

Abstract
Colorectal cancer (CRC) requires massive iron stores, but the complete mechanisms by which CRC modulates local iron handling and metabolically leverages iron are poorly understood. We demonstrate that the liver-derived, endocrine regulator of systemic iron balance, hepcidin, is activated ectopically in CRC. Hepcidin binds to the only known mammalian iron exporter ferroportin, resulting in degradation of ferroportin and intracellular iron trapping. Mice deficient for the hepcidin gene specifically in colon tumor epithelium exhibited significant decreases in tumor number, burden, and size compared to wild-type littermates in a sporadic model of CRC, whereas ferroportin deletion exacerbated these tumor parameters. To further understand the biochemical and metabolic utilization of iron in CRC, we subjected a three-dimensional patient-derived CRC tumor enteroid model to metabolomics and found that iron is prioritized in CRC for the production of nucleotides. These metabolomics findings were recapitulated in our hepcidin/ferroportin mouse CRC models.
Mechanistically, our data suggest that a decrease in mitochondrial function alters nucleotide synthesis following iron chelation. Restoration of nucleotide metabolism with exogenous supplementation of nucleosides led to a partial rescue of growth in patient-derived tumor enteroids and CRC cell lines in the presence of an iron chelator. Moreover, aspartate, a critical metabolite which links mitochondrial respiration and nucleotide synthesis, also partially rescued growth of iron deficient CRC cells. Collectively, these data suggest that ectopic hepcidin in the tumor epithelium establishes an axis to degrade ferroportin and sequester iron in colorectal tumors in order to maintain the nucleotide pool and sustain proliferation.
Introduction
Colorectal cancer (CRC) is the third most common type of cancer and the third leading cause of cancer-related death in the United States (1). A hallmark of CRC is the process by which these tumors deregulate cellular energetics in ways that afford a growth and survival advantage (2). Tremendous attention has been given to understanding how macromolecules, such as glucose, lipids, and proteins, are used differently by CRC cells than adjacent normal cells (3). However, macromolecule production is executed by redundant systems that offer tumors multitude of ways to evolve resistance to therapeutics that target these pathways. Micronutrients are exogenously derived, essential elements that are required by organisms in small quantities for ubiquitous cellular processes (4). Despite the fact that micronutrients are fundamental to cellular metabolism and growth promoting pathways, very little is known about the direct molecular role of micronutrients in cancer cell metabolism.
Iron is a metal micronutrient that is required by most living organisms, from single cell bacteria to complex, multicellular organisms that include humans. On the cellular level, iron orchestrates basic energy metabolism, mitochondrial function, and DNA synthesis, among other functions (5). Interestingly, epidemiological data has correlated iron levels with CRC risk. Individuals with high red meat intake, which contains large quantities of heme iron, and patients with diseases of iron overload, are at an increased risk of developing CRC (6, 7). Moreover, CRC cells sequester massive iron stores relative to adjacent normal cells (8). CRC is the only malignancy that maintains access to two sources of iron acquisition: i) systemic uptake from iron in circulation, and ii) intestinal lumen iron uptake. Accordingly, human colon tumors capitalize on this unique environment by upregulating intestinal iron import from the lumen via overexpression of the apical iron transporter, divalent metal transporter-1 (DMT1) (8, 9). Moreover, CRC patients demonstrated decreased membrane localization of the cellular iron exporter ferroportin in tumors (10). In addition to hyper-activating the many iron-dependent biochemical pathways that require iron, tumors can also use iron in unique, cancer-specific ways, such as activation of STAT3 pathways, WNT signaling, reactive oxygen species, and p53 modulation (6–8, 11, 12). These intratumoral iron stores and iron-dependent oncogenic signaling pathways are essential to CRC growth and survival, as genetic disruption of iron uptake in CRC decreases tumor number, burden, and size (8, 9).
On the systemic level, iron handling is regulated by a liver-derived, endocrine hormone, hepcidin (5). The molecular function of hepcidin is to bind to the only mammalian iron exporter, ferroportin, resulting in ferroportin internalization from the membrane, intracellular degradation, and an increase in intracellular iron concentrations (13). Ferroportin is predominately expressed on cells that regulate systemic iron handling, namely hepatocytes, intestinal enterocytes, and macrophages of the reticuloendothelial system (5). Therefore, in the presence of hepcidin, ferroportin is continually internalized from the membrane and iron mobilization into circulation is limited; in the absence of hepcidin, ferroportin is rapidly stabilized and iron is exported into plasma.
Recent studies have begun to demonstrate ectopic source of hepcidin and ferroportin that play local roles in a range of biological contexts (14–17). In the context of cancer, reports have begun to speculate whether hepcidin and/or ferroportin are utilized to modulate local iron handling inside of tumors (18, 19). Despite these data, two unaddressed paradoxes remain to be elucidated: i) the molecular mechanism by which CRC sequesters massive iron stores, and ii) the fundamental metabolic and biochemical pathways that explain why iron trapping is consistently selected for by CRC cells. We demonstrate in this work that the colon tumor epithelium produces a local hepcidin that controls tumorigenesis. Using patient-derived three-dimensional CRC enteroid models and our genetic mouse CRC models with hepcidin or ferroportin deletion demonstrate that iron deprivation dramatically alters nucleotide production. Moreover, supplementation of exogenous nucleosides can rescue the growth of two- and three-dimensional CRC models in the contexts of iron starvation. Collectively, these findings unveil the basic mechanism by which local iron handling is modulated in CRC and a central role of iron in CRC proliferation by directly maintaining nucleotide metabolism.
Results
Colon tumor epithelium produces a local source of hepcidin
CRC tumor tissue exhibited a significant increase in iron content when compared to adjacent normal tissue (Figure 1A and B). We found that the central, mammalian iron exporter, ferroportin, was undetectable in tumor tissue, but highly present in adjacent normal tissue using samples from a sporadic mouse model of CRC (Figure 1C). Tumor-specific ferroportin protein depletion suggested that this was due to local, intratumoral mechanisms and not endocrine action by circulating, liver-derived ferroportin ligand, hepcidin. Kaplan-Meier survival analysis generated from 530 human CRC biopsies showed that high levels of intratumoral hepcidin expression portend a significant decrease in overall patient survival (Figure 1D). The Cancer Genome Atlas (TCGA) revealed that hepcidin mRNA abundance (encoded by HAMP) was significantly upregulated in a subset of human cancers that include CRC (Extended Figure 1A). Further analysis of a sporadic CRC murine model found that hepcidin expression was not elevated systemically or in multiple extra-colonic tissues relative to wild-type (Extended Figure 1B and C). These findings suggest that specific cancer subsets might select for a local source of hepcidin to control intratumoral iron handling by local ferroportin degradation. An increase in local hepcidin mRNA (Hamp1, abbreviated to Hamp) in CRC was subsequently phenocopied in mouse models of both sporadic and inflammation-associated CRC (Figures 1E and F). To determine the cell type driving intratumoral hepcidin expression in CRC, we first utilized immunohistochemistry approaches and proprietary hepcidin antibodies. However, these antibodies could never be validated in our hands, using primary hepatocytes from Hampfl/fl mice treated with a Cre-expressing adenovirus in vitro or liver samples from mice treated with a low-iron diet (< 5 ppm) for 7 days, despite a significant decrease in Hamp transcript abundance (Extended Figure 2A–C). To circumvent antibody-based approaches, an epithelial enrichment strategy was assessed (Figure 1G). Epcam mRNA, a marker of epithelial cells, was higher in our epithelial fraction (Figure 1H), which confirmed the efficacy of this strategy. In three separate experiments, the Hamp transcript was highest in the tumor epithelial fraction (Figure 1H). These findings were confirmed using fluorescence-activated cell sorting in isolated stromal and epithelial compartments from sporadic CRC tumors (Figure 1I and J). These data collectively demonstrate that the colon tumor epithelium produces a local, extra-hepatic source of hepcidin that correlates with decreased ferroportin protein abundance and portends poor patient survival.
Figure 1. Colorectal cancer produces an ectopic source of epithelial hepcidin that portends decreased patient survival.
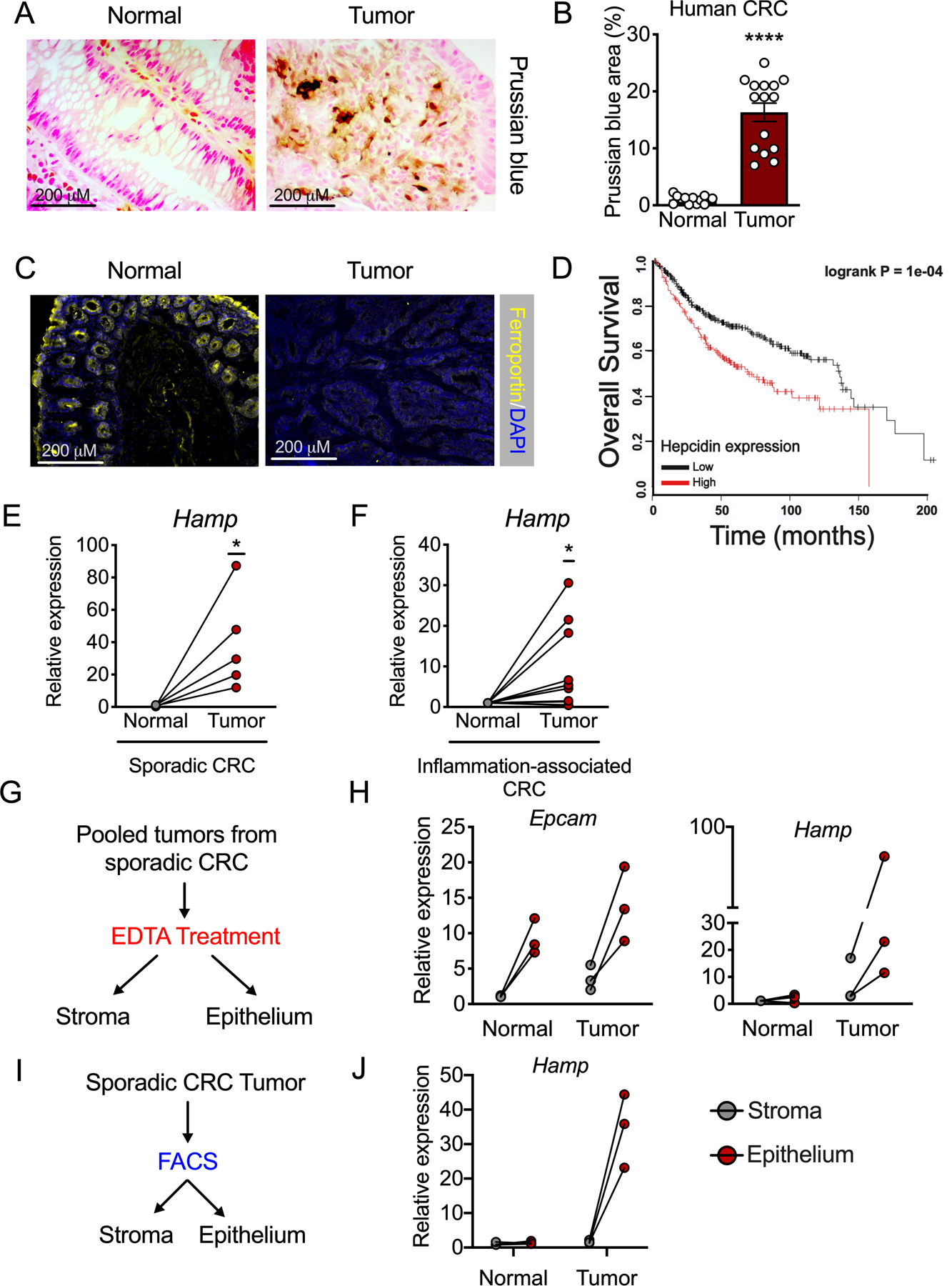
(A) Representative Prussian blue iron staining and (B) Quantification in human colorectal tumor and normal adjacent tissue (Normal N=13 and Tumor N=15 biologically independent samples, p=0.001). (C) Representative staining for ferroportin in colorectal tumor and normal adjacent tissue from a mouse model of sporadic colorectal cancer (N=4 biologically independent normal and tumor samples). (D) Kaplan-Meier survival analysis generated from 530 human CRC biopsies, stratified on high vs. low hepcidin transcript from the median. (E and F) qPCR analysis for hepcidin (Hamp) expression in normal and tumor tissue in sporadic (E) and inflammation-associated (F) mouse models of colorectal cancer (N= 5 and N=12 independent animals, respectively; p=0.046 and p=0.0316, respectively). (G) Experimental design for tumor epithelium vs. stroma enrichment. (H) qPCR analysis for Hamp in epithelium vs stroma compartments of normal and tumor tissue (N=3 independent animals with sporadic colorectal cancer). (I) Experimental design for fluorescence-activated cell sorting gated on live cells and Epcam intensity in tumor stroma (Epcam-) and epithelia (Epcam+) from a mouse model of sporadic colorectal cancer. (J) qPCR analysis for Hamp in epithelium vs stromal compartments of normal and tumor tissue (N=3 biologically independent animals with sporadic colorectal cancer). Data represent the mean ± SEM. Significance was determined by 2-tailed, unpaired (B) or paired (E and F) t test. Survival analysis significance was assessed by Cox proportional hazards regression (D). *p < .05, ****p <.0001.
A local hepcidin/ferroportin axis controls CRC
To determine the functional role of hepcidin in CRC, we deleted Hamp specifically in the colon epithelium (referred to as HampΔCE mice), in a sporadic model of colon cancer (20). This mouse model prevents cell-autonomous action of hepcidin on ferroportin (Figure 2A). Mice deficient for colon epithelial hepcidin displayed decreased tumor number, burden, and size, as compared to littermate controls with loss of APC and intact Hamp (Figure 2B–D). Importantly, the Hamp transcript was significantly elevated in wild-type tumors, a response that was abrogated in the hepcidin-deficient mice, thus confirming that the majority of intratumoral Hamp is produced by the colon tumor epithelium (Figure 2E). There was no observed change in the Ferroportin transcript (Figure 2F). However, antibody staining confirmed that ferroportin expression was markedly decreased in tumors from wild-type mice, while the decrease was blunted in hepcidin deficient mice, suggesting that ferroportin is regulated at the protein level by hepcidin (Extended Figure 2D). The hepcidin-deficient mice were found to have decreased intratumoral iron compared to wild-type mice, further implicating the role of extra-hepatic hepcidin as a critical regulator of intratumoral iron stores (Figure 2G). To investigate whether increased hepcidin expression is sufficient to increase tumorigenesis in CRC, we generated stable CT26 hepcidin overexpressing cells (Figure 2H). Subcutaneous implantation of these cells in vivo in BALB/C mice led to an increased tumor weight by as quickly as 14 days (Figure 2I). To further investigate the hepcidin/ferroportin axis in CRC, a mouse model of ferroportin deletion in the colon epithelium in a sporadic model of colon cancer was generated (referred to as FpnΔCE mice). This model traps iron within the CRC epithelium to mimic hepcidin hyper-activation in CRC (Figure 2J). Mice deficient for colon epithelial ferroportin exhibited increased tumor number, burden, and size, as compared to littermate controls with intact ferroportin (Figures 2K–M). Furthermore, iron stores in both normal and tumor tissue were significantly elevated in the ferroportin-deficient relative to wild-type littermates (Figure 2N). Collectively, these data demonstrate that local colon epithelial hepcidin/ferroportin signaling is central to local iron handling in CRC and this axis controls tumorigenesis and cancer progression.
Figure 2. Local hepcidin/ferroportin signaling is necessary and sufficient for CRC growth.
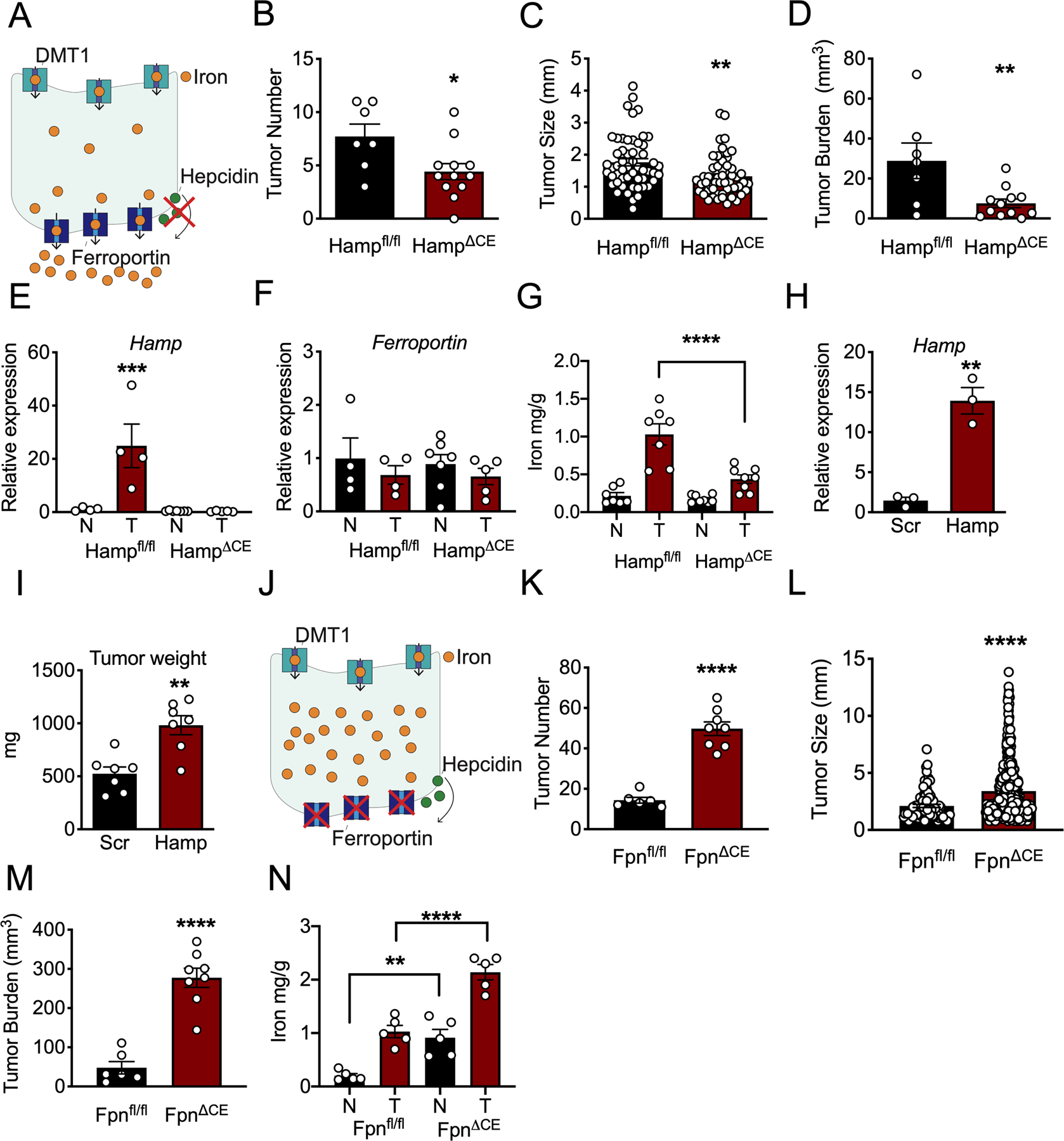
(A) Schematic of tumor iron depletion in sporadic CRC, colon epithelial hepcidin knockout model. (B-D) Tumor parameters of (B) number (Hampfl/fl N=7 and HampΔCE N=12 biologically independent animals, p=0.0237), (C) size (Hampfl/fl N=54 and HampΔCE N=56 biologically independent samples, p=.002), and (D) burden (Hampfl/fl N=7 and HampΔCE N=12 biologically independent animals, p=0.0091) in sporadic CRC, colon epithelial hepcidin (Hamp) knockout model. (E) qPCR analysis in normal and tumor tissue of mice deficient or wild-type for hepcidin hepcidin (Hamp) (p=0.006) and (F) ferroportin (Hampfl/fl N=4 biologically independent normal and tumor samples, HampΔCE N=7 biologically independent normal and 4 biologically independent tumor samples). (G) Iron quantification in normal and tumor tissue from sporadic CRC, mice deficient or wild-type for colon epithelial hepcidin (Hampfl/fl N=7 and HampΔCE N=8 biologically independent normal and tumor samples, p<0.0001). (H) Hamp expression (Scramble N=3 and Hamp N=3 biologically independent cell replicates, p=0.0018) and (I) Tumor weight (Scramble N=7 and Hamp N=7 biologically independent animals, p=0.0014) in mouse colon cancer-derived CT26 cells that stably overexpress hepcidin or a scrambled construct. (J) Schematic of tumor iron sequestration in sporadic colon cancer, colon ferroportin knockout model. (K) Tumor number (Fpnfl/fl N=6 and FpnΔCE N=8 biologically independent animals, p=0.001), (L) size (Fpnfl/fl N=84 and FpnΔCE N=398 biologically independent samples, p=0.001), and (M) burden (Fpnfl/fl N=6 and FpnΔCE N=8 biologically independent animals, p=0.001) in sporadic colon cancer, colon epithelial ferroportin (Fpn) knockout model. (N) Iron quantification in normal and tumor tissue from sporadic CRC, mice deficient or wild-type for colon epithelial ferroportin (Fpnfl/fl N=5 and FpnΔCE N=5 biologically independent normal and tumor samples, normal vs. normal p=0.0038 and tumor vs. tumor p<0.0001). N in figure represents normal adjacent tissue; T in figure represents tumor tissue. Data represent the mean ± SEM. Significance was determined by 2-tailed, unpaired t test (B-D, H-I, and K-M) or 1-way ANOVA with Turkey’s post hoc test (E-G and N). *P <.05, **P <.01, ***P <.001, ****P <.0001 comparing with each genotype or treatment group.
Tumor hypoxia via HIF-2α drives hepcidin expression in CRC
Hepatic hepcidin is canonically regulated via iron sensing machinery. However, a low-iron diet (< 5 ppm) for three months did not change Hamp expression in the colon of mice compared to an iron-replete diet (350 ppm) (Figure 3A). Mice harboring a tamoxifen-inducible Cre recombinase under the control of a colon-specific promoter (CDX2CreER) were bred to mice floxed for APC and p53 and knocked-in for a constitutively active KRASG12D construct in the ROSA26 locus, preceded by a loxP-STOP-loxP cassette (i.e. CDX2CreER;APCfl/fl;KRASG12DLSL;p53fl/fl), referred to as CDX2TripleMutant . CDX2TripleMutant mice were treated with tamoxifen and sacrificed 10 days later. Despite massive colonic dysplasia and activation of canonical oncogenic pathways in vivo, no change in colon Hamp mRNA was observed (Figure 3B). We next developed an in vitro system to investigate this mechanism further by using enteroids, which are three-dimensional intestinal “mini gut” models derived from the intestinal epithelium. Leveraging a library of human CRC biopsies of the Michigan Medicine Translational Tissue Modeling Laboratory (21), we generated a cohort of patient-derived tumor enteroids, cryopreserving a piece of the original tumor tissue at the time of biopsy to compare in vivo to in vitro biology (Figure 3C). Importantly, as was observed in the TCGA, human CRC biopsies exhibited significant activation of the HAMP transcript in tumor tissue relative to paired adjacent normal tissue (Figure 3D). However, HAMP transcript abundance was significantly reduced in vitro when tumor enteroids were generated from these CRC biopsies (i.e. comparing the primary biopsy to enteroids generated from the same tissue) (Figure 3E). Furthermore, there was no difference in HAMP expression between cohorts of enteroids generated from adjacent normal and tumor tissue in CRC (Figure 3F). Collectively, we deduced from these enteroid data that some element of the bona fide tumor microenvironment must be responsible for activating intratumoral hepcidin in CRC.
Figure 3. Hypoxia via HIF-2α activates hepcidin expression in CRC.
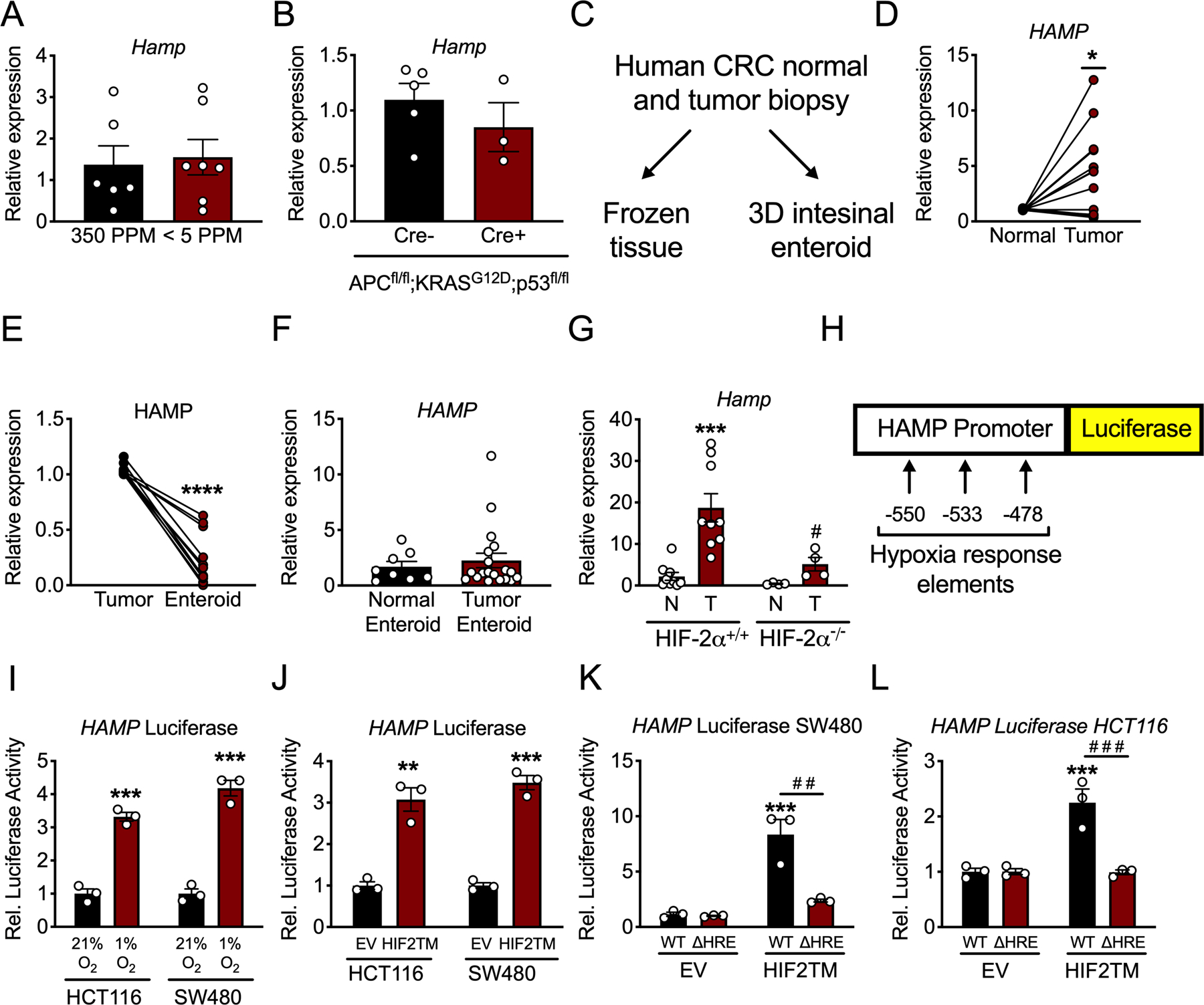
(A) qPCR analysis for hepcidin (Hamp) in the colon of mice that were on iron replete (350 PPM) or low iron (< 5 PPM) diets for three months (350 PPM N=6 and <5 PPM N=7 biologically independent animals). (B) qPCR analysis for Hamp in the colon of mice with inducible, colon epithelial deletion of APC and p53 and activation of KRAS for ten days (Cre- N=5 and Cre+ N=3 biologically independent animals). (C) Schematic for utilization of human CRC patient tumor biopsies. (D) qPCR for HAMP in paired human patient adjacent normal and CRC tumor tissue (N=13 paired, biologically independent samples, p=0.0114). (E) qPCR analysis for HAMP in in vivo tumor tissue and in vitro enteroids generated from the same biopsy tissue (N=11 paired, biologically independent samples, p<0.0001). (F) qPCR analysis for HAMP in entire library of normal and tumor patient-derived enteroids (Normal Enteroid N=8 and Tumor Enteroid N=19 biologically independent samples). (G) qPCR analysis of Hamp from a sporadic model of CRC in mice that were either wild-type for or deficient of HIF-2α (HIF-2α+/+ N=9 and HIF-2α−/− N=4 biologically independent samples from independent animals, HIF-2α+/+ normal vs tumor p=0.0006 and HIF-2α−/− normal vs tumor p=0.0108). (H) Schematic of luciferase reporter construct of 1.7 kb of the human hepcidin promoter, indicating location of hypoxia response elements (HREs). (I) Hepcidin promoter luciferase reporter activity in CRC-derived HCT116 and SW480 cells treated with either normoxia (21% oxygen)/hypoxia (1% oxygen) for 16 hours (HCT116 p=0.0003 and SW480 p=0.0003) or (J) an oxygen stable HIF-2α construct (HIF2TM) (HCT116 p=0.0021 and SW480 p=0.0002) (N=3 biologically independent cell replicates). (K and L) Hepcidin promoter luciferase reporter activity following transfection with HIF2TM as wild-type (WT) or deleted for HREs (ΔHRE) in SW480 (WT EV vs WT HIF2TM p=0.0003 and WT HIF2TM vs ΔHRE HIF2TM p=0.0012) and HCT116 cells (WT EV vs WT HIF2TM p=0.0007 and WT HIF2TM vs ΔHRE HIF2TM p=0.0006) (N=3 biologically independent cell replicates for I-L). Data represent the mean ± SEM. Significance was determined by 2-tailed, unpaired (A-B, F, I-J) or paired (D-E) t test, or by 1-way ANOVA with Tukey’s post hoc (G, K-L). *P < 0.05, ***P < 0.001, and ****P < 0.0001 comparing within a treatment group. # P < 0.05, # # P < 0.05, # # # P < 0.001 comparing between treatment groups.
A hallmark of the tumor microenvironment is hypoxia. Hypoxia promotes the activation of HIFs, which are transcription factors that consist of a heterodimer of an oxygen-sensitive α subunit (HIF-1α, HIF-2α, and HIF-3α) and a constitutively expressed β subunit (ARNT) (22). Previous studies have shown that exclusively HIF-2α, but not HIF-1α, plays an important role in CRC tumorigenesis and progression (23). Interestingly, using a sporadic model of CRC, deletion of HIF-2α in the colon epithelium exhibited significantly lower intratumoral Hamp mRNA as compared to tumors from littermates with intact HIF-2α (Figure 3G). To address whether this was a direct effect by HIF-2α, a luciferase reporter construct consisting of 1.7 kb of the human hepcidin promoter upstream to luciferase was assessed. Of note, there are three canonical hypoxia response elements (HREs) within this region of the human hepcidin promoter (Figure 3H). Incubation of CRC-derived cell lines in hypoxia (1% oxygen) for 16 hours or transfection of an oxygen stable HIF-2α significantly activated the HAMP promoter (Figure 3I and J). Moreover, truncations of the HRE in the luciferase reporter construct blunted activation by HIF-2α (Figure 3K and L). However, treatment of CRC-derived cells or enteroids generated from CDX2TripleMutant mice with FG4592, a chemical activator of HIF or overexpression of an oxygen stable HIF-2α protein in vivo, did not induce endogenous HAMP transcription (Extended Figure 3A–C). Collectively, these data suggest that HIF-2α is necessary but not sufficient to drive endogenous hepcidin in vivo.
We next considered that some additional element of the CRC environment might be necessary for HIF-2α to activate endogenous hepcidin transcription. About 80–90% of CRC patients present with mutation to APC, however, simultaneous deletion of APC and overexpression of HIF-2α in the colon epithelium (i.e. CDX2ER;APCfl/fl;HIF-2αOE) did not induce hepcidin expression (Extended Figure 3D). Furthermore, hepcidin was not modulated by inflammatory stimuli, intestinal-derived bacteria or bacterial metabolites, or changes to DNA methylation and other epigenetic factors, despite the fact that the HAMP promoter is hypomethylated in human CRC (Extended Figure 3E–J). A panel of epigenetic modifiers that included various PARP, histone demethylase, and histone deacetylase inhibitors were not able to induce hepcidin expression when co-treated with FG4592 in HCT116 cells or patient-derived enteroids (Extended Figures 3K and L). Collectively, all of these data demonstrate that HIF-2α is necessary but not sufficient to drive the endogenous hepcidin response in vivo, and that there is likely a yet to be discovered factor that enables HIF-2α sufficiency on endogenous HAMP transcription.
CRC requires iron to maintain nucleotide biogenesis
Overexpression of ferroportin in CRC-derived cells in a doxycycline inducible manner mimic an environment of low intratumoral hepcidin (i.e. unrestricted ferroportin expression). To determine the functional effect of cell-autonomous, physiological iron efflux on CRC cell growth. We attempted to generate doxycycline inducible, ferroportinGFP overexpressing cells (i.e. FPNGFP) in various human and mouse CRC-derived cell lines, as described previously (19). Interestingly and consistently, doxycycline induced FPNGFP was observed in early passages in these cells; however, this response was completely lost in subsequent “late” passages (Extended Figure 4A). This effect was not observed in two normal, non-CRC cell lines (HEK293 and IEC6), which continued to overexpress the FPNGFP construct following doxycycline treatment in late passages (Extended Figure 4B). To assess the overall effect of ferroportin-mediated iron efflux on general cell growth, we utilized our stable, doxycycline inducible normal cells, HEK293 FPNGFP and IEC6 FPNGFP cells. Doxycycline treatment decreased cell growth, a response that was completely rescued by co-treatment with recombinant hepcidin (Extended Figure 4C–H). The precise metabolic and biochemical mechanisms that cancer cells use iron to fuel cellular proliferation remain unknown. Tremendous selective pressure exists in CRC-derived cells against stable doxycycline inducible FPNGFP integration. Therefore, we investigated the role of iron in CRC by modeling a state of intracellular iron depletion in patient-derived tumor enteroids with the well-established, clinically relevant iron chelator deferoxamine (DFO). High concentrations of DFO can lead to cytotoxicity. We sought to establish a mild concentration of DFO that reduced enteroid growth and proliferation without overt cytotoxicity. CRC patient-derived tumor enteroids were treated with DFO at 10 μM and 100 μM for 7 days and compared to vehicle. The 10 μM and 100 μM doses decreased enteroid relative size by 50% and 75%, respectively, which correlated to similar reductions in enteroid proliferation, as marked by Ki67 (Figure 4A–D). CRC patient-derived tumor enteroids were subsequently treated with DFO and subjected the cultures to unbiased metabolomics to uncover metabolic pathways that are dependent on iron. Overall enteroid growth was not affected at this concentration/time point (Extended Figure 5A and B). Interestingly, pyrimidine and purine metabolism were among the most significantly altered metabolic signatures (Figure 4E–G) (Extended Figure 5C). Similar findings were also observed following doxycycline-inducible ferroportin overexpression in HEK293 FPNGFP and IEC6 FPNGFP cells, suggesting that this phenomenon is not unique to DFO administration and that iron is a conserved regulator of nucleotide metabolism in both normal and CRC cells (Extended Figure 6A–C). However, it is likely that iron-dependent nucleotide production is hyper-activated to facilitate growth and proliferation in CRC. Unbiased metabolomics of tumors derived from hepcidin- and ferroportin-deficient mice revealed significant alterations in nucleotide metabolism as well, compared to tumors from mice with intact hepcidin and ferroportin, and these changes matched what was observed in the enteroid metabolomics (Figure 4H). These data demonstrate that iron is prioritized for nucleotide synthesis in CRC in order to sustain proliferation and growth.
Figure 4. Metabolomic profiling in patient-derived tumor enteroids and mouse models reveal an essential role for iron in nucleotide production.

(A-D) Patient-derived CRC tumor enteroids were treated with DFO for 7-days at the indicated concentrations. (A-B) Bright field images (A) and quantification of enteroid size from bright field images (B) (N=3 biologically independent replicates for each treatment, 10 μM p=0.0082 and 100 μM p=0.0015). (C) Immunohistochemistry Ki67 staining (D) and quantification of Ki67+ nuclei (D) (N=3 biologically independent replicates for each treatment, 10 μM p<0.0001 and 100 μM p<0.0001). (E) Pyrimidine and purine metabolite abundance in patient-derived enteroids after 4-days of DFO treatment (10 μM) measured via metabolomics (N=3 biologically independent replicates for each treatment, Thymidine p=0.0024, Thymine p<0.0001, CMP p=0.0015, CDP p=0.0250, dAMP p=0.0274, ATP p=0.0164, Adenine p=0.0243, GMP p=0.0014, IMP p=0.0058, XMP p=0.0059, L-Glutamine p=0.0095, Carbamoyl phosphate p=0.0274, UMP p=0.0022, UDP p=0.0175, Uridine p=0.0019, Uracil p=0.0138, and AICAR p=0.0160). (F G) Schematics of where DFO-mediated iron chelation impinges on pyrimidine and purine synthesis; blue is down regulated, red is upregulated, and black is undetected or unchanged. (H) Nucleotide abundance in tumors from sporadic CRC, colon epithelial hepcidin or ferroportin knockout model deficient or wild-type for colon epithelial hepcidin or ferroportin (Hamp+/+, HampΔCE, Fpn+/+, and FpnΔCE) (Hampfl/fl N=7, HampΔCE N=8, Fpnfl/fl N=5, and FpnΔCE N=5 biologically independent samples, [Hamp] Thymidine p=0.0317, Deoxycytidine p<0.0001, Cytosine p<0.0001, Cytidine 0.0001, pUracil 0..0001, Adenosine p=0.0189, Xantine p=0.0015, Hypoxanthine p==.0010, Guanine p=0.0410, Inosine p<0.0001, and Orotate p=0.0053; [Fpn] Thymine p<0.0001, Deoxycytidine p<0.0001, Cytosine p= 0.0067, Cytidine p<0.0001, Deoxyuridine p=0.0301, Adenosine p<0.0001, Adenine p<0.0001, Guanine p<0.0001, Uridine 5-monophosphate p=0.0075, Uridine p<0.0001, Uracil p<0.0001, Guanosine p<0.0001, Inosine p=.0001, Orotate P<0.0001.). Data represent the mean ± SEM. Significance was determined by 2-tailed, unpaired t test (E and H) or by 1-way ANOVA with Tukey’s post hoc (B and D). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.001 comparing treatment groups. # P < 0.05 is specific for metabolomics changes in tumors from the ferroportin (Fpn) knockout model.
Nucleosides partially rescue CRC growth in presence of DFO
We next analyzed CRISPR gene essentiality data from the Dependency Map (DepMap) to assess the role of intracellular iron in cancer cell proliferation. Knockout of the hepcidin gene HAMP is expected to deplete intracellular iron, but its basal expression is low in most cancer cell lines, including CRC, which is consistent with our observation that HAMP is only induced ectopically in the bona fide in vivo tumor microenvironment. Therefore, we examined knockout of transferrin receptor (encoded by the gene TFRC), which is essential for in vitro iron uptake (24). TFRC is strongly expressed and is essential in the majority of DepMap cancer cell lines (25). Remarkably, we found that the essentiality of TFRC was most highly correlated with nucleotide metabolism genes, followed by genes involved in pathways such as heme synthesis, cholesterol synthesis, the TCA cycle, and transferrin endocytosis, which is known to rely on iron and transferrin (Supplementary Table 1 and 2). This correlation was strongest for genes involved in biosynthesis of purine and pyrimidine bases (Figure 5A). This reveals a tremendously conserved role for iron in nucleotide production across hundreds of discrete cancer cell lines and types.
Figure 5. Nucleoside supplementation restores growth of two- and three-dimensional colorectal cancer models in the presence of DFO.
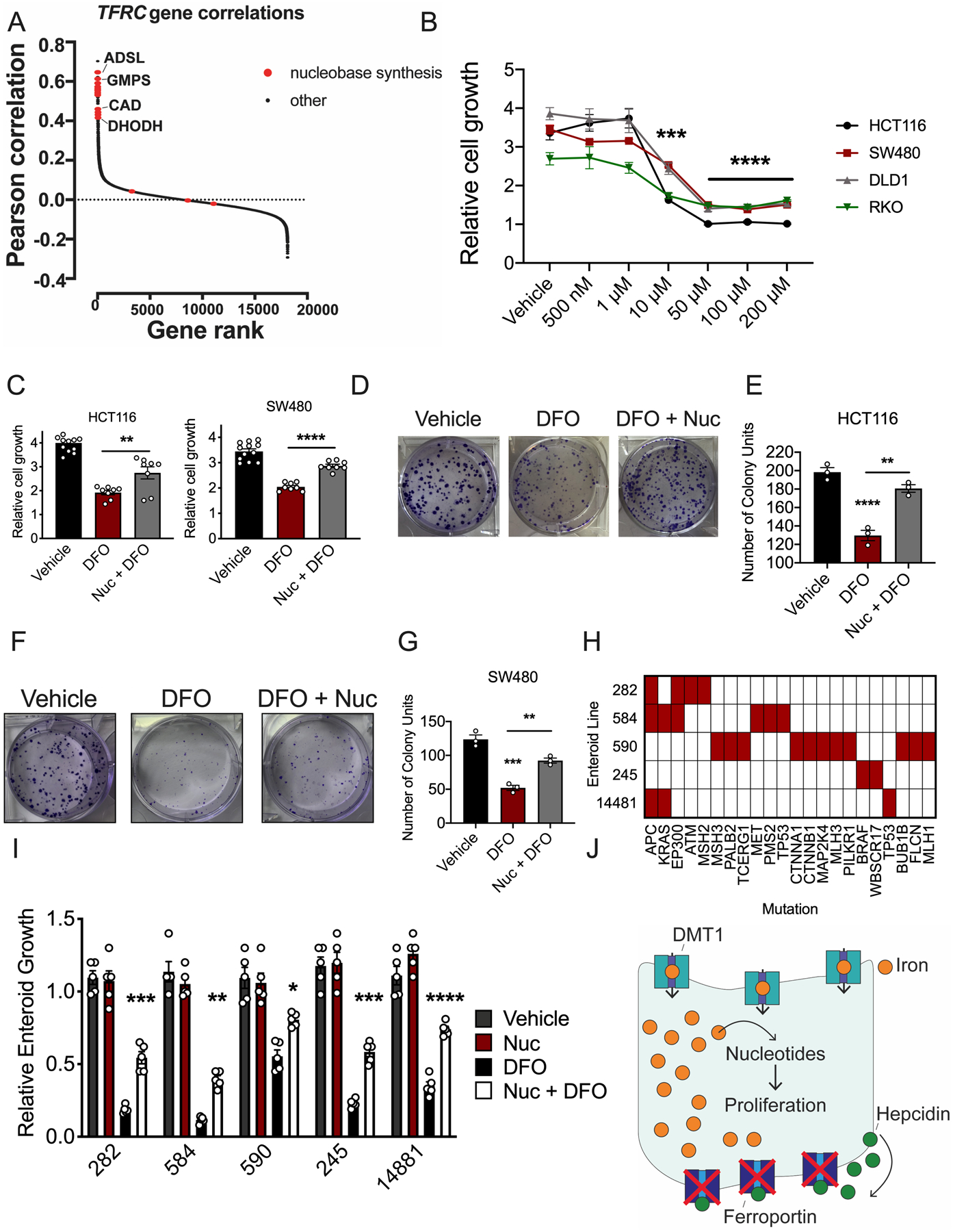
(A) Pearson correlations between essentiality scores in Dependency Map of each gene with TFRC, ranked from highest to lowest. Nucleobase synthesis genes are highlighted in red. (B) DFO dose response in human colorectal cancer (CRC)-derived cell lines 72 hours after treatment, using MTT (Vehicle N=6 and DFO N=4 biologically independent replicates in each cell line, 10μM all cell lines p=0.001, 50μM all cell lines p<0.0001, 100 μM all cell lines p 0.0001, 200μM all cell lines p<0.0001). (C) Nucleoside (100 μM) rescue of DFO (10 μM) growth inhibition in CRC-derived cell lines after 72 hours of co-treatment using MTT (Vehicle N=12 and DFO=8 biologically independent replicates in each cell line, HCT116 p=0.0055 and SW480 p<0.0001. (D) Representative crystal violet staining images (E) and quantification of HCT116 cells pretreated with vehicle or DFO (10 μM) and then treated with vehicle or a nucleoside cocktail for ten days (100 μM) (N=3 biologically independent cell replicates, Vehicle vs DFO p<0.0001, DFO vs Nuc + DFO p=0.0055). (F) Representative crystal violet staining images (G) and quantification of SW480 cells pretreated with vehicle or DFO (10 μM) and then treated with vehicle or a nucleoside cocktail for ten days (100 μM) (N=3 biologically independent cell replicates, Vehicle vs DFO p=0.0001, DFO vs Nuc + DFO p=0.0026). (H) Mutational landscape in small library of patient-derived tumor enteroids. (I) Nucleoside (100 μM) rescue of DFO (10 μM) growth inhibition in patient-derived tumor enteroids 7-days after treatment (N=5 biologically independent replicates in each treatment group, Enteroid 282 p=0.0004, Enteroid 584 p=0.0016, Enteroid 590 p=0.0234, Enteroid 245 p=0.0007, Enteroid 14881 p<0.0001. (J) Schematic of intratumoral iron being utilized for nucleoside production. Data represent the mean ± SEM. Significance was determined by 1-way ANOVA with Tukey’s post hoc (B-C, E, G, I). *P < 0.05, **P < 0.01, ***P < 0.001, ***P < 0.001, and ****P < 0.0001 comparing within cells lines or across treatment groups.
To demonstrate if maintaining nucleotide production is central to iron utilization in CRC, two-dimensional and three-dimensional CRC models were assessed. A DFO dose response in CRC-derived cell lines revealed that treatment with 10 μM DFO lead to an average reduction in cell growth of 50% in all four CRC cell lines assessed (Figure 5B). Importantly, 10 μM DFO was not strong enough to chelate iron from the ubiquitous, intracellular iron storage protein, ferritin, which further suggests this concentration is moderate and does not generate overt cytotoxicity (Extended Figure 7A and B). We therefore sought to rescue DFO-mediated growth inhibition by nucleoside supplementation. Single nucleoside, purine or pyrimidine supplementation alone was insufficient to rescue DFO-mediated growth inhibition (Extended Figure 7C and D). However, supplementation with a cocktail of all four nucleosides partially rescued DFO-mediated growth inhibition in CRC-derived cell lines (Figure 5C–G) (Extended Figure 7F–I). This response was also conserved following ferroportin-mediated iron efflux in IEC6 FPNGFP cells (Extended Figure 7E). To further confirm these observations, we set up a similar rescue experiment in a small library of five CRC patient-derived tumor enteroids. Sequencing revealed that each of the patient tumors used to produce this library were comprised of unique mutational profiles (Figure 5H) (21). Intriguingly, nucleoside supplementation partially rescued DFO-mediated growth inhibition in all five patient-derived enteroids (Figure 5I). These data suggest a conserved role for iron in nucleotide production, irrespective of oncogenic signature or mutational landscape. In total, these data confirm that cancer cells in CRC depend on iron for the production of nucleotides to sustain growth and that restoration of nucleotide metabolism is sufficient to overcome the growth inhibitory effect of DFO-mediated iron depletion (Figure 5J).
Iron depletion alters mitochondrial metabolism and function
Mitochondria are integral in generating critical metabolites used for nucleotide synthesis (26). To assess the impact of iron depletion on mitochondrial function, we measured the oxygen consumption rate (OCR) of mitochondria in iron depleted states via Seahorse. To ensure the OCR represented respiration, we standardized our results to cell number and conducted our analysis at a time point prior to any observable DFO-mediated growth inhibition (Extended Figure 8A). In multiple CRC cell lines, mitochondrial OCR was reduced by DFO-mediated iron chelation (Figure 6A and B) (Extended Figures 8B and C). Moreover, at this early time point, a decrease in NAD+/NADH ratio was observed in iron-deprived cells (Figure 6C–D). The electron transport chain is central to maintaining nucleotide pools via aspartate-dependent nucleotide synthesis. Recent work has demonstrated that cancer cells rely on mitochondrial respiration to sustain proliferation primarily via synthesis of aspartate and subsequent nucleotide production (27–29). Interestingly, metabolomics found a decreased abundance of aspartate in DFO-treated CRC cells patient-derived enteroids, and hepcidin-deficient tumors (Figure 6E–H). Conversely, aspartate concentrations were increased in iron-rich, ferroportin-deficient tumors (Figure 6I). Addition of aspartate was able to partially rescue cells from DFO-mediated growth inhibition (Figure 6J–K). To test the importance of maintaining NAD+/NADH ratio in DFO-mediated growth inhibition, we overexpressed cytosolic or mitochondrial (Mito) NADH-oxidase from Lactobacillus (LbNox), which are not iron-sulfur (Fe-S) clusters (Extended Figure 8D and E). While increased mitochondrial LbNOX expression increased growth relative to vehicle treatment, increased expression was not able to rescue growth inhibition from iron deprivation (Extended Figure 8H and I). NDI1 is a yeast equivalent of mammalian electron transport chain complex I. NDI1 was overexpressed to investigate whether DFO-mediated growth arrest occurs via disruption to Fe-S clusters subunits in complex I. NDI1 was also not able to rescue cells from DFO-mediated growth inhibition but did rescue cells from the complex I inhibitor, phenformin (Extended Figure 8H and I). To evaluate other electron transport chain complexes, a cocktail mix of several antibodies which detect central proteins in electron transport chain were assessed. Western blot analysis showed decreased expression of succinate dehydrogenase (SDH), a Fe-S cluster protein within complex II, which was further confirmed using an additional antibody for SDHB. (Figure 6L and M) (Extended Figure 8J and K). In addition, ferrochelatase was decreased, which is a Fe-S protein critical in heme synthesis (Figure 6L and M) (Extended Figure 8J and K). This suggests that iron chelation may impact mitochondrial respiration at multiple nodes. Taken together, our data demonstrates that iron is central for mitochondrial respiration, downstream nucleotide synthesis, and subsequently CRC cell growth.
Figure 6. DFO-mediated iron depletion alters mitochondrial metabolism.
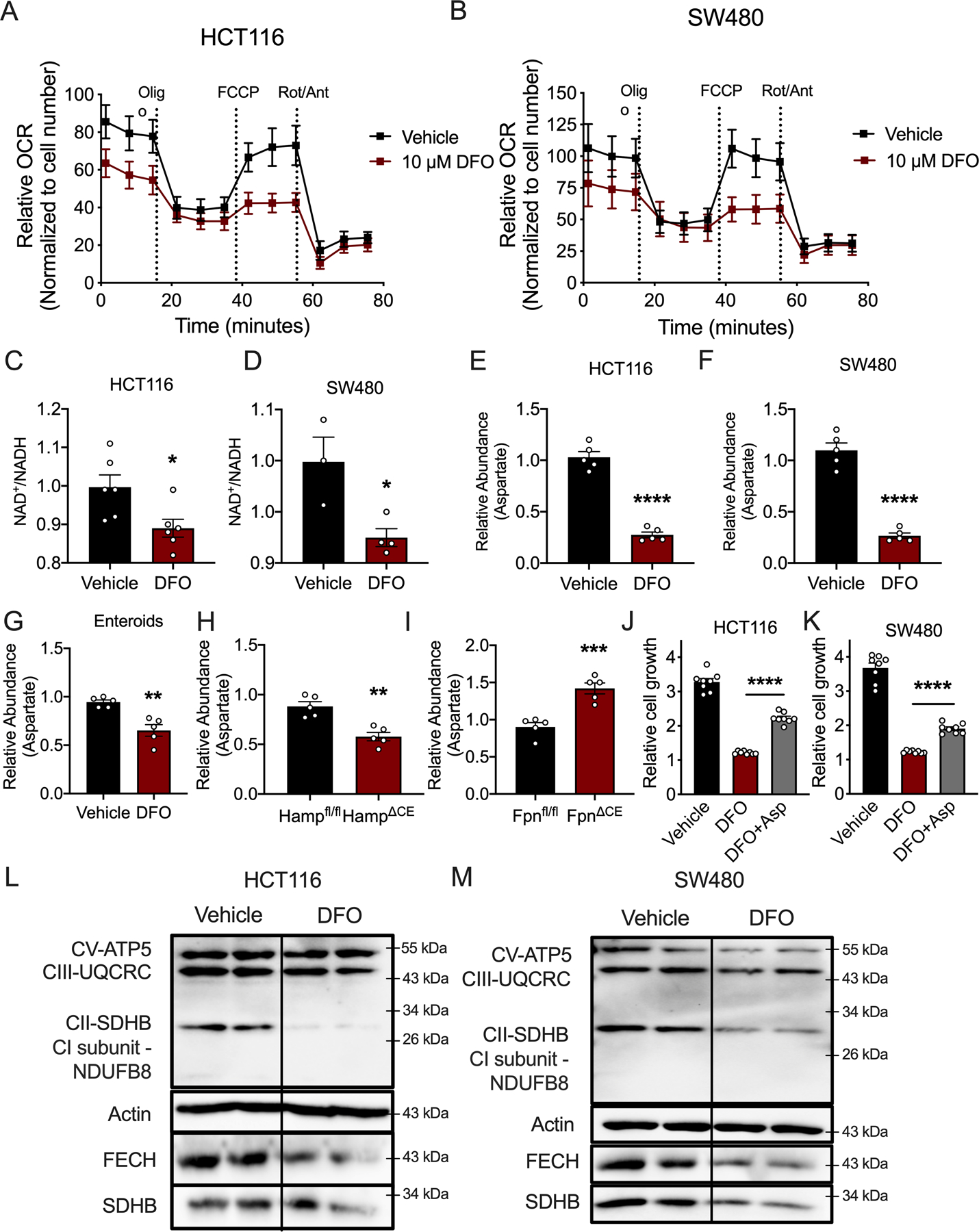
(A) Seahorse analysis of mitochondrial metabolism in HCT116 and (B) SW480 cells 24 hours after DFO administration (10μM) (N=4 biologically independent cell replicates for each treatment condition). (C) NAD+/NADH ratio in HCT116 (N=6 biologically independent cell replicates, p=0.0218) and (D) SW480 (Vehicle N=3 and DFO N=4 biologically independent cell replicates, p=0.0222) treated with DFO (10μM). (E) Aspartate levels in HCT116 cells (N=5 biologically independent cell replicates, p<0.0001) and (F) SW480 cells (N=5 biologically independent cell replicates, p<0.0001) treated with DFO (10μM). (G) Aspartate abundance in patient-derived enteroids treated with DFO (100μM for 72 hours) (N=5 biologically independent replicates, p=0.0019) and (H) sporadic tumors from wild-type or colon-specific hepcidin-deficient mice (Hamp) (N=5 biologically independent samples from independent animals, p=0.0014), and (I) sporadic tumors from wild-type or colon-specific ferroportin-deficient mice (Fpn) (N=5 biologically independent samples from independent animals, p=0.0006). (J) Cell growth in HCT116 cells (N=8 biologically independent cell replicates, p<0.0001) and (K) SW480 cells (N=8 biologically independent cell replicates, p<0.0001) following DFO treatment (10μM) with and without aspartate supplementation (100μM). (L and M) Western blot analysis of mitochondrial enzymes following 24-hours of DFO (10μM) treatment in HCT116 cells (L) and SW480 cells (M). Data represent the mean ± SEM. Significance was determined by 2-tailed, unpaired t test (C-I) or one-way ANOVA followed by Tukey’s post hoc (J and K). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared to vehicle or between genotypes.
Discussion
The present work demonstrates that CRC modulates local, oncogenic iron handling by ectopically expressing the liver-derived, iron-regulatory hormone, hepcidin, in order to downregulate ferroportin. Using a genetic mouse model of sporadic CRC, and a syngeneic model of CRC, we show that CRC-derived hepcidin is necessary and sufficient to drive tumor number, size, and burden. Using unbiased metabolomics on three-dimensional patient-derived CRC enteroids, we unveiled that nucleotide production is central to iron utilization in CRC, and these findings were recapitulated in mouse CRC models of hepcidin or ferroportin deletion. Furthermore, supplementation with exogenous nucleosides is sufficient to rescue growth inhibition caused by DFO-mediated iron chelation, in both two-dimensional and three-dimensional models. In combination with previous studies, this current work produces a model in which HIF-2α is utilized by CRC cells to simultaneously increase intestinal lumen iron uptake via DMT1 overexpression and to decrease iron export by activating autocrine/paracrine hepcidin-mediated ferroportin degradation. Following massive iron sequestration, CRC drives proliferation by prioritizing iron-dependent mitochondrial respiration and downstream nucleotide biosynthesis.
Reports have begun to reveal extra-hepatic, functional sources of hepcidin that act in paracrine and/or autocrine fashions on local ferroportin. This paradigm was catalyzed by reports showing that ferroportin is expressed on cardiomyocytes and it is primarily regulated by a local, cell-autonomous source of hepcidin that maintains mitochondrial activity and contractile function (16). Since then, local, physiological hepcidin/ferroportin signaling has also been described in dendritic cells of the intestinal microenvironment, the epidermis, and pulmonary arterial smooth muscle cells (14, 15, 17). In cancer, a similar model has been proposed, whereby ferroportin protein abundance is reduced in breast cancer cells compared to nonmalignant breast epithelial cells (18). Several additional reports have shown that both breast and prostate cancers can activate their own source of local hepcidin, while investigators have observed increases in hepcidin mRNA and protein in human CRC (30–32). However, these investigations of cancer hepcidin/ferroportin kinetics are mostly correlative and have been limited by a lack of genetic and functional data. Our present work definitively shows in vivo that ectopic hepcidin exerts a functional role in CRC iron trapping, tumorigenesis, growth, and survival. This response appears to be a purely pathological phenomenon because hepcidin was only activated inside bona fide colon tumors and not normal tissue or in vitro cell models, even in contexts of activation of pathways that were shown to drive intraturmoal hepcidin. It remains peculiar as to how in vitro models grow without a functional hepcidin/ferroportin axis to retain iron. Although more studies are needed, the availability of iron could be an important distinguishing factor between in vitro and in vivo tumors. In vivo, the amount of available iron is scarce and the tumor epithelium is competing with other host cells and also with a large diverse microbiome in the lumen.
Another unanswered question in the field of extra-hepatic hepcidin/ferroportin signaling is the transcriptional regulation of hepcidin in non-hepatocytes at large. Reports have implicated a range of intratumoral molecules, including BMP4/7, IL6, Wnt, and GDF15, however, none of these findings have been functionally verified in vivo (30, 31, 33). A hallmark of the tumor microenvironment is hypoxia, as tumors outgrow their blood supply and are challenged by increased metabolic demand and inflammation (22). Interestingly, reports have shown that hypoxia can activate cardiac hepcidin mRNA in rats following 24 hours exposure to 6% oxygen (34). Furthermore, although systemic hypoxia inhibits hepatocyte hepcidin, reports have shown that this mechanism is indirect via erythropoietin action on hepatocytes and that hypoxia signaling actually activates hepcidin in erythropoietin-deficient mice (35). HIF-2α, maintains essential roles in both iron metabolism and CRC progression (8). Using a luciferase reporter construct and mouse models of CRC, the present work demonstrates that HIF-2α is necessary to activate the human hepcidin promoter in vitro and in vivo, and is sufficient in vitro. However, HIF-2α is not sufficient to drive transcription of the endogenous hepcidin gene in CRC-derived cell lines, three-dimensional intestinal mini gut enteroid models, or in mice that overexpress colon epithelial HIF-2α. These findings suggest that there is a cell non-autonomous molecule that is necessary for HIF-2α-mediated activation of hepcidin in CRC. Future work will need to elucidate the complete factor(s) that mediate HIF-2α-mediated transcription of endogenous hepcidin through detailed promoter and epigenetic analysis of hepcidin.
While the growth-promoting role for iron in cancer has been known for years, the precise metabolic and biochemical mechanisms that are used by cancer cells to sustain proliferation have remained elusive. Nucleotides are central to cell growth and replication in both physiological and pathological contexts (14, 36, 37). Central to cancer metabolism is the maintenance of nucleotide levels in order to sustain proliferation, and many canonical oncogenes have been shown to fuel nucleotide synthesis in tumor cells, including those that seem to be responsive to iron (8, 12, 36, 38). However, contrary to a role for iron in the direct regulation of oncogenic proteins that has been previously described, we demonstrate in this work that iron utilization in CRC is prioritized for the direct production of nucleotides. Indeed, we find that across hundreds of cancer cell lines in the Dependency Map the essentiality of de novo nucleotide synthesis genes is highly correlated with essentiality of the transferrin receptor, which mediates iron import. We came to a similar conclusion of a conserved role of iron in nucleotide production with data from our internal library of patient-derived tumor enteroids, all of which depended on iron for nucleotide production and proliferation, despite unique mutational landscapes that are known to drive proliferation. These results solidify that the main metabolic role of iron is in support of nucleotide production for proliferation in cancer.
Importantly, restoration of nucleotide levels is sufficient to overcome the growth inhibitory effect of DFO-mediated iron chelation in two- and three-dimensional models of CRC. This sufficiency illuminates a fundamental role for iron in nucleotide production, not oncogenic protein signaling, as CRC growth is restored despite sustained DFO-mediated inhibition of iron-dependent signaling. These data further suggest that the cancers have evolved roles for iron in regulating oncogenic pathways, such as WNT, p53 and STAT3, might be a secondary event to iron-trapping for nucleotide metabolism (6, 8, 12). Oncogenic proteins are regulated by a number of factors, many of which are mutated in cancer to establish constitutive activation. It is therefore possible that CRC first sequesters iron to maintain the nucleotide pool for proliferation; subsequently, CRC might evolve iron-dependent signaling functions with large intracellular iron levels onboard. Future work will need to tease out the order of these events, the mechanism by which oncogenic proteins evolve to use iron in cancer, and whether the two phenomena are integrated.
Together with this present work, evidence is accumulating to elucidate the mechanism by which iron controls cell proliferation. It has been previously observed that iron chelation results in G1/S phase arrest, the point in the cell cycle when the nucleotide pool mediates duplication of each of the chromosomes in the cell (7). Furthermore, several reports have shown a role for iron in the regulation of the cell cycle family of proteins, CDKs, including in CRC (8, 39). Ribonucleotide reductase is the rate-limiting enzyme involved in the conversion of ribonucleotides into deoxyribonucleotides. Interestingly, the activity of ribonucleotide reductase is dependent on iron since one of its subunits contains a tyrosyl radical that requires iron for its stabilization (40). However, since nucleoside supplementation can partially rescue growth in the presence of DFO, other mechanisms must be mediating growth arrest following a decrease in intratumoral iron. We demonstrate that indeed mitochondrial respiration is lower and electron transport chain is dysregulated following iron chelation. Moreover, aspartate, which is a critical amino acid that integrates mitochondrial respiration, nucleotide synthesis, and cell growth, can partially rescue the growth defects following DFO. SDHB and ferrochelatase were also decreased, which play important roles in nucleotide biosynthesis via the electron transport chain and heme synthesis, respectively. Several Fe-S containing enzymes are directly linked to nucleotide biosynthesis and thus could play a critical role in iron-dependent decrease in nucleotides (41). SDHB and ferrochelatase are Fe-S cluster proteins, which suggests a decrease in Fe-S cluster biogenesis. Future work will need to determine the complete iron-dependent enzymatic map by which nucleotide production is altered.
In conclusion, this work demonstrates that the colon tumor epithelium produces an ectopic, extra-hepatic source of local hepcidin that is necessary and sufficient for CRC growth, and is regulated by HIF-2α. Nucleotide metabolism is central to iron utilization in CRC, as intratumoral iron is utilized to maintain mitochondrial respiration in support of nucleotide synthesis and subsequently CRC proliferation. These data demonstrate the molecular mechanisms by which colon tumors modulate local iron handling, metabolically prioritize iron utilization, and reveal overall vulnerabilities that can be targeted for therapeutic benefit in CRC.
Methods
Animals
For embryonic, colon epithelial-specific disruption of hepcidin, mice floxed for Hamp1 (Hampfl/fl) were crossed to mice harboring Cre recombinase under the control of the CDX2 promoter (CDX2Cre) to generate CDX2Cre; Hampfl/fl mice (HampΔCE) (20). This line was then crossed to APCfl/+ mice to generate a sporadic model of CRC with concomitant hepcidin deletion (i.e. CDX2Cre;APCfl/+;Hampfl/fl mice). Mice were sacked at 4-months of age. Similarly, to achieve colon epithelial-specific disruption of ferroportin, mice floxed for Slc40a1 (Ferroportin) (Fpnfl/fl) were crossed to the colon epithelial-specific Cre and further bred to mice floxed for APC i.e. (CDX2Cre;APCfl/+;Fpn ΔCE mice). All other mouse models (.APCfl/fl;KRASG12DLSL;p53fl/fl, CDX2CreER; HIF-2αOE, CDX2CreER;APCfl/fl;HIF-2αOE, CDX2CreER;APCfl/+;HIF-2αfl/fl) have been previously used in our lab (8, 42, 43). BALB/C mice were purchased from Charles Rivers. All lines are on a C57BL/6J background, except for CDX2Cre;APCfl/+;Fpn fl/fl mice which are a mix between SV129 and C57BL/6J. Littermates wild-type for the gene of interest were used as controls for all animal studies and analysis began on mice that were between 3.5 and 4 months of age for all experiments. Both male and female mice were used. Mice were injected intraperitoneally with tamoxifen (Sigma-Aldrich) at a dose of 100 mg/kg body weight (BW) for 3 consecutive days to ensure Cre-mediated recombination. For the inflammation-associated model of CRC, animals were injected intraperitoneally with 10 mg/ kg azoxythmethane, then cycled on and off 2% (weight/volume) dextran sulfate sodium in their drinking water for 100 days, as previously described (44). For the xenograft tumor model, colon cancer CT26 cells were injected subcutaneously at 6 × 106 cells in BABL/C mice and animals were euthanized 14 days after implantation. All mice were fed ad libitum and maintained under a 12-hour light/12-hour dark cycle. All mice were fed either a standard chow diet (5L0D, PicoLab Laboratory Rodent Diet #3005659–220) or a purified AIN-93G iron-replete (350 ppm, Dyets #115180) or low- iron (<5 ppm, Dyets #115072) diet, where specified. All mice were housed in the Unit for Laboratory Animal Management (ULAM) at the University of Michigan.
Cell culture
Doxycycline-inducible FPN-GFP HEK293 and IEC6 cells were generated previously (45). To generate stable doxycycline- inducible human FPN-GFP colon cancer-derived cell lines, pLenti rtTA3 (Addgene) and pLVX-Tight-Puro hFpn-GFP plasmids were prepared into lentivirus by the University of Michigan Vector Core, coinfected into cells, and selected with 10 μg/ml blasticidin and 1 μg/ml puromycin. Stable, hepcidin overexpressing cell lines were generated by transfecting a mouse hepcidin pLentiLox-Ires construct into CT26 cells and selection with 1 μg/ml puromycin. LbNOX and mito-LbNox plasmids were a generous gift from Dr. Haoqing Yang and the NADH-ubiquinone reductase (NDI1) plasmid was a gift from David Sabatini (Addgene plasmid # 72876). Stable cells were generated similar to that described above. Cell growth analysis was performed using 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Alfa Aesar, 298-93-1) with a 72-hour readout and normalization to Day 0, and colony forming assays were visualized by crystal violet staining after 10 days. The truncated human hepcidin luciferase promoter construct disrupted for hypoxia response elements was generated using primers listed in the Supplementary Table 3. The following concentrations were used for cell treatments: 250 ng/ml doxycycline, 100 μM FG4592 (Selleckchem), 1 mg/ml human recombinant hepcidin (Bachem), 200 μM DFO (Sigma-Aldrich), 10 μM 5AZA (Cayman, Ann Arbor, MI), 10 ng/mL LPS (Sigma-Aldrich), 100 μM Adenosine (A4036–5G, Sigma) 100 μM Cytosine (C3056–1G, Sigma), 100 μM Thymidine (T1895–1G, Sigma), 100 μM Guanosine (G6264–1G, Sigma). HCT116 cells were pretreated with either vehicle or 100 μM FG4592 (Selleckchem) for 16 hours and then were treated with an epigenetic modifier. RT-qPCR was performed 24 hours following treatment of the epigenetic modifier. The following epigenetic modifiers were used: 1 μM AZ6102 (S7767, PeproTech), 5 μM CPI-455 (S8287, Peprotech), 0.2 μM Givinostat (S2170, Peprotech), 1 μM SP2509 (S7680, Peprotech), 1 μM NVP-TNKS656 (S7238, Peprotech), 1 μM I-BET-762 (S7189, Peprotech), 0.2 μM OTX015 (S7360, Peprotech), and 0.2 μM JIB −04 (S7281, Peprotech). AdenoCre and AdenoGFP AAVs were provided by the Viral Vector Core at the University of Michigan. Cells were maintained at 37°C in 5% CO2 and 21% O2. Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS and 1% antibiotic/antimycotic.
Luciferase assay
Cells were lysed in reporter lysis buffer (Promega) and firefly luciferase activity was measured as described previously (45).
Quantitative reverse transcription PCR
mRNA was measured by quantitative reverse transcription PCR (qPCR) (Life Technologies, Thermo Fisher Scientific). The primers used are listed in Supplementary Table 3. Quantification cycle (Cq) values were normalized to β-actin and expressed as the fold change.
Western blot analysis
Lysates were generated as previously described (45). In brief, lysates were separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto nitrocellulose membranes, and probed overnight at 4°C with antibodies against GFP (66002-1-Ig), SDHB (10620-1-AP), FECH (14466-1-AP), or Actin (66009-1) were purchased from Proteintech. Other antibodies used were, Ferroportin (21502, Novus), Ferritin (3998 Cell Signaling, Danvers, MA), and OXPHOS antibody cocktail (Abcam, ab110413). All antibodies were used at a 1:1000 dilution.
Epithelial enrichment
The mice were sacrificed and the tumors were pooled from each respective mouse. All plasticware was precoated with 0.1% bovine serum albumin (BSA) and all steps were carried out on ice unless otherwise specified. The tissue was homogenized with a scalpel and then was incubated in 10 mM DTT for 15 min at room temperature, changing to fresh DTT every 5 min. The tissue was rinsed in DPBS, rinsed once with 8 mM EDTA, and then incubated/rotated in 8 mM EDTA at 4°C for 75 min. The EDTA was removed and the tissue was washed three times with DPBS. The tissue was then “snap-shaken” 10 times to manually separate the colon tumor epithelium. The epithelium-containing supernatant was immediately added to 1.5 ml of cold FBS in a BSA-coated 50-ml tube, and the shaking step was repeated twice more. The epithelium was spun at 40 × g for 2 min at 4°C. The tissue that was not broken free into the supernatant was considered the stromal fraction. The epithelial pellet was washed in DPBS and spun again at 40 × g for 2 min at 4°C. Both cell pellets were then directly resuspended in Trizol for RNA analysis.
Flow Cytometry
Intestinal epithelium and immune cells were isolated and subsequently gated as follows: epithelial cells were gated on single cells, CD45-, EPCAM+ cells; the CD45+ subsets were then further gated with CD11C+, CD11B+, F4/80- as dendritic cells and macrophages were gated with single cells, then CD45+, CD11B+, F4/80+ and Ly6C+ cells. Antibodies and dilutions used were EPCAM(CD326) (BioLegend 118205; 1:100 dilution), CD11c (BioLegend 117305; 1:200 dilution), CD11b (EBioscience 17-0112-83; 1:200), Ly6C (BD Horizon 560594; 1:150 dilution), F4/80 (BD Horizon 563633; 1:100 dilution), and CD45 (Invitrogen 47-0451-82; 1:200 dilution).
Mouse- and patient-derived enteroids
Mouse- and patient-derived colorectal tumor enteroids were generated as previously described (46). Enteroids were co-treated with 100 μM DFO and a 100 μM combination of adenosine, guanosine, cytosine, and thymidine. Enteroids were then imaged after seven days for growth measurement.
Meta-analysis of CRC samples
CRC gene expression data sets along with the patient survival were identified in GEO using the search keywords “colon,” “cancer,” and “microarray” (www.ncbi.nlm.nih.gov/geo/). Only publications providing raw data and clinical survival information and containing at least 30 patients were included. The gene chips were MAS 5.0–normalized in the R statistical environment (www.R-project.org) using the Bioconductor package affy (www.bioconductor.org). Survival analysis using Cox proportional hazards regression was performed as previously described (47). The most reliable probe sets for each gene were selected using Jetset. Kaplan-Meier survival plots were generated using WinSTAT for Excel (Robert K. Fitch Software). Methylation data was generated from the online UALCAN resource (Ualcan.path.uab.edu).
Histology, tissue iron, and immunohistochemistry
Tissue iron detection was performed in formalin-fixed, paraffin-embedded sections stained with Prussian blue and signal was enhanced with 3,3′-Diaminobenzidine tetrahydrochloride (DAB). For mouse tumors iron was detected using Inductively coupled plasma mass spectrometry. For immunohistochemical analysis, frozen sections were probed with polyclonal rabbit anti–ferroportin antibody (Novus 21502, 1:100 dilution), rabbit-anti hepcidin (Abcam 30760, 1:100 dilution) or rabbit-anti Ki67 (Abcam 15580, 1:100 dilution).
Metabolomics on enteroids
Enteroids was treated with DFO in KGM-Gold Keratinocyte Growth Medium (KGMG, Lonza) for 4 days with fresh medium change every other day. Samples were collected by centrifugation and immediately lysed in methanol: water (80:20) at dry-ice temperature for metabolomics analysis. The analysis was performed in biological triplicate. The quantity of the metabolite fraction analyzed was adjusted to the corresponding protein concentration calculated upon processing a parallel sample. Metabolite fractions was processed and analyzed by targeted LC-MS/MS via selected reaction monitoring (SRM), as described previously (48). Processed data was analyzed in Cluster 3.0 and TreeViewer.
DepMap
Corrected and scaled gene essentiality scores for 18,119 genes across 823 cell lines (Project Achilles version 20Q1) were downloaded from the Dependency Map (25). Pearson correlations between TFRC and all other genes were calculated. To determine whether any categories of genes were significantly coessential with TFRC, we performed gene set enrichment analysis (GSEA) with Reactome pathways using the fgsea package in R, retained all pathways with an adjusted p-value of less than 0.01, and ranked them by their normalized enrichment score (49). Correlations were plotted from highest to lowest, and genes involved in the nucleobase biosynthesis Reactome were manually annotated.
Metabolite extraction and bacteria treatment
Metabolites from mouse feces were extracted by methanol: water (80:20) as described previously (50). For treatment with live and heat-killed bacteria, one mouse fecal pellet was suspended in 1 mL of PBS. For heat-killed, this homogenate was boiled for 5 minutes. Either live or heat-killed bacteria were then added to cell culture media at 20 μL/mL.
Seahorse Mitochondria Stress Test
CRC cell lines were seeded at 1.5×104 cells/well in 80 ul/well of normal growth media (DMEM with 25 mM Glucose and 4 mM Glutamine), with or without 10 μM DFO, in an Agilent XF96 V3 PS Cell Culture Microplate (#101085–004), incubated on the bench top at room temperature for 1 hour before incubating at 37°C, 5% CO2 overnight. To hydrate the XF96 FluxPak (#102416–100), 200 uL/well of sterile water was added and the entire cartridge was incubated at 37’C, no CO2 overnight. The following day, one hour prior to running the assay, cells were washed twice with 200 uL/well of assay medium (XF DMEM Base Medium, pH 7.4 (#103575–100) containing 25 mM Glucose (#103577–100), 4 mM Glutamine (#103579–100), and 1 mM Pyruvate (#103578–100). After washing, assay medium, with or without 10 μM DFO, was added to the cell culture plate for a final volume of 180 uL/well. Cells were then incubated at 37’C, no CO2 until analysis. Also one hour prior to the assay, water from the FluxPak hydration was exchanged for 200 uL/well of XF Calibrant (#100840–000) and the cartridge was returned to 37’C, no CO2 until analysis.
Oligomycin (100 uM), FCCP (100 uM), and Rotenone/Antimycin (50 uM) from the XF Cell Mito Stress Test Kit (#103015–100) were re-constituted in assay medium to make the indicated stock concentrations. 20 μL of Oligomycin was loaded into Port A for each well of the FluxPak, 22 μL of FCCP into Port B, and 25 μL of Rotenone/Antimycin into Port C. Port D was left empty. The final FCCP concentration was optimized to achieve maximal respiration in each condition for each cell line.
The Mito Stress Test was conducted on an XF96 Extracellular Flux Analyzer and OCR was analyzed using Wave 2.6 software. Following the assay, OCR was normalized to cell number with the CyQUANT NF Cell Proliferation Assay (C35006) from Thermo Fisher according to manufacturer’s instructions.
NAD/NADH and Hepcidin Assay
NAD/NADH was measured using NAD/NADH-Glo kit (Promega), and serum hepcidin was assessed by ELISA (Intrinsic LifeSciecnes) according to manufacturer’s recommendation.
Statistics
Results are expressed as the mean ± SEM. Significance between 2 groups was tested using a 2-tailed, unpaired or paired t test, as indicated. Significance among multiple groups was tested using a 1-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. A p value of less than 0.05 was considered statistically significant. GraphPad Prism 7.0 was used to conduct the statistical analyses.
Study approval
All animal procedures were approved by the IACUC of the University of Michigan (PRO00010163). Human patient colon cancer tissue was collected according to protocols of the University of Michigan Institutional Review Board (IRB; HUM00064405/0038437/00030020).
Extended Data
Extended Figure 1. Hepcidin expression is increased across many cancer types.

(A) Hepcidin transcript abundance in normal and tumor tissue through The Cancer Genome Atlas. Log2 median centered ratio is plotted (Bladder Normal N=24 and Bladder Tumor N=152; Brain Normal N=10 and Brain Tumor N=547; Colorectal Normal N=22 and Colorectal Tumor N=215; Breast Normal N=61 and Brest Tumor N=532; Cervical Normal N=5 and Cervical Tumor N=155; Head/Neck Normal N=74 and Head/Neck Tumor N=388; Kidney Normal N=329 and Kidney Tumor N=727; Leukemia Normal N=195 and Leukemia Tumor N=197; Liver Normal N=30 Liver Tumor N=138; Lung Normal N=71 and Lung Tumor N=984, Lymphoma Normal N=18 and Lymphoma Tumor N=18, Ovary Normal N=88 and Ovary Tumor N=607; Pancreas Normal N=10 and Pancreas Tumor N=64; and Prostate Normal N=34 and Prostate Tumor N=171 biologically independent samples, Brain p=0.0047. Breast p=0.0009. Colorectal p=0.0006. Kidney p<0.0001, Lung p=0.0092. Ovary p=0.0002. Prostate p=0.0222). (B) qPCR analysis of hepcidin (Hamp) expression levels in tissues/cells of wild-type and colon epithelial-specific APC deficient mice (WT N=4 and APC−/− N=4 biologically independent samples from independent animals in each tissue). (C) Hepcidin concentration in serum of wild-type and colon epithelial-specific APC deficient mice (WT N=4 and APC−/− N=4 biologically independent samples from independent animals). Data represent the mean ± SEM. Significance was determined by 2-tailed, unpaired t test. (A-C) *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared to normal tissue or wild-type animals.
Extended Figure 2. Detection of the hepcidin protein by immunohistochemistry is ineffective.
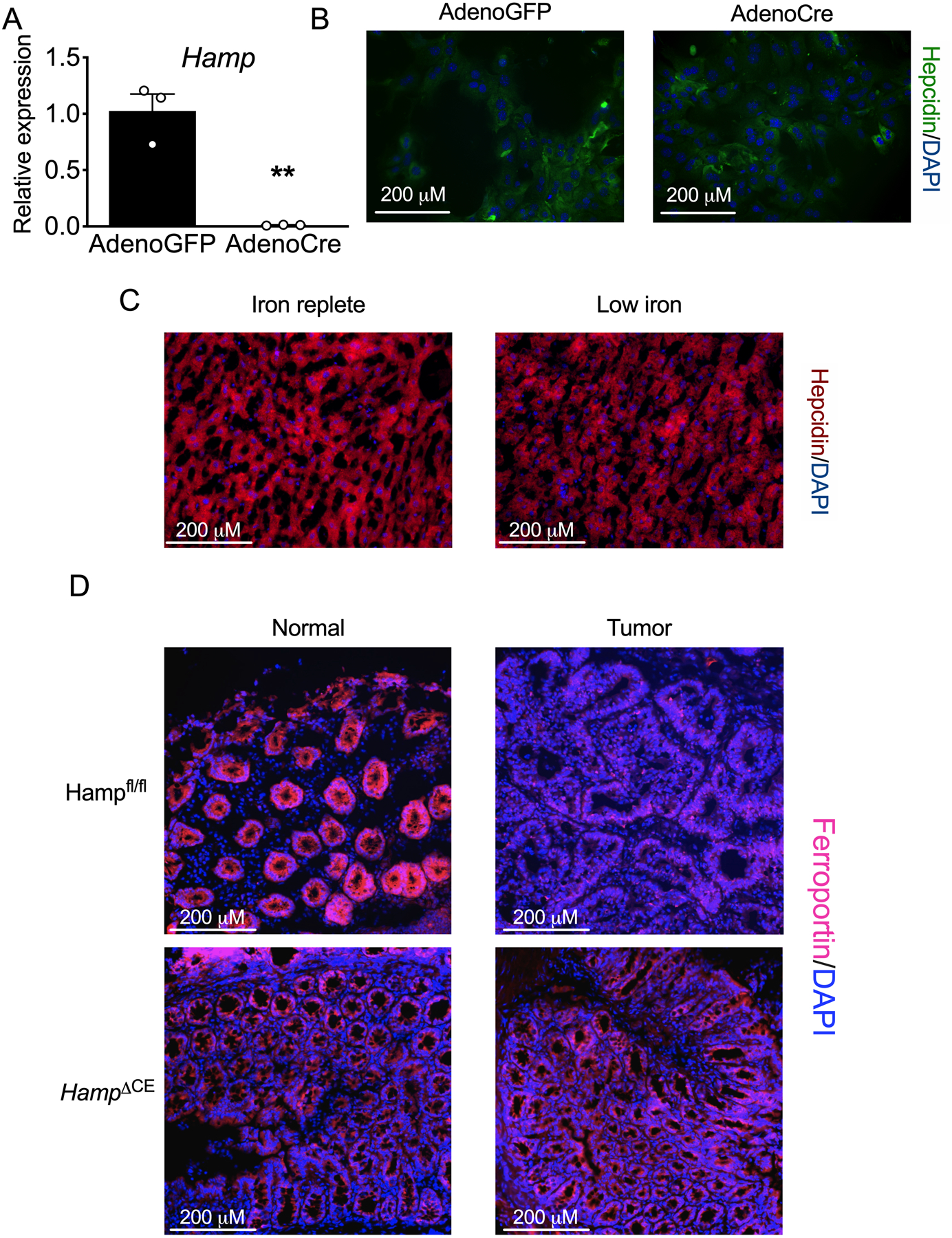
Primary hepatocytes were generated from Hampfl/fl mice and treated in vitro with adenoviruses expressing either GFP or Cre recombinase for 48 hours. These cells were analyzed for hepcidin (Hamp) expression by qPCR analysis (A) and were stained for hepcidin protein (B) (AdenoGFP N=3 and AdenoCre N=3 biologically independent cell replicates, p=0.0025). (C) Representative hepcidin staining in mice that were placed on either an iron replete (350 PPM) or low iron (< 5 PPM) for seven days (N=4 biologically independent samples from independent animals). (D) Ferroportin staining of adjacent normal colon and tumor tissue in a sporadic model of CRC in wild-type and hepcidin-deficient mice (N=3 biologically independent samples from independent animals). Data represent the mean ± SEM. Significance was determined by 2-tailed, unpaired t test (A). **P < 0.01.
Extended Figure 3. Tumoral hepcidin regulation in vitro and in vivo.
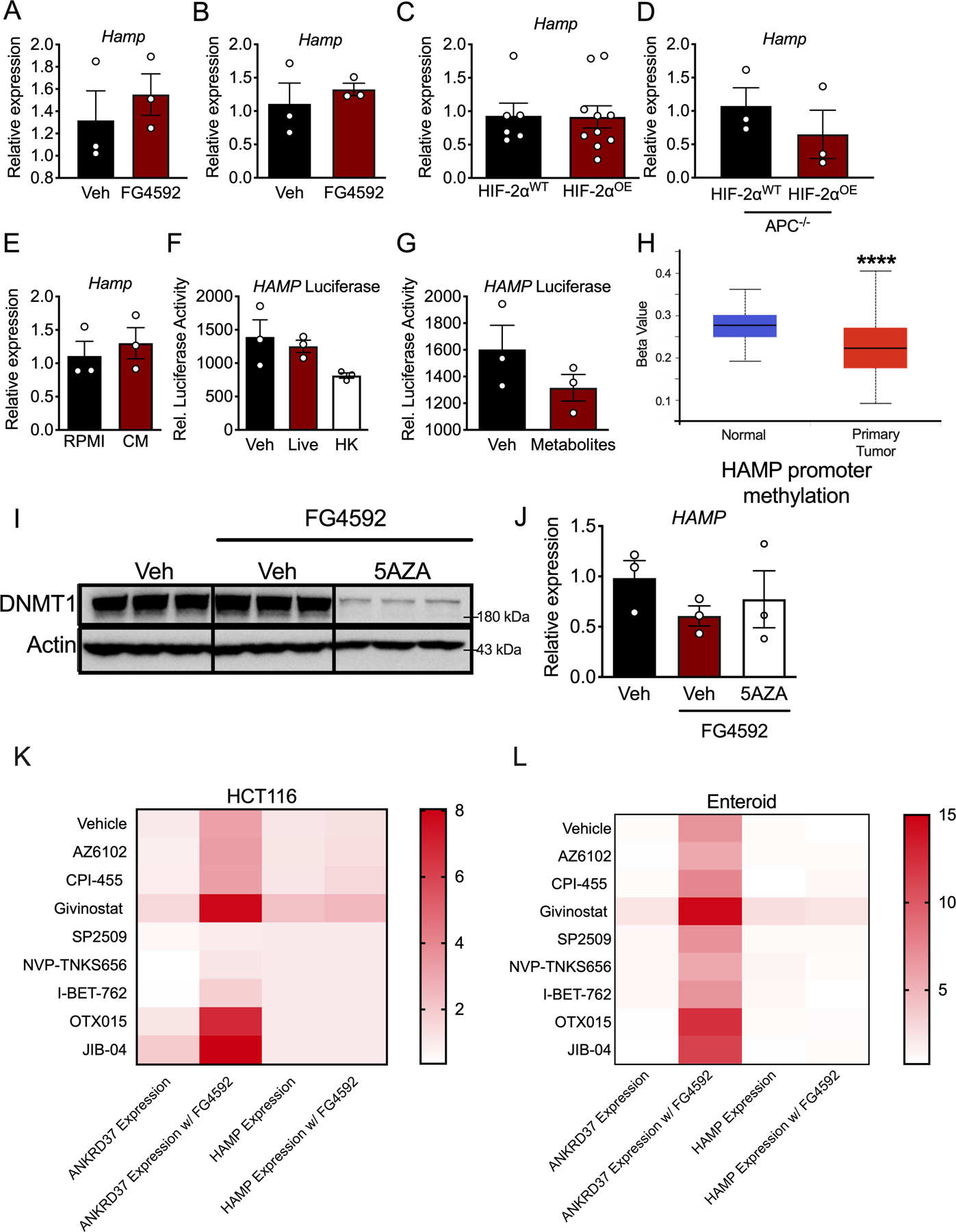
qPCR analysis for hepcidin (Hamp) after treatment with FG4592 (100 μM) or vehicle for 16 hours in (A) HCT116 cells (N=3 biologically independent cell replicates) and (B) enteroids generated from mice with inducible, colon epithelial deletion of APC and p53 and activation of KRAS (N=3 biologically independent samples). (C) qPCR analysis of Hamp in the colon of mice with embryonic, intestinal epithelial-specific overexpression of HIF-2α (HIF-2αOE) compared to wild-type mice (HIF-2αWT) (HIF-2αWT N=6 and HIF-2αOE N=10 biologically independent samples from independent animals). (D) qPCR analysis of Hamp in the colon of colon epithelial-specific HIF-2αWT and HIF-2αOE mice that are also deficient for APC for 30 days HIF-2αWT N=3 and HIF-2αOE N=3 biologically independent samples from independent animals). (E) qPCR analysis of Hamp in HCT116 cells treated for 24 hours with conditioned media (CM) from RAW 264.7 macrophages that had been treated with 10 ng/mL LPS for 16 hours (N=3 biologically independent cell replicates). (F) Relative luciferase activity of the human hepcidin promoter in HCT116 cells treated with vehicle (Veh), live bacteria (Live), or heat-killed bacteria (HK), or (G) vehicle (Veh) or bacteria-derived metabolites (N=3 independent cell replicates). (H) Methylation status of the human hepcidin promoter in human colorectal cancer tissue (Normal N=37 and Tumor N=313). (I) HCT116 cells treated with vehicle (Veh) or 5AZA (10 μM) for 72 hours and then treated with vehicle (Veh) and/or FG4592 (100 μM) for 16 hours and analyzed via Western blot analysis for DNA methyltransferase 1 (DNMT1) and (J) qPCR analysis for HAMP expression (N=3 biologically independent cell replicates). (K) HCT116 cells pretreated with or without FG4592 (100 μM) for 16 hours and then administered a panel of epigenetic modifiers for 24 hours. qPCR was used to measure the expression of HAMP and the HIF-2α target, ANKRD37 (N=3 biologically independent cell replicates for each treatment). (L) Patient-derived enteroids were either pretreated with or without FG4592 (100 μM) for 16 hours and were then administered a panel of epigenetic modifiers for 24 hours. qPCR was used to measure the expression of HAMP and the HIF-2α target, ANKRD37 (N=3 biologically independent samples for each treatment). Data represent the mean ± SEM. Significance was determined by 2-tailed, unpaired t test (A-E, H) or 1-way ANOVA with Tukey’s post hoc (F, J, K-L). ****P < 0.0001.
Extended Figure 4. Colon cancer-derived cell lines are exquisitely sensitive to ferroportin-mediated iron loss.
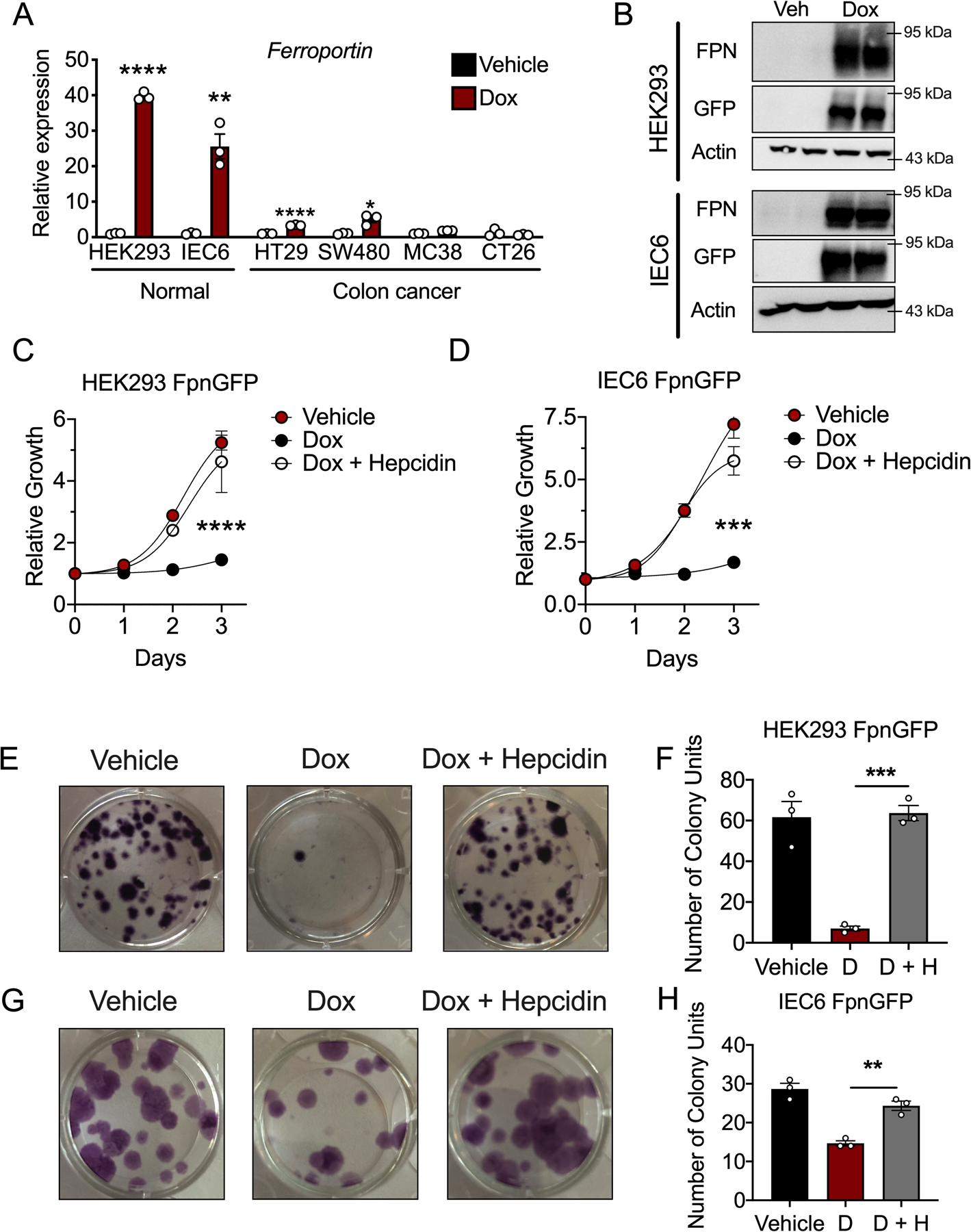
(A) qPCR analysis for the ferroportin transcript in late passages of cells that were made stable for a doxycycline (dox) inducible ferroportinGFP overexpression construct, following 16 hour treatment with dox (250 ng/mL) (N=3 biologically independent cell replicates for each cell line and treatment, HEK293 p=.0001, IEC6 p=0.0021, HT29 p<0.0001, SW480 p=0.0103) (B) Western blot analysis for GFP and ferroportin (FPN) in stable, normal ferroportinGFP overexpressing cell lines following 16 hour treatment with dox (250 ng/mL). (C and D) Cell growth MTT assay in HEK293 ferroportinGFP (C) (N=3 biologically independent cell replicates for each cell line, treatment, and time point, p<0.0001) and (D) IEC6 ferroportinGFP cells (N=3 biologically independent cell replicates for each cell line, treatment, and time point, p=0.0008) treated with vehicle, dox (250 ng/mL), or dox (250 ng/mL) and recombinant hepcidin (1 μg/mL) (E-H) Representative crystal violet staining images (E) and quantification (F) of HEK293 ferroportinGFP (N=3 biologically independent cell replicates for each cell line and treatment, p=0.0005) and IEC6 ferroportinGFP cells, images (G) and quantitation (H) (N=3 biologically independent cell replicates for each cell line and treatment, p=0.0025) ten days following treatment with vehicle, dox (250 ng/mL), or dox (250 ng/mL) and recombinant hepcidin (1 μg/mL) Data represent the mean ± SEM. Significance was determined by 2- tailed, unpaired t test (A) or by 1-way ANOVA with Tukey’s post hoc (C,D,F,H). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 comparing within each cell line or across treatment groups.
Extended Figure 5. Intracellular iron deficiency blunts nucleotide metabolism.

(A) Representative patient-derived CRC tumor enteroids were treated with vehicle or DFO (10 μM) (N=3 biologically independent replicates). (B) Quantification of enteroid growth following 4-days of either vehicle or DFO (10 μM) treatment (N=3 biologically independent replicates). (C) Pathway analysis was conducted on metabolomics data in enteroids after DFO treatment before any decrease in growth was seen. Significance was determined by 2-tailed, unpaired t test.
Extended Figure 6. Intracellular iron deficiency blunts nucleotide metabolism.

(A-B) Heatmap of metabolites of differential abundance in (A) HEK293 and (B) IEC6 doxycycline- inducible ferroportin overexpressing cell lines treated with doxycycline (D) as compared to vehicle (V) for 16 hours (N=3 biologically independent cell replicates, only changes at p < 0.05 compared to V are shown). (C) Pie chart showing the proportion of metabolites of differential abundance in both HEK293 and IEC6 ferroportin overexpressing cells that are involved in nucleotide metabolism.
Extended Figure 7. Moderate doses of DFO do not remove iron from ferritin and individual nucleosides do not rescue DFO-mediated growth inhibition.
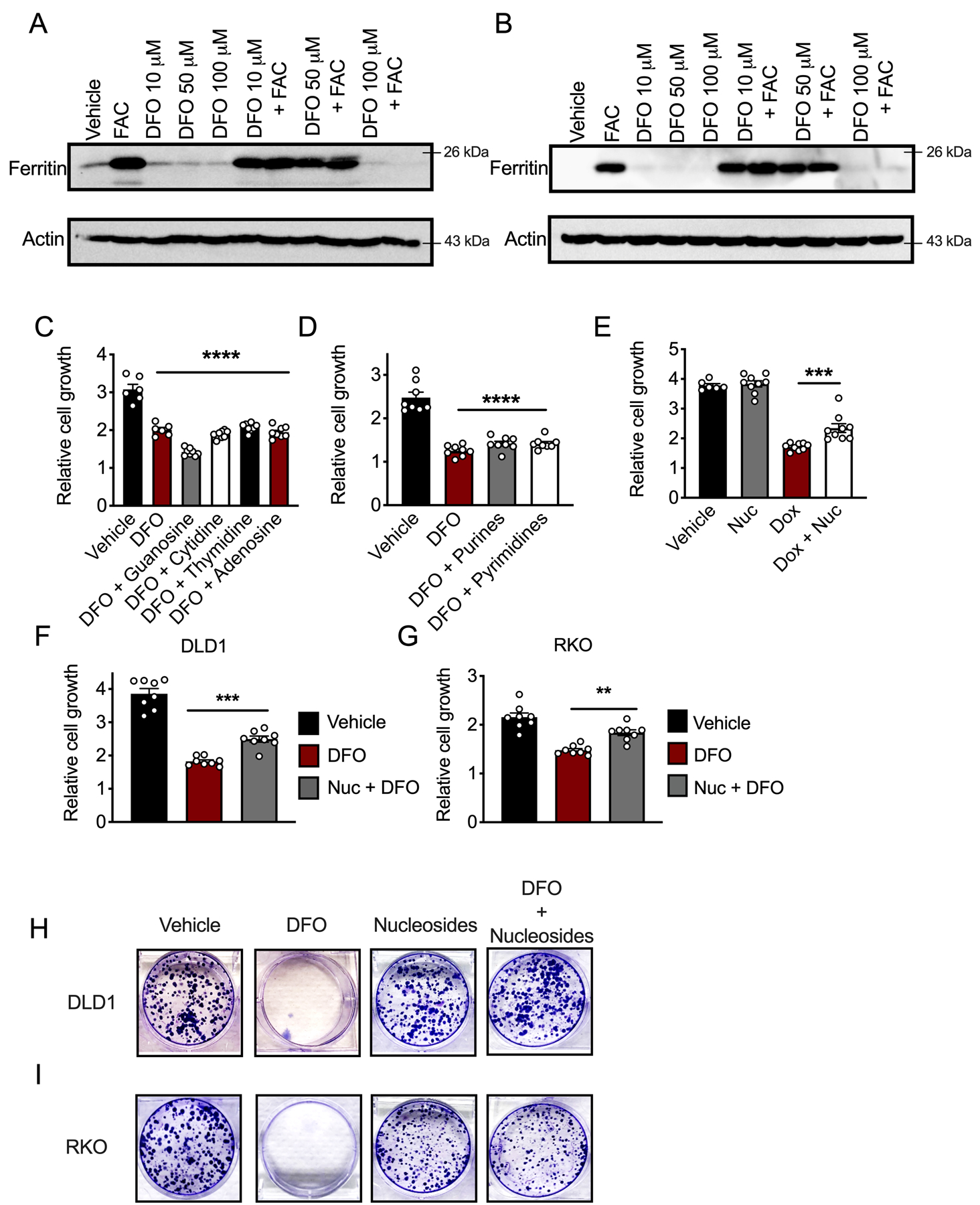
(A and B) Western blot analysis for ferritin after 24 hours pre-loading with ferric ammonium citrate (FAC) (200 μM) and/or overnight DFO, as indicated in (A) HCT116 and (B) SW480 cells. (C and D) Relative growth of HCT116 cells as measured by MTT at 72 hours after treating with DFO (10 μM) and individual nucleosides (100 μM) (C) (Vehicle N=6, DFO N=6, DFO+Nucloeside N=8 biologically independent cell replicates, p<0.0001), or (D) purine or pyrimidine (100 μM) nucleosides (N=8 biologically independent cell replicates for all treatments, p<0.0001. (E) Cell growth MTT assay in IEC6 ferroportinGFP cells treated with vehicle, dox (250 ng/mL), or dox (250 ng/mL) and nucleoside cocktail (100 μM) 72 hours after treatment (Vehicle N=6, Dox N=9, Nuc N=9, and Dox + Nuc N=9 biologically independent cell replicates, p=0.0006). (F and G) Nucleoside (100 μM) rescue of DFO (10 μM) growth inhibition in DLD1 (F) (N=8 biologically independent cell replicates for all treatments, p=0.0007) and RKO (G) (N=8 biologically independent cell replicates for all treatments, p=0.0018 cells) after 72 hours of cotreatment using MTT. (H and I) Representative crystal violet stains of DLD1 or RKO cells that were either pretreated with vehicle or DFO (10 μM) and then given vehicle or a nucleoside cocktail (100 μM) for ten days (N=3 biologically independent cell replicates). Data represent the mean ± SEM. Significance was determined by one-way ANOVA followed by Tukey’s post hoc. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared to vehicle.
Extended Figure 8. Acute DFO insult disrupts mitochondrial metabolism without decreased cell growth.
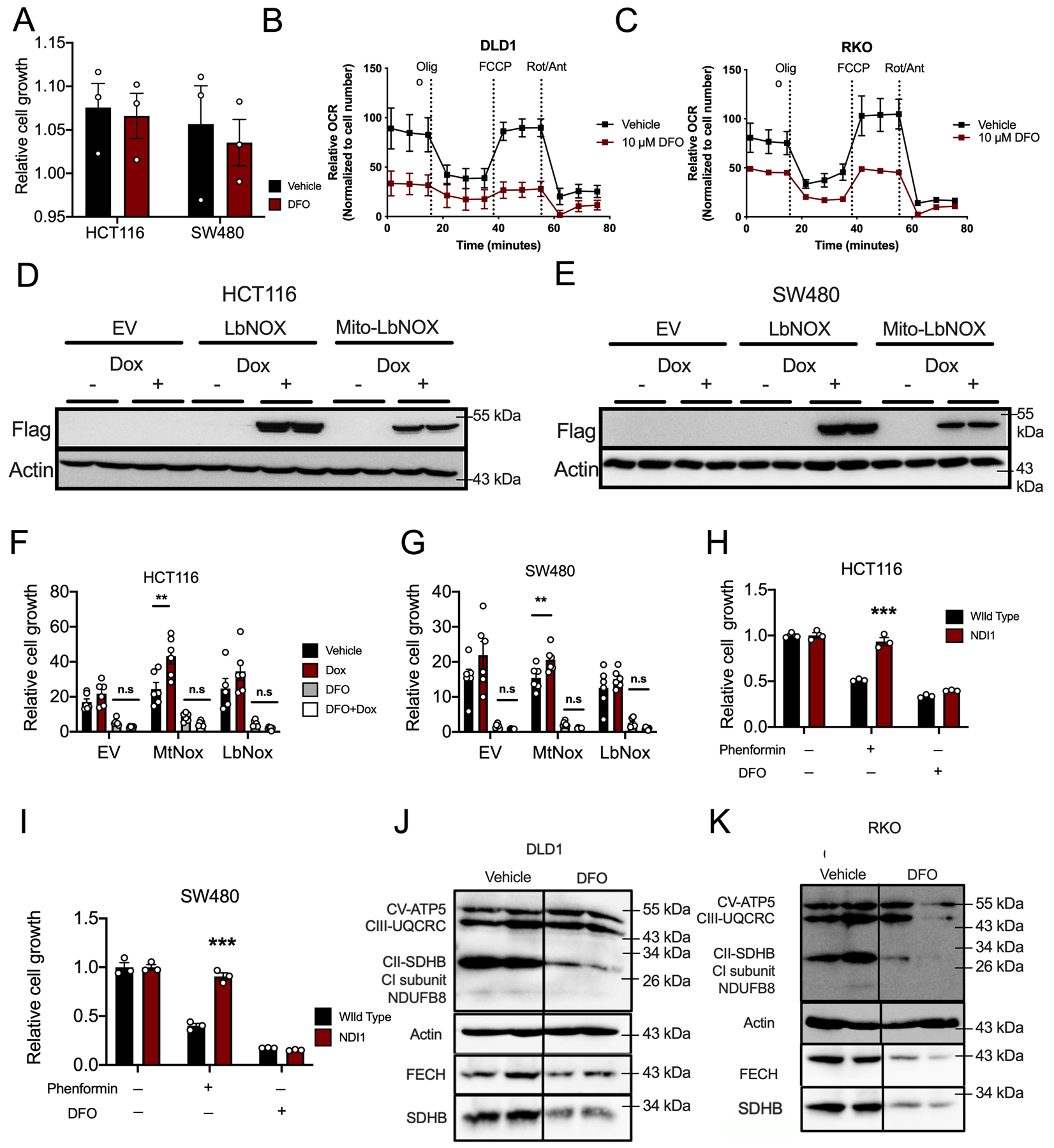
(A) Cell growth assay in HCT116 and SW480 cells 24 hours after DFO treatment (10μM) and assessed by MTT (N=3 biologically independent cell replicates for each treatment and cell line). (B and C) Seahorse analysis of mitochondrial metabolism in DLD1 (B) and RKO (C) cells 24 hours after DFO administration (10μM) (N=4 biologically independent cell replicates for each treatment condition). (D and E) Western blot analysis of doxycycline (Dox)-inducible Lactobacillus NADH-oxidase (LbNox) and mitochondrial NADH-oxidase (Mito-LbNox) in stably generated in HCT116 (D) and SW480 (E) cells. (F and G) MTT cell growth assays in LbNox and Mito-LbNox expressing cells following DFO (10μM) in (F) HCT116 (p=0.014) and (G) SW480 cells (p=0.0076) (N=6 biologically independent cell replicates for each cell line and treatment). (H and I) MTT cell growth assay in cells stably expressing yeast NADH-ubiquinone reductase (NDI1) following DFO (10μM) or phenformin (62.5μM) treatment in (H) HCT116 (p=0.0006) and (I) SW480 (p=0.0003) cells (N=3 biologically independent cell replicates for each cell line and treatment). (J and K) Western blot analysis of mitochondrial enzymes following 24-hours of DFO (10μM) treatment in DLD1 (J) and RKO (K) cells. Data represent the mean ± SEM. Significance was determined by 2-tailed, unpaired t test (A, H-I)) or one-way ANOVA (F-G) followed by Tukey’s post hoc. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared to vehicle.
Supplementary Material
DepMap data analysis
Metabolomics Dataset
Primer Sequences
Acknowledgments
We would like to thank the Shah Lab and Zhaoqi Li for helpful input and suggestions. This work was supported by National Institutes of Health Grants R01CA148828, R01DK095201, and R01CA245546 to Y.M.S. C.A.L. was supported by the NCI (R37CA237421, R01CA248160, R01CA244931, and R01CA215607). C.A.L. and Y.M.S. were supported by University of Michigan Rogel Cancer Center Core Grant (P30CA046592) and University of Michigan Center for Gastrointestinal Research (P30DK034933). Metabolomics studies performed at the University of Michigan were supported by NIH grant DK097153, the Charles Woodson Research Fund, and the UM Pediatric Brain Tumor Initiative. X.X was supported by the National Institutes of Health (P20 GM130422, K01DK114390), a Research Scholar Grant from the American Cancer Society (RSG-18-050-01-NEC), a Research Scholar Award from the American Gastroenterological Association and a Research Program Support Pilot Project Award from UNM comprehensive cancer center (P30CA118100). P.P.H (2T32CA071345-21A1), B.T.D (T32GM007753), A.J.S (F31DK116555), B.C (T32GM007315) and S.A.K (F31CA24745701) were supported by the indicated grants. Enteroid work was done with The Michigan Medicine Translational Tissue Modeling Laboratory a funded initiative with support from the Endowment for Basic Sciences (Center for Gastrointestinal Research, Office of the Dean, Comprehensive Cancer Center, Departments of Pathology, Pharmacology, and Internal Medicine).
Footnotes
Competing Interests
All authors declare no competing interest. PPH is a consultant for Auron Therapeutics. C.A.L. is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting GOT1 as a therapeutic approach (US Patent Numbers: 2012112933-A1, 2015126580-A1, and 20190136238).
Data Availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
- 1.Siegel R, Ma J, Zou Z, and Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 3.Kroemer G, and Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13(6):472–82. [DOI] [PubMed] [Google Scholar]
- 4.Xue X, and Shah YM. Intestinal iron homeostasis and colon tumorigenesis. Nutrients. 2013;5(7):2333–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drakesmith H, Nemeth E, and Ganz T. Ironing out Ferroportin. Cell Metab. 2015;22(5):777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brookes MJ, Boult J, Roberts K, Cooper BT, Hotchin NA, Matthews G, et al. A role for iron in Wnt signalling. Oncogene. 2008;27(7):966–75. [DOI] [PubMed] [Google Scholar]
- 7.Le NT, and Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta. 2002;1603(1):31–46. [DOI] [PubMed] [Google Scholar]
- 8.Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab. 2016;24(3):447–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xue X, Taylor M, Anderson E, Hao C, Qu A, Greenson JK, et al. Hypoxia-inducible factor-2alpha activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res. 2012;72(9):2285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brookes MJ, Hughes S, Turner FE, Reynolds G, Sharma N, Ismail T, et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut. 2006;55(10):1449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixon SJ, and Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10(1):9–17. [DOI] [PubMed] [Google Scholar]
- 12.Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014;7(1):180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–3. [DOI] [PubMed] [Google Scholar]
- 14.Bessman NJ, Mathieu JRR, Renassia C, Zhou L, Fung TC, Fernandez KC, et al. Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science. 2020;368(6487):186–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lakhal-Littleton S, Crosby A, Frise MC, Mohammad G, Carr CA, Loick PAM, et al. Intracellular iron deficiency in pulmonary arterial smooth muscle cells induces pulmonary arterial hypertension in mice. Proc Natl Acad Sci U S A. 2019;116(26):13122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lakhal-Littleton S, Wolna M, Chung YJ, Christian HC, Heather LC, Brescia M, et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malerba M, Louis S, Cuvellier S, Shambat SM, Hua C, Gomart C, et al. Epidermal hepcidin is required for neutrophil response to bacterial infection. J Clin Invest. 2020;130(1):329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinnix ZK, Miller LD, Wang W, D’Agostino R Jr., Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2(43):43ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue D, Zhou CX, Shi YB, Lu H, and He XZ. Decreased expression of ferroportin in prostate cancer. Oncol Lett. 2015;10(2):913–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67(20):9721–30. [DOI] [PubMed] [Google Scholar]
- 21.Dame MK, Attili D, McClintock SD, Dedhia PH, Ouillette P, Hardt O, et al. Identification, isolation and characterization of human LGR5-positive colon adenoma cells. Development. 2018;145(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Triner D, and Shah YM. Hypoxia-inducible factors: a central link between inflammation and cancer. J Clin Invest. 2016;126(10):3689–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue X, Ramakrishnan SK, and Shah YM. Activation of HIF-1alpha does not increase intestinal tumorigenesis. Am J Physiol Gastrointest Liver Physiol. 2014;307(2):G187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112(2):219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49(12):1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahn CS, and Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell. 2015;162(3):552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol. 2018;20(7):782–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, and Sabatini DM. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. 2015;162(3):540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S, Chen Y, Guo W, Yuan L, Zhang D, Xu Y, et al. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signal. 2014;26(11):2539–50. [DOI] [PubMed] [Google Scholar]
- 31.Tesfay L, Clausen KA, Kim JW, Hegde P, Wang X, Miller LD, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. 2015;75(11):2254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward DG, Roberts K, Brookes MJ, Joy H, Martin A, Ismail T, et al. Increased hepcidin expression in colorectal carcinogenesis. World J Gastroenterol. 2008;14(9):1339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanchette-Farra N, Kita D, Konstorum A, Tesfay L, Lemler D, Hegde P, et al. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene. 2018;37(29):4013–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merle U, Fein E, Gehrke SG, Stremmel W, and Kulaksiz H. The iron regulatory peptide hepcidin is expressed in the heart and regulated by hypoxia and inflammation. Endocrinology. 2007;148(6):2663–8. [DOI] [PubMed] [Google Scholar]
- 35.Liu Q, Davidoff O, Niss K, and Haase VH. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest. 2012;122(12):4635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villa E, Ali ES, Sahu U, and Ben-Sahra I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers (Basel). 2019;11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buj R, and Aird KM. Deoxyribonucleotide Triphosphate Metabolism in Cancer and Metabolic Disease. Front Endocrinol (Lausanne). 2018;9:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM, et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun. 2018;9(1):4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nurtjahja-Tjendraputra E, Fu D, Phang JM, and Richardson DR. Iron chelation regulates cyclin D1 expression via the proteasome: a link to iron deficiency-mediated growth suppression. Blood. 2007;109(9):4045–54. [DOI] [PubMed] [Google Scholar]
- 40.Thelander L, Graslund A, and Thelander M. Continual presence of oxygen and iron required for mammalian ribonucleotide reduction: possible regulation mechanism. Biochem Biophys Res Commun. 1983;110(3):859–65. [DOI] [PubMed] [Google Scholar]
- 41.Khodour Y, Kaguni LS, and Stiban J. Iron-sulfur clusters in nucleic acid metabolism: Varying roles of ancient cofactors. Enzymes. 2019;45:225–56. [DOI] [PubMed] [Google Scholar]
- 42.Triner D, Xue X, Schwartz AJ, Jung I, Colacino JA, and Shah YM. Epithelial Hypoxia-Inducible Factor 2alpha Facilitates the Progression of Colon Tumors through Recruiting Neutrophils. Mol Cell Biol. 2017;37(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Solanki S, Devenport SN, Ramakrishnan SK, and Shah YM. Temporal induction of intestinal epithelial hypoxia-inducible factor-2alpha is sufficient to drive colitis. Am J Physiol Gastrointest Liver Physiol. 2019;317(2):G98–G107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Triner D, Devenport SN, Ramakrishnan SK, Ma X, Frieler RA, Greenson JK, et al. Neutrophils Restrict Tumor-Associated Microbiota to Reduce Growth and Invasion of Colon Tumors in Mice. Gastroenterology. 2019;156(5):1467–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwartz AJ, Das NK, Ramakrishnan SK, Jain C, Jurkovic MT, Wu J, et al. Hepatic hepcidin/intestinal HIF-2alpha axis maintains iron absorption during iron deficiency and overload. J Clin Invest. 2019;129(1):336–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dame MK, Jiang Y, Appelman HD, Copley KD, McClintock SD, Aslam MN, et al. Human colonic crypts in culture: segregation of immunochemical markers in normal versus adenoma-derived. Lab Invest. 2014;94(2):222–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gyorffy B, Surowiak P, Budczies J, and Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8(12):e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan M, Breitkopf SB, Yang X, and Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc. 2012;7(5):872–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bennett BD, Yuan J, Kimball EH, and Rabinowitz JD. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat Protoc. 2008;3(8):1299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DepMap data analysis
Metabolomics Dataset
Primer Sequences