

ABSTRACT
The COVID-19 pandemic poses a serious global health threat. The rapid global spread of SARS-CoV-2 highlights an urgent need to develop effective therapeutics for blocking SARS-CoV-2 infection and spread. Stimulator of Interferon Genes (STING) is a chief element in host antiviral defense pathways. In this study, we examined the impact of the STING signaling pathway on coronavirus infection using the human coronavirus OC43 (HCoV-OC43) model. We found that HCoV-OC43 infection did not stimulate the STING signaling pathway, but the activation of STING signaling effectively inhibits HCoV-OC43 infection to a much greater extent than that of type I interferons (IFNs). We also discovered that IRF3, the key STING downstream innate immune effector, is essential for this anticoronavirus activity. In addition, we found that the amidobenzimidazole (ABZI)-based human STING agonist diABZI robustly blocks the infection of not only HCoV-OC43 but also SARS-CoV-2. Therefore, our study identifies the STING signaling pathway as a potential therapeutic target that could be exploited for developing broad-spectrum antiviral therapeutics against multiple coronavirus strains in order to face the challenge of future coronavirus outbreaks.
IMPORTANCE The highly infectious and lethal SARS-CoV-2 is posing an unprecedented threat to public health. Other coronaviruses are likely to jump from a nonhuman animal to humans in the future. Novel broad-spectrum antiviral therapeutics are therefore needed to control known pathogenic coronaviruses such as SARS-CoV-2 and its newly mutated variants, as well as future coronavirus outbreaks. STING signaling is a well-established host defense pathway, but its role in coronavirus infection remains unclear. In the present study, we found that activation of the STING signaling pathway robustly inhibits infection of HCoV-OC43 and SARS-CoV-2. These results identified the STING pathway as a novel target for controlling the spread of known pathogenic coronaviruses, as well as emerging coronavirus outbreaks.
KEYWORDS: HCoV-OC43, SARS-CoV-2, COVID-19, coronavirus, STING, antiviral therapy
INTRODUCTION
Coronaviruses (CoVs), belonging to the Coronaviridae family within the Nidovirales order, have a broad host range from birds to mammals (1–4), but have not yet been found in reptiles and amphibians. The seven strains of human CoVs identified to date fall within two genera. The Alphacoronavirus genus includes HCoV-229E (5) and HCoV-NL63 (6, 7), whereas the Betacoronavirus genus includes HCoV-OC43 (8), HCoV-HKU1 (9), severe acute respiratory syndrome (SARS)-CoV (10, 11), Middle East respiratory syndrome (MERS)-CoV (12), and the newly discovered SARS-CoV-2 (13, 14).
The novel coronavirus SARS-CoV-2 emerged in December 2019 and rapidly swept across the globe, causing a severe respiratory illness, COVID-19, in millions of people (15–20). SARS-CoV-2 has infected over 121 million people worldwide and has claimed 2.7 million lives in the last few months (16, 20). While SARS-CoV-2 infection poses an unprecedented threat to public health, there are only very limited therapeutic options for treating COVID-19 (17, 18). While three vaccines are being rolled out, the virus continues to evolve new variants that are not only more infectious but, in some cases, may have the potential to escape current vaccines. In addition, these vaccines are not likely to be effective against a new CoV. On the other hand, a drug can achieve much quicker responses to new viruses until their specific vaccines can be developed. Therefore, novel broad-spectrum antiviral therapeutics are needed to control future coronavirus outbreaks.
Like all coronaviruses, SARS-CoV-2 is an enveloped virus, with a single-stranded, positive-sense RNA genome (18, 19, 21, 22). It shares 77.2% amino acid sequence identity with SARS-CoV, which was responsible for the SARS epidemic in 2002 to 2003 (13). SARS-CoV-2 is also closely related to MERS-CoV, the agent of Middle East Respiratory Syndrome. SARS-CoV-2 is thought to have jumped from its animal hosts to humans and, subsequently, developed person-to-person transmission, leading to a once-in-a-century pandemic (19, 21, 23). Since SARS-CoV-2 is being introduced to humans for the first time, much remains to be learned about its virology and pathogenic mechanisms (18).
Infection by RNA viruses produces double-stranded RNA (dsRNA) as an intermediate in replication; the dsRNA is sensed by host RNA pattern recognition receptors (PRRs) to stimulate a cascade of signaling pathways that induce transcription of type I/III interferons (IFNs) and other innate proinflammatory cytokines, as well as the oligoadenylate synthetases (OAS)-RNase L and protein kinase R (PKR) pathways (24). IFNs can induce a large number of downstream IFN-stimulated genes (ISGs) to mount an “antiviral state” in the host cell to clear viral infection (23, 25–27). In addition, proinflammatory cytokines and chemokines potentiate the adaptive immune response and recruit a variety of immune cells to further combat virus infection and limit virus spread (23, 25). However, SARS-CoV-2 and many other coronaviruses have evolved multiple mechanisms to evade innate immune responses (15, 23, 25, 26, 28–38). For example, these coronaviruses can encode proteins to target host antiviral pathways, including the IFN signaling, OAS-RNase L, and PKR pathways (24). In addition, coronaviruses replicate inside virus-induced cytosolic double-membrane vesicles (39), which protect the dsRNA from recognition by cytosolic receptors. Furthermore, the viral genomic RNA, with a 5′ cap and 3′ poly(A) sequence, functions as messenger RNA (mRNA) that is translated into 16 nonstructural proteins by the host ribosome machinery to initiate infection immediately after the viral genome reaches the cytoplasm (40). Together, these tactics allow the virus to escape immune destruction and establish rapid and unhindered viral replication in primary infected cells, producing large copy numbers of the virus (23, 25, 26, 28–34). Some of these nonstructural proteins, as well as viral accessory proteins, also serve to dampen the host innate immune response (2, 24).
Although coronaviruses can evade host antiviral pathways, preactivation of these pathways can inhibit coronavirus infection and replication to some degree (32, 41–43). For example, early evidence demonstrated that type I/III IFN pretreatment could inhibit SARS-CoV-2 infection, although the efficiencies vary across studies (25, 38, 44, 45). The fact that IFNs are protective early after infection but later become pathological suggests that eliciting an IFN response at the early stages of infection is critical for blocking SARS-CoV-2 infection, spread, and associated pathogenesis. This notion is supported by SARS-CoV studies showing that failure to elicit an early IFN response correlates with the severity of disease (25). Recently, deficiency in IFN signaling pathways has also been linked to severe COVID-19 (46, 47).
STING is a key mediator of a host antiviral defense pathway (48–50). The canonical role of the STING pathway is to sense damaged DNA and infection by DNA viruses. Recent studies showed that the STING pathway is also involved in sensing RNA virus infection (51). After activation by signals from its upstream nucleic acid sensors, STING recruits TANK binding kinase 1 (TBK1) to phosphorylate itself. The STING-TBK1 complex translocates through the Golgi complex to the perinuclear lysosomal compartments, where it phosphorylates IRF3. Active STING can also stimulate IKK to phosphorylate IκBαB, causing its degradation and NF-κB activation. Activated IRF3 and NF-κB can then translocate into the nucleus to induce transcription of type I IFNs and other inflammatory cytokines, establishing an antiviral state (48, 52, 53). As discussed above, a timely activation of host IFNs and antiviral ISG production is needed to overcome coronavirus immune evasion and inhibit viral infection. We therefore investigated the impact of the STING signaling pathway on coronavirus infection.
In the current study, we discovered that activation of the STING innate immune signaling pathway effectively blocks infection by human coronavirus OC43 (HCoV-OC43) and SARS-CoV-2. We also demonstrated that transcription factor IRF3, the STING downstream innate immune effector, is essential for this anticoronavirus activity. Furthermore, we found that the human STING agonist diABZI (54) can robustly activate human STING to block infection of both HCoV-OC43 and SARS-CoV-2. Therefore, our study identifies the STING signaling pathway as an important therapeutic target that could be specifically activated for hampering infection of SARS-CoV-2 and other coronaviruses.
RESULTS
Activation of STING signaling effectively blocks human coronavirus infection.
Deficiency of type I IFN immunity has been linked to severe COVID-19 disease (46, 47). Because the STING signaling pathway is important for inducing type I/III IFNs and a large number of other antiviral proteins, we hypothesized that activating the STING signaling pathway could inhibit coronavirus infection. To test this, we first examined the antiviral activity of the STING signaling pathway against HCoV-OC43, which belongs to the Betacoronavirus genus along with SARS-CoV-2, SARS-CoV, and MERS-CoV. The human alveolar basal epithelial cell line A549, an established target cell type for HCoV-OC43 infection, was used in this study. In our previous study, we found that STING expression is significantly repressed in A549 cells (Fig. 1A) (55). We have recently developed a method for reactivating STING immunity in STING-silenced cells (55). In a proof-of-principle study, we constructed an adenoviral-associated vector (AAV) encoding a human STING mutant S162A/G230I/Q266I (STINGAII) highly responsive to DMXAA (5,6-dimethylxanthenone-4-acetic acid), a potent mouse STING agonist that does not otherwise activate wild-type human STING (STINGWT) (56–59). We showed that dual treatment of DMXAA and AAV-STINGAII induces robust induction of IFNs, ISGs, and other cytokines specifically in STING-silenced cells, without affecting the function of normal cells that express endogenous STINGWT (55). This method therefore provided an effective approach to specifically activate STING in the target cells of interest. For the current study, the STINGAII AAV virions were used to transduce A549 cells, while STINGWT served as a negative control. At 2 days post AAV transduction, the cells were treated with dimethyl sulfoxide (DMSO) or DMXAA to activate the STING signaling pathway before HCoV-OC43 infection. By performing immunofluorescence (IF) double staining for HCoV-OC43 nucleoprotein (N) protein and STING, we discovered that HCoV-OC43 could efficiently infect DMSO-treated STINGAII or STINGWT cells, but DMXAA-induced STINGAII activation effectively blocked OC43 infection (Fig. 1B).
FIG 1.

DMXAA activation of AAV-encoded human STINGAII inhibits HCoV-OC43 infection. (A) STING is silenced in A549 cells. Whole-cell lysates harvested from A549 and primary human dermal fibroblasts (HDFs) were immunoblotted using the indicated antibodies. GAPDH was used as a loading control. (B) AAV virions encoding STINGWT or STINGAII were used to infect A549 cells. At 2 days postinfection, the cells were treated with DMSO or DMXAA for 3 h and then washed with PBS prior to HCoV-OC43 infection. At 24 h postinfection, cells were immunostained with antibodies against the HCoV-OC43 N protein (OC43 N) and STING and counterstained with DAPI. (C and D) Treatment with IFNs does not significantly inhibit HCoV-OC43 infection but does inhibit NDV-GFP infection in A549 cells. (C) A549 cells were treated with PBS or IFNs for 3 h. The cells were washed with PBS prior to HCoV-OC43 infection. In one of the groups, the infected cells were treated with IFNs for an additional 23 h. At 24 h postinfection, cells were immunostained as in (B). (D) A549 cells were treated with PBS or IFNs for 3 h. The cells were washed with PBS prior to NDV-GFP infection. The IFN treatment was continued for an additional 23 h. At 24 h postinfection, cells were stained with Hoechst 33342. Bar: 50 μm.
In a parallel experiment, we also used type I IFNs to treat A549 cells before and during HCoV-OC43 infection. We observed that IFN treatment did not show any significant inhibitory effect on HCoV-OC43 infection (Fig. 1C). As a positive control for the IFN antiviral activity, we found that IFNs were able to inhibit the infection of a recombinant Newcastle disease virus expressing green fluorescent protein (NDV-GFP) (Fig. 1D).
HCoV-OC43 infection did not stimulate the STING signaling pathway but DMXAA activation of STING effectively inhibits HCoV-OC43 infection.
To rule out the possibility that the AAV vector contributes to the inhibitory effect on HCoV-OC43 infection, we constructed A549 cells stably expressing either STINGWT or STINGAII. In order to more directly compare the effects of IFN treatment and STING pathway activation on HCoV-OC43 infection, we treated A549 cells with phosphate-buffered saline (PBS), IFNs, DMSO, or DMXAA side by side (Fig. 2A and B). Similar to earlier observations, HCoV-OC43 was able to productively infect the stable cells treated by PBS or DMSO (Fig. 2A and B). As visualized by Western blotting of the HCoV-OC43 N protein, extended treatment with IFNs slightly inhibited HCoV-OC43 infection in both stable cell lines (Fig. 2B). However, when A549 cells stably expressing STINGAII were treated with DMXAA, we observed robust inhibition of HCoV-OC43 infection (Fig. 2A and B). On the other hand, DMXAA treatment of STINGWT stable cells did not affect HCoV-OC43 infection (Fig. 2A and B). We also found that, while DMXAA treatment did not introduce any significant cytotoxicity in A549 cells (Fig. 2C), it did trigger marked STING phosphorylation and activation, as indicated by the slow-migrating STING band specifically observed in the cells treated with both STINGAII and DMXAA (Fig. 2B, lane 12). Notably, HCoV-OC43 infection itself did not cause the STING activation as was observed with DMXAA treatment (Fig. 2B, compare lanes 2 to 6 and 8 to 11 with lane 12). The dual STINGAII and DMXAA treatment also induced robust IRF3 degradation, although the underlying mechanism remains to be examined. Together, our data suggest that, while HCoV-OC43 infection does not activate the STING pathway, DMXAA-induced activation of STING signaling can robustly suppress HCoV-OC43 infection.
FIG 2.

DMXAA activation of stably expressed human STINGAII impedes HCoV-OC43 infection. (A) A549 cells stably expressing STINGWT or STINGAII were treated with PBS, IFNs, DMSO, or DMXAA for 3 h. The cells were washed with PBS prior to HCoV-OC43 infection. In one of the groups, the infected cells were treated with IFNs for an additional 23 h. At 24 h postinfection, cells were fixed and immunostained with antibodies against the HCoV-OC43 N protein (OC43 N) and STING and counterstained with DAPI. Bar: 50 μm. (B) Whole-cell lysates harvested from the cells treated as in (A) were immunoblotted using the indicated antibodies. GAPDH was used as a loading control. (C) DMXAA does not show significant cytotoxicity in A549 cells. A549 cells stably expressing STINGWT or STINGAII were treated with the indicated doses of DMSO or DMXAA (stock concentration: 10 mg/ml) for 3 h. The cells were washed with PBS and fed with fresh culture medium. At 27 h posttreatment, cell viability was measured using the CellTiter-GLO 3D cell viability assay.
IRF3, a STING downstream effector, is essential for blocking human coronavirus infection.
The fact that STING activation effectively blocks HCoV-OC43 infection suggests the induction of antiviral activity by IFNs and IFN-stimulated genes (ISGs) downstream of STING can be exploited as a novel strategy to counteract immune escape by coronaviruses. A clear understanding of STING’s downstream effectors will allow us to design strategies to precisely activate these molecules in a tissue-specific and timely manner to inhibit coronavirus infection. After STING is activated by its ligand, such as cGAMP or DMXAA, it recruits TBK1 to activate both IRF3 and NF-κB, which translocate into the nucleus to induce transcription of IFNs and other inflammatory cytokines (48, 52, 53, 60–62). These molecules are secreted from the cell and act in both autocrine and paracrine fashions to activate downstream signaling that elicits ISG expression and the antiviral response (63–65). We therefore applied a CRISPR knockout (KO) approach to determine which downstream effector(s) of the STING signaling pathway are essential for its anticoronavirus activity. To this end, we established CRISPR KO of TBK1, IRF3, and the NF-κB subunit p65 in A549 cells stably expressing STINGAII (Fig. 3A). These cell lines, as well as A549/STINGAII cells stably expressing a guide RNA (sgRNA) targeting firefly luciferase (sgLuc), were treated with DMSO or DMXAA before exposure to HCoV-OC43. We then performed IF double staining and Western blotting for the HCoV-OC43 N protein and STING to determine if CRISPR KO of TBK1, IRF3, and/or p65 ablates the STINGAII/DMXAA antiviral effect (Fig. 3B and C). From these studies, we observed that DMXAA treatment induced a slow-migrating band in the STING blot, which disappeared in TBK1 KO cells, suggesting that this band represents the phosphorylated/activated form of STING (Fig. 3C). This notion was confirmed by immunoblotting using an antibody recognizing the phosphorylated STINGSer366 (Fig. 3C). The phosphorylated STING band is also absent in the p65 KO cells, likely because these cells grew poorly and much less STING is expressed in these cells, making it difficult to detect the shifted STING band (Fig. 3C). More importantly, we found that IRF3 KO significantly suppressed STINGAII/DMXAA-induced antiviral activity, allowing the cells to be infected by HCoV-OC43 to a similar level as observed in the DMSO-treated cells (Fig. 3B and C). In contrast, TBK1 KO and p65 KO cells showed much less effect on STINGAII/DMXAA-induced antiviral activity against HCoV-OC43 (Fig. 3B and C). This study therefore identified IRF3 as the STING downstream effector that plays the most important role in blocking HCoV-OC43 infection.
FIG 3.

The STING-IRF3 signaling axis is critical for blocking HCoV-OC43 infection. (A) CRISPR/Cas9-mediated knockout of p65, IRF3, and TBK1 in A549 cells. A549 cells stably expressing human STINGAII were treated with LentiCRISPR v2 constructs encoding gRNAs targeting the indicated immune effector genes. Whole-cell lysates of A549 parental and the CRIPSR KO cells were immunoblotted using the indicated antibodies to validate the gene KO effect. GAPDH was used as a loading control. (B) A549 cells stably expressing STINGAII and a guide RNA targeting firefly luciferase (sgLuc), IRF3 (sgIRF3), TBK1 (sgTBK1), or p65 (sgp65) were treated with DMSO or DMXAA for 3 h. The cells were washed with PBS prior to HCoV-OC43 infection. At 24 h postinfection, cells were immunostained with antibodies against the HCoV-OC43 N protein (OC43 N) and STING and counterstained with DAPI. Bar: 50 μm. (C) Whole-cell lysates extracted from the cells treated as in (B) were immunoblotted using the indicated antibodies. GAPDH was used as a loading control.
Human STING agonist diABZI effectively inhibits HCoV-OC43 infection.
We next examined whether activating the STING pathway could be explored as a therapeutic approach to block HCoV-OC43 infection. While the STINGAII/DMXAA approach is effective in blocking HCoV-OC43 infection, it is not ideal for use as an antiviral because it requires the expression of the exogenous STINGAII molecule. We therefore tested the idea of using STING agonists that can directly activate the endogenous human STING. 2′3′-Cyclic GMP-AMP (cGAMP) is the natural ligand of human STING; however, it cannot efficiently enter cells. Therefore, we tested two nonnucleotidyl small molecule human STING agonists: amidobenzimidazole (ABZI)-based STING agonist (diABZI) (54) and CAY10748 (66). To rule out off-target effects, we treated A549 cells with each of the human STING agonists and assessed the cell proliferation rate (Fig. 4A). We found that diABZI did not affect the proliferation of A549 cells even at a concentration of 1 μM, but the same concentration of CAY10748 significantly suppressed A549 cell proliferation (Fig. 4A). This is likely due to an off-target effect because there is nearly no STING expressed in A549 cells (Fig. 1A and C). We therefore chose diABZI for the subsequent studies. By testing various concentrations of diABZI, we found that 100 nM diABZI minimally inhibited the proliferation of STINGWT stable cells without affecting the STINGAII stable cells (Fig. 4B). Therefore, this concentration of diABZI was used to treat A549 cells stably expressing STINGWT prior to HCoV-OC43 infection. Compared to DMSO treatment, diABZI treatment nearly completely inhibited HCoV-OC43 infection (Fig. 4C and D). We then used ex vivo cultured human lung explants, which constitute a cell culture system that more closely resembles the physiological environment of the human lung, to confirm the findings. We found that while HCoV-OC43 can infect the cells present in the lung tissue slices incubated with DMSO (Fig. 5), treatment with STING agonist diABZI almost completely blocked HCoV-OC43 infection (Fig. 5).
FIG 4.

Treatment with STING agonist diABZI effectively blocks HCoV-OC43 infection. (A and B) Human STING agonist diABZI is much less toxic than another human STING agonist CAY10748. (A) A549 cells were treated with DMSO or increasing doses of diABZI or CAY10748. At 72 h posttreatment, cell viability was measured using the CellTiter-GLO 3D cell viability assay. (B) A549 cells stably expressing STINGWT or STINGAII were treated with DMSO or increasing doses of diABZI for 3 h. The cells were washed with PBS and fed with fresh culture medium. At 27 h posttreatment, cell viability was measured using the CellTiter-GLO 3D cell viability assay. (C) A549 cells stably expressing STINGWT were treated with DMSO or diABZI for 3 h. The cells were washed with PBS before infection with HCoV-OC43. At 24 h postinfection, cells were immunostained with antibodies against HCoV-OC43 N protein (OC43 N) and STING and counterstained with DAPI. Bar: 50 μm. (D) Whole-cell lysates of the cells treated as in (C) were immunoblotted using the indicated antibodies. GAPDH was used as a loading control.
FIG 5.
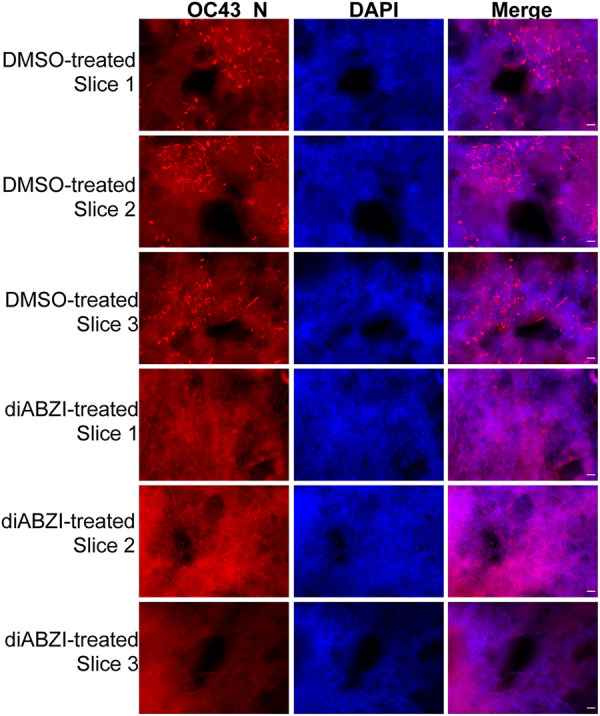
Human STING agonist diABZI can efficiently block HCoV-OC43 infection in human lung tissue slices. Human lung tissue slices were pretreated with DMSO or 1 μM human STING agonist diABZI for 3 h in 200 μl keratinocyte serum-free medium. The slices were treated with HCoV-OC43 virions diluted in 1 ml keratinocyte serum-free medium at 0.5 × 10e6 PFU/ml. The slices were incubated at 33°C in 5% CO2. At 6 days postinfection, slices were immunostained using the antibody against the HCoV-OC43 N protein (OC43 N) and counterstained with DAPI. OC43 N+ bright red dots in the tissues are infected cells. Bar: 50 μm.
Human STING agonist diABZI also blocks SARS-CoV-2 infection.
SARS-CoV-2 has evolved strategies to repress the expression of types I and III IFNs and proinflammatory cytokines/chemokines in infected human lung tissues (15, 25, 35). On the other hand, we have demonstrated that activation of STING signaling induces the expression of IFNs and other ISGs/cytokines (55) and that STING activation effectively blocks HCoV-OC43 infection (Fig. 1 and 5). Because SARS-CoV-2, like HCoV-OC43, belongs to the Betacoronavirus genus, our findings suggest that the STING activation approach could be applied to reignite the host antiviral innate immune response against SARS-CoV-2 infection and spread. We therefore examined the effect of STING activation on SARS-CoV-2 infectibility.
Since A549 cells do not express angiotensin-converting enzyme 2 (ACE2), the cellular receptor for SARS-CoV (Fig. 6A), we used A549 cells stably expressing ACE2 (A549/ACE2) for the SARS-CoV-2 infection experiments (24). These cells were treated with a lentiviral vector to establish stable expression of STINGWT and then cells were treated with DMSO, diABZI, and IFNs. IF staining of the SARS-CoV-2 N protein and STING showed that SARS-CoV-2 could actively infect the A549/ACE2/STINGWT stable cells treated with DMSO, but diABZI treatment markedly reduced the percentage of cells infected by SARS-CoV-2 (Fig. 6B). This finding was confirmed by Western blotting, which showed there was no detectable SARS-CoV-2 N protein in diABZI-treated samples (Fig. 6C). IFN treatment also impeded SARS-CoV-2 infection, although to a lesser extent compared to diABZI (Fig. 6B and C).
FIG 6.

Human STING agonist diABZI can efficiently block SARS-CoV-2 infection. (A) Introducing ACE2 stable expression in A549 cells. Whole-cell lysates harvested from A549 parental or ACE2 stable cells were immunoblotted using the indicated antibodies. GAPDH was used as a loading control. (B) A549/ACE2 cells stably expressing STINGWT were incubated with DMSO or diABZI for 3 h before treatment with SARS-CoV-2. At 24 h postinfection, cells were immunostained with antibodies against SARS-CoV-2 N protein (SARS-CoV-2 N) and STING and counterstained with DAPI. Bar: 50 μm. (C) Whole-cell lysates of the cells treated as in (B) were immunoblotted using the indicated antibodies. GAPDH was used as a loading control.
To validate the finding in a more physiological setting, we examined the impact of STING activation on SARS-CoV-2 infection in human lung tissue slices cultured ex vivo. Compared to OC43, SARS-CoV-2 showed much lower infectivity in the lung slices (Fig. 7). This is likely because the SARS-CoV-N antibody is not as sensitive as the one recognizing the HCoV-OC43 N protein. In addition, for reasons we do not completely understand, SARS-CoV-2 N-positive cells were often observed near the edges of the tissue slices. Nonetheless, treatment with STING agonist diABZI also significantly inhibited SARS-CoV-2 infection in the ex vivo lung tissue slices (Fig. 7, compare rows 4 to 6 with rows 1 to 3). Therefore, these data mirror our earlier findings from the HCoV-OC43 studies and suggest the human STING agonist diABZI is a potential drug for blocking infection of human coronaviruses, including HCoV-OC43 and SARS-CoV-2.
FIG 7.
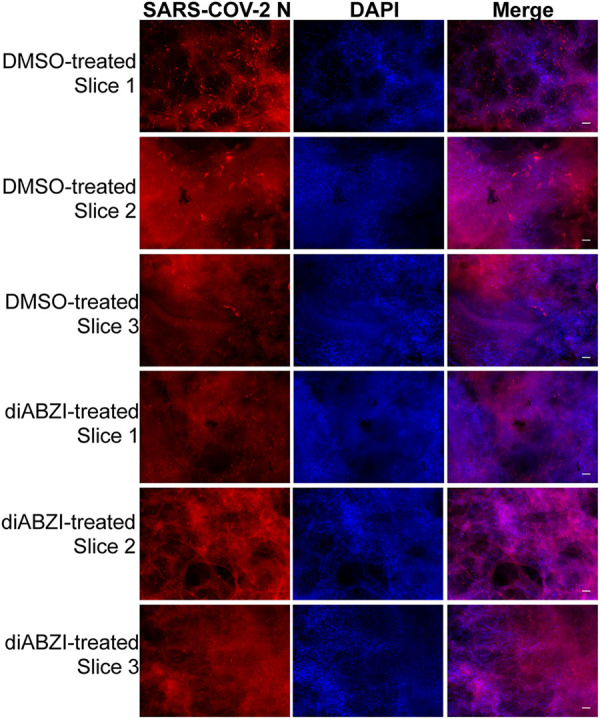
Human STING agonist diABZI can efficiently block SARS-CoV-2 infection in human lung tissue slices. Human lung tissue slices were pretreated with DMSO or 1 μM human STING agonist diABZI for 3 h in 200 μl keratinocyte serum-free medium. The slices were then treated with SARS-CoV-2 virions diluted in 1 ml keratinocyte serum-free medium at 10e6 PFU/ml. The slices were incubated at 37°C in 5% CO2. After 10 days, slices were immunostained using an antibody against the SARS-CoV-2 N protein (SARS-CoV-2 N) and counterstained with DAPI. SARS-CoV-2 N+ bright red dots in the tissues represent infected cells. Bar: 50 μm.
DISCUSSION
Abundant evidence suggests that SARS-CoV-2 and many other coronaviruses have evolved complex molecular mechanisms to suppress host innate immune responses and escape immune eradication (15, 23, 25, 26, 28–39). A lack of an effective antiviral innate immune response during the early phase of infection allows the virus to establish rapid and robust replication in primary infected cells (23, 25, 26, 28–34). However, unbridled viral replication could eventually incite a delayed massive innate inflammatory activation response that can overstimulate pathogenic immune cells to cause life-threatening tissue damage and multiorgan failure (17, 23, 25, 26, 67). Therefore, stimulation of host IFNs and antiviral ISG production during the early phase of viral infection is needed to circumvent the immune evasion mechanism of coronaviruses and inhibit viral infection.
In this current study, we first demonstrated that activating STING signaling by DMXAA and STINGAII dual treatment effectively blocks HCoV-OC43 infection (Fig. 1, Fig. 2). We found that CRISPR KO of the STING downstream effector IRF3 almost completely abolished STING/DMXAA antiviral activity, whereas p65 KO and TBK1 KO only mildly affected the ability of STINGAII/DMXAA to block HCoV-OC43 infection (Fig. 3). This study thus identified IRF3 as a key mediator for blocking HCoV-OC43 infection after STING activation. While the majority of the STING/DMXAA antiviral activity is mediated by IRF3, a small part of this activity is also dependent on NF-κB. TBK1 KO did not completely ablate STING/DMXAA antiviral activity, suggesting that other kinase(s) may be involved. IKKε, a kinase that has been shown to act redundantly with TBK1 to elicit STING-induced NF-κB activation (68), is actively expressed in A549 cells (Fig. 1A, Fig. 3C), suggesting that it may contribute to the TBK1-independent STING antiviral activity.
We also discovered that treatment with IFNs had very little effect on HCoV-OC43 infection, but could moderately repress SARS-CoV-2 infection (Fig. 2, Fig. 6). In contrast, the human STING agonist diABZI could almost completely block the infection of HCoV-OC43 and SARS-CoV-2 both in vitro and ex vivo (Fig. 4, Fig. 7). Therefore, compared to type I IFN pretreatment, temporary stimulation of the STING signaling pathway has demonstrated a much greater potential in blocking coronavirus infection. The fact that diABZI can act directly on endogenous human STING to inhibit HCoV-OC43 and SARS-CoV-2 replication makes it a great therapeutic candidate for impeding the infection of currently known and future emerging coronaviruses.
Our finding that STING activation effectively blocks HCoV-OC43 and SARS-CoV-2 infection suggests that inducing STING downstream IFN- and ISG-mediated antiviral activity can be exploited as a novel strategy to counteract the immune escape mechanism of coronaviruses. However, IFNs and cytokines are protective early in infection but destructive at the later stage of infection (17, 23, 25, 26, 32, 38, 41–47, 67). Therefore, it is important to maintain a delicate balance between antiviral and inflammatory innate immune programs in order to successfully control coronavirus infection without causing inflammatory damage. Although IRF3 is essential for STING downstream antiviral activity (Fig. 3), it is almost completely depleted after STING is activated by either DMXAA or diABZI (Fig. 2, Fig. 4, Fig. 6). A similar observation was made in a previous study, in which STING activation by cyclic dinucleotides also led to IRF3 suppression (69). While the mechanism underlying this phenomenon remains to be investigated, these findings reveal an intrinsic negative-feedback control mechanism of the STING signaling pathway. Our data showed that treating the cells with STING agonists for 3 h was sufficient to cause almost complete inhibition of HCoV-OC43 and SARS-CoV-2 infection. The subsequent shutdown of IRF3 ensures that the activation of STING by its agonists is temporary and does not cause prolonged stimulation of the inflammatory antiviral response. This mechanism therefore affords an opportunity for using STING agonists to achieve disease-stage-specific activation of IFNs, ISGs, and/or cytokines for maximizing anticoronavirus innate immunity while minimizing inflammatory damages.
Taken together, our findings suggest that stimulating STING and downstream innate immune signaling can be exploited as a novel strategy to activate an early and effective host innate immune response for blocking coronavirus infection. Our study also unveils the possibility of applying diABZI as a potential drug for treating infection of HCoV-OC43 and SARS-CoV-2.
In the last two decades, three novel betacoronaviruses, SARS-CoV, MERS-CoV, and SARS-CoV-2, have crossed the species barrier and spilled over to humans to cause highly fatal outbreaks (23). Bats alone harbor more than 400 coronaviruses. Therefore, the spillover is likely to happen again in the future. What we learn from human betacoronaviruses HCoV-OC43 and SARS-CoV-2 will have general implication for developing innovative broad-spectrum antiviral therapeutics against multiple coronavirus strains, allowing us to face the challenge of future coronavirus outbreaks.
MATERIALS AND METHODS
Cell culture.
Vero E6, HEK293T, A549, A549ACE2, and primary human dermal fibroblasts (HDF) (70) were grown in Dulbecco’s modified Eagle medium (DMEM) (Gibco, 11965084) supplemented with 10% fetal calf serum (FCS) (HyClone, SH30071.03) at 37°C in humidified air containing 5% CO2.
Compounds and reagents.
DMXAA (Sigma, D5817, CAS no. 117570-53-3) was purchased from Sigma. Human STING agonist 3 diABZI (Cayman chemical, 28054, CAS no. 2138299-34-8) and CAY10748 (Cayman chemical, 30022, CAS no. 2412902-55-5) were purchased from Cayman Chemical. Human IFN Alpha Hybrid (Universal Type I IFN, PBL, 11200-2) was purchased from PBL Assay Science.
Recombinant plasmid construction.
Adenoviral-associated vector (AAV) production was performed as previously described (55). sgRNAs targeting firefly luciferase (sgLuc), IRF3 (sgIRF3) (71), TBK1 (sgTBK1), and p65 (sgp65) were cloned into the LentiCRISPR-v2 plasmid (addgene, number 52961). The sequence information for sgRNAs is listed in Table 1.
TABLE 1.
DNA oligonucleotides used in this study
| Purpose | Primer name | Sequence |
|---|---|---|
| Target gene | Luciferase (sgLuc) | CTTCGAAATGTCCGTTCGGT |
| IRF3 (sgIRF3) | GAGGTGACAGCCTTCTACCG | |
| TBK1 (sgTBK1) | AAGGATGTTTGCAAGAACA | |
| p65 (sgp65) | GGAAGATCTCATCCCCACC |
Generation of A549 stable cell lines.
To generate A549 cells stably expressing STINGWT or STINGAII, lentivirus was produced by transfecting pLenti-MCPyVEP-STINGWT-IRES-Puro and pLenti-MCPyVEP-STINGAII-IRES-Puro plasmids into HEK293T cells together with psPAX2 and pMD2.G using Lipofectamine 2000 (Invitrogen). A549 cells were transduced with the purified lentiviruses supplemented with polybrene. Starting on day 2 after transduction, cells were selected using 5 μg/ml puromycin for at least 7 days.
To generate A549 cells stably expressing STINGAII and either sgLuc, sgIRF3, sgTBK1, or sgP65, the plasmid pLenti-MCPyVEP-STINGAII-IRES-Zeocin, pLenti-CRISPR-sgLuc, pLenti-CRISPR-sgIRF3, pLenti-CRISPR-sgTBK1, or pLenti-CRISPR-sgp65 was transfected into HEK293T cells together with psPAX2 and pMD2.G using Lipofectamine 2000 (Invitrogen). A549 cells were cotransduced with pLenti-MCPyVEP-STINGAII-IRES-Zeocin lentivirus and either pLenti-CRISPR-sgLuc, pLenti-CRISPR-sgIRF3, pLenti-CRISPR-sgTBK1, or pLenti-CRISPR-sgP65 lentivirus supplemented with polybrene. Starting on day 2 after transduction, cells were selected using 10 μg/ml puromycin for 3 weeks and then maintained in culture medium containing 100 μg/ml zeocin.
Cell proliferation assay.
A549 parental cells or A549 stably expressing STINGWT or STINGAII were seeded at a density of 5,000 cells in 100 μl of medium per well in a 96-well plate. The cells were incubated in humidified air containing 5% CO2 with or without drug using the amount of time as indicated in the figure legends. Cell viability was measured using CellTiter-Glo 3D (Promega) following the manufacturer’s instructions.
Western blot analysis.
The protein samples were resolved on SDS-PAGE gels, transferred onto polyvinylidene difluoride (PVDF) membranes, and immunoblotted using specific primary antibodies as indicated in the figure legends. The primary antibodies used in this study includes anticoronavirus OC43 nucleoprotein protein (OC43 N) (72) (1:4,000, MAB9012, Millipore), anti-SARS-CoV-2 nucleoprotein (SARS-CoV-2 N) (1:4,000, GTX635712, GenTex), anti-STING (1:1,000, 13647S, Cell Signaling Technology), anti-phospho-STING (Ser366) (1:1,000, 50907S, Cell Signaling Technology), anti- IKKε (1:1,000, 3416S, Cell Signaling Technology), anti-GAPDH (1:2,000, 5174S, Cell Signaling Technology), anti-p65 (1:1,000, sc-8008, Santa Cruz Biotechnology), anti-TBK1 (1:1,000, 3013S, Cell Signaling Technology), anti-ACE2 (1:1,000, 21115-1-AP, Proteintech), and anti-IRF3 (1:250, sc-33641, Santa Cruz Biotechnology). The secondary antibodies used were horseradish peroxidase (HRP)-linked anti-rabbit IgG (1:3,000, 7074S, Cell Signaling Technology) and HRP-linked anti-mouse IgG (1:3,000, 7076S, Cell Signaling Technology). Western blots were developed using Western Lightning ECL solution (PerkinElmer) and the images were captured using a Fuji imaging system.
Adenoviral-associated vector production.
Adenoviral-associated vector (AAV) production was performed as previously described (55). HEK293T cells were cultured in 100-mm dishes. The cells were transfected with 5 μg of serotype packaging plasmid, 10 μg of pAdDeltaF6 helper plasmid (University of Pennsylvania Vector Core), and 5 μg of pscAAV carrying the construct of interest. The cell lysates were prepared with three successive freeze-thaw cycles (−80°C/37°C). The cell lysates were then purified using OptiPrep centrifugation at 300,000 × g for 3.5 h at 16°C. Pure gradient fractions were concentrated and desalted using an Amicon Ultra-15 centrifugal concentrator.
Immunofluorescent staining.
Cells were fixed with 3% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min. IF staining was performed as described previously (73). The following primary antibodies were used: anticoronavirus OC43 N protein (1:2,000, MAB9012, Millipore), anti-SARS-CoV-2 nucleoprotein (1:2,000, GTX635712, GenTex), and anti-STING (1:500, 19851-1-AP, Proteintech). The secondary antibodies used were Alexa Fluor 594 goat anti-mouse IgG (Thermo Fisher Scientific) and Alexa Fluor 488 goat anti-rabbit IgG (Thermo Fisher Scientific). All IF images were collected using an inverted fluorescence microscope (IX81; Olympus) connected to a high-resolution charge-coupled-device camera (FAST1394; QImaging). Images were analyzed and presented using SlideBook (version 5.0) software (Intelligent Imaging Innovations, Inc.). The scale bars were added using ImageJ software.
HCoV-OC43 infection.
HCoV-OC43 was purchased from ATCC and amplified in Vero E6 cells. A549 cells were treated with PBS, 500 units/ml of human IFN Alpha Hybrid universal type I IFN (PBL, 11200-2), AAV virions, DMSO, 10 μg/ml DMXAA, or 100 nM human STING agonists, as described in the figure legends. The cells were washed once with PBS and then treated with HCoV-OC43 virions (10e6 PFU/ml) diluted in serum-free DMEM serum-free (at an MOI of 1) for 1 h at 33°C. The cells were then overlaid with DMEM containing 2% FBS and cultured at 33°C for one more day.
Plaque assay.
Confluent Vero E6 cells cultured in 6-well plates were treated with 200 μl of DMEM containing a serial 10-fold dilution of HCoV-OC43 stock for 1 h at 33°C. Inoculum was overlaid with DMEM plus 0.7% agarose and incubated for 7 days at 33°C. Cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet before plaque counting (24).
SARS-CoV-2 infection.
SARS-CoV-2 (USA-WA1/2020 strain) was obtained from BEI and propagated in Vero E6 cells. A549ACE2 cells stably expressing STINGWT were treated with IFNs, DMSO, or human STING agonist diABZI, as described in the figure legends. The cells were treated with SARS-CoV-2 virions diluted in serum-free DMEM (at an MOI of 5) for 1 h at 37°C. The cells were then overlaid with DMEM containing 2% FBS and cultured at 37°C for one more day.
NDV-GFP infection.
Newcastle disease virus (NDV)-GFP was obtained from Luis Martinez-Sobrido (University of Rochester School of Medicine). A549 cells were treated with PBS or 500 units/ml of human IFN Alpha Hybrid universal type I IFN (PBL, 11200-2). The cells were washed once with PBS and then treated with NDV-GFP virions (diluted in serum-free DMEM) at an MOI of 5 for 1 h at room temperature. The cells were then overlaid with DMEM containing 2% FBS and cultured at 37°C for one more day.
HCoV-OC43 and SARS-CoV-2 infection of ex vivo cultured human lung slices.
Normal human lung tissues were obtained from the Penn-CHOP Lung Biology Institute (LBI) at the University of Pennsylvania. Following our previous skin tissue ex vivo culture protocol (70), the tissues were cut into 700-μm-thick slices using a McIlwain tissue chopper. About 12 tissue slices were frozen in 90% FBS and 10% DMSO in each vial in liquid nitrogen for further experiments.
The slices were thawed and washed in PBS twice and then transferred to 200 μl keratinocyte serum-free medium (Gibco, catalog number 17005042) with 1% penicillin-streptomycin. The slices were treated with DMSO or 1 μM human STING agonist diABZI for 3 h. For HCoV-OC43 infection, the slices were treated with HCoV-OC43 virions diluted in 1 ml keratinocyte serum-free medium at 0.5 × 10e6 PFU/ml. The slices were incubated at 33°C in 5% CO2. At 6 days postinfection, slices were immunostained using an antibody against the OC43 N protein (OC43 N) and counterstained with DAPI (4′,6-diamidino-2-phenylindole). For SARS-CoV-2 infection, the slices were treated with SARS-CoV-2 virions diluted in 1 ml keratinocyte serum-free medium at 10e6 PFU/ml. The slices were incubated at 37°C in 5% CO2. At 10 days postinfection, slices were immunostained using an antibody against the SARS-CoV-2 N protein (SARS-CoV-2 N) and counterstained with DAPI.
ACKNOWLEDGMENTS
We thank Courtney E. Comar for technical support, Luis Martinez-Sobrido (University of Rochester School of Medicine) for NDV-GFP, and the members of our laboratories for helpful discussions.
This work was supported by NIH grants R01CA187718, R01AI140442, R21AR074073, R21AI149761, and T32CA115299, an NCI Cancer Center support grant (NCI P30 CA016520), a Penn CFAR pilot award (P30 AI 045008), and the Penn Center for Research on Coronaviruses and Other Emerging Pathogens.
Contributor Information
Jianxin You, Email: jianyou@pennmedicine.upenn.edu.
Tom Gallagher, Loyola University Chicago.
REFERENCES
- 1.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. 2020. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5:536–544. 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7:439–450. 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cui J, Li F, Shi ZL. 2019. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17:181–192. 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Bai R, Teng JL, Tsang CC, Wang M, Zheng BJ, Chan KH, Yuen KY. 2012. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol 86:3995–4008. 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamre D, Procknow JJ. 1966. A new virus isolated from the human respiratory tract. Proc Soc Exp Biol Med 121:190–193. 10.3181/00379727-121-30734. [DOI] [PubMed] [Google Scholar]
- 6.van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, Wertheim-van Dillen PM, Kaandorp J, Spaargaren J, Berkhout B. 2004. Identification of a new human coronavirus. Nat Med 10:368–373. 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fouchier RA, Hartwig NG, Bestebroer TM, Niemeyer B, de Jong JC, Simon JH, Osterhaus AD. 2004. A previously undescribed coronavirus associated with respiratory disease in humans. Proc Natl Acad Sci U S A 101:6212–6216. 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McIntosh K, Dees JH, Becker WB, Kapikian AZ, Chanock RM. 1967. Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc Natl Acad Sci U S A 57:933–940. 10.1073/pnas.57.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, Yuen KY. 2005. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79:884–895. 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976. 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 11.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ, Group SW, SARS Working Group . 2003. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348:1953–1966. 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 12.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 13.Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181:1036–1045. 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Worldometer. COVID-19 coronavirus pandemic. https://www.worldometers.info/coronavirus/.
- 17.Zumla A, Hui DS, Azhar EI, Memish ZA, Maeurer M. 2020. Reducing mortality from 2019-nCoV: host-directed therapies should be an option. Lancet 395:e35–e36. 10.1016/S0140-6736(20)30305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sohrabi C, Alsafi Z, O'Neill N, Khan M, Kerwan A, Al-Jabir A, Iosifidis C, Agha R. 2020. World Health Organization declares global emergency: areview of the 2019 novel coronavirus (COVID-19). Int J Surg 76:71–76. 10.1016/j.ijsu.2020.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fauci AS, Lane HC, Redfield RR. 2020. Covid-19—navigating the uncharted. N Engl J Med 382:1268–1269. 10.1056/NEJMe2002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, Liu L, Shan H, Lei CL, Hui DSC, Du B, Li LJ, Zeng G, Yuen KY, Chen RC, Tang CL, Wang T, Chen PY, Xiang J, Li SY, Wang JL, Liang ZJ, Peng YX, Wei L, Liu Y, Hu YH, Peng P, Wang JM, Liu JY, Chen Z, Li G, Zheng ZJ, Qiu SQ, Luo J, Ye CJ, Zhu SY, Zhong NS. 2020. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382:1708–1720. 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gates B. 2020. Responding to Covid-19—a once-in-a-century pandemic? N Engl J Med 382:1677–1679. 10.1056/NEJMp2003762. [DOI] [PubMed] [Google Scholar]
- 22.Wu YC, Chen CS, Chan YJ. 2020. The outbreak of COVID-19: an overview. J Chin Med Assoc 83:217–220. 10.1097/JCMA.0000000000000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Felsenstein S, Herbert JA, McNamara PS, Hedrich CM. 2020. COVID-19: immunology and treatment options. Clin Immunol 215:108448. 10.1016/j.clim.2020.108448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Renner DM, Comar CE, Whelan JN, Reyes HM, Cardenas-Diaz FL, Truitt R, Tan LH, Dong B, Alysandratos KD, Huang J, Palmer JN, Adappa ND, Kohanski MA, Kotton DN, Silverman RH, Yang W, Morrisey E, Cohen NA, Weiss SR. 2020. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial derived cells and cardiomyocytes. bioRxiv 10.1101/2020.09.24.312553. [DOI] [PMC free article] [PubMed]
- 25.Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, Levantovsky R, Malle L, Moreira A, Park MD, Pia L, Risson E, Saffern M, Salomé B, Esai Selvan M, Spindler MP, Tan J, van der Heide V, Gregory JK, Alexandropoulos K, Bhardwaj N, Brown BD, Greenbaum B, Gümüş ZH, Homann D, Horowitz A, Kamphorst AO, Curotto de Lafaille MA, Mehandru S, Merad M, Samstein RM, Sinai Immunology Review Project . 2020. Immunology of COVID-19: current state of the science. Immunity 52:910–941. 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prompetchara E, Ketloy C, Palaga T. 2020. Immune responses in COVID-19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol 38:1–9. 10.12932/AP-200220-0772. [DOI] [PubMed] [Google Scholar]
- 27.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Wang K, Xing Y, Tu J, Yang X, Zhao Q, Li K, Chen Z. 2014. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell 5:912–927. 10.1007/s13238-014-0104-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maringer K, Fernandez-Sesma A. 2014. Message in a bottle: lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev 25:669–679. 10.1016/j.cytogfr.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YJ, Finkbeiner SR, Weinblatt D, Emmett MJ, Tameire F, Yousefi M, Yang C, Maehr R, Zhou Q, Shemer R, Dor Y, Li C, Spence JR, Stanger BZ. 2014. De novo formation of insulin-producing “neo-beta cell islets” from intestinal crypts. Cell Rep 6:1046–1058. 10.1016/j.celrep.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun L, Xing Y, Chen X, Zheng Y, Yang Y, Nichols DB, Clementz MA, Banach BS, Li K, Baker SC, Chen Z. 2012. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One 7:e30802. 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spiegel M, Pichlmair A, Martinez-Sobrido L, Cros J, Garcia-Sastre A, Haller O, Weber F. 2005. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J Virol 79:2079–2086. 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, Baric RA, Palese P. 2007. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol 81:548–557. 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu X, Pan J, Tao J, Guo D. 2011. SARS-CoV nucleocapsid protein antagonizes IFN-beta response by targeting initial step of IFN-beta induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 42:37–45. 10.1007/s11262-010-0544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hui KPY, Cheung MC, Perera R, Ng KC, Bui CHT, Ho JCW, Ng MMT, Kuok DIT, Shih KC, Tsao SW, Poon LLM, Peiris M, Nicholls JM, Chan MCW. 2020. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: an analysis in ex-vivo and in-vitro cultures. Lancet Respir Med 8:687–695. 10.1016/S2213-2600(20)30193-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acharya D, Liu G, Gack MU. 2020. Dysregulation of type I interferon responses in COVID-19. Nat Rev Immunol 20:397–398. 10.1038/s41577-020-0346-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Péré H, Charbit B, Bondet V, Chenevier-Gobeaux C, Breillat P, Carlier N, Gauzit R, Morbieu C, Pène F, Marin N, Roche N, Szwebel TA, Merkling SH, Treluyer JM, Veyer D, Mouthon L, Blanc C, Tharaux PL, Rozenberg F, Fischer A, Duffy D, Rieux-Laucat F, Kernéis S, Terrier B. 2020. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369:718–724. 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, Guo F, Zhao Z, Zhou Z, Xiang Z, Wang J. 2020. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 11:3810. 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolff G, Limpens R, Zevenhoven-Dobbe JC, Laugks U, Zheng S, de Jong AWM, Koning RI, Agard DA, Grunewald K, Koster AJ, Snijder EJ, Barcena M. 2020. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 369:1395–1398. 10.1126/science.abd3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. 2021. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol 19:155–170. 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sainz B, Jr., Mossel EC, Peters CJ, Garry RF. 2004. Interferon-beta and interferon-gamma synergistically inhibit the replication of severe acute respiratory syndrome-associated coronavirus (SARS-CoV). Virology 329:11–17. 10.1016/j.virol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, Pan P, Wang W, Hu D, Liu X, Zhang Q, Wu J. 2020. Coronavirus infections and immune responses. J Med Virol 92:424–432. 10.1002/jmv.25685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zumla A, Chan JFW, Azhar EI, Hui DSC, Yuen K-Y. 2016. Coronaviruses—drug discovery and therapeutic options. Nat Rev Drug Discov 15:327–347. 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lokugamage KG, Hage A, de Vries M, Valero-Jimenez AM, Schindewolf C, Dittmann M, Rajsbaum R, Menachery VD. 2020. SARS-CoV-2 is sensitive to type I interferon pretreatment. bioRxiv 10.1101/2020.03.07.982264. [DOI] [PMC free article] [PubMed]
- 45.Stanifer ML, Kee C, Cortese M, Zumaran CM, Triana S, Mukenhirn M, Kraeusslich HG, Alexandrov T, Bartenschlager R, Boulant S. 2020. Critical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells. Cell Rep 32:107863. 10.1016/j.celrep.2020.107863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, Ogishi M, Sabli IKD, Hodeib S, Korol C, Rosain J, Bilguvar K, Ye J, Bolze A, Bigio B, Yang R, Arias AA, Zhou Q, Zhang Y, Onodi F, Korniotis S, Karpf L, Philippot Q, Chbihi M, Bonnet-Madin L, Dorgham K, Smith N, Schneider WM, Razooky BS, Hoffmann H-H, Michailidis E, Moens L, Han JE, Lorenzo L, Bizien L, Meade P, Neehus A-L, Ugurbil AC, Corneau A, Kerner G, Zhang P, Rapaport F, Seeleuthner Y, Manry J, Masson C, Schmitt Y, Schlüter A, Le Voyer T, Khan T, Li J, COVID-STORM Clinicians , et al. 2020. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370:eabd4570. 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, Dorgham K, Philippot Q, Rosain J, Beziat V, Manry J, Shaw E, Haljasmagi L, Peterson P, Lorenzo L, Bizien L, Trouillet-Assant S, Dobbs K, de Jesus AA, Belot A, Kallaste A, Catherinot E, Tandjaoui-Lambiotte Y, Le Pen J, Kerner G, Bigio B, Seeleuthner Y, Yang R, Bolze A, Spaan AN, Delmonte OM, Abers MS, Aiuti A, Casari G, Lampasona V, Piemonti L, Ciceri F, Bilguvar K, Lifton RP, Vasse M, Smadja DM, Migaud M, Hadjadj J, Terrier B, Duffy D, Quintana-Murci L, van de Beek D, Roussel L, Vinh DC, Tangye SG, HGID Lab , et al. 2020. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370:eabd4585. 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni G, Ma Z, Damania B. 2018. cGAS and STING: at the intersection of DNA and RNA virus-sensing networks. PLoS Pathog 14:e1007148. 10.1371/journal.ppat.1007148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai X, Chiu YH, Chen ZJ. 2014. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54:289–296. 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 50.Barber GN. 2015. STING: infection, inflammation and cancer. Nat Rev Immunol 15:760–770. 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franz KM, Neidermyer WJ, Tan YJ, Whelan SPJ, Kagan JC. 2018. STING-dependent translation inhibition restricts RNA virus replication. Proc Natl Acad Sci U S A 115:E2058–E2067. 10.1073/pnas.1716937115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Q, Sun L, Chen ZJ. 2016. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17:1142–1149. 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 54.Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, Tran JL, Moore P, Lehmann S, Eberl HC, Muelbaier M, Schneck JL, Clemens J, Adam M, Mehlmann J, Romano J, Morales A, Kang J, Leister L, Graybill TL, Charnley AK, Ye G, Nevins N, Behnia K, Wolf AI, Kasparcova V, Nurse K, Wang L, Puhl AC, Li Y, Klein M, Hopson CB, Guss J, Bantscheff M, Bergamini G, Reilly MA, Lian Y, Duffy KJ, Adams J, Foley KP, Gough PJ, Marquis RW, Smothers J, Hoos A, Bertin J. 2018. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564:439–443. 10.1038/s41586-018-0705-y. [DOI] [PubMed] [Google Scholar]
- 55.Liu W, Kim GB, Krump NA, Zhou Y, Riley JL, You J. 2020. Selective reactivation of STING signaling to target Merkel cell carcinoma. Proc Natl Acad Sci U S A 117:13730–13739. 10.1073/pnas.1919690117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, Rathinam VA, Monks B, Jin T, Xiao TS, Vogel SN, Vance RE, Fitzgerald KA. 2013. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol 190:5216–5225. 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, Hartmann G, Barchet W, Tuschl T, Patel DJ. 2013. Structure-function analysis of STING activation by c[G(2',5')pA(3',5')p] and targeting by antiviral DMXAA. Cell 154:748–762. 10.1016/j.cell.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao P, Zillinger T, Wang W, Ascano M, Dai P, Hartmann G, Tuschl T, Deng L, Barchet W, Patel DJ. 2014. Binding-pocket and lid-region substitutions render human STING sensitive to the species-specific drug DMXAA. Cell Rep 8:1668–1676. 10.1016/j.celrep.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim S, Li L, Maliga Z, Yin Q, Wu H, Mitchison TJ. 2013. Anticancer flavonoids are mouse-selective STING agonists. ACS Chem Biol 8:1396–1401. 10.1021/cb400264n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abe T, Barber GN. 2014. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol 88:5328–5341. 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chiu YH, Zhao M, Chen ZJ. 2009. Ubiquitin in NF-kappaB signaling. Chem Rev 109:1549–1560. 10.1021/cr800554j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon [corrected] geneinduction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 64.Yarilina A, Ivashkiv LB. 2010. Type I interferon: a new player in TNF signaling. Curr Dir Autoimmun 11:94–104. 10.1159/000289199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonizzi G, Karin M. 2004. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25:280–288. 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 66.Xi Q, Wang M, Jia W, Yang M, Hu J, Jin J, Chen X, Yin D, Wang X. 2020. Design, synthesis, and biological evaluation of amidobenzimidazole derivatives as stimulator of interferon genes (STING) receptor agonists. JMed Chem 63:260–282. 10.1021/acs.jmedchem.9b01567. [DOI] [PubMed] [Google Scholar]
- 67.Channappanavar R, Perlman S. 2017. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol 39:529–539. 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D'Silva DB, Moghaddas F, Tailler M, Lawlor KE, Zhan Y, Burns CJ, Wicks IP, Miner JJ, Kile BT, Masters SL, De Nardo D. 2020. TBK1 and IKKepsilon act redundantly to mediate STING-induced NF-kappaB responses in myeloid cells. Cell Rep 31:107492. 10.1016/j.celrep.2020.03.056. [DOI] [PubMed] [Google Scholar]
- 69.Konno H, Konno K, Barber GN. 2013. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155:688–698. 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu W, Yang R, Payne AS, Schowalter RM, Spurgeon ME, Lambert PF, Xu X, Buck CB, You J. 2016. Identifying the target cells and mechanisms of Merkel cell polyomavirus infection. Cell Host Microbe 19:775–787. 10.1016/j.chom.2016.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sali TM, Pryke KM, Abraham J, Liu A, Archer I, Broeckel R, Staverosky JA, Smith JL, Al-Shammari A, Amsler L, Sheridan K, Nilsen A, Streblow DN, DeFilippis VR. 2015. Characterization of a novel human-specific STING agonist that elicits antiviral activity against emerging alphaviruses. PLoS Pathog 11:e1005324. 10.1371/journal.ppat.1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao X, Guo F, Liu F, Cuconati A, Chang J, Block TM, Guo JT. 2014. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc Natl Acad Sci U S A 111:6756–6761. 10.1073/pnas.1320856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu W, Stein P, Cheng X, Yang W, Shao NY, Morrisey EE, Schultz RM, You J. 2014. BRD4 regulates Nanog expression in mouse embryonic stem cells and preimplantation embryos. Cell Death Differ 21:1950–1960. 10.1038/cdd.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]