

ABSTRACT
Noroviruses, members of the Caliciviridae family, are the major cause of epidemic gastroenteritis in humans, causing ∼20 million cases annually. These plus-strand RNA viruses have T=3 icosahedral protein capsids with 90 pronounced protruding (P) domain dimers to which antibodies and cellular receptors bind. In the case of mouse norovirus (MNV), bile salts have been shown to enhance receptor (CD300lf) binding to the P domain. We demonstrated previously that the P domains of several genotypes are markedly flexible and “float” over the shell, but the role of this flexibility was unclear. Recently, we demonstrated that bile causes a 90° rotation and collapse of the P domain onto the shell surface. Since bile binds distally to the P–shell interface, it was not at all clear how it could cause such dramatic changes. Here, we present the near-atomic resolution cryo-electron microscopy (cryo-EM) structure of the MNV protruding domain complexed with a neutralizing Fab. On the basis of previous results, we show here that bile salts cause allosteric conformational changes in the P domain that block antibody recognition of the top of the P domain. In addition, bile causes a major rearrangement of the P domain dimers that is likely responsible for the bile-induced collapse of the P domain onto the shell. In the contracted shell conformation, antibodies to the P1 and shell domains are not expected to bind. Therefore, at the site of infection in the gut, the host’s own bile allows the virus to escape antibody-mediated neutralization while enhancing cell attachment.
IMPORTANCE The major feature of calicivirus capsids is the 90 protruding domains (P domains) that are the site of cell receptor attachment and antibody epitopes. We demonstrated previously that these P domains are highly mobile and that bile causes these “floating” P domains in mouse norovirus (MNV) to contract onto the shell surface. Here, we present the near-atomic cryo-EM structure of the isolated MNV P domain complexed with a neutralizing Fab fragment. Our data show that bile causes two sets of changes. First, bile causes allosteric conformational changes in the epitopes at the top of the P domain that block antibody binding. Second, bile causes the P domain dimer subunits to rotate relative to each other, causing a contraction of the P domain that buries epitopes at the base of the P and shell domains. Taken together, the results show that MNV uses the host’s own metabolites to enhance cell receptor binding while simultaneously blocking antibody recognition.
KEYWORDS: antibodies, bile, cryo-electron microscopy, neutralization, noroviruses
INTRODUCTION
Noroviruses cause ∼20 million cases of epidemic gastroenteritis in humans annually, resulting in more than 70,000 hospitalizations and 570 to 800 deaths in the United States alone (1), and cost approximately US$10 billion each year (2). While infections are not commonly fatal in areas with sufficient health care resources, there are ∼200 million deaths per year in children under the age of 5 years worldwide. The difficulty of controlling the spread of norovirus outbreaks is exacerbated by the highly infectious nature of noroviruses: as few as 10 virions are sufficient to infect an adult (3).
Caliciviruses are T=3 icosahedral particles with 180 copies of the major capsid protein (VP1; ∼58 kDa), which is divided into the N terminus (N), the shell (S), and C-terminal protruding (P) domains (4, 5). The S domain forms a tight shell around the viral RNA genome, while the P domains dimerize to form protrusions on the capsid surface. Each P domain is subdivided into P1 and P2 subdomains, corresponding to the base and the top of the surface protrusions, respectively; the latter contain the binding sites for cellular receptors (6, 7) and some of the neutralizing antibodies (8–10).
We previously determined the cryo-electron microscopy (cryo-EM) structure of mouse norovirus (MNV) virions in phosphate-buffered saline (PBS) at pH 7.4 (5, 10, 11). The P domains were observed to be “floating” >10 Å above the shell. To confirm that MNV was not an anomaly in this regard, the structure of rabbit hemorrhagic disease virus (RHDV) virus-like particles (VLP) was also determined and showed similar features (11). This marked flexibility was also shared with human genotypes, as shown in the cryo-EM structure of human norovirus strain Vietnam026 (GII.10) (12). The latter study also showed that P domain flexibility may play an important role in antibody binding, since a broadly neutralizing antibody (5B18) was observed to bind to the conserved base (P1) domain of GII.10, which is exposed only by extreme flexibility in the P domain. Similarly, epitopes on human genotypes GI.1 (13) and GII.4 (14) are exposed only if the P domain is lifted off the shell. Therefore, we suggested that one biological role of the flexible P domain may be to “confuse” the immune system by presenting a wide array of epitopes exposed only in the expanded, or flexible, state (15).
There is also flexibility within the MNV P domain itself. In the initial crystal structure of the MNV P domain (10), the two loops at the very top of the P2 domain (A′B′ and E′F′) displayed two conformations: a closed structure, where these loops were tightly associated, and an open structure, where the loops were splayed apart. Since mice are the natural animal model for MNV, we developed several monoclonal antibodies (MAbs) from orally infected mice (16) and have determined several structures of the whole virion complexed with fragments of these antibodies. In the ∼13-Å cryo-EM structure of MNV complexed with a neutralizing Fab fragment from antibody A6.2 (5), we found that the crystal structure of Fab A6.2 (17) modeled best onto the open conformation with the hydrophobic heavy-chain CDR3 loop extending into the hydrophobic interface between the A′B′ and E′F′ loops. Subsequent studies on a second neutralizing antibody, 2D3, found that the locations of 2D3 escape mutations were in the heart of the P domain and distal to the A′B′/E′F′ loops and to MAb A6.2 binding contacts (18). Surprisingly, cryo-EM studies demonstrated that both 2D3 and A6.2 bind to the tops of the P domains in very similar ways, yet the 2D3 escape mutations had no effect on A6.2 (5, 10, 17). Therefore, we proposed that the 2D3 escape mutations (D348E and V339I) act in an allosteric-modulator-like manner and affect the epitope structure by altering the structural equilibrium of the P domain.
CD300lf is the cellular receptor for MNV (19, 20). Virus binding to CD300lf is enhanced by serum factors, including bile salts (21). In humans, the major bile salts are taurocholic acid, glycocholic acid, glycochenodeoxycholic acid (GCDCA), and taurochenodeoxycholic acid (TCA), in approximately equal concentrations (21). In the gut, the total concentration of bile is approximately 2.5 to 45 mM (22). The details of the MNV–receptor–bile interactions with isolated P domains have been elucidated by X-ray (21, 23) and cryo-EM structures (24). The bile salts bind in two deep pockets at the P2 domain dimer interface, distant from receptor and antibody binding sites. In both the X-ray and cryo-EM structures, the conformations of the A′B′ and E′F′ loops when bile was bound were nearly identical to the closed apo conformation. The major difference between the apo conformation and the P domain/bile complex is that the C′D′ loop in the bile complex is pointed upward toward the E′F′ loop to create space for the bile salt to bind. What is particularly interesting is that one of the escape mutants to the neutralizing antibodies 2D3 and 4F9 (V339I) lies immediately adjacent to the bile salt binding site. However, whether its mode of escape is related to bile binding or its effects on the P domain structure remains unknown.
The ∼3-Å cryo-EM structures of the entire MNV capsid complexed with bile salts (24) showed that bile binding causes major rearrangements of the MNV capsid (Fig. 1). Bile binding causes the P domain to rotate counterclockwise by ∼90° and collapse onto the shell surface. The major difference between this bile-bound complex and the previous apo X-ray structure (10) of the P domain (in both the open and closed conformations) is that the C′D′ loop turns up when bile binds and makes extensive interactions with the A′B′/E′F′ loops. In this conformation, it seemed unlikely that antibodies could bind to the MNV/bile complex. It was unclear, however, how bile binding so far from the P domain/shell interface could cause such a drastic contraction of the capsid.
FIG 1.

Summary of the MNV capsid structure and the conformational changes induced by bile (24). Shown are cryo-EM structures of the intact MNV capsid in the absence (A and C) and presence (B and D) of the bile salt GCDCA. (A) The mauve arrows show that when bile is added, the entire P domains rotate by ∼90° in the counterclockwise direction. (B) Bile binding changes the surfaces of the tips of the P domains by the movement of the C′D′ loop. (C and D) Bile also causes the entire P domains to contract onto the shell surface by a movement of ∼10 Å (mauve arrow in panel C). The gray arrow in panel C highlights the flexible tether loosely connecting the P domain and shell in the apo capsid structure.
The results presented here clarify the mechanistic consequences of the MNV response to bile binding. A major problem with the previous virion/Fab structures was that the flexibility of the P domain greatly limited the resolution of the antibody/P domain interface. Therefore, we could model the complex only by using crystal structures of the unbound complexes, leaving our models for conformational changes unproven. To solve this remaining issue, we present here the near-atomic resolution cryo-EM structure of the isolated P domain/Fab A6.2 complex to a resolution of 3.2 Å. This structure shows that the Fab binds to the open conformation of the A′B′/E′F′ loops and that, without bile bound, the C′D′ loop points away from the A′B′/E′F′ loops. Therefore, since antibodies and bile recognize markedly different conformations of the P domain dimer, they cannot bind simultaneously to the P domain dimer. This competition was confirmed functionally by binding and neutralization assays. Comparisons of the cryo-EM structures in the presence and absence of bile show that bile causes a rotation of the two P domains within the dimer. This bile-induced rotation alters the surface at the base of the P domain to better complement the top of the shell domain. Therefore, bile buries epitopes throughout the P domain by changing the conformation at the tip of the P domain and causing capsid contraction, thus hiding the epitopes on the P1 and shell domains. This suggests that antibodies made to the virus in the low-bile environment of the tissue may be of limited use once the antibody encounters the virus in the intestinal lumen in the presence of bile. These results may impact the development of vaccines for other viruses that might use host metabolites to escape neutralization.
RESULTS AND DISCUSSION
Bile allosterically blocks antibody binding.
The previous low-resolution cryo-EM structures of the intact MNV/Fab A6.2 complex (5) left a number of questions unanswered about the mechanism of neutralization. To obtain a high-resolution structure for the MNV capsid/A6.2 complex, we initially attempted to use the fact that bile causes contraction of the P domain onto the shell, which results in high-resolution density for the entire P domain. However, after several attempts, it was clear that the Fab was unable to bind to the MNV/bile complex. Therefore, we determined the near-atomic cryo-EM structure of the isolated P domain complexed with Fab A6.2. The P domains form dimers (with 2-fold symmetry) in solution and on the virion, but the EM density was refined without assuming any symmetry (C1). The structure and electron density of the MNV P domain/Fab A6.2 complex are shown in Fig. 2, and statistics are shown in Table 1. As was observed in the cryo-EM structure of intact MNV complexed with Fab A6.2 (5), one Fab is bound to each copy of P domain protein, forming a “Y” structure. While the density for most of the complex was clear and allowed for unambiguous fitting, the density of the constant domains was more diffuse, which is more than likely due to the marked flexibility of the Fab at the elbow region. Similar results were observed in the crystal structure of intact human rhinovirus 14 (HRV14) complexed with neutralizing Fab17 (25). As was predicted in the previous pseudoatomic model built from the ∼13-Å EM density of the MNV/A6.2 complex, the A′B′/E′F′ loops of the P domain are in the open conformation, and the hydrophobic CDR3 loop of the antibody reaches down into the interface between the two loops. Figure 3 shows that the density of the paratope/epitope interface is very well defined and that there are extensive hydrophobic interactions between the heavy-chain CDR3 and the cleft between the A′B′ and E′F′ loops. Previously, we determined the atomic structures of Fab A6.2 (17) and the isolated P domain (10) and used them to interpret the cryo-EM structure of the Fab/MNV virion complex. The position of A6.2 in this pseudoatomic model (5, 10) is very close to that shown in Fig. 3 but is rotated slightly such that fewer light-chain contacts are made. As discussed below, the previous crystal structure of the A′B′/E′F′ loops in the open conformation (10) is only slightly different from the actual structure of the P domain in the Fab/P domain complex determined in this study. Therefore, A6.2 is not inducing conformational changes in the P domain per se but rather is binding to a structure found in solution. In contrast, the heavy-chain CDR3 structure (Fig. 3C) is markedly different from what was observed in the crystal structure of Fab A6.2 alone (17). In the crystal structure of Fab A6.2 alone, the hydrophobic residues fold upon themselves to avoid the aqueous environment. When A6.2 binds to the P domain, the CDR3 loop unfurls to meet the hydrophobic residues in the A′B′/E′F′ cleft. This is similar to the crystal structure of the HRV14/Fab17 complex, where the flexible CDR3 loop changed upon binding but did not induce significant conformational changes in the viral capsid (25).
FIG 2.

Cryo-EM structure of the isolated MNV P domain dimer complexed with neutralizing A6.2 Fab’s. (A) Stereo image of the entire structure and associated cryo-EM envelope. Note that the cryo-EM density at the outermost portion of the Fab constant domains is weak, likely due to the inherent flexibility at the elbow. (B) Stereo image of the structure without density. The two copies of the P domain are color-coded brown and blue, and the Fab light and heavy chains are color-coded green and red, respectively; the two copies of Fab have lighter and darker hues.
TABLE 1.
Data collection and real-space refinement statistics for the MNV P domain/Fab A6.2 complex
| Parametera | Fab complex |
|---|---|
| PDB ID | 7L5J |
| EMDB code | EMD-23187 |
| Data collection | |
| Instrument used | Titan Krios G3i |
| No. of images | 9,681 |
| No. of particles picked | 8,220,523 |
| No. of particles used | 1,825,383 |
| Software | CryoSPARC, v2 |
| Refinement statistics | |
| Resolution (Å) | 3.2 |
| No. of atoms | 11,234 |
| Rwork (%) | 36 |
| CCmask | 0.79 |
| Ramachandran plot (%) | |
| Outliers | 0.07 |
| Allowed | 16.4 |
| Favored | 83.53 |
| Geometry deviation | |
| Bond length (Å) | 0.008 |
| Bond angle (°) | 0.88 |
| B values (Å2) | |
| Min | 67 |
| Max | 310 |
| Mean | 156 |
Rwork, model R-factor; CCmask, density correlation coefficient; Min, minimum; Max, maximum.
FIG 3.

Structure of the complex at the epitope/paratope interface. (A and B) The antibody/P domain interface with (A) and without (B) cryo-EM density. The heavy and light chains are color-coded red and green, respectively. One of the P domain subunits is shown in blue. The hydrophobic heavy-chain CDR3 loop penetrates between the A′B′ and E′F′ loops at the tip of the P domain. (C) Comparison between the crystal structure of Fab A6.2 alone (lighter hues) and the cryo-EM structure presented here. For clarity, the structure of the P domain is not shown here. The arrows indicate how the hydrophobic CDR3 loop “unfurls” and extends as it binds to the hydrophobic gap between the A′B′ and E′F′ loops.
The light-chain and heavy-chain interactions with the P domain are summarized in Tables 2 and 3, respectively. For these calculations, the online PDBePISA application (26) was used. As is quite evident, the heavy chain dominates the interactions, with >6 times as much buried surface area as the light chain. As is also evident, and consistent with Fig. 3, the interface is dominated by hydrophobic interactions. It is interesting to compare the actual contacts with those found in the pseudoatomic model using the 13-Å EM map (17). The paratope/epitope interface in the high-resolution structure presented here has ∼100 Å2 more surface area than our previous pseudoatomic model (5, 10). The increased surface area comes entirely from the heavy chain. This is due to the heavy-chain CDR3 loop extending farther into the A′B′/E′F′ cleft than in our pseudoatomic model.
TABLE 2.
Summary of light-chain contacts
| Light chain |
P domain |
||
|---|---|---|---|
| Residue | Area (Å2) | Residue | Area (Å2) |
| L45 | 10.4 | F297 | 76.1 |
| Y48 | 32.3 | Q298 | 14.4 |
| L53 | 1.1 | L384 | 9.9 |
| A54 | 3.0 | ||
| S55 | 28.0 | ||
| F95 | 10.0 | ||
| Total | 85.0 | 100.4 | |
TABLE 3.
Summary of heavy-chain contacts
| Heavy chain |
P domain |
||
|---|---|---|---|
| Residue | Area (Å2) | Residue | Area (Å2) |
| S30 | 7.1 | Y295 | 46.2 |
| R31 | 65.5 | E296 | 7.6 |
| Y32 | 36.5 | F297 | 56.5 |
| W33 | 54.6 | T301 | 7.0 |
| Q50 | 12.8 | G302 | 23.5 |
| N52 | 5.8 | E303 | 81.3 |
| P53 | 19.8 | S377 | 7.4 |
| H54 | 29.3 | V378 | 32.4 |
| S56 | 6.7 | T379 | 9.0 |
| T57 | 6.4 | A380 | 73.5 |
| L99 | 9.7 | A381 | 31.9 |
| L100 | 39.4 | A382 | 31.0 |
| R101 | 130.8 | S383 | 6.9 |
| Y102 | 94.1 | L384 | 163.3 |
| F103 | 66.1 | D385 | 16.1 |
| Y104 | 41.0 | L386 | 37.0 |
| Y105 | 1.5 | D388 | 20.3 |
| Y107 | 5.6 | ||
| Total | 632.5 | 650.9 | |
Comparisons between the various cryo-EM and X-ray structures show that the P domain exists in multiple conformations in solution and that this equilibrium is influenced by several factors. Figure 4A compares the cryo-EM structure without bile (in dark hues), bound to Fab A6.2, with the open conformation observed in the crystal structure (in light hues) (PDB code 3LQ6) (10). All the loops are essentially in the same conformation, with the A′B′/E′F′ loops splayed apart and the C′D′ loop pointing down, away from the other two loops. It should be noted that the cryo-EM structure of the apo, intact virus is consistent with these structures (24). Figure 4B shows the cryo-EM structure without bile (in dark hues), bound to A6.2, as in Fig. 4A, compared to the crystal structure of the “closed” conformation (in light hues) (PDB code 3LQE) (10). As suggested by the previous MNV/A6.2 modeling (17), it is impossible for A6.2 to bind to the virus in the closed conformation because of predicted clashes between the heavy-chain CDR3 and P domain E′F′ loops. Importantly, without bile bound in the apo, closed X-ray structure, the C′D′ loop points away from the A′B′/E′F′ loops. Finally, Fig. 4C compares the EM structure of the MNV capsid/bile complex (PDB code 6P4J) (24) with that of the A6.2/P domain (no bile) complex presented here. Bile binds at the dimer interface, resulting in a stoichiometry of 2 bile molecules per P domain dimer. The bile interactions with the P domains maintain the 2-fold symmetry of the dimers. When bile binds to the P domain, the C′D′ loop moves toward the A′B′/E′F′ loops and likely stabilizes the closed conformation. Together, these results suggest that the A′B′/E′F′ loops exist in an open and closed conformation equilibrium, and the structure of the apo MNV capsid (24) suggests that the open conformation is favored in solution. When bile binds, this equilibrium shifts toward the closed conformation, and the C′D′ loop moves up toward the A′B′/E′F′ loops to stabilize the closed conformation.
FIG 4.

Comparison of the P domain epitope loop structures under various conditions. (A and B) The A′B′/E′F′ loops in the crystal structure of the P domain alone had two different conformations: open (X-ray apo open) and closed (X-ray apo closed). Those two conformations are compared to the EM (no bile plus Fab A6.2) structure. As shown here, the EM structure of the P domain/Fab A6.2 presented here is very similar to that of the X-ray open conformation, and the CDR3 heavy-chain loop clearly cannot bind to the closed conformation in the X-ray structure of the apo P domain. In all three structures, without bile bound, the C′D′ loop is pointed down. (C) When bile binds, several changes are observed. The A′B′/E′F′ loops adopt the closed conformation, and the C′D′ loop moves up to interact with the E′F′ loop. The receptor, CD300lf, binds between the A′B′ and D′E′ loops, and these two loops separate slightly when bile binds, opening space for the receptor to bind.
The structures of these complexes strongly suggested that bile binding and antibody binding are mutually exclusive. To investigate this, neutralization studies were performed with three different monoclonal antibodies in the presence of increasing concentrations of the bile acids GCDCA and TCA (Fig. 5). Neutralization by all three antibodies is essentially abrogated by 128 μM GCDCA and >512 μM TCA. This finding agrees with previous binding studies showing that TCA binds poorly compared to GCDCA (21) but that at high enough concentrations, TCA binds to the intact capsid (24). TCA likely binds poorly to the P domain compared to GCDCA because the additional sulfate group crowds the deepest recesses of the bile salt binding pocket (21). It could be argued that these results are due to effects of bile salts on cell membranes. Therefore, direct interactions between the P domains and Fab A6.2 were measured using plasmon resonance in the presence of bile salts (Table 4). Both GCDCA and TCA decrease the affinity of Fab A6.2 for the P domain, but TCA is far less efficacious than GCDCA. Therefore, bile binding and antibody binding are mutually exclusive, and the abrogation of antibody binding is due to stabilization of the closed conformation by bile.
FIG 5.
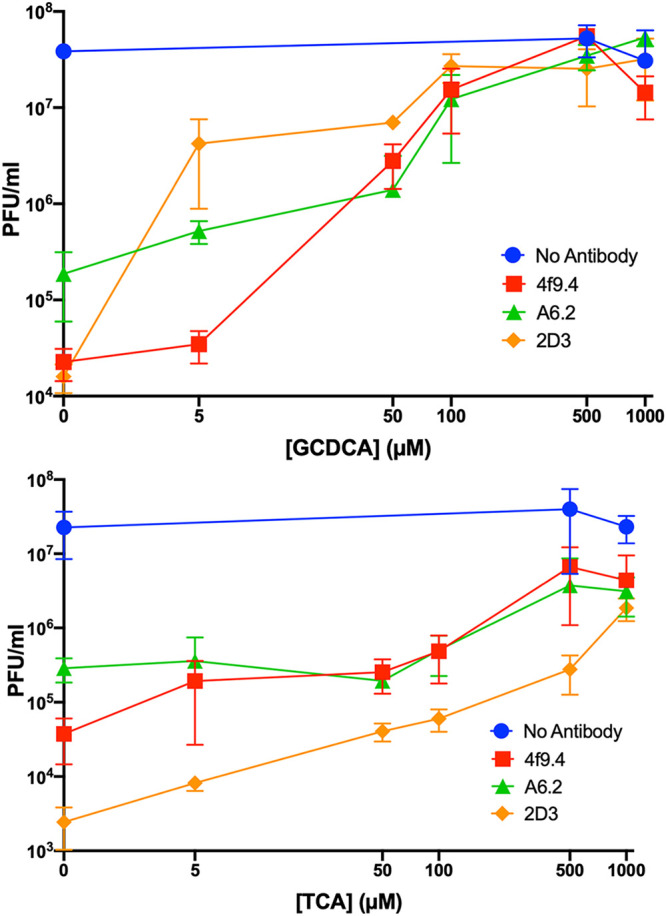
Bile blocks neutralization by three different monoclonal antibodies. In these studies, the virus was incubated with increasing concentrations of GCDCA or TCA in the presence or absence of three different monoclonal antibodies, and the concentration of virus (expressed as PFU per milliliter) in each mixture was determined. Both bile salts abrogated antibody neutralization, but TCA was less efficacious than GCCDA.
TABLE 4.
Effects of bile on the affinity of Fab A6.2 for MNV P domains
| [Bile salts] (μM) | Affinity of Fab A6.2 binding to: |
|
|---|---|---|
| GCDCA | TCA | |
| 0 | 46 nM | 46 nM |
| 5 | 400 nM | 203 nM |
| 10 | 1.2 μM | 337 nM |
| 25 | 35 μM | 770 nM |
Interestingly, one of the escape mutations to antibodies 2D3 and 4F9, V339I (18), lies immediately adjacent to the bile acid binding site. This is an “allosteric” escape in that it is distal to all antibody contact residues. From dynamic simulation calculations, it was apparent that the V339I mutation had long-range effects on the A′B′/E′F′ loops as well the P domain dimer interactions (24). It will be interesting to see whether this mutation shifts the P domain to the closed conformation and/or has any effect on capsid contraction. It may be that this allosteric escape mutation mimics bile acid-induced conformational changes to some degree, thereby decreasing antibody affinity.
Bile rotates the A/B P domain dimers, causing the P domain contraction.
The other major question left unanswered from the intact MNV/bile cryo-EM structure was how bile causes the contraction of the P domain onto the surface of the shell (24). Since bile was not observed to bind at the P domain/shell interface, we suggested that contraction might be due to changes in the character of the solvent (24). Since we now have the P domain structure with or without bile in physiological PBS buffer, we can directly compare the cryo-EM structures so as to better understand how bile binding can cause contraction of the P domain onto the shell (Fig. 6). Since the core of the P1 domain itself (residues 500 to 530) is unaffected by the conformational changes occurring in the loops in the P2 domain, structural alignments were performed using only this region for comparing the structures in the presence and absence of bile. If the relationship between the A and B subunits were unaffected by bile binding, then the P1 domains of the B subunits would be expected to align as well as the fitted P1 domains of the A subunits. However, what is evident from this alignment is that bile causes the A/B dimers to rotate with respect to each other (Fig. 6). Some detailed examples of this rotation are highlighted by green arrows in Fig. 6A and B. Most importantly, when the aligned P domain structure without bile was placed onto the shell using this P1 domain alignment, extensive clashes with the shell were apparent (Fig. 6, orange arrows). Indeed, without bile, there are 5 times more contacts between the B subunit and the shell that are <3.5 Å, and 21 contacts are <2 Å. As shown by the arrows in Fig. 6, bile causes the subunits to rotate and to create an optimal surface for contact with the shell domain. This motion is summarized in the cartoon in Fig. 6C.
FIG 6.

Bile causes a large conformational change in the P domain dimer. (A) Comparison between the EM structures of the P domain with or without GCDCA, where the core (residues 500 to 530) of the P1 domain of subunit A is used for alignment. The apo structures are shown in light hues, and the GCDCA/P domain complex is shown in darker hues. Mauve spheres represent the GCDCA in the bile complex. For clarity, the back portion of the model has been clipped, and therefore, the second molecule of GCDCA is not visible in this view. What is readily apparent in this alignment of the apo structure to the bile-bound structure is that there would be extensive clashes (orange arrows) between the apo B subunit P1 domain and the shell. As indicated by the light blue arrows, when bile is added, the B subunit rotates in a clockwise direction and alleviates the extensive clashes. Green arrows indicate some examples of this movement as bile binds. (B) Closeup of the B subunit/shell interactions in boxed areas of panel A. These extensive clashes between the P1 domain of the apo B subunit and the shell explain why the P domain “floats” above the shell in the absence of bile; the bile-induced rotation and changes in the P1 domain surface are necessary for contraction. (C) Cartoon schematic showing the bile-induced rotation of the B subunit. The color scheme is the same as that in the other panels; the apo and bile-bound B subunits are shown in pink and red, respectively. If the P1 domain of the A subunit of the apo structure is fitted to the bile-bound P domain in the contracted form, the P1 domain of the B subunit has extensive clashes with the shell (orange arrow). When bile binds, the B subunit rotates, alleviates the clashes, and forms a surface complementary to the shell. Only then can the P domain bind to the shell, resulting in the contracted form of the virion.
Another way to demonstrate this rotation is shown in Fig. 7. For these calculations, only the P1 domains were used for structural alignment as described above. To be sure that we were comparing structures determined by using the same conditions and methods, the EM structures and X-ray structures (with or without bile) were compared separately. The backbone atoms of residues 500 to 530 in the A subunits, representing the core of the P1 domain, were aligned in COOT (27), and the resulting distances between the apo and bile-bound structures are shown by red and orange lines for the EM and X-ray structures, respectively (Fig. 7). By selecting the core of the P1 domain, the alignment was not affected by the significant changes in the loops at the top of the P2 domain. If all the P domain distances are plotted here, large deviations in the P2 domain alignments are observed due to C′D′/A′B′/E′F′ loop movement that is unrelated to A/B rotation. As expected, there were no significant differences in the P1 domain itself upon bile binding. Using these structures of the P domains aligned according to the P1 domains of the A subunits, the P1 domains of the B subunits were then compared. If bile did not cause significant changes in the relative orientations of the A/B subunits, then these curves (green and blue) should be comparable to the aligned A subunit P1 domains (red and orange) (Fig. 7). However, this was clearly not the case; some portions of the P1 domains of the B subunits shifted >4 Å upon bile binding. This graphically demonstrates what was observed in Fig. 6. It should be noted that the rotation of the subunits is more than likely governed by the movement in the C′D′ loop, since in this apo crystal structure, the A and B subunits are in open and closed conformations, respectively, but the C′D′ loop is in the down position in both (10). The importance of the C′D′ loop in the A/B rotation is reasonable, since the C′D′ loop comprises ∼400 Å2 of the total ∼1,800-Å2 dimer interface surface area. Future molecular dynamic simulations will focus on the interplay between the C′D′ loop motion, the A/B rotation, and P domain contraction onto the shell surface.
FIG 7.

Graphical representation of the changes in A/B orientation upon bile binding. For these measurements, the apo (EM structure presented here; X-ray structure presented in reference 10) and bile-bound (EM structure presented in reference 24; X-ray structure presented in reference 21) P domain structures were aligned using residues 500 to 530 of the P1 domain core of the A subunit (comparing EM and X-ray structures separately). The EM A and X-ray A lines show the CA distances of the aligned A subunit P1 domains and demonstrate that bile binding does not change the structure of the P1 domain itself. The EM B and X-ray B lines show that if the A subunit P1 domains are aligned, the relative position of the B subunit P1 domain moves by >4 Å in places upon the binding of bile. Therefore, this rotation is observed in both the EM and X-ray structures.
It is noteworthy that these structures are remarkably consistent no matter the source or structural method employed. The A/B rotation upon bile binding is observed in the crystal structures of bacterially expressed P domains (PDB codes 3LQE, 3LQ6, 6H6L, and 6E47). The same rotation is observed when the cryo-EM structure of the bile-bound whole virion (PDB code 6P4J) is compared to the apo (no bile) bacterially expressed P domain complexed with Fab A6.2. The bacterially expressed crystal structure of the bile-bound P domain (6H6L and 6E47) is essentially identical to the whole-virion P domain structure with bile bound (6P4J) and the crystal structure of the apo closed conformation (3LQE) except for the C′D′ loop conformation. This is rather remarkable, since one might expect that crystal packing and bacterial expression would lead to significant differences in P domain conformations. These consistencies lend confidence to the hypothesis that the A/B dimer interface is plastic and is affected by compounds such as bile.
Finally, it is clear that bile causes a complete rearrangement of all currently known epitopes of the P domain; the epitopes at the top of the P domain are no longer recognized by antibodies due to movement of the A′B′, C′D′, and E′F′ loops, and the contraction of the P domain onto the shell buries the P1 and shell epitopes.
Structural model for bile-induced escape from antibody surveillance.
These results, combined with structural studies on MNV over the past 2 decades, point to an unsuspected structural equilibrium that is manipulated by the virus to enhance cell attachment while escaping antibody neutralization. Figure 8 shows an overview of the structural transitions in MNV as the virus switches between receptor- and antibody-binding modes. Structures 1 and 2 represent open and closed conformations, where the A′B′ and E′F′ loops are either splayed apart or tightly associated, respectively. In the original crystal structure of the apo P domain (10), both open and closed conformations were observed, while the open conformation appeared to be favored in the apo MNV EM capsid structure determined in PBS buffer (24). Without bile, the C′D′ loop points down away from the A′B′/E′F′ loops in the apo X-ray (10) and cryo-EM (Fab-bound) structures (Fig. 4) and the intact virion (24). This loop is likely flexible as well, and in the down position, it would not stabilize the closed conformation of the A′B′/E′F′ loops and would allow the open/close transition to occur more freely in solution.
FIG 8.

Overview of the bile-induced switch in the MNV capsid between antibody and receptor recognition based on our current repertoire of structures. In the absence of bile, the P domain floats above the shell, and the A′B′/E′F′ loops are in equilibrium between the open and closed conformations. In both cases, the C′D′ loop points down, away from the A′B′/E′F′ loops. It is more than likely that the C′D′ loop is highly mobile and exists in multiple conformations, including both the up and down positions. However, from the crystal structures of the apo P domain (10), the Fab A6.2/P domain complex (Fig. 4), and the low-resolution structure of the apo MNV virion (24), the C′D′ loop may favor the down conformation in the absence of bile. This structural equilibrium of the C′D′ loop would allow for bile binding as shown in structure 3. Upon binding, bile would shift the structural equilibrium toward the upward conformation of the C′D′ loop, which, in turn, stabilizes the closed conformation of the A′B′/E′F′ loops. Bile binding also causes a rotation of the two P domain subunits that creates a favorable surface for P domain/shell interactions. This results in contraction of the P domain onto the shell, and together, these movements allow the receptor to bind. Alternatively, in the absence of bile, antibodies can bind to the open conformation of the A′B′/E′F′ loops in the floating P domain or to the P1 or shell domains. Importantly, the structures and binding studies presented here suggest that bile and antibodies bind to distinctly different P domain conformations and therefore cannot bind to the P domain at the same time.
MNV conformational changes can proceed in two opposite directions depending on the presence of bile. When the C′D′ loop is in the “up” conformation, there is room for bile to access its binding pocket (Fig. 8, structure 3, arrow 2). This movement of the C′D′ loop upward would lock the A′B′/E′F′ loops into the closed conformation. When bile binds, the A and B subunits in the dimer rotate with respect to each other (arrow 3), changing the surface at the base of the P domain dimer, making it complementary to the surface of the shell. Through the extremely flexible nature of the P/S connecting loop, the entire P domain rotates 90° (arrow 4) and collapses onto the shell surface (arrow 5) (24). Finally, the bile-bound structure slightly opens a crevasse between the A′B′ and D′E′ loops and facilitates the binding of the receptor, CD300lf (Fig. 8, structure 4) (21). In the opposite conformational equilibrium pathway, antibodies recognize the open conformation and bind to the expanded MNV capsid (Fig. 8, structure 5) (5, 10, 18). In addition, the expanded capsid in the absence of bile has greater accessibility for possible antibody binding to the P1 and shell domains. In vivo, which pathway is taken will depend on the relative concentrations and binding affinities of the various bile salt species and antibodies. The extremely high (millimolar) concentrations of bile salts found in the gut likely favor the receptor-binding, antibody escape route, while the low concentrations of bile in serum would favor antibody binding.
Therefore, bile pulls the structural equilibrium toward the receptor-binding conformation and blocks antibodies from binding to the entire P domain. At the tip of the P domain, the A′B′/E′F′ loops close, and none of the neutralizing antibodies tested can bind. Bile also causes contraction of the P domain onto the shell surface and buries the epitopes on the P1 domain and shell (16, 28). As we suggested previously, this contraction may also increase the number of receptor-binding sites in the intact virion (24). Essentially, the virus is using the animal’s own metabolites to thwart antibody recognition while enhancing cell binding. It remains to be seen what parts of this process are shared among noroviruses. However, our data suggest that conditions in the gut (e.g., high bile concentrations) might interfere with the efficacy of the antibody response and thus diminish vaccine efficacy. Further studies are needed to ascertain whether a mutant MNV P domain that mimics the bile-bound, closed conformation could make for a more efficacious vaccine. In the case of human norovirus, bile salts have been observed to stabilize the structures of some of the loops around the histoblood group antigen (HBGA) binding site and to enhance HBGA binding for certain norovirus genotypes (29). However, while bile was found to compensate for HBGA binding differences due to capsid microvariations in GII.2 strains, it did not alter capsid antigenicity (30). Therefore, the data thus far show the importance of bile and P domain plasticity in the noroviruses, but the effects of bile on antigenicity appear to be unique to MNV. More broadly, it is interesting to speculate that other viruses might hijack host metabolites and interfere with vaccine efficacy and adaptive immune memory. If so, such structural information would be critical for improving the immune protection afforded by subunit vaccines.
MATERIALS AND METHODS
Production of antibodies.
Hybridoma cells for antibodies A6.2, 2D3, and 4F9 were grown in Bioreactor 1000 flasks by following previously published protocols (5, 16, 17). The cells were removed from the harvested suspension by centrifugation, and the antibody was precipitated with a 50% saturated solution of ammonium sulfate (final concentration). Since A6.2 is an IgG antibody, it was purified with a protein G column according to the manufacturer’s recommendation. 2D3 and 4F9 are IgA antibodies that do not bind to protein G and were purified by size exclusion chromatography using a Superdex 200 column. All antibodies were stored as ammonium sulfate precipitates (50% saturation).
P domain purification.
For these studies, the entire P domain (residues 225 to 541) from MNV1.CW1 was used. The MNV P domain-containing plasmid (CW1/pmcsg7) (10) was transformed into Rosetta (Novagen) competent cells and plated onto LB plates containing ampicillin and chloramphenicol for selection. One to 2 liters of cells were grown at 37°C to an optical density at 600 nm (OD600) of 0.5 to 0.6 and were chilled to room temperature, and expression was induced with 1 mM (final concentration) isopropyl-β-d-thiogalactopyranoside (IPTG). Protein was expressed for 18 h at room temperature, after which cells were harvested via centrifugation, resuspended in lysis buffer (PBS, 1 mg/ml lysozyme, 1 mM [final concentration] phenylmethylsulfonyl fluoride [PMSF], and 1 tablet of Thermo Scientific Pierce protease inhibitor, EDTA free), and frozen. The thawed mixture was sonicated extensively on ice (temperature held at <12°C) and centrifuged to remove debris. The supernatant was filtered, loaded onto a nickel column, and washed with loading buffer (25 mM K2PO4, 500 mM NaCl, and 25 mM imidazole). Bound protein was eluted using loading buffer with the addition of 300 mM imidazole. The eluted sample was dialyzed extensively against 50 mM Tris-HCl (pH 7.6) prior to further purification using a Mono Q 5/50 GL column (GE Healthcare) preequilibrated with 50 mM Tris-HCl (pH 7.6). The protein was eluted with a NaCl gradient with a buffer containing 50 mM Tris-HCl (pH 7.6) and 1 M NaCl. The P domains eluted at 680 to 700 mM NaCl, and fractions were pooled, examined by SDS-PAGE, and stored at 4°C in 60% saturated ammonium sulfate until further use.
Production and purification of F(ab) and F(ab′).
MAb A6.2 was dialyzed against 100 mM sodium phosphate (pH 7.4) and digested with papain (1:100, wt/wt) in the presence of 25 mM β-mercaptoethanol (β-ME) at 37°C for 17 h. The digestion was quenched by the addition of 50 mM iodoacetamide. The solution was dialyzed against 50 mM Tris (pH 7.6) and loaded onto a Mono Q column preequilibrated with 50 mM Tris (pH 7.6). Fab-containing fragments were collected from the flowthrough, and the Fc and intact antibody were retained under these conditions. A6.2 Fab was stored in 50% ammonium sulfate.
SPR.
The binding of A6.2 Fab to the P domain was measured by surface plasmon resonance (SPR) using a BIAcore 1000 instrument equipped with a nitrilotriacetic acid (NTA) sensor chip essentially as described previously (17). All samples were dialyzed and analyzed using 100 mM sodium phosphate (pH 7.4), 400 mM NaCl, 0.1% bovine serum albumin (BSA), 0.002% (vol/vol) Tween 20 surfactant, and 0.04 mM EDTA, with a corresponding concentration of bile for that experiment. At the start of each run, the nickel was stripped from the NTA chip with 40 μl of 350 mM EDTA, and the strip was recharged with 40 μl of 0.5 mM NiCl2. A 400 nM concentration of the purified His-tagged P domain was immobilized onto the chip, and various concentrations of A6.2 Fab (25 nM to 3.2 μM) were passed over the chip. All experiments were conducted at 25°C with a 40-μl/min flow rate. The analyte (A6.2 Fab) was injected using a 360-s association time at a 20-μl/min flow rate and a 400-s dissociation time. The BIAevaluation package was used for data, and the 1:1 Langmuir model was used to determine dissociation constants.
Neutralization assay.
Neutralization assays were performed using previously published protocols with slight modifications (16). Briefly, 1 × 106 BV-2 cells from spinner cultures were added to each well (10 cm2) of 6-well plates and were allowed to attach for several hours. MNV1 at 1 × 108 PFU/ml was serially diluted with Dulbecco’s modified Eagle’s medium (DMEM), and purified antibodies were added to the various dilutions (final concentrations, 0.8 mg/ml for 4F9, 0.4 mg/ml for 2D3, and 0.1 mg/ml for A6.2). To these solutions, various concentrations of GCDCA or TCA (0 to 1 mM) were added, and the mixtures were incubated for 30 min. A 500-μl volume of each solution was applied to monolayers and allowed to attach for 1 h at room temperature with gentle rocking. After attachment, the solution was removed from wells, and 2 ml of 50% (vol/vol) low-melting-temperature agar/P6 medium (31 mM BSA, 79 mM magnesium chloride hexahydrate, Gibco MEM, 2% nonessential amino acids [NEAA], 0.124 g/liter penicillin, 0.8 g/liter streptomycin sulfate, and 52 mM sodium bicarbonate) was overlaid. Once the agar overlay solidified, 5 ml of medium B (Gibco MEM, 0.1% Pluronic F-68, 1% NEAA, 0.062 g/liter penicillin, 0.4 g/liter streptomycin sulfate, and 26 mM sodium bicarbonate) was added to each well, and the plates were placed in a 37°C incubator under 5% CO2 for 37 h. After this incubation, the medium and agar were removed, and a 0.1% crystal violet–20% ethanol mixture was added in order to visualize plaques. All experiments were performed in triplicate.
Cryo-EM.
The purified MNV P domain was added to purified A6.2 Fab at a 1:2 molar ratio of P domain to antibody. The concentration of the complex for structural work was >1 mg/ml. The complex was separated from unbound species using a Superose 6 size exclusion column. Purified MNV P domain/Fab A6.2 was vitrified as described previously (31) on carbon holey film (R2x1 Quantifoil; Micro Tools GmbH, Jena, Germany) grids. Briefly, grids were cleaned in Gatan Solarus plasma cleaner 950 for 40 s in a hydrogen-oxygen gas mixture. Portions (4 μl) of purified concentrated (∼1-mg/ml) suspensions of the complex were applied to the holey films, blotted with filter paper, and plunged into liquid ethane. An EM-GP2 (Leica) automated plunger was used for vitrification.
The grids were screened for ice and sample quality and were imaged in a Titan Krios G3i microscope (Thermo Fisher). The microscope was equipped with a Gatan BioQuantum K3 imaging filter (Ametek, Inc.) and was operated at 300 keV. A slit width of 20 eV was used for data collection. Images were acquired in EPU (Thermo Fisher) using fast acquisition mode, with beam-image shift used for hole centering instead of stage movement. The direct detector camera (K3; Ametek) operated in super-resolution counted mode, and images were recorded with an overall electron dose of 48 electrons/Å2; the defocus range was −1.5 to −2.5 μm.
The data collection statistics are summarized in Table 1.
Image processing.
Image processing was performed in CryoSPARC, v2.15 (32). A total of 9,681 movies were recorded and were corrected for gain, motion, and contrast transfer function (CTF). Micrographs were binned by 2 during motion correction, and the final pixel size was 0.85 Å on the specimen scale. A total of 8,220,523 particles were picked from the images by the automatic particle picker in CryoSPARC. After several rounds of 2-dimensional (2-D) classification, the best 1,825,383 particles were selected for further processing. Several cycles of uniform 3-D refinement, nonuniform 3-D refinement, and CTF refinement were performed in CryoSPARC, assuming C1 symmetry, and yielded an effective resolution of the P domain/Fab A6.2 complex of 3.2 Å according to a 0.143 FSC gold-standard criterion (33).
Structure refinement.
All structure refinement was performed using PHENIX (34). The initial model used was the pseudoatomic structure of the MNV virion/Fab A6.2 complex that was built using the X-ray structures of the MNV P domain (10) and Fab A6.2 (17). The model was manually fitted into the EM envelope, and several cycles of real-space refinement (rigid body, global minimization, and simulated annealing) in PHENIX and rebuilding in COOT were performed. The final refinement statistics are shown in Table 1.
Data availability.
The coordinates are available in the Protein Data Bank under accession code 7L5J, and the cryo-EM map is available in EMDB (https://www.ebi.ac.uk/pdbe/emdb/index.html/) under accession code EMD-23187.
ACKNOWLEDGMENTS
This work was supported by an NIH grant to T.J.S. and B.M.P. (1R01-AI141465), and A.N.W. was supported by the James W. McLaughlin Fellowship Fund. We acknowledge the support of the Sealy Center for Structural Biology at UTMB.
Contributor Information
Thomas J. Smith, Email: thosmith@utmb.edu.
Colin R. Parrish, Cornell University
REFERENCES
- 1.Hutson AM, Atmar RL, Estes MK. 2004. Norovirus disease: changing epidemiology and host susceptibility factors. Trends Microbiol 12:279–287. 10.1016/j.tim.2004.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartsch SM, O’Shea KJ, Lee BY. 2020. The clinical and economic burden of norovirus gastroenteritis in the United States. J Infect Dis 222:1910–1919. 10.1093/infdis/jiaa292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moe CL, Sobsey MD, Stewart PW, Crawford-Brown D. 1999. Estimating the risk of human calicivirus infection from drinking water. Abstr Int Workshop Human Caliciviruses, Atlanta, GA.
- 4.Prasad BVV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290. 10.1126/science.286.5438.287. [DOI] [PubMed] [Google Scholar]
- 5.Katpally U, Wobus CE, Dryden K, Virgin HW, IV, Smith TJ. 2008. Structure of antibody-neutralized murine norovirus and unexpected differences from viruslike particles. J Virol 82:2079–2088. 10.1128/JVI.02200-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi J-M, Hutson AM, Estes MK, Prasad BVV. 2008. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc Natl Acad Sci U S A 105:9175–9180. 10.1073/pnas.0803275105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan M, Hegde RS, Jiang X. 2004. The P domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J Virol 78:6233–6242. 10.1128/JVI.78.12.6233-6242.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donaldson EF, Lindesmith LC, Lobue AD, Baric RS. 2010. Viral shape-shifting: norovirus evasion of the human immune system. Nat Rev Microbiol 8:231–241. 10.1038/nrmicro2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nilsson M, Hedlund KO, Thorhagen M, Larson G, Johansen K, Ekspong A, Svensson L. 2003. Evolution of human calicivirus RNA in vivo: accumulation of mutations in the protruding P2 domain of the capsid leads to structural changes and possibly a new phenotype. J Virol 77:13117–13124. 10.1128/jvi.77.24.13117-13124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taube S, Rubin JR, Katpally U, Smith TJ, Kendall A, Stuckey JA, Wobus CE. 2010. High-resolution X-ray structure and functional analysis of the murine norovirus 1 capsid protein protruding domain. J Virol 84:5695–5705. 10.1128/JVI.00316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katpally U, Voss NR, Cavazza T, Taube S, Rubin JR, Young VL, Stuckey J, Ward VK, Virgin HW, Wobus CE, Smith TJ. 2010. High-resolution cryo-electron microscopy structures of murine norovirus 1 and rabbit hemorrhagic disease virus reveal marked flexibility in the receptor binding domains. J Virol 84:5836–5841. 10.1128/JVI.00314-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansman GS, Taylor DW, McLellan JS, Smith TJ, Georgiev I, Tame JRH, Park S-Y, Yamazaki M, Gondaira F, Miki M, Katayama K, Murata K, Kwong PD. 2012. Structural basis for broad detection of genogroup II noroviruses by a monoclonal antibody that binds to a site occluded in the viral particle. J Virol 86:3635–3646. 10.1128/JVI.06868-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruoff K, Kilic T, Devant J, Koromyslova A, Ringel A, Hempelmann A, Geiss C, Graf J, Haas M, Roggenbach I, Hansman G. 2019. Structural basis of nanobodies targeting the prototype norovirus. J Virol 93:e02005-18. 10.1128/JVI.02005-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindesmith LC, Mallory ML, Debbink K, Donaldson EF, Brewer-Jensen PD, Swann EW, Sheahan TP, Graham RL, Beltramello M, Corti D, Lanzavecchia A, Baric RS. 2018. Conformational occlusion of blockade antibody epitopes, a novel mechanism of GII.4 human norovirus immune evasion. mSphere 3:e00518-17. 10.1128/mSphere.00518-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith HQ, Smith TJ. 2019. The dynamic capsid structures of the noroviruses. Viruses 11:235. 10.3390/v11030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolawole AO, Xia C, Li M, Gamez M, Yu C, Rippinger CM, Yucha RE, Smith TJ, Wobus CE. 2014. Newly isolated mAbs broaden the neutralizing epitope in murine norovirus. J Gen Virol 95:1958–1968. 10.1099/vir.0.066753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolawole AO, Li M, Xia C, Fischer AE, Giacobbi NS, Rippinger CM, Proescher JB, Wu SK, Bessling SL, Gamez M, Yu C, Zhang R, Mehoke TS, Pipas JM, Wolfe JT, Lin JS, Feldman AB, Smith TJ, Wobus CE. 2014. Flexibility in surface-exposed loops in a virus capsid mediates escape from antibody neutralization. J Virol 88:4543–4557. 10.1128/JVI.03685-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolawole AO, Smith HQ, Svoboda SA, Lewis MS, Sherman MB, Lynch GC, Pettitt BM, Smith TJ, Wobus CE. 2017. Norovirus escape from broadly neutralizing antibodies is limited to allosteric-like mechanisms. mSphere 2:e00334-17. 10.1128/mSphere.00334-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haga K, Fujimoto A, Takai-Todaka R, Miki M, Doan YH, Murakami K, Yokoyama M, Murata K, Nakanishi A, Katayama K. 2016. Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proc Natl Acad Sci U S A 113:E6248–E6255. 10.1073/pnas.1605575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee YC, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, Virgin HW. 2016. Discovery of a proteinaceous cellular receptor for a norovirus. Science 353:933–936. 10.1126/science.aaf1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson CA, Wilen CB, Dai Y-N, Orchard RC, Kim AS, Stegeman RA, Hsieh LL, Smith TJ, Virgin HW, Fremont DH. 2018. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc Natl Acad Sci U S A 115:E9201–E9210. 10.1073/pnas.1805797115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sjövall J. 1959. On the concentration of bile acids in the human intestine during absorption. Bile acids and steroids 74. Acta Physiol Scand 46:339–345. 10.1111/j.1748-1716.1959.tb01763.x. [DOI] [PubMed] [Google Scholar]
- 23.Kilic T, Koromyslova A, Malak V, Hansman GS. 2018. Atomic structure of the murine norovirus protruding domain and soluble CD300lf receptor complex. J Virol 92:e00413-18. 10.1128/JVI.00413-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherman MB, Williams AN, Smith HQ, Nelson C, Wilen CB, Fremont DH, Virgin HW, Smith TJ. 2019. Bile salts alter the mouse norovirus capsid conformation: possible implications for cell attachment and immune evasion. J Virol 93:e00970-19. 10.1128/JVI.00970-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith TJ, Chase ES, Schmidt TJ, Olson NH, Baker TS. 1996. Neutralizing antibody to human rhinovirus 14 penetrates the receptor-binding canyon. Nature 383:350–354. 10.1038/383350a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Koromyslova AD, Devant JM, Kilic T, Sabin CD, Malak V, Hansman GS. 2020. Nanobody-mediated neutralization reveals an Achilles heel for norovirus. J Virol 94:e00660-20. 10.1128/JVI.00660-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kilic T, Koromyslova A, Hansman GS. 2018. Structural basis for human norovirus capsid binding to bile acids. J Virol 93:e01581-18. 10.1128/JVI.01581-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mallory ML, Lindsmith LC, Brewer-Jensen PD, Graham RL, Baric RS. 2020. Bile facilitates human norovirus interactions with diverse histoblood group antigens, compensating for capsid microvariation observed in 2016–2017 GII.2 strains. Viruses 12:989. 10.3390/v12090989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherman MB, Guenther RH, Tama F, Sit TL, Brooks CL, Mikhailov AM, Orlova EV, Baker TS, Lommel SA. 2006. Removal of divalent cations induces structural transitions in red clover necrotic mosaic virus, revealing a potential mechanism for RNA release. J Virol 80:10395–10406. 10.1128/JVI.01137-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. 2017. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14:290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 33.Scheres SH, Chen S. 2012. Prevention of overfitting in cryo-EM structure determination. Nat Methods 9:853–854. 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Afonine PV, Grosse-Kunstleve RW, Adams PD. 2005. The Phenix refinement framework. CCP4 Newsl 42:Contribution 8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The coordinates are available in the Protein Data Bank under accession code 7L5J, and the cryo-EM map is available in EMDB (https://www.ebi.ac.uk/pdbe/emdb/index.html/) under accession code EMD-23187.