides difficile is the leading cause of antibiotic-associated diarrhea and is a significant burden on the community and health care. C. difficile, like all forms of life, incorporates selenium into proteins through a selenocysteine synthesis pathway. The known selenoproteins in C. difficile are involved in a metabolic process that uses amino acids as the sole carbon and nitrogen source (Stickland metabolism). The Stickland metabolic pathway requires the use of two selenium-containing reductases. In this study, we built upon our initial characterization of the CRISPR-Cas9-generated selD mutant by creating a CRISPR-Cas9-mediated restoration of the selD gene at the native locus. Here, we use these CRISPR-generated strains to analyze the importance of selenium-containing proteins on C. difficile physiology. SelD is the first enzyme in the pathway for selenoprotein synthesis, and we found that multiple aspects of C. difficile physiology were affected (e.g., growth, sporulation, and outgrowth of a vegetative cell post-spore germination). Using transcriptome sequencing (RNA-seq), we identified multiple candidate genes which likely aid the cell in overcoming the global loss of selenoproteins to grow in medium which is favorable for using Stickland metabolism. Our results suggest that the absence of selenophosphate (i.e., selenoprotein synthesis) leads to alterations to C. difficile physiology so that NAD+ can be regenerated by other pathways.
IMPORTANCE C. difficile is a Gram-positive, anaerobic gut pathogen which infects thousands of individuals each year. In order to stop the C. difficile life cycle, other nonantibiotic treatment options are in urgent need of development. Toward this goal, we find that a metabolic process used by only a small fraction of the microbiota is important for C. difficile physiology: Stickland metabolism. Here, we use our CRISPR-Cas9 system to “knock in” a copy of the selD gene into the deletion strain to restore selD at its native locus. Our findings support the hypothesis that selenium-containing proteins are important for several aspects of C. difficile physiology, from vegetative growth to spore formation and outgrowth postgermination.
KEYWORDS: Clostridium difficile, Stickland, physiology, selenophosphate
INTRODUCTION
Clostridioides difficile is a major concern as a nosocomial and community-acquired gut pathogen (1). This pathogen has become the most common cause of health care-associated infections in United States hospitals (2, 3). In 2017 in the United States, approximately 223,900 cases of C. difficile infection were identified and nearly 12,800 of those resulted in death. In the same year, it was estimated that C. difficile infections resulted in more than $1 billion in excess health care costs (4). The burden of this pathogen on patients, the community, and health care has led the Centers for Disease Control and Prevention to identify this bacterium as an urgent threat (5).
Key to battling this aggressive pathogen is understanding the basic processes C. difficile uses to complete its life cycle. While our understanding of C. difficile physiology has increased dramatically in the last decade, specifically in toxin production/regulation, sporulation, and germination, our understanding of metabolic processes is lacking (6–11). A recent review by Neumann-Schaal et al. nicely discussed the known metabolic processes involved in energy generation in C. difficile (12). C. difficile has multiple metabolic pathways that overlap to ensure generation of key metabolites. The Wood-Ljungdahl pathway was recently found to be not as active in C. difficile as in other acetogens, but this pathway can be used in conjunction with butyrate formation to regenerate NAD+ in the absence of Stickland metabolism (13). Carbon metabolism includes breaking down sugars such as glucose and mannitol to generate pyruvate and acetyl coenzyme A (acetyl-CoA) for glycolysis and the tricarboxylic acid cycle, although the latter is incomplete. Pyruvate is a key metabolite in many different central carbon metabolism and fermentation pathways (12). It can be utilized by pyruvate formate-lyase to generate CO2 for carbon fixation (14, 15), and it can be degraded to acetyl-CoA to generate butyrate (12). Pyruvate is also used in fermentation pathways to generate propionate via the reductive branch of Stickland metabolism. Fermentation pathways also include Stickland metabolism which contributes to electron bifurcation and the membrane spanning Rnf complex to generate a sodium/proton gradient for substrate-level phosphorylation. While it appears that many of the metabolic processes in C. difficile have been elucidated, how these processes interact when others are impaired has yet to be studied (12).
Stickland metabolism is a primary source of energy for a small group of anaerobic bacteria which use amino acids as their sole carbon and nitrogen source (i.e., C. difficile, Clostridium sporogenes, and Clostridium sticklandii). The main goal of this metabolic pathway is to generate NAD+ and a small amount of ATP for the cell (16–19). In the oxidative branch, an amino acid, most frequently isoleucine, leucine, or valine (ILV), is decarboxylated or deaminated and generates products for other metabolic processes plus NADH (17, 19, 20). In the reductive branch, d-proline or glycine are deaminated or reduced by their respective reductases (proline reductase, PrdB, and glycine reductase, GrdA) to regenerate NAD+ to be reused by the cell (17, 18, 21, 22). Recently, Stickland metabolism was suggested as a significant contributor during C. difficile infection in a murine model. Proline and hydroxyproline were found to be the most abundant molecules at the start of infection; 5-aminovalerate, a product of the proline reductase in Stickland metabolism, is an abundant molecule toward the end of infection (23, 24).
Both the proline and glycine reductases are selenoproteins (20, 25). Selenoproteins are made through the incorporation of selenium, as selenocysteine, during protein synthesis. Selenocysteine is generated through a synthesis pathway where inorganic phosphate reacts with hydrogen selenide to generate selenophosphate by the selenophosphate synthetase, SelD. Through the use of a selenocysteinyl-tRNA (Sec) synthase, SelA, selenophosphate is incorporated into serine-charged tRNAs to generate selenocysteine. The selenocysteine-specific elongation factor, SelB, recognizes an in-frame stop codon followed by a selenocysteine insertion sequence (SECIS). This recognition results in a halt in translation to allow for the incorporation of the selenocysteine into the protein sequence (26).
We hypothesized that if we eliminated the global production of selenoproteins, the two Stickland reductases would not be generated and the resulting strain would be incapable of performing Stickland metabolism. Previously, we generated a CRISPR-Cas9 genome editing tool for use in C. difficile (27). In that work, we created a C. difficile ΔselD strain and analyzed the growth phenotype of this strain compared to that of the wild-type parent and the mutant complemented with a wild-type selD allele in trans. We showed that C. difficile R20291 ΔselD (KNM6) had no growth defect in rich brain heart infusion supplemented with yeast extract (BHIS medium) but did have a slight growth defect in tryptone-yeast extract (TY) medium, which is a peptide-rich medium and should encourage the cells to use Stickland metabolism for growth (27). Here, we build upon our prior work by using the CRISPR-Cas9 system to restore the selD gene at its native locus. Using this selD-restored strain, we sought to further characterize this mutant throughout different lifecycles to determine the global role of selenoproteins on C. difficile physiology. We find that loss of selenophosphate/Stickland metabolism reprograms C. difficile metabolism to pathways that can regenerate NAD+ from NADH.
RESULTS
Complementation in trans results in growth differences at different hydrogen levels.
When originally characterizing the C. difficile KNM6 (ΔselD) mutant strain, we found that the complementing plasmid would not restore the mutant to growth comparable to that of wild-type, in either TY or TY supplemented with glucose (TYG) medium, despite the use of a fragment upstream of the selDAB operon that should contain the native promoter region (Fig. 1A). However, as published in the prior work, we were able to complement the phenotype, and this complementation was dependent on the abundance of hydrogen gas in the anaerobic chamber. When monitored using a COY Anaerobic Monitor (CAM-12), a 4% hydrogen level did not permit the complementing plasmid to restore the growth phenotype to wild-type levels (Fig. 1A). However, when the hydrogen level was lowered to ∼1.7%, we observed complementation in TY medium (Fig. 1B). This observation was interesting and suggests that hydrogen abundance in the anaerobic chamber influences C. difficile physiology or the ability of selD, when expressed from a multicopy plasmid, to function within C. difficile. However, to fully characterize the C. difficile ΔselD mutant, we wanted to avoid this problem altogether by restoring the CRISPR-Cas9-mediated deletion with selD at its native locus.
FIG 1.
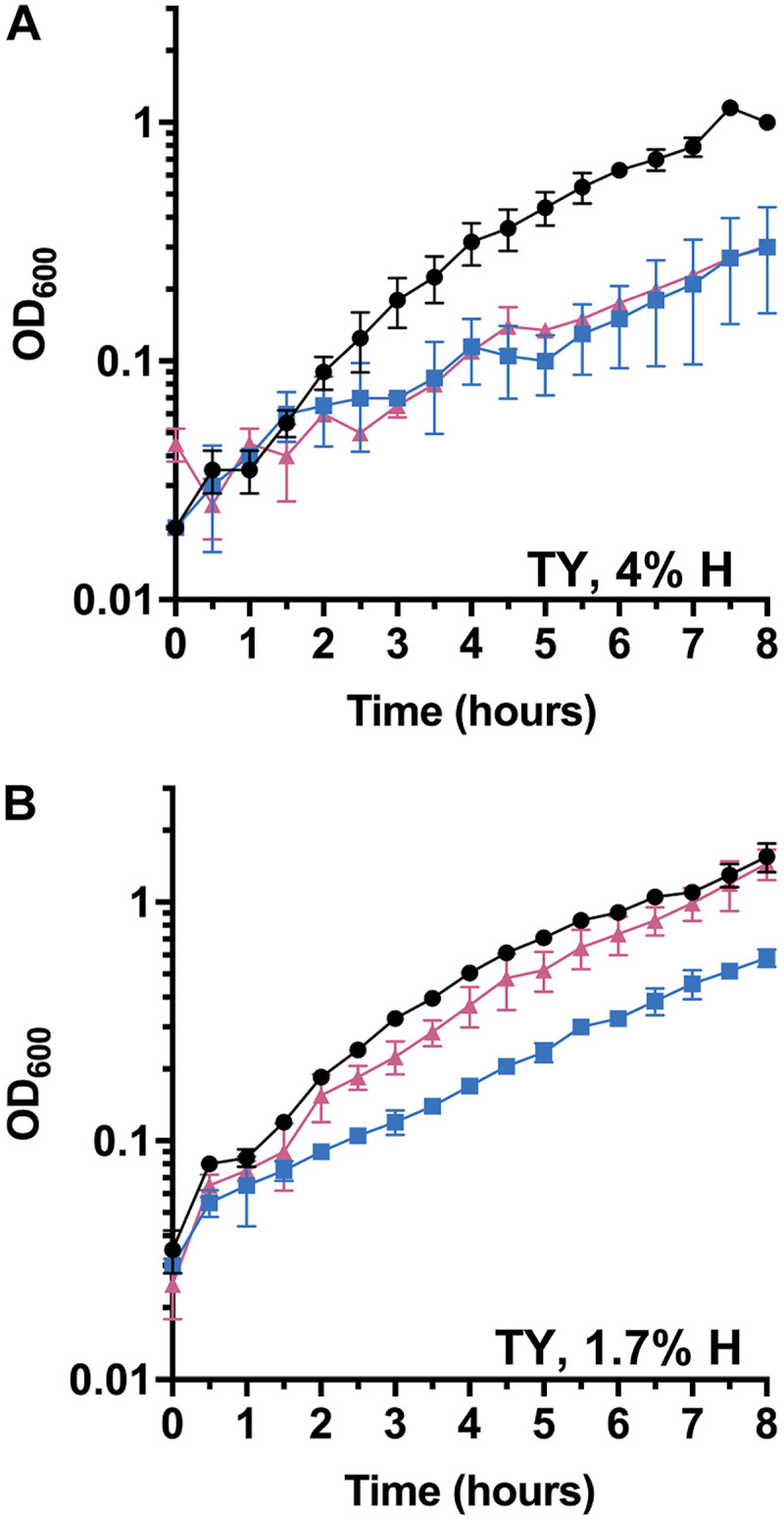
Growth curves with complementing plasmid. C. difficile R20291 pJS116 (wild-type, empty vector) (●), C. difficile KNM6 pJS116 (ΔselD/empty vector) (■), and C. difficile KNM6 pKM142 (ΔselD::selD+/pselD) (▲) were grown in TY medium at 4% hydrogen (A) and 1.7% hydrogen (B), and growth was monitored over an 8 h period. Data points represent the average from two independent experiments and error bars represent the standard deviation from the mean.
Generation of a C. difficile restored selD+ strain by CRISPR-Cas9 genome editing.
Our previously developed tool was used to generate a ΔselD strain and was limited to this type of mutation. By modifying the existing CRISPR-Cas9 plasmid, we have improved the functionality of this tool to include insertions within the genome. Recently, Muh et al. developed a C. difficile CRISPRi tool which included dCas9 to be under the conditional expression of a xylose-inducible promoter system (28). We replaced the previous tetracycline-inducible promoter system, which was shown to have uncontrolled expression of Cas9, with the xylose-inducible promoter. We replaced the homology region and gRNA target sequences in our new CRISPR-Cas9 gene editing plasmid. The targeting region is 26 bp away from the deletion site to reintroduce selD at its native locus (Fig. 2A). Restoration-plasmid-containing C. difficile ΔselD was passaged on xylose-containing agar medium. Using this strategy, we generated a C. difficile selD-restored strain (KNM9; ΔselD::selD+) (Fig. 2B). The restoration was performed once and the efficiency was low (8% to obtain a mixed colony which was passaged once on TY-xylose or BHIS agar to obtain a restoration efficiency of 94% or 88%, respectively).
FIG 2.

Insertion by C. difficile CRISPR-Cas9 genome editing tool and the slight growth defect of a C. difficile ΔselD strain. (A) Graphical representations of the three strains used in this study. Shown as a triangle is the target site of the gRNA, and the line indicates the site of the deletion. (B) DNA was isolated from C. difficile R20291 (wild type), KNM6 (ΔselD), and KNM9 (ΔselD::selD+). The region surrounding the selD gene was amplified from the chromosome, and the resulting DNA was separated on an agarose gel. A clean deletion of selD is indicated by a faster-migrating DNA band while wild type and the insertion mutation (restoration) are indicated by a slower-migrating DNA band. (C and D) C. difficile R20291 (wild type) (●), C. difficile KNM6 (ΔselD) (■), and C. difficile KNM9 (ΔselD::selD+) (▲) were grown in (■) BHIS medium (C) and TY medium (D) at 1.7% hydrogen, and growth was monitored over a 24-h period. Data points represent the average from three independent experiments and error bars represent the standard deviation from the mean.
C. difficile ΔselD strain has a growth defect in peptide-rich medium.
Because we generated a selD-restored strain, we repeated the growth experiments to confirm that the restored strain would complement the phenotype. When the C. difficile wild-type (R20291), ΔselD (KNM6), and ΔselD::selD+ (KNM9) strains were grown in rich BHIS medium, we again saw no difference between the growth of these three strains over 24 h (Fig. 2C). When these three strains were grown in TY (Fig. 2D) or TYG (see Fig. S1 in the supplemental material), we observed a growth defect of the ΔselD strain compared to the growth of wild-type or ΔselD::selD+ (Fig. 2D and Fig. S1). Again, there was no observable difference whether or not glucose was supplemented in the medium on the growth of these strains. For this reason, TY medium was used for most of the subsequent experiments. The growth data are consistent with our previous findings along with the additional finding that the ΔselD mutant strain grows to similar levels as those of the wild-type and restored strains in TY medium at 24 h (Fig. 2D). We will note that these and all subsequent experiments were performed at ∼1.7% hydrogen and will discuss our rationale for this later in the manuscript.
Sporulation.
To understand how the absence of selenoproteins affects C. difficile spore formation, we determined the sporulation frequency in C. difficile R20291, KNM6 (ΔselD), and KNM9 (ΔselD::selD+) (29). As a negative control, we generated a deletion of spo0A in C. difficile R20291 using CRISPR-Cas9 (Fig. S2). Compared to the C. difficile R20291, wild-type, strain, the C. difficile ΔselD mutant produced fewer spores in both BHIS and TY media (Fig. 3A). Restoration of the selD gene resulted in a return of sporulation to wild-type levels. These results indicate that the absence of selenoproteins/selenophosphate has a mild effect on C. difficile sporulation.
FIG 3.
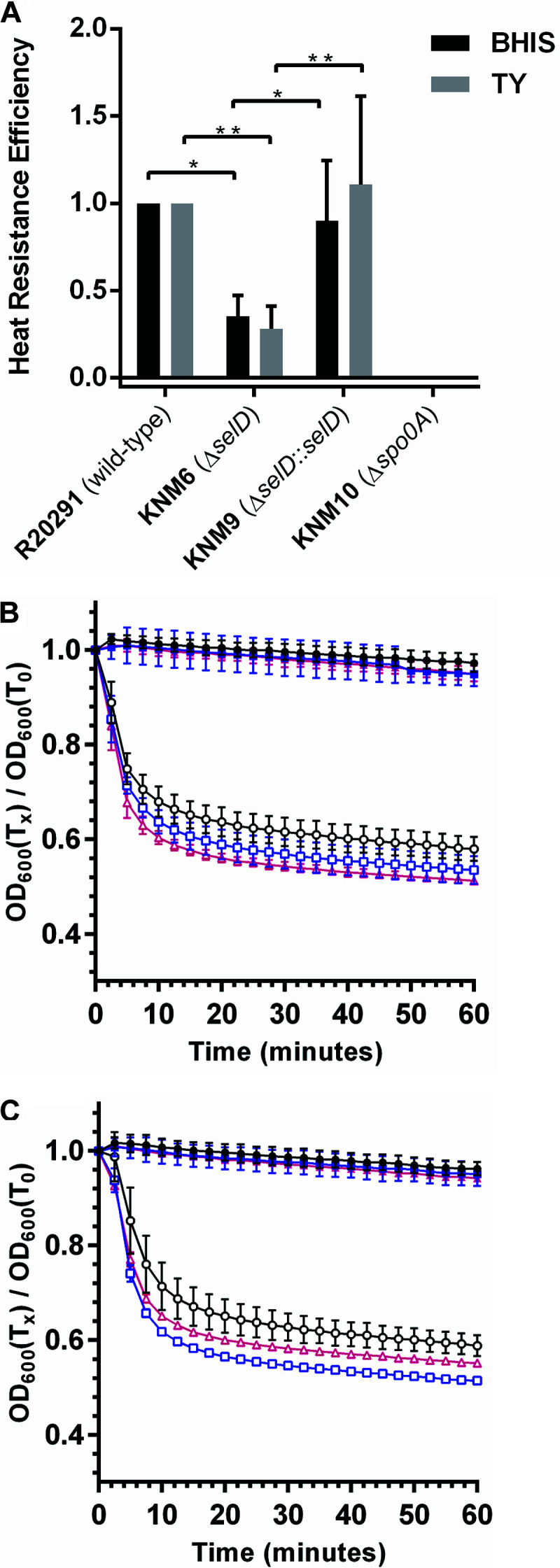
selD plays a significant role in sporulation and has no significant effect on germination of C. difficile spores. (A) C. difficile R20291 (wild-type), C. difficile KNM6 (ΔselD), C. difficile KNM9 (ΔselD::selD+), and C. difficile KNM10 (Δspo0A) strains were allowed to sporulate for 48 h. Cells were harvested and separated into heat-treated and untreated aliquots. The heat-treated samples were heated at 65°C for 40 min. Both heat-treated and untreated samples were serially diluted in PBS and plated onto BHIS-TA. CFU were enumerated and efficiencies were calculated. The data represent the averages from three biological replicates, and error bars represent the standard error of the mean. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple-comparison test (*, P ≤ 0.05; **, P ≤ 0.01), and the C. difficile KNM10 (Δspo0A) strain was excluded from statistical analysis. (B and C) Purified C. difficile R20291 (wild-type) (circles), C. difficile KNM6 (ΔselD) (squares), and C. difficile KNM9 (ΔselD::selD+) (triangles) spores were suspended in (B) BHIS (squares) or TY (C) (triangles) medium supplemented with 10 mM taurocholate (open symbols) or without taurocholate (closed symbols). The change in OD600 during germination was measured over time at 37°C. The data represent the average of three biological replicates and error bars represent the standard deviation from the mean.
Selenoprotein synthesis has no effect on C. difficile spore germination.
Due to the mild defect in sporulation, we then wanted to determine whether the ΔselD strain had any defect in germination. Strains were grown and allowed to sporulate on BHIS medium to minimize any effects from both growth and sporulation defects. The spores were purified and analyzed for germination using the optical density assay. When C. difficile wild-type (R20291), ΔselD (KNM6), and ΔselD::selD+ (KNM9) spores were suspended in rich BHIS medium supplemented with 10 mM taurocholate (TA), rapid germination occurred (Fig. 3B). Since the mutant phenotype is apparent in TY medium, we germinated spores in this medium supplemented with 10 mM TA and measured the drop in optical density. Similar to those in rich medium, the spores rapidly germinated in TY medium (Fig. 3C). These results suggest that the selenophosphate synthetase gene, selD, plays no significant role in the early events during spore germination.
Significant delay in outgrowth of ΔselD germinated spores in peptide-rich medium.
Because we observed a defect in growth of the C. difficile KNM6 (ΔselD) strain, we hypothesized that the strain would have a deficiency in outgrowth from spores. Purified spores from wild-type C. difficile R20291, KNM6 (ΔselD), and KNM9 (ΔselD::selD+) were germinated for 10 min at 37°C in rich BHIS medium to ensure that the germination conditions would not affect outgrowth of a vegetative cell from the germinated spore. After washing of the germinated spores in either BHIS or TY medium, the germinated spores were resuspended in the anaerobic chamber in prereduced BHIS or TY medium. We then monitored the optical densities over a 24-h period (Fig. 4). When cells were allowed to outgrow in rich BHIS medium, there was a nonsignificant delay in growth in the C. difficile KNM6 (ΔselD) strain of approximately 30 min compared to the growth of the wild-type and restored strains (Fig. 4A). On the other hand, when cells were allowed to outgrow in peptide-rich TY medium, C. difficile KNM6 (ΔselD) had an observed outgrowth defect compared to the growth of the wild-type and restored strains (Fig. 4B).
FIG 4.

selD plays a role in outgrowth of C. difficile R20291 spores. Purified C. difficile R20291 (wild-type) (●), C. difficile KNM6 (ΔselD) (■), and C. difficile KNM9 (ΔselD::selD+) (▲) spores were germinated and then resuspended in BHIS (A) or TY (B) medium, and the OD600 was measured over an 8-h period and then again at 24 h. Data points represent the average from three independent experiments and error bars represent the standard deviation from the mean. Statistical significance was determined using a two-way ANOVA with Tukey’s multiple-comparison test (*, P ≤ 0.05). Only time points in which comparisons between R20291 versus KNM6 and KNM6 versus KNM9 were both significant are labeled.
RNA-seq comparison of wild-type versus ΔselD strains.
If the hypothesis that Stickland metabolism is a primary source of energy for C. difficile is true, we wondered why the growth defect in peptide-rich medium was not more severe compared to that in wild-type and restored strains. We then hypothesized that the C. difficile KNM6 (ΔselD) strain was able to compensate for the loss of global selenoprotein synthesis through upregulation of other metabolic pathways. To determine which pathways were differentially expressed, we performed RNA-seq on the wild-type C. difficile R20291 and C. difficile KNM6 (ΔselD) strains during exponential growth in TY medium supplemented with glucose. At the time of performing these experiments, we were not aware of whether glucose would have an effect on gene expression and, therefore, decided to include this carbohydrate in the medium. We performed these experiments at both ∼1.7% and 4% hydrogen levels to determine whether atmospheric hydrogen abundance affects C. difficile physiology.
First, we noticed when comparing expression of genes in wild-type cells grown at low and high hydrogen levels, the cells had significant downregulation of ribosomal proteins at high hydrogen. This suggests that the cells perceive high hydrogen as a stressful condition. Due to this finding, we chose to exclude highly up- or downregulated gene expression due to hydrogen levels when comparing wild-type and mutant expression levels. In other words, we excluded expression of genes which had significant expression changes in either low or high hydrogen levels of wild-type cultures. After this exclusion, we obtained a narrower list for comparison of wild-type and ΔselD strains at low hydrogen levels and further determined their known or hypothesized function and to what pathway each gene belongs (Table 1) (30, 31). As expected, many of the genes are involved in metabolism: 36% of genes which were upregulated and 22% of genes which were downregulated (Fig. 5). In both cases, there were large numbers of genes of unknown function, more than 25% both up- and downregulated. Other than those involved in metabolism and unknown functions, the largest group of genes which contained upregulated expression in the ΔselD strain were characterized as transferases. Although, many of these genes also fit into another KEGG pathway, i.e., metabolism. Another group of genes which were significant were those of transporters or involved in secretion, likely increasing the cell’s import and export systems to compensate for this growth impairment. The largest group of genes which were downregulated, besides those involved in metabolism or unknown functions, belonged to secretion systems or hydrolases. Again, the cell was likely increasing certain pathways or import/export systems to allow energy to be utilized elsewhere.
TABLE 1.
Differentially expressed genes in the C. difficile ΔselD strain compared to those in the wild-type strain
| Name | Fold change | Annotation or KEGG function | KEGG pathway |
|---|---|---|---|
| Upregulated | |||
| CDR20291_0963 | 70.544 | Alginate O-acetyltransferase complex protein | Unclassified metabolism |
| CDR20291_0962 | 44.982 | Putative uncharacterized protein | Unknown |
| CDR20291_0440 | 17.707 | Cell surface protein (putative hemagglutinin/adhesin) | Unknown |
| CDR20291_1747 | 12.712 | Putative conjugative transposon regulatory protein | Unknown |
| cysM | 8.129 | Putative O-acetylserine sulfhydrylase | Energy and amino acid metabolism |
| cysA | 5.460 | Serine O-acetyltransferase | Energy and amino acid metabolism |
| mtlF | 4.901 | Phosphotransfer system (PTS), mannitol-specific IIA component | Carbohydrate metabolism, transferases |
| mtlD | 4.753 | Mannitol-1-phosphate 5-dehydrogenase | Carbohydrate metabolism, oxidoreductases |
| CDR20291_1260 | 4.374 | Putative membrane protein | Unknown |
| mtlR | 3.886 | Mannitol operon transcriptional antiterminator | Transcription factors |
| CDR20291_2627 | 3.031 | Cytosine permease | Transporters |
| mtlA | 2.984 | PTS system, mannitol-specific IIBC component | Carbohydrate metabolism, transferases |
| CDR20291_2626 | 2.978 | Putative carbon-nitrogen hydrolase | Unknown |
| CDR20291_0474 | 2.623 | Putative exported protein | Unknown |
| ribH | 2.430 | 6,7-Dimethyl-8-ribityllumazine synthase (riboflavin synthase beta chain) | Metabolism of cofactors and vitamins, transferases |
| CDR20291_1593 | 2.319 | Putative arsenical pump membrane protein | Unknown |
| ribD | 2.232 | Diaminohydroxyphosphoribosylaminopyrimidine deaminase/5-amino-6-(5-phosphoribosylamino) uracil reductase (riboflavin biosynthesis protein) | Metabolism of cofactors and vitamins, cell community, oxidoreductases, hydrolases |
| fdhA | 2.185 | l-Seryl-tRNA (Ser) seleniumtransferase | Metabolism of other amino acids, translation, transferases |
| modA | 2.176 | Molybdate transport system substrate-binding protein | Membrane transport, transporters |
| CDR20291_0182 | 2.171 | Putative membrane-associated metalloprotease | Unknown |
| CDR20291_2397 | 2.167 | TetR-family transcriptional regulator | Unknown |
| CDR20291_0146 | 2.153 | Putative riboflavin transporter | Unknown |
| CDR20291_0178 | 2.152 | Cyclopropane-fatty-acyl-phospholipid synthase | Unclassified metabolism, transferases |
| CDR20291_2191 | 2.145 | Putative pilin protein, general secretion pathway protein G | Membrane transport, secretion system |
| CDR20291_1446 | 2.124 | Prophage antirepressor-related protein | Unknown |
| CDR20291_2012 | 2.100 | ABC-2 type transport system ATP-binding protein | Transporters |
| CDR20291_1405 | 2.089 | Putative polysaccharide deacetylase | Unknown |
| ribB | 2.083 | Riboflavin synthase | Metabolism of cofactors and vitamins, transferases |
| CDR20291_0763 | 2.073 | Aconitate hydratase | Carbohydrate metabolism, lyases |
| glyA | 2.069 | Glycine hydroxymethyltransferase | Carbohydrate, energy, amino acid, other amino acid, and cofactors and vitamin metabolism, transferases |
| iorB | 2.027 | Indolepyruvate ferredoxin oxidoreductase, beta subunit | Unclassified metabolism, oxidoreductases |
| CDR20291_1816 | 2.007 | Putative lipoprotein | Unknown |
| CDR20291_0765 | 2.001 | MarR family transcriptional regulator | Unknown |
| Downregulated | |||
| nfo | 2.042 | Deoxyribonuclease IV | Replication and repair, hydrolases |
| CDR20291_1819 | 2.090 | Putative phage-related deoxycytidylate deaminase (putative late competence protein) | Nucleotide metabolism, secretion systems, hydrolases |
| CDR20291_0177 | 2.112 | Putative oxidoreductase, NAD/FAD binding subunit | Unknown |
| CDR20291_3153 | 2.114 | Type IV pilus assembly protein | Secretion systems |
| CDR20291_1319 | 2.257 | Putative phage shock protein | Unknown |
| CDR20291_1685 | 2.274 | Putative exonuclease | Unknown |
| CDR20291_0175 | 2.333 | Anaerobic carbon-monoxide dehydrogenase catalytic subunit | Energy metabolism, xenobiotics biodegradation and metabolism, oxidoreductases |
| CDR20291_2875 | 2.424 | Conserved hypothetical protein | Unknown |
| CDR20291_2931 | 2.599 | Putative amino acid permease | Unknown |
| CDR20291_2077 | 3.005 | Putative sodium-dicarboxylate symporter | Unknown |
| CDR20291_2932 | 3.055 | Putative glutamine amidotransferase | Peptidases |
| selD | 49.582 | Selenide, water dikinase | Metabolism of other amino acids, transferases |
FIG 5.

Distribution of functions of up- and downregulated genes from RNA-seq. Pie charts of the KEGG functional pathways of genes which were upregulated (A) and downregulated (B) from RNA-seq analysis. Percentages of each function are shown in the legends.
Validation of RNA-seq by quantitative RT-PCR.
We then chose genes which we have highlighted as well as others of interest to validate the RNA-seq results by qRT-PCR. C. difficile R20291 and C. difficile KNM6 strains were grown as previously described for RNA-seq, DNA was depleted, and cDNA libraries were generated. Using the housekeeping gene, rpoB, as an internal normalization control, we determined the fold change of transcripts in the KNM6 (ΔselD) strain compared to that of transcripts in the R20291 (wild-type) strain at both 4% and 1.7% hydrogen levels. The target genes CDR20291_0962 and CDR20291_0963 had transcript levels which were interesting at 1.7% hydrogen percentage since it appeared that, in one biological replicate, the genetic switch was likely turned on while other biological replicates had fold change values close to one and were likely turned in the off position (Fig. 6A and B). One target gene, mtlF, which is part of the mannitol utilization pathway, had a slightly increased fold change in transcript levels at both 4% and 1.7% by qRT-PCR, and the trend correlated with the results found from RNA-seq (Fig. 6C). Mannitol utilization may be a large factor helping the strain grow in the absence of selenoproteins.
FIG 6.

Fold change of transcript levels in C. difficile KNM6 (ΔselD) compared to that in C. difficile R20291. RNA was extracted from C. difficile R20291 and KNM6 (ΔselD) strains grown to an OD600 of 0.6 in TYG medium, DNA was depleted, and cDNA was synthesized. Quantitative reverse transcription-PCR was performed to determine the fold change of transcript levels in the KNM6 (ΔselD) strain compared to that in the wild-type R20291 strain at 4% and 1.7% hydrogen levels. Fold changes for the following genes are shown: CDR20291_0962 (A), CDR20291_0963 (B), mtlF (C), prdB (D), grdB (E), CDR20291_2627 (F), and CDR20291_2932 (G). Data represent the average from three independent experiments run in technical triplicate and error bars represent the standard deviation from the mean. Technical replicate outliers were excluded. *, P < 0.05. **, P < 0.01. NS, not significant.
Since we hypothesized Stickland metabolism to be impaired in the mutant strain, we chose to analyze the proline reductase gene, prdB, and the glycine reductase gene, grdB. The fold change in expression of prdB was higher than was seen in RNA-seq (Fig. 6D) but due to variance in the samples did not meet the threshold for significance. However, there is a clear trend for higher expression under these conditions. In qRT-PCR, the transcription of grdB was downregulated in the mutant compared to that of the wild type (3.6- and 4.4-fold downregulated in 4% and 1.7% hydrogen, respectively) (Fig. 6E). On the other hand, the transcription of grdB was slightly upregulated in RNA-seq (1.3- and 2-fold upregulated in 4% and 1.7% hydrogen, respectively).
Finally, we validated two uncharacterized genes. CDR20291_2627, a putative hemagglutinin/adhesion, was upregulated in the RNA-seq at both 4% hydrogen and 1.7% hydrogen (Fig. 6F). We also observed upregulation of this gene in the qRT-PCR validations. Again, these did not meet the threshold for significance. Additionally, CDR20291_2932, a putative glutamine deaminase, was slightly downregulated, and we see this trend by qRT-PCR (Fig. 6G).
DISCUSSION
It is becoming increasingly appreciated that Stickland metabolism and Stickland substrates are important for C. difficile pathogenesis. In the oxidative branch of Stickland metabolism, several different amino acids can be deaminated or decarboxylated. In the reductive branch, either proline or glycine is used by its respective reductase. The proline reductase, PrdB, converts proline and NADH to 5-aminovalerate and NAD+ while the glycine reductase, GrdA, converts glycine, NADH, and ADP to acetate, ATP, and NAD+. The activity of the PrdB and GrdA proteins is dependent on the incorporation of a modified amino acid, selenocysteine.
Incorporation of selenium into proteins is governed by the SelD, SelA, and SelB proteins. The first step in this pathway is the generation of selenophosphate from hydrogen selenide by the selenophosphate synthetase, SelD. We hypothesized that a mutation in C. difficile selD would significantly alter C. difficile physiology by blocking the total incorporation of selenium into protein as an amino acid, a selenocysteine, or part of selenium-containing cofactors. Surprisingly, we found that plasmid-based complementation of the selD deletion led to an odd observation, that complementation only occurred at low hydrogen percentage (1.7%) in our anaerobic chamber. At higher hydrogen abundance (4%), we could not complement our mutant.
To address this, we used our CRISPR/Cas9 genome editing tool to restore the selD gene at its native locus. By targeting the Cas9 nuclease to a region just outside the deleted region, we were able to isolate a restored strain. This C. difficile ΔselD::selD+ strain grew similarly to the wild-type strain. We then analyzed the physiology of the strains. We observed no differences in germination between the wild-type, mutant, and restored strains, but the ΔselD strain had a mild decrease in spore formation. However, we observed a significant delay during the outgrowth of germinated C. difficile spores in peptide-rich medium (TY). This highlights the importance of selenophosphate during the early stages of return to vegetative growth.
To understand the overall impact of SelD on C. difficile physiology, we determined the global expression differences between the wild-type and mutant strains at low and high hydrogen percentages. First, we observed that fdhA was upregulated in the selD mutant strain. fdhA encodes the selenocysteinyl-tRNA synthetase that charges serine-charged tRNA with selenophosphate to generate selenocysteine-charged tRNA. This suggests that in the absence of selenoprotein, C. difficile senses this loss and attempts to increase the rate of selenocysteine-charged tRNA.
On first glance, CDR20291_0963 and CDR20291_0962 are the two most highly upregulated genes in the ΔselD strain. CDR20291_0963 is uncharacterized in C. difficile but has high similarity to the AlgI protein, an alginate O-acetyltransferase, characterized in Pseudomonas aeruginosa. Whether this protein has the same function in C. difficile is yet to be determined. The other gene, CDR20291_0962, is an uncharacterized gene of no known function. When run through NCBI BLAST and the Conserved Domain Database (32, 33), this protein sequence has a DHHW motif which is common among bacteria. What we found interesting is that both of these genes have recently been characterized to contain a RecV-targeted genetic switch upstream, and their altered expression is likely unrelated to the selD mutation but, rather, due to the switch being in the “on” position (34).
Interestingly, every gene in the operon mtlARFD had upregulated expression in the ΔselD strain. The expression levels varied between almost 3- to 5-fold upregulation compared to that of the wild type. We hypothesize that mannose metabolism may play a larger role in a strain lacking selenoproteins. In this pathway, mannitol is taken into the cell and broken down to mannitol-1-phosphate by MtlF and MtlA. Then, MtlD uses NAD+ to convert mannitol-1-phosphate into fructose-6-phosphate plus NADH to then be used for glycolysis (35–38). Prior work by Teschner and Garel has shown that the Escherichia coli MtlD can run this reaction in reverse to generate NAD+ instead of consuming it (39).
Besides mannose metabolism, cysteine and methionine metabolism appeared to be increased by the upregulation of cysM and cysA. It is interesting that these genes are utilizing H2S produced by C. difficile to generate l-cysteine and acetate (40, 41). Acetate is also a final product in the reduction of glycine by Stickland metabolism (17). It is possible that the cell is generating acetate through this method in order to utilize the molecule in other metabolic pathways.
Another metabolic process which had multiple genes upregulated was for riboflavin synthesis: ribD, a deaminase and reductase gene; ribH, a lumazine synthase gene; and ribB, a riboflavin synthase gene. Riboflavin is an essential cofactor which can be broken down into flavins (flavin mononucleotide [FMN]) and flavin adenine dinucleotide (FAD) to be used by the cell. Riboflavin can also be taken up by the environment if present (42). A gene proposed to be a riboflavin transporter, CDR20291_0146, was also upregulated from our RNA-seq analysis. This essential cofactor is likely being used in multiple processes, and its synthesis or uptake is being upregulated to feed these metabolic pathways.
Of those genes which had downregulated expression, there are a few worth noting. Multiple genes involved in transport or utilization of amino acids were downregulated, possible due to the decrease in the dependence on Stickland metabolism to provide NAD+ for the cell to generate energy. CDR20291_2931, a putative amino acid permease gene for transport of amino acids into the cell, and CDR20291_2932, a putative glutamine amidotransferase gene for transforming glutamine into generating an available carbon-nitrogen group, are both proposed to be involved in the utilization of amino acids (43, 44). As well, a putative sodium-dicarboxylate symporter gene, CDR20291_2077, could also be involved in amino acid uptake into the cell. The downregulation of these three genes indicates that the cell is moving its resources away from the degradation of amino acids as a source of energy.
Overall, our data highlight the importance of selenophosphate for C. difficile physiology. The absence of selenophosphate leads to the rerouting of C. difficile metabolism so that the cell is less dependent on Stickland metabolism of amino acids and the regeneration of NAD+ by the reductive branch of Stickland. However, what was surprising was the effect of atmospheric chamber conditions (H2 percentage) on C. difficile physiology. Further work should be done to understand how hydrogen abundance influences C. difficile physiology, potentially in autotrophic metabolism, and when reporting on C. difficile physiology, the concentration of hydrogen should be reported.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
C. difficile strains were routinely grown in an anaerobic atmosphere (1.7% to 4% H2, 5% CO2, 85% N2) at 37°C in brain heart infusion supplemented with 5 g/liter yeast extract and 0.1% l-cysteine (BHIS), as described previously (8, 45–47), or TY medium (3% tryptone, 2% yeast extract) (48). Hydrogen levels were determined by a COY anaerobic monitor (CAM-12). For conjugation experiments, cells were plated on TY medium for Bacillus subtilis-based conjugations. Where indicated, growth was supplemented with taurocholate (TA; 0.1% wt/vol), thiamphenicol (10 μg/ml), d-cycloserine (250 μg/ml), xylose (1% wt/vol), and/or glucose (1% wt/vol) as needed. Induction of the CRISPR-Cas9 system was performed on TY agar plates supplemented with thiamphenicol (10 μg/ml) and xylose (1% wt/vol). E. coli strains were routinely grown at 37°C in LB medium. Strains were supplemented with chloramphenicol (20 μg/ml) as needed. B. subtilis BS49 was routinely grown at 37°C in LB broth or on LB agar plates. Strains were supplemented with chloramphenicol (2.5 μg/ml) and/or tetracycline (5 μg/ml).
Plasmid construction and molecular biology.
To construct the CRISPR-Cas9 selD complementing plasmid, the previously published CRISPR-Cas9 pyrE targeting plasmid, pJK02 (27), was modified by replacing traJ with oriT Tn916 for B. subtilis conjugation by amplification from pJS116 using primers 5′Tn916ori and 3′Tn916ori. The resulting fragments was introduced into pJK02 by Gibson assembly at the ApaI site and transformed into E. coli DH5α to generate pKM126. Next, the donor region to be used for homology directed repair was PCR amplified from C. difficile R20291 genomic DNA using primers 5′selD_comp and 3′selD_comp 2 where this fragment contains a 500-bp upstream homology arm and a 500-bp downstream homology arm surrounding selD. The resulting fragment was cloned by Gibson assembly into pKM126 at the NotI and XhoI restriction sites and transformed into E. coli DH5α to generate pKM181. Lastly, the gBlock for selD-targeting sgRNA, CRISPR_selD_comp2, was introduced by Gibson assembly into the KpnI and MluI restriction sites and transformed into E. coli DH5α resulting in pKM183. To improve efficiency of the CRISPR-Cas9 editing system and provide more control of Cas9 expression, the tetracycline-inducible system was replaced with the xylose-inducible system (28). To do this, the xylose-inducible promoter was PCR amplified from pIA33 using primers 5′selDcomp_HR_xylR 2 and 3′cas9_Pxyl 2, inserted by Gibson assembly into pKM183 at the XhoI and PacI restriction sites, and transformed into E. coli DH5α to generate pKM194.
To construct the CRISPR-Cas9 spo0A deletion mutant, the gBlock for spo0A targeting sgRNA, CRISPR_spo0A_2, was introduced by Gibson assembly into the KpnI and MluI restriction sites in pKM197 (49) and transformed into E. coli DH5α resulting in pKM213. The homology arms to be used for homology directed repair were PCR amplified from C. difficile R20291 genomic DNA using primers 5′spo0A_UP and 3′spo0A_UP for the 500-bp upstream arm and primers 5′spo0A_DN and 3′spo0A_DN for the 500-bp downstream arm. The resulting fragments were then cloned by Gibson assembly at the NotI and XhoI restriction sites and transformed into E. coli DH5α resulting in pKM215.
Conjugation for CRISPR-Cas9 plasmid insertion.
CRISPR-Cas9 plasmid pKM194 was transformed into B. subtilis BS49 to be used as a donor for conjugation with C. difficile KNM6. Likewise, the pKM215 plasmid was transformed into B. subtilis BS49 to be used as a donor for conjugation with C. difficile R20291. C. difficile R20291 or KNM6 was grown anaerobically in BHIS broth overnight. This was then diluted in fresh prereduced BHIS broth and grown anaerobically for 4 h. Meanwhile, B. subtilis BS49 containing the CRISPR plasmids was grown aerobically at 37°C in LB broth supplemented with tetracycline and chloramphenicol for 3 h. One hundred microliters of each culture was plated on TY agar medium. After 24 h, the growth was harvested by suspending in 2 ml prereduced BHIS broth. A loopful of this suspended growth was spread onto several BHIS agar plates supplemented with thiamphenicol, kanamycin, and d-cycloserine. C. difficile transconjugants were screened for the presence of Tn916 using tetracycline resistance, as described previously. Thiamphenicol-resistant, tetracycline-sensitive transconjugants were selected and used for further experiments.
Induction of the CRISPR-Cas9 system and isolating mutants.
The C. difficile KNM6 strain containing the selD-targeting plasmid, pKM194, was streaked onto TY agar medium supplemented with thiamphenicol and xylose for induction. This was then passaged a second time on the same medium. After isolating colonies on BHIS supplemented with xylose, DNA was extracted and tested for the insertion by PCR amplification of the selD region using primers 5′selD and 3′selD. From this, one mixed colony out of 12 samples was isolated. This mixed colony was then passaged on TY agar medium supplemented with thiamphenicol and xylose. Colonies were isolated on BHIS agar medium supplemented with xylose, DNA was extracted, and isolates were tested by PCR amplification again as above. From this, 14 out of 15 colonies had insertions. Confirmed restored strains were passaged 3 times in BHIS liquid medium in order to lose the CRISPR-Cas9 plasmid. After pick and patch on BHIS agar with and without thiamphenicol, loss of plasmid was confirmed by PCR amplification of a portion of cas9 using primer set 5′tetR_CO_Cas9 and 3′COcas9 (975) and the gRNA using primer set 5′gdh and 3′gRNA 2.
The C. difficile R20291 strain containing the spo0A-targeting plasmid, pKM215, was streaked onto TY agar medium supplemented with thiamphenicol and xylose for induction. This was then passaged a second time on the same medium. After isolating colonies on BHIS supplemented with xylose, DNA was extracted and tested for the insertion by PCR amplification of the spo0A region using primers 5′spo0A_del and 3′spo0A_del. All tested colonies (36 total) were mutants. Two isolates were passaged twice in BHIS liquid medium in order to lose the CRISPR-Cas9 plasmid. After pick and patch on BHIS agar with and without thiamphenicol, loss of plasmid was confirmed by PCR amplification of a portion of cas9 using primer set 5′tetR_CO_Cas9 and 3′COcas9 (975) and the gRNA using primer set 5′gdh and 3′gRNA 2.
Sporulation and heat resistance assay.
To determine differences in sporulation efficiencies between the C. difficile R20291 (wild-type), KNM6 (ΔselD), KNM9 (ΔselD::selD+), and KNM10 (Δspo0A) strains, sporulation and heat resistance were determined as described previously (29). Briefly, C. difficile R20291, KNM6, KNM9, and KNM10 strains were spread onto BHIS agar medium supplemented with taurocholate. From this, colonies were restreaked onto either BHIS or TY agar medium, making a lawn on the plate. After 48 h of growth, half of the plate was harvested and mixed into 600 μl of prereduced phosphate-buffered saline (PBS). Then, 300 μl of the sample was transferred to a separate tube and heat treated at 65°C in a heat block for 30 min, inverting the tube every 10 min to ensure even heating. Both the untreated and the heat-treated samples were serially diluted in PBS and plated onto BHIS agar medium supplemented with taurocholate. CFU were counted 22 h after plating. Heat resistance was calculated by dividing the CFU for the heat-treated sample by the CFU for the untreated sample, and the average was calculated for each strain.
Spore purification.
Spores were purified from C. difficile R20291, KNM6, and KNM9 strains as previously described (46, 50, 51). Briefly, spores were streaked onto BHIS agar medium (20 to 30 plates) and allowed to sporulate for 5 to 6 days before each was scraped into microcentrifuge tubes containing 1 ml of sterile dH2O and kept at 4°C overnight. The spores and debris mixture was washed five times in sterile dH2O by centrifuging for 1 min at 14,000 × g per wash. Spores were combined into 2- ml aliquots in sterile dH2O, layered on top of 8 ml of 50% sucrose, and centrifuged at 4,000 × g for 20 min. The supernatant containing vegetative cells and cell debris was discarded and the spores were resuspended in sterile dH2O and washed five more times as before. The spores were stored at 4°C until use.
Germination assay.
Purified C. difficile R20291, KNM6, and KNM9 spores were first heat activated at 65°C for 30 min and then placed on ice until use. To compare germination between the three strains, the optical density at 600 nm (OD600) was measured over time in different media. Germination was carried out in clear Falcon 96-well plates at 37°C in a final volume of 100 μl and final concentrations of 10 mM taurocholate and 1× BHIS or TY. Spores were added to a final OD600 of 0.5 and germination was analyzed for 1 h using a plate reader (Spectramax M3 plate reader; Molecular Devices, Sunnyvale, CA) (50, 52).
Outgrowth assays.
Purified C. difficile R20291, KNM6, and KNM9 spores were heat activated at 65°C for 30 min and then placed on ice until use. Spore samples were washed one time with dH2O and then spores were added to a final optical density (OD600) of 0.5 in 8 ml of BHIS medium supplemented with taurocholate (10 mM final concentration). The spores were incubated in germination solution in a hot water bath at 37°C for 10 min and then immediately placed on ice. In all subsequent steps, the germinated spores were kept on ice or at 4°C and washed once with BHIS or TY medium, and supernatant was removed. The germinated spores were passed into the anaerobic chamber without using vacuum (flooded chamber with gas) and each was resuspended in 20 ml of prereduced BHIS or TY medium. The outgrowth was monitored by measuring OD600 over time.
RNA processing.
C. difficile R20291 (wild type) and KNM6 (ΔselD) were grown to an OD600 of 0.6 in TYG medium at low (1.7%) and high (4%) hydrogen levels. At that time, RNA was extracted as described using the FastRNA Blue kit (MP Biologicals). DNA was depleted using the TURBO DNA-free kit (Invitrogen), repeating the steps in the kit three times to achieve complete depletion. rRNA was then depleted using the Ribo-Zero rRNA removal kit for bacteria (Illumina). Enrichment of mRNA and generation of cDNA libraries were completed using the TruSeq Stranded mRNA library prep kit (Illumina).
RNA-seq.
cDNA libraries were submitted for Illumina high output single-end 50 sequencing at the Tufts Genomic Core. Reads were assembled using the DNASTAR SeqMan NGen 15 program in a combined assembly noting replicates. Raw expression data between wild-type and mutant strains were then normalized to rpoB and then quantified using DNASTAR ArrayStar 15, assemblies were normalized by assigned reads per kilobase of template per million mapped reads (RPKM), experiments’ values were capped at a minimum of 1, and all genes were normalized by calibration to the median expression value of rpoB. Fold change was determined four ways: wild type at 4% hydrogen to wild type at 1.7% hydrogen, wild type at 1.7% to wild type at 4%, wild type at 4% to mutant at 4%, and wild type at 1.7% to mutant at 1.7%. Expression which was >2-fold up- or downregulated from that of the wild type at the different hydrogen levels was excluded when comparing the respective wild-type to mutant comparisons. From this, any gene expression which was >2-fold up- or downregulated was considered significant. As a final step, we excluded any genes which had little coverage or low reads visualized using DNASTAR GenVision.
Quantitative RT-PCR.
C. difficile R20291 (wild type) and KNM6 (ΔselD) were grown, RNA was extracted, and DNA was depleted the same as described above. Fifty ng of total RNA was used in cDNA synthesis using the SuperScript III first-strand synthesis system (Thermo Scientific) according to the protocol, including controls for each sample without the presence of reverse transcriptase. Primers to be used for quantitative reverse transcription-PCR (qRT-PCR) were designed using the Primer Express 3.0 software (Applied Biosystems) and efficiencies were validated prior to use. cDNA samples were then used as templates for qPCRs to amplify CDR20291_0963, CDR20291_0962, prdB, grdB, mtlF, CDR20291_2627, and CDR20291_2932 (primers are listed in Table S3 in the supplemental material) using PowerUp SYBR green Master Mix (Applied Biosystems) and a QuantStudio 6 Flex real-time PCR machine (Applied Biosystems). Reactions were performed in a final volume of 10 μl including 1 μl of undiluted cDNA sample and a 500 nM concentration of each primer. Reactions were run in technical triplicate of each biological triplicate for both wild-type and mutant samples at each hydrogen level. Outliers of technical replicate samples were omitted from analysis. Results were calculated using the comparative cycle threshold method (53), in which the amount of target mRNA was normalized to that of an internal control (rpoB).
Statistical analysis.
C. difficile R20291 pJS116 (wild-type, empty vector), C. difficile KNM6 pJS116 (ΔselD/empty vector), and C. difficile KNM9 (ΔselD::selD+/pselD) strains were grown in biological duplicate. C. difficile R20291, KNM6, and KNM9 strains were grown in biological triplicate.
C. difficile R20291, KNM6, KNM9, and KNM10 strains were allowed to sporulate in biological triplicate. Statistical significance to determine differences in sporulation was determined using a two-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001), and the C. difficile KNM10 (Δspo0A) strain was excluded from statistical analysis. C. difficile R20291, KNM6, and KNM9 spores were purified in biological triplicate where each replicate was grown/sporulated and purified separately to be used for germination and outgrowth assays. C. difficile R20291, KNM6, and KNM9 spores were germinated and growth was monitored over time in biological triplicate to determine outgrowth. Statistical significance between strains during outgrowth was determined using a two-way ANOVA with Tukey’s multiple-comparison test (P ≤ 0.05). Only time points where both comparisons of R20291 versus KNM6 and KNM6 versus KNM9 are statistically significant with P ≤ 0.05 are reported.
RNA was extracted from C. difficile R20291 and KNM6 strains for RNA-seq in biological duplicate for each strain. RNA was extracted from C. difficile R20291 and KNM6 strains for qRT-PCR in biological triplicate and each was run in technical triplicate as well. A t test for two samples assuming unequal variances was performed on R20291 and KNM6 qPCR samples. In each experiment, data represent averages of each of the indicated replicates and error bars represent the standard deviation from the mean. Technical replicate outliers were excluded from qRT-PCR analysis.
ACKNOWLEDGMENTS
We thank Craig Ellermeier at the University of Iowa for the generous gift of the xylose-inducible promoter. We also thank members of the Sorg lab, Leif Smith, and members from Leif Smith’s lab at Texas A&M University for their helpful comments and suggestions during the preparation of the manuscript.
This project was supported by awards 5R01AI116895 and 1U01AI124290 to J.A.S. from the National Institute of Allergy and Infectious Diseases.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material is available online only.
jb.00008-21-s0001.pdf (1.1MB, pdf)
jb.00008-21-s0002.xlsx (258.5KB, xlsx)
Contributor Information
Joseph A. Sorg, Email: jsorg@bio.tamu.edu.
Tina M. Henkin, Ohio State University
REFERENCES
- 1.Poutanen SM, Simor AE. 2004. Clostridium difficile-associated diarrhea in adults. CMAJ 171:51–58. 10.1503/cmaj.1031189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 3.Smits WK, Lyras D, Lacy DB, Wilcox MH, Kuijper EJ. 2016. Clostridium difficile infection. Nat Rev Dis Primers 2:16020. 10.1038/nrdp.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.CDC. 2019. Antibiotic resistance threats in the United States, 2019. U.S. Department of Health and Human Services, CDC. [Google Scholar]
- 5.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18:247–263. 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179. 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 8.Francis MB, Allen CA, Shrestha R, Sorg JA. 2013. Bile acid recognition by the Clostridium difficile germinant receptor, CspC, is important for establishing infection. PLoS Pathog 9:e1003356. 10.1371/journal.ppat.1003356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL. 2007. Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 66:206–219. 10.1111/j.1365-2958.2007.05906.x. [DOI] [PubMed] [Google Scholar]
- 10.Dembek M, Barquist L, Boinett CJ, Cain AK, Mayho M, Lawley TD, Fairweather NF, Fagan RP. 2015. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. mBio 6:e02383. 10.1128/mBio.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fimlaid KA, Bond JP, Schutz KC, Putnam EE, Leung JM, Lawley TD, Shen A. 2013. Global analysis of the sporulation pathway of Clostridium difficile. PLoS Genet 9:e1003660. 10.1371/journal.pgen.1003660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neumann-Schaal M, Jahn D, Schmidt-Hohagen K. 2019. Metabolism the Difficile way: the key to the success of the pathogen Clostridioides difficile. Front Microbiol 10:219. 10.3389/fmicb.2019.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gencic S, Grahame DA. 2020. Diverse energy-conserving pathways in Clostridium difficile: growth in the absence of amino acid Stickland acceptors and the role of the Wood-Ljungdahl pathway. J Bacteriol 202. 10.1128/JB.00233-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinske C, Sawers RG. 2016. Anaerobic formate and hydrogen metabolism. EcoSal Plus 7. 10.1128/ecosalplus.ESP-0011-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ragsdale SW. 1997. The eastern and western branches of the Wood/Ljungdahl pathway: how the east and west were won. Biofactors 6:3–11. 10.1002/biof.5520060102. [DOI] [PubMed] [Google Scholar]
- 16.Nisman B, Raynaud M, Cohen GN. 1948. Extension of the Stickland reaction to several bacterial species. Arch Biochem 16:473. [PubMed] [Google Scholar]
- 17.Stickland LH. 1935. Studies in the metabolism of the strict anaerobes (genus Clostridium): the oxidation of alanine by Cl. sporogenes. IV. The reduction of glycine by Cl. sporogenes. Biochem J 29:889–898. 10.1042/bj0290889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stickland LH. 1935. Studies in the metabolism of the strict anaerobes (genus Clostridium): the reduction of proline by Cl. sporogenes. Biochem J 29:288–290. 10.1042/bj0290288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stickland LH. 1934. Studies in the metabolism of the strict anaerobes (genus Clostridium): the chemical reactions by which Cl. sporogenes obtains its energy. Biochem J 28:1746–1759. 10.1042/bj0281746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouillaut L, Self WT, Sonenshein AL. 2013. Proline-dependent regulation of Clostridium difficile Stickland metabolism. J Bacteriol 195:844–854. 10.1128/JB.01492-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stadtman TC. 1956. Studies on the enzymic reduction of amino acids: a proline reductase of an amino acid-fermenting Clostridium, strain HF. Biochem J 62:614–621. 10.1042/bj0620614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadtman TC, Elliott P. 1957. Studies on the enzymic reduction of amino acids. II. Purification and properties of D-proline reductase and a proline racemase from Clostridium sticklandii. J Biol Chem 228:983–997. 10.1016/S0021-9258(18)70675-3. [DOI] [PubMed] [Google Scholar]
- 23.Jenior ML, Leslie JL, Young VB, Schloss PD. 2017. Clostridium difficile colonizes alternative nutrient niches during infection across distinct murine gut microbiomes. mSystems 2. 10.1128/mSystems.00063-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jenior ML, Leslie JL, Young VB, Schloss PD. 2018. Clostridium difficile alters the structure and metabolism of distinct cecal microbiomes during initial infection to promote sustained colonization. mSphere 3. 10.1128/mSphere.00261-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson S, Calos M, Myers A, Self WT. 2006. Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. J Bacteriol 188:8487–8495. 10.1128/JB.01370-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Self WT. 2010. Specific and nonspecific incorporation of selenium into macromolecules. Comprehensive Natural Products II: Chemistry and Biology: Amino Acids, Peptides and Proteins 5:121–148. [Google Scholar]
- 27.McAllister KN, Bouillaut L, Kahn JN, Self WT, Sorg JA. 2017. Using CRISPR-Cas9-mediated genome editing to generate C. difficile mutants defective in selenoproteins synthesis. Sci Rep 7:14672. 10.1038/s41598-017-15236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muh U, Pannullo AG, Weiss DS, Ellermeier CD. 2019. A xylose-inducible expression system and a CRISPRi-plasmid for targeted knock-down of gene expression in Clostridioides difficile. J Bacteriol 201. 10.1128/JB.00711-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen A, Fimlaid KA, Pishdadian K. 2016. Inducing and quantifying Clostridium difficile spore formation. Methods Mol Biol 1476:129–142. 10.1007/978-1-4939-6361-4_10. [DOI] [PubMed] [Google Scholar]
- 30.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. 2016. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44:D457–462. 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Geer LY, Bryant SH. 2017. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45:D200–D203. 10.1093/nar/gkw1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Sekulovic O, Mathias Garrett E, Bourgeois J, Tamayo R, Shen A, Camilli A. 2018. Genome-wide detection of conservative site-specific recombination in bacteria. PLoS Genet 14:e1007332. 10.1371/journal.pgen.1007332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behrens S, Mitchell W, Bahl H. 2001. Molecular analysis of the mannitol operon of Clostridium acetobutylicum encoding a phosphotransferase system and a putative PTS-modulated regulator. Microbiology (Reading) 147:75–86. 10.1099/00221287-147-1-75. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe S, Hamano M, Kakeshita H, Bunai K, Tojo S, Yamaguchi H, Fujita Y, Wong SL, Yamane K. 2003. Mannitol-1-phosphate dehydrogenase (MtlD) is required for mannitol and glucitol assimilation in Bacillus subtilis: possible cooperation of mtl and gut operons. J Bacteriol 185:4816–4824. 10.1128/jb.185.16.4816-4824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang M, Gu L, Cheng C, Ma J, Xin F, Liu J, Wu H, Jiang M. 2018. Recent advances in microbial production of mannitol: utilization of low-cost substrates, strain development and regulation strategies. World J Microbiol Biotechnol 34:41. 10.1007/s11274-018-2425-8. [DOI] [PubMed] [Google Scholar]
- 38.Byer T, Wang J, Zhang MG, Vather N, Blachman A, Visser B, Liu JM. 2017. MtlR negatively regulates mannitol utilization by Vibrio cholerae. Microbiology (Reading) 163:1902–1911. 10.1099/mic.0.000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teschner W, Serre MC, Garel JR. 1990. Enzymatic properties, renaturation and metabolic role of mannitol-1-phosphate dehydrogenase from Escherichia coli. Biochimie 72:33–40. 10.1016/0300-9084(90)90170-l. [DOI] [PubMed] [Google Scholar]
- 40.Pye VE, Tingey AP, Robson RL, Moody PC. 2004. The structure and mechanism of serine acetyltransferase from Escherichia coli. J Biol Chem 279:40729–40736. 10.1074/jbc.M403751200. [DOI] [PubMed] [Google Scholar]
- 41.Rabeh WM, Cook PF. 2004. Structure and mechanism of O-acetylserine sulfhydrylase. J Biol Chem 279:26803–26806. 10.1074/jbc.R400001200. [DOI] [PubMed] [Google Scholar]
- 42.Vitreschak AG, Rodionov DA, Mironov AA, Gelfand MS. 2002. Regulation of riboflavin biosynthesis and transport genes in bacteria by transcriptional and translational attenuation. Nucleic Acids Res 30:3141–3151. 10.1093/nar/gkf433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mantsala P, Zalkin H. 1984. Glutamine nucleotide sequence of Saccharomyces cerevisiae ADE4 encoding phosphoribosylpyrophosphate amidotransferase. J Biol Chem 259:8478–8484. 10.1016/S0021-9258(17)39755-7. [DOI] [PubMed] [Google Scholar]
- 44.Massiere F, Badet-Denisot MA. 1998. The mechanism of glutamine-dependent amidotransferases. Cell Mol Life Sci 54:205–222. 10.1007/s000180050145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen CA, Babakhani F, Sears P, Nguyen L, Sorg JA. 2013. Both fidaxomicin and vancomycin inhibit outgrowth of Clostridium difficile spores. Antimicrob Agents Chemother 57:664–667. 10.1128/AAC.01611-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Francis MB, Allen CA, Sorg JA. 2015. Spore cortex hydrolysis precedes dipicolinic acid release during Clostridium difficile spore germination. J Bacteriol 197:2276–2283. 10.1128/JB.02575-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sorg JA, Dineen SS. 2009. Laboratory maintenance of Clostridium difficile. Curr Protoc Microbiol Chapter 9:Unit9A 1. [DOI] [PubMed] [Google Scholar]
- 48.Dupuy B, Sonenshein AL. 1998. Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol 27:107–120. 10.1046/j.1365-2958.1998.00663.x. [DOI] [PubMed] [Google Scholar]
- 49.Bhattacharjee D, Sorg JA. 2020. Factors and conditions that impact electroporation of Clostridioides difficile strains. mSphere 5. 10.1128/mSphere.00941-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shrestha R, Cochran AM, Sorg JA. 2019. The requirement for co-germinants during Clostridium difficile spore germination is influenced by mutations in yabG and cspA. PLoS Pathog 15:e1007681. 10.1371/journal.ppat.1007681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Francis MB, Sorg JA. 2016. Detecting cortex fragments during bacterial spore germination. J Vis Exp 54146. 10.3791/54146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shrestha R, Sorg JA. 2018. Hierarchical recognition of amino acid co-germinants during Clostridioides difficile spore germination. Anaerobe 49:41–47. 10.1016/j.anaerobe.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]