

Abstract
Hidradenitis Suppurativa (HS) is a chronic, relapsing, and remitting inflammatory disease of the skin with significant heritability and racial disposition. The pathogenesis of HS remains enigmatic, but occlusion of the terminal hair follicle and dysregulation of the local innate immune response may contribute to pathogenesis. Genetic predisposition might also contribute to disease susceptibility and phenotypic heterogeneity since mutations in γ-secretase have been found to underly a minor but characteristic subset HS patients. In this review, we synthesized the current data on γ-secretase in HS, evaluated its importance in the context of disease pathobiology, and discuss avenues of future studies.
Keywords: Hidradenitis Suppurativa, γ-secretase, Genetics
Introduction
Hidradenitis Suppurativa (HS), also known as acne inversa, is a chronic, relapsing inflammatory disease of the skin characterized by painful acne-like lesions, nodules, abscesses, sinus tracts, and scar formation primarily in intertriginous regions (i.e. axillae, submammary folds, groin). HS is associated with a high comorbidity burden and the lowest quality of life among any dermatologic condition, yet remains under-recognized and poorly understood (Reddy et al., 2019). Global incidences of HS vary by country. In the United States the incidence of HS is rising. HS is reported in individuals of all age groups, races, and genders but shows a predilection toward African Americans and women. Registry studies estimate the prevalence of HS at 0.3%, 0.22% and 0.09% in individuals of African, biracial and Caucasian descent, respectively. Furthermore, among these groups, the prevalence peaks between 20–40 years of age and declines after 50 (Garg et al., 2017a, Garg et al., 2017b, Sabat et al., 2020). Despite this demonstrated need, the pathogenesis of HS remains poorly studied.
Studies report that 30–42% of HS patients report a positive family history of the disease, which points towards a potential genetic etiology. A recent Dutch twin cohort study found a narrow-sense heritability of 77% for HS (van Straalen et al., 2020). Furthermore, a minority of these patients across multiple ethnicities have been found to exhibit a monogenic form of the disease that is associated with heterozygous mutations in the γ-secretase complex (Ingram, 2016). There is an increased incidence of HS in the setting of other genetic inflammatory syndromes, and multiple syndromic forms of HS have been identified such as PASH (pyoderma gangrenosum, acne, and HS) and Dowling-Degos disease (DDD), many of which have been tracked to specific mutations in a handful of candidate genes (Scheinfeld, 2013). The sporadic form of HS, in contrast to the familial form, appears to encompass the majority of disease burden (60–70%) and is thought to be driven by a polygenic architecture (Jfri et al., 2019). Several unique phenotypes have even been identified in both familial and sporadic HS, and in certain endemic populations (such as males of Asian ancestry) with clear evidence of heritability (Pink et al., 2012, Wang et al., 2010, Xu et al., 2016). Studies have found associations between environmental factors and HS, which suggests a multi-factorial etiology. Whether specific genetic variations increase susceptibility to developing HS in the presence of specific environmental triggers remains an open question and suggests the existence of previously undescribed genetic risk factors.
The paucity of HS genome-wide association studies (GWASs) have made systematic dissection of HS pathophysiology challenging from a genetic level and due to its inflammatory nature some have turned to immune-profiling for insights (Gudjonsson et al., 2020, Lowe et al., 2020). Furthermore, a more complete understanding of genetic features underlying HS may help to develop a more nuanced classification system with better prognostic value, improve patient management and identify key candidate therapeutics. To date no genotype-phenotype correlation has been established, but combined genetic and immunological studies could bridge the gap (Frew et al., 2019). Although a minority of the total HS patients exhibit family history, clinical, genetic and molecular studies in familial cohorts harboring γ-secretase mutations began to define pathological mechanisms involved in the etiology of HS. Subsequent studies in laboratory animals further identify molecular mechanisms involved in HS. Together, GWASs and laboratory studies have shown feasibility in dissecting potential mechanisms of HS pathology. In this review, we synthesize the current information on γ-secretase genetics underlying a subpopulation of patients with HS and evaluate its importance in the context of disease pathobiology and future research.
Mutations in γ-secretase demonstrate clinical significance in a subset of HS patients
The γ-secretase complex is a heterogenous transmembrane protease complex composed of the catalytic presenilin-1/2 (PSEN1/PSEN2) and co-factor subunits presenilin-enhancer-2 (PSENEN), Nicastrin (NCSTN), and anterior pharynx defective 1 (APH1A/APH1B). It functions to cleave over 70 type I membrane proteins such as cadherins, notch, and amyloid precursor protein (APP) (Merilahti et al., 2017). Dysfunctional γ-secretase-APP axis is well-known in the development of Alzheimer’s disease; however, epidemiological studies to-date have not identified an increased risk of Alzheimer’s among HS patients with γ-secretase complex mutations or overlapping pathogenic variants between the two disease populations (Garg and Strunk, 2017, Theut Riis et al., 2017). Alzheimer’s and HS associated γ-secretase mutations may have distinct functional outcomes with regards to downstream signaling and efficacy in cleaving different substrates. More specifically, one could hypothesize that HS-associated γ-secretase mutations have no affect on the ability of γ-secretase to cleave APP or that these mutations are found in isoforms not expressed in the brain.
In 2006, Gao et al. identified a putative risk locus within 1p21.1–1q25.3, a >900 gene region, in a four-generation Chinese family using genome-wide linkage scan (GWLS) (Gao et al., 2006). This was further narrowed to a >200 gene region within 1q21.3–1q23.2 in a follow-up Chinese case report. In a 2010 GWLS, Wang et. al. identified γ-secretase mutations in a cohort of 6 Han Chinese families with an autosomal dominant transmission pattern that harbored separate heterogenous rare variants in NCSTN, PSEN1, or PSENEN- which localized to the 1q23.2 locus (Wang et al., 2010). Gao et. al. and Wang et. al. represented two of four genetic studies employing a genome-wide approach in HS kindreds to date. The final two identified putative risk loci at 1q23.2 (NCSTN) in an Iranian family, and both chromosome 19 and 6q25.1–25.2 in a number of European families, respectively (Faraji Zonooz et al., 2016, Irwin McLean et al., 2006). In addition, a handful of other studies probing African American, Indian, Japanese, British, and French families identified γ-secretase mutations that co-segregated with a disease phenotype (Ratnamala et al., 2016, Takeichi et al., 2019). The remainder of mutations were identified via targeted sequencing; overall, 50 single-nucleotide polymorphisms (SNPs) associated with HS have been identified in Chinese (23), French (3), British (3), Thai (3), African American (1) encompassing the NCSTN, PSEN1, and PSENEN genes (Table 1), 23 of which were determined to be “likely pathogenic” by American College of Medical Genetics (ACMG) criteria (Frew et al., 2017). The locations of these mutations in γ-secretase protein domains are shown in Figure 1. Current population data indicate that such heterozygous, nonsynonymous γ-secretase mutations are rarely found in healthy controls and demonstrate high penetrance in affected pedigrees (Wang et al., 2010). Interestingly, linkage disequilibrium was identified in 12 pairs of variants, and two specific mutations, NCSTN-R117X and -Q568X, were each found in families from different races (Frew et al., 2019, Li A. et al., 2018).
Table 1:
Identified mutations in HS patients in NCSTN, PSENEN, and PSEN1.
| NCSTN - 1q23.2 | |||||||
|---|---|---|---|---|---|---|---|
| DNA change | Amino Acid Change | Mutation Type | Ethnic Origin (# of families) | Familial (F) or Case (C), | Isolated HS or Syndrome/ Associated Conditions | Method of Sequencing (*indicates the use of controls) | |
| 1 | c.97G>A | p.Gly33Arg | Missense | Japanese (1)(Takeichi et al., 2020) | F | Isolated HS | Whole Exome Sequencing |
| 2 | c.223G>A | p.Val75Ile | Missense | Chinese (1) (Zhang et al., 2013) | F | Isolated HS | Targeted Sequencing* |
| 3 | c.210_21 1delAG | p.Thr70fsX 18 | Truncating | Chinese (1) (Liu et al., 2011) | F | Isolated HS | Whole Exome Sequencing* |
| 4 | c.218delC Exon 4 | p.P73LFs*15 | Frameshift | Chinese (1) (Wu et al., 2018) | F | Isolated HS | Targeted Sequencing* |
| 5 | c.278del C | p.P93LFSX15 | Frameshift | Chinese (1) (Li C. et al., 2018) | C | SAPHO | Whole Exome Sequencing* |
| 6 | c.344_35 1del | p.Thr115As n*20 | Truncating | N/A (3) (Duchatelet et al., 2015) | C | PASH | Targeted Sequencing |
| 7 | c.349C>T | p.Arg117X | Truncating | Chinese (1) (Wang et al., 2010), Caucasian (1),(Liu M. et al., 2016) African American (1)(Chen et al., 2015), Japanese(1) | F (all) | Isolated HS (all) | GWLS* Targeted Sequencing Targeted Sequencing Targeted Sequencing |
| 8 | c.477 C>A | p.C159X | Truncating | Chinese(Savva et al., 2013) (1) | F | Isolated HS | Targeted Sequencing* |
| 9 | c.487delC | p.Gln163Se rfsX39 | Truncating | French (3)(Miskinyte et al., 2012) | F | Isolated HS | Targeted Sequencing* |
| 10 | c.497C>A | p.Ser166X | Truncating | Chinese(Ma et al., 2014) | F | Isolated HS | Targeted Sequencing |
| 11 | c.553G>A | p.Asp185Asn | Missense | British (1) (Pink et al., 2013) | C | Isolated HS | N/A |
| 12 | c.582+1d elG | Splice site | Splice site | Japanese (1) (Nomura et al., 2013) | F | Isolated HS | Targeted Sequencing* |
| 13 | c.617C>A | p.Ser206X | Truncating | Chinese(Shi et al., 2018) | F | Isolated HS | Targeted Sequencing* |
| 14 | c.632C>G | p.Pro211Arg | Missense | Chinese (1) (Li et al., 2011) | F | Isolated HS | Targeted Sequencing* |
| 15 | c.647A>C | p.Gln216Pro | Missense | Chinese (1)(Zhang et al., 2013) | F | Isolated HS | Targeted Sequencing* |
| 16 | c.687insC C | p.Cys230ProfsX31 | Frameshift | Indian (1)(Li et al., 2011) | F | HS + Acne Conglobata (AC) | Targeted Sequencing* |
| 17 | c.887A>G | p.Pro296Arg | Missense | Chinese (1)(Xu et al., 2016) | F | Isolated HS | Targeted Sequencing |
| 18 | c.944C>T | p.Ala315Val | Missense | Chinese (1)(Zhang et al., 2016) | F | Isolated HS | Targeted Sequencing |
| 19 | c.978delG | p.M326IfsX30 | Truncating | Singaporean (Haines et al., 2012) | F | Isolated HS | Targeted Sequencing* |
| 20 | c.996+7G >A | Splice site | Splice site | Mixed - European (1)(Pink et al., 2012) | F | Isolated HS | Targeted Sequencing* |
| 21 | c.1101+1 G>A | Splice site | Splice site | Mixed- European (2) (Pink et al., 2011) | F | Isolated HS | Targeted Sequencing* |
| 22 | c.1101+1 0A>G | Splice site | Splice site | British (1)(Pink et al., 2012) | F | Isolated HS | Targeted Sequencing* |
| 23 | c.1125+1 G>A | Splice site | Splice site | British (1)(Pink et al., 2011) | F | Isolated HS | Targeted Sequencing* |
| 24 | c.1180- 5C>G | Splice site | Splice site | British (1)(Ingram et al., 2013) | F (1) S (2) | Isolated HS | Targeted Sequencing |
| 25 | c.1229C> T | p.A410V | Missense | Caucasian(Liu M. et al., 2016) | F | Isolated HS | Targeted Sequencing |
| 26 | c.1258C> T | p.Gln420X | Truncating | Singaporean (Haines et al., 2012) Chinese(Jiao et al., 2013, Yang et al., 2015) | F | Isolated HS | Targeted Sequencing* |
| 27 | c.1258C> T | p.Arg429X | Truncating | Japanese(Nishimori et al., 2017) | S | Isolated HS | Targeted Sequencing |
| 28 | c.1300C> T | p.Arg434X | Truncating | French (1)(Miskinyte et al., 2012) | F | Isolated HS | Targeted Sequencing |
| 29 | c.1352+1 G>A | Splice site | Splice site | Chinese (1)(Liu et al., 2011) | F | Isolated HS | Targeted Sequencing* |
| 30 | c.1551+1 G>A | Splice site | Splice site | Chinese (1)(Wang et al., 2010) | F | Isolated HS | GWLS* |
| 31 | c.1635C>G | p.Ala486 Thr517del | Truncating | Iranian (1)(Faraji Zonooz et al., 2016) | F | PASH | GWLS* |
| 32 | c.1695T>G | p.Tyr565X | Truncating | Chinese (1)(Li et al., 2011) | F | Isolated HS | Targeted Sequencing* |
| 33 | c.1702C>T | p.Gln568X | Truncating | Caucasian (1), Japanese (1)(Nomura et al., 2014) | F | Isolated HS | Targeted Sequencing* |
| 34 | c.1752delG | p.Glu584As pfxX44 | Truncating | Chinese (1)(Wang et al., 2010) | F | Isolated HS | GWLS* |
| 35 | c.1768A>G | p.Ser590Al afsX3 | Truncating | French(Miskinyte et al., 2012) | F | Isolated HS | Targeted Sequencing* |
| 36 | c.1799delTG | p.Leu600X | Truncating | Indian (1)(Li et al., 2011) | F | HS + Acne Conglobata | Targeted Sequencing |
| 37 | c.1912_1 915delCA GT | p.S500fs p.S638fs | Frameshift | Dutch (Vossen et al., 2020) | F | Isolated HS | Whole Genome & Targeted Sequencing |
| PSENEN - 19q13.12 | |||||||
| 1 | c.168T>G | p.Tyr56X | Truncating | Ashkenazi Jewish (4)(Pavlovsky et al., 2018) | F | DDD | Targeted Sequencing* |
| 2 | c.167- 2A>G | Splice Site | Splice Site | Chinese(Zhou et al., 2016) | F | DDD | Targeted Sequencing* |
| 3 | c.194T>G | p.Leu65Arg | Missense | Chinese(Zhou et al., 2016) | F | DDD | Targeted Sequencing* |
| 4 | c.66delG | p.Phe23LeufsX46 | Truncating | Chinese(Liu Y. et al., 2016, Wang et al., 2010) | F | Isolated HS | Targeted Sequencing |
| 5 | c.66_67insG | p.Phe23Val fsX98 | Truncating | British (1) (Pink et al., 2011) | F | Isolated HS | Targeted Sequencing |
| 6 | c.279delC | p.Phe94Ser fsX51 | Truncating | Chinese(Pink et al., 2012) | F | Isolated HS | Targeted Sequencing |
| 7 | c.84_85insT | p.L28FfsX93 | Insertion | Thai (2)(Wenrui et al., 2018) | F | DDD | Targeted Sequencing* |
| 8 | c.62– 1G>C(Ralser et al., 2017) | Exon 2 | Splice Site | Indian (2)(Ralser et al., 2017) | F | DDD | Targeted Sequencing* |
| 9 | g.1412T>C | Splice Site | Splice Site | French(Ralser et al., 2017) | F | DDD | Targeted Sequencing* |
| 10 | c.35T>A | p.Leu12X | Truncating | German (2)(Ralser et al., 2017) | F | DDD | Targeted Sequencing* |
| 11 | c.115C>T | p.Arg39X | Truncating | German (1)(Ralser et al., 2017) | F | DDD | Targeted Sequencing* |
| PSEN1 – 14q 24.2 | |||||||
| 1 | c.725delC | p.Phe242L eufsX11 | Truncating | Chinese (3)(Wang et al., 2010) | F | Isolated HS | GWLS* |
| 2 | c.837+16 G>T | Splice site | Splice site | Chinese(Lazic et al., 2012) | Case | Isolated HS | Targeted Sequencing |
| 3 | c.953A>G | p.Glu318Gly | Missense | British (3)(Ingram et al., 2013) | F | Isolated HS | Targeted Sequencing* |
Figure 1,
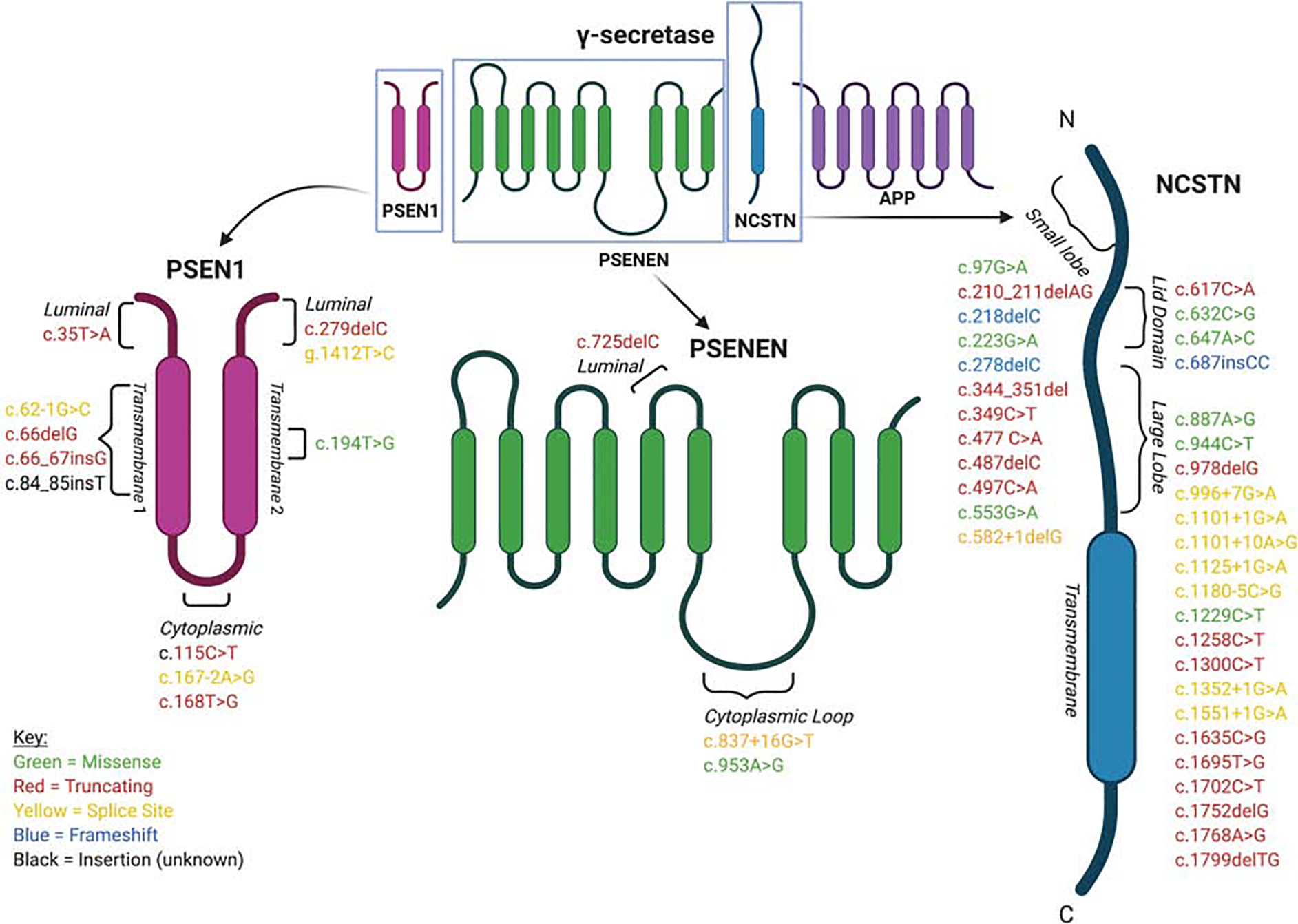
The locations of confirmed mutations in γ-secretase protein domains
Most of these γ-secretase-mutation-positive patients, are identified in families, often with multiple affected family members. Of note, classification terms such as ‘familial’, ‘typical’, ‘atypical’, ‘syndromic’. ‘sporadic’, are unreconciled and require further validation (Frew et al., 2019). The majority of these patients are found in particular demographics (e.g. male, Asian) and observed to have severe, widespread, treatment-resistant, anatomically atypical, or syndromic disease with superimposed comorbidities such as acne conglobata, pyoderma gangrenosum, and hyperpigmentation, among others (Pink et al., 2013). Comparisons against existing HS classification systems demonstrate that γ-secretase mutation positive patients, compared with sporadic HS patients, fit best with the categories of LC2 or “follicle-centered”, atypical, nodular, and scarring folliculitis using the Canoui, Naasan, Martorell-Calatayud, and van der Zee classification systems, respectively (Canoui-Poitrine et al., 2013, Ingram and Piguet, 2013, Martorell et al., 2020, Naasan and Affleck, 2015, van der Zee and Jemec, 2015, Xu et al., 2016). However, poor interrater reliability and the lack of validation limit the utility of these classification systems (van Straalen et al., 2018).
In the Alzheimer’s IDENTITY trial, semagacestat, a γ-secretase inhibitor, was administered but resulted in unspecified skin toxicity in a large portion of patients (Henley et al., 2014). More striking is that in a subsequent study of patients with desmoid tumor niragacestat, another γ-secretase inhibitor, 12/17 exhibited adverse skin toxicities. 6/7 evaluated by dermatology exhibited new-onset, recurring follicular and cystic lesions with surrounding inflammation in intertriginous areas, strongly resembling the HS phenotype (O’Sullivan Coyne et al., 2018). Biopsies of two patients showed inflamed follicular cysts, confirming pathology localized to the hair follicle. These lesions then resolved upon halting of treatment. These patients had no personal or family history of HS or its commonly cited comorbidities, suggesting that targeted γ-secretase inhibition can induce HS-like lesions, which supports the findings from genetic studies identifying loss-of-function mutations in components of the γ-secretase complex.
γ-secretase dysfunction leads to defective terminal hair follicle homeostasis
Occlusion of the follicular infundibulum, due to mechanisms including hyperkeratosis and disrupted epithelial differentiation, is considered the initiating event in HS pathogenesis (Prens and Deckers, 2015), though some believe subclinical inflammation may precede or even contribute to the occlusion (Frew et al., 2018). Several studies suggest that γ-secretase may play a key role in occlusion.
Developmentally, the absence of γ secretase in mice is known to convert hair follicles into epidermal cysts by altering the differential fate of outer root sheath cells (Pan et al., 2004). Several studies have linked impaired functionality of γ secretase to the formation of HS-like lesions in mice. Conditional knock-out of γ secretase components results in many histopathologic features of HS (He et al., 2018, Kamp et al., 2011, Pan et al., 2004).
In vitro, haploinsufficiency of NCSTN in keratinocyte cell lines upregulated the expression of type I interferon genes (Cao et al., 2019). In molecular studies of familial HS, NCSTN deficiency has been found to impact keratinocyte differentiation and proliferation through several candidate pathways (He et al., 2019, He et al., 2018, Xiao et al., 2016). Six patients with HS and DDD, a hair follicle-centered pigmentary disorder, were found to possess PSENEN mutations co-segregating with the unique phenotype and were histopathologically distinguished from PSENEN-mutation positive DDD-only patients by the presence of follicular hyperkeratosis (Ralser et al., 2017), suggesting a potential link between gene dysfunction and keratinocyte proliferation. Interestingly, a study of hair follicle keratinocytes from 18 HS patients (7 with family history, 11 without) found that they released greater pro-inflammatory cytokines IL-1β, IP-10, and CCL5 when stimulated in vitro, leading the authors to implicate an intrinsic pro-inflammatory keratinocyte phenotype in HS (Hotz et al., 2016). In addition, a systematic review encompassing immunohistochemical data from ~500 HS patients demonstrated the localization of IL-1β, IL-22, IL-36, IL-37 with keratinocytes and highlighted the intimate relationship between the pro-inflammatory milieu and dysregulated hyperkeratosis (Frew et al., 2018). The abovementioned data on NCSTN and PSENEN suggest γ-secretase dysfunction may be linked to HS-associated follicular disruption by mechanisms localized to the keratinocyte. Importantly, the γ-secretase inhibitor-induced, pathologically-confirmed folliculo-cystic lesions in healthy individuals regressed after cessation, which supports a role for γ-secretase as a potential target for treatment in at least a subset of patients.
The predominance of loss-of-function mutations implicate haploinsufficiency as a likely mechanism of γ-secretase-induced disease in familial HS (Wang et al., 2010, Yang et al., 2015). However, the presence of missense mutations in both sporadic (4) and familial (6) cases, as well as conflicting results from translational biology may implicate altered functional enzymatic activity. Loss of a single PSEN1 allele in mice does not produce skin disorders, and only occurs with more severe reduction in presenilin expression. WT mice treated with a γ-secretase inhibitor, which maintained levels of γ-secretase but specifically inhibited its enzymatic activity, produced similar epidermal abnormalities to Ncstn +/− mice, including follicular hyperkeratosis and inclusion cyst formation (Li et al., 2007). Another study of Ncstn −/− mice and Ncstn −/−; Psen1 −/− mice found that both developed follicular inclusion cysts compared with wild-type, but the double-knockout mice developed these lesions earlier, and this was dependent on the level of γ-secretase (O’Brien and Wong, 2011). In vitro study of human tissue from HS patients harboring γ-secretase mutations found that membrane expression of γ-secretase was unchanged despite reduction in cellular protein expression (Table 1) (Pink et al., 2016), which may be due to physiologic post-transcriptional selection of <5% of fully assembled complexes that are then localized to the membrane (Yang et al., 2018). It seems likely that patients with only a partial loss of function may still produce enough amounts of functional protein to support normal physiology above a certain threshold. Any reduction in the level of functional protein, γ-secretase activity or increasing the threshold may subsequently elicit a clinical phenotype (Melnik and Plewig, 2013). A recent study identified a new NCSTN mutation causing HS in a Dutch family. The associated immunobiological functions of NCSTN and its co-expressed genes Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT) and Peroxisome Proliferator Activated Receptor Delta (PPARD) link genetics to the most common environmental and metabolic HS risk factors, smoking and obesity (Vossen et al., 2020). This begs the question, how do environmental factors increase the risk of developing HS in those that harbor γ-secretase mutations?
Emerging studies have provided import data supporting this stance. A systematic review and in silico analysis of 34 HS γ-secretase-mutations predicted structural alterations in substrate recruitment sites, catalytic domains, and post-translational modifications, consistent with altered enzymatic activity and substrate processing (Li A. et al., 2018). A second, more extensive in silico analysis bolstered these results by showing that 39 pathogenic familial HS associated γ-secretase mutations underwent significant structural changes in known sites of substrate binding and cleavage, either through nonsense mediated decay (23) or altered binding affinity (16) (Frew and Navrazhina, 2019). Such changes were found to be distinct from those found in Alzheimer’s associated γ-secretase mutations HS-associated γ-secretase (Frew and Navrazhina, 2019, Li A. et al., 2018). One studied HS PSEN1 mutation was found to affect the opposite side of the transmembrane-5 domain from the affected sites of reported Alzheimer’s mutations (Frew and Navrazhina, 2019). Such mechanistic differences, as well as the myriad of γ-secretase substrates, may shed light on the lack of co-occurrence between the familial forms Alzheimer’s and HS despite overlapping loci.
γ-secretase may act through multiple secondary pathways such as Notch, PI3K, and EGFR
Isolation of γ-secretase-dependent pathways specific to HS genesis is complicated by the large number of known γ-secretase substrates, the pleiotropy of its components, and the lack of a reliable animal model for in vivo study. Thus, while the following pathways are the most well-described, many likely remain undiscovered.
The Notch pathway has gained attention in HS due to its role in maintaining the hair follicle stem cell pool, functional regulatory T cells (Treg) in the hair follicle and promoting antimicrobial defenses at the epidermis (Sabat et al., 2020). In the skin, notch normally maintains stemness in the hair follicle stem cells and disruption of signaling leads to aberrant differentiation and proliferation of keratinocytes and their precursors. Treg cells are required for development and maintenance of the hair follicle (Ali et al., 2017), as well as immunological balance in the skin, both of which notch signaling supports. Lastly, studies have shown an essential role for notch in supporting T cell derived IL-22, which maintains the skin microbiome (Sabat et al., 2020). These roles might explain why, γ-secretase mutations that influence notch signaling can elicit the diverse aberrations seen in HS skin lesions (e.g. follicular cystic formation, inflammatory immune cell infiltration, and altered skin microbiota).
Notch 1–4 are well-characterized targets of γ-secretase, and controlled disruption of Notch pathway components in mice results in epidermal and follicular aberrations that resemble histopathological findings in HS (Pink et al., 2012). While some Notch molecules are abnormally expressed in HS tissue and HaCaT cells with γ-secretase mutations (Li A. et al., 2018, Xiao et al., 2016), minimal evidence exists that indicates Notch aberrations are specific to HS or of sufficient statistical significance to be considered risk-associated loci for disease development (Frew et al., 2019). Functional assessment of four NCSTN missense mutations found that three maintained downstream Notch signaling while the fourth did not, casting doubt on the assumption that Notch-dependent pathways drive monogenic HS (Zhang and Sisodia, 2015). In silico and gene expression analyses of identified pathogenic mutations have failed to identify Notch as a specific marker of HS (Blok et al., 2016, Frew and Navrazhina, 2019), and genotype-phenotype correlation revealed no significance between impact on notch signaling and the HS phenotype (Frew et al., 2019). A recent study demonstrated that mRNA levels of NCSTN, Notch, and PI3K/AKT are overexpressed in lesional HS skin versus controls and there is no association between positive family history and mRNA levels (Hessam et al., 2020). The lack of direct evidence from animal models or human studies makes the role of Notch in HS controversial, suggesting that other pathways play a role in the molecular pathogenesis of HS.
Abnormalities in the phosphoinositide-3-kinases (PI3K) and endothelial growth-factor receptor (EGFR) pathways have previously been linked to epidermal and follicular dysfunction (Zhang et al., 2007), and emerging studies suggest that these pathways interact with microRNAs to play a role in familial HS pathogenesis. NCSTN knockdown in HaCaT cells led to decreased keratinocyte miRNA-100–5p, a microRNA that was previously found to be downregulated in familial HS patients, which then resulted in increased PI3K and keratinocyte hyperproliferation (He et al., 2019, Xiao et al., 2016). He et al. found that NCSTN mutations lead to reduced miR-30a-3p levels, which increases RAB31 expression due to diminished negative regulation, and this increase in RAB31 accelerates the degradation of activated EGFR on keratinocytes, leading to abnormal differentiation (He et al., 2018). In silico assessment of pathogenic γ-secretase mutations found that HS-associated ERBB4, SCN1B, and TIE1 were differentially expressed and that this was specific to HS when compared with other inflammatory dermatoses to account for background cutaneous inflammation (Frew and Navrazhina, 2019).
Questioning the role of γ-secretase: Future Work
Many HS experts cite the poor understanding of disease pathobiology as a significant bottleneck for HS management and a critical area for future work (Hoffman et al., 2017). Despite the myriad of discovered variants, only a minority (<5%) of HS patients have been found to harbor the monogenic γ-secretase-mutation-associated familial HS phenotype, far fewer than even the 30–40% reporting family history. A recent key study of predominantly Caucasian cohort of 188 HS patients found that just 6.4% had mutations in γ-secretase (Duchatelet et al., 2020). Overall, the majority of HS patients studied to date are found negative for γ-secretase-mutations when assessed by targeted sequencing (Frew et al., 2017, Ingram et al., 2013, Pink et al., 2012). While many pathogenic variants co-segregate with the HS phenotype in familial kindreds, others do not and indicate a benign nature (Al-Ali et al., 2010, Jarvik and Browning, 2016, Nomura et al., 2014). The sole whole-genome expression profiling study done on HS patients found no difference in whole-blood mRNA expression in NCSTN, PSEN1, or PSENEN between HS and healthy controls, though a small sample size was studied and no validation was performed (Blok et al., 2016). Most of the disease burden is in sporadic HS (60–70%), yet few studies have been performed in this population robust enough to probe its polygenic architecture and identify low to moderate impact variants and their attributable risks.
The view that HS has a polygenic foundation has subsequently gained traction, supported by strong, well-documented associations with other chronic inflammatory disorders including inflammatory bowel disease, spondyloarthropathy, lupus, and pyoderma (Deckers et al., 2017, van der Zee et al., 2016, Vekic et al., 2016). Numerous genes besides γ-secretase components have also been identified to associate with HS including connexin-26, fibroblast growth factor receptor, and inositol polyphosphate-5-phosphate (Tricarico et al., 2019), albeit with variable phenotypes. The racial predisposition toward African Americans is also important; given that disparate risks in immune-mediated disease development and variable responses to treatment of such conditions can, at least in part, be traced to ancestral heterogeneity (Nedelec et al., 2016), similar assessments in HS, particularly large-scale, hypothesis-free approaches such as GWAS, may be worthwhile.
A handful of studies have employed this approach with promising results. A pharmacogenomics GWAS study of the Pioneer I and II trials found a single variant in BCL2 that associated with response to adalimumab in HS patients in a TNF-dependent matter localized to the follicular unit (Liu et al., 2019). Sequence investigation of the IL12RB1 receptor subunit gene identified two haplotype groups associated with significant differences in age at disease presentation, stage of disease, and number of skin areas (Giatrakos et al., 2013). Similar analysis of the TNF gene found significant association between SNPs of the promoter region and susceptibility to HS, disease course, and response to TNF antagonists (Savva et al., 2013). Study of two independent cohorts (total n = 261) showcased that high copy number (>6) of the defensin (DEFB) cluster was associated with a markedly increased odds ratio (6.72 after meta-analysis, P <0.0001) for HS development and fewer than 6 copies was linked with earlier onset, fewer skin localizations, and less frequent purulence (Giamarellos-Bourboulis et al., 2016).
Nonetheless, the several identified HS mutations in NCSTN, PSENEN, and PSEN1, many of which were determined to be causative in familial HS, and their demonstrated relevance at the clinical and pathobiological levels advocate for continued investigation into γ-secretase. The establishment of guidelines for conducting the necessary multi-institutional studies, particularly genotype-phenotype analysis and exome sequencing of affected kindreds, representative of the broader HS population has already been undertaken and are steps in the right direction (Byrd et al., 2019). Conducting larger, prospective studies of familial HS patients that include clinical data collection for rigorous phenotyping will provide more data to establish a reliable, unbiased classification system. HS remains a clinical diagnosis with only anecdotal evidence for the use of biomarkers, histopathologic findings, and objective diagnostics. Yet when approached clinically, the lack of awareness, embarrassment in discussion, low socioeconomic status among patients, lack of follow-up due to increased use of emergency and inpatient care, and dearth of HS specialists in the US all serve as barriers to obtaining accurate clinical information from HS patients (Hoffman et al., 2017). At the experimental level, establishing relevant animal disease models, designing translational studies aimed at distinguishing among the many contributing mechanisms to HS, and performing functional validation of identified variants are key tasks in this process.
In conclusion, here we review the available literature on γ-secretase in HS and evaluate its evidence in the context of clinical, epidemiologic, pathobiological, and molecular studies. The release of ENCODE 3 and its associated tools poise future studies in HS to uncover important genetic and epigenetic features that may further clarify the etiologies of HS (Moore et al., 2020). Studying the γ-secretase complex as well as the greater genetic architecture of HS will allow for markedly improved and individualized treatment for individuals with this debilitating disease.
Acknowledgements:
We thank all laboratory members for their help and encouragement. This study was supported by Henry Ford Immunology Program grants (T71016 and T71017) to QSM and LZ, by the National Institutes of Health (NIH) grants R61AR076803, R01AR063611, and R01AR069681 to QSM, RO1AR072046 to LZ.
Disclosure of Potential Conflict of Interest:
IH: Grants/Research Funding: Pfizer Inc., Bayer, Incyte; Consultant Fees: Incyte, Pfizer, UCB, Boehringer Ingelheim, Clarify Medical, Jansen; Sub-Investigator Fee: Chemocentyx; Principal Investigator: Lenicura; Advisory Board: AbbVie; President: HS Foundation
The remaining authors declare that they have no relevant conflicts of interest.
Footnotes
Data availability statement
There is no Dataset related to this article
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Al-Ali FM, Ratnamala U, Mehta TY, Naveed M, Al-Ali MT, Al-Khaja N, et al. Hidradenitis suppurativa (or Acne inversa) with autosomal dominant inheritance is not linked to chromosome 1p21.1–1q25.3 region. Experimental dermatology 2010;19(9):851–3. [DOI] [PubMed] [Google Scholar]
- Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K, et al. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell 2017;169(6):1119–29.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blok JL, Li K, Brodmerkel C, Jonkman MF, Horvath B. Gene expression profiling of skin and blood in hidradenitis suppurativa. The British journal of dermatology 2016;174(6):1392–4. [DOI] [PubMed] [Google Scholar]
- Byrd AS, Dina Y, Okoh UJ, Quartey QQ, Carmona-Rivera C, Williams DW, et al. Specimen Collection for Translational Studies in Hidradenitis Suppurativa. Scientific reports 2019;9(1):12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, Viallette C, Gabison G, Poli F, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. The Journal of investigative dermatology 2013;133(6):1506–11. [DOI] [PubMed] [Google Scholar]
- Cao L, Morales-Heil DJ, Roberson EDO. Nicastrin haploinsufficiency alters expression of type I interferon-stimulated genes: the relationship to familial hidradenitis suppurativa. Clin Exp Dermatol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Mattei P, You J, Sobreira NL, Hinds GA. gamma-Secretase Mutation in an African American Family With Hidradenitis Suppurativa. JAMA dermatology 2015;151(6):668–70. [DOI] [PubMed] [Google Scholar]
- Deckers IE, Benhadou F, Koldijk MJ, Del Marmol V, Horvath B, Boer J, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: Results from a multicenter cross-sectional study. Journal of the American Academy of Dermatology 2017;76(1):49–53. [DOI] [PubMed] [Google Scholar]
- Duchatelet S, Miskinyte S, Delage M, Ungeheuer MN, Lam T, Benhadou F, et al. Low Prevalence of GSC Gene Mutations in a Large Cohort of Predominantly Caucasian Patients with Hidradenitis Suppurativa. The Journal of investigative dermatology 2020;140(10):2085–8.e14. [DOI] [PubMed] [Google Scholar]
- Duchatelet S, Miskinyte S, Join-Lambert O, Ungeheuer MN, Frances C, Nassif A, et al. First nicastrin mutation in PASH (pyoderma gangrenosum, acne and suppurative hidradenitis) syndrome. The British journal of dermatology 2015;173(2):610–2. [DOI] [PubMed] [Google Scholar]
- Faraji Zonooz M, Sabbagh-Kermani F, Fattahi Z, Fadaee M, Akbari MR, Amiri R, et al. Whole Genome Linkage Analysis Followed by Whole Exome Sequencing Identifies Nicastrin (NCSTN) as a Causative Gene in a Multiplex Family with gamma-Secretase Spectrum of Autoinflammatory Skin Phenotypes. The Journal of investigative dermatology 2016;136(6):1283–6. [DOI] [PubMed] [Google Scholar]
- Frew JW, Hawkes JE, Krueger JG. A systematic review and critical evaluation of immunohistochemical associations in hidradenitis suppurativa. F1000Research 2018;7:1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew JW, Hawkes JE, Sullivan-Whalen M, Gilleaudeau P, Krueger JG. Inter-Relater Reliability of Phenotypes, and Exploratory Genotype- Phenotype Analysis in Inherited Hidradenitis Suppurativa. The British journal of dermatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew JW, Navrazhina K. In silico Analysis of Gamma-Secretase-Complex Mutations in Hidradenitis Suppurativa Demonstrates Disease-Specific Substrate Recognition and Cleavage Alterations. Frontiers in medicine 2019;6:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew JW, Vekic DA, Woods J, Cains GD. A systematic review and critical evaluation of reported pathogenic sequence variants in hidradenitis suppurativa. Br J Dermatol 2017;177(4):987–98. [DOI] [PubMed] [Google Scholar]
- Gao M, Wang PG, Cui Y, Yang S, Zhang YH, Lin D, et al. Inversa acne (hidradenitis suppurativa): a case report and identification of the locus at chromosome 1p21.1–1q25.3. The Journal of investigative dermatology 2006;126(6):1302–6. [DOI] [PubMed] [Google Scholar]
- Garg A, Kirby JS, Lavian J, Lin G, Strunk A. Sex- and Age-Adjusted Population Analysis of Prevalence Estimates for Hidradenitis Suppurativa in the United States. JAMA dermatology 2017a;153(8):760–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Lavian J, Lin G, Strunk A, Alloo A. Incidence of hidradenitis suppurativa in the United States: A sex- and age-adjusted population analysis. Journal of the American Academy of Dermatology 2017b;77(1):118–22. [DOI] [PubMed] [Google Scholar]
- Garg A, Strunk A. Risk of Alzheimer’s disease is not increased among patients with hidradenitis suppurativa: A retrospective population-based cohort analysis. J Am Acad Dermatol 2017;77(1):176–7. [DOI] [PubMed] [Google Scholar]
- Giamarellos-Bourboulis EJ, Platzer M, Karagiannidis I, Kanni T, Nikolakis G, Ulrich J, et al. High Copy Numbers of beta-Defensin Cluster on 8p23.1, Confer Genetic Susceptibility, and Modulate the Physical Course of Hidradenitis Suppurativa/Acne Inversa. The Journal of investigative dermatology 2016;136(8):1592–8. [DOI] [PubMed] [Google Scholar]
- Giatrakos S, Huse K, Kanni T, Tzanetakou V, Kramer M, Grech I, et al. Haplotypes of IL-12Rbeta1 impact on the clinical phenotype of hidradenitis suppurativa. Cytokine 2013;62(2):297–301. [DOI] [PubMed] [Google Scholar]
- Gudjonsson JE, Tsoi LC, Ma F, Billi AC, van Straalen KR, Vossen ARJV, et al. Contribution of plasma cells and B cells to hidradenitis suppurativa pathogenesis. JCI Insight 2020;5(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines R, Common J, Teo D, Tang M, Lane E. sequencing of the γ-secretase complex in Singaporean patients with acne inversa reveals a novel mutation in nicastrin, but suggests other mechanisms must be present: p-15. British Journal of Dermatology 2012;166(4):e33. [Google Scholar]
- He Y, Li C, Xu H, Duan Z, Liu Y, Zeng R, et al. AKT-dependent hyperproliferation of keratinocytes in familial hidradenitis suppurativa with NCSTN mutation: A potential role of defective miR-100–5p. The British journal of dermatology 2019. [DOI] [PubMed] [Google Scholar]
- He Y, Xu H, Li C, Zhang X, Zhou P, Xiao X, et al. Nicastrin/miR-30a-3p/RAB31 Axis Regulates Keratinocyte Differentiation by Impairing EGFR Signaling in Familial Acne Inversa. The Journal of investigative dermatology 2018. [DOI] [PubMed] [Google Scholar]
- Henley DB, Sundell KL, Sethuraman G, Dowsett SA, May PC. Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Current medical research and opinion 2014;30(10):2021–32. [DOI] [PubMed] [Google Scholar]
- Hessam S, Gambichler T, Skrygan M, Scholl L, Sand M, Meyer T, et al. Increased expression profile of NCSTN, Notch and PI3K/AKT3 in hidradenitis suppurativa. Journal of the European Academy of Dermatology and Venereology : JEADV 2020. [DOI] [PubMed] [Google Scholar]
- Hoffman LK, Ghias MH, Garg A, Hamzavi IH, Alavi A, Lowes MA. Major gaps in understanding and treatment of hidradenitis suppurativa. Seminars in cutaneous medicine and surgery 2017;36(2):86–92. [DOI] [PubMed] [Google Scholar]
- Hotz C, Boniotto M, Guguin A, Surenaud M, Jean-Louis F, Tisserand P, et al. Intrinsic Defect in Keratinocyte Function Leads to Inflammation in Hidradenitis Suppurativa. The Journal of investigative dermatology 2016;136(9):1768–80. [DOI] [PubMed] [Google Scholar]
- Ingram JR. The Genetics of Hidradenitis Suppurativa. Dermatol Clin 2016;34(1):23–8. [DOI] [PubMed] [Google Scholar]
- Ingram JR, Piguet V. Phenotypic heterogeneity in hidradenitis suppurativa (acne inversa): classification is an essential step toward personalized therapy. The Journal of investigative dermatology 2013;133(6):1453–6. [DOI] [PubMed] [Google Scholar]
- Ingram JR, Wood M, John B, Butler R, Anstey AV. Absence of pathogenic gamma-secretase mutations in a South Wales cohort of familial and sporadic hidradenitis suppurativa (acne inversa). The British journal of dermatology 2013;168(4):874–6. [DOI] [PubMed] [Google Scholar]
- Irwin McLean W, Wood P, Irvine AD, Von Der Werth J. Mapping of two genetic loci for autosomal dominant hidradenitis suppurativa. Experimental dermatology 2006;15(6):479-. [Google Scholar]
- Jarvik GP, Browning BL. Consideration of Cosegregation in the Pathogenicity Classification of Genomic Variants. American journal of human genetics 2016;98(6):1077–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jfri AH, O’Brien EA, Litvinov IV, Alavi A, Netchiporouk E. Hidradenitis Suppurativa: Comprehensive Review of Predisposing Genetic Mutations and Changes. Journal of cutaneous medicine and surgery 2019:1203475419852049. [DOI] [PubMed] [Google Scholar]
- Jiao T, Dong H, Jin L, Wang S, Wang J. A novel nicastrin mutation in a large Chinese family with hidradenitis suppurativa. The British journal of dermatology 2013;168(5):1141–3. [DOI] [PubMed] [Google Scholar]
- Kamp S, Fiehn AM, Stenderup K, Rosada C, Pakkenberg B, Kemp K, et al. Hidradenitis suppurativa: a disease of the absent sebaceous gland? Sebaceous gland number and volume are significantly reduced in uninvolved hair follicles from patients with hidradenitis suppurativa. The British journal of dermatology 2011;164(5):1017–22. [DOI] [PubMed] [Google Scholar]
- Lazic T, Li Q, Frank M, Uitto J, Zhou LH. Extending the phenotypic spectrum of keratitis-ichthyosis-deafness syndrome: report of a patient with GJB2 (G12R) Connexin 26 mutation and unusual clinical findings. Pediatric dermatology 2012;29(3):349–57. [DOI] [PubMed] [Google Scholar]
- Li A, Peng Y, Taiclet LM, Tanzi RE. Analysis of Hidradenitis Suppurativa-Linked Mutations in Four genes and the Effects of PSEN1-P242LfsX11 on Cytokine and Chemokine Expression in Macrophages. Human molecular genetics 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xu H, Wang B. Is SAPHO Syndrome Linked to PASH Syndrome and Hidradenitis Suppurativa by Nicastrin Mutation? A Case Report. The Journal of rheumatology 2018;45(11):1605–7. [DOI] [PubMed] [Google Scholar]
- Li CR, Jiang MJ, Shen DB, Xu HX, Wang HS, Yao X, et al. Two novel mutations of the nicastrin gene in Chinese patients with acne inversa. The British journal of dermatology 2011;165(2):415–8. [DOI] [PubMed] [Google Scholar]
- Li T, Wen H, Brayton C, Das P, Smithson LA, Fauq A, et al. Epidermal growth factor receptor and notch pathways participate in the tumor suppressor function of gamma-secretase. The Journal of biological chemistry 2007;282(44):32264–73. [DOI] [PubMed] [Google Scholar]
- Liu M, Davis JW, Idler KB, Mostafa NM, Okun MM, Waring JF. Genetic analysis of NCSTN for potential association with hidradenitis suppurativa in familial and nonfamilial patients. The British journal of dermatology 2016;175(2):414–6. [DOI] [PubMed] [Google Scholar]
- Liu M, Degner J, Georgantas RW, Nader A, Mostafa NM, Teixeira HD, et al. A GENETIC VARIANT IN THE BCL2 GENE ASSOCIATES WITH ADALIMUMAB RESPONSE IN HIDRADENITIS SUPPURATIVA CLINICAL TRIALS AND REGULATES EXPRESSION OF BCL2. The Journal of investigative dermatology 2019. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gao M, Lv YM, Yang X, Ren YQ, Jiang T, et al. Confirmation by exome sequencing of the pathogenic role of NCSTN mutations in acne inversa (hidradenitis suppurativa). The Journal of investigative dermatology 2011;131(7):1570–2. [DOI] [PubMed] [Google Scholar]
- Liu Y, Miao T, Ma J, Shao L, Luo S, Li Y, et al. PSENEN c.66delG in sporadic acne inversa. European journal of dermatology : EJD 2016;26(3):298–9. [DOI] [PubMed] [Google Scholar]
- Lowe MM, Naik HB, Clancy S, Pauli M, Smith KM, Bi Y, et al. Immunopathogenesis of hidradenitis suppurativa and response to anti-TNF-α therapy. JCI Insight 2020;5(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Yu Y, Yu G, Zhang F. Identification of one novel mutation of the NCSTN gene in one Chinese acne inversa family. Dermatologica Sinica 2014;32(2):126–8. [Google Scholar]
- Martorell A, Jfri A, Koster SBL, Gomez-Palencia P, Solera M, Alfaro-Rubio A, et al. Defining hidradenitis suppurativa phenotypes based on the elementary lesion pattern: results of a prospective study. Journal of the European Academy of Dermatology and Venereology : JEADV 2020;34(6):1309–18. [DOI] [PubMed] [Google Scholar]
- Melnik BC, Plewig G. Impaired Notch-MKP-1 signalling in hidradenitis suppurativa: an approach to pathogenesis by evidence from translational biology. Experimental dermatology 2013;22(3):172–7. [DOI] [PubMed] [Google Scholar]
- Merilahti JAM, Ojala VK, Knittle AM, Pulliainen AT, Elenius K. Genome-wide screen of gamma-secretase-mediated intramembrane cleavage of receptor tyrosine kinases. Molecular biology of the cell 2017;28(22):3123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskinyte S, Nassif A, Merabtene F, Ungeheuer MN, Join-Lambert O, Jais JP, et al. Nicastrin mutations in French families with hidradenitis suppurativa. The Journal of investigative dermatology 2012;132(6):1728–30. [DOI] [PubMed] [Google Scholar]
- Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020;583(7818):699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naasan H, Affleck A. Atypical hidradenitis suppurativa. Clin Exp Dermatol 2015;40(8):891–3. [DOI] [PubMed] [Google Scholar]
- Nedelec Y, Sanz J, Baharian G, Szpiech ZA, Pacis A, Dumaine A, et al. Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell 2016;167(3):657–69.e21. [DOI] [PubMed] [Google Scholar]
- Nishimori N, Hayama K, Kimura K, Fujita H, Tadashi T. 054 A novel γ-secretase gene mutation in a Japanese family with hidradenitis. Journal of Investigative Dermatology 2017;137(10):S201. [Google Scholar]
- Nomura Y, Nomura T, Sakai K, Sasaki K, Ohguchi Y, Mizuno O, et al. A novel splice site mutation in NCSTN underlies a Japanese family with hidradenitis suppurativa. The British journal of dermatology 2013;168(1):206–9. [DOI] [PubMed] [Google Scholar]
- Nomura Y, Nomura T, Suzuki S, Takeda M, Mizuno O, Ohguchi Y, et al. A novel NCSTN mutation alone may be insufficient for the development of familial hidradenitis suppurativa. Journal of dermatological science 2014;74(2):180–2. [DOI] [PubMed] [Google Scholar]
- O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annual review of neuroscience 2011;34:185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan Coyne G, Woodring TS, Lee CR, Chen AP, Kong HH. Hidradenitis Suppurativa-Like Lesions Associated with Pharmacologic Inhibition of Gamma-Secretase. The Journal of investigative dermatology 2018;138(4):979–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Lin MH, Tian X, Cheng HT, Gridley T, Shen J, et al. gamma-secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Developmental cell 2004;7(5):731–43. [DOI] [PubMed] [Google Scholar]
- Pavlovsky M, Sarig O, Eskin-Schwartz M, Malchin N, Bochner R, Mohamad J, et al. A phenotype combining hidradenitis suppurativa with Dowling–Degos disease caused by a founder mutation in PSENEN. British Journal of Dermatology 2018;178(2):502–8. [DOI] [PubMed] [Google Scholar]
- Pink AE, Dafou D, Desai N, Holmes O, Hobbs C, Smith CH, et al. Hidradenitis suppurativa: haploinsufficiency of gamma-secretase components does not affect gamma-secretase enzyme activity in vitro. The British journal of dermatology 2016;175(3):632–5. [DOI] [PubMed] [Google Scholar]
- Pink AE, Simpson MA, Brice GW, Smith CH, Desai N, Mortimer PS, et al. PSENEN and NCSTN mutations in familial hidradenitis suppurativa (Acne Inversa). The Journal of investigative dermatology 2011;131(7):1568–70. [DOI] [PubMed] [Google Scholar]
- Pink AE, Simpson MA, Desai N, Dafou D, Hills A, Mortimer P, et al. Mutations in the gamma-secretase genes NCSTN, PSENEN, and PSEN1 underlie rare forms of hidradenitis suppurativa (acne inversa). The Journal of investigative dermatology 2012;132(10):2459–61. [DOI] [PubMed] [Google Scholar]
- Pink AE, Simpson MA, Desai N, Trembath RC, Barker JNW. gamma-Secretase mutations in hidradenitis suppurativa: new insights into disease pathogenesis. The Journal of investigative dermatology 2013;133(3):601–7. [DOI] [PubMed] [Google Scholar]
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: An update. J Am Acad Dermatol 2015;73(5 Suppl 1):S8–11. [DOI] [PubMed] [Google Scholar]
- Ralser DJ, Basmanav FB, Tafazzoli A, Wititsuwannakul J, Delker S, Danda S, et al. Mutations in gamma-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. The Journal of clinical investigation 2017;127(4):1485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnamala U, Jhala D, Jain NK, Saiyed NM, Raveendrababu M, Rao MV, et al. Expanding the spectrum of gamma-secretase gene mutation-associated phenotypes: two novel mutations segregating with familial hidradenitis suppurativa (acne inversa) and acne conglobata. Experimental dermatology 2016;25(4):314–6. [DOI] [PubMed] [Google Scholar]
- Reddy S, Strunk A, Garg A. Comparative Overall Comorbidity Burden Among Patients With Hidradenitis Suppurativa. JAMA dermatology 2019;155(7):797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabat R, Jemec GBE, Matusiak Ł, Kimball AB, Prens E, Wolk K. Hidradenitis suppurativa. Nat Rev Dis Primers 2020;6(1):18. [DOI] [PubMed] [Google Scholar]
- Savva A, Kanni T, Damoraki G, Kotsaki A, Giatrakou S, Grech I, et al. Impact of Toll-like receptor-4 and tumour necrosis factor gene polymorphisms in patients with hidradenitis suppurativa. The British journal of dermatology 2013;168(2):311–7. [DOI] [PubMed] [Google Scholar]
- Scheinfeld N Diseases associated with hidranitis suppurativa: part 2 of a series on hidradenitis. Dermatology online journal 2013;19(6):18558. [PubMed] [Google Scholar]
- Shi TW, Bai N, Zhang JA, Lu F, Chen XB, Kong XD, et al. Mutations in the gamma-secretase genes PSEN1, PSENEN, and NCSTN in a family with acne inversa. European journal of dermatology : EJD 2018;28(3):374–6. [DOI] [PubMed] [Google Scholar]
- Takeichi T, Matsumoto T, Nomura T, Takeda M, Niwa H, Kono M, et al. A novel NCSTN missense mutation in the signal peptide domain causes hidradenitis suppurativa, which has features characteristic of an autoinflammatory keratinization disease. The British journal of dermatology 2019. [DOI] [PubMed] [Google Scholar]
- Takeichi T, Matsumoto T, Nomura T, Takeda M, Niwa H, Kono M, et al. A novel NCSTN missense mutation in the signal peptide domain causes hidradenitis suppurativa, which has features characteristic of an autoinflammatory keratinization disease. The British journal of dermatology 2020;182(2):491–3. [DOI] [PubMed] [Google Scholar]
- Theut Riis P, Egeberg A, Gislason GH, Jemec GB. Patients with hidradenitis suppurativa have no increased risk of Alzheimer disease. The British journal of dermatology 2017;177(1):273–5. [DOI] [PubMed] [Google Scholar]
- Tricarico PM, Boniotto M, Genovese G, Zouboulis CC, Marzano AV, Crovella S. An Integrated Approach to Unravel Hidradenitis Suppurativa Etiopathogenesis. Front Immunol 2019;10:892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zee HH, Horvath B, Jemec GBE, Prens EP. The Association between Hidradenitis Suppurativa and Crohn’s Disease: in Search of the Missing Pathogenic Link. The Journal of investigative dermatology 2016;136(9):1747–8. [DOI] [PubMed] [Google Scholar]
- van der Zee HH, Jemec GB. New insights into the diagnosis of hidradenitis suppurativa: Clinical presentations and phenotypes. J Am Acad Dermatol 2015;73(5 Suppl 1):S23–6. [DOI] [PubMed] [Google Scholar]
- van Straalen K, Verhagen T, Horvath B, Ardon C, Vossen A, Driessen R, et al. Poor inter-rater reliability of hidradenitis suppurativa phenotypes by Canoui-Poitrine et al. Experimental dermatology: WILEY 111 RIVER ST, HOBOKEN 07030–5774, NJ USA; 2018. p. 31-. [DOI] [PubMed] [Google Scholar]
- van Straalen KR, Prens EP, Willemsen G, Boomsma DI, van der Zee HH. Contribution of Genetics to the Susceptibility to Hidradenitis Suppurativa in a Large, Cross-sectional Dutch Twin Cohort. JAMA dermatology 2020;156(12):1359–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekic DA, Frew JW, Woods J, Cains GD. Adopting the orphan: The importance of recognising hidradenitis suppurativa as a systemic auto-inflammatory disease. The Australasian journal of dermatology 2016;57(1):69–70. [DOI] [PubMed] [Google Scholar]
- Vossen A, van Straalen KR, Swagemakers SMA, de Klein J, Stubbs AP, Venter DJ, et al. A novel nicastrin mutation in a three-generation Dutch family with hidradenitis suppurativa: a search for functional significance. J Eur Acad Dermatol Venereol 2020;34(10):2353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Yang W, Wen W, Sun J, Su B, Liu B, et al. Gamma-secretase gene mutations in familial acne inversa. Science 2010;330(6007):1065. [DOI] [PubMed] [Google Scholar]
- Wenrui L, Haoxiang X, Yanyan H, Lin L, Chengrang L. Comorbidities or Different Entities? Phenotype Variability Associated with PSENEN Mutations. British Journal of Dermatology 2018. [DOI] [PubMed] [Google Scholar]
- Wu C, Yang J, Zhang S, Li J, Jin H, Zhang X. A novel NCSTN gene mutation in a Chinese family with acne inversa. Molecular genetics and genomics : MGG 2018. [DOI] [PubMed] [Google Scholar]
- Xiao X, He Y, Li C, Zhang X, Xu H, Wang B. Nicastrin mutations in familial acne inversa impact keratinocyte proliferation and differentiation through the Notch and phosphoinositide 3-kinase/AKT signalling pathways. Br J Dermatol 2016;174(3):522–32. [DOI] [PubMed] [Google Scholar]
- Xu H, Xiao X, Hui Y, Zhang X, He Y, Li C, et al. Phenotype of 53 Chinese individuals with nicastrin gene mutations in association with familial hidradenitis suppurativa (acne inversa). The British journal of dermatology 2016;174(4):927–9. [DOI] [PubMed] [Google Scholar]
- Yang G, Zhou R, Zhou Q, Guo X, Yan C, Ke M, et al. Structural basis of Notch recognition by human gamma-secretase. Nature 2018. [DOI] [PubMed] [Google Scholar]
- Yang JQ, Wu XJ, Dou TT, Jiao T, Chen XB, Min M, et al. Haploinsufficiency caused by a nonsense mutation in NCSTN underlying hidradenitis suppurativa in a Chinese family. Clin Exp Dermatol 2015;40(8):916–9. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wang L, Chen L, Ren W, Mei A, Chen X, et al. Two novel mutations of the NCSTN gene in Chinese familial acne inverse. Journal of the European Academy of Dermatology and Venereology : JEADV 2013;27(12):1571–4. [DOI] [PubMed] [Google Scholar]
- Zhang S, Meng J, Jiang M, Zhao J. Characterization of a Novel Mutation in the NCSTN Gene in a Large Chinese Family with Acne Inversa. Acta dermato-venereologica 2016;96(3):408–9. [DOI] [PubMed] [Google Scholar]
- Zhang X, Sisodia SS. Acne inversa caused by missense mutations in NCSTN is not fully compatible with impairments in Notch signaling. The Journal of investigative dermatology 2015;135(2):618–20. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Wang R, Liu Q, Zhang H, Liao FF, Xu H. Presenilin/gamma-secretase-dependent processing of beta-amyloid precursor protein regulates EGF receptor expression. Proceedings of the National Academy of Sciences of the United States of America 2007;104(25):10613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Wen GD, Soe LM, Xu HJ, Du J, Zhang JZ. Novel Mutations in PSENEN Gene in Two Chinese Acne Inversa Families Manifested as Familial Multiple Comedones and Dowling-Degos Disease. Chinese medical journal 2016;129(23):2834–9. [DOI] [PMC free article] [PubMed] [Google Scholar]