

ABSTRACT
Kaposi’s sarcoma-associated herpesvirus (KSHV) is an important oncogenic virus previously shown to be neurotropic, but studies on neuronal cell infection and pathogenesis are still very limited. Here, we characterized the effects of KSHV infection on neuronal SH-SY5Y cells by the recombinant virus rKSHV.219, which expresses both green fluorescent protein (GFP) and red fluorescent protein (RFP) to reflect the latent and lytic phases of infection. We demonstrated that infected cells have a higher growth rate and that KSHV infection can be sustained. Interestingly, the infected cells can transition spontaneously back and forth between lytic and latent phases of infection, producing progeny viruses but without any adverse effects on cell growth. In addition, transcriptome analysis of viral and cellular genes in latent and lytic cells showed that unlike other infected cell lines, the latently infected cells expressed both latent and most, but not all, of the lytic genes required for infectious virion production. The viral genes uniquely expressed by the lytic cells were mainly involved in the early steps of virus binding. Some of the cellular genes that were deregulated in both latently and lytically infected cells are involved in cell adhesion, cell signal pathways, and tumorigenesis. The downregulated cellular CCDN1, PAX5, and NFASC and upregulated CTGF, BMP4, YAP1, LEF1, and HLA-DRB1 genes were found to be associated with cell adhesion molecules (CAMs), hippo signaling, and cancer. These deregulated genes may be involved in creating an environment that is unique in neuronal cells to sustain cell growth upon KSHV infection and not observed in other infected cell types.
IMPORTANCE Our study has provided evidence that neuronal SH-SY5Y cells displayed unique cellular responses upon KSHV infection. Unlike other infected cells, this neuronal cell line displayed a higher growth rate upon infection and can spontaneously transition back and forth between latent and lytic phases of infection. Unlike other latently infected cells, a number of lytic genes were also expressed in the latent phase of infection in addition to the established latent viral genes. They may play a role in deregulating a number of host genes that are involved in cell signaling and tumorigenesis in order to sustain the infection and growth advantages for the cells. Our study has provided novel insights into KSHV infection of neuronal cells and a potential new model for further studies to explore the underlying mechanism in viral and host interactions for neuronal cells and the association of KSHV with neuronal diseases.
KEYWORDS: Kaposi’s sarcoma-associated herpesvirus, cellular response, latent infection, lytic infection, neuronal cell infection
INTRODUCTION
Kaposi’s sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV8), belongs to the human gammaherpesvirus family. It is the etiological agent of several human tumors, Kaposi’s sarcoma (KS), the rare AIDS-related B cell neoplasm primary effusion lymphoma (PEL), and a subset of lymphoproliferative multicentric Castleman’s disease (MCD) (1–4). Recently, KSHV was also found to associate with an inflammatory cytokine syndrome (KSHV inflammatory cytokine syndrome [KICS]) that is characterized by elevated viral interleukin-6 (vIL-6) and KSHV loads (5).
Similar to other human herpesviruses, KSHV exhibits two distinct patterns of infection, latent and lytic, and the capacity to shift between them in response to poorly characterized external stimulation (6). Upon infection, KSHV initially establishes latency in infected cells, where a very limited number of viral genes are expressed to promote cellular survival, maintenance of latency, and immune evasion (7–9). In KSHV infection, the latent repertoire includes open reading frame 71 (ORF71) (viral FLICE-inhibitory protein [vFLIP]), ORF72 (v-cyclin), ORF73 (latency-associated nuclear antigen [LANA]), K12 (kaposin), and various viral microRNAs (miRNAs) (10–14). These latency genes are clustered in one gene locus and are transcribed as a polycistronic mRNA, while miRNAs and kaposin are transcribed as independent genes (13, 15). KSHV LANA functions to maintain the viral episomal DNA during cell division by tethering the episome to chromatin, ensuring coordinated episomal replication and partitioning into daughter cells (16). LANA also interacts with the replication transcription activator (RTA) promoter and represses RTA-mediated lytic gene activation during latency (17, 18).
During latent infection, KSHV evades immune surveillance and establishes a lifelong persistent infection (7–9). The latent phase of infection is reversible. Reactivation of latent KSHV to productive lytic replication occurs due to stimulation such as hypoxia, oxidative stress, and inflammatory cytokines or via nonspecific chemical activation. These stimuli result in the expression of RTA, which in turn drives the gene expression of the rest of the KSHV genome. RTA triggers transcriptional activation by binding directly to an RTA response element (RRE) in the promoters of viral genes or indirectly through interactions with host cellular DNA binding factors like AP-1 and Oct (19). RTA also functions as an E3 ubiquitin ligase to induce the ubiquitination and degradation of interferon (IFN) regulatory factor 7 (IRF7), a key modulator of type I IFN production (20). In addition, it was previously shown that RTA not only induces the degradation of the transcriptional repressor K-RBP (K-RTA binding protein) but also targets several other cellular transcription repressors and viral proteins for proteasomal processing (21, 22).
KSHV has a limited tissue tropism and host range in vivo (23). Humans are the only natural host of KSHV, and studies of the viral replicative cycle and pathogenesis in vivo were hampered by the lack of an animal model that faithfully recapitulates human pathogenesis. Nevertheless, KSHV can infect a variety of human and nonhuman cell types in vitro, including B cells, epithelial cells, endothelial cells, fibroblasts, and macrophages (24). While most herpesviruses exhibit some degree of neurotropism, little is known about KSHV infection of neuronal cells or potential neuropathogenesis. Recently, we detected KSHV latent infection in neurons and oligodendrocytes in parenchymal brain tissues of individuals infected by KSHV, and primary human neurons can be infected by KSHV in vitro. Our data suggest that the central nervous system (CNS) and neuronal cells are potential sites for latent KSHV infection in vivo (25). In addition, it has also been shown that KSHV can infect the neuronal cell line SH-SY5Y in vitro (26). These limited data thus support the potential neurotropism of KSHV.
In this study, we further characterize KSHV infection in bone marrow-derived neuroblastoma SH-SY5Y cells and the effects of infection. We confirmed that SH-SY5Y cells can be infected by KSHV, but in addition, we show that infected cells demonstrate accelerated growth properties and can transition between both lytic and latent phases of viral replication without cell death. Furthermore, we have used transcriptome sequencing (RNAseq) to comparatively analyze cellular and viral gene expression and define pathways that are associated with differential phenotypes resulting from viral infection. Consistent with the unique phenotypes, our study shows that gene expression modulation in SH-SY5Y cells infected with KSHV is distinct from that in endothelial cells or B cells, suggesting that SH-SY5Y cells could be an interesting model to further investigate whether this unique aspect of KSHV infection is specific to SH-SY5Y cells or may be common to other neuronal cells upon infection.
RESULTS
Bone marrow-derived neuroblastoma SH-SY5Y cells can be infected by KSHV, and the infected cells displayed spontaneous lytic viral replication and higher growth rates.
The neuroblastoma cell line SH-SY5Y, previously suggested to be susceptible to KSHV infection (25), was more extensively characterized upon infection with KSHVBC3 and rKSHV.219. At a multiplicity of infection (MOI) of 0.5, SH-SY5Y cells infected with either KSHVBC3 or rKSHV.219 showed the typical punctate nuclear immunohistochemistry (IHC) staining pattern with anti-LANA antibody (Ab). This pattern is similar to that observed in chronically infected BC3 cells (Fig. 1A). Similarly, SH-SY5Y cells infected with rKSHV.219 were monitored for up to 2 weeks by fluorescence microscopy for the expression of the virally encoded fluorescent reporter that indicates either latent (green fluorescent protein-positive [GFP+]) or lytic (GFP+/red fluorescent protein-positive [RFP+]) replication patterns. After de novo infection of SH-SY5Y cells, KSHV latency was evident by day 2 postinfection, as indicated by the expression of GFP (Fig. 1B). Interestingly, cells expressing RFP were also detected at 2 days postinoculation despite the absence of 12-O-tetradecanoylphorbol-13-acetate (TPA) or sodium butyrate (NaB) induction. This suggests that infected SH-SY5Y cells can undergo spontaneous lytic reactivation. The intensity of RFP signals was initially weak but increased at 4 days postinfection (dpi). Overall, the fluorescence intensity of both GFP and RFP increased over time (Fig. 1B), indicating a robust infection. To confirm the fluorescence evidence suggesting rKSHV.219 infection of SH-SY5Y cells, IHC staining of KSHV LANA was conducted. The infected cells displayed the characteristic punctate nuclear LANA staining (Fig. 1C), as observed in KSHVBC3 infections. rKSHV.219-infected SH-SY5Y cells (KSHV SH-SY5Y cells) were passaged in vitro for more than 20 passages and maintained both latent and lytic infections, as evidenced by reporter gene expression, without apparent cell death. This phenotypic finding is distinct from those for virtually all other KHSV infection models, which typically undergo latent infection with low-level spontaneous lytic reactivation leading to cell death upon virus production.
FIG 1.
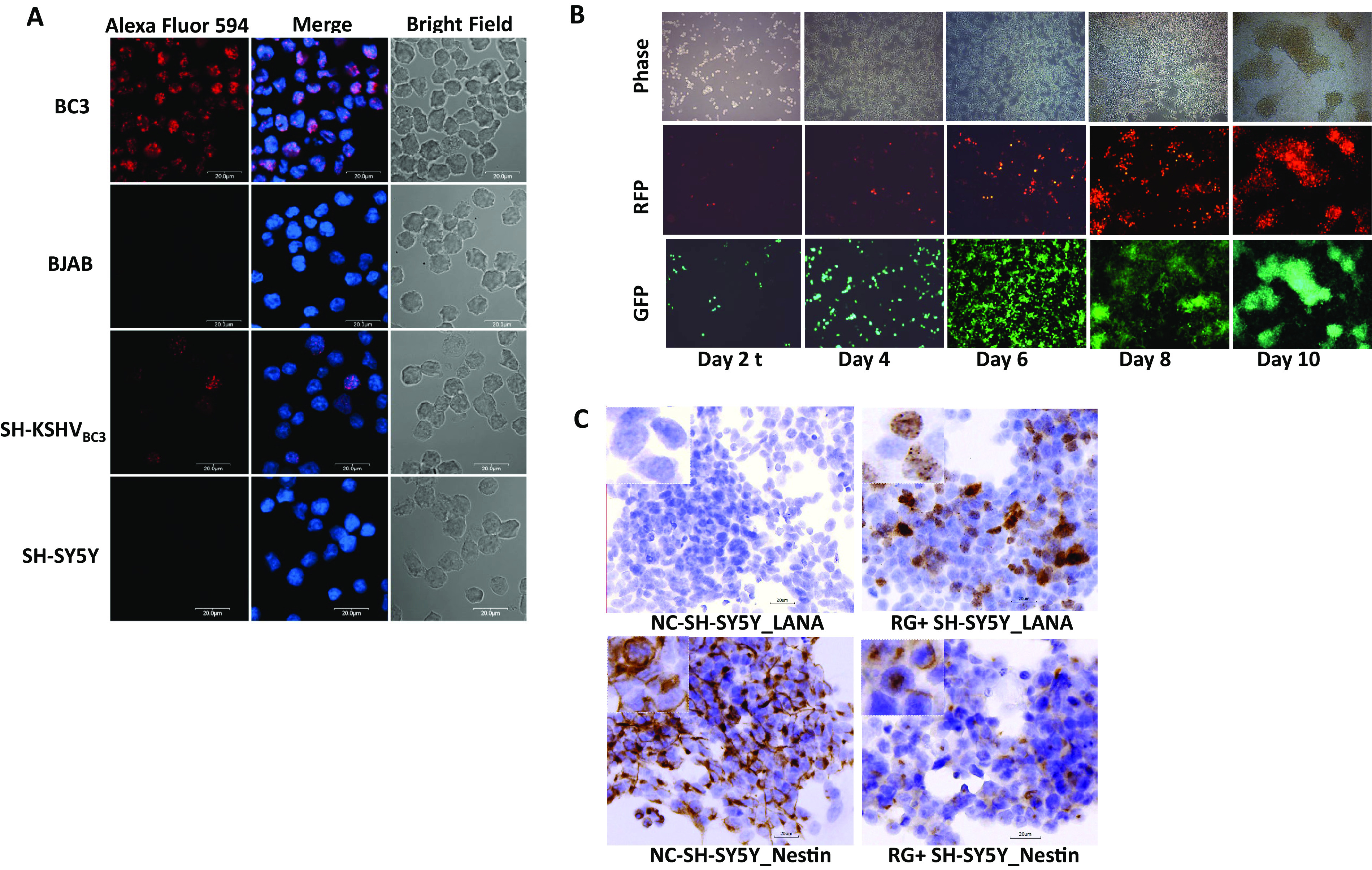
Bone marrow-derived neuroblastoma SH-SY5Y cells can be infected by KSHV derived from BC3 and Vero 219 cells. (A) SH-SY5Y cells were infected by KSHV (KSHVBC3) generated from the BC3 cell line. The BC3 cell line contained the KSHV genome and was cultured in DMEM with 10% FBS. The supernatant was collected at day 5 after passaging, and SH-SY5Y cells were infected with KSHVBC3 at an MOI of 0.5. rKSHV.219 generations (see Materials and Methods) and infection of SH-SY5Y cells at an MOI of 0.5 are shown. Images were taken by a fluorescence microscope at day 2 postinfection. (B) Fluorescence microscope images of SH-SY5Y cells infected by rKSHV.219 at days 2, 4, 6, 8, and 10 postinfection showing both latent infection with GFP and lytic infection with RFP. Images were taken from infected SH-SY5Y cells from day 2 up to day 10 postinfection, as indicated. (C) LANA and nestin protein expression in KSHV-infected RG+ SH-SY5Y cells at day 5 postinfection using IHC. Higher-magnification (×400) images are shown in the top left boxes.
Interestingly, monitoring the cell growth of KSHV-infected SH-SY5Y cells showed that the infected cultures replicated more rapidly than uninfected SH-SY5Y cells; the cell number of the KSHV SH-SY5Y cells was 11.7 times higher than the number of uninfected cells at 4 dpi, even though the same numbers of cells were seeded at day 0, and the increase was exponential over time and peaked at 10 dpi (Fig. 2A). To confirm that the increasing numbers of cells are due to the replication of the infected cells and that the infection can be sustained, SH-SY5Y cells were infected by rKSHV.219 at day 0 and labeled with eFluor 670 to monitor cell division and proliferation by flow cytometry on different days after infection. The forward and side scatter were used to evaluate cell replication using the gating strategy shown in Fig. S1 in the supplemental material. A decrease in the eFluor 670 signal with time should correlate with the increase in GFP signals due to the division of infected cells. GFP-positive SH-SY5Y cells with eFluor 670 staining were detected 24 h after infection and were maintained for up to 7 days. As expected, there was an increasing number of GFP-positive cells with time, whereas the eFluor 670 signal of the infected cells decreased proportionally to the increase in GFP+ cells, demonstrating that the infected cells were actively replicating and that the infection was sustained (Fig. 2B).
FIG 2.

Analysis of KSHV-infected SH-SY5Y cells by flow cytometry. (A) The replication of rKSHV.219-infected SH-SY5Y cells and uninfected control (NC) cells from day 0 up to day 10 after infection was monitored by flow analysis. (B) The division of rKSHV.219-infected SH-SY5Y cells was monitored from day 1 up to day 7 after infection. The fluorescence-based proliferation dye eFluor 670 was used to indicate that infected lytic SH-SY5Y cells were actively undergoing replication. Orange represents RG+ SH-SY5Y cells upon KSHV infection. Gray represents uninfected cells added as an internal control for autofluorescence. The x axis represents GFP, and the y axis represents the proliferation eFluor 670 dye. (C) The replication of sorted rKSHV.219-infected GFP+ SY-SY5Y cells and uninfected control cells was analyzed from day 0 up to day 10 after infection. (D) Replication of sorted rKSHV.219-infected RG+ SY-SY5Y cells and uninfected control cells from day 0 up to day 10 after infection.
Since the infected KSHV SH-SY5Y cell cultures appeared to contain both GFP+ (latent) and RFP+ (lytic) cells, they were further sorted to separate the GFP+ from GFP- and RFP-double-positive (RG+) cells and analyzed. Since the mixed infected culture (unsorted KSHV SH-SY5Y cells) demonstrated a higher growth rate than the uninfected culture, the growth rates of the sorted GFP+ and RG+ populations were also analyzed and compared with that of the uninfected cells. Interestingly, both infected SH-SY5Y GFP+ and RG+ cells displayed similar higher growth rates than the uninfected cells over the course of 10 days (Fig. 2C and D), indicating that KSHV latent or lytic infection conferred accelerated growth to the infected SH-SY5Y cells.
Viral DNA and gene expression levels are significantly increased in infected SH-SY5Y cells with spontaneous lytic activation of KSHV.
To determine whether KSHV virions were being produced from the RG+ SH-SY5Y cell population by the spontaneous lytic activation of KSHV, viral DNA was extracted from the culture supernatant, and cell lysates and KSHV ORF26 copy numbers were quantified by real-time PCR. The presence of infectious viruses (103 tissue culture infective doses [TCID]/ml) could also be detected in the culture supernatant by day 3 after infection. In parallel, viral DNA was also quantified for the latent GFP+ cell population. As expected, the KSHV viral DNA copy number increased with time (Fig. 3A) in the culture supernatant of the lytic RG+ cells. Even though GFP+ cells displayed a slight increase of the viral DNA copy number, it was 3-fold lower than that of lytic RG+ cells on day 7. The quantity of cell-associated KSHV DNA in cell lysates from RG+ cells (12 copies/cell) was also higher than that in GFP+ cells (4 to 5 copies/cell) throughout 7 days of culture (Fig. 3B). Unlike conventional latent infection of other cell types, infections of SH-SY5Y cells that expressed only the GFP+ marker nevertheless produced a detectable quantity of viral progeny, although the level of production was not equivalent to the levels produced continuously in RG+ cells (Fig. 3A). It is possible that there is a minor cell population that has gone through some lytic viral replication. Indeed, our following RNAseq findings confirmed that the GFP+ SH-SY5Y cells exhibited some lytic gene expression and differed from the expression patterns previously observed for latent infection of other cell lines (27). Taken together, our results showed that KSHV-infected SH-SY5Y cells displayed more rapid growth and spontaneous lytic activation with modest viral production, in contrast to other KSHV-infected cell lines, which go through apoptosis after lytic induction and the production of a burst of viral progeny (28, 29). Moreover, lytic infection of neuronal SH-SY5Y cells by KSHV prolonged survival and led to the expression of viral genes and proteins.
FIG 3.
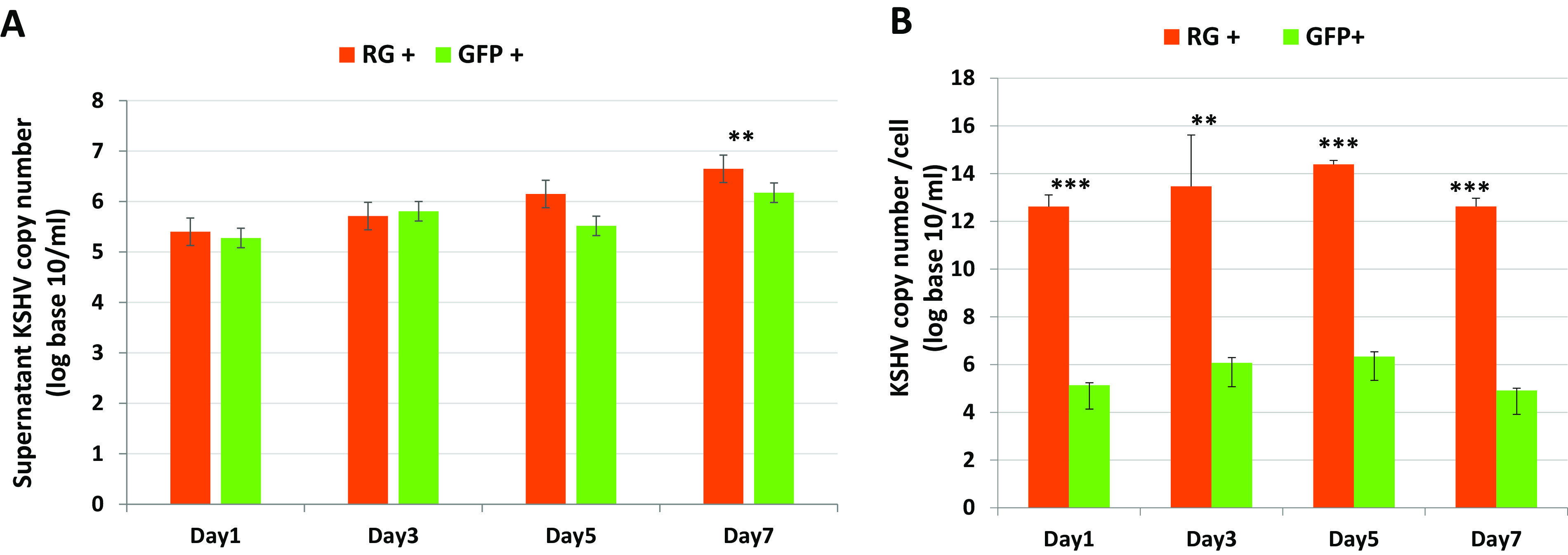
Sorted lytic and latent KSHV-infected SH-SY5Y cells produce various amounts of KSHV upon culturing. (A) Comparisons of rKSHV.219 DNA copy numbers from the supernatants of infected GFP+ SH-SY5Y and RG+ SH-SY5Y cells at the indicated time points after sorting. (B) Comparisons of rKSHV.219 DNA copy numbers from cell lysates of infected GFP+ SH-SY5Y and RG+ SH-SY5Y cells at the indicated time points after sorting. The results are presented as the means ± SD from 3 independent experiments. Student’s t test was used to test for significant differences. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
KSHV-infected SH-SY5Y cells showed a transition of phenotype between lytic (RG+) and latent (GFP+) infection.
KSHV-infected RG+ phenotype SH-SY5Y cells contain both GFP+ and RFP+ cells. To determine whether the latent or lytic phenotype of infected cells was a stable property, individual infected cells were isolated in soft agar and monitored for fluorescent marker expression over time. Infected SH-SY5Y cells could be readily cloned, formed colonies in soft agar, and supported the assessment of GFP and/or RFP expression over time in progeny cells (Fig. S2). Interestingly, fluorescent reporter expression by RG+ cells was found to change during the analyzed period. The RG+ cells in a single colony could maintain their phenotype, or they could change to GFP+ only, and subsequently become double positive again. A representative example of two independent cloning experiments is shown in Fig. 4A. These results suggest that KSHV-infected SH-SY5Y cells can cycle between lytic and latent phases of infection or that infection in this cell type produces near equilibrium between latency and lytic infection.
FIG 4.
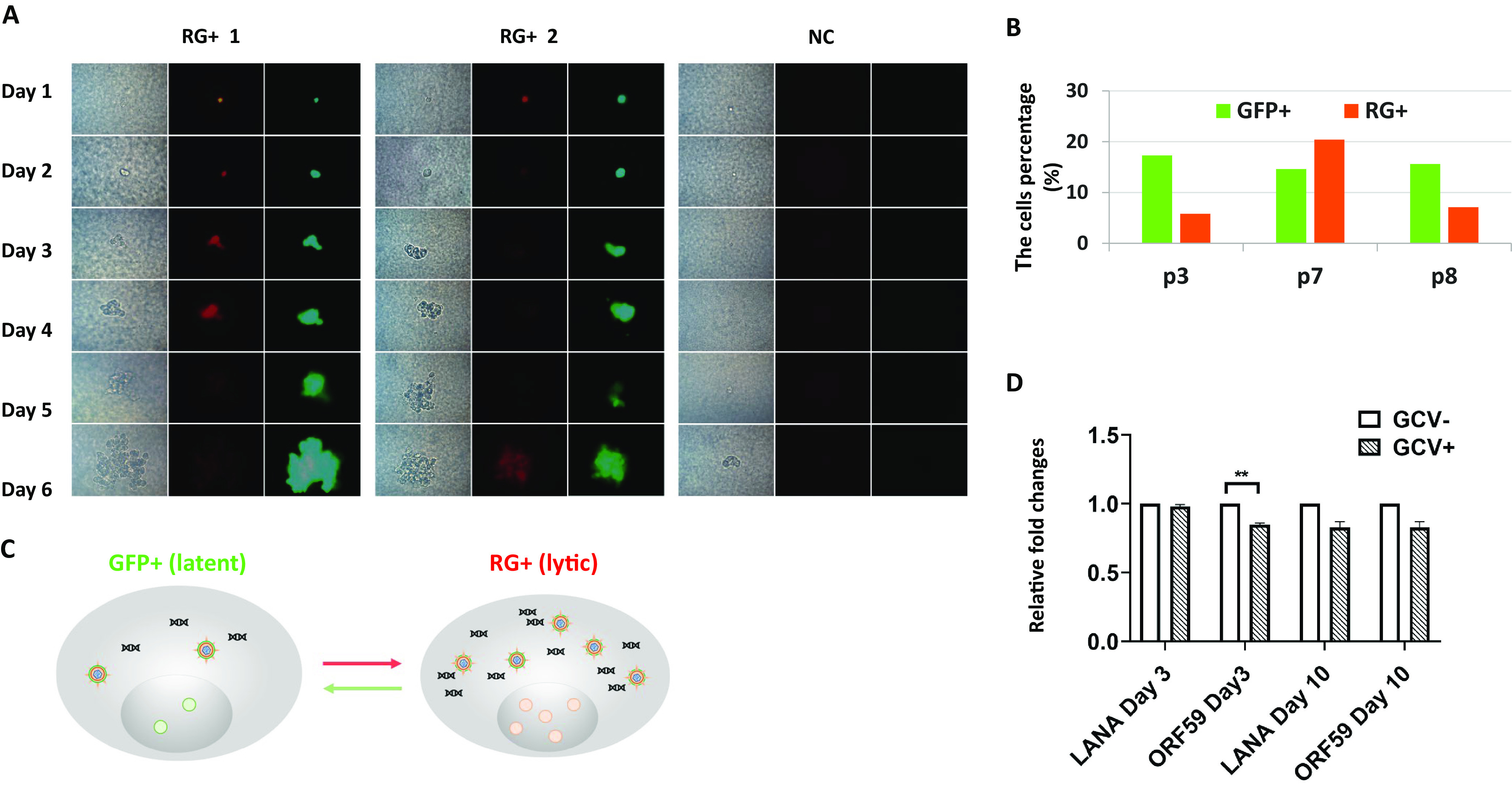
Single-cell cloning showed that KSHV-infected SH-SY5Y cells can cycle between lytic and latent phases of infection. (A) A single infected cell colony formed at day 1 up to day 6 after cloning showed cycling between latent and lytic phases of infection. RG+ 1 and RG+ 2 are images taken from two independent experiments, and NC represents uninfected control cells. (B) Percentages of GFP+ (latent marker) and RFP+ (lytic marker) cells in rKSHV.219-infected cell cultures upon passaging (passage 3 [p3], p7, and p8) as determined by flow cytometry. The established cells of GFP+ SH-SY5Y and RG+ SH-SY5Y cells were passaged every 5 days when the cells reached 90% confluence. (C) Model showing the transition of rKSHV.219-infected SH-SY5Y cells between the lytic phase as indicated by RFP and latency as indicted by GFP. (D) Sorted infected GFP+ SH-SY5Y cells were cultured in the presence or absence of GCV, and the cells were harvested at days 3 and 10. The mRNA was extracted from the harvested cells, and RT-qPCR was carried out to quantify the expression of LANA and ORF59. The experiments were repeated three times, and the results are presented as relative fold changes normalized to the gene expression levels in the absence of GCV (**, P < 0.01 by paired Student’s t test).
Furthermore, to confirm that infected SH-SY5Y cells going through latent and lytic phases of infection are both continuously present in the infected culture, they were sorted into the two GFP+ and RG+ populations and then passaged and monitored (Fig. 4B). Interestingly, the number of cells expressing GFP and RFP varied in both populations. For example, the percentage of RG+ cells was highest in passage 7 compared to passages 3 and 8. This is not surprising because the switch between lytic and latent infection is dynamic and varies upon passaging in culture. This, together with the single-cell cloning results, supports the notion that lytic viral replication in RG+ cells can be repressed to revert to latency.
To confirm that the infected cells indeed go through lytic replication without induction, as shown by the switch from GFP+ to RG+, ganciclovir (GCV), which inhibits lytic viral gene expression, was added to inhibit lytic viral gene expression, and reverse transcription-quantitative PCR (RT-qPCR) was carried out with the RNA extracted from treated and nontreated cells to quantify latent (LANA) and lytic (ORF59 [DNA polymerase processivity factor]) gene expression levels. The results showed that ORF59 gene expression was reduced (P < 0.01) in the presence of GCV on day 3 after treatment compared with nontreated cells. On day 10, there were reductions of both LANA and ORF59 compared with the levels in nontreated cells, but they were not statistically significant. This was expected since prolonged GCV treatment will lead to some inhibition of the expression of most viral genes.
SH-SY5Y cell authenticity can be confirmed by STR (short tandem repeat) typing and expression of the neuronal cell marker nestin.
In order to validate that the SH-SY5Y cells are permissive to KSHV infection and exclude the possibility that they were contaminated by other cell types such as 293T cells, which are susceptible to KSHV infection, STR typing was used to confirm our SH-SY5Y cells’ authenticity. The results showed that our SH-SY5Y cell profile matched the ATCC STR database exactly for SH-SY5Y cells, where 8 core loci, TH01, D5S818, D13S317, D7S820, D16S359, CSF1PO, vWA, and TPOX, were expressed at levels similar to those in the database (Table S2). Additionally, undifferentiated neuronal cell marker nestin gene expression could be detected in both uninfected and infected SH-SY5Y cells (Fig. 1C). IHC staining showed that uninfected SH-SY5Y cells expressed high levels of nestin; however, the infected RG+ SH-SY5Y cells maintained nestin expression but at a reduced level. It is likely that some cellular gene expression in infected SH-SY5Y cells, such as nestin, can be suppressed upon KSHV infection, which was confirmed by the RNAseq analysis described below.
RNAseq and analysis.
Given the ability of KSHV-infected SH-SY5Y cells to transition between lytic and latent infections, it was of interest to determine which viral and cellular genes are involved in the two phases of infection. This was accomplished by RNAseq followed by comparative transcriptomic analyses. Figure 5A shows the flowchart of the analysis. Extracted RNA from sorted KSHV-infected RG+ and GFP+ populations yielded 14 million and 17 million reads, respectively, that aligned to the human genome hg19 reference (Fig. 5B). Consistent with its higher KSHV genome content, the RG+ population had the highest number of reads aligned to the KSHV reference (42,564 reads), whereas fewer reads, 2,556, mapped to KSHV in the GFP+ population. There were no reads aligned to KSHV in the uninfected control (negative-control [NC]) population. Viral gene analysis was compared only between infected GFP+ cells and RG+ cells.
FIG 5.
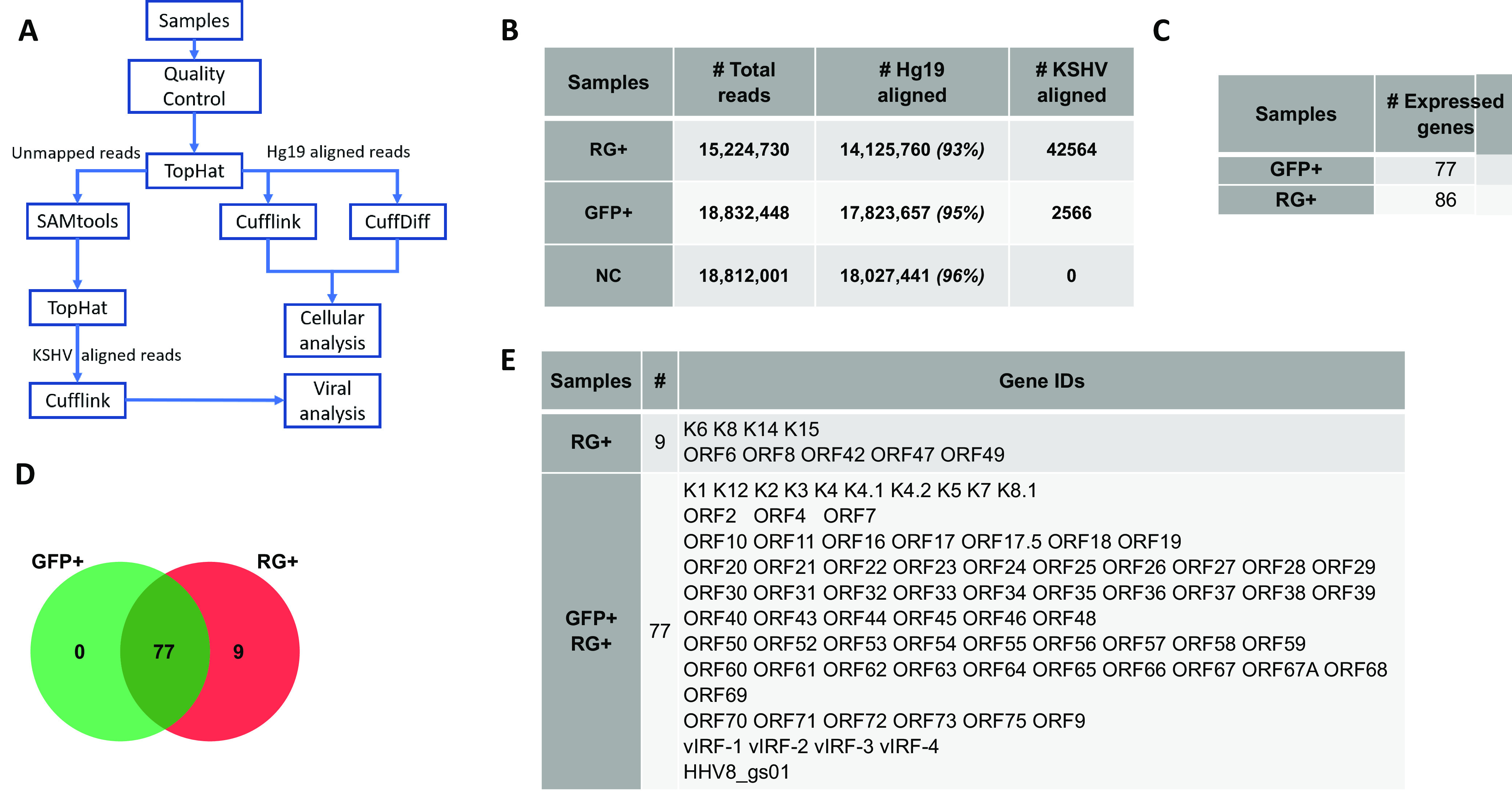
Flowchart of RNAseq analysis and KSHV-expressed viral genes across two groups of rKSHV.219-infected GFP+ and RG+ cells. (A) Flowchart of RNAseq analysis. (B) Total reads from RNAseq across two groups of GFP+ SH-SY5Yand RG+ SH-SY5Y cells (hg19 was used as the reference alignment). (C) Number of viral genes expressed across the two groups of GFP+ and RG+ infected cell populations. (D) Venn diagram showing intersecting genes of the GFP+ and RG+ infected cell populations. (E) Viral genes associated with RG+ infected cells and overlapping genes between RG+ and GFP+ infected cell populations.
Viral gene expression and classified functions.
The viral genes across the two infected populations were first analyzed for uniquely or commonly expressed genes. As expected, the RG+ population expressed a higher number of viral genes than the GFP+ population. The Venn diagram in Fig. 5D shows that despite differences in levels of expression, there were 77 common KSHV genes shared between the GFP+ and RG+ populations. Nine unique genes were expressed only in RG+ cells (Fig. 5C to E): the majority of them were viral structural genes involved in the packaging and assembly of KSHV, which appear to determine the RG+ phenotype.
The KSHV gene expression levels, fold changes, and functional assignments of the expressed viral genes were further analyzed (Fig. 6). Genes with the same function were grouped. Figure 6A and B show results for the genes associated with latency, expression/replication, and structural proteins, whereas Fig. 6C shows results for KSHV genes that encode immunomodulation and have unknown functions. The FPKM (fragments per kilobase of exon per million reads) values for each viral gene detected in GFP+ and RG+ transcriptomes are shown. The pairwise fold changes of viral gene expression between GFP+ and RG+ cells are listed. Our results are consistent with KSHV latent infection of other cell types in that fewer genes were expressed in GFP+ cells than in RG+ cells (10–12, 14). Figure 6A shows the expression of four upregulated latent genes, K12, ORF71, ORF72, and ORF73, that may play a role in maintaining the viral episome, promoting infected cell survival, and suppressing lytic replication in latently infected SH-SY5Y cells since the expression of these four genes was reduced in the RG+ group, with the largest reduction being 3.9-fold for ORF73 (LANA).
FIG 6.

KSHV gene expression levels and functions across two groups of GFP+ and RG+ infected cells individually and intergroup comparison of RG+/GFP+ infected cells. (A) Heat map showing latency functionality and expression and replication genes. (B) Structural genes. (C) Immunomodulation genes and genes with unknown functionality. Clustering of genes was based on KSHV genes with ≥10 reads in at least one sample, fold changes value of exclusively expressed genes in RG+ designated as infinity (inf). Each cluster with ≥5 genes was then annotated with the most enriched function. Gene expression is shown as an FPKM log2 heat map and FPKM log2 exact values for GFP+ and RG+ infected cell populations individually, and the FPKM log2 scale is shown in the bottom, from 0 (white) to >18 (dark gray). Data from the intergroup comparison of fold changes of gene expression are shown as a log2 fold change heat map and fold change log2 values, and the fold change scale was from −6 (dark blue) to +6 (dark red).
There were four KSHV expression/replication genes expressed at higher levels (>1.5-fold) in the RG+ than in the GFP+ population: K8, ORF6, ORF49, and ORF70. Three of these, K8, ORF6, and ORF49, were exclusively expressed in the RG+ group. The K8 gene encodes the immediate early basic leucine zipper-containing protein, K-bZIP. ORF6 encodes a single-stranded DNA binding protein that participates in origin-dependent replication, and ORF49 is the herpesvirus BBRF1 homolog expressed at the late stage of infection. A previous study reported that virus-like particles (VLPs) produced by 293/ΔBBRF1 cells are defective infectious particles devoid of infectious DNA, suggesting that BBRF1 is incorporated into the mature virion and serves a function immediately after infection, and a new round of viral transcription can be initiated (30). ORF70 is an early lytic gene that encodes thymidylate synthase and was expressed at a level 2.4-fold higher in RG+ than in GFP+ cells. Only two expression/replication genes were downregulated (≥1.5-fold) in RG+ cells. They were OFR30, which encodes viral transcription factors required for the expression of late genes, and ORF44, which is a DNA replication protein (helicase/primase subunit).
Among the structural genes (Fig. 6B), there were four that were expressed at a higher level (>1.5-fold) in RG+ than in GFP+ cells; three out of four, ORF8, ORF42, and ORF47, were exclusively expressed in RG+ cells. The fourth, ORF65, was expressed at 1.6-fold-higher levels in RG+ cells. ORF8 encodes glycoprotein B (gB), ORF42 encodes a tegument protein, ORF47 encodes glycoprotein L (gL), and ORF65 encodes a capsid protein. All four genes are expressed in the late stages of infection. Five genes were expressed in RG+ cells at a lower level than in GFP+ cells. These were ORF28, ORF31, ORF52, ORF53, and ORF75. The ORF28 gene encodes glycoprotein H (gH), which is the homolog of Epstein-Barr virus (EBV) gp150 known to induce the shielding of B cell surface markers and contributes to antigen presentation during productive infection (31). The ORF31 gene encodes a viral preinitiation complex (vPIC) subunit, and ORF52 (tegument protein) is required for the production of progeny viruses (32). ORF53 encodes the envelope glycoprotein (gN), and ORF75 encodes a tegument protein.
Among the immunomodulation genes (Fig. 6C), five genes, K4.1, K6, K7, K14, and K15, were expressed at >1.5-fold-higher levels in RG+ cells than in GFP+ cells. Three of them, K6, K14, and K15, were exclusively expressed in RG+ cells The K6 gene encodes the lytic protein vMIP-1, which may be involved in angiogenesis (33). The K14 gene encodes the OX-2 membrane glycoprotein homolog vOX-2. The K15 gene encodes the latency-associated membrane protein (LAMP). HHV8-gs01 is unclearly characterized. Three genes, K2, ORF4, and vIRF-1, were expressed at levels ≥1.5-fold lower in RG+ than in GFP+ cells. The K2 gene encodes vIL-6, ORF4 encodes the KSHV complement control protein (KCP), and vIRF-1 encodes viral interferon regulatory factor 1, which has high homology with cellular IRF. The vIRF-1 gene has been shown to cause transformation and inhibit tumor necrosis factor alpha (TNF-α)-mediated apoptosis (34). Overall, as expected, lytic gene expression was upregulated in RG+ cells, with some genes being significantly upregulated versus cells in apparent latency. In contrast, a number of lytic gene RNAs were absent or downregulated in GFP+ cells. There were 9 out of 75 lytic genes (12%) that were absent in the GFP+ group; these were K6, K8, K14, K15, ORF6, ORF8, ORF42, ORF47, and ORF49. It is perhaps not surprising that five of these, K14, K15, ORF8, ORF42, and ORF47, are membrane glycoproteins involved in the regulation of viral entry, which would be inopportune to have on the surface of a cell attempting to avoid immune recognition during latency.
Cellular gene and pathway analysis.
Using the reads aligned to the hg19 genome and transcriptome, the gene expression patterns in uninfected SH-SY5Y, RG+, and GFP+ populations were defined. A gene with an abundance value of >1 FPKM in any population was defined as an expressed gene. Based on this, we obtained 19,327 genes that were expressed in at least one of the cell populations. Significantly differentially expressed (DEGs) genes in the infected populations were obtained by comparing the genes with a false discovery rate (FDR) of <5% to the NC population. Genes with a ≥2-fold change were defined as upregulated genes, and the ones with fold changes of <0.5 were defined as downregulated genes. Genes with fold changes in the range of 0.5 to 2 were filtered out as unaltered. The results show the number of significantly up-/downregulated genes in each population compared to the NC population (Fig. 7A). Venn diagrams of the samples and the up- and downregulated genes common to both lytically and latently infected cells are shown in Fig. 7B and C, respectively.
FIG 7.
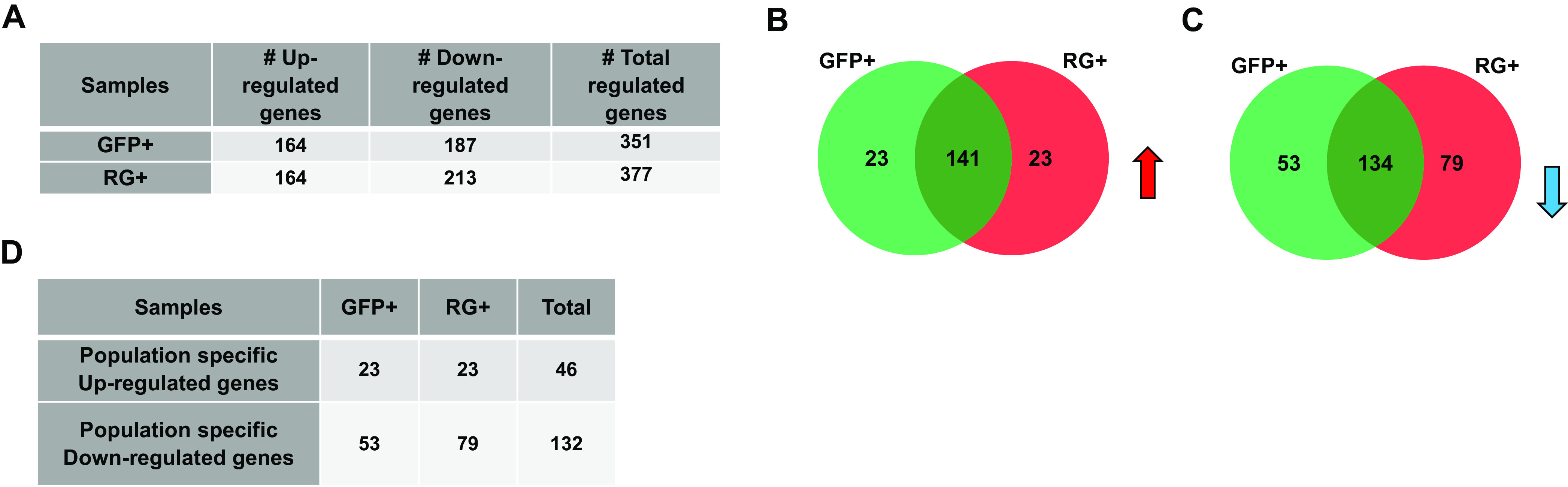
Significantly differentially expressed cellular genes across GFP+ and RG+ KSHV-infected cells. (A) Numbers of up-/downregulated genes across GFP+ and RG+ rKSHV.219-infected cells. (B) Venn diagram showing upregulated intersecting genes across two groups of GFP+ and RG+ infected cells (indicated by the red upward arrow). (C) Venn diagram showing downregulated intersecting genes across two groups of GFP+ and RG+ infected cells (indicated by the blue downward arrow). (D) Numbers of unique up-/downregulated genes associated with specific phenotypes of GFP+ and RG+ infected cells.
There were 23 uniquely upregulated and 53 downregulated genes in GFP+ cells, and there were 23 uniquely upregulated and 79 downregulated genes in RG+ cells (Fig. 7D). All the up-/downregulated genes are listed in Fig. S3 and S4. The majority of the differentially expressed genes were transcriptional regulators that control cell growth and the cell cycle, neuronal differentiation, transformation, and tumorigenesis. In support of our results from IHC staining of the neuronal marker nestin (Fig. 1C), the nestin gene expression level was found to be ∼6-fold lower in infected cells than in uninfected cells (data not shown). With 351 and 377 significantly differentially expressed genes in the GFP+ and RG+ populations, respectively (Fig. 7A and B), we analyzed each set of significantly dysregulated genes using DAVID to identify cellular pathways that are consistent with the set of differentially regulated genes. There were five Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that were associated with the shared differentially regulated genes in the two infected groups. There were also eight pathways that might be associated with the GFP+ phenotype uniquely and two for the RG+ phenotype (Fig. S5). The pathways unique to the GFP+ cells are of special interest since they are presumed to participate in maintaining latency. Indeed, three of the eight unique pathways for the GFP+ phenotype were linked to viral diseases and cancers (Fig. 8). Interestingly, common genes were found to be involved in two (hsa05416 and hsa05200) of the three pathways (Fig. 8A and B). For example, CCND1, involved in both pathways, is central in connecting with other cellular factors such as MAPK10, BMP4, GLI3, LEF1, and PDGFRB, proteins known to be involved in a variety of cellular process such as proliferation, differentiation, and transcriptional regulation as well as neuronal cell differentiation (35). The third GFP+ phenotype-associated pathway (hsa05169) involved genes such as CD40 (Fig. 8C and D), an immune-costimulatory protein found in cancer cells, and TNFAIP3, a tumor necrosis factor-inducible zinc finger-deubiquitinating enzyme associated with EBV infection and other human diseases (36).
FIG 8.
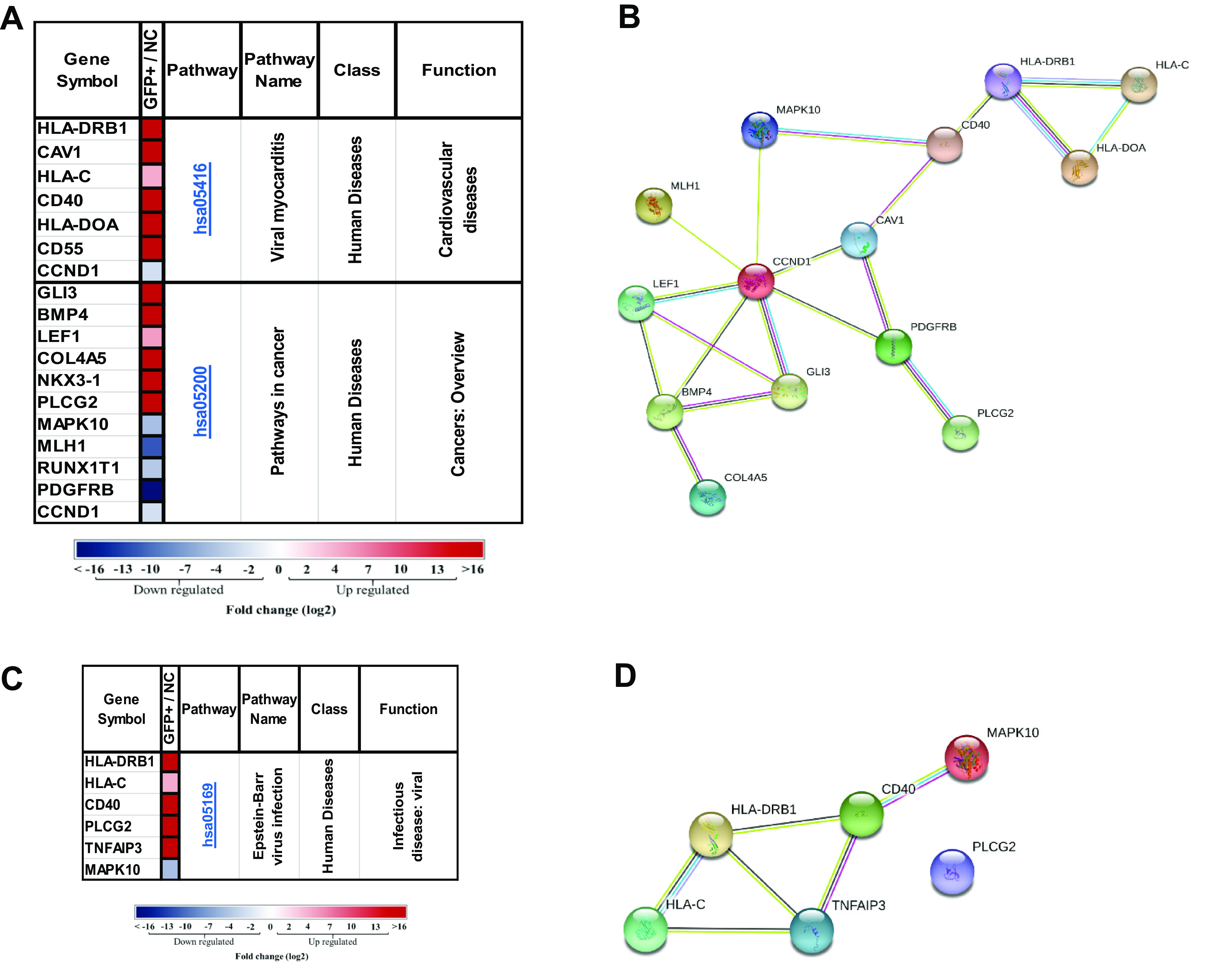
Viral myocarditis, cancer, and Epstein-Barr virus infection KEGG pathways were involved in the GFP+ group of KSHV infection. (A) Significantly differentially expressed genes (DEGs) of the GFP+ infected cell population in the hsa05416 and hsa05200 KEGG pathways (gene fold changes [GFP+/NC] are illustrated on a log2 scale from dark blue to dark red). (B) Protein-protein interactions of DEGs in the GFP+ infected cell population and the hsa05416 and hsa05200 KEGG pathways. (C) Significant DEGs of the GFP+ infected cell population in the hsa05169 KEGG pathway (gene fold changes [GFP+/NC] are illustrated on a log2 scale from dark blue to dark red). (D) Protein-protein interactions of DEGs in GFP+ cells and the hsa05169 KEGG pathway. Colored nodes designate query proteins and the first shell of interactions, white nodes designate the second shell of interactions, the known interactions determined experimentally are shown as a pink line, interactions from a curated database are shown as a turquoise line, and predicted interactions are shown in lines with other colors.
Among the 5 KEGG pathways that were found to be common between the GFP+ and RG+ phenotypes, gene ontogeny functional analysis demonstrated that three (hsa04514, hsa04390, and hsa05202) have shared genes (Fig. 9A). Interestingly, they are involved in cell adhesion, hippo signaling, and transcriptional deregulation in cancers. For example, NFASC (Fig. 9B), which exhibited maximal linkage with other genes in the pathways, encodes neurofascin, a neuronal cell adhesion molecule (CAM) associated with cell migration, adhesion, neurite outgrowth, neuronal differentiation (35), tumor progression, and metastasis (37).
FIG 9.
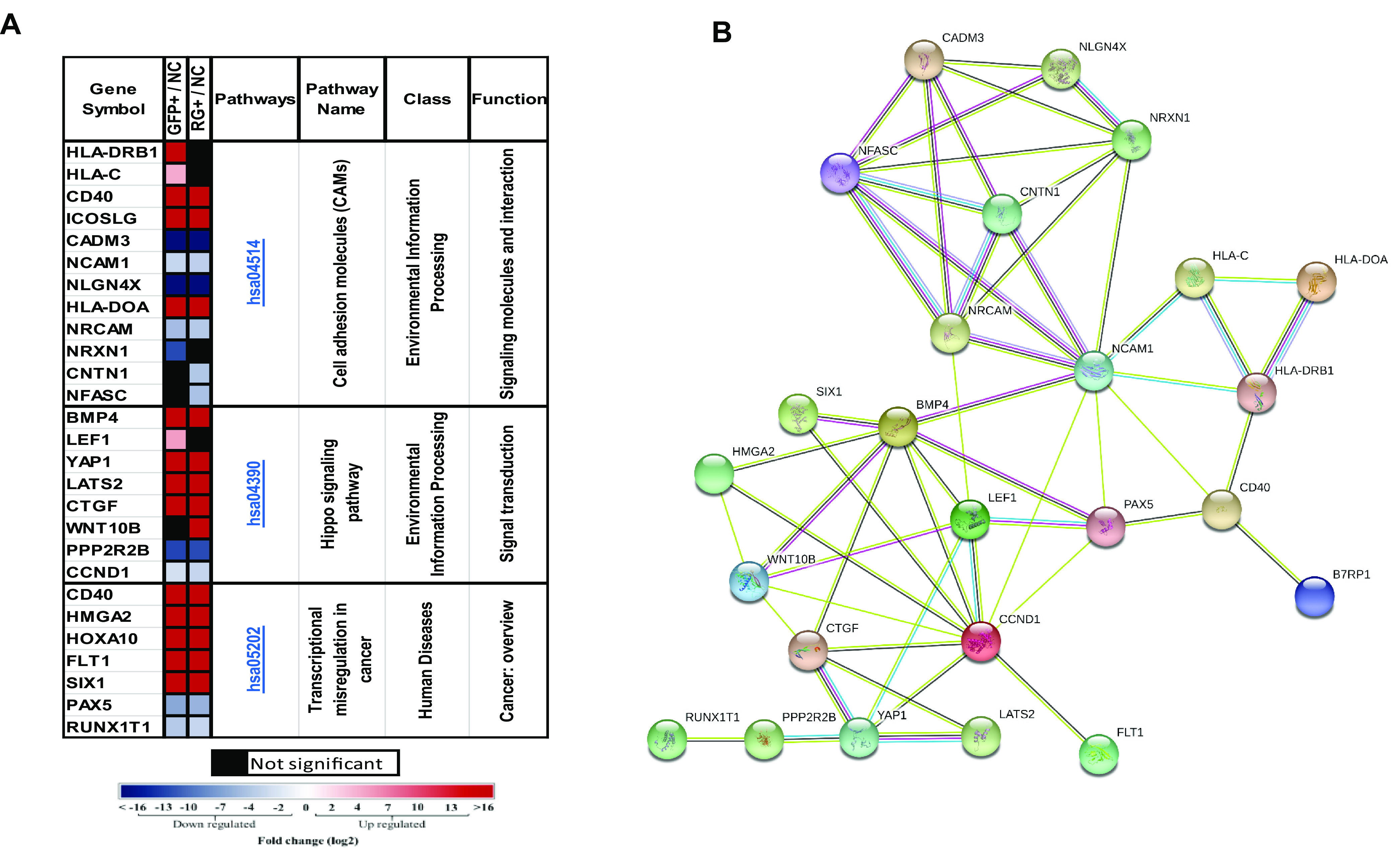
Cell adhesion molecules, hippo signaling pathway, and transcriptional deregulation KEGG pathways predicted to be involved in GFP+ and RG+ rKSHV.219-infected populations. (A) Significantly differentially expressed genes (DEGs) of GFP+ and RG+ infected cell populations in the hsa04514, hsa04390, and hsa05202 KEGG pathways (gene fold changes of GFP+/NC and RG+/NC are illustrated on a log2 scale from dark blue to dark red). (B) Protein-protein interactions of DEGs in infected populations and in the hsa04514, hsa04390, and hsa05202 KEGG pathways. Colored nodes designate query proteins and the first shell of interactions, white nodes designate the second shell of interactions, known interactions determined experimentally are shown as a pink line, interactions from a curated database are shown as a turquoise line, and predicted interactions are shown in lines with other colors.
Finally, we compared significantly regulated genes in the GFP+ and RG+ populations with significantly regulated genes from infected TIME (hTert-immortalized microvascular endothelial) cells (38). Figure S6A shows the number of common significantly regulated genes between the groups. There are 44 common genes across all groups. Figure S6B shows their log2 fold changes. Thirteen significantly regulated genes were found in common between TIME and GFP+ cells, and 14 genes were found between RG+ and TIME cells. Figure S6C shows the fold changes of these 27 genes.
DISCUSSION
A number of human herpesviruses have been shown to be neurotropic, especially alphaherpesviruses such as herpes simplex virus (HSV), which is known to establish latency in the central nervous system (CNS), and it can be reactivated to lytic replication to cause disease (39). KSHV has a wide host range in vitro; it can infect endothelial cells, epithelial cells, B cells, macrophages, and the neuroblastoma cell line SH-SY5Y (26, 40–43). Our laboratory has previously shown that KSHV can establish infection in parenchymal cells in various brain tissues in HIV-positive individuals (25). The infection was found to be latent, as indicated by the expression of LANA, and the majority of infected cells were neurons, demonstrating that KSHV can also be neurotropic and that the CNS can be a reservoir for latent infection. Here, we confirm that SH-SY5Y cells can be infected by KSHV and further demonstrate that the infected cells displayed a higher growth rate. The infection can be sustained long term, and the infected cells can spontaneously transition between latent and lytic phases of the KSHV infection cycle.
SH-SY5Y cells were originally derived from a bone marrow biopsy specimen taken from a patient with neuroblastoma; it has the ability to differentiate along the neuronal lineage and is often used as an in vitro model of neuronal function and differentiation. It has been widely used as a neuronal model for a number of viruses, including Zika virus, West Nile virus (WNV), and the alphaherpesviruses, etc. (44–46). Furthermore, the SH-SY5Y cell line has been used as a model to study neurodegeneration (47, 48). Thus, it can be a useful model to further investigate KSHV infection and cellular responses upon infection, as reported in this study.
A unique feature of KSHV-infected SH-SY5Y cells is the ability of the infected cells to sustain infection and continue to transition between latency and spontaneous lytic reactivation to generate infectious virions upon long-term culture and passaging without cell death. This has not been observed with most infected cells in vitro. This is also in contrast to a report by Jha et al. suggesting that KSHV infection of SH-SY5Y cells was predominantly lytic (26). It is possible that this was due to differences in culture conditions since we can observe lytically infected cells as early as 2 days postinfection, and the transition back to the latent phenotype was observed only upon longer culturing and passaging. In fact, most other infected cells types have been reported to go through a brief lytic phase but will quickly become latent; spontaneous lytic reactivation occurred in only a small fraction (1 to 5%) of the KSHV latently infected cells since lytic induction of viral replication was shown to be cytotoxic and led to apoptosis (49–52). Mixed lytic and latent gene expression upon infection is unusual but has been observed in infected primary dermal microvascular endothelial cultures. Furthermore, the infection could not be sustained long term in culture, in contrast to what we observed here with SH-SY5Y cells (53).
At this point, it is not clear what causes infected SH-SY5Y cells to go through spontaneous lytic activation and what suppresses the expression to return to the latent phase of infection. Lytically infected SH-SY5Y cells did not exhibit any cytopathic effect (CPE) or cell death. The lytic replication of most latently infected cells in vitro was usually induced by chemicals such as TPA and sodium butyrate, which reactivate KSHV via the protein kinase C (PKC) pathway or by inhibiting the histone deacetylase (HDAC) associated with epigenetic modification (18). The mechanism involved in controlling the transition of infected SH-SY5Y cells is unclear; one possible reason is that RG+ SH-SY5Y cells can transition back to the latent phase, possibly involving paracrine and other unknown factors, and it will provide us with an opportunity to identify distinct key viral and cellular genes and the potential pathways that may be involved in determining the GFP+ latent and RG+ lytic phases of KSHV infection (18). It is possible that this is a mechanism of neuronal cells to suppress viral infection to avoid apoptosis and for the virus to sustain and escape the host immune response but can be periodically reactivated to produce infectious virions, which is well documented for HSV-1 infection of neurons (54). Here, we propose a model of a phenotypic transition between lytic RG+ and latent GFP+ and vice versa. Cells presumed to be latently infected, by virtue of the initial expression of GFP only, were also documented to go through an apparent lytic replication phase (RFP+) and either remain GFP+/RFP+ or return to GFP+ only over time (Fig. 4C).
Another unique feature is that KSHV infection of SH-SY5Y cells can be sustained long term, unlike most in vitro infections of other cell types, where the virus will be lost upon passaging (55). The infection can lead to a higher growth rate of the infected cells. This suggests that the viral genes and/or cellular genes that are deregulated upon infection can further enhance the replication of the already immortalized SH-SY5Y cell line. Indeed, our transcription analyses of infected cells displaying both latent and lytic infection phenotypes show common up- or downregulated genes that could be involved in transcriptional regulation, transformation, and cellular signaling pathways.
For the viral genes, we found that there were 77 commonly expressed viral genes for both GFP+ latent and RG+ lytic infection phenotypes that could be involved in infection and affect cell growth. These genes were expressed during different phases of viral infection, and they are known to be involved in viral latency, immunoregulation, transcriptional regulation, as well as viral morphogenesis. Our transcriptome analyses also showed that all the viral genes expressed in the latent phases were also expressed in the lytic phase, except for nine viral genes that were expressed exclusively by the lytic phenotype. Five of these genes (K14, K15, ORF8, ORF42, and ORF47) encode proteins that are involved in the regulation of viral entry. This is different from other KSHV latently infected cells, where only the latency-associated genes ORF71 to -73, K12 (kaposin), vIRF-3, and 18 mature miRNAs are expressed (56). Since all the genes found to associate with the latent phenotype were also found to associate with the lytic phenotype, this suggests that viral gene expression in latent cells was suppressed or aborted late in infection, with some of the required genes (such as the nine viral genes) being suppressed to prevent the virus from completing its replicative cycle. Indeed, it has been shown that abortive lytic reactivation short of expressing the full set of viral genes required for virion production and cell lysis can occur (57–59). The 77 viral genes expressed in GFP+ cells that were in common with the RG+ cells also suggest that these infected cells were not necessarily truly latent and differed from the classical form of latency. This may reflect either cells that have gone through abortive lytic gene expression or simply cells that have lost RFP gene expression. The RFP gene is expressed using the KSHV PAN promoter, and it has been shown to have limitations in reflecting truly lytic viral replication (60). However, the expression of 77 but not all the lytic genes required by KSHV replication supports the notion of abortive lytic replication, reflected by the lack of expression of the RFP gene. Thus, RFP and GFP, while having limitations, are still useful markers for rKSHV.219 to monitor latent and lytic phases of infection. It is also interesting that the dramatic differences in the viral gene reads between the RG+ and GFP+ cells (42,564 versus 2,566) suggest that a large number of other viral genes involved in the latent phenotype were also induced with the lytic phenotype, in addition to the nine unique genes associated with lytic viral replication. It is unlikely that our analyzed transcriptomes of the GFP+ latent cells were contaminated with those of RG+ lytic phenotypes because RNA was quickly extracted shortly from the sorted cells for RNAseq analysis. In addition, RNAseq data showed that none of the nine unique genes associated with the lytic phenotype are found in the cells associated with the latent phenotype. It is not clear at this point which specific viral genes are involved in the deregulation of cellular genes to lead to enhanced cell replication, and this will need to be further investigated.
For the cellular genes, our transcriptome data revealed a number of differentially expressed cellular genes for the two cell populations. There were 275 deregulated cellular genes shared between the latent and lytic phenotypes upon infection. Some of these deregulated genes are involved in common cellular pathways that could affect cell growth. Some specific upregulated genes are CTGF, BMP4, YAP1, LEF1, and HLA-DRB1, and some specific downregulated genes are CCDN1, PAX5, and NFASC. These deregulated genes are linked in common pathways and could work coordinately to regulate biological processes such as cell adhesion, proliferation, differentiation, transcriptional regulation, and oncogenesis. One such pathway is the hippo signal transduction pathway, another is the pathway involved in transcriptional deregulation in cancers, and a third one involves the regulation of cell adhesion molecules (CAMs).
There were 76 cellular genes specifically deregulated in the cells with the latent phenotype. Interestingly, some of these 76 genes are linked and associated with three cellular pathways. Two of the pathways are involved in viral diseases and cancers, and the third was associated with EBV infection. In the latent GFP+ cells, the downregulated CCND1 and MAPK10 genes can be linked to the upregulated genes GLI3, BMP4, and LEF1 to participate in the same pathway involved in EBV infection and may be involved in tumorigenesis. These affected cellular pathways together with the effects of viral genes can subsequently lead to a more rapid replication phenotype of KSHV-infected cells. Further studies to decipher their functions will provide a better understanding of not only how KSHV leads to transformation but also the potential roles that these genes may play in oncogenesis.
In summary, we have demonstrated that KSHV infection of SH-SY5Y cells exhibited several unique characteristics, including sustained maintenance of viral infection, the ability to survive infection, and rapid proliferation of infected cells, all resulting in lifelong infection and survival within the host. Binding of KSHV to the host cell membrane and subsequent signal induction are two early steps of the KSHV life cycle. This is then followed by the deregulation of the signaling of CAMs, hippo signaling, and tumorigenesis pathways through the crucial cellular factors expressed differentially in RG+ and GFP+ cells. Unlike conventional latent infection, the GFP+ latent cell population shared a substantial number of lytic viral genes with the RG+ lytic cells, but the deregulated cellular genes play a role in aborting lytic infection to enable RG+ lytic cells to transition back to latency to escape the immune response, leading to persistent KSHV infection. Further studies will be needed to identify the specific viral and cellular genes and the underlying mechanism involved in KSHV neuronal infection and neuropathogenesis, and the findings will be helpful for designing preventive strategies or therapeutics to eliminate KSHV infection.
MATERIALS AND METHODS
Cell line culture and STR typing.
SH-SY5Y cells were originally isolated from a bone marrow biopsy specimen taken from a 4-year-old female with neuroblastoma epithelium-like cells (61). The SH-SY5Y neuroblastoma cell line (ATCC CRL2266) and HEK293T cells (ATCC CCL-2) were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 50 mg/ml streptomycin, and 50 U of penicillin. Vero 219 cells (ATCC CCL-81) were grown in DMEM supplemented with 10% FBS and 6 μg/ml puromycin. The cells were maintained at 37°C with 5% CO2.
STR testing sample preparation was carried out according to the ATCC’s instructions. Briefly, SH-SY5Y cells were cultured in a T25 flask at 37°C with 5% CO2. The cells were then trypsinized and centrifuged at 125 × g, and the cell pellet was resuspended in a small volume of phosphate-buffered saline (PBS). The cells were counted and diluted to 1 × 106 cells/ml, and 40 μl of the cell suspension was spotted onto a sample collection card, air dried at room temperature for 15 min, and sent to the ATCC for STR analysis.
Recombinant KSHV (rKSHV.219) generation and titration.
The generation of recombinant fluorescence-labeled infectious rKSHV.219 was carried out by propagating latent virus in Vero 219 cells and then inducing lytic replication (49). Vero 219 cells in T75 flasks were infected with a nonpropagating (in human cells) baculovirus that expresses KSHV RTA (Bac50) for 4 h in 3.5 ml serum-free DMEM. After 4 h, the Bac50-containing supernatant was removed, and DMEM with 1.25 mM sodium butyrate (NaB) and 6 μg/ml puromycin was added. The sodium butyrate-containing medium was removed 30 h later and replaced with fresh DMEM. After 72 h, the virus-containing supernatant was collected and filtered through a 0.45-μm low-protein-binding filter. To determine the titer of harvested rKSHV.219, HEK293T cells were infected with serial dilutions of the viral supernatants (4−1 to 4−7); the number of GFP-positive cells was counted, and the virus titer was calculated using the Reed-Muench method (62).
Infection of SH-SY5Y cells with rKSHV.219.
SH-SY5Y cells at 1 × 105 cells/ml were seeded in a volume of 1 ml in a 12-well plate. The cells were infected with rKSHV.219 at a multiplicity of infection (MOI) of 0.5 on the following day. The culture plate was centrifuged at 300 × g for 20 min at 25°C to promote viral contact with target cells. Infected cultures were maintained for 2 weeks and passaged to maintain ∼70 to 80% confluent growth. The infection phenotype was monitored by fluorescence microscopy (Nikon Eclipse TE300) for GFP/RFP expression. Fluorescence microscopic images were digitally acquired using the TE300 microscope with NIS-Elements-inverted software (Nikon, USA).
Flow cytometry and cell sorting.
Infection of SH-SY5Y cells by rKSHV.219 was monitored by flow cytometry for GFP/RFP expression using a FACSCalibur instrument and CellQuest Pro software (BD Biosciences). To determine the percentage of lytically induced cells expressing RFP/GFP or to separate RFP-negative (RFP−)/GFP+ and RFP−/GFP− SH-SY5Y cells, the cells were sorted and analyzed using the FACSAria II cell sorter (BD Biosciences) and FACSDiva software (version 8.0.1). The gating parameters are shown in Fig. S1 in the supplemental material.
Soft agar assay.
To monitor individual phenotypically distinct cells over time, cells were cultured in semisolid medium. Briefly, a bottom soft agar layer of 4 ml of warm 0.7% agar and 10% FBS was used for each 60-mm culture dish. After the agar solidified, uninfected 3 × 103 SH-SY5Y cells and rKSHV.219-infected GFP+ SH-SY5Y cells or RFP+/GFP+ SH-SY5Y cells (abbreviated RG+ here) were resuspended in 4 ml 0.35% agarose in DMEM containing 10% FBS and added on top of the bottom soft agar layer. The plates were incubated at 37°C with 5% CO2, and the cells were supplemented with soft agar every 3 to 4 days. Colonies that formed were stained with 0.2% Nitro Blue Tetrazolium (Sigma-Aldrich, St. Louis, MO, USA). Individually marked colonies were observed for GFP and RFP expression over time and photographed daily by inverted fluorescence microscopy up to day 6. Each experiment was conducted in triplicate, and data were obtained from an average of three independent experiments.
Ganciclovir treatment of rKSHV.219-infected SH-SY5Y cells.
KSHV-infected SH-SY5Y cells were passaged and then sorted by fluorescence-activated cell sorter (FACS) analysis to obtain only GFP+ SH-SY5Y cells. The sorted cells were shown to be more than 99% pure GFP+ SH-SY5Y cells and free of any RFP+/GFP+ SH-SY5Y cells. The sorted GFP+ SH-SY5Y cells were then seeded at 1 × 105 cells/ml into a 12-well plate, and the cells were treated with ganciclovir (GCV) (catalog no. G2536; Sigma-Aldrich) at a concentration of 80 μM on the following day. The fluorescence intensity was observed every other day under a fluorescence microscope. This concentration of GCV was selected because no cellular toxicity was observed at this concentration, based on an experiment testing various concentrations of GCV for cellular toxicity (data not shown). RG+ SH-SY5Y cells were collected on day 3 and day 10 of GCV treatment; cells with no GCV treatment were used as the negative control (NC). RNA extractions from the harvested cells were carried out according to the miRNeasy Mini handbook (Qiagen). The KSHV latent LANA and lytic ORF59 (KSHV DNA polymerase processivity factor) gene expression levels were quantified by RT-qPCR using the QuantStudio 3 system (Applied Biosystems). Each PCR mixture included 100 ng of the cDNA template and 0.5 μM each forward and reverse primers with 2× iTaq universal SYBR green supermix (catalog no. 1725121; Bio-Rad), adding nuclease-free water to 20 μl. The RT-qPCR program was set at 95°C for 5 min, 40 cycles of 95°C for 15 s and 60°C for 1 min, and then 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. The primers used are listed in Table S1.
Quantification of intracellular and extracellular KSHV genomes.
For the quantification of intracellular KSHV DNA copy numbers, infected cells were harvested and washed twice with PBS to remove residual virions. Cellular DNA was extracted using the Gentra Pugene kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Extracellular KSHV virions in the culture supernatant were extracted from the cell culture supernatant. Clarified supernatants from the infected cells were first treated with DNase I (Invitrogen, Thermo Fisher Scientific, Warrington, UK) at 37°C to remove residual extracellular viral and cellular DNA. Virion-associated DNA, which is protected from DNase I by the viral envelope, was then extracted by phenol-chloroform followed by ethanol precipitation. Intracellular and extracellular viral DNA copy numbers were determined by real-time PCR using a KSHV ORF26 amplicon and TaqMan universal master mix (Thermo Fisher Scientific, Warrington, UK) with quantification on a QuantStudio 3 real-time PCR detection system (Thermo Fisher Scientific). The reactions were performed in optical 96-well plates at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. All reactions were run in triplicates. A plasmid, pCR2.1ORF-26, was used as a KSHV amplification standard. Tenfold serial dilutions from 1.48 × 106 copies/μl to 1.48 copies/μl of the plasmid were used to generate a standard curve. The primers and probes used in this study are listed in Table S1.
Immunohistochemistry.
Immunohistochemical staining was used to detect viral latency-associated nuclear antigen (LANA) protein and nestin expression in 1 × 105 SH-SY5Y cells infected with rKSHV.219 at an MOI of 0.5 and grown directly on poly-l-lysine-coated chamber slides. Uninfected SH-SY5Y cells were used as the NC. At 4 days postinfection, each well of the chamber slide was washed with PBS gently and then fixed for 20 min with 4% paraformaldehyde. Chamber slides were washed 3 times with PBS, and endogenous peroxidase was quenched with 3% H2O2 for 30 min. After removing residual peroxide with distilled water, wells were blocked with 2.5% normal horse serum (NHS) for 10 min at room temperature. The primary mouse anti-LANA antibody (NCL-HHV8-LANA; Leica) was diluted 1:200, and antinestin antibody10C2 (catalog no. ab22035) was diluted 1:100 and incubated overnight at 4°C. The following day, after washing off excess primary antibody, a horseradish peroxidase (HRP)-conjugated anti-mouse secondary Ab (catalog no. K4001; Dako) was added, and the mixture was incubated for 2 h at room temperature, followed by 3 washes with PBS to remove residual Ab. The slides were then treated with DAB (3,3’-diaminobenzidine) chromogen (catalog no. K3468; Dako) and developed for 3 min at room temperature for chromogen deposition. A light Harris hematoxylin counterstain was used to reveal cellular morphology.
RNAseq.
RNA extractions from recombinant rKSHV.219-infected and noninfected SH-SY5Y cells were carried out according to the miRNeasy Mini handbook (Qiagen). The infected samples were sorted by flow cytometry and divided into 2 groups, GFP+ only and RFP+/GFP+ (RG+), and uninfected cells were used as NCs. Quality control of the reads was carried out using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and Trimmomatic tools (63) to filter out low-quality reads as defined by an average Phred (Q) score of <20 per base, a >1% probability of an incorrect base, or a read length of <35 bases. Qualified reads were aligned against the hg19 genome using the TopHat tool (http://ccb.jhu.edu/software/tophat/index.shtml). Aligned reads were used for further cellular analysis. Reads not mapped to hg19 were converted to paired-end reads in fastq files using SAMtools and aligned against the KSHV genome (GenBank accession no. NC_009333.1) for viral analysis. Using the Cufflinks tool (http://cufflinks.cbcb.umd.edu), the aligned reads (TopHat output) were used to calculate the expression levels as FPKM (fragments per kilobase of exon per million reads) values. Differentially expressed genes, and fold changes for each, among the phenotypically segregated populations were obtained using the CuffDiff tool. Genes with significant differential expression were defined as those with a false discovery rate (FDR) of <5%. Significantly dysregulated pathways were analyzed using the DAVID tool (http://david.ncifcrf.gov/) according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (64). For the populations, the included significantly differentially expressed genes were analyzed using the DAVID tool (http://david.ncifcrf.gov/). Significant KEGG pathways (http://www.genome.jp/kegg/) were defined as those with an FDR of <0.05 (Benjamini-Hochberg-adjusted P value [65]).
Statistical analysis.
Statistical analysis was performed using SPSS version 17.0 (SPSS Inc., Chicago, IL, USA). Independent t tests were used for data measurement. Error bars represent standard deviations (SD). A P value of <0.05 was considered statistically significant.
Data availability.
All RNAseq data have been uploaded to the GEO database with the accession number GSE173342.
ACKNOWLEDGMENTS
We thank Dirk Anderson at the Flow Cytometry Service Center for his help with flow cytometry and Danielle Shea for help with the data analysis.
This study is supported in part by the NIH/NCI (R01 CA75903, CA239591, and U54 CA190155), the FIC (D43 TW010354), and the NIGMS (P30 GM103509).
Footnotes
Supplemental material is available online only.
Contributor Information
Charles Wood, Email: cwood1@unl.edu.
Jae U. Jung, Lerner Research Institute, Cleveland Clinic
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–1869. 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Ganem D. 1997. KSHV and Kaposi’s sarcoma: the end of the beginning? Cell 91:157–160. 10.1016/S0092-8674(00)80398-0. [DOI] [PubMed] [Google Scholar]
- 3.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120:939–949. 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 5.Uldrick TS, Wang V, O’Mahony D, Aleman K, Wyvill KM, Marshall V, Steinberg SM, Pittaluga S, Maric I, Whitby D, Tosato G, Little RF, Yarchoan R. 2010. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without multicentric Castleman disease. Clin Infect Dis 51:350–358. 10.1086/654798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boshoff C, Chang Y. 2001. Kaposi’s sarcoma-associated herpesvirus: a new DNA tumor virus. Annu Rev Med 52:453–470. 10.1146/annurev.med.52.1.453. [DOI] [PubMed] [Google Scholar]
- 7.Aneja KK, Yuan Y. 2017. Reactivation and lytic replication of Kaposi’s sarcoma-associated herpesvirus: an update. Front Microbiol 8:613. 10.3389/fmicb.2017.00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renne R, Lagunoff M, Zhong W, Ganem D. 1996. The size and conformation of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J Virol 70:8151–8154. 10.1128/JVI.70.11.8151-8154.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhatt AP, Damania B. 2013. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front Immunol 3:401. 10.3389/fimmu.2012.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cotter MA, II, Robertson ES. 1999. The latency-associated nuclear antigen tethers the Kaposi’s sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254–264. 10.1006/viro.1999.9999. [DOI] [PubMed] [Google Scholar]
- 11.Guasparri I, Keller SA, Cesarman E. 2004. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med 199:993–1003. 10.1084/jem.20031467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Lee HR, Amatya R, Jung JU. 2015. Multi-step regulation of innate immune signaling by Kaposi’s sarcoma-associated herpesvirus. Virus Res 209:39–44. 10.1016/j.virusres.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. 1998. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J Virol 72:8309–8315. 10.1128/JVI.72.10.8309-8315.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valiya Veettil M, Dutta D, Bottero V, Bandyopadhyay C, Gjyshi O, Sharma-Walia N, Dutta S, Chandran B. 2014. Glutamate secretion and metabotropic glutamate receptor 1 expression during Kaposi’s sarcoma-associated herpesvirus infection promotes cell proliferation. PLoS Pathog 10:e1004389. 10.1371/journal.ppat.1004389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadler R, Wu L, Forghani B, Renne R, Zhong W, Herndier B, Ganem D. 1999. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J Virol 73:5722–5730. 10.1128/JVI.73.7.5722-5730.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uppal T, Banerjee S, Sun Z, Verma SC, Robertson ES. 2014. KSHV LANA—the master regulator of KSHV latency. Viruses 6:4961–4998. 10.3390/v6124961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guito J, Lukac DM. 2012. KSHV Rta promoter specification and viral reactivation. Front Microbiol 3:30. 10.3389/fmicb.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye F, Lei X, Gao S-J. 2011. Mechanisms of Kaposi’s sarcoma-associated herpesvirus latency and reactivation. Adv Virol 2011:193860. 10.1155/2011/193860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Persson LM, Wilson AC. 2010. Wide-scale use of Notch signaling factor CSL/RBP-Jkappa in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J Virol 84:1334–1347. 10.1128/JVI.01301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu Y, Wang SE, Hayward GS. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59–70. 10.1016/j.immuni.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 21.Yang Z, Yan Z, Wood C. 2008. Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J Virol 82:3590–3603. 10.1128/JVI.02229-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu Y, Hayward GS. 2010. The ubiquitin E3 ligase RAUL negatively regulates type I interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33:863–877. 10.1016/j.immuni.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bechtel JT, Liang Y, Hvidding J, Ganem D. 2003. Host range of Kaposi’s sarcoma-associated herpesvirus in cultured cells. J Virol 77:6474–6481. 10.1128/jvi.77.11.6474-6481.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gjyshi O, Bottero V, Veettil MV, Dutta S, Singh VV, Chikoti L, Chandran B. 2014. Kaposi’s sarcoma-associated herpesvirus induces Nrf2 during de novo infection of endothelial cells to create a microenvironment conducive to infection. PLoS Pathog 10:e1004460. 10.1371/journal.ppat.1004460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tso FY, Sawyer A, Kwon EH, Mudenda V, Langford D, Zhou Y, West J, Wood C. 2017. Kaposi’s sarcoma-associated herpesvirus infection of neurons in HIV-positive patients. J Infect Dis 215:1898–1907. 10.1093/infdis/jiw545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jha HC, Mehta D, Lu J, El-Naccache D, Shukla SK, Kovacsics C, Kolson D, Robertson ES. 2015. Gammaherpesvirus infection of human neuronal cells. mBio 6:e01844-15. 10.1128/mBio.01844-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carroll PA, Brazeau E, Lagunoff M. 2004. Kaposi’s sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology 328:7–18. 10.1016/j.virol.2004.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Austgen K, Oakes SA, Ganem D. 2012. Multiple defects, including premature apoptosis, prevent Kaposi’s sarcoma-associated herpesvirus replication in murine cells. J Virol 86:1877–1882. 10.1128/JVI.06600-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore PS. 2007. Chapter 30: KSHV manipulation of the cell cycle and apoptosis. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 30.Feederle R, Shannon-Lowe C, Baldwin G, Delecluse HJ. 2005. Defective infectious particles and rare packaged genomes produced by cells carrying terminal-repeat-negative Epstein-Barr virus. J Virol 79:7641–7647. 10.1128/JVI.79.12.7641-7647.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gram AM, Oosenbrug T, Lindenbergh MF, Bull C, Comvalius A, Dickson KJ, Wiegant J, Vrolijk H, Lebbink RJ, Wolterbeek R, Adema GJ, Griffioen M, Heemskerk MH, Tscharke DC, Hutt-Fletcher LM, Wiertz EJ, Hoeben RC, Ressing ME. 2016. The Epstein-Barr virus glycoprotein gp150 forms an immune-evasive glycan shield at the surface of infected cells. PLoS Pathog 12:e1005550. 10.1371/journal.ppat.1005550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Avey D, Fu B, Wu JJ, Ma S, Liu X, Zhu F. 2016. Kaposi’s sarcoma-associated herpesvirus inhibitor of cGAS (KicGAS), encoded by ORF52, is an abundant tegument protein and is required for production of infectious progeny viruses. J Virol 90:5329–5342. 10.1128/JVI.02675-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A 93:14862–14867. 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo T, Park J, Lee D, Hwang SG, Choe J. 2001. Viral interferon regulatory factor 1 of Kaposi’s sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J Virol 75:6193–6198. 10.1128/JVI.75.13.6193-6198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samatov TR, Wicklein D, Tonevitsky AG. 2016. L1CAM: cell adhesion and more. Prog Histochem Cytochem 51:25–32. 10.1016/j.proghi.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Giulino L, Mathew S, Ballon G, Chadburn A, Barouk S, Antonicelli G, Leoncini L, Liu YF, Gogineni S, Tam W, Cesarman E. 2011. A20 (TNFAIP3) genetic alterations in EBV-associated AIDS-related lymphoma. Blood 117:4852–4854. 10.1182/blood-2010-10-310995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, Massague J. 2014. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156:1002–1016. 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sychev ZE, Hu A, DiMaio TA, Gitter A, Camp ND, Noble WS, Wolf-Yadlin A, Lagunoff M. 2017. Integrated systems biology analysis of KSHV latent infection reveals viral induction and reliance on peroxisome mediated lipid metabolism. PLoS Pathog 13:e1006256. 10.1371/journal.ppat.1006256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steiner I, Kennedy PG, Pachner AR. 2007. The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol 6:1015–1028. 10.1016/S1474-4422(07)70267-3. [DOI] [PubMed] [Google Scholar]
- 40.Coleman CB, Nealy MS, Tibbetts SA. 2010. Immature and transitional B cells are latency reservoirs for a gammaherpesvirus. J Virol 84:13045–13052. 10.1128/JVI.01455-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kikuta H, Itakura O, Taneichi K, Kohno M. 1997. Tropism of human herpesvirus 8 for peripheral blood lymphocytes in patients with Castleman’s disease. Br J Haematol 99:790–793. 10.1046/j.1365-2141.1997.4653269.x. [DOI] [PubMed] [Google Scholar]
- 42.Dollery SJ, Santiago-Crespo RJ, Kardava L, Moir S, Berger EA. 2014. Efficient infection of a human B cell line with cell-free Kaposi’s sarcoma-associated herpesvirus. J Virol 88:1748–1757. 10.1128/JVI.03063-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Staskus KA, Sun R, Miller G, Racz P, Jaslowski A, Metroka C, Brett-Smith H, Haase AT. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. J Virol 73:4181–4187. 10.1128/JVI.73.5.4181-4187.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeh JX, Park E, Schultz KLW, Griffin DE. 2019. NF-κB activation promotes alphavirus replication in mature neurons. J Virol 93:e01071-19. 10.1128/JVI.01071-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Braz-De-Melo HA, Pasquarelli-do-Nascimento G, Correa R, das Neves Almeida R, de Oliveira Santos I, Prado PS, Picolo V, de Bem AF, Pizato N, Magalhaes KG. 2019. Potential neuroprotective and anti-inflammatory effects provided by omega-3 (DHA) against Zika virus infection in human SH-SY5Y cells. Sci Rep 9:20119. 10.1038/s41598-019-56556-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Natekar JP, Rothan HA, Arora K, Strate PG, Kumar M. 2020. Cellular microRNA-155 regulates virus-induced inflammatory response and protects against lethal West Nile virus infection. Viruses 12:9. 10.3390/v12010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen S, Cai F, Wang J, Yang Z, Gu C, Wang G, Mao G, Yan J. 2019. Salidroside protects SHSY5Y from pathogenic alphasynuclein by promoting cell autophagy via mediation of mTOR/p70S6K signaling. Mol Med Rep 20:529–538. 10.3892/mmr.2019.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hearst SM, Walker LR, Shao Q, Lopez M, Raucher D, Vig PJ. 2011. The design and delivery of a thermally responsive peptide to inhibit S100B-mediated neurodegeneration. Neuroscience 197:369–380. 10.1016/j.neuroscience.2011.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vieira J, O’Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 50.Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest 113:124–136. 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2:342–346. 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 52.Wang C-Y, Sugden B. 2004. New viruses shake old paradigms. J Clin Invest 113:21–23. 10.1172/JCI20662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciufo DM, Cannon JS, Poole LJ, Wu FY, Murray P, Ambinder RF, Hayward GS. 2001. Spindle cell conversion by Kaposi’s sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J Virol 75:5614–5626. 10.1128/JVI.75.12.5614-5626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shipley MM, Mangold CA, Kuny CV, Szpara ML. 2017. Differentiated human SH-SY5Y cells provide a reductionist model of herpes simplex virus 1 neurotropism. J Virol 91:e00958-17. 10.1128/JVI.00958-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chandriani S, Ganem D. 2007. Host transcript accumulation during lytic KSHV infection reveals several classes of host responses. PLoS One 2:e811. 10.1371/journal.pone.0000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. 2003. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol Mol Biol Rev 67:175–212. 10.1128/mmbr.67.2.175-212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scholz BA, Harth-Hertle ML, Malterer G, Haas J, Ellwart J, Schulz TF, Kempkes B. 2013. Abortive lytic reactivation of KSHV in CBF1/CSL deficient human B cell lines. PLoS Pathog 9:e1003336. 10.1371/journal.ppat.1003336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goncalves PH, Ziegelbauer J, Uldrick TS, Yarchoan R. 2017. Kaposi sarcoma herpesvirus-associated cancers and related diseases. Curr Opin HIV AIDS 12:47–56. 10.1097/COH.0000000000000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Myoung J, Ganem D. 2011. Infection of primary human tonsillar lymphoid cells by KSHV reveals frequent but abortive infection of T cells. Virology 413:1–11. 10.1016/j.virol.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellison TJ, Kedes DH. 2014. Variable episomal silencing of a recombinant herpesvirus renders its encoded GFP an unreliable marker of infection in primary cells. PLoS One 9:e111502. 10.1371/journal.pone.0111502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ross RA, Spengler BA, Biedler JL. 1983. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J Natl Cancer Inst 71:741–747. [PubMed] [Google Scholar]
- 62.Jeffery HC, Wheat RL, Blackbourn DJ, Nash GB, Butler LM. 2013. Infection and transmission dynamics of rKSHV.219 in primary endothelial cells. J Virol Methods 193:251–259. 10.1016/j.jviromet.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanehisa M, Goto S, Kawashima S, Nakaya A. 2002. The KEGG databases at GenomeNet. Nucleic Acids Res 30:42–46. 10.1093/nar/30.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57:289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S6; Table S1 to S3. Download JVI.00003-21-s0001.pdf, PDF file, 840 KB (839.5KB, pdf)
Data Availability Statement
All RNAseq data have been uploaded to the GEO database with the accession number GSE173342.