

ABSTRACT
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of the ongoing coronavirus disease 2019 (COVID-19) pandemic. While previous studies have shown that several SARS-CoV-2 proteins can antagonize the interferon (IFN) response, some of the mechanisms by which they do so are not well understood. In this study, we describe two novel mechanisms by which SARS-CoV-2 blocks the IFN pathway. Type I IFNs and IFN-stimulated genes (ISGs) were poorly induced during SARS-CoV-2 infection, and once infection was established, cells were highly resistant to ectopic induction of IFNs and ISGs. Levels of two key IFN signaling pathway components, Tyk2 and STAT2, were significantly lower in SARS-CoV-2-infected cells. Expression of nonstructural protein 1 (NSP1) or nucleocapsid in the absence of other viral proteins was sufficient to block IFN induction, but only NSP1 was able to inhibit IFN signaling. Mapping studies suggest that NSP1 prevents IFN induction in part by blocking IRF3 phosphorylation. In addition, NSP1-induced depletion of Tyk2 and STAT2 dampened ISG induction. Together, our data provide new insights into how SARS-CoV-2 successfully evades the IFN system to establish infection.
IMPORTANCE SARS-CoV-2 is the causative agent of COVID-19, a serious disease that can have a myriad of symptoms from loss of taste and smell to pneumonia and hypercoagulation. The rapid spread of SARS-CoV-2 can be attributed in part to asymptomatic transmission, where infected individuals shed large amounts of virus before the onset of disease. This is likely due to the ability of SARS-CoV-2 to effectively suppress the innate immune system, including the IFN response. Indeed, we show that the IFN response is efficiently blocked during SARS-CoV-2 infection, a process that is mediated in large part by nonstructural protein 1 and nucleocapsid. Our study provides new insights on how SARS-CoV-2 evades the IFN response to successfully establish infection. These findings should be considered for the development and administration of therapeutics against SARS-CoV-2.
KEYWORDS: NSP1, SARS-CoV-2, type I interferons, innate immunity
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a recently emerged coronavirus responsible for coronavirus disease 2019 (COVID-19), a disease associated with an ongoing pandemic that has caused over two million deaths. The virus, first reported in the city of Wuhan, China in 2019 (1, 2), rapidly spread across the globe, infecting over a hundred million people and causing one of the worst public health disasters in recent history. Though genetically related to SARS-CoV (3), SARS-CoV-2 is much more efficient in spreading from person to person, including from asymptomatic carriers, making it extremely difficult to contain (4, 5). While the virus was reported to infect people of all age groups, the death rate is especially high among the elderly population. Although a number of vaccine candidates have recently been approved for use, effective antiviral strategies to alleviate severe disease in COVID-19 patients are limited.
The interferon (IFN) system is a critical arm of the mammalian innate immune response against viruses. Double-stranded RNA produced during viral replication can be sensed by cytoplasmic and endosomal pattern recognition receptors (e.g., RIG-I, MDA5, or Toll-like receptors), leading to induction of multiple IFNs (6). The secreted IFNs bind to cell surface receptors that signal through a number of intracellular transducers to drive expression of IFN-stimulated genes (ISGs), resulting in an antiviral state (7). However, most viral pathogens have evolved mechanisms to evade or counteract the IFN system (8). With respect to coronaviruses, several virus-encoded proteins are known to inhibit IFN induction and/or signaling (reviewed in references 9 and 10). A series of recent studies suggest that more than half of viral proteins encoded by SARS-CoV-2 can block the IFN response (11–22). However, the data from these reports are not consistent with each other (11), and the mechanisms involved in SARS-CoV-2-mediated suppression of the IFN response are still poorly understood.
In this study, we used a systematic approach to investigate how SARS-CoV-2 suppresses the host IFN response during infection. Consistent with recent findings from other groups (20, 23), we observed that SARS-CoV-2-infected cells do not mount a robust IFN response. SARS-CoV-2-infected cells were also resistant to IFN induction by Sendai virus, and ISG expression in response to IFN-α treatment was suppressed, indicating that viral determinants actively block both IFN induction and signaling. Analyses of how individual viral protein expression affected the innate immune response revealed that while both nonstructural protein 1 (NSP1) and nucleocapsid protein (N) suppressed IFN induction, only NSP1 significantly inhibited downstream IFN signaling. Of note, strong suppression of IRF3 phosphorylation and depletion of Tyk2 and STAT2, critical components of IFN signaling, were observed during SARS-CoV-2 infection. The depletion of Tyk2 and STAT2 may be the result of NSP1-mediated global reduction of translation. Taken together, our data indicate that SARS-CoV-2 NSP1 and N are strong antagonists of the host IFN response.
RESULTS
SARS-CoV-2 blocks IFN induction.
To understand how the host cell responds to SARS-CoV-2 during infection, we assessed the transcriptional induction of Ifnb mRNA and secretion of IFN-β from ACE2-expressing HEK 293T (HEK 293T-ACE2) cells using quantitative real-time PCR (qRT-PCR) and enzyme-linked immunosorbent assay (ELISA), respectively. While robust replication of the virus was observed in HEK 293T-ACE2 cells, similar to that in Vero E6 cells, by 24 and 48 hpi (Fig. 1A), no significant increase in Ifnb mRNA (Fig. 1B) or secreted IFN-β (Fig. 1C) was observed in response to infection. Conversely, infection of these cells with Sendai virus (SeV) resulted in increased secretion of IFN-β into culture medium. In contrast, cells which were first infected with SARS-CoV-2 secreted very low levels of IFN-β in response to SeV infection (Fig. 1C). Similarly, no induction of the ISG IFIT1 was observed in SARS-CoV-2-infected cells, and significantly lower levels were induced upon challenge with SeV or poly(I·C) (Fig. 1D and E). These results are in agreement with a recent study describing human airway epithelial cell cultures infected with SARS-CoV-2 (23). Next, we examined the ability of SARS-CoV-2 to infect cells in which type I IFN had been induced by SeV either 8 h preinfection or 16 h postinfection (hpi). Induction of IFNs by SeV pretreatment significantly reduced SARS-CoV-2 replication (Fig. 1F). In contrast, addition of SeV to cells already infected with SARS-CoV-2 had little effect, indicating that this coronavirus actively blocks IFN induction (Fig. 1F). Of note, similar amounts of intracellular SeV RNA were detected from mock- and SARS-CoV-2-infected cells after 16 h of SeV infection (Fig. 1G), indicating that SARS-CoV-2-infected cells are as permissive for SeV infection as mock-infected cells. The suppression of IFN induction during SARS-CoV-2 infection was further evident from the lack of phosphorylation of IRF3 (Fig. 1H) and absence of SeV-induced-IRF3 transport into the nucleus (Fig. 1I).
FIG 1.

SARS-CoV-2 blocks IFN induction. (A) Vero E6 and HEK 293T-ACE2 cells were infected with SARS-CoV-2 (multiplicity of infection [MOI] = 1), and total RNA was harvested at 0, 8, 16, 24, and 48 h postinfection (hpi). Viral RNA level was measured by qRT-PCR, normalized to the ACTB mRNA level, and expressed as fold values relative to mock-infected cells. (B) HEK 293T-ACE2 cells were infected with SARS-CoV-2 (MOI = 1), and total RNA was harvested at 24 and 48 hpi. Ifnb level was measured by qRT-PCR, normalized to ACTB mRNA level, and expressed as fold values relative to mock-infected cells. (C) Mock- or SARS-CoV-2-infected (30 hpi) HEK 293T-ACE2 cells were challenged with 50 hemagglutination units (HAU)/ml Sendai virus (SeV) for 16 h. IFN-β in cell culture supernatants was measured by ELISA. (D and E) HEK 293T-ACE2 cells were infected with SARS-CoV-2 (MOI = 1). After 24 h, the cells were transfected with IFIT1 firefly luciferase reporter and control Renilla reporter plasmids and then challenged with 100 HAU/ml of SeV (D) or 2 μg/ml of poly(I·C) (E) for 16 h. Firefly and Renilla luciferase activities were measured in cell lysates, after which the IFIT1 reporter luciferase activity was normalized against Renilla reporter values and further normalized to the activity in uninduced mock-infected cells. (F) HEK 293T-ACE2 cells infected with SeV (50 HAU/ml) for 8 h preinfection (pretreatment) or 16 h postinfection (posttreatment) were subsequently infected with SARS-CoV-2 (MOI = 1). Total RNA was harvested 48 hpi. SARS-CoV-2 genomic RNA was measured by qRT-PCR, normalized to ACTB mRNA level, and expressed as fold values relative to mock-infected cells. (G) HEK 293T cells were infected with SARS-CoV-2 (MOI = 1). After 24 h, cells were infected with SeV (50 HAU/ml) for 16 h, after which total RNA was harvested. SeV genomic RNA was measured by qRT-PCR, normalized to ACTB mRNA level, and expressed as fold value relative to mock-infected cells. (H) HEK 293T-ACE2 cells were infected with SARS-CoV-2 (MOI = 1) for 16 h and then mock infected or infected with 50 HAU/ml of SeV for 8 h. Cell lysates were subjected to immunoblotting using antibodies against spike, IRF3, phospho-IRF3, and β-actin. (I) A549 cells were electroporated with plasmid encoding ACE2 and then infected with SARS-CoV-2 (MOI = 1) for 16 h, followed by challenge with 50 HAU/ml of SeV for 8 h. Cells were fixed, and spike and IRF3 localization was determined by indirect-immunofluorescence analysis. Data are means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01.
SARS-CoV-2 NSP1 and N block IFN induction.
To determine which viral proteins were responsible for antagonizing type I IFN induction, we constructed expression plasmids for all known proteins of SARS-CoV-2 with C-terminal 3× FLAG tags using cDNA generated from infected-cell lysates. NSP10 was tagged with 3× FLAG on its N terminus due to the instability of the C-terminally tagged version. The expression and molecular weights of most sequence-verified constructs were determined by immunoblotting (Fig. 2A). Expression of NSP6 and ORF10 was verified by immunoprecipitation coupled with immunoblotting (Fig. 2B), whereas expression of NSP3 (>250 kDa) was confirmed by indirect immunofluorescence (IF) (Fig. 2C).
FIG 2.
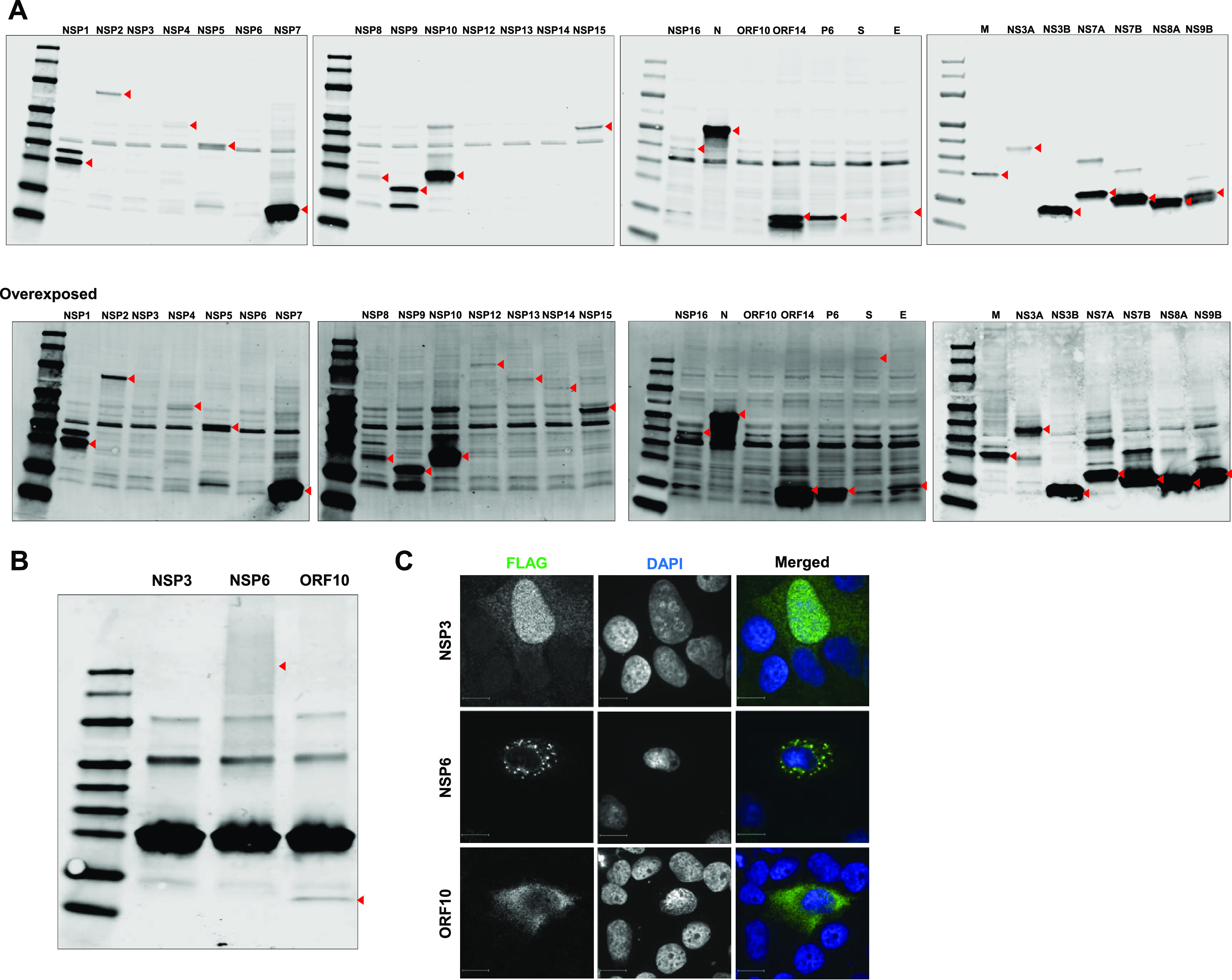
Expression of SARS-CoV-2 proteins. (A) Huh7 cells were transfected with plasmids encoding the indicated viral proteins with 3× FLAG tags. After 24 h, cell lysates were subjected to immunoblotting with antibodies against FLAG. The positions of the FLAG-tagged viral proteins are indicated with red arrowheads. (B) HEK 293T cells were transfected with plasmids encoding FLAG-tagged SARS-CoV-2 NSP3, NSP6, and ORF10, for which protein expression was not confirmed by Western blotting. Cells were harvested 24 h posttransfection, immunoprecipitated with anti-FLAG antibody, and subjected to Western blot analysis with immunostaining with antibody against FLAG. (C) Huh7 cells were transfected with NSP3, NSP6, and ORF10 for 48 h, fixed, and imaged by IF microscopy after staining using antibody against FLAG.
The effect of SARS-CoV-2 proteins on SeV-induced IFN induction was assessed using an IFN-β luciferase reporter assay and ELISA. Expression of the N and NSP1 proteins reduced IFN-β reporter activity by 70% and 30%, respectively (Fig. 3A). Secretion of IFN-β was also significantly impaired in cells expressing these viral proteins (Fig. 3B). As the antiviral transcription factors IRF3 and NF-κB are activated during the induction of IFNs (6), we assessed whether N or NSP1 affected their activities. Whereas both viral proteins inhibited IRF3-dependent transcription by >50%, NF-κB reporter activity was more strongly affected by NSP1 (64% reduction) (Fig. 3C and D). We next examined whether localization of IRF3 was affected in SeV-infected cells expressing N protein or NSP1. IRF3 did not translocate to nuclei of NSP1-expressing cells, which is consistent with the observation that this viral protein inhibits IFN induction (Fig. 3E and F). Conversely, nuclear accumulation of IRF3 was not significantly affected in cells expressing N protein or the control protein NS3A (Fig. 3E and F). Mapping studies using overexpression of IFN induction pathway components suggest that one critical step targeted by NSP1 is the phosphorylation of IRF3 (Fig. 3G). Specifically, NSP1-mediated reduction in IRF3 signaling was rescued by expression of a constitutively active form of IRF3 (IRF3-5D). We tested how expression of an NSP1 mutant (KH164AA) that is unable to suppress translation (24) affected induction of type I IFNs. The KH164AA mutant was unable to abrogate IFN-β reporter activity (Fig. 4A and B), and this phenotype corresponded to its inability to block IRF3 nuclear accumulation (Fig. 4C and D). These data strongly suggest that NSP1-mediated suppression of IFN production is linked to its ability to shut down host translational machinery and nuclear transport of IRF3.
FIG 3.

SARS-CoV-2 NSP1 and N block IFN induction. (A) HEK 293T cells were transfected with plasmids encoding the indicated viral proteins, IFN-β firefly luciferase reporter and control Renilla reporter. Twenty-four hours later cells were infected with 100 HAU/ml of SeV for 16 h, after which firefly and Renilla luciferase activities were measured in cell lysates. IFN-β reporter activity was normalized against Renilla reporter values, and the data are presented as fold activity relative to a pcDNA empty vector control. (B) HEK 293T cells were transfected with plasmids encoding NSP1, NS3A, or N and 24 h later challenged with 100 HAU/ml of Sendai virus for 16 h. The culture supernatants were harvested, and IFN-β levels were determined by ELISA. (C and D) HEK 293T cells were transfected with plasmids encoding NSP1 or N proteins and firefly luciferase under the control of IRF3 (C)- or NF-κB (D)-responsive promoters as well as a constitutively expressed Renilla luciferase reporter. Twenty-four hours later, cells were challenged with 100 HAU/ml of SeV for 16 h, after which firefly and Renilla luciferase activities were measured in cell lysates. The firefly luciferase activity was normalized against Renilla luciferase values, and the data are presented as fold activity relative to a pcDNA empty vector control. (E and F) A549 cells were transfected with plasmids encoding NSP1, N, or NS3A and 24 h later challenged with 100 HAU/ml of SeV for 8 h. The cells were then processed for indirect immunofluorescence using antibodies against FLAG and IRF3. The cytoplasmic and nuclear IRF3 signal were quantitated using Volocity software (n = 30). (G) HEK 293T cells were transfected with plasmids encoding NSP1 or N and RIG-I, IKKε, TBK1, IRF3, or IRF3-5D, IRF3-promoter firefly luciferase reporter, and constitutively expressed Renilla luciferase reporter. Samples were harvested at 24 h posttransfection, after which firefly and Renilla luciferase activities were measured in cell lysates. The firefly luciferase activity was normalized against Renilla luciferase values, and the data are presented as fold activity relative to a pcDNA empty vector control. Data are means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
FIG 4.

Translational shut off by NSP1 and TRIM25-independent mechanism by N blocks IFN induction. (A) HEK 293T cells were transfected with wither empty vector (pcDNA) and plasmids encoding wild-type NSP1 or mutant NSP1-KH164AA for 24 h. Cell lysates were then subjected to immunoblotting with antibodies against FLAG. (B) HEK 293T cells were transfected with empty vector (pcDNA) or plasmids encoding wild-type NSP1 or mutant NSP1-KH164AA, IFN-β-responsive firefly luciferase reporter, and control Renilla reporter. After 24 h, the cells were challenged with 100 HAU/ml of SeV for 16 h and then harvested for luciferase assays. The firefly reporter activities were normalized against Renilla reporter values, and the data are presented as fold activity relative to uninduced pcDNA empty vector control. (C and D) A549 cells were transfected with plasmid encoding wild-type NSP1, mutant (KH164AA) NSP1, or NS7B (negative control) for 24 h, after which cells were challenged with 100 HAU/ml of SeV for 8 h. Samples were then processed for indirect immunofluorescence microscopy using antibodies against FLAG and IRF3. The fluorescent intensities in the nucleus and cytoplasm were measured using Volocity software (n = 20). (E) HEK 293T cells were transfected with plasmids encoding SARS-CoV-2 N or NS3A proteins or empty vector (pcDNA). After 48 h, cell lysates were immunoprecipitated with anti-FLAG antibody and then subjected immunoblot analysis using antibodies against FLAG, TRIM25, and β-actin. (F) HEK 293T cells were transfected with a plasmid encoding FLAG-TRIM25 or empty vector (pcDNA) for 48 h, after which cell lysates were subjected to immunoblot analysis using antibodies against TRIM25 and β-actin. (G) HEK 293T-ACE2 cells were transfected with a plasmid encoding FLAG-tagged TRIM25 or empty vector (pcDNA) for 48 h. Cells were then infected with SARS-CoV-2 (MOI = 1) for 48 h, after which total RNA was harvested and subjected to qRT-PCR to quantify viral genomic RNA, which was normalized to ACTB mRNA level and expressed as fold values relative to pcDNA empty vector-transfected cells. (H) HEK 293T cells were transfected with plasmids encoding GST-tagged human RIG-I CARD domains (GST-h2CARD) or GST alone, together with HA-tagged ubiquitin (HA-Ub), V5-tagged human TRIM25 (hTRIM25-V5), and the indicated FLAG-tagged viral proteins. Clarified whole-cell lysates were subjected to GST pulldown (IP: GST), followed by immunoblot analysis with anti-GST, anti-HA, anti-V5, and anti-FLAG antibodies. Influenza A virus (IAV) NS1 served as a positive control for blocking TRIM25-mediated ubiquitination of the RIG-I CARD domains. Data are means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further understand how N protein affected IFN induction, we probed for an interaction between SARS-CoV-2 N protein and TRIM25. Previously it was reported that the N protein of the closely related coronavirus SARS-CoV inhibits TRIM25-mediated ubiquitination of RIG-I (25), an essential step in RIG-I activation. Stable interaction between N protein and TRIM25 was observed by coimmunoprecipitation (Fig. 4E), and the importance of TRIM25 in restricting SARS-CoV-2 replication was demonstrated by overexpression of TRIM25 (Fig. 4F and G). However, N protein did not impair RIG-I ubiquitination by TRIM25 (Fig. 4H), and thus, the mechanism of N-mediated suppression of IFN-β induction remains to be elucidated.
SARS-CoV-2 blocks ISG induction.
To investigate how SARS-CoV-2 affects the signaling arm of the IFN response, we first assessed the induction of ISGs in SARS-CoV-2-infected HEK 293T-ACE2 cells. While robust viral replication was observed in these cells (Fig. 1A), no significant induction of the ISG Ifit1 mRNA was detected (Fig. 5A). Conversely, treatment of HEK 293T-ACE2 cells with IFN-α strongly upregulated Ifit1 expression, indicating the presence of a functional IFN signaling pathway in these cells. Next, we examined the sensitivity of SARS-CoV-2 to pre- and postinfection treatment with IFN-α, IFN-γ, and IFN-λ. Consistent with recent reports (20, 23, 26, 27), replication of SARS-CoV-2 was strongly inhibited by pretreatment with IFN-α, whereas posttreatment had only a moderate effect (Fig. 5B). Interestingly, neither pre- nor posttreatment of cells with IFN-γ reduced viral replication, whereas IFN-λ inhibited virus replication but not to the same degree as IFN-α. These results were further confirmed by IFN-stimulated response element (ISRE) promoter reporter assays, in which strong inhibition was observed in SARS-CoV-2-infected cells (Fig. 5C). In contrast, IFN-γ activation site (GAS) promoter activity was not inhibited in SARS-CoV-2-infected cells treated with IFN-γ (Fig. 5D). We next examined how SARS-CoV-2 affected levels of cellular proteins involved in type I IFN signaling. Immunoblot analysis revealed that levels of STAT2 and Tyk2 were significantly reduced during infection, whereas IFNAR1, Jak1, and STAT1 levels were largely unaffected (Fig. 5E and F).
FIG 5.
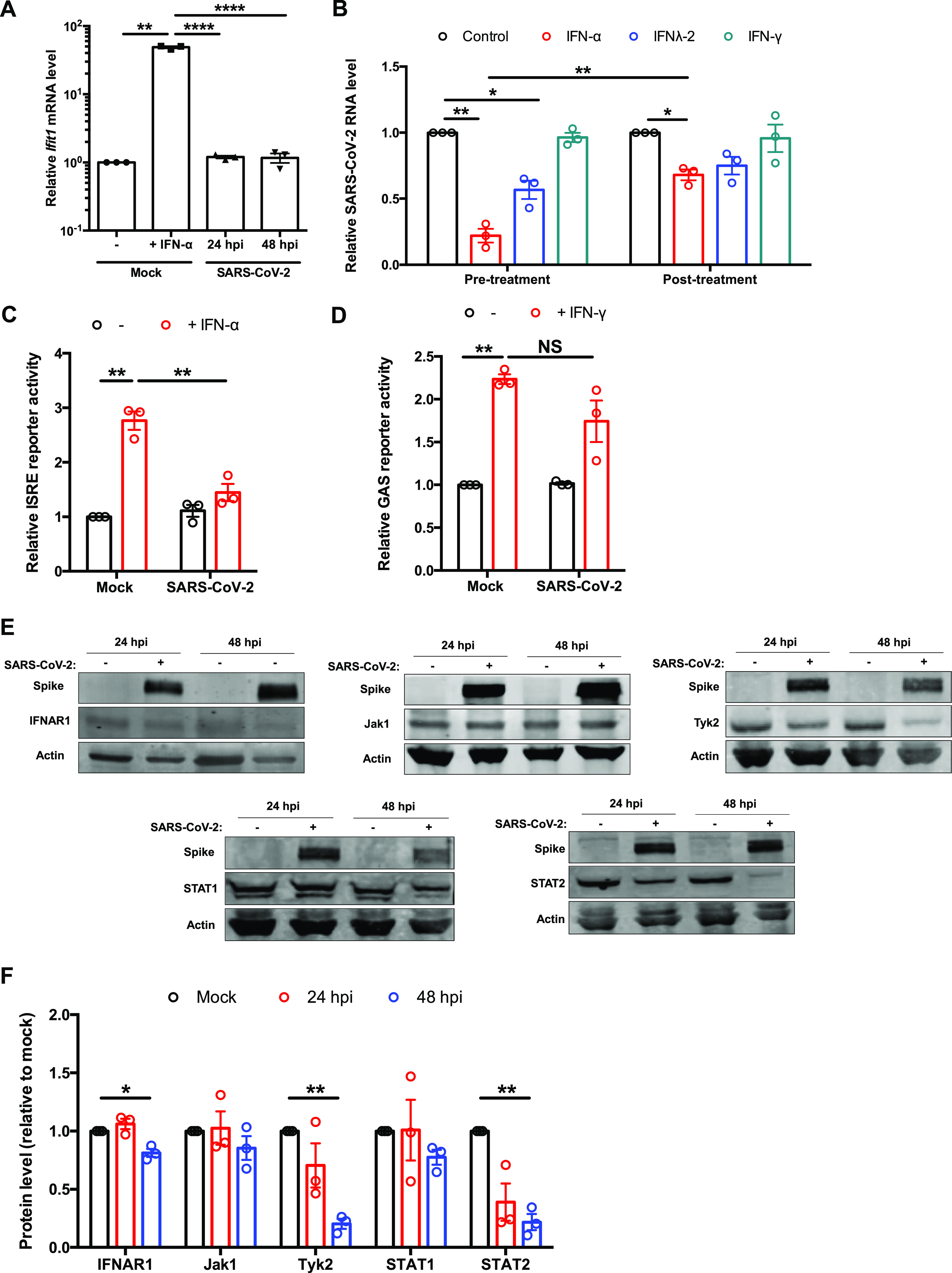
SARS-CoV-2 blocks ISG induction. (A) HEK 293T-ACE2 cells were infected with SARS-CoV-2 (MOI = 2), and total RNA was harvested at 24 and 48 hpi. Mock-infected cells were treated with IFN-α (100 U/ml) for 16 h as a control. Ifit1 level was measured by qRT-PCR and normalized to the ACTB mRNA level. (B) HEK 293T-ACE2 cells were treated with IFN-α (100 U/ml), IFN-λ (100 ng/ml), or IFN-γ (10 U/ml) for 6 h preinfection (pretreatment) or 16 hpi (posttreatment) of SARS-CoV-2 (MOI = 1). Total RNA was harvested 48 hpi, and viral genomic RNA was measured by qRT-PCR. All values are expressed as fold values relative to mock-infected samples. (C) HEK 293T-ACE2 cells were infected with SARS-CoV-2 (MOI = 1), transfected 24 h later with ISRE firefly luciferase reporter and control Renilla reporter plasmids, and then induced with 100 U/ml of IFN-α for 16 h. The samples were harvested and processed by luciferase assay. The ISRE reporter activity was normalized against Renilla reporter values, which were further normalized to values for the uninduced mock-infected cells. (D) HEK 293T-ACE2 cells were infected with SARS-CoV-2 (MOI = 1), transfected 24 h later with IFN-γ-responsive (GAS) firefly luciferase reporter and control Renilla reporter, and induced with 10 U/ml of IFN-γ. After 16 h, luciferase activities were measured, and GAS-dependent luciferase activities were normalized against Renilla reporter values; the data are presented as fold activity relative to mock samples. (E and F) HEK 293T-ACE2 cells were infected with SARS-CoV-2 (MOI = 1), and cell lysates collected 24 and 48 hpi were subjected to immunoblotting using antibodies against spike, IFNAR1, Jak1, Tyk2, STAT1, STAT2, and β-actin. The intensities of the protein bands were measured using Image Studio software, normalized to β-actin level, and expressed as fold values relative to mock-infected controls. Data are means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; NS, not significant.
Next, infected cells were treated with IFN-α or IFN-γ for 2 h, after which localization of STAT1 and STAT2 was examined by confocal microscopy. Compared to uninfected cells, STAT2 fluorescence was greatly diminished in infected cells, and nuclear translocation of the protein in response to IFN treatment was not evident (Fig. 6A). In contrast, neither the STAT1 fluorescent signal nor its nuclear translocation was noticeably affected by infection (Fig. 6B). To determine how SARS-CoV-2-mediated STAT2 depletion occurred, we treated infected cells with inhibitors of proteasome (epoxomicin)- and lysosome (bafilomycin A1)-dependent degradation. Epoxomicin treatment partially rescued STAT2 levels, suggesting that depletion of this transcription factor during SARS-CoV-2 infection is mediated at least in part by proteasomal degradation (Fig. 6C and D). As SARS-CoV-2 is known to suppress host cell translation (28), we assessed whether the loss of STAT2 and potentially other antiviral host factors was due to this effect. When green fluorescent protein (GFP)-expressing cells were infected with SARS-CoV-2, there was no reduction in GFP mRNA but a 30% decrease in protein levels was observed (Fig. 6E to G). Treatment of uninfected cells with cycloheximide resulted in reduction of STAT2 and Tyk2 protein levels comparable to those seen during infection (Fig. 6H and I), suggesting that virus-mediated suppression of host cell translation is important for blocking IFN signaling.
FIG 6.
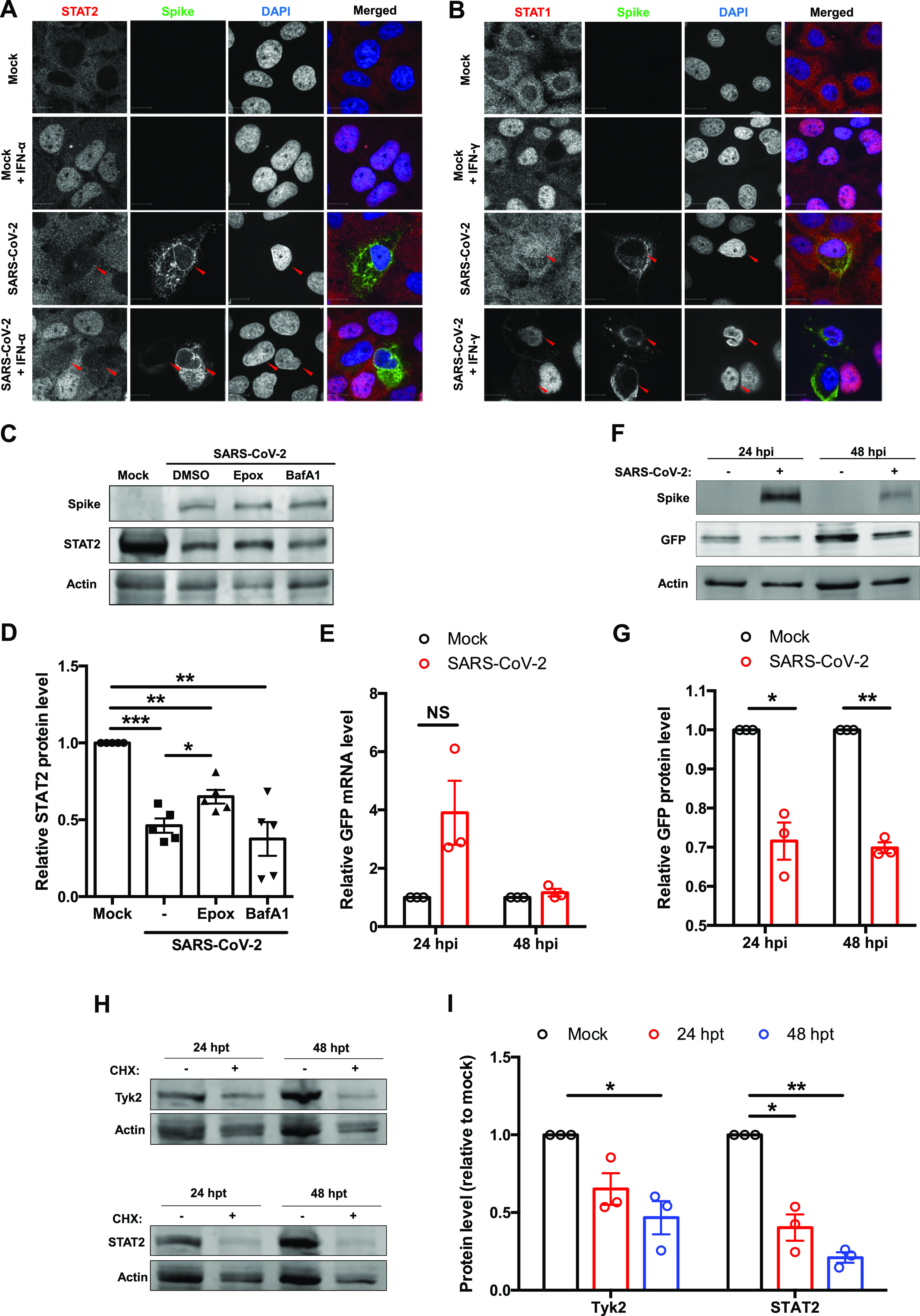
Host cell protein expression during SARS-CoV-2 infection. (A and B) Huh7 cells were infected with SARS-CoV-2 (MOI = 0.5) and at 46 hpi treated with IFN-α (100 U/ml) for 2 h. Cells were then processed for indirect immunofluorescence using antibodies against spike and STAT2 (A) or STAT1 (B). (C and D) HEK 293T-ACE2 cells were infected with SARS-CoV-2 (MOI = 2) and at 16 hpi treated with either dimethyl sulfoxide (DMSO), 100 μM epoxomicin, or bafilomycin A1 for 36 h. Cell lysates were subjected to immunoblotting using antibodies against spike, STAT2, and β-actin. The intensities of the protein bands were measured using Image Studio software, normalized to β-actin level, and expressed as fold values relative to uninfected controls (n = 5). (E to G) HEK 293T-ACE2 cells were transduced with a lentivirus encoding AcGFP for 4 h and then infected with SARS-CoV-2 (MOI = 1). Total RNA and proteins were extracted at 24 and 48 hpi, and relative GFP transcript (normalized to ACTB mRNA) and protein (normalized to β-actin) levels were determined by qRT-PCR and immunoblotting, respectively. (H and I) HEK 293T cells were treated with 100 μM cycloheximide for 24 or 48 h, after which cell lysates were subjected to immunoblotting using antibodies against STAT2, Tyk2, and β-actin. The intensities of the protein bands were measured using Image Studio software, normalized to β-actin levels, and expressed as fold values relative to uninfected control. Data are means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant.
SARS-CoV-2 NSP1 blocks IFN signaling.
To identify the viral proteins that inhibit IFN signaling, we performed ISRE reporter assays in cells expressing individual SARS-CoV-2 proteins. NSP1 was the only viral protein that significantly suppressed ISRE reporter activity (Fig. 7A). To examine the mechanism behind this phenomenon, we again employed the NSP1 mutant KH164AA, which is unable to inhibit host cell translation. The mutant NSP1 did not block ISRE-dependent transcription (Fig. 7B), which is consistent with a role for viral translational suppression in preventing IFN induction (12, 24, 29). Levels of STAT2 in cells expressing NSP1 were also examined by confocal microscopy. Compared to signal in cells expressing SARS-CoV-2 NS7B or the NSP1 KH164AA mutant, STAT2 signal was significantly reduced in cells expressing wild-type NSP1 (Fig. 7C and D). These results were consistent with immunoblotting data, in which levels of ectopically expressed STAT2-GFP were reduced by wild-type NSP1 but not by the KH164AA mutant (Fig. 8A and B). Furthermore, in cells coexpressing Renilla luciferase and wild-type NSP1, a 40% reduction in luciferase reporter activity was observed (Fig. 8C). Similarly, in cells expressing GFP and NSP1, there was no significant reduction in mRNA level but a 40 to 45% reduction in protein level of GFP was observed (Fig. 8D to F), demonstrating an inhibitory effect of NSP1 on overall protein production. NSP1-mediated depletion of STAT2 could be partially rescued by treatment with epoxomicin (Fig. 8G and H), indicating that proteasome-mediated degradation plays a role in STAT2 depletion. However, although the difference was statistically insignificant, epoxomicin treatment also increased STAT2 protein levels in control groups (Fig. 8G and H), indicating that STAT2 is normally turned over via the proteasomal machinery. Our data are consistent with a scenario where virus-induced global translational shutdown leads to depletion of critical host antiviral factors with short half-lives, such as STAT2 and Tyk2, that are needed for induction of ISGs.
FIG 7.
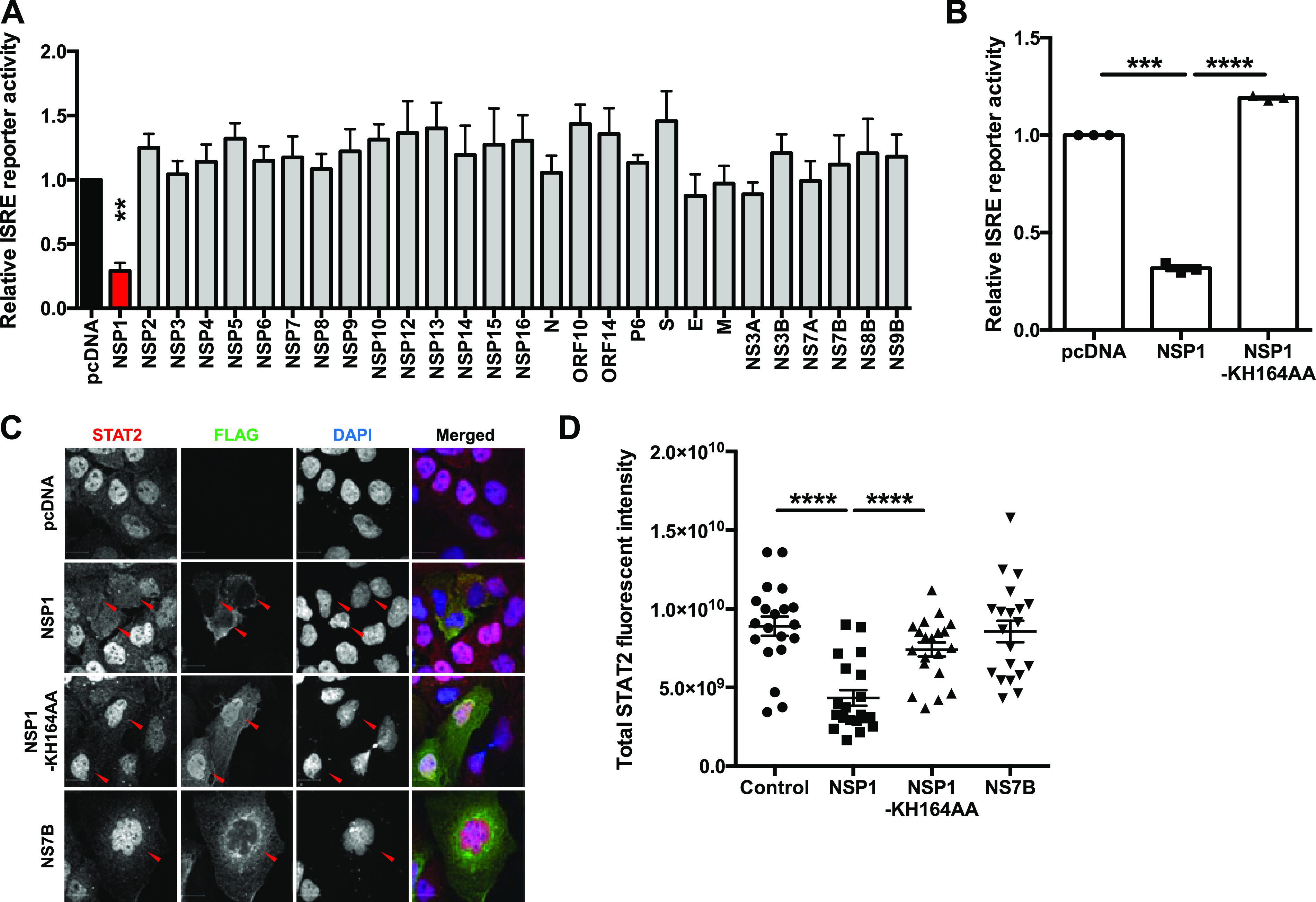
SARS-CoV-2 NSP1 blocks IFN signaling. (A) HEK 293T cells were transfected with plasmids encoding the indicated viral proteins, ISRE firefly luciferase reporter, and control Renilla luciferase reporter. Twenty-four hours later, cells were induced with 100 U/ml of IFN-α for 16 h. Firefly and Renilla reporter activity was measured by luciferase assay. The ISRE reporter activity was normalized against Renilla reporter values, and the data are presented as fold activity relative to the pcDNA empty vector control. (B) HEK 293T cells were transfected with pcDNA carrying the indicated proteins, ISRE firefly luciferase reporter, and control Renilla reporter. The cells were induced 24 h later with 100 U/ml of IFN-α for 16 h, and then firefly and Renilla luciferase activities were measured. The ISRE reporter activity was normalized against Renilla reporter values, and the data are presented as fold activity relative to the pcDNA empty vector control. (C and D) Huh7 cells were transfected with plasmids encoding the indicated SARS-CoV-2 proteins. Twenty-four hours later, cells were induced with 100 U/ml of IFN-α for 2 h and then processed for indirect immunofluorescence microscopy with antibodies against FLAG and STAT2. The total fluorescent intensity of STAT2 was measured using Volocity software (n = 20). Data are means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
FIG 8.
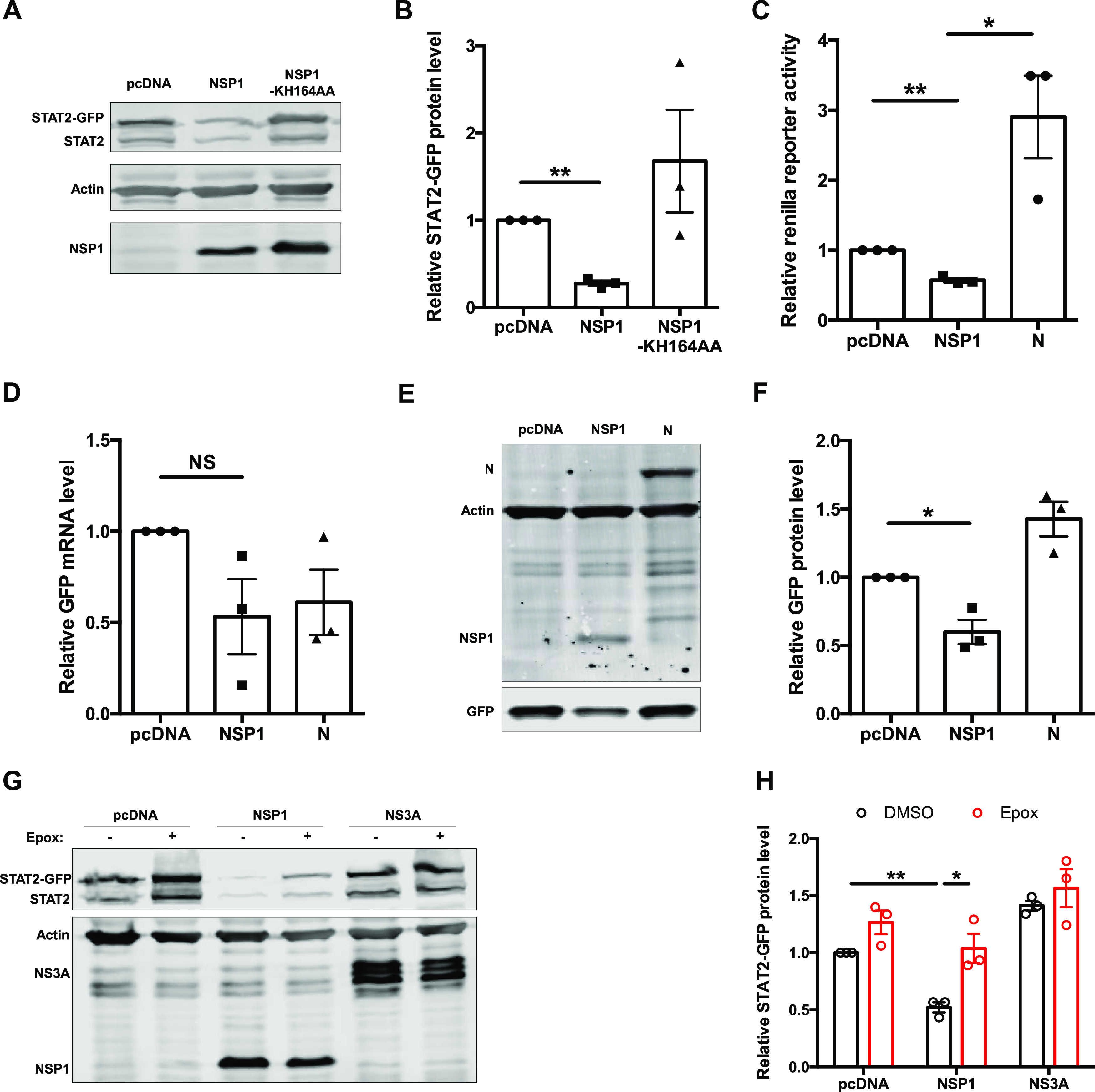
Host cell protein expression in SARS-CoV-2 NSP1-transfected cells. (A and B) HEK 293T cells were transfected with plasmids encoding wild-type or mutant (KH164AA) NSP1 proteins and STAT2-GFP for 24 h, after which lysates were subjected to immunoblotting with antibodies against FLAG, STAT2, and β-actin. The intensities of the bands were calculated using Image Studio software, normalized to β-actin levels, and expressed as fold values compared to empty vector-transfected samples. (C) HEK 293T cells were transfected with plasmids encoding NSP1 or N protein or empty vector (pcDNA) and Renilla luciferase reporter. After 24 h, cell lysates were subjected to luciferase assay. The data are presented as fold activity relative to the empty vector control. (D to F) HEK 293T cells were transduced with lentivirus encoding AcGFP for 4 h and then transfected with plasmids encoding NSP1 or N protein for 24 h. Total RNA and protein were harvested, and GFP transcript (normalized to ACTB mRNA) and protein (normalized to β-actin) levels were determined by qRT-PCR and immunoblotting, respectively. (G and H) HEK 293T cells were transfected with plasmids encoding NSP1 or NS3A and STAT2-GFP. After 24 h, cells were treated with either DMSO or 100 μM epoxomicin for 24 h. Cell lysates were then processed by immunoblotting with antibodies against FLAG, STAT2, and β-actin. Data are means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; NS, not significant.
DISCUSSION
In this study, we systematically examined the interaction between SARS-CoV-2 and the IFN system in human cells. SARS-CoV-2-infected cells poorly induced IFNs and ISGs in our experiments, which is in agreement with recent findings from other groups (20, 23). Using SeV as an IFN agonist, we confirmed that the poor type I IFN production in infected cells was indeed due to an active block in the pathway, which was further corroborated by reduced IRF3 activation and nuclear transport in infected cells even after activation with SeV.
To identify the viral determinants behind this phenotype, we cloned all known viral proteins from viral cDNA as 3× FLAG-tagged constructs. Using these constructs in IFN-β reporter assays, we identified the role of NSP1 and N protein in reducing transcription of type I IFNs. Interestingly, we did not observe any reduction in IFN induction by any other proteins, including NSP3, NSP6, NSP13, NSP14, NSP15, P6, NS3B, NS9B, NS8, and M protein, that have been implicated in blocking IFN induction by other groups (13, 15–18, 20, 21). These inconsistencies may be due to the use of codon-optimized versions of the plasmids in other studies and/or the difference in protein tags used and their positions in the viral proteins. The expression levels of codon-optimized versions of viral protein constructs would certainly be higher than those in this study or perhaps even during infection, both of which use bona fide codons in the viral genome. Thus, whether all the reported proteins expressed from codon-optimized plasmid actually play roles in blocking IFN production during infection requires further analyses.
While both N and NSP1 reduced signaling through IRF3 and NF-κB, only expression of NSP1 significantly reduced IRF3 translocation into the nucleus. The inhibitory step of NSP1 in the IFN induction pathway was mapped to phosphorylation of IRF3, an observation not reported before. These results are consistent with our observations in infected cells and suggest that NSP1 likely blocks type I IFN induction at multiple steps. Interestingly, the mutant NSP1 (KH164AA) that cannot block translation was also unable to block IRF3 nuclear translocation or IFN production. This is either due to the depletion of key factors for IRF3 phosphorylation by translational shutoff or due to the mutation on NSP1 resulting in pleotropic effects. In fact, previous mutagenesis studies on SARS-CoV NSP1 demonstrated that most mutations result in altering more than one function of the protein (30). While N protein of both SARS-CoV and SARS-CoV-2 interacts with the E3 ubiquitin ligase TRIM25 (25), the SARS-CoV-2 N protein does not inhibit the RIG-I ubiquitination as a means to block IFN induction. As we did not observe a significant reduction in IRF3 nuclear transport or IRF3 reporter activity in mapping experiments, the mechanism of action of N protein is unclear at the moment and requires further investigation.
SARS-CoV-2-infected cells also failed to induce ISGs, indicating that the virus actively blocks IFN signaling. This was further supported by our observation that while the virus was sensitive to pretreatment of cells with type I and III IFNs, infected cells were highly resistant to IFN treatment. Furthermore, SARS-CoV-2 infection suppressed ISRE reporter activity in response to IFN-α treatment but did not inhibit GAS reporter activity following addition of IFN-γ. These observations are consistent with earlier reports that while SARS-CoV-2 infection does not activate IFN, the virus is sensitive to type I and III IFNs (20, 23). To better understand this phenomenon, we examined the levels of the type I IFN signaling components IFNAR1, Jak1, Tyk2, STAT1, and STAT2 during SARS-CoV-2 infection. IFNAR1 and Jak1 were largely unaffected, but there was significant loss of STAT2 and Tyk2, which could explain the defect in type I and type III IFN signaling. Our data suggest that the NSP1-dependent inhibition of host cell translation in infected cells results in the depletion of proteins with shorter half-lives, such as Tyk2 and STAT2, making these cells impervious to IFN signaling. Reduction in Tyk2 protein levels could explain the decrease in STAT1 phosphorylation observed during SARS-CoV-2 infection (20, 21, 31).
Together, our findings extend those of others (11–22) which indicate that SARS-CoV-2 uses multiple strategies to interfere with the production of type I IFNs and their downstream signaling. Once established, SARS-CoV-2 infection is largely impervious to IFN agonists and IFN treatment. NSP1 is an important viral determinant that blocks IFN induction, in part by blocking IRF3 phosphorylation and nuclear transport. Moreover, by interfering with host cell translation, NSP1 causes depletion of labile antiviral factors, including Tyk2 and STAT2. Further studies will focus on elucidating the precise mechanisms by which NSP1 blocks phosphorylation of IRF3 as well as how N protein blocks IFN induction.
MATERIALS AND METHODS
Cell culture and virus infection.
A549, Vero E6, HEK 293T, and Huh7 cells from the American Type Culture Collection (Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 100 U/ml penicillin and streptomycin, 1 mM HEPES (Gibco), 2 mM glutamine (Gibco), and 10% heat-inactivated fetal bovine serum (FBS) at 37°C in 5% CO2. SARS-CoV-2 (hCoV-19/Canada/ON-VIDO-01/2020; GISAID accession no. EPI_ISL_425177) was kindly provided by Darryl Falzarano (Vaccine and Infectious Disease Organization, Saskatoon, Canada). HEK 293T-ACE2 cells were developed by electroporating a plasmid encoding human ACE2 (32) (Addgene plasmid no. 1786; a gift from Hyeryun Choe) into HEK 293T cells. The cells were passaged six times in culture, surface-stained for ACE2 (goat anti-ACE2; AF933-SP; R&D Systems), and the highest 2% of cells expressing ACE2 flow were sorted from the bulk population. Virus culture and experiments were performed according to level 3 containment procedures. Virus stocks were generated and titrated (by plaque assay) in Vero E6 cells. Sendai virus (Cantell strain no. 10100774) was purchased from Charles River.
Plasmids and transfection.
SARS-CoV-2 protein plasmids were generated from cDNA template prepared from RNA isolated from SARS-CoV-2-infected Vero E6 cells by reverse transcription and PCR. Viral-gene-specific cDNAs, including C-terminal FLAG tag cassettes, were cloned between NheI and FseI restriction sites in pCDNA 3.1(−) 3× FLAG plasmid. Due to the instability of the C-terminally tagged NSP10 construct in bacteria, a 3× FLAG sequence was added in frame to the N-terminal region of the protein. The primers used for cloning are listed in Table 1. All constructs were verified by Sanger sequencing.
TABLE 1.
Primers used for cloning
| PCR target | Primer sequence (5′→3′) |
|---|---|
| NSP1 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGGAGAGCCTTGTCCCTGGTTTC |
| NSP1 FseI rev | CGCCGGGCCGGCCCCCTCCGTTAAGCTCACGCATGAG |
| NSP2 PasI for | TAGCCCCAGGGGCCACCATGGCATACACTCGCTATGTCGATAAC |
| NSP2 FseI rev | CGCCGGGCCGGCCACCGCCTTTGAGTGTGAAGGTATT |
| NSP3 PasI for | CTAGCCCCAGGGGCCACCATGGCACCAACAAAGGTTACTTTTGGT |
| NSP3 FseI rev | CGCCGGGCCGGCCACCACCCTTAAGTGCTATCTTTGT |
| NSP4 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGAAAATTGTTAATAATTGGTTGAAG |
| NSP4 FseI rev | CGCCGGGCCGGCCCTGCAAAACAGCTGAGGTGATAGA |
| NSP5 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGAGTGGTTTTAGAAAAATGGCATTC |
| NSP5 FseI rev | CGCCGGGCCGGCCTTGGAAAGTAACACCTGAGCATTG |
| NSP6 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGAGTGCAGTGAAAAGAACAATCAAG |
| NSP6 FseI rev | CGCCGGGCCGGCCCTGTACAGTGGCTACTTTGATACA |
| NSP7 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGTCTAAAATGTCAGATGTAAAGTGC |
| NSP7 FseI rev | CGCCGGGCCGGCCTTGTAAGGTTGCCCTGTTGTCCAG |
| NSP8 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGGCTATAGCCTCAGAGTTTAGTTCC |
| NSP8 FseI rev | CGCCGGGCCGGCCCTGTAATTTGACAGCAGAATTGGC |
| NSP9 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGAATAATGAGCTTAGTCCTGTTGCA |
| NSP9 FseI rev | CGCCGGGCCGGCCTTGTAGACGTACTGTGGCAGCTAA |
| NSP10 NheI for | AAGCTGGCTAGCCCCAGGGGCCACCATGGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGACTACAAGGATGACGAGACAAGGGCCGGCCCGGCGGGAGCGGCGGGGCTGGTAATGCAACAGAAGTGCCT |
| NSP10 FseI rev | AAGCTTGGATCCTACTGAAGCATGGGTTCGCGGAGTTG |
| NSP12 PasI for | CTAGCCCCAGGGGCCACCATGTCAGCTGATGCACAATCGTTCCTAAACCGGGTTTGCGGTGTAAGTGCAGCC |
| NSP12 FseI rev | CGCCGGGCCGGCCCTGTAAGACTGTATGCGGTGTGTA |
| NSP13 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGGCTGTTGGGGCTTGTGTTCTTTGC |
| NSP13 FseI rev | CGCCGGGCCGGCCTTGTAAAGTTGCCACATTCCTACG |
| NSP14 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGGCTGAAAATGTAACAGGACTCTTT |
| NSP14 FseI rev | CGCCGGGCCGGCCCTGAAGTCTTGTAAAAGTGTTCCA |
| NSP15 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGAGTTTAGAAAATGTGGCTTTTAAT |
| NSP15 FseI rev | CGCCGGGCCGGCCTTGTAATTTTGGGTAAAATGTTTC |
| NSP16 PasI for | CTAGCCCCAGGGGCCACCATGTCTAGTCAAGCGTGGCAACCGGGT |
| NSP16 FseI rev | CGCCGGGCCGGCCGTTGTTAACAAGAACATCACTAGA |
| Nucleocapsid NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGTCTGATAACGGACCCCAAAATCAGCGAAAT |
| Nucleocapsid FseI rev | CGCCGGGCCGGCCAGCCTGAGTTGAGTCAGCACTGCTC |
| ORF10 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGGGCTATATAAACGTTTTCGCT |
| ORF10 FseI rev | CGCCGGGCCGGCCTGTGAGATTAAAGTTAACTACATC |
| ORF14 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGCTGCAATCGTGCTACAACTTC |
| ORF14 FseI rev | CGCCGGGCCGGCCATCTGTCAAGCAGCAGCAAAGCAA |
| P6 NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGTTTCATCTCGTTGACTTTCAG |
| P6 FseI rev | CGCCGGGCCGGCCATCAATCTCCATTGGTTGCTCTTC |
| S NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGTTTGTTTTTCTTGTTTTATTG |
| S FseI rev | CGCCGGGCCGGCCTGTGTAATGTAATTTGACTCCTTT |
| E NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGTACTCATTCGTTTCGGAAGAG |
| E FseI rev | CGCCGGGCCGGCCGACCAGAAGATCAGGAACTCTAGA |
| M NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGGCAGATTCCAACGGTACTATT |
| M FseI rev | CGCCGGGCCGGCCCTGTACAAGCAAAGCAATATTGTC |
| NS3A NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGGATTTGTTTATGAGAATCTTC |
| NS3A FseI rev | CGCCGGGCCGGCCCAAAGGCACGCTAGTAGTCGTCGT |
| NS3B NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGATGCCAACTATTTTCTTTGCT |
| NS3B FseI rev | CGCCGGGCCGGCCCACTATTGTAAGGTATACAATAGT |
| NS7A NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGAAAATTATTCTTTTCTTGGCA |
| NS7A FseI rev | CGCCGGGCCGGCCTTCTGTCTTTCTTTTGAGTGTGAA |
| NS7B NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGATTGAACTTTCATTAATTGAC |
| NS7B FseI rev | CGCCGGGCCGGCCGGCGTGACAAGTTTCATTATGATC |
| NS8B NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGAAATTTCTTGTTTTCTTAGGA |
| NS8B FseI rev | CGCCGGGCCGGCCGATGAAATCTAAAACAACACGAAC |
| NS9B NheI for | AGCTGGCTAGCCCCAGGGGCCACCATGGACCCCAAAATCAGCGAAATG |
| NS9B FseI rev | CGCCGGGCCGGCCTTTTACCGTCACCACCACGAATTC |
| NSP1-KH164AA fusion for | TTTCAAGAAAACTGGAACACTGCAGCAAGCAGTGGTGTTACCCGTGAA |
| NSP1-KH164AA fusion rev | TTCACGGGTAACACCACTGCTTGCTGCAGTGTTCCAGTTTTCTTGAAA |
For indirect immunofluorescence analysis, the appropriate expression plasmids were transfected into A549 cells or Huh7 cells using TransIT-LT1 (Mirus). For luciferase reporter assays in A549 or HEK 293T cells, plasmid transfection was performed using Lipofectamine 2000 (Invitrogen).
Antibodies and compounds.
The following antibodies were purchased from the indicated sources: mouse anti-SARS-CoV/SARS-CoV-2 spike antibody (1A9) (GTX632604) from GeneTex; rabbit anti-STAT2 (human, sc‐476), rabbit anti-STAT1 (sc‐346), and mouse anti-Jak1 (sc-1677) from Santa Cruz; mouse anti‐FLAG (F3165), rabbit anti-glutathione S-transferase (GST) (G7781), and mouse anti‐β‐actin (A3853) from Sigma-Aldrich; goat anti-GFP (ab6673), rabbit anti-Tyk2 (ab223733), rabbit anti-IFNAR1 (ab124764), and rabbit anti-TRIM25 (ab167154) from Abcam; rabbit anti-IRF-3 (no. 11904) and rabbit anti-phospho-IRF-3 (no. 4947) from Cell Signaling; mouse horseradish peroxidase (HRP)-conjugated anti-hemagglutinin (HA) (A01244) from GenScript; and mouse anti-V5 (R96025) from Invitrogen. The proteasome inhibitor epoxomicin, the lysosome inhibitor bafilomycin A1, and the translation inhibitor cycloheximide were purchased from Sigma‐Aldrich. High-molecular-weight (HMW) poly(I·C) (no. tlrl-pic) was purchased from InvivoGen.
Immunoblotting.
HEK 293T-ACE2 cells (3 × 105) were seeded into 12-well plates and were infected the next day with SARS-CoV-2 or transfected with appropriate expression plasmids. At designated time points postinfection or posttransfection, cells were washed twice with phosphate-buffered saline (PBS) before being lysed with 2× SDS sample buffer with β-mercaptoethanol (2%). The samples were incubated at 98°C for 10 min to denature proteins, which were then separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes for immunoblotting. The membranes were incubated with blocking solution (5% bovine serum albumin [BSA; Sigma-Aldrich] in PBS–0.05% Tween 20) for 30 min before exposure to primary antibodies diluted in blocking solution for 90 min. Following three washes with PBS–0.05% Tween 20 for 10 min each, the blots were incubated with secondary antibodies in blocking solution for 60 min. The blots were washed three times with PBS–0.05% Tween 20 and once with PBS and then imaged with an Odyssey infrared imaging system. Quantification of proteins was performed using Odyssey Image Studio Lite software version 5.2.
Immunoprecipitation.
HEK 293T cells were transfected with indicated plasmids using Lipofectamine 2000 (Life Technologies) for 24 h according to the manufacturer’s protocol. The cells were pelleted and resuspended in immunoprecipitation (IP) buffer (150 mM NaCl, 50 mM Tris [pH 7.5], 1% Triton X-100, 1 mM NaF, 1 mM dithiothreitol [DTT], and protease inhibitor cocktail [Roche]). The supernatant was clarified by centrifugation at 16,000 × g for 15 min. Aliquots of the cell lysate were incubated with anti-FLAG M2 magnetic beads (Millipore) or glutathione Sepharose 4B resin (GE Healthcare) at 4°C for 2 h. After washes with IP buffer, SDS sample buffer was added to the beads and boiled, and the proteins were resolved by SDS-PAGE.
Confocal microscopy.
Huh7 or A549 cells on coverslips were fixed for 10 min at room temperature with 4% electron microscopy-grade paraformaldehyde (Electron Microscope Sciences) in PBS and permeabilized with 0.5% Triton X-100 in PBS. Following three washes with PBS, the samples were incubated in blocking buffer at room temperature for 30 min. Primary antibodies were added in blocking buffer and incubated at room temperature for 45 min. After three washes in PBS, samples were incubated with secondary antibodies (1:1,000; Invitrogen) in blocking buffer and DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich) (1 μg/ml) for 45 min at room temperature. Coverslips were washed three times with PBS and once with deionized water and mounted on microscope slides using Prolong Gold antifade mounting reagent (Life Technologies). Samples were imaged using an Olympus IX-81 spinning-disk confocal microscope equipped with a 60× PlanApo N oil objective. Images were analyzed using Volocity 6.2.1 software (PerkinElmer).
qRT-PCR.
Total RNA was isolated from cells using the RNA NucleoSpin kit (Macherey Nagel) and reverse transcribed using random primers (Invitrogen) and Improm-II reverse transcriptase (Promega) at 42°C for 1.5 h to generate cDNAs. The cDNAs were diluted 1:5 with water, and 5% volume was mixed with the appropriate primers (Integrated DNA Technologies) and PerfecTa SYBR green SuperMix with low ROX (Quanta Biosciences) and amplified for 40 cycles (30 s at 94°C, 40 s at 55°C, and 20 s at 68°C) in a Bio-Rad CFX96 qRT-PCR machine. The gene targets and primer sequences are listed in Table 2. The cycle threshold (CT) values were normalized using ACTB mRNA as the internal control. The ΔΔCT values were determined using control samples as the reference value. Relative levels of mRNAs were calculated using the formula 2−ΔΔCT.
TABLE 2.
Primers used for PCR
| Gene target | Primer sequence (5′→3′) |
|---|---|
| SARS-CoV-2 spike | Forward: CCTACTAAATTAAATGATCTCTGCTTTACT |
| Reverse: CAAGCTATAACGCAGCCTGTA | |
| Ifnb | Forward: TAGCACTGGCTGGAATGAGA |
| Reverse: TCCTTGGCCTTCAGGTAATG | |
| Actb | Forward: CCTGGCACCCAGCACAAT |
| Reverse: GCCGATCCACACGGAGTACT | |
| Ifit1 | Forward: AGAAGCAGGCAATCACAGAAAA |
| Reverse: CTGAAACCGACCATAGTGGAAAT | |
| SeV HN | Forward: AAAATTACATGGCTAGGAGGGAAAC |
| Reverse: GTGAATGGAATGGTTGTGACTCTTA |
Luciferase reporter assay.
HEK 293T cells seeded in 12-well plates were transfected with the following promoter reporter (firefly luciferase) constructs: ISG56:pGL3B/561 (gift from Ganes Sen, Lerner Research Institute, Cleveland, OH, USA), IFN‐β:p125‐luc and IRF3:p55‐CIB‐Luc (provided by T. Taniguchi, University of Tokyo, Japan), ISRE:pGL4 ISRE (Promega), NF‐κB:pNF‐kB‐Luc (Stratagene), or GAS:pGAS‐Luc plasmid (Stratagene) together with the Renilla luciferase:pRL‐TK (Promega) construct as a transfection control. At indicated time points, cells were washed once with PBS and then lysed in 200 μl luciferase lysis buffer (0.1% [vol/vol] Triton X-100, 25 mM glycylglycine [pH 7.8], 15 mM MgSO4, 4 mM EGTA, and 1 mM dithiothreitol), after which the samples were used immediately or stored at −80°C until use. For luciferase assays, the samples were thawed, and 50 μl of each was aliquoted into white 96-well microplates (Greiner Bio-one) in duplicates for both firefly and Renilla luciferase assays. The firefly luciferase substrate d-luciferin (Gold Biotechnology USA) was prepared at a final concentration of 70 μM in luciferase assay buffer (25 mM glycylglycine [pH 7.8], 15 mM K2PO4, [pH 7.8], 15 mM MgSO4, 4 mM EGTA, 1 mM DTT, and 2 mM ATP), and 50 μl was added to each well and incubated in the absence of light for 5 min. Luciferase activity measured using a Synergy HTX plate reader (Biotek). For Renilla luciferase measurements, the substrate coelenterazine (Gold Biotechnology USA) was prepared at a final concentration of 1.4 μM in luciferase assay buffer (25 mM glycylglycine [pH 7.8], 15 mM K2PO4, [pH 7.8], 15 mM MgSO4, and 4 mM EGTA). Fifty microliters was added to each well, and luciferase activity was measured using a Synergy HTX plate reader (Biotek).
ELISA.
Levels of human IFN-β in the cell culture supernatant were measured using a Quantikine human IFN-β immunoassay kit (R&D Systems, Inc.) according to the manufacturer’s instructions. The total fluorescence was measured using a Synergy HTX plate reader (Biotek).
Statistical analyses.
All statistical analyses were performed using GraphPad Prism software. Paired Student's t test was performed for pairwise statistical comparison, while one-way analysis of variance (ANOVA) was used for comparison of multiple samples. The means and standard errors of the means are shown in all bar and line graphs.
ACKNOWLEDGMENTS
This work was funded by the grants from the Canadian Institutes of Health Research (CIHR) to T.C.H. (OV3-172302), D.H.E. (OV3-172302), K.E.M. (PJT 159442), and A.K. (OV3-172302), the Li Ka Shing Institute of Virology to T.C.H., and the Canada Excellence Research Chair to L.K.M. The funders had no role in study design, data collection and interpretation, or decision to submit the work for publication.
We thank V. Mancinelli and E. Reklow for technical support.
A.K., R.I., and T.C.H. conceived the research and drafted the manuscript. A.K., R.I., T.S., J.C., J.L.-O., N.F., M.E., D.E., L.P.N., and A.F.-L. carried out the experiments. A.K., R.I., and T.S. created the figures. All authors discussed the results and contributed to the revision of the final manuscript.
We declare no competing financial interests.
Contributor Information
Anil Kumar, Email: anil.kumar@usask.ca.
Tom C. Hobman, Email: tom.hobman@ualberta.ca.
Tom Gallagher, Loyola University Chicago.
REFERENCES
- 1.Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, Zhao Y, Li Y, Wang X, Peng Z. 2020. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323:1061. 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team . 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. 2020. The proximal origin of SARS-CoV-2. Nat Med 26:450–452. 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie M, Chen Q. 2020. Insight into 2019 novel coronavirus—an updated interim review and lessons from SARS-CoV and MERS-CoV. Int J Infect Dis 94:119–124. 10.1016/j.ijid.2020.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arons MM, Hatfield KM, Reddy SC, Kimball A, James A, Jacobs JR, Taylor J, Spicer K, Bardossy AC, Oakley LP, Tanwar S, Dyal JW, Harney J, Chisty Z, Bell JM, Methner M, Paul P, Carlson CM, McLaughlin HP, Thornburg N, Tong S, Tamin A, Tao Y, Uehara A, Harcourt J, Clark S, Brostrom-Smith C, Page LC, Kay M, Lewis J, Montgomery P, Stone ND, Clark TA, Honein MA, Duchin JS, Jernigan JA, Public Health–Seattle and King County and CDC COVID-19 Investigation Team . 2020. Presymptomatic SARS-CoV-2 Infections and Transmission in a Skilled Nursing Facility. N Engl J Med 382:2081–2090. 10.1056/NEJMoa2008457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fensterl V, Chattopadhyay S, Sen GC. 2015. No love lost between viruses and interferons. Annu Rev Virol 2:549–572. 10.1146/annurev-virology-100114-055249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mesev EV, LeDesma RA, Ploss A. 2019. Decoding type I and III interferon signalling during viral infection. Nat Microbiol 4:914–924. 10.1038/s41564-019-0421-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Sastre A. 2017. Ten strategies of interferon evasion by viruses. Cell Host Microbe 22:176–184. 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park A, Iwasaki A. 2020. Type I and type III interferons—induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 27:870–878. 10.1016/j.chom.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sa Ribero M, Jouvenet N, Dreux M, Nisole S. 2020. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog 16:e1008737. 10.1371/journal.ppat.1008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia H, Shi PY. 2020. Antagonism of type I interferon by severe acute respiratory syndrome coronavirus 2. J Interferon Cytokine Res 40:543–548. 10.1089/jir.2020.0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banerjee AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, Bhat P, Ollikainen N, Quinodoz SA, Loney C, Thai J, Miller ZD, Lin AE, Schmidt MM, Stewart DG, Goldfarb D, De Lorenzo G, Rihn SJ, Voorhees RM, Botten JW, Majumdar D, Guttman M. 2020. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183:1325–1339.E21. 10.1016/j.cell.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, Geurink PP, Wilhelm A, van der Heden van Noort GJ, Ovaa H, Muller S, Knobeloch KP, Rajalingam K, Schulman BA, Cinatl J, Hummer G, Ciesek S, Dikic I. 2020. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587:657–662. 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, Kratzat H, Hayn M, Mackens-Kiani T, Cheng J, Straub JH, Sturzel CM, Frohlich T, Berninghausen O, Becker T, Kirchhoff F, Sparrer KMJ, Beckmann R. 2020. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 369:1249–1255. 10.1126/science.abc8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li JY, Liao CH, Wang Q, Tan YJ, Luo R, Qiu Y, Ge XY. 2020. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res 286:198074. 10.1016/j.virusres.2020.198074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang HW, Zhang HN, Meng QF, Xie J, Li Y, Chen H, Zheng YX, Wang XN, Qi H, Zhang J, Wang PH, Han ZG, Tao SC. 2020. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol 17:998–1000. 10.1038/s41423-020-0514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, Guo F, Zhao Z, Zhou Z, Xiang Z, Wang J. 2020. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 11:3810. 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuen CK, Lam JY, Wong WM, Mak LF, Wang X, Chu H, Cai JP, Jin DY, To KK, Chan JF, Yuen KY, Kok KH. 2020. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect 9:1418–1428. 10.1080/22221751.2020.1780953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acharya D, Liu G, Gack MU. 2020. Dysregulation of type I interferon responses in COVID-19. Nat Rev Immunol 20:397–398. 10.1038/s41577-020-0346-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, Cupic A, Makio T, Mei M, Moreno E, Danziger O, White KM, Rathnasinghe R, Uccellini M, Gao S, Aydillo T, Mena I, Yin X, Martin-Sancho L, Krogan NJ, Chanda SK, Schotsaert M, Wozniak RW, Ren Y, Rosenberg BR, Fontoura BMA, Garcia-Sastre A. 2020. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci U S A 117:28344–28354. 10.1073/pnas.2016650117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, Menachery VD, Rajsbaum R, Shi PY. 2020. Evasion of type I interferon by SARS-CoV-2. Cell Rep 33:108234. 10.1016/j.celrep.2020.108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mu J, Fang Y, Yang Q, Shu T, Wang A, Huang M, Jin L, Deng F, Qiu Y, Zhou X. 2020. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov 6:65. 10.1038/s41421-020-00208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanderheiden A, Ralfs P, Chirkova T, Upadhyay AA, Zimmerman MG, Bedoya S, Aoued H, Tharp GM, Pellegrini KL, Manfredi C, Sorscher E, Mainou B, Lobby JL, Kohlmeier JE, Lowen AC, Shi PY, Menachery VD, Anderson LJ, Grakoui A, Bosinger SE, Suthar MS. 2020. Type I and type III IFN restrict SARS-CoV-2 infection of human airway epithelial cultures. J Virol 94:e00985-20. 10.1128/JVI.00985-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narayanan K, Huang C, Lokugamage K, Kamitani W, Ikegami T, Tseng CT, Makino S. 2008. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J Virol 82:4471–4479. 10.1128/JVI.02472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu Y, Li W, Gao T, Cui Y, Jin Y, Li P, Ma Q, Liu X, Cao C. 2017. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J Virol 91:e02143-16. 10.1128/JVI.02143-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mantlo E, Bukreyeva N, Maruyama J, Paessler S, Huang C. 2020. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antiviral Res 179:104811. 10.1016/j.antiviral.2020.104811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felgenhauer U, Schoen A, Gad HH, Hartmann R, Schaubmar AR, Failing K, Drosten C, Weber F. 2020. Inhibition of SARS-CoV-2 by type I and type III interferons. J Biol Chem 295:13958–13964. 10.1074/jbc.AC120.013788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schubert K, Karousis ED, Jomaa A, Scaiola A, Echeverria B, Gurzeler LA, Leibundgut M, Thiel V, Muhlemann O, Ban N. 2020. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol 27:959–966. 10.1038/s41594-020-0511-8. [DOI] [PubMed] [Google Scholar]
- 29.Kamitani W, Narayanan K, Huang C, Lokugamage K, Ikegami T, Ito N, Kubo H, Makino S. 2006. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci U S A 103:12885–12890. 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jauregui AR, Savalia D, Lowry VK, Farrell CM, Wathelet MG. 2013. Identification of residues of SARS-CoV nsp1 that differentially affect inhibition of gene expression and antiviral signaling. PLoS One 8:e62416. 10.1371/journal.pone.0062416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee AC, Zhang X, Wang Y, Hu B, Huang X, Yuen TT, Cai JP, Zhou J, Yuan S, Zhang AJ, Chan JF, Yuen KY. 2020. Attenuated interferon and proinflammatory response in SARS-CoV-2-infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis 222:734–745. 10.1093/infdis/jiaa356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]