

Abstract
Background:
Preferentially expressed antigen in melanoma (PRAME) is a cancer-testis antigen overexpressed in many human malignancies while poorly expressed or absent in healthy tissues, making it a good target for anticancer immunotherapy. Development of an effective off-the-shelf adoptive T-cell therapy for patients with relapsed or refractory solid tumors and hematological malignancies expressing PRAME antigen requires the identification of MHC class I and II PRAME antigens recognized by the TAA-T cell product. We therefore set out to extend the repertoire of HLA-restricted PRAME peptide epitopes beyond the few already characterized.
Methods:
Peptide libraries of 125 overlapping 15-mer peptides spanning the entire PRAME protein sequence were used to identify HLA class I and class II-restricted epitopes. We also determined the HLA-restriction of the identified epitopes.
Results:
PRAME-specific T-cell products were successfully generated from PBMCs of 12 healthy donors. Ex-vivo expanded T-cells were polyclonal, consisting of both CD4+ and CD8+ T-cells which elicited anti-tumor activity in vitro. Nine MHC class I-restricted PRAME epitopes were identified (seven novel and two previously described). We also characterized sixteen 15-mer peptide sequences confirmed as CD4-restricted epitopes.
Conclusion:
TAA T-cells derived from healthy donors recognize a broad range of CD4+ and CD8+ HLA-restricted PRAME epitopes, which could be used to select suitable donors for generating off-the-shelf TAA-specific T-cells.
Keywords: PRAME, T-cell epitope, off-the-shelf immunotherapy
INTRODUCTION
Preferentially expressed antigen of melanoma (PRAME) is a cancer-testis antigen overexpressed in a variety of human malignancies, including lymphoid and myeloid malignancies and solid tumors, while being poorly expressed in healthy adult tissues except for testis, endometrium and at very low levels in ovaries and adrenal glands [1–9]. PRAME is a dominant repressor of retinoic acid receptor (RAR) signaling and plays a role in cancer cell proliferation and survival [10, 11]. PRAME has been reported to induce cytotoxic T cell- mediated immune responses, suggesting that PRAME may be an effective target for anticancer immunotherapy [3, 12–15].
Studies have shown that autologous multi tumor-associated antigen (TAA)-specific T-cell infusions can safely induce disease stabilization or maintain durable remissions in patients with relapsed or refractory solid tumors and leukemias/lymphomas [16–19]. However, there are many challenges for manufacturing autologous T-cell products including the time and cost to generate products, the inability to generate a product from lymphopenic patients, and the heterogeneity of such T-cell products [20, 21]. Off-the-shelf third-party T-cell products derived from healthy donors represent an alternative T-cell source for TAA-T cell products. Third-party virus-specific T-cells partially matched through HLA class I and II with the recipient have proven to be safe and effective in the prophylaxis and treatment of viral infections after hematopoietic stem cell transplant [22–24]. Partially matched T-cells from an off-the-shelf cell bank may, therefore, be an efficacious source of TAA-T cells that could increase the number of cancer patients benefiting from adoptive T-cell therapy.
Development of an effective off-the-shelf adoptive T-cell therapy for patients with relapsed or refractory malignancies expressing PRAME antigen depends on the identification of PRAME antigens recognized by the TAA-T cell product. PRAME peptide epitopes have not been studied extensively except for human histocompatibility leukocyte antigen (HLA)-A*02 restricted cytotoxic T lymphocyte epitopes [25–27]. Our goal was to broaden the repertoire of known HLA- restricted PRAME peptides and their HLA-restricted alleles to facilitate the use of PRAME specific T-cells for the majority of patients in an off-the-shelf setting. Here, we show that PRAME-specific T-cells recognize a broad number of CD4+ and CD8+ restricted epitopes and show that these allogeneic off-the-shelf PRAME-specific T-cells (TAA-T) kill PRAME+ tumor cells in vitro unlike T-cells that showed no specificity to PRAME antigen.
MATERIALS AND METHODS
Samples
Healthy donor buffy coats were obtained from the Gulf Coast Regional Blood Center, Houston, TX. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Lymphoprep (STEMCELL Technologies, Cambridge, MA) and frozen down in cryopreservation medium [50% RPMI 1640 medium, 10% dimethyl sulfoxide (DMSO) and 40% fetal bovine serum (FBS)]. HLA typing of the healthy donor PBMCs was performed by The Sequencing Center, Fort Collins, CO.
Generation of PRAME-specific T-cell products
PRAME-specific T-cell products were generated from total PBMCs by previously established protocol [28]. Briefly, mature dendritic cells (DC), generated by adherence, were used as antigen-presenting cells. Non-adherent cells were stimulated with PRAME peptide-pulsed (200 ng/200 uL/5 x 106 cells), irradiated (25 Gy) DC at an effector-to-target ratio of 10:1 and cultured in complete media consisting of 50% Click’s medium (Irvine Scientific, Santa Ana, CA), 40% RPMI (GE Healthcare, Logan, UT), 10% human AB serum (Gemini BioProducts, West Sacramento, CA), and supplemented with 2 mM GlutaMax (Gibco, Grand Island, NY). Second and third restimulations of T-cells were carried out weekly with PRAME peptide-pulsed, irradiated DC at an effector-to-target ratio of 20:1. On day 28, cells were harvested and evaluated for antigen specificity and functionality.
Anti-IFN-γ enzyme-linked immunospot (ELISpot) assay
PRAME specificity of each T-cell product was evaluated by stimulating expanded cells with PRAME PepMix (JPT Peptide Technology, Berlin, Germany) and measuring IFN-γ production by ELISpot assay. T-cells were plated at 1x105 cells/well with no peptide, PRAME PepMix (200 ng/well), actin PepMix (the irrelevant antigen used as a negative control), and Staphylococcal enterotoxin B (SEB), a superantigen used as a positive control. Spot-forming cells (SFCs) were enumerated by Zellnet Consulting (Fort Lee, NJ).
Cytotoxicity Assay
Cytolytic activity of the tumor antigen (PRAME)-specific T-cells versus the Wilms tumor cell lines Wit49 (donated by Kentsis Research Lab, Sloan Kettering Institute, New York, NY) and 17.94 (DSMZ, Braunschweig, Germany) was tested in a calcein AM cytotoxicity assay. Target cells were resuspended in complete medium at a final concentration of 106/mL and incubated with 10 µM calcein-AM (ThermoFisher, Waltham, MA) for 30 minutes at 37°C in 5% CO2. After 2 washes in complete medium, cells were resuspended at 105 cells/mL. Partially HLA-matched healthy donor derived PRAME-specific T-cells versus T-cells with irrelevant specificity (i.e.non-specific T-cells (NSTs)), were co-cultured with tumor cells at effector to target (E:T) ratios of 40:1, 20:1 and 10:1 and 5:1 in triplicate in 96-well round bottom plate (Corning, NY). PRAME-specific T-cell and NST [phytohaemmaglutinin (PHA) blasts or PBMCs] effector cells were plated at appropriate ratios and incubated at 37°C in 5% CO2 for 4 hours. After incubation, 75 µL of each supernatant was harvested and transferred into new plates. Samples were measured using a microplate spectrofluorometer (excitation filter: 485 + 9 nm; emission filter: 515 + 9 nm) with data expressed as arbitrary fluorescent units (AFU). Percent lysis was calculated according to the formula [(test release – spontaneous release)/(maximum release – spontaneous release)] x 100. Spontaneous release represents calcein release from target cells in medium alone, and maximum release represents calcein release from target cells lysed in medium plus 2% Triton X-100, plated in triplicate at least. Background from media was subtracted from all the values.
Immunophenotyping
PRAME-specific T-cell products were phenotyped by extracellular antibody staining with anti- CD3, CD4, CD8, CD14, CD16, CD 19, CD56, CD45RO, CD62L, CCR7 (Miltenyi Biotec, Bergisch Gladbach, Germany; BioLegend, San Diego, CA) and acquired on a CytoFLEX cytometer (Beckman Coulter, Brea, CA). The data was analyzed using FlowJo X software (FlowJo LLC, Ashland, OR).
PRAME Peptides
The PRAME peptide library consisting of 125 15-mer peptides overlapping by 11 amino acids and spanning the entire sequence of PRAME protein was designed to identify HLA class I and HLA class II-restricted epitopes (A&A Labs, San Diego, CA). 23 peptide pools comprising 10–12 peptides were prepared so that each 15-mer peptide was included in only two pools (Supplementary Figure S1). To determine minimal PRAME epitopes, additional 9-mer peptides overlapping by 8 amino acids spanning immunogenic 15-mer peptides were obtained from A&A Labs, San Diego, CA. All peptides were reconstituted at 10 µg/µl in DMSO and stored at −80°C until further use.
Epitope mapping
T-cell response to pools of peptide libraries and individual PRAME peptides were determined by IFN-γ ELISpot assay. ELISpot assay was performed as previously described. 1×105 T-cells/well were plated alone, with actin (negative control), SEB (positive control), each peptide pool (1µg/well) and individual peptide (10µg/well). IFN-γ spot-forming cells (SFC) were enumerated by Zellnet Consulting (Fort Lee, NJ). Responses that were at least 10 SFC/1x105 T-cells or greater than two-fold the background level of nonstimulated T-cells or T-cells stimulated with actin were considered positive responses.
HLA restriction of individual peptides that showed specificity by ELISpot was determined by intracellular cytokine staining, measuring IFN-γ and TNF-α release. T-cells were stimulated for 6h with PRAME PepMix or individual PRAME peptides (200 ng/peptide/well) in the presence of anti-CD28 and CD49d antibodies (BD Biosciences, San Jose, CA, USA) and Brefeldin A (Golgiplug, BD Biosciences). Controls (unstimulated T-cells, actin, SEB) were included in each experiment. Intracellular IFN-γ (BioLegend, San Diego, CA) and TNF-α (Miltenyi Biotec, Bergisch Gladbach, Germany) staining were performed on fixed and permeabilized cells (Cytofix/Cytoperm, BD Biosciences). Data was acquired with a CytoFLEX cytometer (Beckman Coulter, Brea, CA), and analyzed using FlowJo Flow Cytometry software (FlowJo LLC, Ashland, OR). Minimal epitopes recognized by HLA I-restricted cell lines were determined by IFN-γ ELISpot assay. To confirm the restricted HLA allele, we used anti-human HLA class I B35 antigen monoclonal antibody in ELISpot plate (MyBioSource, San Diego, CA).
Immunofluorescence
The coverslips were seeded with tumor cells and pancreatic cells at 2.5e5/500 µl per well and left overnight for the confluency to reach 50–70%. Once the confluency was achieved, the cells were rinsed in 1X PBS, followed by fixation with 4% PFA for 15 minutes at room temperature and permeabilization for 20 minutes in 0.1%Triton X-100. Nonspecific binding was blocked with 2% BSA for an hour at room temperature. Primary anti-PRAME antibody (Sigma, St. Louis, MO) was diluted 1:30 in 0.1% BSA in PBS and incubated overnight at 4°C. After the overnight incubation, the slides were rinsed and incubated with an appropriate secondary antibody diluted 1:100 in 0.1% BSA in PBS for an hour at room temperature (Donkey Anti-Rabbit Alexa Flour 568, Abcam, Cambridge, MA). Prolong Gold Mounting media with DAPI was used to counterstain the nuclei and mount the slides. All images were taken 24 hours after mounting of slides and imaged using Olympus BX53 microscope.
Data analysis
Results were evaluated using descriptive statistics (medians and ranges). The Student t test was used to test for significance. Data analysis was performed in GraphPad Prism (GraphPad Software, La Jolla, CA).
RESULTS
PRAME-specific T-cells can be expanded from healthy donors
PRAME-specific T-cells were expanded from twelve healthy donor PBMCs. Priming and restimulations of PBMCs were carried out with PRAME-pulsed, irradiated DC. We harvested a median of 452 × 106 T-cells (range 161 × 106–17.5 × 108 T-cells) by day 21 of culture (Figure 1A) which would meet cell dose for use in our proposed clinical trial using off-the-shelf PRAME-specific T cells.
Figure 1.
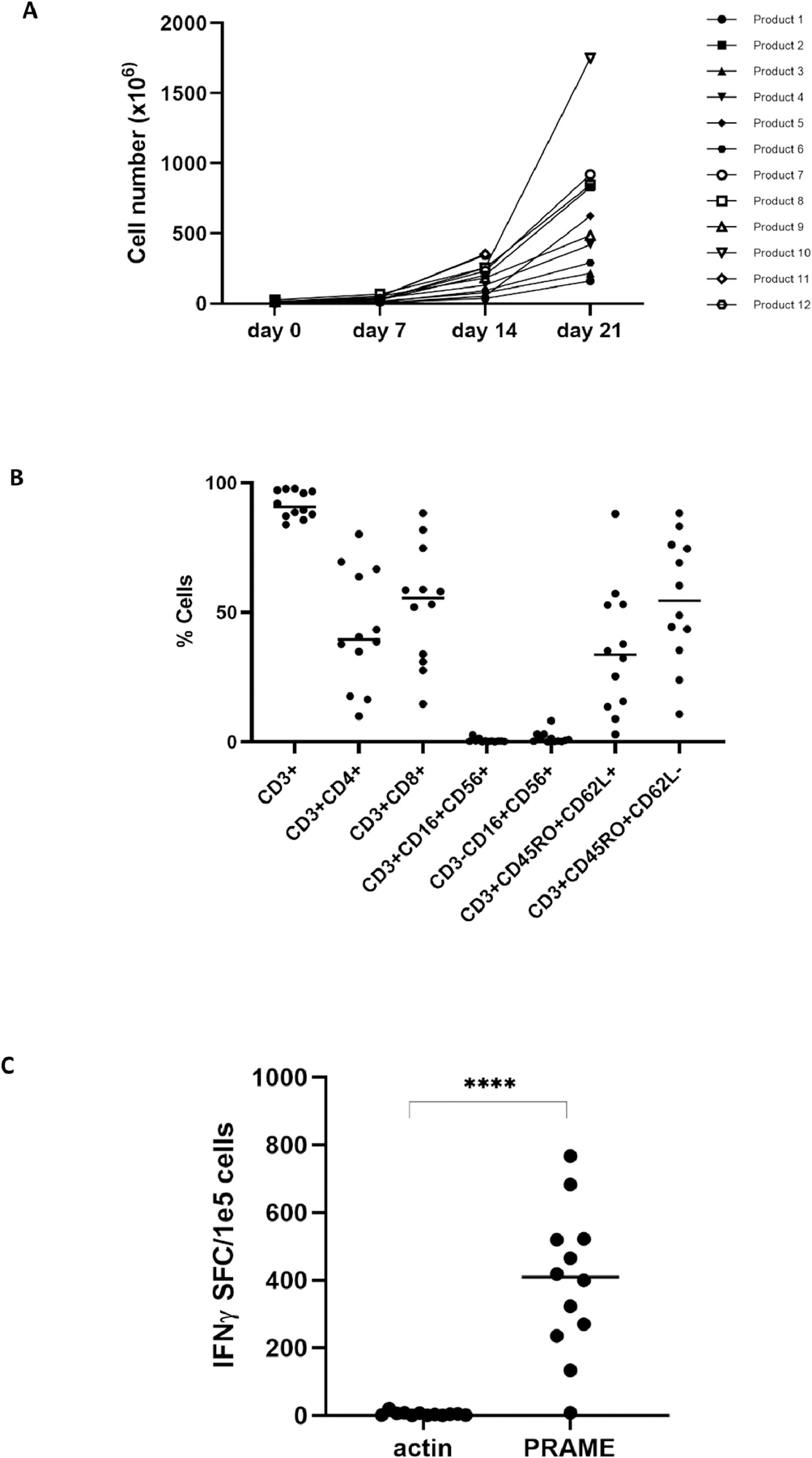
PRAME-specific T-cells can be expanded from healthy donors.
(A) Total cell number of PRAME-specific T-cells generated from 12 healthy donors. PBMCs were primed with PRAME-pulsed DCs on day 0 and restimulated on day 7 and day 14. (B) Phenotyping analysis of PRAME-specific T-cell products, assessed by flow cytometry, shows a mixture of both CD4+ and CD8+ T-cells with a balanced predominance of central memory (CD3+CD45RO+CD62L+) and effector memory (CD3+CD45RO+CD62L-) phenotype. There was no outgrowth of NK (CD3-CD16+CD56+) or NKT (CD3+CD16+CD56+) cells. (C) The specificity of the T-cell product for the PRAME antigen was measured by IFN-γ ELISpot assay.
Phenotyping of expanded T-cells showed a median CD3+ content of 90.75% (range 83.9–97.8%) with a mixture of CD4+ T-cells (median 39.6%, range 9.9–80.2%) and CD8+ T-cells (55.6%, 14.5–88.3%). There was no outgrowth of natural killer (NK) cells or natural killer T (NKT) cells. B cells and dendritic cells accounted for less than 2% of final products, meeting clinical release criteria for TAA-T products to be used in clinical trial. Expanded T-cells were composed of both central memory (median 33.7%, 2.88–88.0%) and effector memory (54.7%, 10.7–88.3%) T-cells (Figure 1B). PRAME antigen specificity was evaluated using IFN-γ ELISpot assay. Eleven T-cell products demonstrated response to PRAME (median 405.48 SFC/1×105 cells, range 8–762.5) while median actin (negative control) was 4.0 SFC/1×105 cells (range 0.5–20) (p-value <0.0001) (Figure 1C).
PRAME-specific T-cells elicit antitumor activity against partially HLA-matched solid tumor cell lines
To evaluate the in vitro anti-tumor activity of healthy donor-derived T-cells targeting PRAME, in a “third party setting”, T-cells were cocultured with the Wilms tumor cell line(s) Wit49 and WT 17.94 known to express PRAME (Figure 2A). Tumor cell lines were matched in at least one HLA-antigen (range 3–9) with the T-cell products (Table S1). HLA class I (A, B, C) and HLA class II (DP, DQ, DR) groups were included in the analysis. Specifically, TAA-T #1 was matched to 17.94 at 9 HLA alleles and to Wit49 at 4 HLA alleles. TAA-T #2 was matched to 17.94 at 4 HLA alleles; but due to more limited TAA-T cell numbers, Wit49 was not included in the TAA-T #2 cytotoxicity assays. TAA-T #3 was matched to 17.94 and Wit49 at 3 HLA alleles each. Figure 2B–I shows the results of three TAA-T cell products which showed polyclonality (Figure 2B) and specificity for PRAME (Figure 2C). These T-cell products were tested for cytolytic activity against the two Wilms tumor cell lines Wit49 and WT 17.94. Specific tumor recognition and killing occurred even with single HLA class I matched targets (Figures 2D–H). As control for nonspecific lysis or allogeneic reactivity, T-cells with irrelevant specificity (Nonspecific T cells (NSTs)) from the same donor were used in all experiments and were not able to kill the tumor cells (Figures 2D–H). Moreover, TAA-T were not able to kill autologous PHA blasts/PBMCs (Figure 2I) demonstrating a lack of autoreactivity in vitro. In sum, healthy donor-derived off-the-shelf PRAME-specific T-cells showed specific killing against PRAME+ tumor cell lines, with notable minimal cytotoxicity by NSTs derived from the same donor to these tumor cell lines and absence of autoreactivity in vitro, thereby demonstrating the potential for the use of these T-cell products in the off-the-shelf setting.
Figure 2.
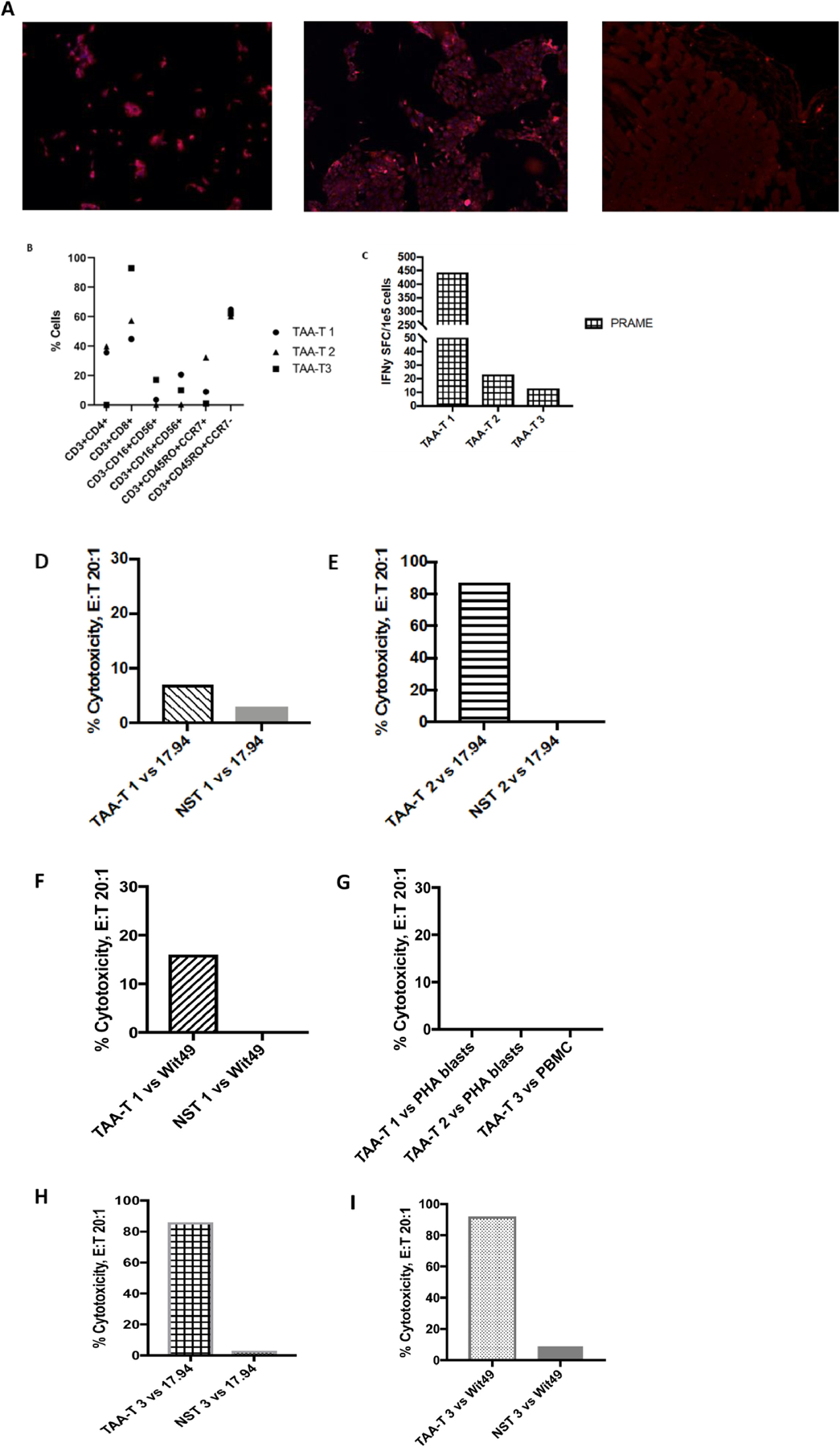
Off-the-Shelf TAA-T cells are cytolytic against Wilms Tumor cell lines
(A) Wilms tumor cell lines 17.94 (left) and Wit49 (middle) stained positive for PRAME by immunofluorescence, compared to pancreatic cells, negative control (right). (B) TAA-T products are polyclonal and demonstrate cytokine secretion upon stimulation with PRAME as measured by flow cytometry and intracellular cytokine staining. (C) TAA-T products demonstrate specificity for PRAME as measured by IFN-γ ELISpot assay (background response to actin subtracted from final results). (D)-(I) PRAME specific TAA-T products demonstrate antigen-specific cytotoxicity to Wilms tumor cell lines 17.94 (D)(F)(G) and Wit49 (F)(H) with absent non-specific activity to autologous PHA blasts or PBMCs (I).
Novel CD8-restricted T-cell PRAME epitopes identified in healthy donor-derived T cell products
In order to develop a third party PRAME-specific T cell bank, it is important to identify epitopes recognized in the context of MHC class I and class II. In order to map the class I-restricted responses, we used a mapping grid [29] consisting of 23 mini-pools comprising 125 individual 15-mer peptides spanning the entire PRAME protein sequence to identify PRAME epitopes eliciting a response by ex-vivo expanded T-cells (Figure S1). IFN-γ production by PRAME- sensitized T-cells in response to stimulation with mini-pools was measured by ELISpot assay. In an example of one T-cell product, dominant responses were observed for 16 mini-pools (1, 2, 3, 4, 5, 6, 7, 8, 12, 14, 15, 16, 17, 18, 19, and 23) (Figure 3A). T-cell responses to single 15-mer peptides selected from the grid and their neighboring peptides were determined by IFN-γ ELISpot assay (Figure 3B). Single peptides 18, 19, 35, 36, 37, 43, 44, 48, 49, 51, 124, and 125 were identified as immunogenic (Figure 3C). HLA restriction of these peptides was evaluated with intracellular IFN-γ and TNF-α cytokine staining. CD8+ T-cells released IFN-γ and TNF-α in response to overlapping 15-mer peptides: 36 ASLYSFPEPEAAQPM, 37 SFPEPEAAQPMTKKR, 43 EQPFIPVEVLVDLFL, and 44 IPVEVLVDLFLKEGA, indicating that these peptides were HLA class I-restricted epitopes (Figure 3D). As shown in Figure 3E, CD4+ T-cells did not release IFN-γ and TNF-α in response to these peptides. The minimal 9-mer epitopes overlapping by 8 amino acids spanning the 15-mer peptides were determined by IFN-γ ELISpot. CD8+ T-cells secreted IFN-γ upon stimulation with epitopes FPEPEAAQP and VEVLVDLFL (Figure 3F). We applied an algorithm (NetMHC [www.cbs.dtu.dk/services/NetMHCpan/]) to determine the restricted HLA allele of FPEPEAAQP and VEVLVDLFL epitopes. The algorithm predicted strong binding to HLA-B*35:03 for FPEPEAAQP and weak binding for HLA-B*38:01 for VEVLVDLFL. We further tested to confirm the HLA-B*35 restriction of FPEPEAAQP using an anti-HLA-B*35 antibody. T-cell product alone showed specificity for FPEPEAAQP while products with anti-HLA-B*35 showed decreased specificity (Figure 3G). The complete data of novel CD8+-restricted T-cell epitopes identified in PRAME is summarized in Table 1.
Figure 3.
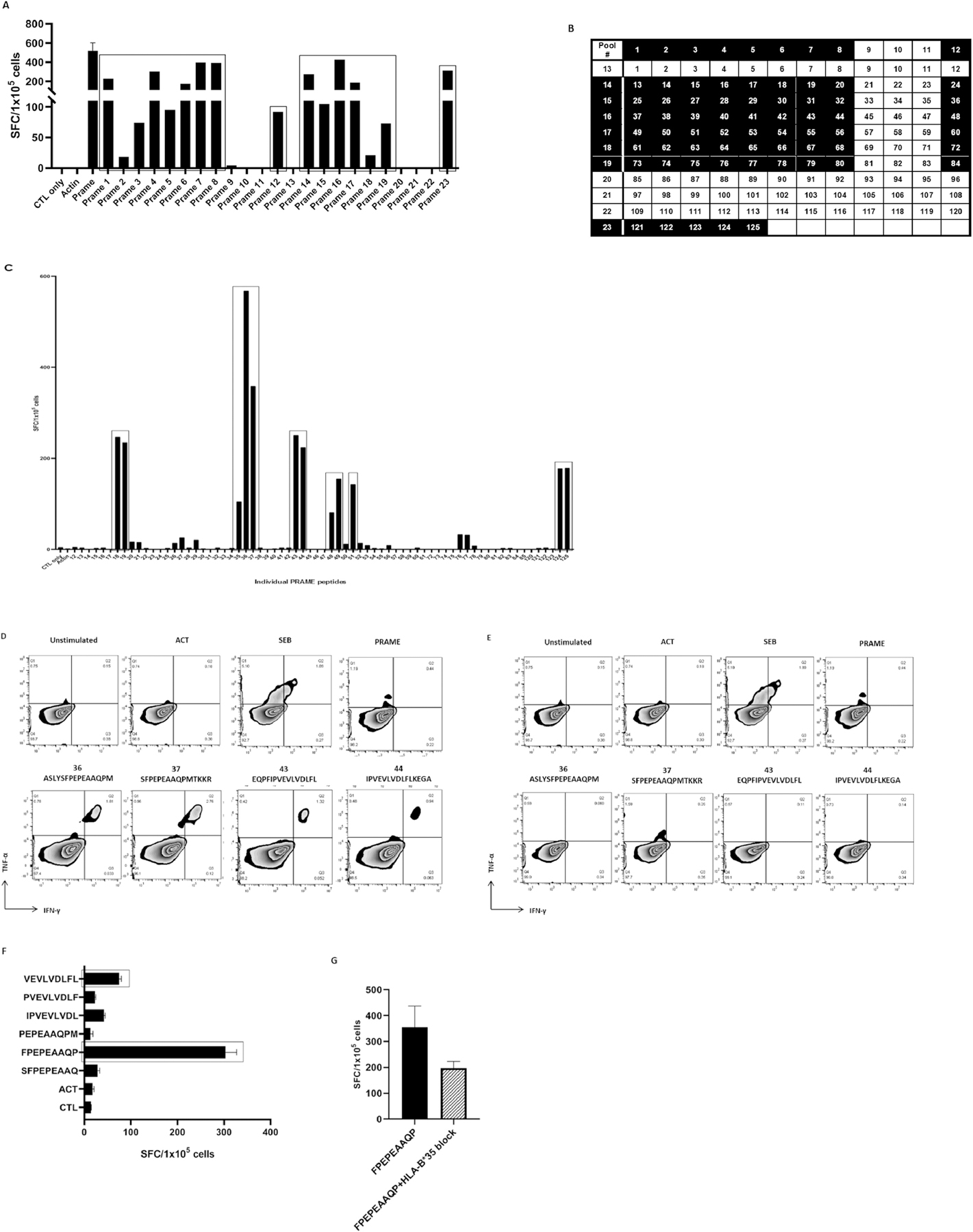
Identification of CD8-restricted T-cell epitopes
(A) IFN-γ production by T-cells in response to PRAME peptide pool stimulation. (B)(C) T-cell responses to single PRAME peptides, present in the mini-pools, were evaluated by IFN-γ ELISpot assay. (D) HLA-I-restricted epitopes (36, 37, 43, and 44) were determined by measuring IFN-γ and TNF-α release by CD8+ T-cells. (E) CD4+ T-cells showed no specificity for HLA-I-restricted epitopes (36, 37, 43, 44). (F) The minimal 9-mer peptides FPEPEAAQP and VEVLVDLFL were determined by IFN-γ ELISpot assay. (G) FPEPEAAQP HLA restriction was confirmed using anti-HLA-B*35 antibody.
Table 1.
Peptide sequences of CD8-restricted T-cell epitopes identified in PRAME.
| Peptide sequence | Amino acid location | HLA-A | HLA-B | HLA-C |
|---|---|---|---|---|
| AGQSLLKDE | 29–37 | 01:01; 01:01 | 08:01; 18:01 | 06:02; 07:01 |
| AWPFTCLPL | 75–83 | 24:02; 26:01 | 15:08; 44:03 | 01:02; 16:01 |
| WPFTCLPLG | 76–84 | 24:02; 26:01 | 15:08; 44:03 | 01:02; 16:01 |
| SGNRASLYS | 137–145 | 02:01; 24:02 | 35:01; 35:03 | 04:01; 04:01 |
| LYSFPEPEA* | 143–151 | 02:01; 68:01 | 38:01; 44:02 | 05:01; 12:03 |
| FPEPEAAQP | 146–154 | 24:357; 26:01 | 35:03; 38:01 | 04:01; 12:03 |
| VEVLVDLFL | 175–183 | 24:357; 26:01 | 35:03; 38:01 | 04:01; 12:03 |
| EKVKRKKNV | 197–205 | 01:01; 01:01 | 08:01; 18:01 | 06:02; 07:01 |
| KVKRKKNVL | 198–206 | 01:01; 01:01 | 08:01; 18:01 | 06:02; 07:01 |
| RKKNVLRLC | 201–209 | 02:01; 24:02 | 35:01; 35:03 | 04:01; 04:01 |
| NLTHVLYPV* | 435–443 | 02:01; 26:01 | 51:01; 51:01 | 05:01; 16:02 |
Previously described HLA class I-restricted PRAME epitopes. Red: strong binder (<2). Green: weak binder (2–10).
Broad CD4 specific activity in donor PRAME-specific T-cells
We next analyzed the breadth of CD4-restricted epitopes recognized by PRAME-specific T-cells using the same approach as above. A representative example is shown in Figure 4. The T-cell product recognized 4 mini pools: 2, 11, 17, and 18 (Figure 4A). Testing of the single peptides and their neighboring peptides revealed recognition of single peptides 50 EKVKRKKNVLRLCCK, 70 SPEKEEQYIAQFTSQ, and 71 EEQYIAQFTSQFLSL (Figure 4B, 4C). CD4+ T-cells released IFN-γ and TNF-α in response to 15-mer peptides 50 EKVKRKKNVLRLCCK and 71 EEQYIAQFTSQFLSL indicating that these peptides were HLA class II-restricted epitopes (Figure 4D). CD8+ T-cells showed no specificity to peptides 50 and 71 (Figure 4E). The complete data of CD4+-restricted T-cell epitopes identified in PRAME is summarized in Table 2.
Figure 4.
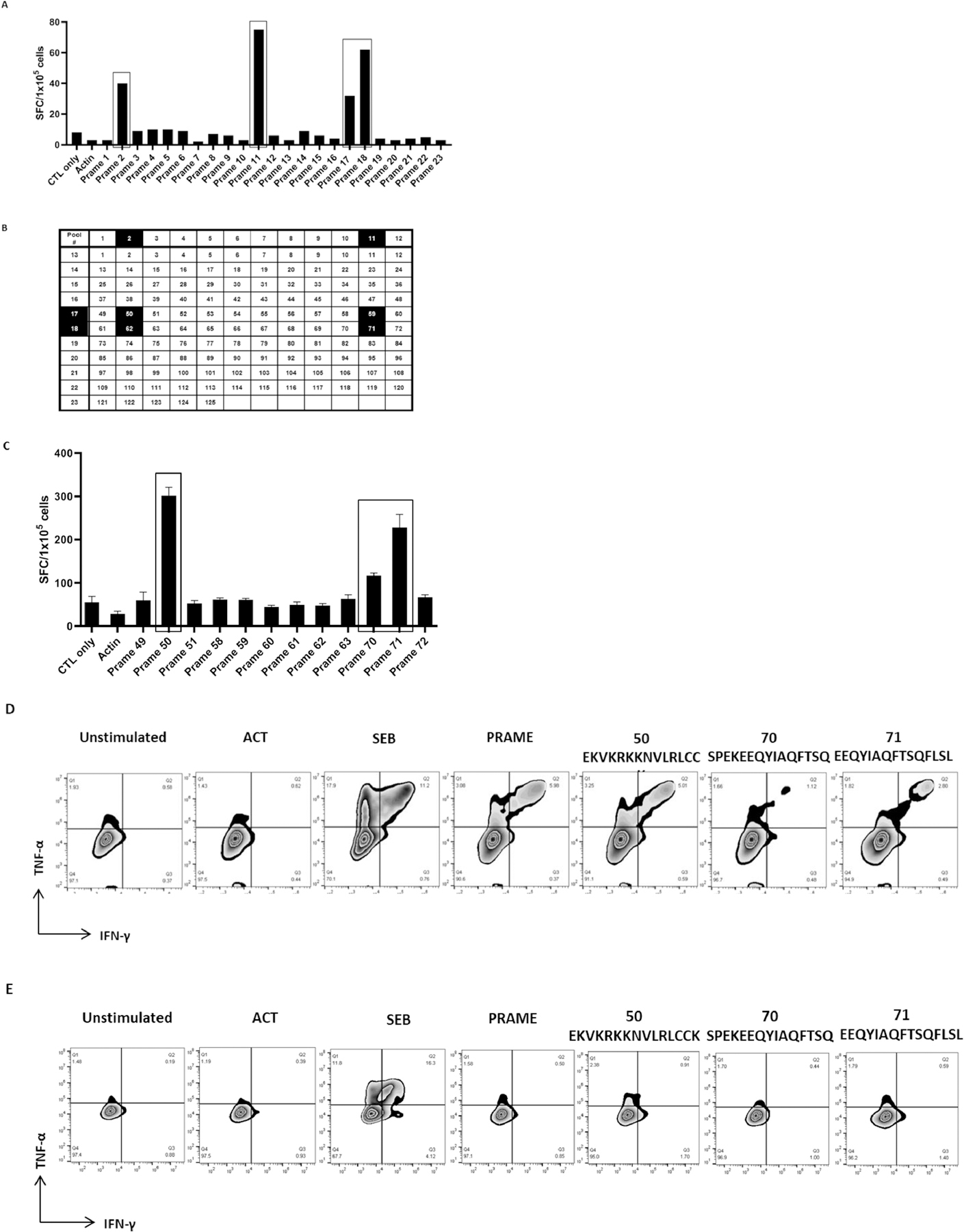
Identification of CD4-restricted T-cell epitopes
(A) IFN-γ production by T-cells in response to PRAME peptide pool stimulation. (B)(C) T-cell responses to single PRAME peptides, present in the mini-pools 2, 11, 17, and 18, were evaluated by IFN-γ ELISpot assay. (D) HLA-II-restricted epitopes (50 and 71) were determined by measuring IFN-γ and TNF-α release by CD4+ T-cells. (E) CD8+ T-cells did not release IFN-γ and TNF-α in response to class II-restricted epitopes.
Table 2.
Peptide sequences of CD4-restricted T-cell epitopes identified in PRAME
| Peptide sequence | Amino acid location | HLA-DRB1 | HLA-DRB3 | HLA-DRB4 | HLA-DRB5 | HLA-DQA1 | HLA-DQB1 | HLA-DPA1 | HLA-DPB1 |
|---|---|---|---|---|---|---|---|---|---|
| RLWGSIQSRYISMSV | 5–19 | 01:01; 11:01 | 02:02 | 01:01; 05:01 | 05:01; 05:01 | 01:03; 01:03 | 04:01; 04:02 | ||
| TSPRRLVELAGQSLL | 21–35 | 01:01; 15:01 | 01:01 | 01:01; 01:02 | 05:01; 06:02 | 01:03; 01:03 | 04:01; 04:01 | ||
| RLVELAGQSLLKDEA | 25–39 | 01:01; 15:01 | 01:01 | 01:01; 01:02 | 05:01; 06:02 | 01:03; 01:03 | 04:01; 04:01 | ||
| PFTCLPLGVLMKGQH | 77–91 | 07:01; 11:03 | 2:02 | 01:01 | 02:01; 05:01 | 02:01; 03:01 | 01:03; 02:02 | 01:01; 04:02 | |
| LPLGVLMKGQHLHLE | 81–95 | 07:01; 11:03 | 2:02 | 01:01 | 02:01; 05:01 | 02:01; 03:01 | 01:03; 02:02 | 01:01; 04:02 | |
| 01:01; 11:01 | 02:02 | 01:01; 05:01 | 05:01; 05:01 | 01:03; 01:03 | 04:01; 04:02 | ||||
| 08:01; 08:10 | 04:01; 06:01 | 03:01; 04:02 | 01:03; 01:03 | 04:01; 04:01 | |||||
| 01:85; 14:07 | 01:01 | 01:03; 01:02 | 05:01; 01:02 | 01:03; 01:02 | 04:01; 01:01 | ||||
| VLMKGQHLHLETFKA | 85–99 | 08:01; 08:10 | 04:01; 06:01 | 03:01; 04:02 | 01:03; 01:03 | 04:01; 04:01 | |||
| DVLLAQEVRPRRWKL | 105–119 | 01:85; 14:07 | 01:01 | 01:03; 01:02 | 05:01; 01:02 | 01:03; 01:02 | 04:01; 01:01 | ||
| DELFSYLIEKVKRKK | 188–202 | 07:01; 11:03 | 2:02 | 01:01 | 02:01; 05:01 | 02:01; 03:01 | 01:03; 02:02 | 01:01; 04:02 | |
| SYLIEKVKRKKNVLR | 193–207 | 07:01; 11:03 | 2:02 | 01:01 | 02:01; 05:01 | 02:01; 03:01 | 01:03; 02:02 | 01:01; 04:02 | |
| 08:01; 08:10 | 04:01; 06:01 | 03:01; 04:02 | 01:03; 01:03 | 04:01; 04:01 | |||||
| EKVKRKKNVLRLCCK | 197–211 | 08:01; 08:10 | 04:01; 06:01 | 03:01; 04:02 | 01:03; 01:03 | 04:01; 04:01 | |||
| 04:02; 15:02 | 01:02 | 01:03; 03:01 | 03:02; 06:01 | 01:03; 02:01 | 04:01; 17:01 | ||||
| CCKKLKIFAMPMQDI | 209–223 | 01:85; 14:07 | 01:01 | 01:03; 01:02 | 05:01; 01:02 | 01:03; 01:02 | 04:01; 01:01 | ||
| AMPMQDIKMILKMVQ | 217–231 | 01:85; 14:07 | 01:01 | 01:03; 01:02 | 05:01; 01:02 | 01:03; 01:02 | 04:01; 01:01 | ||
| QDIKMLKMVQLDSI | 221–235 | 01:85; 14:07 | 01:01 | 01:03; 01:02 | 05:01; 01:02 | 01:03; 01:02 | 04:01; 01:01 | ||
| SPYLGQMINLRRLLL | 253–267 | 01:85; 14:07 | 01:01 | 01:03; 01:02 | 05:01; 01:02 | 01:03; 01:02 | 04:01; 01:01 | ||
| 01:01; 15:01 | 01:01 | 01:01; 01:02 | 05:01; 06:02 | 01:03; 01:03 | 04:01; 04:01 | ||||
| EEQYIAQFTSQFLSL | 281–295 | 04:02; 15:02 | 01:02 | 01:03; 03:01 | 03:02; 06:01 | 01:03; 02:01 | 04:01; 17:01 | ||
| LERLAYLHARLRELL | 457–471 | 01:85; 14:07 | 02:01 | 01:01; 01:04 | 05:01; 05:03 | 02:01; 02:01 | 10:01; 14:01 |
Red: strong binder (<2). Green: weak binder (2–10).
T-cell epitopes elicit both CD4+ and CD8+ T-cell responses
We have also shown that some PRAME peptides are able to simultaneously induce MHC class I-restricted CD8+ and MHC class II-restricted CD4+ T-cell responses. As shown in Figure 5, peptide 7 (RLVELAGQSLLKDEA) activated both CD4+ and CD8+ T-cells in vitro.
Figure 5.
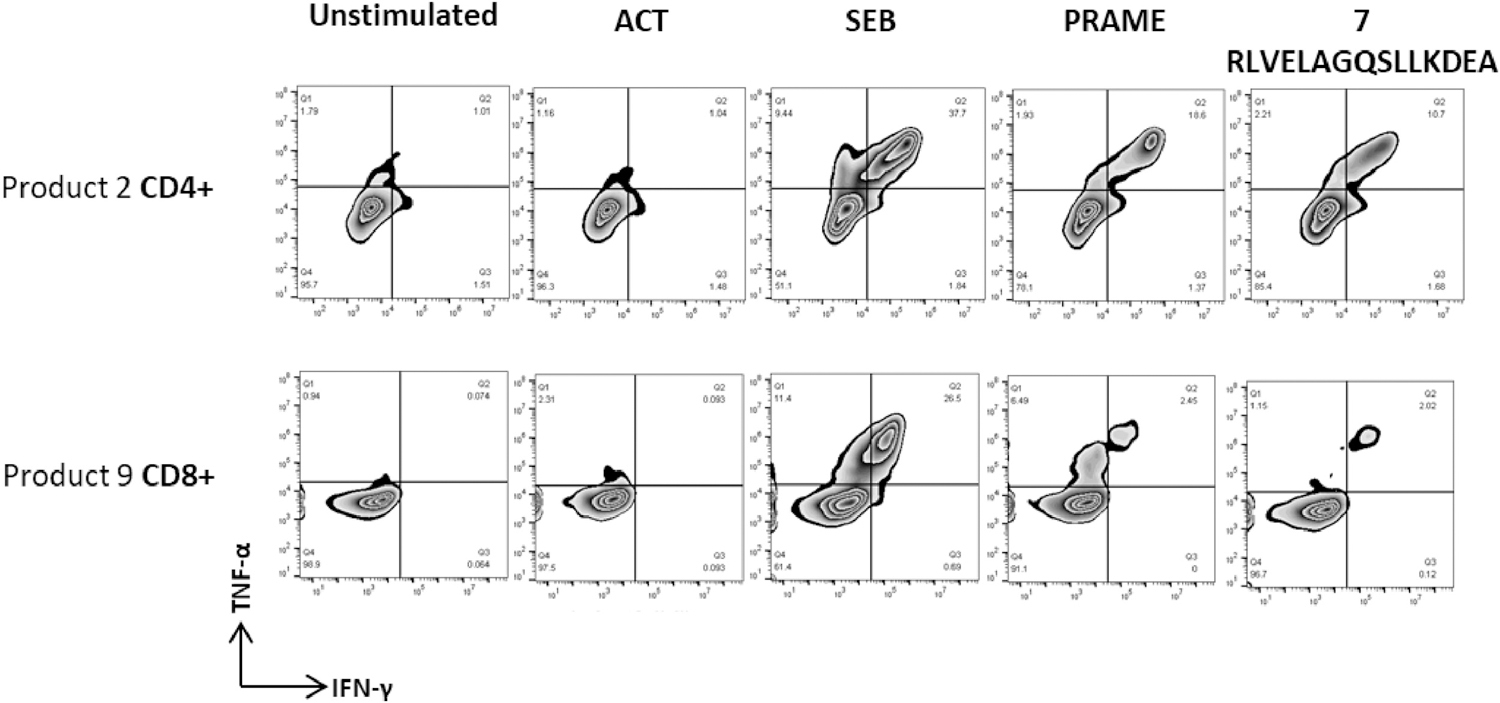
T-cell epitopes elicit both CD4+ and CD8+ T-cell responses
Cytotoxic (CD8+) and helper (CD4+) T-cell response to PRAME epitope RLVELAGQSLLKDEA. Results show that this peptide activated both CD8+ and CD4+ T-cells.
DISCUSSION
PRAME is highly expressed in a wide range of cancers, including melanoma, leukemias, sarcoma, renal cell cancer, Wilms tumor, non-small cell lung cancer (NSCLC), neuroblastoma, breast cancer, and multiple myeloma and PRAME expression has been associated with poor prognosis in a multitude of solid tumors [4, 7, 30, 31]. PRAME expression is minimal in healthy tissues such as the gonads, adrenal glands, bone marrow, and brain with highest expression in the testes [30]. Consequently, numerous clinical trials are currently evaluating PRAME-specific cancer vaccines and adoptive T cell therapies. However, the PRAME-specific T-cell therapies being evaluated thus far have all utilized a patient-specific approach which is limited by the time it takes to manufacture these autologous products and the inherently inadequate anti-tumor T-cell response detected in cancer patients with relapsed refractory disease [18]. Third-party antigen-specific T-cells have demonstrated safety and efficacy in the virus setting since off-the- shelf allogeneic virus specific T-cells (VST) have been effective for the treatment of life threatening viral diseases in immunocompromised patients [22, 32]. Our group and others have shown that matching third-party VST products with virus specific activity through at least one shared HLA allele has been crucial for demonstrating virus-specific T-cell efficacy in vivo [33–35]. Several studies have shown that high-avidity PRAME-specific cytotoxic T lymphocytes can be successfully isolated from healthy donors [13, 28, 36]. Therefore, we posited that this third-party approach could be expanded to the cancer setting to facilitate the rapid treatment of patients who have an inherently inadequate anti-tumor T-cell response. In this study, we generated PRAME-specific T-cells from healthy donors and identified novel CD8+ and CD4+ restricted PRAME epitopes important for the generation of an off-the-shelf third party T-cell bank. Further, we showed that these third party, partially HLA-matched T-cell products killed partially HLA matched tumor cell lines in vitro. While cytotoxicity was based on the bulk T cell product, the enhanced cytolytic activity compared to non-specific T cell products is an important proof-of-principle to demonstrate the anti-tumor potency of these products which will be used going forward in our proposed clinical trial evaluating third party PRAME-specific T cells for the treatment of patients with solid tumors.
Patients with relapsed or refractory (r/r) Wilms tumor have particularly poor outcomes, with less than 10% of patients with anaplastic histology and r/r disease surviving at 5 years despite aggressive, toxic therapies [37, 38]. High dose chemotherapy followed by autologous stem cell transplant has been shown to improve outcomes in a subset of these patients, supporting the role of the immune system [39]. In a phase I clinical trial, tumor-associated antigen (TAA)- specific T-cells (TAA-T) specific for Wilms tumor 1 (WT1), PRAME, and survivin were generated from 15 patients with r/r solid tumors, 7 of which had Wilms tumor. This was shown to be safe at the maximum dose level tolerated with antigen spreading identified to support anti-tumor immunity post infusion [18]. While autologous products demonstrate an excellent safety profile, patients are often subjected to salvage therapies over the 4 to 8 weeks of waiting period for product generation. We have shown that PRAME-specific T-cells generated from healthy donors demonstrate tumor-specific cytotoxicity in vitro to partially HLA-matched tumor cell lines (Figure 2). Hence, these data support the ability to treat solid tumor patients with an off-the-shelf, partially HLA-matched allogeneic product for “on demand” treatment.
The development of a third party PRAME specific T-cell therapeutic for the treatment of WT would therefore overcome the inherent and practical limitations of using autologous patient-derived products. Based on the experience from the virus specific cell setting, effectiveness of a third party tumor-associated antigen (TAA) specific T-cell therapy are dependent on a comprehensive identification of the epitopes within the tumor-associated protein(s) of interest. We and others have shown that rigorous culture conditions facilitate the identification of the functional T-cell clones critical for anti-viral and anti-tumor responses in vitro [40, 41]. The importance of such T-cell epitope identification extends beyond the manufacture of a robust third party T-cell bank. For example, T-cells specific for identified epitopes can be tracked using multimers both in vitro and in vivo, and even in situ [42]. Moreover, identification of the TCR responding to these specific epitopes [43] allows the use of TCR sequencing to track unique T-cell clones that may contribute to the anti-tumor response, and the construction of an engineered αβTCR for gene-modified T-cell therapies targeting specific individual TAA epitopes [44]. Finally, identifying the breadth of the epitope specific T-cell response is also a critical step for effective vaccine design [45].
To date, several T-cell epitopes targeting PRAME have been described [25, 27, 46]. In this study, we describe a further series of novel class I and II epitopes specific for PRAME using our ex vivo expansion protocol [28]. We confirmed predicted class I epitopes from the 15-mer pools using specifically manufactured 9-mer peptides and identified epitopes spanned the entire sequence of PRAME protein (Figure 6). Elicited central memory and effector memory T-cell responses suggest potential activity against PRAME-expressing targets as was evidenced by the ability of these allogeneic healthy donor-derived T-cells to kill partially HLA matched tumor cells unlike T cells that were not specific for PRAME. We also identified PRAME epitopes that elicited CD4+ and CD8+ T-cell responses simultaneously which is important for an effective cytotoxic T-cell response and persistence of adoptively transferred T-cells in vivo [47, 48]. However, we were not able to confirm the exact MHC-class I epitope response possibly due to the fact that we are observing both CD8+ and CD4+ responses to these epitopes. Future experiments however, will explore the cytolytic activity of selected and sorted epitope-specific T cells to PRAME+ tumor lines.
Figure 6.
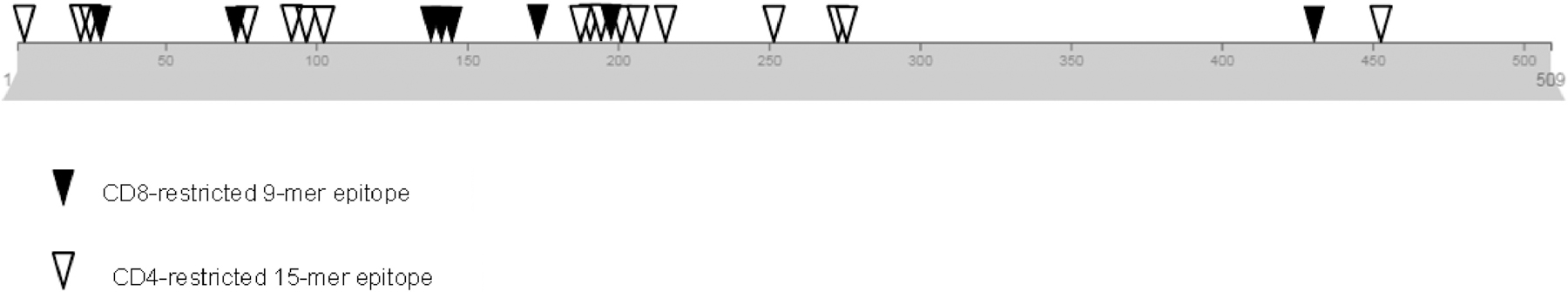
PRAME protein diagram with identified epitopes
The locations of CD4 and CD8-restricted epitopes identified within PRAME protein are shown.
In summary, in this proof of principle study we have demonstrated how a third party tumor antigen specific T-cell product targeting PRAME could be applied to the solid tumor setting. The identification of novel class I and class II HLA-restricted PRAME-specific T-cell epitopes which are represented in populations from different geographic regions (allelefrequencies.net), allows us to pick the optimal T-cell donor product for any given patient by ensuring that epitope specific responses are restricted to HLA allele or alleles shared between donor and recipient. We identified the AWPFTCLPL peptide that prediction algorithms suggest is an HLA-A*24-restricted T-cell epitope. The gene frequency of HLA-A*24 (A*24:02) is high in Asian and Hispanic populations. We also identified peptide KVKRKKNVL which is an HLA-B*08 (B*08:01)-restricted T-cell epitope. The HLA-B*08:01 allele is highly prevalent in individuals of Caucasian ancestry. It is also common in Asian/Pacific Islander and African American populations [49]. Therefore, defining PRAME-specific T-cells beyond HLA-A*02-restricted epitopes could be useful when developing T-cell therapeutics for worldwide application. Moreover, creating off-the-shelf products has many potential advantages since such products are readily available for the treatment of patients with aggressive disease or for patients where an autologous product cannot be manufactured. Finally, T-cell products derived from healthy donors may be more reliably expanded to specific quality and potency specifications to facilitate the manufacture of large quantities of tumor-specific T-cells for the treatment of a broad number of solid tumors that express cancer testis antigens such as PRAME.
Supplementary Material
Figure S1. Identification of T-cell epitopes
(A) Peptide libraries of 15-mer peptides overlapping by 11 amino acids spanning the entire sequence of the PRAME protein consisting of 509 amino acids. The sequence of the first 2 15- mers with 11 amino acid overlap is illustrated. (B) 23 peptide pools comprising 10–12 peptides were prepared so that each 15-mer peptide was included in only two pools.
Table S1. HLA matching of third-party TAA-T products HLA types of Wilms tumor cell lines (17.94, Wit49) and partially matched TAA-T cell products are shown.
Acknowledgments
We thank the staff of the Center for Cancer and Immunology Research at Children’s National Hospital. This work was supported by a grant from the National Institutes of Health (1P01 CA225618–01A1 to C.M.B and P.J.K.) and was supported by Ben’s Run and by the Board of Visitors of the Children’s National Health System.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Potential Conflicts of Interest
C.M.B. is on the advisory board for Cellectis and is on the scientific advisory boards for Catamaran Bio and Mana Therapeutics with stock and/or ownership, is on the Board of Directors for Caballeta Bio with stock options and has stock in Neximmune and Torque Therapeutics. P.J.H and C.R.Y.C. are co-founders of Mana Therapeutics and PJH is on the board of directors of Mana Therapeutics. PJH is on the scientific advisory board of Cellevolve.
M.D.K. is on a scientific advisory panel for Gilead Sciences. C.M.B., C.R.Y.C., P.J.H. and M.S. have filed a patent application based on the findings in this paper. The other authors declare that there are no conflicts of interests.
REFERENCES
- 1.Boon K, et al. , Comparison of medulloblastoma and normal neural transcriptomes identifies a restricted set of activated genes. Oncogene, 2003. 22(48): p. 7687–7694. [DOI] [PubMed] [Google Scholar]
- 2.Epping M, et al. , PRAME expression and clinical outcome of breast cancer. British Journal of Cancer, 2008. 99(3): p. 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ikeda H, et al. , Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity, 1997. 6(2): p. 199–208. [DOI] [PubMed] [Google Scholar]
- 4.Oberthuer A, et al. , The tumor-associated antigen PRAME is universally expressed in high-stage neuroblastoma and associated with poor outcome. Clinical Cancer Research, 2004. 10(13): p. 4307–4313. [DOI] [PubMed] [Google Scholar]
- 5.Steinbach D, et al. , PRAME gene expression in childhood acute lymphoblastic leukemia. Cancer Genetics and Cytogenetics, 2002. 138(1): p. 89–91. [DOI] [PubMed] [Google Scholar]
- 6.Szczepanski M, et al. , PRAME expression in head and neck cancer correlates with markers of poor prognosis and might help in selecting candidates for retinoid chemoprevention in pre-malignant lesions. Oral Oncology, 2013. 49(2): p. 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan P, et al. , Expression and prognostic relevance of PRAME in primary osteosarcoma. Biochemical and Biophysical Research Communications, 2012. 419(4): p. 801–808. [DOI] [PubMed] [Google Scholar]
- 8.van Baren N, et al. , PRAME, a gene encoding an antigen recognized on a human melanoma by cytolytic T cells, is expressed in acute leukaemia cells. British Journal of Haematology, 1998. 102(5): p. 1376–1379. [DOI] [PubMed] [Google Scholar]
- 9.Yang L, et al. , PRAME Gene Copy Number Variation Is Related to Its Expression in Multiple Myeloma. DNA and Cell Biology, 2017. 36(12): p. 1099–1107. [DOI] [PubMed] [Google Scholar]
- 10.Epping MT, et al. , The human tumor antigen repressor of retinoic acid PRAME is a dominant receptor signaling. Cell, 2005. 122(6): p. 835–847. [DOI] [PubMed] [Google Scholar]
- 11.Yin B, PRAME: From diagnostic marker and tumor antigen to promising target of RNAi therapy in leukemic cells. Leukemia Research, 2011. 35(9): p. 1159–1160. [DOI] [PubMed] [Google Scholar]
- 12.Griffioen M, et al. , Detection and functional analysis of CD8+ T cells specific for PRAME: a target for T-cell therapy. Clin Cancer Res, 2006. 12(10): p. 3130–6. [DOI] [PubMed] [Google Scholar]
- 13.Matko S, et al. , PRAME peptide-specific CD8(+) T cells represent the predominant response against leukemia-associated antigens in healthy individuals. Eur J Immunol, 2018. 48(8): p. 1400–1411. [DOI] [PubMed] [Google Scholar]
- 14.Rezvani K, et al. , Functional leukemia-associated antigen-specific memory CD8+ T cells exist in healthy individuals and in patients with chronic myelogenous leukemia before and after stem cell transplantation. Blood, 2003. 102(8): p. 2892–900. [DOI] [PubMed] [Google Scholar]
- 15.Rezvani K, et al. , Ex vivo characterization of polyclonal memory CD8(+) T-cell responses to PRAME-specific peptides in patients with acute lymphoblastic leukemia and acute and chronic myeloid leukemia. Blood, 2009. 113(10): p. 2245–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapuis AG, et al. , Transferred WT1-Reactive CD8(+) T Cells Can Mediate Antileukemic Activity and Persist in Post-Transplant Patients. Science Translational Medicine, 2013. 5(174). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapuis AG, et al. , Transferred melanoma-specific CD8(+) T cells persist, mediate tumor regression, and acquire central memory phenotype. Proceedings of the National Academy of Sciences of the United States of America, 2012. 109(12): p. 4592–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hont AB, et al. , Immunotherapy of Relapsed and Refractory Solid Tumors With Ex Vivo Expanded Multi-Tumor Associated Antigen Specific Cytotoxic T Lymphocytes: A Phase I Study. Journal of Clinical Oncology, 2019. 37(26): p. 2349–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams KM, et al. , Complete Remissions Post Infusion of Multiple Tumor Antigen Specific T Cells for the Treatment of High Risk Leukemia and Lymphoma Patients after HCT. Blood, 2017. 130: p. 1.28684444 [Google Scholar]
- 20.Bach PB, Giralt SA, and Saltz LB, FDA Approval of Tisagenlecleucel Promise and Complexities of a $475 000 Cancer Drug. Jama-Journal of the American Medical Association, 2017. 318(19): p. 1861–1862. [DOI] [PubMed] [Google Scholar]
- 21.Roddie C, et al. , Manufacturing chimeric antigen receptor T cells: issues and challenges. Cytotherapy, 2019. 21(3): p. 327–340. [DOI] [PubMed] [Google Scholar]
- 22.Leen AM, et al. , Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood, 2013. 121(26): p. 5113–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leen AM, et al. , Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med, 2006. 12(10): p. 1160–6. [DOI] [PubMed] [Google Scholar]
- 24.Withers B, et al. , Third-Party Donor Virus-Specific T Cells Are Efficacious in the Treatment of Refractory Viral Infection Following Allogeneic HSCT, but May Not Persist Post-Infusion. Blood, 2015. 126(23). [Google Scholar]
- 25.Kessler JH, et al. , Efficient identification of novel HLA-A*0201-presented cytotoxic T lymphocyte epitopes in the widely expressed tumor antigen PRAME by proteasome-mediated digestion analysis. Journal of Experimental Medicine, 2001. 193(1): p. 73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kessler JH and Melief CJM, Identification of T-cell epitopes for cancer immunotherapy. Leukemia, 2007. 21(9): p. 1859–1874. [DOI] [PubMed] [Google Scholar]
- 27.Quintarelli C, et al. , High-avidity cytotoxic T lymphocytes specific for a new PRAME-derived peptide can target leukemic and leukemic-precursor cells. Blood, 2011. 117(12): p. 3353–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber G, et al. , Generation of multi-leukemia antigen-specific T cells to enhance the graft-versus-leukemia effect after allogeneic stem cell transplant. Leukemia, 2013. 27(7): p. 1538–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanley PJ, et al. , Functionally active virus-specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T-cell populations in cord blood and will target a range of viral epitopes. Blood, 2009. 114(9): p. 1958–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Epping MT and Bernards R, A causal role for the human tumor antigen preferentially expressed antigen of melanoma in cancer. Cancer Research, 2006. 66(22): p. 10639–10642. [DOI] [PubMed] [Google Scholar]
- 31.Toledo SRC, et al. , Insights on PRAME and osteosarcoma by means of gene expression profiling. Journal of Orthopaedic Science, 2011. 16(4): p. 458–466. [DOI] [PubMed] [Google Scholar]
- 32.Tzannou I, et al. , Off-the-Shelf Virus-Specific T Cells to Treat BK Virus, Human Herpesvirus 6, Cytomegalovirus, Epstein-Barr Virus, and Adenovirus Infections After Allogeneic Hematopoietic Stem-Cell Transplantation. Journal of Clinical Oncology, 2017. 35(31): p. 3547–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haque T, et al. , Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood, 2007. 110(4): p. 1123–1131. [DOI] [PubMed] [Google Scholar]
- 34.Keller MD and Bollard CM, Virus-specific T-cell therapies for patients with primary immune deficiency. Blood, 2020. 135(9): p. 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Reilly RJ, et al. , Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transplantation, 2016. 51(9): p. 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quintarelli C, et al. , Cytotoxic T lymphocytes directed to the preferentially expressed antigen of melanoma (PRAME) target chronic myeloid leukemia. Blood, 2008. 112(5): p. 1876–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dome JS, et al. , Advances in Wilms Tumor Treatment and Biology: Progress Through International Collaboration. Journal of Clinical Oncology, 2015. 33(27): p. 2999–U94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullen EA, et al. , Impact of Surveillance Imaging Modality on Survival After Recurrence in Patients With Favorable-Histology Wilms Tumor: A Report From the Children’s Oncology Group. Journal of Clinical Oncology, 2018. 36(34): p. 3396–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ha TC, et al. , An international strategy to determine the role of high dose therapy in recurrent Wilms’ tumour. European Journal of Cancer, 2013. 49(1): p. 194–210. [DOI] [PubMed] [Google Scholar]
- 40.Hanley PJ, et al. , CMV-specific T cells generated from naïve T cells recognize atypical epitopes and may be protective in vivo. Sci Transl Med, 2015. 7(285): p. 285ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Gal FA, et al. , Tissue homing and persistence of defined antigen-specific CD8(+) tumor-reactive T-cell clones in long-term melanoma survivors. Journal of Investigative Dermatology, 2007. 127(3): p. 622–629. [DOI] [PubMed] [Google Scholar]
- 42.Abdelaal HM, Cartwright EK, and Skinner PJ, Detection of Antigen-Specific T Cells Using In Situ MHC Tetramer Staining. International Journal of Molecular Sciences, 2019. 20(20): p. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shao HW, et al. , Identification of peptide-specific TCR genes by in vitro peptide stimulation and CDR3 length polymorphism analysis. Cancer Letters, 2015. 363(1): p. 83–91. [DOI] [PubMed] [Google Scholar]
- 44.Rego RT, Morris EC, and Lowdell MW, T-cell receptor gene-modified cells: past promises, present methodologies and future challenges. Cytotherapy, 2019. 21(3): p. 341–357. [DOI] [PubMed] [Google Scholar]
- 45.Oka Y, et al. , Wilms’ Tumor Gene 1 (WT1) Peptide Vaccine Therapy for Hematological Malignancies: From CTL Epitope Identification to Recent Progress in Clinical Studies Including a Cure-Oriented Strategy. Oncology Research and Treatment, 2017. 40(11): p. 682–690. [DOI] [PubMed] [Google Scholar]
- 46.Kawahara M, et al. , Identification of HLA class I-restricted tumor-associated antigens in adult T cell leukemia cells by mass spectrometric analysis. Experimental Hematology, 2006. 34(11): p. 1496–1504. [DOI] [PubMed] [Google Scholar]
- 47.Castellino F and Germain RN, Cooperation between CD4(+) and CD8(+) T cells: When, where, and how. Annual Review of Immunology, 2006. 24: p. 519–540. [DOI] [PubMed] [Google Scholar]
- 48.Schoenberger SP, et al. , T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature, 1998. 393(6684): p. 480–3. [DOI] [PubMed] [Google Scholar]
- 49.Maiers M, Gragert L, and Klitz W, High resolution HLA alleles and haplotypes in the US population. Human Immunology, 2006. 67(10): p. S16–S16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Identification of T-cell epitopes
(A) Peptide libraries of 15-mer peptides overlapping by 11 amino acids spanning the entire sequence of the PRAME protein consisting of 509 amino acids. The sequence of the first 2 15- mers with 11 amino acid overlap is illustrated. (B) 23 peptide pools comprising 10–12 peptides were prepared so that each 15-mer peptide was included in only two pools.
Table S1. HLA matching of third-party TAA-T products HLA types of Wilms tumor cell lines (17.94, Wit49) and partially matched TAA-T cell products are shown.