

ABSTRACT
Acinetobacter baumannii is a poorly understood bacterium capable of life-threatening infections in hospitals. Few antibiotics remain effective against this highly resistant pathogen. Development of rationally designed antimicrobials that can target A. baumannii requires improved knowledge of the proteins that carry out essential processes allowing growth of the organism. Unfortunately, studying essential genes has been challenging using traditional techniques, which usually require time-consuming recombination-based genetic manipulations. Here, we performed saturating mutagenesis with dual transposon systems to identify essential genes in A. baumannii, and we developed a CRISPR interference (CRISPRi) system for facile analysis of these genes. We show that the CRISPRi system enables efficient transcriptional silencing in A. baumannii. Using these tools, we confirmed the essentiality of the novel cell division protein AdvA and discovered a previously uncharacterized AraC family transcription factor (ACX60_RS03245) that is necessary for growth. In addition, we show that capsule biosynthesis is a conditionally essential process, with mutations in late-acting steps causing toxicity in strain ATCC 17978 that can be bypassed by blocking early-acting steps or activating the BfmRS stress response. These results open new avenues for analysis of essential pathways in A. baumannii.
IMPORTANCE New approaches are urgently needed to control A. baumannii, one of the most drug-resistant pathogens known. To facilitate the development of novel targets that allow inhibition of the pathogen, we performed a large-scale identification of genes whose products the bacterium needs for growth. We also developed a CRISPR-based gene knockdown tool that operates efficiently in A. baumannii, allowing rapid analysis of these essential genes. We used these methods to define multiple processes vital to the bacterium, including a previously uncharacterized gene regulatory factor and export of a protective polymeric capsule. These tools will enhance our ability to investigate processes critical for the essential biology of this challenging hospital-acquired pathogen.
KEYWORDS: Acinetobacter baumannii, CRISPR, Tn-seq, essential genes, gene knockdown
INTRODUCTION
The Gram-negative bacterium Acinetobacter baumannii is among the most difficult to treat pathogens causing diseases in hospitals. A. baumannii is an important cause of pneumonia and bloodstream infections and is associated with outbreaks in health care environments. The microorganism has rapidly evolved resistance to a wide variety of antimicrobials, leaving few therapeutic options for infected patients. Some strains show resistance to all available antibiotics, including carbapenems and the last-line polymyxins, rendering them exceedingly difficult, if not impossible, to treat (1, 2). There is an urgent need for new antimicrobials that can target the pathogen.
Devising novel strategies to target and attack A. baumannii requires that we understand proteins that have essential cell functions. Unfortunately, much remains unknown about fundamental processes in Acinetobacter. The genus has diverged from other Gammaproteobacteria and lacks sequence orthologs of several important proteins involved in key pathways, including cell wall synthesis, cell division, stress responses, and transcriptional regulation (3, 4). Orphan or hypothetical proteins may have evolved to mediate these processes in unique ways in Acinetobacter. A number of such proteins were recently identified through functional genomics examination of transposon mutant drug susceptibility phenotypes (5). While this work established functional connections between important biological pathways and uncharacterized proteins, it was designed for analysis of mostly nonessential genes. Phenotypes linked to essential genes, including those required by the organism for growth in standard nutrient medium, have yet to be systematically examined in A. baumannii, although they will likely provide much needed insights into pathways enabling growth, stress resistance, and persistence.
Although a number of tools allow genetic analysis in A. baumannii (6, 7), studying essential genes is usually an inefficient process. Such genes are typically analyzed by using conditional alleles, such as temperature-sensitive mutants or variants in which transcription control elements have been substituted by an inducible promoter (8). Engineering mutants in this fashion, however, is time-consuming and not amenable to scaling for high-throughput analysis. An alternative approach uses the highly transformable, nonpathogenic relative Acinetobacter baylyi to examine terminal phenotypes after direct allelic exchange of essential gene deletions (9, 10). The approach enables a direct view into the consequences of complete gene loss, but such deletions do not allow tunability of gene expression, and analogies with A. baumannii must be verified.
CRISPR interference (CRISPRi) is a recently described method for easily programmable gene knockdown in a variety of organisms (11). The most widely used CRISPRi systems employ a nuclease-deficient variant of the Streptococcus pyogenes Cas9 enzyme (dCas9) that is guided to a target gene by a single guide RNA (sgRNA), a hybrid molecule that incorporates Cas9 scaffolding and DNA targeting sequences (12). Recognition of target DNA depends on both the sgRNA and a protospacer adjacent motif (PAM) bordering the DNA sequence being targeted. The bound dCas9-sgRNA complex represses expression of the target gene by sterically hindering transcriptional initiation or elongation (12). A mobilizable CRISPRi system has been developed for use with several pathogens, including A. baumannii (13), but the reported knockdown efficiency (∼10-fold) is likely to be insufficient for analysis of highly expressed genes in the organism.
In this article, we present a comprehensive set of candidate essential genes in A. baumannii identified through high-density transposon mutagenesis and the development of a CRISPRi system for efficient analysis of these genes. We used these tools to confirm the essentiality of a recently identified orphan protein functioning in cell division, AdvA, as well as a previously uncharacterized protein belonging to the AraC family of transcriptional activators. In addition, we demonstrate the conditional essentiality of late steps in the biosynthesis of capsular polysaccharides. This work identifies new sites of vulnerability specific to A. baumannii and lays the foundation for future large-scale studies of essential gene function in the pathogen.
RESULTS
Identification of candidate essential genes in Acinetobacter baumannii by Tn-seq.
To determine gene essentiality across the A. baumannii genome, we performed saturating random transposon mutagenesis in strain ATCC 17978 and identified mutations that allow colonies to form on rich medium. To this end, we used two independent transposition systems that enable random insertional mutagenesis at high efficiency at different sites in the genome: a Himar1 mariner transposon system (5), which generates insertions at TA dinucleotides (14, 15), and a Tn10-altered target specificity (Tn10-ATS) system, which generates insertions effectively at random due to relaxed site specificity (14, 16). We generated two separate, highly saturated mutant libraries from ∼550,000 and ∼300,000 mutant colonies with each system, respectively (see Materials and Methods). We next used massively parallel sequencing of transposon-genome junctions (Tn-seq) to identify the location of transposon insertions within the libraries. Genomic DNA immediately adjacent to transposon insertions was amplified, enumerated by the Illumina platform, and mapped within the chromosome. With the mariner library, insertions were mapped to 174,238 unique TA sites out of a total of 269,711 possible sites (see Materials and Methods), equivalent to 64.6% of sites hit. With the Tn10-ATS library, insertions were mapped to 98,462 unique chromosomal positions. A total of 368,173 mutants with distinct chromosomal transposon insertions were represented across both libraries, equivalent to an average of one insertion approximately every 10 bp.
To identify essential genes, these Tn-seq data sets were analyzed independently using Bayesian and Gumbel methods with the TRANSIT software package (17). These methods identify genes with unusually long, consecutive stretches of potential insertion sites lacking insertions. The probability of long gaps occurring by chance is then calculated by a Bayesian (with mariner) or non-Bayesian (with Tn10-ATS) analysis of the Gumbel distribution (17, 18). With the mariner library, 392 genes passed the posterior probability threshold for essentiality, and 79 genes were called “uncertain” due to having a probability of essentiality not exceeding this threshold (see Data Set S1 in the supplemental material). With Tn10-ATS, 476 genes were called essential. Genes predicted to have high probability of essentiality with both transposition systems represent the most reliable candidates for being essential in the organism, so we analyzed the overlap between hits with both systems, including the essential and uncertain calls with mariner and the essential calls with Tn10-ATS. This identified 372 genes as hits with both systems (Fig. 1), and we define these 372 genes as the candidate essential gene set in A. baumannii (Data Set S1). This set of genes corresponded well with candidate essential genes identified in previous Tn-seq studies with the same strain (19) and with the unrelated, multidrug-resistant (MDR) strain AB5075 (20), with 72% (267 genes) showing essentiality across all three studies (see Fig. S1 in the supplemental material).
FIG 1.

Candidate essential genes in A. baumannii identified by Tn-seq with two transposon systems. The Venn diagram shows candidate essential genes identified in ATCC 17978 by mariner transposition (471 genes called essential or uncertain [blue circle]) or by the Tn10-ATS system (476 genes called essential [gray circle]). The intersection of the genes sets had 372 genes, representing the candidate essential gene set using both systems.
CRISPR interference system for gene knockdown in A. baumannii.
To facilitate the analysis of the candidate essential genes, we developed a CRISPRi system in A. baumannii. The system comprises an anhydrotetracycline (aTc)-inducible dcas9 gene inserted at single copy in the chromosomal attTn7 site downstream of the glmS locus (21), and a constitutive sgRNA module via a high-copy-number plasmid derived from pWH1266 (22) (Fig. 2A). In an initial test of gene knockdown with the CRISPRi system in strain ATCC 17978, we targeted the constitutive β-lactamase ADC, levels of which can be determined by measuring the rate of hydrolysis of its specific chromogenic substrate nitrocefin (23). We designed an sgRNA containing 24 nucleotides (nt) targeting the nontemplate (NT) strand of adc starting 90 bp downstream of its predicted transcription start site (TSS) (Fig. 2B; see Table S1 in the supplemental material) (24). We observed reduction of β-lactamase levels by almost 2-fold in the absence of dcas9 induction with this construct compared to a nontargeting control plasmid (Fig. 2C), similar to the low levels of repression in the absence of inducer seen with other CRISPRi platforms (25, 26). To induce dcas9 and effect gene silencing with the CRISPRi system, we have found that aTc concentrations between 50 and 200 ng/ml result in robust knockdown with targeting strains, while having minimal effect on growth with control strains. After dcas9 was induced by aTc (100 ng/ml) for 2 h, β-lactamase synthesis decreased by approximately 30-fold compared to that of the control, approaching the background level seen with deletion of the adc gene (Fig. 2C). As expected, adc knockdown by CRISPRi increased susceptibility to ampicillin, a substrate of the ADC enzyme (23, 27). dcas9 induction completely blocked growth of the strain harboring the adc-targeting sgRNA in the presence of a dose of the drug that was subinhibitory with control cells, with partial growth inhibition observed in the absence of induction (Fig. 2D). These results show that efficient gene knockdown can be achieved in A. baumannii, enabling investigation of gene-phenotype relationships.
FIG 2.

CRISPRi system for efficient knockdown of gene expression in A. baumannii. (A) The CRISPRi system comprises an aTc-inducible dcas9 (driven by the tet promoter) at the chromosomal attTn7 locus, and a high-copy-number plasmid-based constitutive sgRNA (driven by the J23119 promoter). oripWH, pWH1266 origin of replication; oripBR, pBR322 origin of replication. (B to D) Efficient CRISPRi knockdown of the adc β-lactamase. (B) The diagram shows the position in the adc locus targeted by sgRNAadc (vertical arrow) relative to the predicted adc mRNA TSS (arrowhead) (24). (C) β-Lactamase was measured in sonicates of the indicated strain grown with or without aTc (100 ng/ml). Bars show mean ± standard deviation (SD) (n = 2). (D) adc knockdown enhances susceptibility to ampicillin (AMP). YDA004 (ATCC 17978 tetP-dcas9) with sgRNAadc or control plasmid was cultured in microplates in the presence or absence of aTc (100 ng/ml) and/or AMP (16 μg/ml), and growth was monitored by OD measurements. Symbols indicate geometric mean, and area-filled dotted bands indicate SD (n = 3). Where not visible, the SD is within the confines of the symbol. “control” refers to pYDE007 (a nontargeting control plasmid).
We next used CRISPRi to examine essential genes in A. baumannii. We focused our analysis on genes that have evolved in a unique manner in this microorganism compared to other bacteria, as the encoded proteins represent potential targets for selectively inhibiting A. baumannii. We first focused on AdvA, a newly identified protein that has an essential function in cell division and lacks close orthologs in most bacteria outside Acinetobacter (5). The protein is connected by its drug susceptibility phenotypic signature to proteins that coordinate chromosome segregation with cell division (5). Alongside AdvA, we analyzed the highly conserved initiator of cell division, FtsZ. Viable transposon insertions were almost completely undetectable in ftsZ, as predicted for this essential gene, and were detected only within a narrow, central region of advA, in agreement with previous findings with lower-density mutant banks (Fig. 3A) (5). These insertions likely contributed to conflicting AdvA essentiality calls (mariner predicted essential and Tn10-ATS predicted nonessential [Data Set S1]). The region permissive of transposon mutation may represent a nonessential linker between two essential AdvA domains. Consistent with this notion, the permissive region was downstream of a domain having predicted structural homology with two-component system sensor kinases (5). With a CRISPRi guide targeting ftsZ (NT strand, starting 79 bp downstream of the predicted TSS [28]) (Table S1), uninduced cells grew rapidly as short rods characteristic of WT A. baumannii, while dcas9 induction with aTc blocked growth and resulted in a filamentous morphology after 3 h (Fig. 3B and C). We have shown previously using a conditional allele that advA deficiency inhibits growth (5). To confirm this phenotype by CRISPRi, we designed an sgRNA construct that targeted the NT strand of its coding region starting 132 bp downstream of the nearest predicted TSS (Fig. 3A; Table S1). The targeted region was at least 60 bp away from the predicted TSS of the divergently transcribed neighboring gene, serB, outside the region bound by the initial RNA polymerase (RNAP) complex (−55 to +20 from a TSS); therefore, this site should not block serB transcription (12). In agreement with this prediction, quantitative reverse transcription-PCR (qRT-PCR) analysis showed that advA transcriptional repression by this sgRNA was efficient and specific. Compared to that in the control (nontargeting) strain, advA mRNA levels were lowered approximately 7-fold absent dcas9 induction and 200-fold with dcas9 induction (Fig. 3D); in contrast, serB transcript levels were not significantly affected with or without dcas9 induction (Fig. 3D). As with ftsZ, this advA-targeting sgRNA resulted in growth inhibition (Fig. 3E) and striking filamentation after 3 h of dcas9 induction (Fig. 3F). While growth inhibition occurred less rapidly and at a higher cell density than with ftsZ, advA knockdown cultures were completely blocked for growth when back diluted to a lower cell density in fresh induction medium (Fig. 3E). With the same level of dcas9 induction, no significant effect on growth, morphology, or advA transcription was observed with a strain having the nontargeting control sgRNA (see Fig. S2A to C in the supplemental material). CRISPRi targeting of advA also caused a growth defect in M9 minimal medium containing either glucose-Casamino Acids or succinate, indicating that the phenotype was not dependent on rich broth medium (Fig. S2D). These results support our previous genetic analysis demonstrating an essential role for AdvA in A. baumannii cell replication (5).
FIG 3.
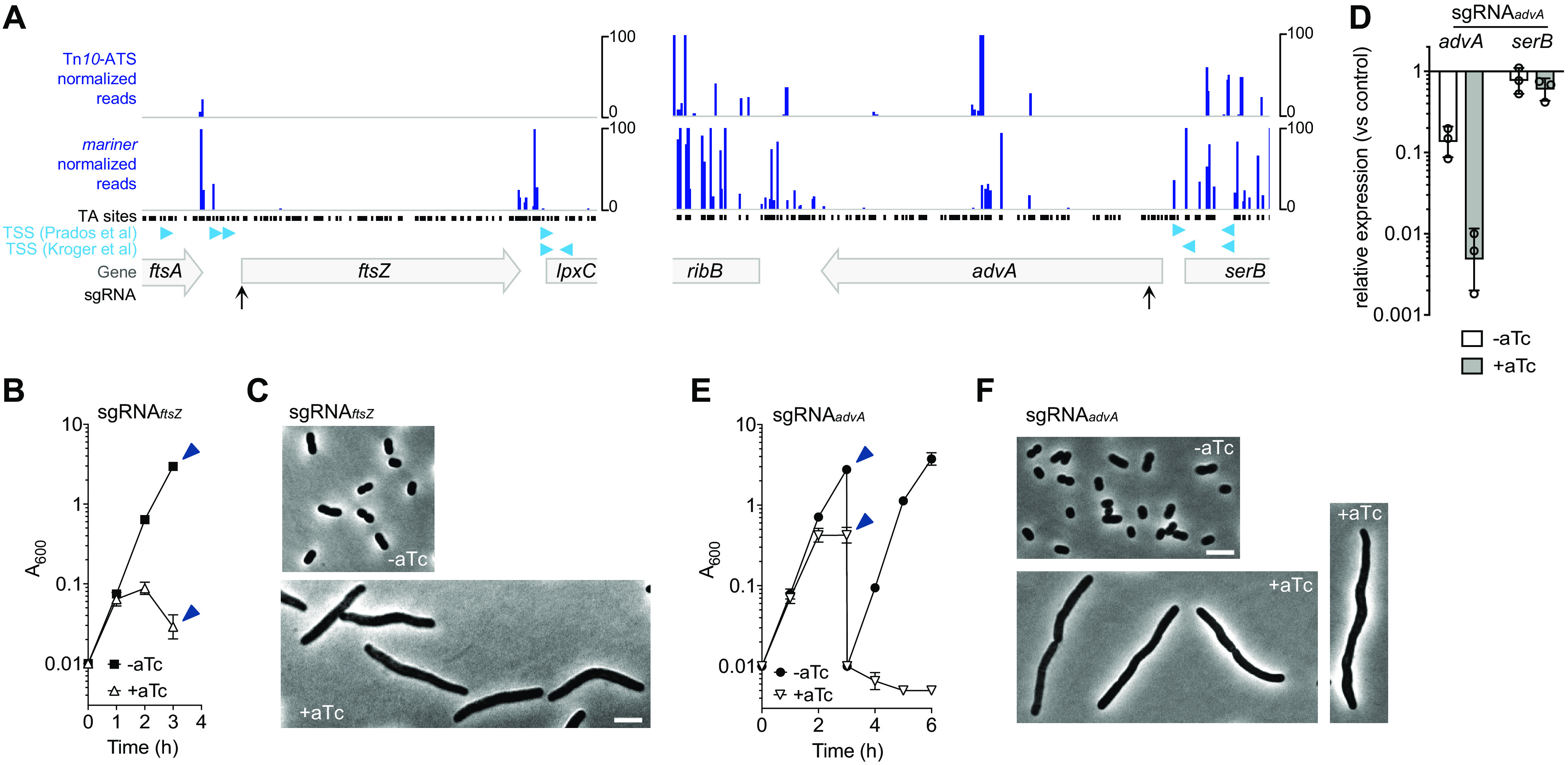
CRISPRi knockdown of ftsZ and advA blocks growth and cell division. (A) Transposon mutations in ftsZ and in most regions of advA were not detectable within Tn-seq pools. Normalized Tn-seq read counts were plotted according to transposon insertion position in mariner and Tn10-ATS libraries. Black points show locations of potential mariner insertion sites (TA dinucleotides). Predicted TSSs are as described in references 24 (Kroger et al.) and 28 (Prados et al.). Positions targeted by sgRNAs are indicated as in Fig. 2. (B) YDA004 with sgRNAftsZ was cultured with or without 200 ng/ml aTc, and growth was monitored. Data points show geometric mean A600 ± SD (n = 3). At 3 h, samples were collected (arrowheads) and imaged by phase-contrast microscopy (C). (D) Efficient and specific CRISPRi knockdown of advA. YDA004 cells with sgRNAadvA or the nontargeting control were cultured for 2 h with aTc (200 ng/ml) or no inducer, and advA and serB transcription levels relative to that in the untreated control were analyzed via qRT-PCR. Bars show geometric mean ± SD (n = 3). In unpaired t tests comparing sgRNAadvA with the control, advA transcription showed a significant decrease with (P = 0.008) or without (P = 0.01) aTc; serB transcription showed no significant change (P > 0.07). (E and F) AdvA is essential in LB. Growth (E) and morphology (from samples indicated by arrowheads [F]) were analyzed as in panels B and C. Data points show geometric mean A600 ± SD (n = 3). Scale bars = 5 μm.
ACX60_RS03245, encoding a predicted transcription factor, is an essential gene.
Transcriptional regulation in A. baumannii shows some unusual features, including a small number of sigma factors (4), a large number of transcriptional regulators that jointly control virulence and antibiotic resistance (23, 29, 30), and a reliance on the RNA-binding protein Hfq for growth (31, 32). Given these features and its divergence from other Gammaproteobacteria, we predicted that the pathogen may encode unidentified essential regulators of transcription. As shown in Table S2 in the supplemental material, we identified 13 candidate essential regulators from our Tn-seq data sets. These included the housekeeping sigma factor (rpoD), the heat shock sigma factor (rpoH) (33), and hfq, as expected; three loci encoding proteins with LexA or Cro/CI homology that were internal to prophages (14) and likely suppress lytic phage replication; and several previously uncharacterized putative transcription factors belonging to the TetR or AraC family (Table S2). In addition, we identified envZ and ompR, which form a two-component system that is nonessential in strain AB5075 (34), as candidate essential transcriptional regulators in ATCC 17978. Our Tn-seq data are thus able to define candidate regulatory proteins that may exert control over essential aspects of A. baumannii growth.
We focused our analysis on ACX60_RS03245 (here referred to as RS03245), a previously uncharacterized candidate essential gene encoding a predicted AraC family transcription factor. RS03245 is conserved across A. baumannii isolates (35) and is also a candidate essential hit in previous Tn-seq analyses (19, 20). AraC family regulators typically control catabolism of sugars and amino acids, stress responses, and production of virulence factors (36, 37), and it is unusual for a protein of this family to be essential in rich medium. The domain architecture of the RS03245-encoded protein is similar to that of other members of the AraC regulator family (see Fig. S3A in the supplemental material). In addition, structural homology modeling (38) predicted a relationship with CdpR, a nonessential AraC family regulator that controls quorum sensing and virulence in Pseudomonas aeruginosa (39), despite low sequence identity (22%) (Fig. S3B and C). Transposon insertions were undetectable in the RS03245 locus (Fig. 4A), consistent with the encoded protein playing an important role in controlling processes essential for A. baumannii growth.
FIG 4.
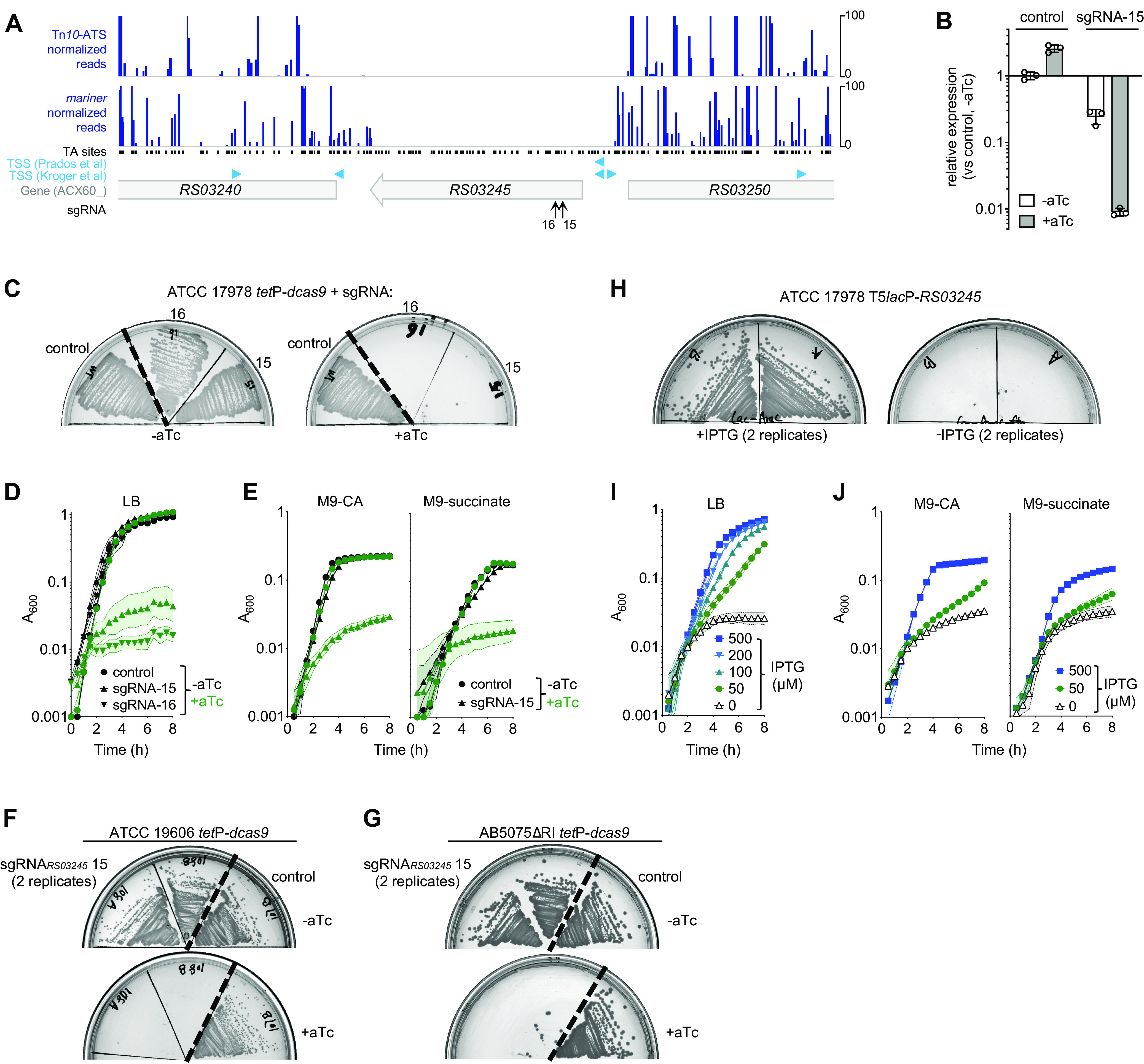
ACX60_RS03245 (RS03245) is an essential predicted transcription factor. (A) Transposon mutations within RS03245 were not detectable in Tn-seq pools. Tn-seq read counts and annotated sites in the RS03245 locus region are shown as in Fig. 3. Predicted TSSs are as described in references 24 (Kroger et al.) and 28 (Prados et al.). (B) Efficient CRISPRi knockdown of RS03245. YDA004 cells with sgRNA targeting RS03245 (sgRNA-15) or control plasmid were cultured for 2 h without or with aTc (50 ng/ml), and RS03245 expression relative to the untreated control was analyzed via qRT-PCR. Bars show geometric mean ± SD (n = 3). P < 0.0008 in unpaired t tests comparing sgRNA-15 versus control under each treatment condition. (C to G) CRISPRi knockdown of RS03245 blocks growth in different media and with multiple strain backgrounds. YDA004 with the indicated sgRNA or control plasmid was grown on LB agar (C) or the indicated liquid medium in microplates (D and E) without or with aTc (200 ng/ml in panel C and 100 ng/ml in panels D and E). Data points show geometric mean A600 ± SD (n ≥ 2). ATCC 19606 tetP-dcas9 (F) or AB5075 ΔRI tetP-dcas9 (G) harboring sgRNA-15 or control plasmid was streaked on LB agar without or with aTc (200 ng/ml) and imaged after overnight incubation. (H to J) Bacteria containing IPTG-regulated RS03245 depend on IPTG for growth. JBA58 (T5lacP-RS03245) was grown on LB agar medium without or with IPTG (1 mM) (H) or in microplates with the indicated liquid medium having graded concentrations of IPTG (I and J). Data points show geometric mean ± SD (n ≥ 3).
We used CRISPRi to examine the essentiality of RS03245 predicted by Tn-seq. Knockdown of RS03245 was highly efficient using an sgRNA targeting the 5′ end of the coding region near the predicted TSS (sgRNA-15) (Fig. 4A; Table S1). In the absence of dcas9 induction, RS03245 transcript levels were decreased 4-fold compared to the nontargeting control, while induction for 2 h caused transcript levels to decrease by more than 100-fold (Fig. 4B). When targeting RS03245 with sgRNA-15 or with a second, different sgRNA (sgRNA-16) (Fig. 4A; Table S1), CRISPRi induction blocked growth on solid (Fig. 4C) and liquid (Fig. 4D) LB media. Growth was also blocked in M9 minimal medium containing either glucose-Casamino Acids or succinate (Fig. 4E; Fig. S3D), indicating that RS03245 essentiality was a general phenotype not specifically depending on rapid growth in rich medium. The inducer concentrations used in the experiments described above (aTc at 50 to 200 ng/ml) were likely at or near saturation, as a titration experiment showed that a dose of inducer as low as 1.56 ng/ml was sufficient to inhibit colony formation on LB agar by sgRNA-15 (Fig. S3E). These results underline the sensitivity of A. baumannii to depletion of the RS03245 protein.
We confirmed the above phenotypes by (i) using CRISPRi in two different strain backgrounds and (ii) analyzing RS03245 essentiality with a completely different genetic approach (a conditional allele) that does not depend on the CRISPRi machinery. First, we moved the tetP-dcas9 module to the attTn7 site of A. baumannii strains ATCC 19606 and AB5075 ΔRI, a derivative of AB5075 lacking two large resistance islands (40). dcas9 induction in the presence of an RS03245-targeting guide (sgRNA-15) but not the control construct completely blocked colony formation on LB agar medium with both strain backgrounds (Fig. 4F and G), indicating that RS03245 has an important function independent of strain background. This is supported by the finding that RS03245 was an essential gene candidate with AB5075 based on Tn-seq (20). Second, we engineered a derivative of ATCC 17978 in which RS03245 expression was IPTG (isopropyl-β-d-thiogalactopyranoside) dependent. This was accomplished by replacing the RS03245 promoter with a lacIq-T5lacP control module using homologous recombination. This mutant (JBA58) depended on IPTG for growth on LB agar (Fig. 4H), in liquid LB (Fig. 4I), as well as in minimal M9 medium (Fig. 4J), with growth increasing with increasing IPTG concentration (Fig. 4I and J). These phenotypes closely resembled the effects of CRISPRi knockdown of RS03245. Introducing a constitutive copy of RS03245 (as fusion to either green fluorescent protein [GFP] or 3×FLAG epitope) into JBA58 restored the ability to grow in the absence of IPTG (Fig. S3F), indicating that the IPTG-dependent growth phenotypes could be attributed solely to control of RS03245 expression. Together, these results establish RS03245, encoding a predicted AraC family transcription factor, as a novel essential gene in A. baumannii.
Conditional essentiality of late-stage capsule biosynthesis proteins.
The consequences of blocking synthesis of capsule, a key virulence factor, on the physiology of A. baumannii are incompletely understood (3). Based on bioinformatics analyses and homology with well-studied systems in other organisms (41, 42), capsule biosynthesis across diverse A. baumannii isolates is mediated by a Wzy-dependent pathway in which activated sugars (Fig. 5A, steps encoded by genes shaded purple and blue) are utilized by sequential glycosyltransferases (Fig. 5A, encoded by genes shaded green) to build an oligosaccharide repeat unit on an undecaprenyl phosphate (Und-P) lipid carrier (43). The repeat units are then flipped, polymerized, and exported to the surface (3) (Fig. 5A, steps encoded by genes in orange). In addition to preventing the formation of structural capsule, defects in this pathway occurring after the initial glycosyltransferase step (Fig. 5A, pink) may have toxic consequences if they generate stalled intermediates that sequester Und-P, which is essential to peptidoglycan synthesis (3, 44, 45). In our high-density Tn-seq analysis, 7 out of 9 genes encoding enzymes predicted to act after the ItrA initiating glycosyltransferase were candidate essential genes (gtr7, gtr8, wzx, wzy, wza, wzb, and wzc [Data Set S1]). This analysis is consistent with the model that lesions in late steps in capsule synthesis acting after a committed step are lethal in ATCC 17978 due to the generation of dead-end intermediates.
FIG 5.
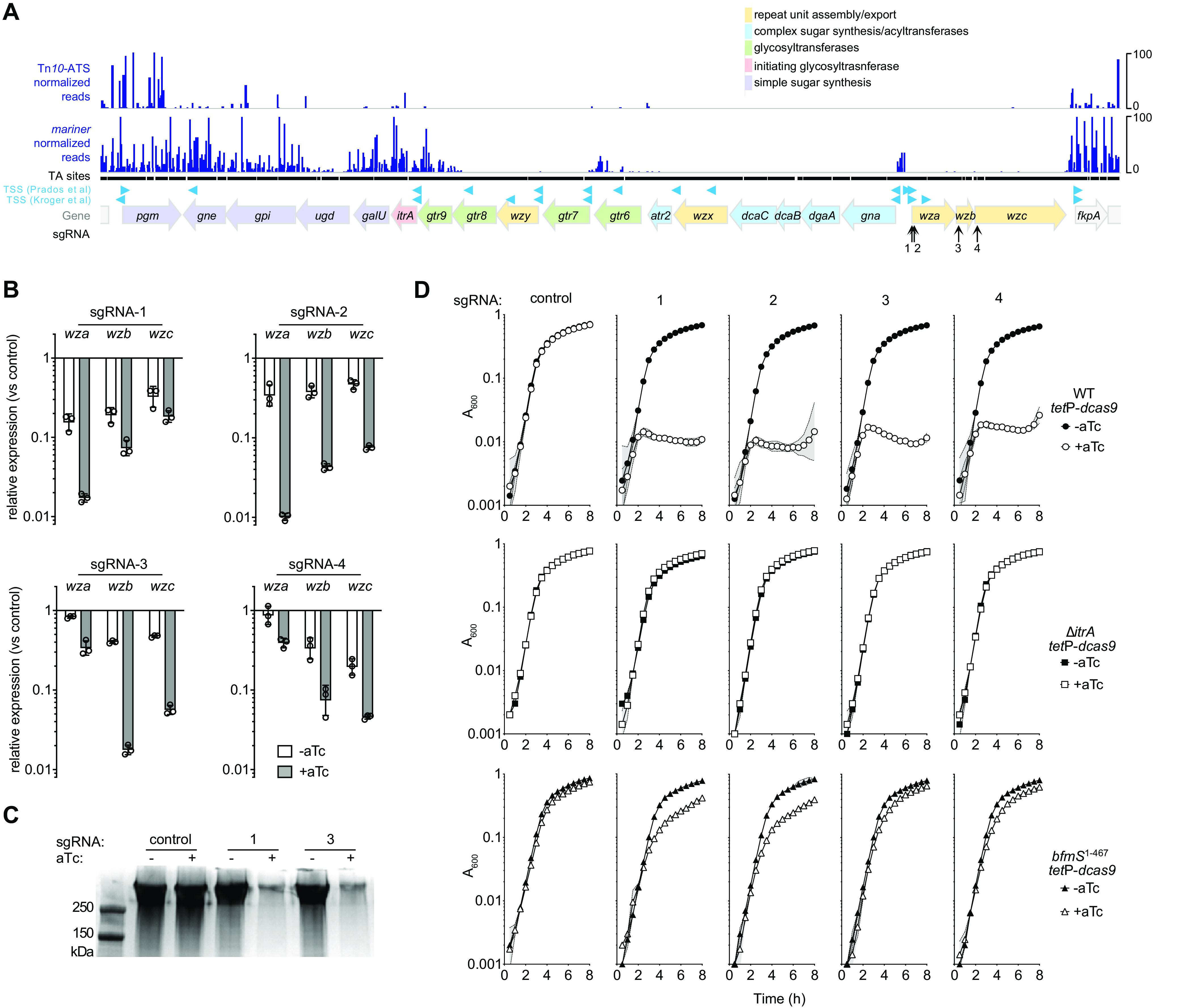
Conditional essentiality of capsule biosynthesis. (A) Location and Tn-seq read abundance of insertions within the ATCC 17978 K locus. Tn-seq read counts and annotated sites are shown as in Fig. 3; black points indicating individual TA sites appear merged due to the wide view (∼25 kb). Predicted TSSs are as described in references 24 (Kroger et al.) and 28 (Prados et al.). Several putative antisense promoters identified in reference 23 are not shown. (B) CRISPRi in the ATCC 17978 ΔitrA background with the indicated sgRNA targeting different points within the wza-wzb-wzc operon efficiently silences the cotranscribed genes. wza, wzb, and wzc transcript levels after growth with 0 or 200 ng/ml aTc were analyzed via qRT-PCR. Bars show geometric mean fold change versus untreated control ± SD (n = 3). In unpaired t tests comparing the control to each sgRNA with aTc treatment, P < 0.005 with all probes and sgRNAs; without aTc, P < 0.04 with all probes and sgRNAs, except for wza and sgRNA-4 (P = 0.51). (C) CRISPRi knockdown of wza-wzb-wzc inhibits production of capsule in WT ATCC 17978. Capsular polysaccharides were analyzed by the SDS-PAGE–alcian blue method from bacteria harboring the indicated sgRNAs and grown with 0 or 200 ng/ml aTc. (D) CRISPRi knockdown of late-stage capsule export blocks growth in the ATCC 17978 WT (top), but growth inhibition is suppressed with itrA deletion (middle) or bfmS mutation (bottom). Strains containing the indicated sgRNA were cultured with 0 or 200 ng/ml aTc in microplates. Data points show geometric mean A600 ± SD (n ≥ 2). The ΔitrA tetP-dcas9 strain was JBA9; the bfmS1–467 tetP-dcas9 strain was JBA8.
We used CRISPRi to test this model and assess whether blocking of the early step, by ItrA, in which Und-P acceptors are likely dedicated to capsule, can relieve toxicity. First, we examined the knockdown efficiency of sgRNAs that target the late-stage capsule export module encoded by the cotranscribed genes wza-wzb-wzc (Fig. 5A). We designed 4 distinct sgRNA constructs targeting different positions within this operon (Fig. 5A, vertical arrows; Table S1). While CRISPRi has known polar effects (46), and these sgRNAs are predicted to modulate expression of the entire wza-wzb-wzc operon, each gene encodes a part of the same complex dedicated to capsule export and high-level polymerization (47). Blocking operon transcription by CRISPRi should thus enable targeted examination of this process. To measure the efficiency of transcriptional interference by the 4 sgRNAs, we took advantage of the acapsular ATCC 17978 ΔitrA strain background (48) (see Table S4 in the supplemental material). As we will describe in more detail below, this mutant allows normal growth despite blocking late stages of capsule synthesis, consistent with the proposed model. In the presence of dcas9 induction, each sgRNA was effective at repressing the wza-wzb-wzc operon in this strain, with differences in the level of repression of each individual gene dependent on the site of dCas9 delivery (Fig. 5B). With each sgRNA, the magnitude of the repression was greatest for genes whose 5′ regions were directly targeted (Fig. 5B). Repression of operon genes downstream of the target site was weaker but significant (Fig. 5B, sgRNAs 1 to 3). With sgRNAs 3 and 4, which targeted the interior of the operon, genes upstream of the target site were also repressed, albeit more weakly than the targeted gene (Fig. 5B); this has been referred to as reverse polarity (25, 46). In the absence of inducer, minimal repression (6-fold or lower) was seen with the sgRNAs (Fig. 5B). No significant effect on transcription was observed with the parallel nontargeting control in the absence or presence of inducer (see Fig. S4A in the supplemental material). Using two of the sgRNAs (1 and 3), we verified that production of capsular polysaccharides by wild-type (WT) cells was also efficiently blocked after CRISPRi induction but not in the absence of inducer (Fig. 5C) (Materials and Methods). Thus, CRISPRi targeting of the wza-wzb-wzc operon results in efficient blockade of capsule synthesis, with different patterns of repression of the encoding genes depending on the sgRNA used.
Consistent with these results and our Tn-seq analysis, each of the four sgRNAs targeting wza-wzb-wzc blocked growth of WT ATCC 17978 only when dcas9 was induced (Fig. 5D, circles). In contrast to the situation with ATCC 17978, several other A. baumannii strains, including AB5075, tolerate mutation of late-stage capsule synthesis genes without showing defective in vitro growth (49, 50). Consistent with these findings, CRISPRi knockdown of wza-wzb-wzc in AB5075 ΔRI did not impede growth, despite dCas9 induction and regardless of the site of its recruitment within the operon (Fig. S4C and D; Table S1). Targeting wza-wzb-wzc by CRISPRi resulted in the expected rough, translucent colony morphology associated with capsule loss, consistent with efficient knockdown (Fig. S4E). CRISPRi of the essential gene RS03245 blocked AB5075 ΔRI growth when cultured in parallel on the same induction medium, as expected, serving as a positive control (Fig. S4E). These results support our previous observation that the lethal effect of late-stage capsule assembly block is strain dependent (3).
To test the model that late-step capsule synthesis defects in ATCC 17978 can be tolerated when the predicted committed step in Und-P usage is prevented (by blocking ItrA), we repeated the CRISPRi induction experiment using the isogenic ATCC 17978 ΔitrA mutant. Strikingly, CRISPRi blocking of wza-wzb-wzc had no effect on growth in the absence of itrA (Fig. 5D, squares), despite transcriptional interference being highly effective under these conditions (Fig. 5B). The ability of the ΔitrA mutation to suppress the nonviability caused by late-stage capsule block was confirmed by isolating a ΔitrA Δwzc double mutant strain. Unlike the ATCC 17978 Δwzc single mutant which could not be successfully isolated from an ItrA+ background in the absence of compensatory mutations (48), the Δwzc mutant was easily isolated in a ΔitrA background, and the resulting double mutant had WT growth kinetics (Fig. S4B). Together these results indicate that the essentiality of late steps in capsule assembly can be bypassed when flux into the pathway is prevented.
We used CRISPRi to confirm an additional pathway allowing bypass of lethality associated with capsule production defects. Our previous work identified bfmS as a site of suppressor mutations that allowed growth of an ATCC 17978 Δwzc strain (48). BfmS is part of the BfmRS two-component system, and null mutations in bfmS cause augmented expression of envelope stress response genes and genes determining synthesis of envelope structures, including capsule and Und-P (23). These changes may enhance how cells cope with deleterious dead-end intermediates associated with capsule production defects. We used CRISPRi to confirm the suppressive interaction between bfmS and late-stage capsule block. After moving the CRISPRi system to an ATCC 17978 BfmS− strain background (bfmS1–467) (Table S4) (23, 48), guide RNAs 1 to 4 were tested for ability to inhibit growth in the absence and presence of dcas9 induction. As predicted and in contrast to WT (BfmS+) bacteria, bfmS1–467 cells tolerated CRISPRi targeting of wza-wzb-wzc despite dcas9 induction, although partial growth inhibition was observed compared to the control sgRNA (Fig. 5D, triangles). These results are consistent with envelope stress response activation providing a second pathway to allow toleration of late-stage capsule synthesis defects.
DISCUSSION
In this study, we have defined a candidate essential gene set in A. baumannii, and we have established a CRISPRi tool for analysis of these genes. Our essential gene search was comprehensive and utilized two different global transposition systems based on unrelated classes of transposase enzymes. This approach provided a high level of saturation of the genome and allowed us to build a consensus set of candidate essential genes. This candidate essential gene set can guide the selection of targets in future studies aimed at dissecting the A. baumannii essential genome.
To target essential genes, we developed a CRISPRi knockdown system for A. baumannii. The system was controllable and efficient, with low-level (∼2- to 7-fold) decreases observed in the absence of dcas9 induction by aTc and effective shutdown of gene expression (up to 200-fold inhibition) with induction. Knockdown is likely to be titratable using dilutions of aTc, as indicated by the relationship of aTc concentration with colony formation by cells containing RS03245-targeting sgRNA (Fig. S3E). CRISPRi knockdown was as effective as classical allelic replacement techniques in allowing analysis of conditional growth phenotypes and was functional in multiple strain backgrounds (Fig. 4). The efficiency of our system is an advancement over a previous system that showed ∼10-fold knockdown with A. baumannii (13). The reasons for this difference are not clear but may relate to different promoter strengths driving dcas9 and sgRNA expression and/or different sgRNA copy number (on a high-copy-number plasmid versus on the chromosome). By using CRISPRi in combination with libraries of diverse guides, the platform described here should facilitate large-scale examination of terminal and hypomorphic phenotypes linked to essential genes (12, 25). Such studies have the potential to illuminate essential protein function and inform novel antimicrobial target development and would complement previous functional genomics studies that focused largely on nonessential genes in A. baumannii (5). In addition, the CRISPRi system may be useful for extended studies such as those in animal infection models. The stability of phenotypes generated by prolonged CRISPRi challenge is unclear, however, and may depend on the frequency at which escape variants arise. In this regard, with both the CRISPRi and conditional allele approaches targeting essential genes, colonies representing potential escape variants appeared on solid medium at low frequency (Fig. 4C, G, and H).
Using the essential gene candidates and the knockdown approach developed in this study, we analyzed a number of unique essential proteins in A. baumannii. These proteins represent a set of potential targets for narrow-spectrum therapies against the pathogen. We confirmed that AdvA, a unique cell division protein, is required for A. baumannii growth. We also identified a novel, essential predicted transcription factor, RS03245. Part of the core genome (35), RS03245 is one of 33 genes encoding AraC family transcription factors in A. baumannii but was the only one determined to be essential in this and previous Tn-seq studies (see Table S3 in the supplemental material). RS03245 depletion was associated with growth inhibition without a drop in optical density (OD), indicating that deficiency of the transcription factor does not cause lysis. The degree to which bacterial viability is affected by RS03245 deficiency and the mechanism of growth inhibition remain to be defined. By analogy with most AraC family proteins, which are transcriptional activators (36), RS03245 likely enhances transcription of genes contributing to one or more essential pathways, the identity of which we are currently investigating. In addition, we demonstrated the conditional essentiality of proteins that assemble and export capsule. Late steps in capsule biosynthesis are essential in ATCC 17978, unless suppressed by one of at least two pathways controlled by itrA or bfmS. This conditional essentiality mirrors that seen with defects in Wzy-dependent synthesis of capsule in Streptococcus pneumoniae (51, 52) and Escherichia coli (53), as well as of O antigen in E. coli (54). It also parallels the conditional toxicity of lipooligosaccharide (LOS) synthesis lesions in A. baumannii, in which defects in late-acting synthesis steps are lethal unless flux into the pathway is reduced (55–57). The molecular basis for toxicity and for the differential ability of A. baumannii strains to cope with capsule synthesis defects (3) is unclear. Understanding these processes in future studies may inform strategies to attack the pathogen’s protective envelope.
In summary, we have identified candidate essential genes in A. baumannii and developed a CRISPRi tool that facilitated rapid validation of the essentiality of several candidates of unique importance to the pathogen. The study of these critical proteins has the potential to inform strategies for attacking A. baumannii selectively. This work opens new avenues for research into the pathogen’s dependence on transcriptional control and envelope homeostasis for growth and survival. More broadly, the CRISPRi platform developed here should enhance both targeted and large-scale analyses of gene function and genetic interactions in the microorganism.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and antibiotics.
The bacterial strains used in this work are described in Table S4 in the supplemental material. A. baumannii strains were derivatives of ATCC 17978, unless otherwise noted. Bacteria were cultured in lysogeny broth (LB) (10 g/liter tryptone, 5 g/liter yeast extract, 10 g/liter NaCl), unless otherwise noted. M9 minimal medium (58) contained 0.4% glucose and 0.2% Casamino Acids or 0.2% succinate. Cultures were incubated at 37°C in flasks with shaking or in tubes on a roller drum. Where indicated, growth was in 96-well microplates incubated with shaking in a plate reader (Epoch 2 or Synergy H2M; Biotek). Growth was monitored by measuring absorbance at 600 nm (A600). LB agar was supplemented with antibiotics (carbenicillin [Cb] at 50 to 100 μg/ml with all strains except A. baumannii AB5075 ΔRI at 1,600 μg/ml, kanamycin [Km] at 10 to 20 μg/ml, or gentamicin [Gm] at 10 or 40 μg/ml) or sucrose as needed (Sigma-Aldrich).
Construction of transposon mutant libraries.
Mutagenesis with the mariner and Tn10-ATS transposon systems was performed by electroporation with pDL1100 and pDL1073, respectively (5, 14). Transformed cells were spread on membrane filters, allowed to recover on solid SOC, and enriched after transfer to selective solid LB-Km medium as described previously (5, 14). Colonies were lifted from filters by agitation in sterile phosphate-buffered saline (PBS), mixed with sterile glycerol (10%), aliquoted, and stored at −80°C. With mariner, colonies from 10 previously constructed mariner subpools (5), as well as 28 additional subpools constructed for this study, were analyzed in aggregate, representing a total of approximately 550,000 mutant colonies. With Tn10-ATS, colonies from 11 previously constructed subpools (14), as well as 12 additional subpools constructed for this study, were analyzed in aggregate, representing approximately 300,000 mutant colonies in total.
Tn-seq Illumina library preparation and sequencing.
Genomic DNA was extracted from samples (Qiagen DNeasy kit) and quantified by a SYBR green microtiter assay. Transposon-adjacent DNA was tagmented and amplified for Illumina sequencing using a modified Nextera DNA Library Prep method as described previously (14). Samples were multiplexed, reconditioned, and size selected (250 or 275 to 600 bp; Pippin HT) before sequencing (single-end 50 bp) using custom primers (5, 14) on a HiSeq2500 with high-output V4 chemistry at the Tufts University Genomics Core Facility.
Tn-seq data analysis.
Sequencing reads were quality filtered and clipped of adapters with BBDuk, collapsed with fastx_collapser, and mapped to the A. baumannii chromosome (NZ_CP012004) with Bowtie (5). To process the mariner data set into an input file for TRANSIT analysis, the coordinates of TA sites in the NZ_CP012004 genome that can be uniquely mapped to (i.e., not part of repeat regions) were identified, and mapped reads were tabulated according to these sites in wig format using custom python scripts. With the Tn10-ATS data set, mapped reads were tabulated in a wig file containing all chromosome coordinates using python. Read counts were normalized across all subpools within a data set (mariner or Tn10-ATS) by the TTR method with the TRANSIT software package (17), merged into a single wig file for each data set, and scaled such that median read coverages at nonzero insertion sites were similar between data sets. To determine gene essentiality with TRANSIT, the mariner data set was analyzed by the Gumbel method (parameters: ignore C-terminal 10%, sample size of 10,000, burn-in of 500, trim of 1, minimum read of 5), and Tn10-ATS was analyzed by the Tn5gaps method (parameters: ignore C-terminal 10%, minimum read of 5) (17). Orthologs in CP000521 (the ATCC 17978 genome file used in reference 19) and CP008706 (the AB5075-UW genome used in reference 20) were matched to NZ_CP012004 genes by using Mauve to identify positional orthologs (59) and by Boundary-Forest Clustering (60). Relationships of essential genes were analyzed via Venn diagrams using BioVenn (61). The Integrative Genomics Viewer (62) was used to visualize normalized Tn-seq read counts and unique TA insertion sites along the chromosome coordinates.
Molecular cloning and mutant construction.
The oligonucleotide primers and plasmids used in this study are listed in Table S5 and Table S4, respectively, in the supplemental material. All constructs containing cloned PCR products were verified by sequencing (Genewiz). A mini-Tn7 element containing dcas9 was constructed by PCR amplifying the tetR-tetP-dcas9-rrnB T1-T7 Te fragment from pdCas9 bacteria (Addgene 44249; a gift from Stanley Qi) with primers containing SpeI and PstI sites, cloning in the HincII site of pUC18, and subcloning in the SpeI and PstI sites of pUC18T-mini-Tn7 T-Gm (63) to generate pYDE009 (Cbr Gmr). The mini-Tn7 element of pYDE009 was moved into A. baumannii by four-parental mating (48, 64) using Vogel Bonner medium with Gm at 10 μg/ml (ATCC 17978 and AB5075 ΔRI) or 40 μg/ml (ATCC 19606), creating YDA004, JBA163, and JBA106, respectively. Integration at the attTn7 locus was confirmed by PCR (21, 65). A plasmid for sgRNA expression in A. baumannii was constructed by replacing the BamHI-SalI fragment internal to tetA in shuttle vector pWH1266 (22) with a PCR product having the same restriction sites and J23119, sgRNA, and terminator from pgRNA-bacteria (Addgene 44251; a gift from Stanley Qi), generating pYDE007 (Cbr). To construct a Kmr derivative of pYDE007, the EcoRI-PstI fragment containing bla was replaced with a PCR product containing the kanamycin resistance gene from pDL1100 and the same restriction sites, generating pJE53 (Kmr). pYDE0007 and pJE53 contain the nontargeting guide sequence AACTTTCAGTTTAGCGGTCT, derived from the mRFP (red fluorescent protein) guide in pgRNA-bacteria. These plasmids served as nontargeting controls in CRISPRi experiments.
Plasmids encoding sgRNAs for targeted CRISPRi were constructed by PCR amplification of the gRNA scaffold region of pYDE007, using a forward primer containing a targeting sequence and SpeI site at its 5′ end, a reverse primer with ApaI in its 5′ end, and OneTaq PCR master mix (NEB). Reverse primers for most sgRNAs also contained a unique KpnI or BglII site to assist clone identification by restriction fragment analysis. PCR products were digested with SpeI and ApaI and cloned by replacing the SpeI-ApaI guide fragment in pYDE007. sgRNA plasmids were introduced into YDA004, JBA163, and JBA106 via electroporation. The sgRNA targeting sequences were the 23 or 24 bases 5′ to PAM sites selected based on (i) proximity to the TSS preceding each target gene, (ii) targeting the NT strand, and (iii) having a 12-nt seed region found only once in the genome (12, 66).
A strain containing a conditional allele of RS03245 was constructed as follows. As the 3′ homology arm, we PCR amplified the lacIq-T5lacP-RS03245 sequence from pJE10 (RS03245-gfp cloned in lacIq-T5lacP vector pYDE152) using primers containing SphI and NotI sites and cloned the product in pUC18, generating pJE41. As the 5′ homology arm, approximately 1 kb of sequence upstream of RS03245 was PCR amplified with primers having SphI and SalI sites and cloned in pUC18, generating pJE40. Clones were joined and subcloned in pJB4648 by 3-way ligation, generating pJE44. pJE44 was delivered into A. baumannii ATCC 17978 by electroporation, and counterselection allelic exchange (48) was used to isolate JBA58, in which the native RS03245 promoter is replaced with lacIq-T5lacP. To construct complementing plasmids, RS03245 translationally fused to GFP or a 3×FLAG epitope was subcloned between the BamHI and SphI sites internal to tetA in pWH1266, generating pJE51 and pJE50, respectively. In each, RS03245 is expressed by a constitutive tet promoter.
A ΔitrA Δwzc mutant was constructed by introducing pEGE76 (an allelic exchange vector with Δwzc::Gmr) (48) into EGA295 (a ΔitrA single mutant) by electroporation and using counterselection allelic exchange to isolate JBA48 (ΔitrA Δwzc Gmr).
β-Lactamase assays.
Overnight bacterial cultures were back diluted to an A600 of 0.025 and grown to an A600 of 0.05, and cultures were divided, with one group receiving aTc (100 ng/ml). After 2 additional hours of growth, cells were harvested by centrifugation, washed, and resuspended in ice-cold 0.1 M phosphate buffer (pH 7). Periplasmic contents were liberated by ultrasonication using a Branson high-intensity cuphorn sonifier chilled to 4°C (4 cycles of 1 min on at 50% output and 1 min off). Extracts were clarified by centrifugation and diluted with 0.1 M phosphate buffer (pH 7), and the protein concentration was determined via Bradford assay (Pierce). β-Lactamase content was measured in 250-μl reaction mixtures containing 10 μg total protein and 20 μg/ml nitrocefin in 0.1 M phosphate buffer (pH 7) by reading absorbance at 486 nm every minute for 15 min at room temperature in microplates (Biotek Synergy H1M). β-Lactamase activity was calculated as initial reaction rate (Vmax) × dilution factor.
Microscopy.
Bacteria were immobilized on agarose pads (1% in PBS) and imaged via a 100×, 1.3 phase-contrast objective on a Leica AF6000 microscope.
Analysis of capsular polysaccharide.
Overnight bacterial cultures were diluted to an OD of 0.025, grown to an OD of 0.05, and divided into groups receiving 0 or 200 ng/ml aTc. Cultures were grown for 4 additional hours. Polysaccharides were isolated from whole-cell lysates with slight variations from previously described methods (48, 67). Cells were pelleted, frozen at −80°C, and resuspended with 60 mM Tris (pH 8) buffer containing 10 mM MgCl2 and 50 μM CaCl2. Lysonase (Novagen) was added at 0.5 μl per 100-μl suspension, and samples were incubated at 37°C for 30 min followed by vortexing. SDS was added to 0.5%, and samples were incubated at 37°C for 15 min. Two microliters of proteinase K (NEB) was added, and samples were incubated at 56°C for 1 h. SDS sample buffer was added to a 1× concentration, and samples were boiled for 5 min. Samples were separated on 4 to 20% Bio-Rad TGX Tris-glycine gels and stained overnight with alcian blue. Gels were imaged via white light transillumination with a ChemiDoc MP (Bio-Rad).
qRT-PCR gene expression analysis.
Overnight bacterial cultures were diluted to an OD of 0.025, grown to an OD of 0.05, and divided into aTc-treated or untreated groups. After additional growth (for 2 h with all sgRNAs except sgRNA-1, which was cultured independently and analyzed at 4 h), samples were combined with 1 volume of ice-cold ethanol-acetone and frozen at −80°C. Samples were thawed and washed with Tris-EDTA (TE), followed by RNA extraction (RNeasy kit; Qiagen) and DNase treatment (DNA-free kit; Ambion). RNA was reverse transcribed using Superscript II reverse transcriptase (Invitrogen). cDNA was diluted and used as the template with the Power-Up SYBR green master mix (Applied Biosystems) in a StepOnePlus system according to the manufacturer’s instructions for two-step reverse transcription-PCR (RT-PCR). Primers for quantitative PCR (qPCR) were designed using PrimerQuest (IDT). Assay efficiency was assessed by generating a standard curve with a dilution series of cDNA and was determined to be >92% with each target. Controls lacking reverse transcriptase were performed to confirm lack of signal from residual genomic DNA. Gene expression levels were quantified by using the threshold cycle (2−ΔΔCT) method with rpoC as an endogenous control (23).
Data availability.
All sequence data can be found in the NCBI Sequence Read Archive under accession code PRJNA668602.
ACKNOWLEDGMENTS
This work was supported by Northeastern University startup funds and NIAID awards U01AI124302 and R21AI128328.
We thank Defne Surujon for bioinformatics analysis, Stanley Qi for plasmid gifts, Colin Manoil for strain AB5075 ΔRI, Win Chai for use of microscopy equipment, Elizabeth Schwartz for technical assistance, and members of the Geisinger lab for helpful discussions.
Footnotes
Supplemental material is available online only.
jb.00565-20-s0001.pdf (5.2MB, pdf)
jb.00565-20-s0002.xlsx (1.8MB, xlsx)
Contributor Information
Edward Geisinger, Email: e.geisinger@northeastern.edu.
George O'Toole, Geisel School of Medicine at Dartmouth.
REFERENCES
- 1.Nowak J, Zander E, Stefanik D, Higgins PG, Roca I, Vila J, McConnell MJ, Cisneros JM, Seifert H,. MagicBullet Working Group WP4. 2017. High incidence of pandrug-resistant Acinetobacter baumannii isolates collected from patients with ventilator-associated pneumonia in Greece, Italy and Spain as part of the MagicBullet clinical trial. J Antimicrob Chemother 72:3277–3282. 10.1093/jac/dkx322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qureshi ZA, Hittle LE, O'Hara JA, Rivera JI, Syed A, Shields RK, Pasculle AW, Ernst RK, Doi Y. 2015. Colistin-resistant Acinetobacter baumannii: beyond carbapenem resistance. Clin Infect Dis 60:1295–1303. 10.1093/cid/civ048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geisinger E, Huo W, Hernandez-Bird J, Isberg RR. 2019. Acinetobacter baumannii: envelope determinants that control drug resistance, virulence, and surface variability. Annu Rev Microbiol 73:481–506. 10.1146/annurev-micro-020518-115714. [DOI] [PubMed] [Google Scholar]
- 4.Robinson A, Brzoska AJ, Turner KM, Withers R, Harry EJ, Lewis PJ, Dixon NE. 2010. Essential biological processes of an emerging pathogen: DNA replication, transcription, and cell division in Acinetobacter spp. Microbiol Mol Biol Rev 74:273–297. 10.1128/MMBR.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geisinger E, Mortman NJ, Dai Y, Cokol M, Syal S, Farinha A, Fisher DG, Tang AY, Lazinski DW, Wood S, Anthony J, van Opijnen T, Isberg RR. 2020. Antibiotic susceptibility signatures identify potential antimicrobial targets in the Acinetobacter baumannii cell envelope. Nat Commun 11:4522. 10.1038/s41467-020-18301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biswas I. 2015. Genetic tools for manipulating Acinetobacter baumannii genome: an overview. J Med Microbiol 64:657–669. 10.1099/jmm.0.000081. [DOI] [PubMed] [Google Scholar]
- 7.Tucker AT, Nowicki EM, Boll JM, Knauf GA, Burdis NC, Trent MS, Davies BW. 2014. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. mBio 5:e01313-14. 10.1128/mBio.01313-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong SM, Akerley BJ. 2003. Inducible expression system and marker-linked mutagenesis approach for functional genomics of Haemophilus influenzae. Gene 316:177–186. 10.1016/s0378-1119(03)00762-5. [DOI] [PubMed] [Google Scholar]
- 9.Bailey J, Cass J, Gasper J, Ngo ND, Wiggins P, Manoil C. 2019. Essential gene deletions producing gigantic bacteria. PLoS Genet 15:e1008195. 10.1371/journal.pgen.1008195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallagher LA, Bailey J, Manoil C. 2020. Ranking essential bacterial processes by speed of mutant death. Proc Natl Acad Sci U S A 117:18010–18017. 10.1073/pnas.2001507117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tarasava K, Oh EJ, Eckert CA, Gill RT. 2018. CRISPR-enabled tools for engineering microbial genomes and phenotypes. Biotechnol J 13:e1700586. 10.1002/biot.201700586. [DOI] [PubMed] [Google Scholar]
- 12.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters JM, Koo BM, Patino R, Heussler GE, Hearne CC, Qu J, Inclan YF, Hawkins JS, Lu CHS, Silvis MR, Harden MM, Osadnik H, Peters JE, Engel JN, Dutton RJ, Grossman AD, Gross CA, Rosenberg OS. 2019. Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nat Microbiol 4:244–250. 10.1038/s41564-018-0327-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geisinger E, Vargas-Cuebas G, Mortman NJ, Syal S, Dai Y, Wainwright EL, Lazinski D, Wood S, Zhu Z, Anthony J, van Opijnen T, Isberg RR. 2019. The landscape of phenotypic and transcriptional responses to ciprofloxacin in Acinetobacter baumannii: acquired resistance alleles modulate drug-induced SOS response and prophage replication. mBio 10:e01127-19. 10.1128/mBio.01127-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lampe DJ, Grant TE, Robertson HM. 1998. Factors affecting transposition of the Himar1 mariner transposon in vitro. Genetics 149:179–187. 10.1093/genetics/149.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleckner N, Bender J, Gottesman S. 1991. Uses of transposons with emphasis on Tn10. Methods Enzymol 204:139–180. 10.1016/0076-6879(91)04009-d. [DOI] [PubMed] [Google Scholar]
- 17.DeJesus MA, Ambadipudi C, Baker R, Sassetti C, Ioerger TR. 2015. TRANSIT—a software tool for Himar1 TnSeq analysis. PLoS Comput Biol 11:e1004401. 10.1371/journal.pcbi.1004401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang N, Ozer EA, Mandel MJ, Hauser AR. 2014. Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. mBio 5:e01163-14. 10.1128/mBio.01163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallagher LA, Ramage E, Weiss EJ, Radey M, Hayden HS, Held KG, Huse HK, Zurawski DV, Brittnacher MJ, Manoil C. 2015. Resources for genetic and genomic analysis of emerging pathogen Acinetobacter baumannii. J Bacteriol 197:2027–2035. 10.1128/JB.00131-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar A, Dalton C, Cortez-Cordova J, Schweizer HP. 2010. Mini-Tn7 vectors as genetic tools for single copy gene cloning in Acinetobacter baumannii. J Microbiol Methods 82:296–300. 10.1016/j.mimet.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Hunger M, Schmucker R, Kishan V, Hillen W. 1990. Analysis and nucleotide sequence of an origin of DNA replication in Acinetobacter calcoaceticus and its use for Escherichia coli shuttle plasmids. Gene 87:45–51. 10.1016/0378-1119(90)90494-c. [DOI] [PubMed] [Google Scholar]
- 23.Geisinger E, Mortman NJ, Vargas-Cuebas G, Tai AK, Isberg RR. 2018. A global regulatory system links virulence and antibiotic resistance to envelope homeostasis in Acinetobacter baumannii. PLoS Pathog 14:e1007030. 10.1371/journal.ppat.1007030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kroger C, MacKenzie KD, Alshabib EY, Kirzinger MWB, Suchan DM, Chao TC, Akulova V, Miranda-CasoLuengo AA, Monzon VA, Conway T, Sivasankaran SK, Hinton JCD, Hokamp K, Cameron ADS. 2018. The primary transcriptome, small RNAs and regulation of antimicrobial resistance in Acinetobacter baumannii ATCC 17978. Nucleic Acids Res 46:9684–9698. 10.1093/nar/gky603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo BM, Marta E, Shiver AL, Whitehead EH, Weissman JS, Brown ED, Qi LS, Huang KC, Gross CA. 2016. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165:1493–1506. 10.1016/j.cell.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fontana J, Dong C, Ham JY, Zalatan JG, Carothers JM. 2018. Regulated expression of sgRNAs tunes CRISPRi in E. coli. Biotechnol J 13:e1800069. 10.1002/biot.201800069. [DOI] [PubMed] [Google Scholar]
- 27.Bou G, Martinez-Beltran J. 2000. Cloning, nucleotide sequencing, and analysis of the gene encoding an AmpC beta-lactamase in Acinetobacter baumannii. Antimicrob Agents Chemother 44:428–432. 10.1128/aac.44.2.428-432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prados J, Linder P, Redder P. 2016. TSS-EMOTE, a refined protocol for a more complete and less biased global mapping of transcription start sites in bacterial pathogens. BMC Genomics 17:849. 10.1186/s12864-016-3211-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gebhardt MJ, Gallagher LA, Jacobson RK, Usacheva EA, Peterson LR, Zurawski DV, Shuman HA. 2015. Joint transcriptional control of virulence and resistance to antibiotic and environmental stress in Acinetobacter baumannii. mBio 6:e01660-15. 10.1128/mBio.01660-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gebhardt MJ, Shuman HA. 2017. GigA and GigB are master regulators of antibiotic resistance, stress responses, and virulence in Acinetobacter baumannii. J Bacteriol 199:e00066-17. 10.1128/JB.00066-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma A, Dubey V, Sharma R, Devnath K, Gupta VK, Akhter J, Bhando T, Verma A, Ambatipudi K, Sarkar M, Pathania R. 2018. The unusual glycine-rich C terminus of the Acinetobacter baumannii RNA chaperone Hfq plays an important role in bacterial physiology. J Biol Chem 293:13377–13388. 10.1074/jbc.RA118.002921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuo HY, Chao HH, Liao PC, Hsu L, Chang KC, Tung CH, Chen CH, Liou ML. 2017. Functional characterization of Acinetobacter baumannii lacking the RNA chaperone Hfq. Front Microbiol 8:2068. 10.3389/fmicb.2017.02068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou YN, Kusukawa N, Erickson JW, Gross CA, Yura T. 1988. Isolation and characterization of Escherichia coli mutants that lack the heat shock sigma factor sigma 32. J Bacteriol 170:3640–3649. 10.1128/jb.170.8.3640-3649.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tipton KA, Rather PN. 2017. An ompR-envZ two-component system ortholog regulates phase variation, osmotic tolerance, motility, and virulence in Acinetobacter baumannii strain AB5075. J Bacteriol 199:e0075-16. 10.1128/JB.00705-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casella LG, Weiss A, Perez-Rueda E, Ibarra JA, Shaw LN. 2017. Towards the complete proteinaceous regulome of Acinetobacter baumannii. Microb Genom 3:mgen000107. 10.1099/mgen.0.000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. 1997. AraC/XylS family of transcriptional regulators. Microbiol Mol Biol Rev 61:393–410. 10.1128/.61.4.393-410.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin RG, Rosner JL. 2001. The AraC transcriptional activators. Curr Opin Microbiol 4:132–137. 10.1016/s1369-5274(00)00178-8. [DOI] [PubMed] [Google Scholar]
- 38.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao J, Yu X, Zhu M, Kang H, Ma J, Wu M, Gan J, Deng X, Liang H. 2016. Structural and molecular mechanism of CdpR involved in quorum-sensing and bacterial virulence in Pseudomonas aeruginosa. PLoS Biol 14:e1002449. 10.1371/journal.pbio.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallagher LA, Lee SA, Manoil C. 2017. Importance of core genome functions for an extreme antibiotic resistance trait. mBio 8:e01655-17. 10.1128/mBio.01655-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kenyon JJ, Hall RM. 2013. Variation in the complex carbohydrate biosynthesis loci of Acinetobacter baumannii genomes. PLoS One 8:e62160. 10.1371/journal.pone.0062160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh JK, Adams FG, Brown MH. 2018. Diversity and function of capsular polysaccharide in Acinetobacter baumannii. Front Microbiol 9:3301. 10.3389/fmicb.2018.03301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitfield C. 2006. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu Rev Biochem 75:39–68. 10.1146/annurev.biochem.75.103004.142545. [DOI] [PubMed] [Google Scholar]
- 44.Jorgenson MA, Kannan S, Laubacher ME, Young KD. 2016. Dead-end intermediates in the enterobacterial common antigen pathway induce morphological defects in Escherichia coli by competing for undecaprenyl phosphate. Mol Microbiol 100:1–14. 10.1111/mmi.13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yother J. 2011. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu Rev Microbiol 65:563–581. 10.1146/annurev.micro.62.081307.162944. [DOI] [PubMed] [Google Scholar]
- 46.Peters JM, Silvis MR, Zhao D, Hawkins JS, Gross CA, Qi LS. 2015. Bacterial CRISPR: accomplishments and prospects. Curr Opin Microbiol 27:121–126. 10.1016/j.mib.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collins RF, Beis K, Dong C, Botting CH, McDonnell C, Ford RC, Clarke BR, Whitfield C, Naismith JH. 2007. The 3D structure of a periplasm-spanning platform required for assembly of group 1 capsular polysaccharides in Escherichia coli. Proc Natl Acad Sci U S A 104:2390–2395. 10.1073/pnas.0607763104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geisinger E, Isberg RR. 2015. Antibiotic modulation of capsular exopolysaccharide and virulence in Acinetobacter baumannii. PLoS Pathog 11:e1004691. 10.1371/journal.ppat.1004691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tipton KA, Chin CY, Farokhyfar M, Weiss DS, Rather PN. 2018. Role of capsule in resistance to disinfectants, host antimicrobials, and desiccation in Acinetobacter baumannii. Antimicrob Agents Chemother 62:e01188-18. 10.1128/AAC.01188-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Russo TA, Luke NR, Beanan JM, Olson R, Sauberan SL, MacDonald U, Schultz LW, Umland TC, Campagnari AA. 2010. The K1 capsular polysaccharide of Acinetobacter baumannii strain 307-0294 is a major virulence factor. Infect Immun 78:3993–4000. 10.1128/IAI.00366-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.James DB, Yother J. 2012. Genetic and biochemical characterizations of enzymes involved in Streptococcus pneumoniae serotype 2 capsule synthesis demonstrate that Cps2T (WchF) catalyzes the committed step by addition of beta1–4 rhamnose, the second sugar residue in the repeat unit. J Bacteriol 194:6479–6489. 10.1128/JB.01135-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xayarath B, Yother J. 2007. Mutations blocking side chain assembly, polymerization, or transport of a Wzy-dependent Streptococcus pneumoniae capsule are lethal in the absence of suppressor mutations and can affect polymer transfer to the cell wall. J Bacteriol 189:3369–3381. 10.1128/JB.01938-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ranjit DK, Young KD. 2016. Colanic acid intermediates prevent de novoshape recovery of Escherichia coli spheroplasts, calling into question biological roles previously attributed to colanic acid. J Bacteriol 198:1230–1240. 10.1128/JB.01034-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jorgenson MA, Young KD. 2016. Interrupting biosynthesis of O antigen or the lipopolysaccharide core produces morphological defects in Escherichia coli by sequestering undecaprenyl phosphate. J Bacteriol 198:3070–3079. 10.1128/JB.00550-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richie DL, Takeoka KT, Bojkovic J, Metzger LEt, Rath CM, Sawyer WS, Wei JR, Dean CR. 2016. Toxic accumulation of LPS pathway intermediates underlies the requirement of LpxH for growth of Acinetobacter baumannii ATCC 19606. PLoS One 11:e0160918. 10.1371/journal.pone.0160918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei JR, Richie DL, Mostafavi M, Metzger LEt, Rath CM, Sawyer WS, Takeoka KT, Dean CR. 2017. LpxK is essential for growth of Acinetobacter baumannii ATCC 19606: relationship to toxic accumulation of lipid A pathway intermediates. mSphere 2:e00199-17. 10.1128/mSphere.00199-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang G, Baidin V, Pahil KS, Moison E, Tomasek D, Ramadoss NS, Chatterjee AK, McNamara CW, Young TS, Schultz PG, Meredith TC, Kahne D. 2018. Cell-based screen for discovering lipopolysaccharide biogenesis inhibitors. Proc Natl Acad Sci U S A 115:6834–6839. 10.1073/pnas.1804670115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 59.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Surujonu D, Bento J, van Opijnen T. 2020. Boundary-Forest Clustering: large-scale consensus clustering of biological sequences. bioRxiv 10.1101/2020.04.28.065870. [DOI]
- 61.Hulsen T, de Vlieg J, Alkema W. 2008. BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9:488. 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 64.Carruthers MD, Nicholson PA, Tracy EN, Munson RS, Jr.. 2013. Acinetobacter baumannii utilizes a type VI secretion system for bacterial competition. PLoS One 8:e59388. 10.1371/journal.pone.0059388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jacobs AC, Thompson MG, Black CC, Kessler JL, Clark LP, McQueary CN, Gancz HY, Corey BW, Moon JK, Si Y, Owen MT, Hallock JD, Kwak YI, Summers A, Li CZ, Rasko DA, Penwell WF, Honnold CL, Wise MC, Waterman PE, Lesho EP, Stewart RL, Actis LA, Palys TJ, Craft DW, Zurawski DV. 2014. AB5075, a highly virulent isolate of Acinetobacter baumannii, as a model strain for the evaluation of pathogenesis and antimicrobial treatments. mBio 5:e01076-14. 10.1128/mBio.01076-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guzzo M, Castro LK, Reisch CR, Guo MS, Laub MT. 2020. A CRISPR interference system for efficient and rapid gene knockdown in Caulobacter crescentus. mBio 11:e02415-19. 10.1128/mBio.02415-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hitchcock PJ, Brown TM. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol 154:269–277. 10.1128/JB.154.1.269-277.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All sequence data can be found in the NCBI Sequence Read Archive under accession code PRJNA668602.