

ABSTRACT
The intestinal protozoan Cryptosporidium is a leading cause of diarrheal disease and mortality in young children. There is currently no fully effective treatment for cryptosporidiosis, which has stimulated interest in anticryptosporidial development over the last ∼10 years, with numerous lead compounds identified, including several tRNA synthetase inhibitors. Here, we report the results of a dairy calf efficacy trial of the methionyl-tRNA (Cryptosporidium parvum MetRS [CpMetRS]) synthetase inhibitor 2093 and the spontaneous emergence of drug resistance. Dairy calves experimentally infected with Cryptosporidium parvum initially improved with 2093 treatment, but parasite shedding resumed in two of three calves on treatment day 5. Parasites shed by each recrudescent calf had different amino acid-altering mutations in the gene encoding CpMetRS (CpMetRS), yielding either an aspartate 243-to-glutamate (D243E) or a threonine 246-to-isoleucine (T246I) mutation. Transgenic parasites engineered to have either the D243E or T246I CpMetRS mutation using CRISPR/Cas9 grew normally but were highly 2093 resistant; the D243E and T246I mutant-expressing parasites, respectively, had 2093 half-maximal effective concentrations (EC50s) that were 613- and 128-fold that of transgenic parasites with wild-type CpMetRS. In studies using recombinant enzymes, the D243E and T246I mutations shifted the 2093 IC50 >170-fold. Structural modeling of CpMetRS based on an inhibitor-bound Trypanosoma brucei MetRS crystal structure suggested that the resistance mutations reposition nearby hydrophobic residues, interfering with compound binding while minimally impacting substrate binding. This is the first report of naturally emerging Cryptosporidium drug resistance, highlighting the need to address the potential for anticryptosporidial resistance and establish strategies to limit its occurrence.
KEYWORDS: Cryptosporidium, anticryptosporidial, cryptosporidiosis, drug resistance evolution, drug resistance mechanisms, methionyl-tRNA synthetase
INTRODUCTION
Infection of small intestinal epithelial cells with Cryptosporidium parasites causes the diarrheal illness cryptosporidiosis, which in humans is mostly due to either Cryptosporidium hominis or Cryptosporidium parvum (1–4). Symptoms of cryptosporidiosis last 12 days on average in immunocompetent people but are often prolonged and potentially fatal in malnourished children and immunocompromised people, such as those with AIDS and transplant recipients (1). In addition to being a prominent cause of chronic diarrhea in AIDS patients, cryptosporidiosis was a leading cause of life-threatening diarrhea in children under 2 years of age in a landmark case-control study recently conducted at seven high-burden sites in Africa and the Indian subcontinent (5, 6). Cryptosporidium infections are also strongly associated with childhood malnutrition, growth stunting, and delayed cognitive development (7, 8).
Despite the large public health burden, no Cryptosporidium vaccine exists and treatments for cryptosporidiosis remain inadequate. Nitazoxanide (NTZ) is the only FDA-approved drug for cryptosporidiosis and is reliably efficacious only for immunocompetent adults, for whom it shortens the duration of diarrhea by 1 to 2 days (9). Unfortunately, nitazoxanide is no more effective than a placebo in HIV-positive people, and data for young malnourished children are limited to small studies in which it appears at best modestly efficacious (10, 11).
The need for improved anticryptosporidials motivated multiple drug development efforts employing both phenotypic and target-based methods over the last decade. These efforts have led to the identification of numerous promising lead compounds and two preclinical candidates that are poised for human studies (i.e., a Novartis phosphatidyl-inositol-4-kinase inhibitor and the 6-carboxamide benzoxaborole AN7973) (12–20). There is the potential to realize an enormous public health benefit with intelligent deployment of improved anticryptosporidial drugs.
In many ways, the anticryptosporidial field evokes the situation for malaria shortly before the introduction of chloroquine in 1945, a time at which there was nearly a blank slate for treating malaria and the inevitability of drug resistance was not appreciated. Yet, chloroquine resistance first became evident for malaria around 1957, just 12 years after its release (21). Based on the hard lessons from malaria chloroquine resistance and antimicrobial resistance in general, combination therapy is now the standard for antimalarial development, as well as for treatment of numerous other infectious diseases for which the risk of resistance is high (e.g., HIV and tuberculosis) (22). This history stresses the need to understand the potential for Cryptosporidium drug resistance and how to best deploy new anticryptosporidials. To date, the possibility of anticryptosporidial resistance remains completely unexamined.
Aminoacyl-tRNA synthetase (aaRS) inhibitors are protein synthesis inhibitors with promise as potential treatments for bacterial, fungal, and parasitic infections. aaRS enzymes catalyze the formation of tRNAs charged with their corresponding amino acids that serve as the substrates for protein synthesis. Two aaRS inhibitors are already approved for human use: the topical antistaphylococcal antibiotic mupirocin, a natural product that inhibits isoleucyl-tRNA synthetase (23), and tavaborole, a leucyl-tRNA synthetase (LeuRS) inhibitor that is approved for topical treatment of onychomycosis (24, 25). Beyond these topical agents, several aaRS inhibitors are in clinical trials as systemic antimicrobials, including a methionyl-tRNA synthetase (MetRS) inhibitor for treatment of Clostridium difficile infections (26), a LeuRS inhibitor for treatment of Gram-negative bacterial infections (27, 28), and a LeuRS inhibitor for treatment of multidrug-resistant (MDR) tuberculosis (29, 30).
aaRS inhibitors figure prominently within the anticryptosporidial developmental pipeline (31). Halofuginone, a prolyl-tRNA synthetase inhibitor, is approved for prevention and treatment of C. parvum infection in dairy cattle in Europe, but it cannot be used in humans due to a narrow safety window (32); other prolyl-tRNA synthetase inhibitors are under study (16). Phenylalanyl-tRNA synthetase (PheRS) and lysyl-tRNA synthetase (LysRS) inhibitors with promising activity in several mouse models of infection are also under development (14, 33). We previously reported that a MetRS inhibitor, 2093, potently inhibits the Cryptosporidium MetRS enzyme and is efficacious against C. parvum in two murine models of infection, with no signs of toxicity (34).
MetRSs are categorized into two forms based on sequence similarity: type I MetRSs (MetRS1), which are homologous to the human mitochondrial MetRS, MetRSs of most Gram-positive bacteria, and those of some parasites, including Trypanosoma species, and type II MetRSs (MetRS2), which are homologous to the human cytoplasmic MetRS and those found in most Gram-negative bacteria (35). Some Gram-positive bacteria, such as Bacillus anthracis and some Streptococcus pneumoniae strains, have both a MetRS1 and a MetRS2. C. parvum and C. hominis each have a single MetRS gene that encodes a protein homologous to MetRS1 enzymes. We identified the anticryptosporidial activity of 2093 and related compounds by screening MetRS1 inhibitors under development for treatment of Gram-positive bacterial infections and trypanosomes (34).
We now report results of a 2093 drug efficacy trial performed in C. parvum-infected dairy calves and the spontaneous emergence of MetRS inhibitor resistance. We identified two mutant parasite strains with different single amino acid substitutions in the C. parvum MetRS (CpMetRS). We confirmed C. parvum resistance to 2093 and further investigated the mechanism of resistance using a combination of CRISPR/Cas9 genome editing to modify the gene for C. parvum MetRS (CpMetRS), in vitro enzymatic assays performed with recombinant mutant enzymes, and structural modeling. This study is the first report of emergent Cryptosporidium drug resistance and, therefore, highlights a dire need to anticipate the potential for Cryptosporidium drug resistance and establish strategies to mitigate its development.
RESULTS
Relapse of C. parvum infection during MetRS inhibitor treatment of infected dairy calves.
Neonatal dairy calves are highly susceptible to C. parvum infection and develop a self-resolving diarrheal illness that closely resembles cryptosporidiosis in young children. Experimentally infected calves therefore provide a natural clinical model of C. parvum infection to determine both the microbiological and clinical efficacy of drug leads (36, 37). Based on promising results in both ifn-γ−/− mice and NOD SCID gamma mice (34), we selected the MetRS inhibitor 2093 for testing in the calf model. We previously showed in a cell-based assay that the rate of C. parvum elimination by 2093 is maximized by exposure to concentrations at or above three times the 90% effective concentration (EC90) (34). A preliminary pharmacokinetics (PK) study performed on uninfected calves demonstrated plasma and fecal levels in excess of three times the EC90 for over 24 h following a single oral dose of 10 mg per kg of body weight (see Fig. S1a in the supplemental material). To ensure 2093 levels exceeding three times the EC90 despite possible reduced absorption and accelerated intestinal transit due to diarrhea, a dose of 15 mg per kg every 12 h was chosen for the efficacy study.
Bull Holstein calves were infected with ∼5 × 107 C. parvum (Bunch Grass Farm Iowa isolate) at 24 to 48 h of age. 2093 or vehicle control treatments were begun on day 2 of infection, the time typically corresponding to the onset of diarrhea in this model (19, 37), and continued for 7 days (Fig. 1a). Figure 1b shows the rate of oocyst shedding in the feces versus time for individual calves. 2093 initially appeared effective, with progressive elimination of oocyst shedding from all treated calves during the first 4 days of infection. Total oocyst shedding, calculated as the area under the curve (AUC) for each animal, was reduced nearly 2 logs during the first 4 days of compound treatment (Fig. 1c), and both diarrhea and dehydration were negligible despite severe diarrhea in control animals (Fig. 1d and e). However, two of three 2093-treated calves relapsed, with a progressive rise in fecal oocysts beginning on day 4 of treatment (i.e., experiment day 6), followed by increased diarrhea and dehydration. The remaining calf appeared to be cured (Fig. 1b). Recrudescence could not be attributed to inadequate dosing or altered drug levels in the presence of Cryptosporidium infection and diarrhea, since both fecal and plasma levels were well in excess of three times the EC90 beginning within 5 h of initiating treatment (Fig. S1b). We cannot exclude the possibility that the concentration of 2093 in solution was inadequate in the gut lumen, since our method quantified both dissolved and undissolved 2093 in the feces. Nonetheless, these data suggested potential emergence of 2093-resistant parasites. It was impossible to isolate viable oocysts and test directly for drug resistance in vitro, because recrudescent C. parvum shedding was not recognized by quantitative PCR (qPCR) until after sample freezing and euthanization of the animals.
FIG 1.

Resurgent C. parvum shedding occurred during compound 2093 treatment of infected dairy calves. (a) Summary of neonatal calf model and compound 2093 clinical efficacy study. Bull Holstein calves were infected within 48 h of birth by oral administration of ∼5 × 107 C. parvum oocysts (Bunch Grass Farms Iowa isolate). Fecal oocyst shedding was quantified daily using qPCR. Vehicle or 2093 (15 mg per kg every 12 h) was administered orally on study days 2 to 8. (b) Oocyst shedding versus time for 2093-treated and control calves. The colored box highlights the timing of treatment, and oocyst shedding is plotted for individual animals (n = 3 for each group). Note that 2 animals in the control group died on day 5 due to unrelated causes (one due to congenital bladder obstruction and one due to pneumonia). LOD, limit of detection for the qPCR assay. Samples with negative PCR results are plotted at the LOD. (c) Total oocyst shedding per gram of dried feces during study days 2 to 6. The area under the curve (AUC) in excess of the qPCR LOD is plotted for each calf. Lines show means and standard deviations. *, P = 0.005 (unpaired two-tailed t test) for reduced oocyst shedding in 2093-treated animals up to the point of recrudescence in two of the three treated calves. (d and e) Fecal consistency (d) and dehydration versus time (e) for 2093-treated and control calves. Data are the means and standard errors (n = 3 except for n = 1 in the case of the vehicle group after day 5).
MetRS gene coding mutations in shed oocysts from 2093-treated calves.
Mutation of the target gene is one potential mechanism of acquired drug resistance, so the known target of 2093 provided an opportunity to investigate this possibility. For this, we used total DNA isolated from the feces of each vehicle- and 2093-treated calf on experiment days 1 and 9 as a PCR template and Sanger sequenced the CpMetRS genomic locus. Predictably, no PCR product could be obtained from the cured calf on day 9. The genomic CpMetRS sequences on day 1 of the experiment were identical to the C. parvum Iowa strain gene sequence for all calves, but on day 9 each of the two relapsed calves had a distinct coding single nucleotide variant (SNV) in the CpMetRS gene (Fig. S2). These variants were not present in the two control calves on day 9. The identified CpMetRS mutations were predicted to result in substitutions at amino acids 243 and 246 of the CpMetRS enzyme: aspartate 243 mutated to glutamate (D243E), and threonine 246 mutated to isoleucine (T246I) (Fig. S2a). 2093 treatment in the calf model therefore appeared to select for Cryptosporidium strains with two independent CpMetRS mutations that potentially conferred drug resistance.
CpMetRS coding mutations confer parasite resistance to inhibition by 2093.
We used a recently described method for CRISPR/Cas9 genome editing in C. parvum (38) to determine the effects of the amino acid-altering CpMetRS mutations that were identified in the calf drug efficacy trial on parasite sensitivity to 2093. This method combines in vitro electroporation of a plasmid encoding the guide RNA and Cas9 enzyme and a linear repair cassette, followed by infection of ifn-γ−/− mice and in vivo selection of transgenic parasites with paromomycin. We engineered four transgenic C. parvum lines by replacing the parental CpMetRS locus with a transgene encoding wild-type CpMetRS or CpMetRSs with the D243E mutation, the T246I mutation, or both mutations in combination. The repair cassette was also designed to insert sequences for the enolase 3′ untranslated region (UTR) for transcriptional termination of CpMetRS and a fused nanoluciferase (Nluc)-neomycin phosphotransferase (Neo) under the control of a constitutively active enolase promoter located just after the CpMetRS stop codon (Fig. 2a). For each mutant parasite line, CpMetRS was recoded from bp 728 to 1686 to prevent homology-directed repair without insertion of the selection marker. The recoded sequence also altered the guide RNA (gRNA) recognition region to prevent repeated Cas9-mediated DNA cleavage (Fig. 2b). Each transgenic parasite line was generated without difficulty, and each was passaged once in NOD SCID gamma mice under paromomycin selection to remove potential parental contaminants before subsequent experiments. PCR using flanking primers demonstrated insertion of the repair cassettes at the endogenous locus, with amplification of the 1,211-bp and 2,945-bp bands predicted for the parental wild-type (WT; Iowa) and transgenic parasite lines, respectively (Fig. 2c). Sanger sequencing confirmed the successful introduction of each desired mutation (Fig. 2d).
FIG 2.
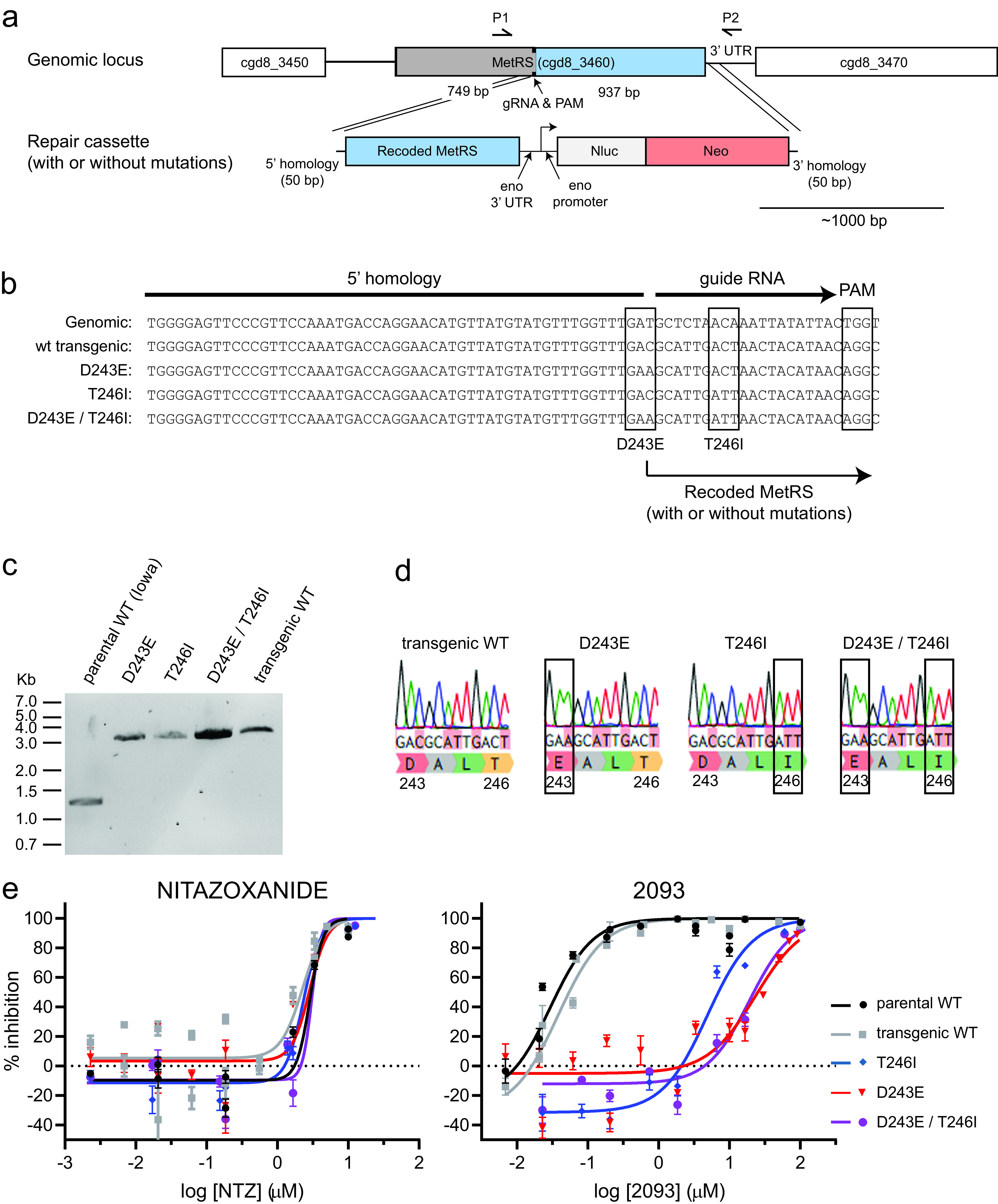
CRISPR/Cas9-engineered MetRS mutations confer C. parvum resistance to 2093. (a) Strategy used to modify the C. parvum MetRS genomic locus by CRISPR/Cas9 gene editing. Each DNA fragment is drawn to scale with the ∼1,000-bp scale bar shown. (b) Aligned sequences showing the wild-type locus, WT transgenic locus, and intended mutations, along with the PAM CRISPR/Cas9 cut site and guide RNA. (c) PCR confirmation of the genomic insertion using primers P1 and P2 shown in panel a. The anticipated PCR product sizes for the parental and transgenic strains were 1,211 bp and 2,945 bp, respectively. (d) Sanger sequencing chromatograms confirming introduction of each desired mutation. (e) Susceptibility of parental and transgenic C. parvum isolates to nitazoxanide (NTZ; negative control) and 2093. Data are combined from at least two biological replicate experiments per strain. Curves were calculated using all data points, but only every other dose is plotted in order to reduce clutter on the graphs.
We next purified oocysts for each transgenic C. parvum line from mouse feces, and the in vitro growth rates and drug susceptibility of each parasite line were assessed using a high-content microscopy-based assay for parasite development (39). There were no significant differences in growth of the transgenic lines relative to Iowa strain C. parvum, as measured at 48 h postinfection (Fig. S3). The CpMetRS mutations had no effect on C. parvum susceptibility to the unrelated growth inhibitor nitazoxanide (NTZ; negative control). On the other hand, the MetRS coding mutations reduced susceptibility to 2093 (Fig. 2e). The 2093 half-maximal effective concentrations (EC50s) for the D243E and T246I mutant MetRS C. parvum strains were 613- and 128-fold higher than those of the transgenic WT control, respectively (EC50transgenic wt = 0.038 μM, EC50D243E = 23.4 μM, and EC50T246I = 4.89 μM). 2093 resistance of the D243E/T246I double mutant was like that of the D243E single mutant. These data confirmed that the MetRS mutations selected in the calf model conferred C. parvum 2093 resistance, while not affecting susceptibility to nitazoxanide.
Enzymatic properties of mutant CpMetRS enzymes.
The wild-type and two mutant CpMetRS genes were cloned, and the proteins were overexpressed in Escherichia coli and purified for enzymatic studies. Enzymatic activity was confirmed in the aminoacylation assay (34), which detects the esterification of radiolabeled methionine to the tRNA substrate. In order to measure the Michaelis-Menten constants for the enzymes, an ATP:PPi exchange assay was used, as previously explained (40). First, for the wild-type enzyme, the Km values for methionine and ATP were 31.3 and 1,623 μM, respectively. These results are within a factor of ∼2 of previously published results (34). The Km values for the two mutant enzymes were considerably lower than for the WT enzyme (Table 1), and the kcat/Km values were higher, indicating greater catalytic efficiency. Next, the 50% inhibitory concentration (IC50) and Ki values of 2093 were determined for the three enzymes (Table 1). 2093 was extremely potent for the wild-type CpMetRS, with a Ki of 0.0017 nM. The IC50s of 2093 against both mutant enzymes were greater than the top dilutions tested in two assays (>10 μM and >30 μM), indicating a >170-fold upward shift in the IC50 values.
TABLE 1.
Kinetic parameters and sensitivity to 2093 inhibition of wild-type and mutant (T246I and D243E) CpMetRS enzymes
| Enzyme | Met titration |
ATP titration |
IC50 (nM) for 2093 | Ki (nM) for 2093 | ||||
|---|---|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat /Km (μM−1 s−1) | Km (μM) | kcat (s−1) | kcat /Km(μM−1 s−1) | |||
| WT | 31.3 | 30.6 | 0.98 | 1,623 | 99.0 | 0.064 | 174.1 ± 23.3 (n = 2) | 0.0017 ± 0.0003 (n = 2) |
| T246I mutant | 2.93 | 116.8 | 39.8 | 410.3 | 111.2 | 0.271 | >10,000, >30,000 | >31.8, >99.9 |
| D243E mutant | 6.94 | 158.6 | 22.8 | 890.6 | 138.9 | 0.156 | >10,000, >30,000 | >33.3, >99.9 |
Mechanism of compound 2093 resistance.
We previously solved multiple crystal structures of the Trypanosoma brucei MetRS (TbMetRS) with inhibitors bound, including with 2093 (PDB code 6CML), which demonstrated that they inhibit enzymatic activity by competing with substrate binding (41–44). The CpMetRS and TbMetRS are 76% identical (19 of 25 amino acids) within the inhibitor binding pocket (34), which presented the opportunity to further examine the mechanism of CpMetRS resistance to 2093. We generated a CpMetRS model structure based on the T. brucei structures using I-TASSER (45) and then docked compound 2093 into the model (Fig. 3). The predicted mode of binding of 2093 to C. parvum MetRS shows the 2-chloro,4-methoxy-benzyl positioned in the enlarged methionine substrate pocket and the 5-Cl-imidazo[4,5b]pyridine in the “auxiliary pocket” formed upon inhibitor binding (41, 42) (also explained in Materials and Methods). D243 forms a salt bridge with H63. This salt bridge is at 3.8 Å from both C-6 and C-7 of the imidazo[4,5-b]pyridine bicyclic system. We hypothesize that the D243E mutation disrupts the side wall of the auxiliary pocket by virtue of its larger size, which would lead to less favorable binding of the inhibitor. T246 is within van der Waals distance of the F242 phenyl ring. The resistance mutation T246I probably leads to a clash with the phenyl ring and thereby causes local unfolding of the α-helix it belongs to, thereby disrupting the back wall of the auxiliary pocket.
FIG 3.

2093 docked into a comparative model of CpMetRS. 2093 is coded with C (cyan), N (blue), and O (red). The resistance mutations D243 and T246 are labeled. D243 forms a salt bridge with H63. This salt bridge is at 3.8 Å from both C-6 and C-7 of the imidazo[4,5-b]pyridine bicyclic system. T246 is within van der Waals distance of the F242 phenyl ring. 2093 is also shown as a stick diagram for reference.
DISCUSSION
This study is important because it highlights the likelihood of Cryptosporidium drug resistance, which has thus far been neglected during anticryptosporidial drug development. Resistance to the CpMetRS inhibitor 2093 arose after 4 days of drug exposure in the neonatal dairy calf model despite plasma and fecal concentrations well above the in vitro EC90 for 2093 (Fig. S1b). Two amino acid-altering SNVs in CpMetRS emerged independently, a D243E mutation and a T246I mutation. CRISPR/Cas9 genome editing showed that these mutations confer high-level C. parvum resistance to 2093, while not significantly affecting parasite growth or susceptibility to nitazoxanide. In studies using recombinant enzymes, the D243E and T246I mutations shift the 2093 IC50 >170-fold and moderately reduce the Km for methionine (5× to 10×). Modeling of CpMetRS based on a high-resolution crystal structure of TbMetRS (PDB code 4EG5, chain B) suggests that the resistance mutations reposition nearby hydrophobic residues and likely interfere with inhibitor binding while minimally impacting substrate binding. These mutations likely have little impact on Cryptosporidium fitness since they did not affect the in vitro asexual growth rate. Our data provide genetic confirmation that CpMetRS is the molecular target responsible for the anticryptosporidial activity of 2093 and its analogs and, more importantly, demonstrate rapid emergence of drug-resistant C. parvum mutants.
Drug resistance in our study resulted from two coding single nucleotide alterations in CpMetRS. It is uncertain if parasites harboring these mutations were preexisting in the large population of parasites used to initiate infection or arose by spontaneous mutation in the presence of selective drug pressure. We lack critical data, such as the spontaneous mutation rate of Cryptosporidium and the total burden of parasites present in the intestinal lumen, that are needed to model when the mutations developed. Suggesting a mechanism of spontaneous mutation during our experiment, the MetRS SNVs identified in this study are not reported in the 53 Cryptosporidium genomes currently available in CryptoDB (https://cryptodb.org/cryptodb/). There are also no MetRS inhibitors in agricultural use to which the C. parvum isolate used in our calf study may have been previously exposed.
Evolution of antimicrobial resistance through spontaneous mutation is favored by the presence of large numbers of organisms, which could provide an explanation for outgrowth of resistant strains in the calf model despite its previously documented efficacy in murine models. For example, Gram-negative bacterial resistance with specific target mutations developed in 3 of 14 people after only 1 day of treatment in a phase II human trial of the LeuRS inhibitor GSK2251052 (AN3365) for complicated urinary tract infections (27). This occurred despite an inability to select for GSK2251052 resistance in mice, presumably due to lower numbers of organisms in the mouse model. The possibility of C. parvum MetRS mutations was not assessed in mice, although parasites in 2093-treated ifn-γ−/− mice persisted at a low level after treatment (34). In our calf study, recrudescence occurred while the animals were still receiving 2093. Peak oocyst shedding in each control calf ranged from 107 to 108 oocysts per gram of feces. This number is consistent with previous studies of C. parvum-infected calves (19, 37), and based on the approximate volume of feces produced by a calf with cryptosporidiosis, on the order of 1010 to 1011 oocysts are shed during a typical 10-day experiment using this model (19, 37, 46). If we look to another apicomplexan parasite, Plasmodium falciparum, the estimated number of C. parvum organisms in the calf model is in line with the burden of organisms during acute falciparum malaria (estimated at 108 to 1012 [47]), a scenario in which treatment failure due to newly derived resistance during treatment with a single drug, e.g., with atovaquone, is well documented (48). Up to 1 in 105 malaria parasites develop resistance to atovaquone (49), virtually guaranteeing the development of resistance on monotherapy. We also note that the simplistic view of microbial evolution described above ignores the potential for sexual recombination within the human host, a life cycle feature of Cryptosporidium species that distinguishes it from Plasmodium. Sexual recombination could therefore provide an additional mechanism to generate genetic diversity in the face of selective pressure, which could accelerate the evolution of drug resistance. A key unknown for cryptosporidiosis is the number of organisms during a human infection, which may of course differ for different host groups (e.g., parasite numbers might be greater in AIDS patients than in immunocompetent hosts).
The types and numbers of mutations required to confer antimicrobial resistance are specific characteristics of different drug classes and targets and together with fitness costs resulting from resistance mutations determine the propensity for emergence of resistance to a given drug (22). For example, S. aureus resistance to rifampin typically results from one of several single amino acid mutations in the RNA polymerase β subunit (RpoB) that emerge readily (50, 51); as a result, rifampin is not useful as monotherapy for Staphylococcus aureus infections. Studies with a first-generation MetRS inhibitor, REP8839, showed resistance frequency rates for S. aureus in the same range as rifampin (10−7 to 10−8 over a 48-h exposure) (52), suggesting a low barrier to resistance. The first-step REP8839 Staphylococcus resistance SNVs occurred in close proximity to the corresponding CpMetRS SNVs identified in this study (52), and the effects of these SNVs on enzyme kinetics with the natural substrates (methionine and ATP) were small for Staphylococcus as well as for Cryptosporidium. These findings raise concerns about developing MetRS inhibitors or other enzyme inhibitors that are vulnerable to resistance by subtle mutations in Cryptosporidium’s genome. The use of MetRS inhibitors as part of combination therapy may still be a consideration given their in vivo activity. Furthermore, later-generation MetRS inhibitors have been developed with much lower resistance frequency rates in S. aureus (on the order of 10−10) (F. Buckner and E. Fan, unpublished data), suggesting the potential for discovering MetRS inhibitors with higher barriers to resistance in Cryptosporidium.
There are several limitations to our study, which was initially designed as a drug efficacy trial and not intended to be a comprehensive study of Cryptosporidium drug resistance. Key factors likely to affect emergence of MetRS inhibitor resistance and the general likelihood of Cryptosporidium resistance developing via target mutations remain unknown, including knowledge of Cryptosporidium’s rate of spontaneous mutation, the impact of sexual recombination in the presence of drug pressure, and the effect of target mutations on parasite fitness. Furthermore, our data only address the CpMetRS target mutations that spontaneously emerged in the calf model. Alternative mechanisms of drug resistance, such as altered expression of MDR efflux pumps or other compensatory alterations in gene expression, remain completely unexplored. Such resistance mechanisms may operate independently during evolution of drug resistance or may mediate drug tolerance that enables parasite persistence while more resistant mutants evolve.
Despite these limitations, our experience with 2093 in the dairy calf model has important implications for anticryptosporidial drug development. Ideally, the likelihood of Cryptosporidium resistance to different chemical classes or inhibitors of different targets should be assessed at early stages of development and the information used to help prioritize or deprioritize different drug leads within the anticryptosporidial pipeline. Furthermore, it is likely that except for compounds or targets with exceptionally high barriers to resistance, drug-drug or drug-potentiator combinations will be needed to mitigate the risk of resistance, as is now standard for tuberculosis (53), HIV (54), and anticancer therapies (22, 55). A lack of experimental tools to study anticryptosporidial resistance may account for the absence of prior studies. We are currently using 2093 as a positive-control compound to develop practical methods (i.e., not requiring dairy calves) to evolve Cryptosporidium drug resistance, but the lack of simple methods to cultivate large numbers of Cryptosporidium in vitro poses a significant technical challenge. If successful, we hope to use these methods to aid anticryptosporidial development by determining the relative propensities of different compound classes to induce resistance and studying the effects of drug-drug combinations.
In summary, these data demonstrate spontaneous emergence of drug-resistant C. parvum during a dairy calf clinical efficacy trial of the CpMetRS inhibitor 2093. Resistance was mediated by CpMetRS mutations coding for altered amino acids that appear to reduce compound binding with minimal effect on substrate binding. Rapid evolution of resistance was likely favored by the large burden of C. parvum in the dairy calf model; it remains unknown if other mechanisms mediating tolerance or resistance occurred that might have enabled parasite persistence and thereby facilitated emergence of highly resistant CpMetRS mutants. Our experience heightens concern about the inevitability of Cryptosporidium drug resistance. The anticryptosporidial field is poised for introduction of improved treatments in the next several years. With the goal of understanding how to best deploy these new treatments, we are now developing methods to systematically study anticryptosporidial drug resistance and identify approaches to impede its occurrence.
MATERIALS AND METHODS
Neonatal calf model of cryptosporidiosis.
Animal studies were approved by the University of Vermont (UVM) Institutional Animal Care and Use Committee (IACUC). The University of Vermont has an Animal Welfare Assurance with the Office of Laboratory Animal Welfare (OLAW) of the National Institutes of Health and is registered as a research institution by the U.S. Department of Agriculture. The work was performed in compliance with the Guide for the Care and Use of Laboratory Animals (56) and with the Animal Welfare Act and its associated regulations (USDA-APHIS “Blue Book” [www.aphis.usda.gov/animal-welfare]).
Holstein bull calves were acquired at birth from Green Mountain Dairy (Sheldon, VT), given synthetic colostrum with 200 g of IgG (Land O’Lakes, Ardent Hills, MO) and bovine coronavirus and Escherichia coli antibodies (First Defense Bolus; Immucell Corporation, Portland, ME) within 2 h of birth, and transported to UVM. Uninfected animals were group housed and infected at 24 to 48 h of age during an interruption in bottle feeding by oral administration of ∼5 × 107 viable C. parvum Iowa isolate oocysts (Bunch Grass Farms, Deary, ID) suspended in 10 ml of deionized water. Animals were moved to individual raised pens immediately after infection. Animals with severe diarrhea and other symptoms were supported with oral electrolytes or intravenous fluids and flunixin meglumine (Banamine; Merck) as needed. 2093 was suspended for dosing in 10 ml of 60% Phosal 53 MCT (Lipoid), 30% polyethylene glycol 400 (PEG 400), and 10% ethanol by volume. Doses were prepared fresh each day from solid 2093 and squirted into the calves’ mouths during interruptions in bottle feeding. For PK studies, fecal samples were collected at the indicated times by manual anal stimulation. For treatment efficacy studies, clinical evaluations were performed by a blinded observer at least twice daily at feeding times. Diarrhea, dehydration, and overall health (i.e., interest in feeding, playfulness, and alertness) were scored according to a previously published 3-point scale with 1 defined as normal and 3 severely abnormal (37). Overall health data are not reported, as scoring was confounded by unrelated complications that necessitated euthanasia of two control calves on day 5 (one with pneumonia and one with a congenital bladder obstruction). Daily fecal samples were obtained from collection bins located under each pen. Fecal samples used for parasite quantification were dried at 90°C until a stable weight was reached, and C. parvum abundance per gram of fecal dry matter was measured using a previously validated qPCR assay (primers listed in Table S1) (57). The lower limit of detection for this qPCR assay is ∼100 oocysts/g of dried feces.
Pharmacological methods.
Serum samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS), using compound spiked into control serum as a standard. Fecal compound 2093 was measured by homogenization of feces in phosphate-buffered saline (PBS; 0.1 g/ml) in a polypropylene tube and then further dilution prior to addition of an internal standard (enalapril) and acetonitrile protein precipitation. The supernatant was transferred to a fresh tube and dried using a SpeedVac. Samples were then resuspended and analyzed by LC-MS/MS.
PCR analysis of CpMetRS in shed oocysts.
Oocyst DNA was prepared from fecal samples as described above. The CpMetRS gene was Sanger sequenced on day 3 (3 vehicle calves and 3 treatment calves) and on day 9 (single surviving vehicle calf and 2 treatment calves that showed a relapse). The day 9 samples from the treatment group that showed point mutations were then amplified from genomic DNA, gel purified using the QIAquick gel extraction kit (Qiagen, Netherlands), cloned into the AVA0421 plasmid (58), and sequence verified.
Cell-based C. parvum growth inhibition assay.
A previously established high-content microscopy assay was used to measure activity of 2093 against the parental and transgenic C. parvum strains when grown in the human colon cancer cell line HCT-8 (ATCC; CCL-244) (39). HCT-8 cells were grown in RPMI 1640 medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 120 U/ml of penicillin, and 120 μg/ml of streptomycin (ATCC) at 37°C to near confluence in clear-bottomed 384-well plates in 50 μl per well. C. parvum oocysts were induced to excyst by treatment with 10 mM hydrochloric acid (10 min at 37°C) and then 2 mM sodium taurocholate in PBS (10 min at 16°C), washed and resuspended in the above-mentioned medium, and then added to cell monolayers at a concentration of 5.5 × 103 per well. Compounds were added 3 h after infection, and assay plates were incubated for 48 h postinfection at 37°C under 5% CO2. Wells were then washed three times with PBS containing 111 mM d-galactose, fixed with 4% formaldehyde in PBS for 15 min at room temperature (RT), permeabilized with 0.25% Triton X-100 (10 min at 37°C), washed three times with PBS with 0.1% Tween 20, and blocked with 4% bovine serum albumin (BSA) in PBS for 2 h at 37°C. Parasitophorous vacuoles were stained with 1.33 μg/ml of fluorescein-labeled Vicia villosa lectin (Vector Laboratories) diluted in 1% BSA in PBS with 0.1% Tween 20 (1 h at 37°C), followed by the addition of Hoechst 33258 (AnaSpec) at a final concentration of 0.09 mM diluted in water (15 min at 37°C). Wells were then washed five times with PBS containing 0.1% Tween 20. A Nikon Eclipse TE2000 epifluorescence microscope with an EXi Blue fluorescence microscopy camera (Qimaging, Canada) with a 20× objective (0.45 numerical aperture) was used to capture images. Nucleus and parasite images were exported separately as .tif files and were analyzed using macros developed on the ImageJ platform (National Institutes of Health) to determine the numbers of parasites and host cells (39).
Generation of transgenic C. parvum by CRISPR/Cas9 genome editing.
Transgenic C. parvum were generated using CRISPR/Cas9 genome editing and a previously reported method employing in vitro electroporation of excysted sporozoites followed by infection of ifn-γ−/− mice and paromomycin selection (38). Mouse experiments for generation of transgenic C. parvum were approved by the University of Vermont Institutional Animal Care and Use Committee.
To generate the Cas9/guide RNA plasmid, the CpMetRS guide sequence was cloned into the BbsI restriction site of the plasmid Aldo-Cas9-ribo. For the repair construct, a synthetic 999-bp fragment of the recodonized version of the CpMetRS (starting from amino acid number 247) along with the 3′ UTR sequence of the enolase gene (cgd5_1960) was purchased (GenScript, NJ). The synthetic construct was PCR amplified to introduce desired mutations and the 5′ homology region was introduced as an overhang in the forward primer. The enolase promoter-nluc-neomycin cassette was amplified from the vector Cplic3HAENNE provided by the Striepen lab (University of Pennsylvania) and the 3′ homology region was added at the end of this construct as a primer overhang. A 40-bp sequence homology was introduced in the two above-mentioned fragments during the PCR, and they were ligated with the NEBuilder HiFi DNA assembly master mix (New England BioLabs; catalog number E5520S). The whole cassette was then amplified with the end primer set. Each recodonized construct was cloned into pCR4Blunt-TOPO vector (Invitrogen) and sequence verified. To transfect C. parvum, the repair cassette for each mutant was PCR amplified from this vector using the end primer set. All PCR primers are included in Table S1.
Cryptosporidium parvum Iowa strain oocysts were purchased from Bunch Grass Farm (Deary, ID), excysted by treatment with 10 mM hydrochloric acid (10 min at 37°C) and then 2 mM sodium taurocholate in PBS (10 min at 16°C), and electroporated with Cas9/guide plasmid and repair DNA template using a Lonza Nucleofector 4D electroporator and the previously reported protocol (38). C57BL/6 ifn-γ−/− mice (Jackson Laboratory) aged 4 to 6 weeks (n = 2) were infected by injection of transfected sporozoites into an externalized loop of small intestine, and transgenic parasites were selected for by inclusion of paromomycin (Gemini Bio; 400-155P, 20 g/liter) in the mouse drinking water. Fecal samples were collected from cages and luminescence measurements were performed as described previously (38). Mouse feces containing transgenic parasites were then passaged by infection of NOD SCID gamma mice (Jackson Laboratory), which become chronically infected with C. parvum (15). Oocysts were purified from these mice for subsequent experiments.
Primers complementary to the 5′ homologous CpMetRS region and the CpMetRS 3′ UTR (primers P1 and P2 in Table S1) were used for PCR on fecal DNA from the infected NOD-SCID gamma mice to confirm the correct 5′ and 3′ integration events following homologous recombination. For this, total fecal DNA was purified using an E.Z.N.A. stool DNA kit (Omega Bio-Tek) according to the manufacturer’s pathogen detection protocol, except for inclusion of six freeze-thaw cycles in kit lysis buffer as a first step. Sanger sequencing was used to confirm incorporation of the desired mutations, and parasites were purified for use in dose-response assays by sucrose flotation followed by cesium chloride purification using a previously described protocol (59). Mouse fecal pellets were collected every 2 h for oocyst purification, which enhanced parasite viability by preventing desiccation.
Recombinant CpMetRS expression and in vitro enzymatic studies.
The expression and purification of the CpMetRS proteins were performed as previously described (58), with a few changes as follows. PA0.5G noninduction medium was prepared with 10× metal mix and 100 μg/ml each of l-methionine and 17 amino acids. Cultures were then incubated overnight at 37°C. ZYP-5052 medium was prepared with ampicillin at 100 μg/ml, carbenicillin at 100 μg/ml, and chloramphenicol at 34 μg/ml and incubated for 24 to 48 h at room temperature. The protein/Ni2+ mixture was incubated overnight at 4°C. Wash buffer contained 50 mM imidazole and 8 washes were performed, while elution buffer had 250 mM imidazole and eluted proteins were concentrated down to ∼100 μl using Amicon Ultra 0.5-ml centrifugal filters (Amicon, Darmstadt, Germany).
Enzyme kinetics and 2093 Ki values for CpMetRS WT, CpMetRS 74-9 (T246I) and CpMetRS 76-9 (D243E) were measured using an ATP:PPi exchange assay performed at room temperature (40). For Ki measurements, 2093 was preincubated for 5 min in a 96-well plate with 30 nM CpMetRS, 50 mM l-methionine, 2.5 mM NaPPi, 3 μCi of [32P]tetrasodium pyrophosphate (NEX019005MC; Perkin-Elmer), 2.5 mM dithiothreitol (DTT), 0.1 mg/ml of BSA, 0.2 mM spermine, 25 mM HEPES-KOH (pH 7.9), 10 mM MgCl2, 50 mM KCl, and 2% dimethyl sulfoxide (DMSO). Reactions were started with addition of 2.5 mM ATP; after 20 min, 5 μl of the reaction mixture (in duplicate) was quenched into a MultiScreenHTS Durapore 96-well filter plate (MSHVN4B50; Millipore Sigma) containing 200 μl of 10% charcoal in 0.5% HCl and 50 μl of wash buffer (1 M HCl with 200 mM NaPPi). The filter plates were washed three times with 200 μl of wash buffer on a vacuum manifold. The plates were dried for 1 h at RT, and then 25 μl of scintillation cocktail was added. Counts per minute were quantified on a MicroBeta2 scintillation counter (Perkin-Elmer). Percent inhibition was calculated by subtracting the background wells (containing all assay reagents except ATP, l-methionine, and compound) and comparing this value to the high control wells (containing all assay reagents without compound). The IC50 values were calculated by nonlinear regression methods using the Collaborative Drug Database (Burlingame, CA). Ki values were calculated from the IC50s that were shifted above the enzyme concentration (30 nM) using the Cheng-Prusoff equation as follows: IC50 = (1 + [Met]/KmMet)(1 + KmATP/[ATP])Ki (40). The Km values for l-methionine were determined using the assay conditions described above for each CpMetRS enzyme (30 nM) without compounds and with 5 μCi of [32P]tetrasodium pyrophosphate, 2.5 mM ATP, and 2.5 mM NaPPi, while titrating l-methionine. The reaction was quenched as described above at 0, 4, 8, 12, 16, and 20 min. Similarly, the Km values for ATP were measured by using 1 mM l-methionine and 2.5 mM NaPPi while titrating ATP. The Km and maximum rate of metabolism (Vmax) values were calculated using GraphPad Prism (v6.0). The kcat is equal to the Vmax divided by the enzyme concentration (30 nM). All IC50 and Km determination assays were performed twice to ensure reproducibility.
Comparative modeling of CpMetRS and docking of compound 2093.
A three-dimensional (3D) model for UniProt amino acid sequence Q5CVN0 was constructed with I-TASSER (60). A user-specified template was provided using the crystal structure of Trypanosoma brucei MetRS in complex with inhibitor 1312 [2-((3-((3,5-dichlorobenzyl)amino)propyl)amino)quinolin-4(1H)-one, PDB: 4eg5, chain B] to ensure a conformation of the protein that possesses the “auxiliary pocket” seen with dozens of inhibitors of that type, not only with T. brucei MetRS (41, 42) but also with the congeners of Brucella melitensis (61) and Staphylococcus aureus (unpublished data; PDB codes 4QRD and 4QRE). The overall sequence identity between the template and target was 37% and the similarity 53%; the sequence identity in the expected binding site is 76%. Because I-TASSER does not take into account the presence of the ligand manual rotamer, adjustment for five amino acid residues in the ligand binding site was necessary (residues Y24, H63, T185, T242, and R221) so that they were the same as in the T. brucei MetRS complex.
Subsequently, 2093 was docked in the 3D model of C. parvum MetRS with the Monte Carlo algorithm implemented in QXP+ (version 2016) (62), allowing for full flexibility of the ligand and the amino acid residues in direct contact.
Data analysis, statistical methods, and figure preparation.
Data were analyzed using GraphPad Prism version 7.01, and graphs were then exported as .eps files. The area under the curve (AUC) for oocyst shedding in calf feces was calculated for each animal using a plot of log10-transformed fecal oocyst shedding per gram of fecal dry matter versus time from days 2 to 6, with a baseline of 2 (log10 = 100). The P value for oocyst shedding was determined using an unpaired 2-tailed t test. Oocyst growth index data were assessed for normality using the D’Agostino and Pearson normality test. Because the measured growth indices for the D243E/T246I double mutant cell line failed the normality test, the mean growth indices of each cell line were compared to that of Iowa strain parasites using the nonparametric Kruskal-Wallis and Dunn’s multiple-comparison tests. Dose-response curves and half-maximal effective concentrations (EC50s) were determined by nonlinear regression using the equation for log (inhibitor) versus response curve with variable slope with the top response constrained to 100%. Figures were prepared using Adobe Illustrator CS5.
Data and material availability.
All data reported here are presented in the text and/or supplemental material. The plasmids and genetically modified C. parvum strains are available from C.D.H. under a material transfer agreement (MTA) with the University of Vermont Larner College of Medicine. The plasmids for expression of recombinant CpMetRS proteins are available from F.S.B. under an MTA with the University of Washington.
ACKNOWLEDGMENTS
We are grateful to Colin McMartin for providing us with the QXP+ software.
This work was supported by PATH with funding from the UK government (grant 300341-111) and by National Institutes of Health grant R01 AI143951 (C.D.H.).
M.M.H., conceptualization, methodology, validation, formal analysis, visualization, and writing (original draft, review, and editing); E.E.S., conceptualization, methodology, and formal analysis; R.K.M.C., conceptualization, writing (review and editing), and funding acquisition; J.R.G., methodology, validation, formal analysis, writing (original draft, review, and editing), and visualization; E.L.D.H., conceptualization, writing (review and editing), and funding acquisition; P.M., methodology, validation, and writing (review and editing); A.M., methodology, validation, formal analysis, and writing (original draft); R.M.R., methodology, validation, and formal analysis; J.E.T., conceptualization, methodology, and writing (review and editing); C.L.M.J.V., methodology, writing (original draft, review, and editing), and visualization; A.S., conceptualization, methodology, and writing (review and editing); Z.Z., methodology; D.M.O., methodology and formal analysis; D.W.G., methodology and formal analysis; E.F., writing (review and editing), supervision, and funding acquisition; F.S.B., conceptualization, methodology, writing (original draft, review, and editing), supervision, and funding acquisition; C.D.H., conceptualization, methodology, formal analysis, visualization, writing (original draft, review, and editing), supervision, and funding acquisition.
We declare no competing interests.
Footnotes
Supplemental material is available online only.
aac.00023-21-s000s1.pdf (287.8KB, pdf)
Contributor Information
Frederick S. Buckner, Email: FBuckner@medicine.washington.edu.
Christopher D. Huston, Email: christopher.huston@uvm.edu.
REFERENCES
- 1.Chen XM, Keithly JS, Paya CV, LaRusso NF. 2002. Cryptosporidiosis. N Engl J Med 346:1723–1731. 10.1056/NEJMra013170. [DOI] [PubMed] [Google Scholar]
- 2.Bouzid M, Hunter PR, Chalmers RM, Tyler KM. 2013. Cryptosporidium pathogenicity and virulence. Clin Microbiol Rev 26:115–134. 10.1128/CMR.00076-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouzid M, Tyler KM, Christen R, Chalmers RM, Elwin K, Hunter PR. 2010. Multi-locus analysis of human infective Cryptosporidium species and subtypes using ten novel genetic loci. BMC Microbiol 10:213. 10.1186/1471-2180-10-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Checkley W, White AC, Jaganath D, Arrowood MJ, Chalmers RM, Chen X-M, Fayer R, Griffiths JK, Guerrant RL, Hedstrom L, Huston CD, Kotloff KL, Kang G, Mead JR, Miller M, Petri WA, Priest JW, Roos DS, Striepen B, Thompson RCA, Ward HD, Van Voorhis WA, Xiao L, Zhu G, Houpt ER. 2015. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet Infect Dis 15:85–94. 10.1016/S1473-3099(14)70772-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acácio S, Biswas K, O’Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Platts-Mills JA, Juma J, Kabir F, Nkeze J, Okoi C, Operario DJ, Uddin J, Ahmed S, Alonso PL, Antonio M, Becker SM, Blackwelder WC, Breiman RF, Faruque ASG, Fields B, Gratz J, Haque R, Hossain A, Hossain MJ, Jarju S, Qamar F, Iqbal NT, Kwambana B, Mandomando I, McMurry TL, Ochieng C, Ochieng JB, Ochieng M, Onyango C, Panchalingam S, Kalam A, Aziz F, Qureshi S, Ramamurthy T, Roberts JH, Saha D, Sow SO, Stroup SE, Sur D, Tamboura B, Taniuchi M, Tennant SM, Toema D, Wu Y, Zaidi A, Nataro JP, Kotloff KL, Levine MM, Houpt ER, et al. 2016. Use of quantitative molecular diagnostic methods to identify causes of diarrhoea in children: a reanalysis of the GEMS case-control study. Lancet 388:1291–1301. 10.1016/S0140-6736(16)31529-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korpe PS, Haque R, Gilchrist C, Valencia C, Niu F, Lu M, Ma JZ, Petri SE, Reichman D, Kabir M, Duggal P, Petri WA. 2016. Natural history of cryptosporidiosis in a longitudinal study of slum-dwelling Bangladeshi children: association with severe malnutrition. PLoS Negl Trop Dis 10:e0004564. 10.1371/journal.pntd.0004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khalil IA, Troeger C, Rao PC, Blacker BF, Brown A, Brewer TG, Colombara DV, De Hostos EL, Engmann C, Guerrant RL, Haque R, Houpt ER, Kang G, Korpe PS, Kotloff KL, Lima AAM, Petri WA, Platts-Mills JA, Shoultz DA, Forouzanfar MH, Hay SI, Reiner RC, Mokdad AH. 2018. Morbidity, mortality, and long-term consequences associated with diarrhoea from Cryptosporidium infection in children younger than 5 years: a meta-analyses study. Lancet Glob Health 6:e758–e768. 10.1016/S2214-109X(18)30283-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abubakar I, Aliyu SH, Arumugam C, Hunter PR, Usman NK. 2007. Prevention and treatment of cryptosporidiosis in immunocompromised patients. Cochrane Database Syst Rev 2007(1):CD004932. 10.1002/14651858.CD004932.pub2. [DOI] [PubMed] [Google Scholar]
- 10.Amadi B, Mwiya M, Musuku J, Watuka A, Sianongo S, Ayoub A, Kelly P. 2002. Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: a randomised controlled trial. Lancet 360:1375–1380. 10.1016/S0140-6736(02)11401-2. [DOI] [PubMed] [Google Scholar]
- 11.Amadi B, Mwiya M, Sianongo S, Payne L, Watuka A, Katubulushi M, Kelly P. 2009. High dose prolonged treatment with nitazoxanide is not effective for cryptosporidiosis in HIV positive Zambian children: a randomised controlled trial. BMC Infect Dis 9:195. 10.1186/1471-2334-9-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love MS, Beasley FC, Jumani RS, Wright TM, Chatterjee AK, Huston CD, Schultz PG, McNamara CW. 2017. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Negl Trop Dis 11:e0005373. 10.1371/journal.pntd.0005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo F, Zhang H, McNair NN, Mead JR, Zhu G. 2018. The existing drug vorinostat as a new lead against cryptosporidiosis by targeting the parasite histone deacetylases. J Infect Dis 217:1110–1117. 10.1093/infdis/jix689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baragana B, et al. 2019. Lysyl-tRNA synthetase as a drug target in malaria and cryptosporidiosis. Proc Natl Acad Sci U S A 116:7015–7020. 10.1073/pnas.1814685116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jumani RS, Bessoff K, Love MS, Miller P, Stebbins EE, Teixeira JE, Campbell MA, Meyers MJ, Zambriski JA, Nunez V, Woods AK, McNamara CW, Huston CD. 2018. A novel piperazine-based drug lead for cryptosporidiosis from the Medicines for Malaria Venture Open-Access Malaria Box. Antimicrob Agents Chemother 62:e01505-17. 10.1128/AAC.01505-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain V, Yogavel M, Kikuchi H, Oshima Y, Hariguchi N, Matsumoto M, Goel P, Touquet B, Jumani RS, Tacchini-Cottier F, Harlos K, Huston CD, Hakimi M-A, Sharma A. 2017. Targeting prolyl-tRNA synthetase to accelerate drug discovery against malaria, leishmaniasis, toxoplasmosis, cryptosporidiosis, and coccidiosis. Structure 25:1495–1505.e1496. 10.1016/j.str.2017.07.015. [DOI] [PubMed] [Google Scholar]
- 17.Lee S, Ginese M, Beamer G, Danz HR, Girouard DJ, Chapman-Bonofiglio SP, Lee M, Hulverson MA, Choi R, Whitman GR, Ojo KK, Arnold SLM, Van Voorhis WC, Tzipori S. 2018. Therapeutic efficacy of bumped kinase inhibitor 1369 in a pig model of acute diarrhea caused by Cryptosporidium hominis. Antimicrob Agents Chemother 62:e00147-18. 10.1128/AAC.00147-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manjunatha UH, Vinayak S, Zambriski JA, Chao AT, Sy T, Noble CG, Bonamy GMC, Kondreddi RR, Zou B, Gedeck P, Brooks CF, Herbert GT, Sateriale A, Tandel J, Noh S, Lakshminarayana SB, Lim SH, Goodman LB, Bodenreider C, Feng G, Zhang L, Blasco F, Wagner J, Leong FJ, Striepen B, Diagana TT. 2017. A Cryptosporidium PI(4)K inhibitor is a drug candidate for cryptosporidiosis. Nature 546:376–380. 10.1038/nature22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lunde CS, Stebbins EE, Jumani RS, Hasan MM, Miller P, Barlow J, Freund YR, Berry P, Stefanakis R, Gut J, Rosenthal PJ, Love MS, McNamara CW, Easom E, Plattner JJ, Jacobs RT, Huston CD. 2019. Identification of a potent benzoxaborole drug candidate for treating cryptosporidiosis. Nat Commun 10:2816. 10.1038/s41467-019-10687-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang W, Choi R, Hulverson MA, Zhang Z, McCloskey MC, Schaefer DA, Whitman GR, Barrett LK, Vidadala RSR, Riggs MW, Maly DJ, Van Voorhis WC, Ojo KK, Fan E. 2017. 5-Aminopyrazole-4-carboxamide-based compounds prevent the growth of Cryptosporidium parvum. Antimicrob Agents Chemother 61:e00020-17. 10.1128/AAC.00020-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Packard RM. 2014. The origins of antimalarial-drug resistance. N Engl J Med 371:397–399. 10.1056/NEJMp1403340. [DOI] [PubMed] [Google Scholar]
- 22.Hughes D, Andersson DI. 2015. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet 16:459–471. 10.1038/nrg3922. [DOI] [PubMed] [Google Scholar]
- 23.Nakama T, Nureki O, Yokoyama S. 2001. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. J Biol Chem 276:47387–47393. 10.1074/jbc.M109089200. [DOI] [PubMed] [Google Scholar]
- 24.Rock FL, Mao W, Yaremchuk A, Tukalo M, Crépin T, Zhou H, Zhang Y-K, Hernandez V, Akama T, Baker SJ, Plattner JJ, Shapiro L, Martinis SA, Benkovic SJ, Cusack S, Alley MRK. 2007. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 316:1759–1761. 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 25.Elewski BE, Aly R, Baldwin SL, González Soto RF, Rich P, Weisfeld M, Wiltz H, Zane LT, Pollak R. 2015. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies. J Am Acad Dermatol 73:62–69. 10.1016/j.jaad.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 26.Nayak SU, Griffiss JM, Blumer J, O'Riordan MA, Gray W, McKenzie R, Jurao RA, An AT, Le M, Bell SJ, Ochsner UA, Jarvis TC, Janjic N, Zenilman JM. 2017. Safety, tolerability, systemic exposure, and metabolism of CRS3123, a methionyl-tRNA synthetase inhibitor developed for treatment of Clostridium difficile, in a phase 1 study. Antimicrob Agents Chemother 61:e02760-16. 10.1128/AAC.02760-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Dwyer K, Spivak AT, Ingraham K, Min S, Holmes DJ, Jakielaszek C, Rittenhouse S, Kwan AL, Livi GP, Sathe G, Thomas E, Van Horn S, Miller LA, Twynholm M, Tomayko J, Dalessandro M, Caltabiano M, Scangarella-Oman NE, Brown JR. 2015. Bacterial resistance to leucyl-tRNA synthetase inhibitor GSK2251052 develops during treatment of complicated urinary tract infections. Antimicrob Agents Chemother 59:289–298. 10.1128/AAC.03774-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez V, Crépin T, Palencia A, Cusack S, Akama T, Baker SJ, Bu W, Feng L, Freund YR, Liu L, Meewan M, Mohan M, Mao W, Rock FL, Sexton H, Sheoran A, Zhang Y, Zhang Y-K, Zhou Y, Nieman JA, Anugula MR, Keramane EM, Savariraj K, Reddy DS, Sharma R, Subedi R, Singh R, O’Leary A, Simon NL, De Marsh PL, Mushtaq S, Warner M, Livermore DM, Alley MRK, Plattner JJ. 2013. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob Agents Chemother 57:1394–1403. 10.1128/AAC.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Hernandez V, Rock FL, Choi W, Mak YSL, Mohan M, Mao W, Zhou Y, Easom EE, Plattner JJ, Zou W, Pérez-Herrán E, Giordano I, Mendoza-Losana A, Alemparte C, Rullas J, Angulo-Barturen I, Crouch S, Ortega F, Barros D, Alley MRK. 2017. Discovery of a potent and specific M. tuberculosis leucyl-tRNA synthetase inhibitor: (S)-3-(aminomethyl)-4-chloro-7–(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656). J Med Chem 60:8011–8026. 10.1021/acs.jmedchem.7b00631. [DOI] [PubMed] [Google Scholar]
- 30.Palencia A, Li X, Bu W, Choi W, Ding CZ, Easom EE, Feng L, Hernandez V, Houston P, Liu L, Meewan M, Mohan M, Rock FL, Sexton H, Zhang S, Zhou Y, Wan B, Wang Y, Franzblau SG, Woolhiser L, Gruppo V, Lenaerts AJ, O’Malley T, Parish T, Cooper CB, Waters MG, Ma Z, Ioerger TR, Sacchettini JC, Rullas J, Angulo-Barturen I, Pérez-Herrán E, Mendoza A, Barros D, Cusack S, Plattner JJ, Alley MRK. 2016. Discovery of novel oral protein synthesis inhibitors of Mycobacterium tuberculosis that target leucyl-tRNA synthetase. Antimicrob Agents Chemother 60:6271–6280. 10.1128/AAC.01339-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jumani RS, Hasan MM, Stebbins EE, Donnelly L, Miller P, Klopfer C, Bessoff K, Teixeira JE, Love MS, McNamara CW, Huston CD. 2019. A suite of phenotypic assays to ensure pipeline diversity when prioritizing drug-like Cryptosporidium growth inhibitors. Nat Commun 10:1862. 10.1038/s41467-019-09880-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim Y-J, Lee H-K, Cortese JF, Wirth DF, Dignam JD, Rao A, Yeo C-Y, Mazitschek R, Whitman M. 2012. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol 8:311–317. 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vinayak S, Jumani RS, Miller P, Hasan MM, McLeod BI, Tandel J, Stebbins EE, Teixeira JE, Borrel J, Gonse A, Zhang M, Yu X, Wernimont A, Walpole C, Eckley S, Love MS, McNamara CW, Sharma M, Sharma A, Scherer CA, Kato N, Schreiber SL, Melillo B, Striepen B, Huston CD, Comer E. 2020. Bicyclic azetidines cure infection with the diarrheal pathogen Cryptosporidium by inhibiting parasite phenylalanyl-tRNA synthetase. Sci Transl Med 12:eaba8412. 10.1126/scitranslmed.aba8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buckner FS, Ranade RM, Gillespie JR, Shibata S, Hulverson MA, Zhang Z, Huang W, Choi R, Verlinde CLMJ, Hol WGJ, Ochida A, Akao Y, Choy RKM, Van Voorhis WC, Arnold SLM, Jumani RS, Huston CD, Fan E. 2019. Optimization of methionyl tRNA-synthetase inhibitors for treatment of Cryptosporidium infection. Antimicrob Agents Chemother 63:e02061-18. 10.1128/AAC.02061-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gentry DR, Ingraham KA, Stanhope MJ, Rittenhouse S, Jarvest RL, O’Hanlon PJ, Brown JR, Holmes DJ. 2003. Variable sensitivity to bacterial methionyl-tRNA synthetase inhibitors reveals subpopulations of Streptococcus pneumoniae with two distinct methionyl-tRNA synthetase genes. Antimicrob Agents Chemother 47:1784–1789. 10.1128/aac.47.6.1784-1789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zambriski JA, Nydam DV, Bowman DD, Bellosa ML, Burton AJ, Linden TC, Liotta JL, Ollivett TL, Tondello-Martins L, Mohammed HO. 2013. Description of fecal shedding of Cryptosporidium parvum oocysts in experimentally challenged dairy calves. Parasitol Res 112:1247–1254. 10.1007/s00436-012-3258-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stebbins E, Jumani RS, Klopfer C, Barlow J, Miller P, Campbell MA, Meyers MJ, Griggs DW, Huston CD. 2018. Clinical and microbiologic efficacy of the piperazine-based drug lead MMV665917 in the dairy calf cryptosporidiosis model. PLoS Negl Trop Dis 12:e0006183. 10.1371/journal.pntd.0006183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, Cipriano MJ, Striepen B. 2015. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature 523:477–480. 10.1038/nature14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bessoff K, Sateriale A, Lee KK, Huston CD. 2013. Drug repurposing screen reveals FDA-approved inhibitors of human HMG-CoA reductase and isoprenoid synthesis that block Cryptosporidium parvum growth. Antimicrob Agents Chemother 57:1804–1814. 10.1128/AAC.02460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Green LS, Bullard JM, Ribble W, Dean F, Ayers DF, Ochsner UA, Janjic N, Jarvis TC. 2009. Inhibition of methionyl-tRNA synthetase by REP8839 and effects of resistance mutations on enzyme activity. Antimicrob Agents Chemother 53:86–94. 10.1128/AAC.00275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koh CY, Kim JE, Shibata S, Ranade RM, Yu M, Liu J, Gillespie JR, Buckner FS, Verlinde CLMJ, Fan E, Hol WGJ. 2012. Distinct states of methionyl-tRNA synthetase indicate inhibitor binding by conformational selection. Structure 20:1681–1691. 10.1016/j.str.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koh CY, Kim JE, Wetzel AB, de van der Schueren WJ, Shibata S, Ranade RM, Liu J, Zhang Z, Gillespie JR, Buckner FS, Verlinde CLMJ, Fan E, Hol WGJ. 2014. Structures of Trypanosoma brucei methionyl-tRNA synthetase with urea-based inhibitors provide guidance for drug design against sleeping sickness. PLoS Negl Trop Dis 8:e2775. 10.1371/journal.pntd.0002775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang W, Zhang Z, Barros-Álvarez X, Koh CY, Ranade RM, Gillespie JR, Creason SA, Shibata S, Verlinde CLMJ, Hol WGJ, Buckner FS, Fan E. 2016. Structure-guided design of novel Trypanosoma brucei methionyl-tRNA synthetase inhibitors. Eur J Med Chem 124:1081–1092. 10.1016/j.ejmech.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Z, Barros-Álvarez X, Gillespie JR, Ranade RM, Huang W, Shibata S, Molasky NMR, Faghih O, Mushtaq A, Choy RKM, de Hostos E, Hol WGJ, Verlinde CLMJ, Buckner FS, Fan E. 2020. Structure-guided discovery of selective methionyl-tRNA synthetase inhibitors with potent activity against Trypanosoma brucei. RSC Med Chem 11:885–895. 10.1039/D0MD00057D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nydam DV, Wade SE, Schaaf SL, Mohammed HO. 2001. Number of Cryptosporidium parvum oocysts or Giardia spp cysts shed by dairy calves after natural infection. Am J Vet Res 62:1612–1615. 10.2460/ajvr.2001.62.1612. [DOI] [PubMed] [Google Scholar]
- 47.White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. 2014. Malaria. Lancet 383:723–735. 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- 48.Looareesuwan S, Viravan C, Webster HK, Kyle DE, Hutchinson DB, Canfield CJ. 1996. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am J Trop Med Hyg 54:62–66. 10.4269/ajtmh.1996.54.62. [DOI] [PubMed] [Google Scholar]
- 49.Rathod PK, McErlean T, Lee PC. 1997. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc Natl Acad Sci U S A 94:9389–9393. 10.1073/pnas.94.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woodford N, Ellington MJ. 2007. The emergence of antibiotic resistance by mutation. Clin Microbiol Infect 13:5–18. 10.1111/j.1469-0691.2006.01492.x. [DOI] [PubMed] [Google Scholar]
- 51.Aubry-Damon H, Soussy CJ, Courvalin P. 1998. Characterization of mutations in the rpoB gene that confer rifampin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 42:2590–2594. 10.1128/AAC.42.10.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ochsner UA, Young CL, Stone KC, Dean FB, Janjic N, Critchley IA. 2005. Mode of action and biochemical characterization of REP8839, a novel inhibitor of methionyl-tRNA synthetase. AAC 49:4253–4262. 10.1128/AAC.49.10.4253-4262.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dye C. 2009. Doomsday postponed? Preventing and reversing epidemics of drug-resistant tuberculosis. Nat Rev Microbiol 7:81–87. 10.1038/nrmicro2048. [DOI] [PubMed] [Google Scholar]
- 54.Panel on Antiretroviral Guidelines for Adults and Adolescents. 2019. What to start: initial combination regimens for the antiretroviral-naive patient. https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv/what-start-initial-combination-regimens-antiretroviral-naive.
- 55.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. 2013. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13:714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 56.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 57.Parr JB, Sevilleja JE, Samie A, Amidou S, Alcantara C, Stroup SE, Kohli A, Fayer R, Lima AAM, Houpt ER, Guerrant RL. 2007. Detection and quantification of Cryptosporidium in HCT-8 cells and human fecal specimens using real-time polymerase chain reaction. Am J Trop Med Hyg 76:938–942. 10.4269/ajtmh.2007.76.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi R, Kelley A, Leibly D, Hewitt SN, Napuli A, Van Voorhis W. 2011. Immobilized metal-affinity chromatography protein-recovery screening is predictive of crystallographic structure success. Acta Crystallogr Sect F Struct Biol Cryst Commun 67:998–1005. 10.1107/S1744309111017374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pawlowic MC, Vinayak S, Sateriale A, Brooks CF, Striepen B. 2017. Generating and maintaining transgenic Cryptosporidium parvum parasites. Curr Protoc Microbiol 46:20B.2.1–20B.2.32. 10.1002/cpmc.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. 2015. The I-TASSER Suite: protein structure and function prediction. Nat Methods 12:7–8. 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ojo KK, Ranade RM, Zhang Z, Dranow DM, Myers JB, Choi R, Nakazawa Hewitt S, Edwards TE, Davies DR, Lorimer D, Boyle SM, Barrett LK, Buckner FS, Fan E, Van Voorhis WC. 2016. Brucella melitensis methionyl-tRNA-synthetase (MetRS), a potential drug target for brucellosis. PLoS One 11:e0160350. 10.1371/journal.pone.0160350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McMartin C, Bohacek RS. 1997. QXP: powerful, rapid computer algorithms for structure-based drug design. J Comput Aided Mol Des 11:333–344. 10.1023/A:1007907728892. [DOI] [PubMed] [Google Scholar]