

Abstract
Background:
Late-life cognitive function is heterogeneous, ranging from no decline to severe dementia. Prior studies of cognitive trajectories have tended to focus on a single measure of global cognition or individual tests scores, rather than considering longitudinal performance on multiple tests simultaneously.
Objective:
The current study aimed to examine cognitive trajectories from two independent datasets to assess whether similar patterns might describe longitudinal cognition in the decade preceding death, as well as what participant characteristics were associated with trajectory membership.
Methods:
Data were drawn from autopsied longitudinally followed participants of two cohorts (total N=1346), community-based cohort at the University of Kentucky Alzheimer’s Disease Research Center (n=365) and National Alzheimer’s Coordinating Center (n=981). We used group-based multi-trajectory models (GBMTM) to identify cognitive trajectories over the decade before death using Mini-Mental State Exam, Logical Memory-Immediate, and Animal Naming performance. Multinomial logistic and Random Forest analyses assessed characteristics associated with trajectory groups.
Results:
GBMTM identified four similar cognitive trajectories in each dataset. In multinomial models, death age, Braak neurofibrillary tangles (NFT) stage, TDP-43, and α-synuclein were associated with declining trajectories. Random Forest results suggested the most important trajectory predictors were Braak NFT stage, cerebral atrophy, death age, and brain weight. Multiple pathologies were most common in trajectories with moderate or accelerated decline.
Conclusion:
Cognitive trajectories associated strongly with neuropathology, particularly Braak NFT stage. High frequency of multiple pathologies in trajectories with cognitive decline suggests dementia treatment and prevention efforts must consider multiple diseases simultaneously.
Keywords: Neurodegenerative disorders, dementia, neuropsychological tests, cognitive decline, trajectories
INTRODUCTION
Cognitive impairment and dementia are associated with multiple brain pathologies in elderly persons [1–3], particularly accumulation of tau neurofibrillary tangles (NFTs) with amyloid-β (Aβ) plaques, α-synuclein, and TAR-DNA binding protein 43 kDa (TDP-43) [3, 4]. Additionally, infarctions and other cerebrovascular pathologies are prevalent and deleterious for cognition [5, 6]. Although prior studies have characterized cognitive status before death related to specific neurodegenerative diseases, fewer studies have evaluated trajectories of cognitive decline in the presence of multiple pathologies [4, 7–10].
Group-based trajectory models (GBTM) are a specialized application of finite mixture modeling developed to identify longitudinal patterns and distinctive trajectories [11, 12]. GBTM allows visualization of cognitive trajectories, as well as classification of similar individuals into clinically meaningful groups [11]. Group-based multi-trajectory modelling (GBMTM), an extension of GBTM, identifies shared trajectories across multiple outcomes of interest [13] (e.g., cognitive function as measured by multiple cognitive tests). Prior studies seeking to identify distinct patterns of cognition [7, 14–17] have relied on either cognitive test scores that are examined one test at a time [14], or on a summary global cognition score derived from all tests [8]. Here, we used GBMTM to identify cognitive trajectories based simultaneously on three cognitive tests, representing global cognition, episodic memory, and category fluency.
Autopsied research volunteers from the University of Kentucky Alzheimer’s Disease Research Center (UK-ADRC), as well as a separate sample of autopsied research volunteers from various ADRCs contributing data to the National Alzheimer’s Coordinating Center (NACC) Neuropathology Data Set, were included in the current study. The National Institute on Aging funds all ADRCs. While UK-ADRC research participants were mostly recruited from the community, many ADRCs recruit from memory disorders clinics. We examined cognitive trajectories to assess whether similar patterns might describe longitudinal cognition in the decade preceding death, as well as what characteristics were associated with trajectory group membership.
Methods
Study participants (UK-ADRC)
Data were drawn from the community-based cohort study of aging and dementia at the UK-ADRC.[18] Included participants were enrolled from 1989–2017 and were ≥ age 55 years at baseline (the usual age of eligibility for this cohort is age 70 and over). Inclusion criteria were available cognitive test data (see “Neuropsychological battery test scores“), Alzheimer’s disease (AD) pathologies (Braak NFT stage, Aβ plaque rating), α-synuclein, and TDP-43 proteinopathies. We excluded participants with brain cancer, Down syndrome, frontotemporal lobar degeneration, and other rare dementia syndromes (given small numbers of cases for comparison between the datasets). FTLD cases are rare in old age, as were in the present sample, as in other community-based cohorts [19, 20].
Study participants (NACC)
Data were drawn from the NACC Uniform Data Set (UDS), and Neuropathology Data Set (NP), comprising participants enrolled at ADRCs throughout the United States (UK-ADRC data was used as a separate comparison sample). NACC maintains multicenter databases comprising standardized ADRC data protocols. Twenty-six ADRCs contributed data to both NACC UDS and NP through the September 2019 data freeze (https://www.alz.washington.edu/), when our data were extracted. To generate an independent dataset comparable to UK-ADRC, we included participants based on the same criteria as above.
Neuropsychological battery test scores
At each clinical evaluation, participants were administered a battery of cognitive tests. To study their cognitive trajectories, we included tests measuring global cognition (Mini-Mental State Examination; MMSE) [21], episodic memory (Wechsler Memory Scale-Revised [WMS-R] Logical Memory Story A) [22], and category verbal fluency (Animal Naming Test) [23] as these were consistently measured across all participants.
The MMSE is frequently used to evaluate global cognition in older adults; scores range from 0–30 [21]. Logical Memory measures the total number of story units recalled verbatim from a narrated short story; scores range from 0–25 [22]. In the Animal Naming Test, participants name as many animals as they can in 60 seconds [24]. We considered MMSE <27, Logical Memory <9, and Animal Naming <12 as abnormal scores, these scores representing 1.5 SD below the mean performance among the cognitively normal NACC participants over age 55 (n = 17,873 across 39 centers located throughout the US).
In March 2015, the NACC UDS changed to Version 3.0, wherein the MMSE was replaced by the Montreal Cognitive Assessment (MoCA) [25], and WMS-R Logical Memory IA-Immediate was replaced by Craft Story 21 Recall-Immediate [26]. NACC provides harmonized data crosswalks to researchers that bridge these scores [26]. Monsell et al. reported that the new tests (Version 3.0) were well correlated with the previous tests (Version 2.0) [26]. Hence, we used the provided conversion tables that allow scores on the new tests to be converted to equivalent scores on the previous tests. We harmonized scores for all NACC participants from March 2015 onwards. UK-ADRC continued to obtain the MMSE, and so those scores were used, while the harmonized Logical Memory scores were used.
Cognitive status
Participants were evaluated clinically for cognitive impairment at each visit [27, 28]. We used the last visit clinical diagnosis to define cognitive status of the participants as normal cognition, impaired cognition (but not MCI; presence of medical comorbidities), MCI, or dementia [29].
Neuropathological assessment
Details of neuropathological assessment at UK-ADRC [30, 31] and NACC [32] have been described previously. Aβ was considered present when the CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) ratings for Diffuse plaques or Neuritic plaques were at least sparse [33] (i.e., we dichotomized as Sparse/Moderate/Frequent vs None, because the presence of any brain amyloid is considered at least low level of Alzheimer’s disease neuropathologic change (ADNC) according to the National Institute on Aging-Alzheimer’s Association criteria [33]. Braak NFT stages were dichotomized into an indicator for high Braak NFT stage V-VI vs. I-IV. TDP-43 proteinopathy was considered present if TDP-43 inclusion bodies were detected in the hippocampus, whereas α-synuclein proteinopathy was considered present when Lewy bodies were detected in the brain stem, neocortex, or the medial temporal lobe [32, 34]. We did not use Braak staging for α-synuclein pathology because it was developed for Parkinson’s disease, which is rare in both cohorts.
Cerebrovascular pathology included atherosclerosis severity at Circle of Willis (all vessels ≥50% vs. <50% occluded); any infarcts/lacunes (yes vs. no); brain arteriolosclerosis (moderate/severe vs. none/mild).
Additionally, cerebral amyloid angiopathy was categorized as moderate/severe vs. none/mild [32]; Cerebral atrophy was classified moderate/severe vs. none/mild; Both right and left hippocampi were evaluated for hippocampal sclerosis (HS) in UK-ADRC cases; presence of HS on either side was considered as HS. For NACC, HS was considered present if right and/or left HS was reported, but not all ADRCs assess both sides of the hippocampal formation [32].
Analyses and statistical methods
All analyses were first performed for UK-ADRC data, and then the same analyses were applied to the NACC data to attempt to replicate the results. We used GBMTM [35, 36] to estimate latent trajectories in the decade before death and compared the trajectories and group membership characteristics to evaluate whether the trajectories were similar despite differences in recruitment and population characteristics.
To fit the GBMTM, we first fit separate GBTM for each test; we fit three, four, and five group models to determine the best-fitting number of trajectories. Four trajectories were selected for each of the three tests based on the Bayesian Information Criterion (BIC). An selection criterion was mean maximum posterior probability in all trajectory groups being > 0.7, meaning on average every participant assigned to a trajectory has >70% probability of membership [11]. Age at death, sex, and education were included in the GBTM to account for their influence on group membership, but neuropathology and clinical diagnoses were not included.
Once we determined the best fitting number of trajectories for each measure, we fit a single GBMTM with four latent groups. Trajectory membership is probabilistic and based on the participant’s performance on all three tests simultaneously. Each participant has an estimated probability of membership in each trajectory group, with a total probability equal to 1.0. Maximum probability assignment was used to determine membership for post-hoc analyses. Further, once optimal GBMTM models were selected, we assessed trajectory face validity by examining the longitudinal mean scores of the participants assigned to each trajectory group.
Multinomial logistic regression was used to estimate the association of demographic characteristics and neuropathology with trajectory membership, with No Decline as the reference. Adjusted odds ratios (aOR) with 95% confidence intervals (CI) were obtained from the model, which included variables of interest: age at death, sex, education, APOE ε4 (indicator for any ɛ4 alleles vs. none), and indicators for the presence of Braak NFT stage V/VI, Aβ, TDP-43, atherosclerosis, arteriolosclerosis, α-synuclein and HS. While analyzing NACC data, indicator variables for ADRC were included as a fixed effect to account for center effects.
We quantified importance of all available variables of interest (Supplementary Table 1) in explaining overall trajectory group membership. Random forest (RF) and bagging ensemble algorithm [37], which is a reliable variable selection method and produces unbiased variable importance [38] were then applied. As a sensitivity analysis, we repeated the analyses on the subgroups of participants who began the follow-up interval with normal cognition.
PROC TRAJ was used to estimate GBTM and GBMTM [35]; PROC LOGISTIC was used to fit the multinomial logistic regression (SAS:9.4®). RF was conducted using the cforest function in the “party” R package [37]. The reported results for multinomial and RF analysis are based on multiple imputation of missing neuropathological data (Supplementary Table 2). The imputation was conducted by chained random forest using imputation with predictive mean matching with 5-iterations and 100 trees. The imputation was conducted using the “missRanger” R package [39]. Complete case analysis results are available in the supplementary material. Significance level was set at 0.05.
Results
Participants’ characteristics
UK-ADRC included 365 autopsied participants (Supplementary Figure 1): mean (SD) age at death was 87.0 (8.0) years; educational attainment was 15.6 (3.0) years; median annual visit number was 9.9 (IQR: 5–14 visits); majority were female (n=228, 62.5%), and White race (n=354, 97.0%). Among autopsied NACC participants (n=981): mean age at death was 80.7 (9.6) years; education was 15.4 (3.1) years; median annual 5.0 visits (IQR: 3–7 visits); majority were male (n=527, 53.7%); and White race (n=911, 92.9%). A smaller proportion of UK-ADRC participants carried the APOE-ε4 allele (36.2% vs 45.8%) or had a dementia diagnosis at the time of death (56.2% vs 82.1%) versus NACC (Supplementary Table 1).
Cognitive trajectories
Participants in the UK-ADRC and NACC overall showed similar cognitive trajectories (Figure 1): we labeled the trajectories as “No Decline” (mean test scores remained normal during follow-up); “Mild Decline” (no decline in global cognition, slow decline in memory and fluency); “Moderate Decline” (decline from normal to abnormal global cognition, memory, and fluency); and “Accelerated Decline” (decline from abnormal to severe impairment in global cognition, memory, and fluency). Figure 1 and Supplementary Figure 2 (participants who started follow-up with normal cognition) show the observed means (dashed lines) and the estimated means (solid lines) with 95% CI for each trajectory.
Figure 1:
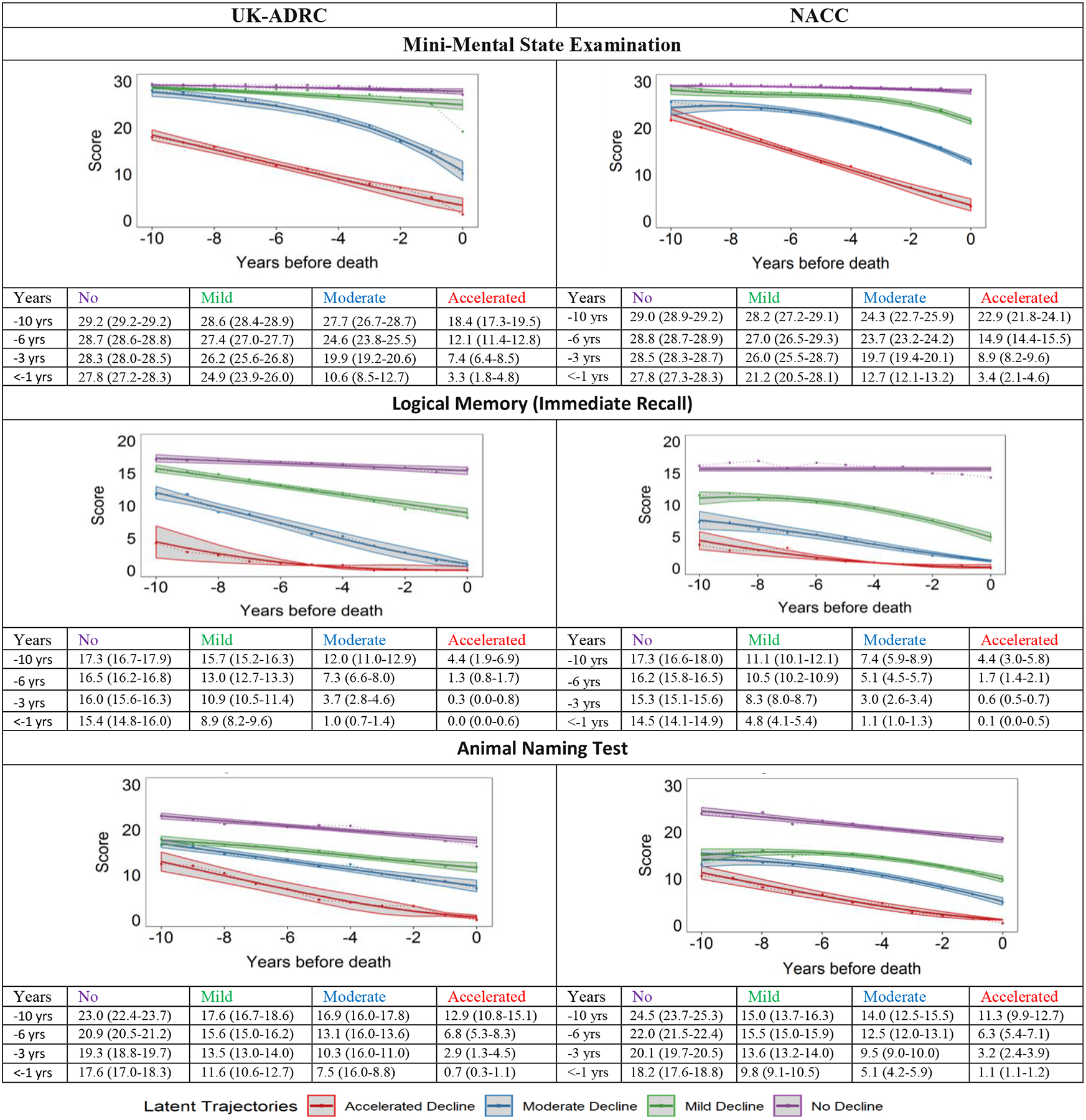
Group-based multi-trajectory modeling was used to identify end-of-life latent cognitive trajectories in autopsied UK-ADRC and NACC research volunteers. Within each study sample, there is a 1:1 correspondence of group membership in the plots.
Trajectory groups: No Decline (purple), Mild Decline (green), Moderate decline (blue) and Accelerated Decline (red).Shaded areas are 95% CI. The tables present test scores 10, 6, 3, and in < 1 year before death. UK-ADRC, University of Kentucky-Alzheimer’s Disease Center; NACC, National Alzheimer’s Coordinating Center
Cognitive trajectories in the UK-ADRC
The No Decline group, comprising 27.9% of UK-ADRC participants (Figure 1), had better mean cognitive scores throughout follow-up than the other groups across all tests. Mean MMSE scores remained relatively stable, while mean Logical Memory and Animal Naming scores showed a slight decline but remained normal throughout follow-up. The Mild Decline group (29.6%) declined marginally in the MMSE and Animal Naming trajectories, but the group was distinct from No Decline due to decreasing mean Logical Memory scores about 7–8 years before death. Moderate Decline (25.8%) started with normal mean MMSE scores but rapidly declined 6 to 7 years before death, while the Logical Memory trajectory started in the normal range and dropped to abnormal. However, Animal Naming scores were relatively better preserved. Accelerated Decline (16.7%) had abnormal scores 10 years before death. This group had low scores in all three cognitive scores, but Logical Memory scores were most affected.
Among UK-ADRC participants who started follow-up with normal cognition (n=228) (Supplementary Figure 2), trajectory patterns were slightly different. MMSE trajectory for Accelerated Decline started with >26 mean MMSE and declined rapidly about 8 years before death. However, the mean Logical Memory and Animal Naming scores at baseline were 10.5 and 16.1 respectively and declined rapidly, about 6 years before death. The Moderate Decline group also experienced decline in Logical Memory and Animal Naming scores about 6 years before death.
Table 1 presents participant characteristics by trajectories. Compared to the other groups, persons in the Accelerated Decline group on average died earlier, majority were female, diagnosed with dementia at the last visit (98.4%), and had higher proportions of APOE ε4 allele (55.7%), Braak NFT stage V/VI (90.2%), TDP-43 proteinopathy (60.9%), HS (45.9%), moderate/severe cerebral amyloid angiopathy (42.6%), and moderate/severe cerebral atrophy (67.2%). The Mild Decline and No Decline groups comparatively had a lower burden of APOE ε4 allele, proteinopathies, cerebral atrophy, and HS than the Moderate and Accelerated groups. Among the participants who began follow-up with normal cognition (n=228), those assigned to the Accelerated Decline and Moderate Decline groups were older than the No Decline and Mild Decline groups, and the burden of proteinopathies was higher (Supplementary Table 3).
Table 1:
UK-ADRC Participant Characteristics by Trajectory Group (N=365)
| Characteristics | No Decline | Mild Decline | Moderate Decline | Accelerated Decline |
|---|---|---|---|---|
| 102 (27.9) | 108 (29.6) | 94 (25.8) | 61 (16.7) | |
| Age at death, y mean (SD) | 87.1 (6.1) | 89.0 (6.7) | 87.6 (8.8) | 82.4 (9.6) |
| Female sex | 60 (58.8) | 64 (59.3) | 60 (63.8) | 44 (72.1) |
| Race (White) | 102 (100.0) | 101 (93.5) | 91 (96.8) | 60 (98.4) |
| Education, y mean (SD) | 16.7 (2.6) | 15.7 (2.8) | 15.1 (3.1) | 14.4 (3.4) |
| APOE ε4 allele (≥1 allele) | 28 (27.5) | 32 (29.6) | 38 (40.4) | 34 (55.7) |
| Dementia | 10 (9.8) | 47 (43.5) | 88 (93.6) | 60 (98.4) |
| Whole brain, weight (g) mean (SD) | 1212.1 (134.2) | 1164.9 (141.9) | 1113.7 (174.3) | 1056.1 (136.6) |
| Aβ Plaques | 81 (79.4) | 92 (85.2) | 85 (90.4) | 59 (96.7) |
| α-synuclein | 24 (23.5) | 27 (25.0) | 32 (34.0) | 29 (47.5) |
| TDP-43 inclusion bodies | 20 (19.6) | 35 (31.0) | 48 (50.0) | 39 (60.9) |
| V to VI | 15 (14.7) | 38 (36.2) | 67 (71.3) | 55 (90.2) |
| Moderate/Severe | 8 (7.8) | 20 (18.5) | 49 (52.1) | 41 (67.2) |
| Hippocampal Sclerosis | 9 (8.8) | 16 (14.8) | 37 (39.4) | 28 (45.9) |
| Moderate/severe | 15 (14.7) | 25 (23.2) | 23 (24.5) | 26 (42.6) |
| ≥ 50% Occluded | 54 (52.9) | 71 (65.7) | 56 (59.6) | 37 (60.7) |
| Moderate/Severe | 26 (25.5) | 28 (25.9) | 23 (24.5) | 13 (21.3) |
| No | 44 (43.1) | 55 (50.9) | 41 (43.6) | 24 (39.3) |
Mean (SD) or proportion as shown. SD, standard deviation; Abbreviations: UK-ADRC, University of Kentucky-Alzheimer’s Disease Research Center;. APOE, Apolipoprotein; Aβ, Amyloid-β; TDP-43, transactive response DNA binding protein 43; NFT, Neurofibrillary tangles; Missing data are reported in Supplementary Table 2.
Multinomial logistic regression estimated associations between participant characteristics and trajectory membership (Table 3). With a 5-year increase in age at death, participants were less likely to be in the Accelerated Decline group (aOR 0.68, 95% CI 0.51–0.92). Braak NFT stage V/VI was strongly associated with higher odds of belonging to the Accelerated Decline (aOR 43.95, 95% CI 12.00–163.98), Moderate Decline (aOR 17.69, 95% CI 7.69–44.10), and Mild Decline (aOR 3.58, 95% CI 1.66–7.70) group membership compared to No Decline. Presence of TDP-43 proteinopathy had higher odds of being in the Accelerated Decline group (aOR 3.52, 95% CI 1.04–12.87). While HS was significantly associated with group membership, this association was not significant in complete case analyses (Supplementary Table 6). There was no significant association of α-synuclein, atherosclerosis, or APOE ε4 with group membership.
Table 3:
Multinomial logistic regression was used to estimate adjusted odds ratios (aOR) of membership in a group with cognitive decline vs. no decline within cohorts. Results are based on models fully adjusted for all variables listed.
| Variable | Accelerated vs No | Moderate vs No | Mild vs No |
|---|---|---|---|
| UK-ADRC (n=365) | aOR (95%CI) | ||
| Age at death (5-yr increase) | 0.68 (0.51–0.92) | 1.14 (0.89–1.46) | 1.24 (0.99–1.54) |
| Sex | 1.98 (0.75–5.19) | 0.80 (0.38–1.70) | 0.71 (0.38–1.33) |
| Education | 0.71 (0.61–0.83) | 0.77 (0.68–0.88) | 0.83 (0.73–0.93) |
| APOE ε4 allele ≥1 vs 0 | 1.17 (0.47–2.94) | 0.91 (0.33–1.76) | 0.84 (0.42; 1.75) |
| (V to VI) vs (I to IV) | 43.95 (12.00–163.98) | 17.69 (7.63–44.10) | 3.58 (1.66–7.70) |
| TDP-43 Yes vs No | 3.52 (1.04–12.87) | 1.53 (0.55–4.14) | 1.51 (0.63–3.63) |
| Aβ Yes vs No | 1.10 (0.16–7.54) | 0.84 (0.28–2.53) | 1.20 (0.52–2.77) |
| α-synuclein Yes vs No | 1.61 (0.52–3.98) | 1.50 (0.67–3.15) | 1.14 (0.56–2.20) |
| >50% vs <50% Occluded | 2.03 (0.80–5.54) | 1.14 (0.66–2.75) | 1.54 (0.84–2.93) |
| Mod/Severe vs Mild/None | 0.74 (0.28–2.19) | 0.96 (0.33–1.74) | 0.98 (0.40–1.82) |
| HS Yes vs No | 8.78 (2.25–33.28) | 5.96 (1.89–20.86) | 1.34 (0.43–4.03) |
| NACC (n=981) | aOR (95%CI) | ||
| Age at death (5yr increase) | 0.57 (0.48–0.67) | 0.69 (0.60–0.79) | 0.88 (0.77–1.00) |
| Sex | 1.14 (0.65–2.02) | 0.79 (0.49–1.29) | 0.79 (0.50–1.25) |
| Education | 0.82 (0.74–0.89) | 0.81 (0.75–0.89) | 0.86 (0.80–0.92) |
| APOE ε4 allele | 1.70 (0.95–3.06) | 2.17 (1.31–3.58) | 1.55 (0.96–2.50) |
| (V to VI) vs (I to IV) | 26.18 (12.07–56.82) | 14.48 (8.38–25.02) | 3.93 (2.38–6.50) |
| TDP-43 Yes vs No | 4.32 (2.07–8.99) | 3.36 (1.75–6.44) | 2.23 (1.19–4.18) |
| Aβ Yes vs No | 0.65 (0.19–2.22) | 1.53 (0.62–3.82) | 0.96 (0.51–1.83) |
| α-synuclein Yes vs No | 2.54 (1.37–4.68) | 2.23 (1.30–3.82) | 2.52 (1.51–4.21) |
| >50% vs <50% occluded | 1.65 (0.90–3.03) | 1.02 (0.61–1.72) | 0.80 (0.50–1.30) |
| Mod/Severe vs Mild/None | 3.05 (1.69–5.49) | 1.92 (1.18–3.12) | 1.75 (1.11–2.75) |
| HS Yes vs No | 2.41 (0.95–6.16) | 2.56 (1.11–5.93) | 1.37 (0.60–3.13) |
No Decline group was the reference; Abbreviations: aOR, adjusted odds ratio; 95%CI, 95 % confidence intervals; APOE, Apolipoprotein E; Aβ, Amyloid-β; TDP, transactive response DNA binding protein; NFT, Neurofibrillary tangle; HS, Hippocampal Sclerosis.
Cognitive trajectories in NACC
On average, individuals in all NACC trajectory groups died younger (~6 years) than UK-ADRC participants. Estimated cognitive trajectories in NACC were similar in shape to those in UK-ADRC (Figure 1): No Decline (16.0%), Mild Decline (31.3%), Moderate Decline (38.3%), and Accelerated Decline (14.4%) groups, but the distribution of membership differed. In addition, estimated mean Logical Memory and Animal Naming scores were lower at the beginning of follow-up compared to the UK-ADRC participants. Participant characteristics of the NACC trajectory groups are presented in Table 2 and Supplementary Table 4 (participants starting as normal).
Table 2:
NACC Participant Characteristics by Trajectory Group (N=981)
| Variable | No Decline | Mild Decline | Moderate Decline | Accelerated Decline |
|---|---|---|---|---|
| 157(16.0) | 307(31.3) | 376 (38.3) | 141(14.4) | |
| Age at death, y, mean (SD) | 85.1 (8.3) | 82.2 (9.5) | 79.1 (9.1) | 76.9 (10.2) |
| Female sex | 69 (44.0) | 134 (43.7) | 174 (46.3) | 77 (54.6) |
| Race (White) | 147 (93.6) | 288 (93.8) | 353 (93.9) | 123 (87.2) |
| Education, y, mean (SD) | 16.5 (2.9) | 15.4 (3.1) | 15.1 (3.1) | 15.3 (3.3) |
| APOE ε4 allele (≥1 allele) | 41 (26.1) | 129 (42.0) | 215 (57.2) | 64 (45.4) |
| Dementia | 28 (17.8) | 261 (85.0) | 375 (99.7) | 141 (100.0) |
| Whole brain, weight (g) mean (SD) | 1226.4 (137.8) | 1193.6 (164.5) | 1136.3 (148.6) | 1029.2 (190.7) |
| Aβ Plaques | 129 (82.2) | 278 (90.6) | 367 (97.6) | 136 (96.5) |
| α-synuclein | 31 (19.8) | 124 (40.4) | 157 (41.8) | 68 (48.2) |
| TDP-43 inclusion bodies | 20 (12.7) | 70 (22.8) | 121 (32.2) | 52 (36.9) |
| V to VI | 33 (21.0) | 170 (55.4) | 318 (84.6) | 124 (87.9) |
| Moderate/Severe | 28 (19.1) | 99 (32.3) | 189 (50.3) | 106 (75.2) |
| Hippocampal Sclerosis | 11 (7.0) | 32 (10.4) | 73 (19.4) | 28 (19.9) |
| Moderate/Severe | 25 (16.5) | 88 (28.9) | 173 (45.3) | 70 (49.3) |
| ≥ 50% Occluded | 62 (39.5) | 105 (34.2) | 132 (35.1) | 63 (44.7) |
| Moderate/Severe | 65 (41.4) | 168 (54.7) | 214 (56.0) | 92 (65.3) |
| Yes | 57 (36.9) | 106 (34.5) | 123 (32.7) | 35 (24.8) |
Mean (SD) or proportion as shown. SD, standard deviation; Abbreviations: NACC, National Alzheimer’s Coordinating Center. APOE, Apolipoprotein E; Aβ, Amyloid-β; TDP, transactive response DNA binding protein; NFT, Neurofibrillary tangles; Missing data are reported in Supplementary Table 2.
Based on multinomial logistic regression (Table 3), a 5-year increase in age at death was associated with lower odds of Accelerated Decline (aOR 0.57, 95% CI 0.48–0.67) and Moderate Decline (aOR 0.69, 95% CI 0.60–0.79) membership versus No Decline. Braak NFT stage V/VI (aOR 26.18, 95% CI 12.07–56.82) and TDP-43 pathology (aOR 4.32, 95% CI 2.07–8.99) were associated with Accelerated Decline. The Accelerated Decline (aOR 2.54, 95% CI 1.37–4.68), Moderate Decline (aOR 3.36, 95% CI 1.75–6.44), and Mild Decline (aOR 2.23, 95% CI, 1.19–4.18) groups were associated with higher odds of having α-synuclein compared to the No Decline. Moderate/severe arteriolosclerosis was associated with higher odds of membership in the Accelerated Decline (aOR 3.05, 95% CI 1.69–5.49), Moderate Decline (aOR 1.92, 95% CI 1.18–3.12), and Mild Decline (aOR 1.75, 95% CI 1.11–2.75). Complete case analyses are presented in Supplementary Table 5.
Distribution of multiple pathologies by trajectory groups
Figure 2 shows the frequencies of AD neuropathologic change (ADNC) and comorbid brain pathologies by trajectory groups. Over 80% of UK-ADRC cohort and >86% of NACC cohort brains had ADNC pathology with at least one comorbid pathology. The Moderate Decline and Accelerated Decline groups had higher frequencies of quadruple misfolded proteins (QMP) i.e. presence of all four misfolded proteins [1], as well as the presence of TDP-43 with cerebrovascular pathologies. The presence of ≥ 2 proteinopathies was also largely accompanied by moderate/severe cerebrovascular pathologies. While among participants who began as cognitively normal (Supplementary Figure 3), the Accelerated Decline groups did not have any participants with pure quadruple misfolded proteins, but was accompanied with CVD pathologies.
Figure 2:
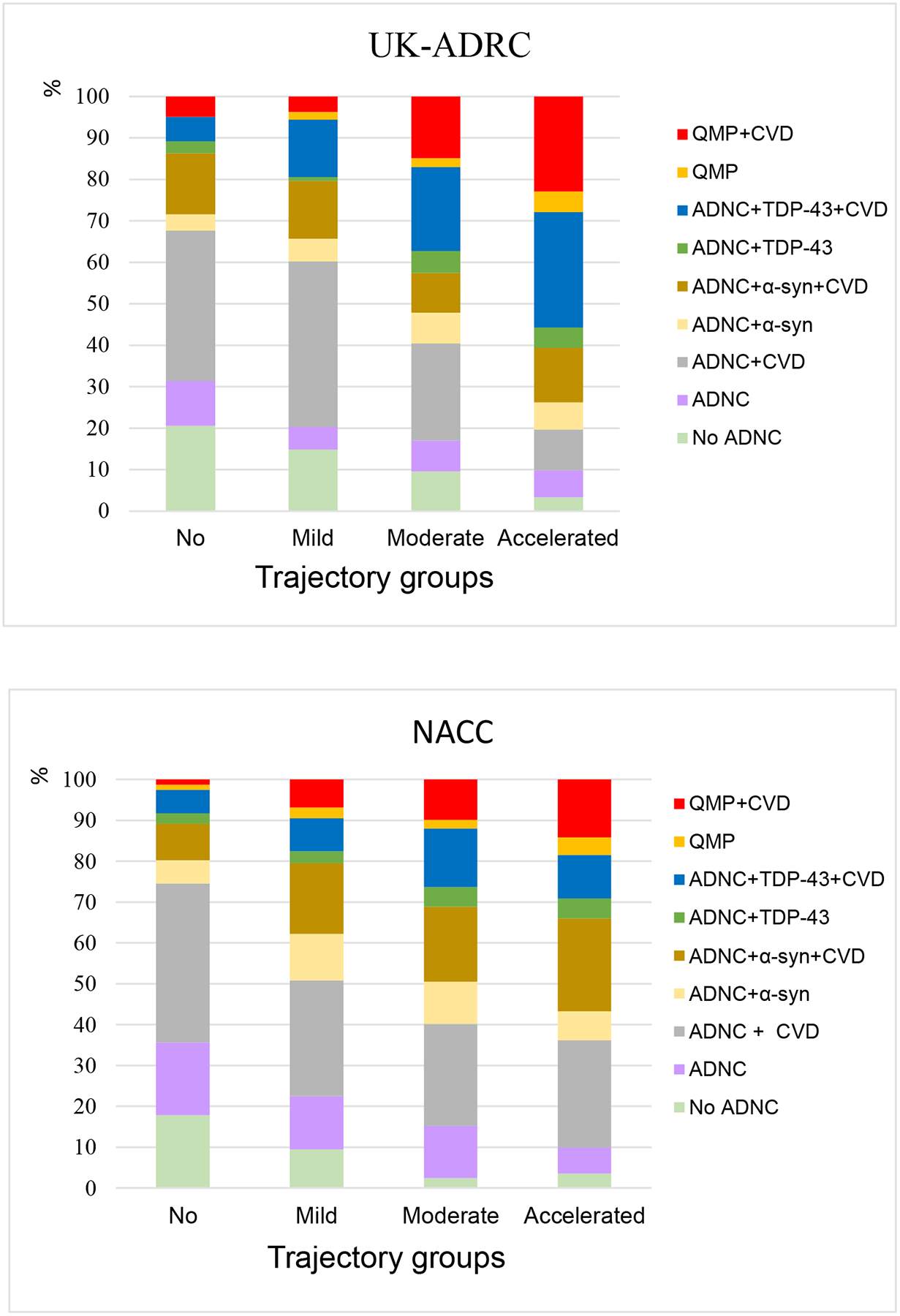
Distribution of neuropathology combinations by trajectory groups.
Abbreviations: ADNC, Alzheimer’s disease neuropathologic change; No ADNC, Tau alone or Tau +CVD or Tau +TDP-43±CVD or Tau +α-synuclein ±CVD; Aβ, Amyloid-β; TDP, transactive response DNA binding protein; QMP, quadruple misfolded proteins, α-syn, α-synuclein; CVD, presence of at least one of the three: Atherosclerosis(>50% Occluded), Arteriosclerosis (Moderate/Severe) and presence of Infarcts/Lacunes.
By contrast, 13.2% (48/365) of the UK-ADRC participants and 7.2% (71/981) of the NACC participants met criteria for ‘No ADNC’ (i.e., tau with absence of any Aβ). With one exception, these ‘No ADNC’ cases can also be classified as possible Primary Age-Related Tauopathy (PART)[40]. In the UK-ADRC cohort, 48/48 ‘No ADNC’ cases had Braak NFT stage I-IV. Of these, n=2 were in the Accelerated Decline group, n=9 in Moderate Decline, n=16 in Mild Decline, and n=21 in the No Decline. In NACC, 70/71 ‘No ADNC’ cases met criteria for possible PART. Of these, n=5 were in the Accelerated Decline group, n=9 in Moderate Decline, n=29 in Mild Decline, and n=28 in the No Decline group (Figure 2).
In the RF analysis, all 16 predictors (Supplementary Table 1) were evaluated to assess their relative importance in classifying participants into trajectory groups (Figure 3). For UK-ADRC participants, the five most important variables were Braak NFT stage, cerebral atrophy, HS, brain weight, and age at death. Similarly, for NACC cases, Braak NFT stage, age at death, cerebral atrophy, brain weight, and α-synuclein were most important.
Figure 3:
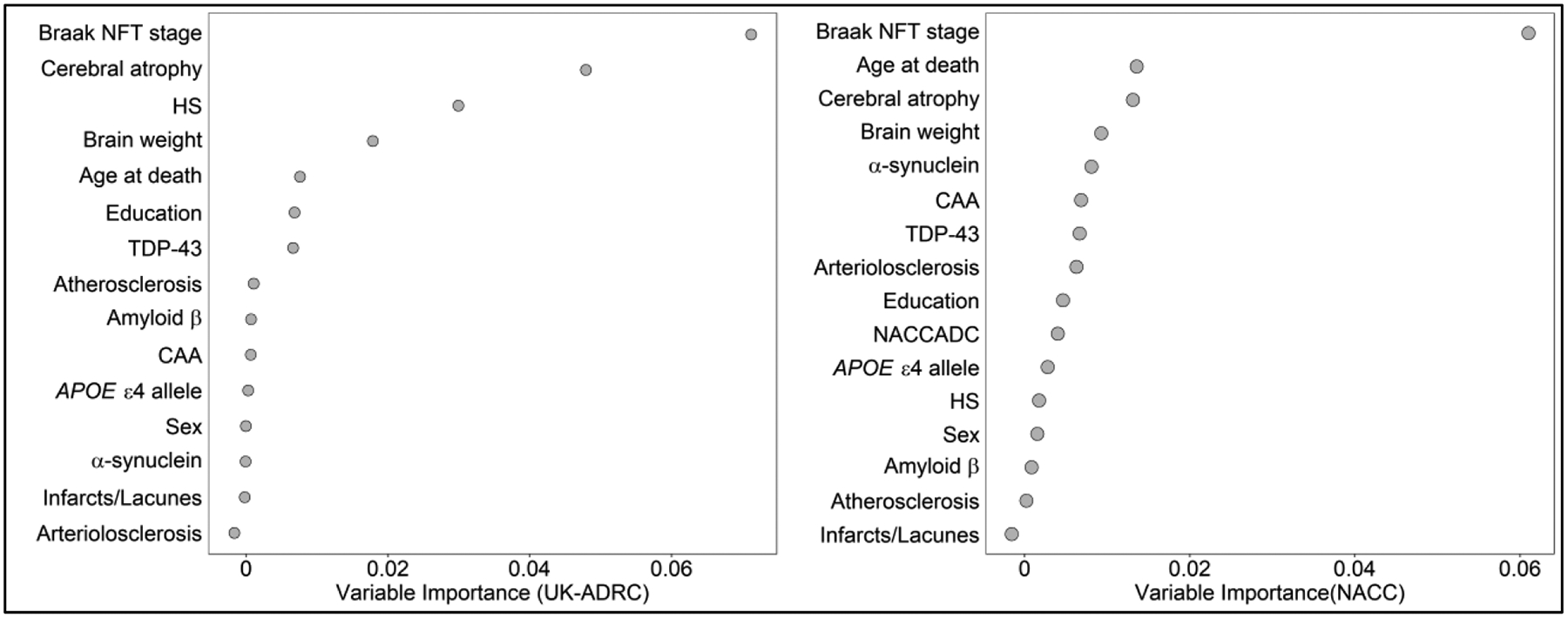
Random Forest results indicate strength of association for each variable in with overall trajectory membership within each cohort.
Variables ranked based on Mean Decrease Accuracy.
Abbreviations: NFT, Neurofibrillary tangle; APOE, Apolipoprotein E; Aβ, Amyloid-β; TDP-43, transactive response DNA binding protein; HS, Hippocampal Sclerosis; CAA, Cerebral amyloid angiopathy.
Discussion
We estimated cognitive trajectories among ADRC volunteers in their last decade of life based on longitudinal patterns of three cognitive test scores, considered simultaneously. GBMTM models identified four trajectories (we labeled as: No, Mild, Moderate, and Accelerated Decline) in both the UK-ADRC and NACC datasets. Although the NACC participants died younger and had, generally, worse cognitive status compared to the UK-ADRC participants, the trajectories during end of life, and the underlying pathologies, were quite similar.
The GBMTM approach allowed us to account for how longitudinal performance on each test was related to longitudinal performance on the other two tests. Importantly, the results have good face validity, which was assessed by mean scores in each trajectory groups (e.g., participants assigned to the No Decline group should have observed scores indicating normal cognition).
One of the strengths of the GBMTM method is the ability to characterize patterns of variation in longitudinal outcomes. In both cohorts, although the Moderate and the Accelerated Decline groups had a pronounced decline in the test scores before death, the trajectory patterns were dissimilar. Mean test scores in the Accelerated Decline group were lower at the start and showed a constant decline, and almost 100% of participants had dementia diagnoses. Accelerated Decline was associated with proportionally greater burden of proteinopathies and cerebrovascular pathologies than the other trajectory groups. The Moderate Decline trajectory scores rapidly decreased starting about 8 years before death, and >90% carried a dementia diagnosis. However, looking at the individual tests, the Logical Memory and Animal Naming scores were low a decade before death, whereas the Mild Decline group participants showed decline only in the last 4–5 years before death. These findings suggest that GBMTM models may be useful in recognizing the subpopulations of older adults that show varied patterns of cognitive performance and potentially disease burden.
Consistent with previous studies, neocortical tau proteinopathy (the pathology found in Braak NFT stages V/VI) was strongly associated with cognitive decline [31, 41]. Results from both the multinomial logistic and RF analyses emphasized the importance of Braak NFT stages in trajectory membership. However, point estimates from the multinomial model should be interpreted with caution due to the wide confidence intervals, which arose primarily due to sparse cells in the Braak NFT stages I/II/III/IV in the Accelerated and the Moderate Decline groups. Even so, we consider the association very strong. The possible PART cases (Braak NFT stages I-IV and no amyloid) were mostly in the No Decline and the Mild Decline group, emphasizing the role of high Braak NFT stages (V-VI) in cognitive decline [40, 42].
Also consistent with previous studies was the lack of a strong association between amyloid-β (in the absence of high Braak NFT stages) and cognitive trajectories [43]. Amyloid plaques were present in all trajectory groups and did not predict group membership in the multinomial analysis, and the RF analysis also showed amyloid was not important for group membership. Although the APOE ε4 carrier proportions were >40% in the Moderate and Accelerated decline groups, after controlling for the other proteinopathies there was no association with trajectory groups, except with the Moderate Decline group in the NACC cohort. The association between APOE ε4 and late-life cognitive decline appears to be mediated primarily by the relationship between APOE and ADNC, and once ADNC affects cognition, the association between APOE and cognition is diminished [44].
TDP-43 proteinopathy was prevalent in Accelerated and Moderate Decline groups and was strongly associated with group membership. TDP-43 proteinopathy has a strong association with cognitive impairment [1, 3, 45], and is associated independently with cognitive decline in the presence or absence of comorbid ADNC [4, 45]. Presence of α-synuclein proteinopathy was strongly associated with group membership among the NACC participants but not among the UK-ADRC participants, perhaps due to age differences in the two cohorts, given that participants with α-synuclein proteinopathy die at a relatively younger age [46]. Moderate/severe atherosclerosis and arteriolosclerosis were also strongly associated with the Accelerated Decline group. Furthermore, moderate/severe cerebral atrophy was proportionally higher in Accelerated and Moderate Decline and was one of the five of the most important variables in the RF analysis. The confluence of proteinopathies, age at death, cerebrovascular pathologies, cerebral atrophy, HS, and brain weight appeared to play roles in the slopes of the trajectories.
This study has several strengths. First was the availability of longitudinal follow-up with both clinical and neuropathological data. Second, we were generally able to replicate UK-ADRC results with NACC data collected from different ADRCs. Third, careful assessment of missing data and performing multiple imputation increased the validity of our findings. Additionally, we performed a sensitivity analysis in participants who started follow-up with clinically normal cognition, allowing a basis for clinical inference with respect to a presumed normal baseline. Although there were differences in the cohorts in terms of age at death, proportions of APOE ε4 allele, TDP-43, α-synuclein, cerebrovascular diseases, and hippocampal sclerosis, multiple comorbidities were prevalent in Moderate and Accelerated Decline groups from both cohorts.
The study has some limitations. There is possible misclassification of trajectory group membership due to missing data and the fact that group membership is probabilistic. Additionally, while we used three tests to measure cognition, these were selected due to data availability, but this prevented analysis by cognitive domains. Genetic data were limited to APOE genotype. In addition, our results have limited generalizability, as our data were restricted to primarily white, well-educated and autopsied participants. Future studies are needed that focus on living populations with more demographically diverse research volunteers. Finally, residual center effects may persist despite covariate adjustment for center; however, prior research has shown good to excellent agreement in neuropathologic ratings across ADRCs [33].
In conclusion, this study provides evidence that older adults follow distinct trajectories of cognitive performance during end of life. The relationship between trajectory groups and cognitive performance correlated with both number of proteinopathies and burden of cerebrovascular pathology in the brain. Despite the younger age at death of the NACC participants compared to the UK-ADRC participants, strikingly similar neuropathologic profiles featuring multiple pathologies were associated with trajectories. Thus, high burden of complex neuropathologies is not exclusively a phenomenon of extreme old age (as might be the case if we only saw these associations in the UK-ADRC data), and prevention and treatment strategies focused on a single disease may fail to decrease dementia burden in the population.
Supplementary Material
ACKNOWLEDGMENTS:
We are extremely grateful to the many research volunteers, clinicians, and other colleagues who provide and organize these data.
The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Thomas Wisniewski, MD), P30 AG013854 (PI Robert Vassar, PhD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG005131 (PI James Brewer, MD, PhD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P30 AG049638 (PI Suzanne Craft, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), P50 AG047270 (PI Stephen Strittmatter, MD, PhD).
FUNDING:
UK-ADRC is funded by grant P30 AG028383 from the NIA (PI Linda Van Eldik, PhD). This work was also partially supported by NIA grant R01 AG038651 and NICHD grant R01 HD064993.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Contributor Information
Shama D. Karanth, Department of Epidemiology, University of Kentucky, Lexington, KY 40536, USA; Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
Frederick A. Schmitt, Department of Neurology, University of Kentucky, Lexington, KY 40536, USA; Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
Peter T. Nelson, Department of Pathology, University of Kentucky, Lexington, KY 40536, USA; Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
Yuriko Katsumata, Department of Biostatistics, University of Kentucky, Lexington, KY 40536, USA; Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
Richard J. Kryscio, Department of Statistics, University of Kentucky, Lexington, KY 40536, USA; Department of Biostatistics, University of Kentucky, Lexington, KY 40536, USA; Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
David W. Fardo, Department of Biostatistics, University of Kentucky, Lexington, KY 40536, USA; Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
Jordan P. Harp, Department of Neurology, University of Kentucky, Lexington, KY 40536, USA; Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
Erin L. Abner, Department of Epidemiology, University of Kentucky, Lexington, KY 40536, USA; Department of Biostatistics, University of Kentucky, Lexington, KY 40536, USA; Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
References
- [1].Karanth S, Nelson PT, Katsumata Y, Kryscio RJ, Schmitt FA, Fardo DW, Cykowski MD, Jicha GA, Van Eldik LJ, Abner EL (2020) Prevalence and Clinical Phenotype of Quadruple Misfolded Proteins in Older Adults. JAMA Neurology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Davis DG, Poduska JW, Patel E, Mendiondo MS, Markesbery WR (2010) Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain pathology (Zurich, Switzerland) 20, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A, Hurtig HI, Siderowf A, Grossman M, McMillan CT, Miller B, Duda JE, Irwin DJ, Wolk D, Elman L, McCluskey L, Chen-Plotkin A, Weintraub D, Arnold SE, Brettschneider J, Lee VM, Trojanowski JQ (2018) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141, 2181–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wilson RS, Capuano AW, Bennett DA, Schneider JA, Boyle PA (2016) Temporal course of neurodegenerative effects on cognition in old age. Neuropsychology 30, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kapasi A, DeCarli C, Schneider JA (2017) Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 134, 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Abner EL, Nelson PT, Kryscio RJ, Schmitt FA, Fardo DW, Woltjer RL, Cairns NJ, Yu L, Dodge HH, Xiong C, Masaki K, Tyas SL, Bennett DA, Schneider JA, Arvanitakis Z (2016) Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement 12, 882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pietrzak RH, Lim YY, Ames D, Harrington K, Restrepo C, Martins RN, Rembach A, Laws SM, Masters CL, Villemagne VL, Rowe CC, Maruff P (2015) Trajectories of memory decline in preclinical Alzheimer’s disease: results from the Australian Imaging, Biomarkers and Lifestyle Flagship Study of ageing. Neurobiol Aging 36, 1231–1238. [DOI] [PubMed] [Google Scholar]
- [8].Wilson RS, Wang T, Yu L, Bennett DA, Boyle PA (2020) Normative Cognitive Decline in Old Age. Annals of Neurology 87, 816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Boyle PA, Yang J, Yu L, Leurgans SE, Capuano AW, Schneider JA, Wilson RS, Bennett DA (2017) Varied effects of age-related neuropathologies on the trajectory of late life cognitive decline. Brain 140, 804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilson RS, Leurgans SE, Boyle PA, Schneider JA, Bennett DA (2010) Neurodegenerative basis of age-related cognitive decline. Neurology 75, 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nagin DS (1999) Analyzing developmental trajectories: A semiparametric, group-based approach. Psychological Methods 4, 139–157. [DOI] [PubMed] [Google Scholar]
- [12].Shearer DM, Thomson WM, Broadbent JM, McLean R, Poulton R, Mann J (2016) High-risk glycated hemoglobin trajectories established by mid-20s: findings from a birth cohort study. BMJ Open Diabetes Research & Care 4, e000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nagin DS, Jones BL, Passos VL, Tremblay RE (2018) Group-based multi-trajectory modeling. Stat Methods Med Res 27, 2015–2023. [DOI] [PubMed] [Google Scholar]
- [14].Baker E, Iqbal E, Johnston C, Broadbent M, Shetty H, Stewart R, Howard R, Newhouse S, Khondoker M, Dobson RJB (2017) Trajectories of dementia-related cognitive decline in a large mental health records derived patient cohort. PLoS One 12, e0178562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zahodne LB, Schupf N, Brickman AM, Mayeux R, Wall MM, Stern Y, Manly JJ (2016) Dementia Risk and Protective Factors Differ in the Context of Memory Trajectory Groups. J Alzheimers Dis 52, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ding X, Charnigo RJ, Schmitt FA, Kryscio RJ, Abner EL (2019) Evaluating trajectories of episodic memory in normal cognition and mild cognitive impairment: Results from ADNI. PLoS One 14, e0212435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lai D, Xu H, Koller D, Foroud T, Gao S (2016) A MULTIVARIATE FINITE MIXTURE LATENT TRAJECTORY MODEL WITH APPLICATION TO DEMENTIA STUDIES. Journal of applied statistics 43, 2503–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schmitt FA, Nelson PT, Abner E, Scheff S, Jicha GA, Smith C, Cooper G, Mendiondo M, Danner DD, Van Eldik LJ, Caban-Holt A, Lovell MA, Kryscio RJ (2012) University of Kentucky Sanders-Brown healthy brain aging volunteers: donor characteristics, procedures and neuropathology. Curr Alzheimer Res 9, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Onyike CU, Diehl-Schmid J (2013) The epidemiology of frontotemporal dementia. International Review of Psychiatry 25, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nag S, Yu L, Boyle PA, Leurgans SE, Bennett DA, Schneider JA (2018) TDP-43 pathology in anterior temporal pole cortex in aging and Alzheimer’s disease. Acta Neuropathol Commun 6, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12, 189–198. [DOI] [PubMed] [Google Scholar]
- [22].Wechsler D (1987) Manual for the Wechsler memory scale-revised. San Antonio, TX: Psychological Corporation. [Google Scholar]
- [23].Weintraub S, Salmon D, Mercaldo N, Ferris S, Graff-Radford NR, Chui H, Cummings J, DeCarli C, Foster NL, Galasko D, Peskind E, Dietrich W, Beekly DL, Kukull WA, Morris JC (2009) The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer disease and associated disorders 23, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Morris JC, Heyman A, Mohs RC, Hughes JP, van Belle G, Fillenbaum G, Mellits ED, Clark C (1989) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 39, 1159–1165. [DOI] [PubMed] [Google Scholar]
- [25].Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H (2005) The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53, 695–699. [DOI] [PubMed] [Google Scholar]
- [26].Monsell SE, Dodge HH, Zhou XH, Bu Y, Besser LM, Mock C, Hawes SE, Kukull WA, Weintraub S (2016) Results From the NACC Uniform Data Set Neuropsychological Battery Crosswalk Study. Alzheimer Dis Assoc Disord 30, 134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Abner EL, Kryscio RJ, Cooper GE, Fardo DW, Jicha GA, Mendiondo MS, Nelson PT, Smith CD, Van Eldik LJ, Wan L, Schmitt FA (2012) Mild cognitive impairment: statistical models of transition using longitudinal clinical data. Int J Alzheimers Dis 2012, 291920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Abner EL, Nelson PT, Schmitt FA, Browning SR, Fardo DW, Wan L, Jicha GA, Cooper GE, Smith CD, Caban-Holt AM, Van Eldik LJ, Kryscio RJ (2014) Self-reported head injury and risk of late-life impairment and AD pathology in an AD center cohort. Dement Geriatr Cogn Disord 37, 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, Ramos EM, Kukull WA (2006) The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 20, 210–216. [DOI] [PubMed] [Google Scholar]
- [30].Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR (2006) Neuropathologic substrate of mild cognitive impairment. Arch Neurol 63, 38–46. [DOI] [PubMed] [Google Scholar]
- [31].Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, Abner EL, Markesbery WR (2007) Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. Journal of neuropathology and experimental neurology 66, 1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Besser LM, Kukull WA, Teylan MA, Bigio EH, Cairns NJ, Kofler JK, Montine TJ, Schneider JA, Nelson PT (2018) The Revised National Alzheimer’s Coordinating Center’s Neuropathology Form-Available Data and New Analyses. J Neuropathol Exp Neurol 77, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on A, Alzheimer’s A (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta neuropathologica 123, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, Bayston A, Beach TG, Blanc F, Bohnen N, Bonanni L, Bras J, Brundin P, Burn D, Chen-Plotkin A, Duda JE, El-Agnaf O, Feldman H, Ferman TJ, Ffytche D, Fujishiro H, Galasko D, Goldman JG, Gomperts SN, Graff-Radford NR, Honig LS, Iranzo A, Kantarci K, Kaufer D, Kukull W, Lee VMY, Leverenz JB, Lewis S, Lippa C, Lunde A, Masellis M, Masliah E, McLean P, Mollenhauer B, Montine TJ, Moreno E, Mori E, Murray M, O’Brien JT, Orimo S, Postuma RB, Ramaswamy S, Ross OA, Salmon DP, Singleton A, Taylor A, Thomas A, Tiraboschi P, Toledo JB, Trojanowski JQ, Tsuang D, Walker Z, Yamada M, Kosaka K (2017) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jones BL, Nagin DS (2007) Advances in Group-Based Trajectory Modeling and an SAS Procedure for Estimating Them. Sociological Methods & Research 35, 542–571. [Google Scholar]
- [36].Jones BL, Nagin DS, Roeder K (2001) A SAS Procedure Based on Mixture Models for Estimating Developmental Trajectories. Sociological Methods & Research 29, 374–393. [Google Scholar]
- [37].Breiman L (2001) Random forests. Machine learning 45, 5–32. [Google Scholar]
- [38].Strobl C, Boulesteix A-L, Zeileis A, Hothorn T (2007) Bias in random forest variable importance measures: Illustrations, sources and a solution. BMC Bioinformatics 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mayer M, missRanger: Fast Imputation of Missing Values., https://CRAN.R-project.org/package=missRanger,
- [40].Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, Kofler J, Kukull WA, Mackenzie IR, Masliah E, McKee A, Montine TJ, Murray ME, Neltner JH, Santa-Maria I, Seeley WW, Serrano-Pozo A, Shelanski ML, Stein T, Takao M, Thal DR, Toledo JB, Troncoso JC, Vonsattel JP, White CL 3rd, Wisniewski T, Woltjer RL, Yamada M, Nelson PT (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Tredici KD, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kövari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG (2012) Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. Journal of Neuropathology & Experimental Neurology 71, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Abner EL, Kryscio RJ, Schmitt FA, Fardo DW, Moga DC, Ighodaro ET, Jicha GA, Yu L, Dodge HH, Xiong C, Woltjer RL, Schneider JA, Cairns NJ, Bennett DA, Nelson PT (2017) Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 81, 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kövari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. Journal of neuropathology and experimental neurology 71, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yu L, Boyle P, Schneider JA, Segawa E, Wilson RS, Leurgans S, Bennett DA (2013) APOE epsilon4, Alzheimer’s disease pathology, cerebrovascular disease, and cognitive change over the years prior to death. Psychol Aging 28, 1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C, Coyle-Gilchrist ITS, Chui HC, Fardo DW, Flanagan ME, Halliday G, Hokkanen SRK, Hunter S, Jicha GA, Katsumata Y, Kawas CH, Keene CD, Kovacs GG, Kukull WA, Levey AI, Makkinejad N, Montine TJ, Murayama S, Murray ME, Nag S, Rissman RA, Seeley WW, Sperling RA, White Iii CL, Yu L, Schneider JA (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142, 1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nelson PT, Kryscio RJ, Jicha GA, Abner EL, Schmitt FA, Xu LO, Cooper G, Smith CD, Markesbery WR (2009) Relative preservation of MMSE scores in autopsy-proven dementia with Lewy bodies. Neurology 73, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.