

Abstract
The technique of colour EM that was recently developed enabled localisation of specific macromolecules/proteins of interest by the targeted deposition of diaminobenzidine (DAB) conjugated to lanthanide chelates. By acquiring lanthanide elemental maps by energy‐filtered transmission electron microscopy (EFTEM) and overlaying them in pseudo‐colour over the conventional greyscale TEM image, a colour EM image is generated. This provides a powerful tool for visualising subcellular component/s, by the ability to clearly distinguish them from the general staining of the endogenous cellular material. Previously, the lanthanide elemental maps were acquired at the high‐loss M4,5 edge (excitation of 3d electrons), where the characteristic signal is extremely low and required considerably long exposures. In this paper, we explore the possibility of acquiring the elemental maps of lanthanides at their N4,5 edge (excitation of 4d electrons), which occurring at a much lower energy‐loss regime, thereby contains significantly greater total characteristic signal owing to the higher inelastic scattering cross‐sections at the N4,5 edge. Acquiring EFTEM lanthanide elemental maps at the N4,5 edge instead of the M4,5 edge, provides ∼4× increase in signal‐to‐noise and ∼2× increase in resolution. However, the interpretation of the lanthanide maps acquired at the N4,5 edge by the traditional 3‐window method, is complicated due to the broad shape of the edge profile and the lower signal‐above‐background ratio. Most of these problems can be circumvented by the acquisition of elemental maps with the more sophisticated technique of EFTEM Spectrum Imaging (EFTEM SI). Here, we also report the chemical synthesis of novel second‐generation DAB lanthanide metal chelate conjugates that contain 2 lanthanide ions per DAB molecule in comparison with 0.5 lanthanide ion per DAB in the first generation. Thereby, fourfold more Ln3+ per oxidised DAB would be deposited providing significant amplification of signal. This paper applies the colour EM technique at the intermediate‐loss energy‐loss regime to three different cellular targets, namely using mitochondrial matrix‐directed APEX2, histone H2B‐Nucleosome and EdU‐DNA. All the examples shown in the paper are single colour EM images only.
Keywords: colour EM, DAB, DDD, direct detection device, EFTEM, intermediate loss, lanthanide DAB, low loss, spectrum imaging
1. INTRODUCTION
The Electron Microscope (EM) has gained immense popularity within the scientific community since its invention,1 partly because in addition to providing for the acquisition of high resolution images, it also has the ability to simultaneously investigate the chemical/elemental composition of the sample. The elemental composition of the sample is primarily obtained either by Energy Dispersive X‐ray (EDX) or Electron Energy Loss Spectroscopy (EELS), with the latter having higher sensitivity and resolution.2, 3, 4 The EELS technique has been applied in materials science to map elements with single atom sensitivity5, 6, 7 and in biological science to detect and quantify many endogenous elements.8, 9, 10, 11 The EELS technique can be applied either in the Transmission Electron Microscopy (TEM) mode, generally referred to as the Energy Filtered TEM (EFTEM)12, 13, 14, 15, 16 or in the Scanning Transmission Electron Microscopy (STEM) mode, referred to as STEM‐EELS or EELS Spectrum‐Imaging.17, 18, 19, 20, 21, 22 Although EFTEM mode has lower sensitivity than STEM‐EELS, it provides larger fields of view, at least an order of magnitude larger, typically 105–107 pixels in comparison to 103–105 pixels in STEM‐EELS.10, 17 For certain biological applications, a more encompassing field of view is just as important as either resolution or sensitivity, as is the case for the application of colour EM electron probes to simultaneously label multiple cellular proteins/organelles in cells.23, 24, 25 In the method that we developed, the localisation of the multiple targeted molecules was achieved by the sequential deposition of specific lanthanide chelates conjugated to diaminobenzidine, which were selectively oxidised by orthogonal photosensitisers/peroxidases.23 The core‐loss or high‐loss (M4,5 edge) elemental map/maps of the lanthanides obtained by the EFTEM mode were then overlaid in pseudo‐colour onto a conventional electron micrograph to create colour EM images.23, 26, 27
The M4,5 core‐loss edge for the lanthanides occurs at an energy loss > 800 eV. At these energy loss regions, the elemental signal is an extremely small fraction of the total incident beam,3 requiring exposure times exceeding several minutes to acquire an elemental map. Such long exposures lead to poor image quality primarily due to specimen drift,28 which can be somewhat offset, by taking a series of EFTEM images at shorter exposure times and then using drift correction to align them into a single image.28, 29, 30, 31, 32 In additional to the aforementioned drift correction strategy, we demonstrated that using a Direct Detection Devices (DDD) instead of the traditional phosphor‐based scintillator imaged by a photosensitive Charge Coupled Device (CCD) to obtain EFTEM maps provided significant improvements to signal‐to‐noise (SNR) and spatial resolution.30 The proof of concept of the inherent advantages of directly detecting electrons for spectroscopy versus photoconversion in a scintillator was first demonstrated by Egerton, but he had to limit routine use in this mode due to the excessive beam damage.33 It was only recently that beam‐hardened CMOS Direct Detection Devices (DDD) able to operate continuously for months on a 300 KV TEM without significant beam damage were invented.34, 35, 36, 37, 38, 39, 40, 41 Recently, other labs have also started to use DDD, primarily for STEM‐EELS, to improve the spectral resolution and SNR.42, 43, 44
However, in spite of these advances in EFTEM elemental mapping, we are fundamentally limited by the total intensity available at the high‐energy loss regions, which decreases exponentially with the increase in energy loss, in the so‐called Power Law form.3 Alternatively, lanthanides also have a relatively strong N4,5 edge which occur at much lower‐energy loss regions, at 99 and 195 eV, in comparison to the M4,5 edge onset at 832 and 1588 eV for La and Lu, respectively.45 It has been suggested in the ‘EELS Atlas’, that for lanthanides the N4,5 edge can also be used for elemental mapping and microanalysis.45 In continuation of our work of acquiring EFTEM elemental maps on the DDD, this paper discusses the advantages and pitfalls of obtaining elemental maps at the lower‐energy loss region (N4,5 edge) for biological samples specifically labelled with lanthanide conjugated diaminobenzidine. Technically, the energy loss region up to 50 eV is considered low‐loss region, and the energy loss region > 50 eV is considered high‐loss region.2 In this report to make a clear distinction between the M4,5 and N4,5 edge of the Lanthanides, we will refer to the M4,5 edge as the high‐loss (HL) region and the N4,5 edge as the intermediate‐loss (IL) region. To facilitate these studies and future applications of colour‐EM, we also report the synthesis and application of a novel second‐generation series of lanthanide chelates of DAB (Ln2‐DAB) that deposit more metal upon DAB (photo)oxidation.
2. MATERIALS AND METHODS
2.1. Materials
Reagents were obtained from Sigma‐Aldrich (St. Louis, MO, USA). Solvents and cell culture reagents were obtained from Thermo Fisher Scientific (Pittsburgh, PA, USA) except where noted. Reactions were monitored by LC‐MS (Ion Trap XCT with 1100 LC, Agilent, Santa Clara, CA, USA) using an analytical Luna C18(2) reverse‐phase column (Phenomenex, Torrance, CA, USA), acetonitrile/H2O (with 0.05% v/v CF3CO2H) linear gradients, 1 mL/min flow, and ESI positive or negative ion mode. Compounds were purified by silica gel column chromatography or alternatively by preparative HPLC using the above gradients and semipreparative Luna C18(2) columns at 3.5 mL/min. UV−Vis absorption spectra were recorded on a Shimadzu UV‐2700 (Kyoto, Japan) spectrophotometer. A second generation of the lanthanide chelates of the DAB (Ln2‐DAB) was chemically synthesised (see Figure 1), which has four times the metal/lanthanide per DAB than the first generation (Ln‐DAB2). For the detailed description of the chemical synthesis of Ln2‐DAB, please refer to the supplementary section.
FIGURE 1.
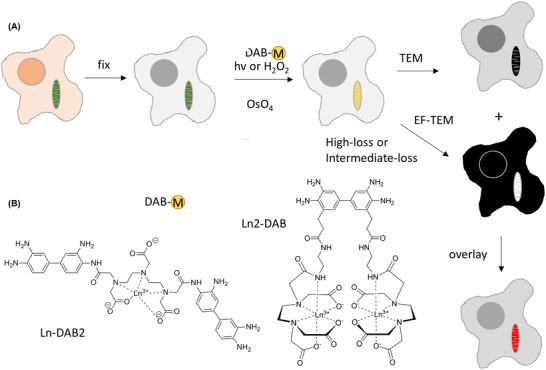
(A) Scheme of process for obtaining colour‐EM images of cells. Cells containing mitochondria labelled with a photosensitiser or peroxidase are fixed, incubated with a metal chelate of DAB, DAB‐M and either irradiated or incubated with H2O2, respectively. Following osmification, embedding and sectioning, corresponding TEM and EF‐TEM images are collected at element‐distinctive high‐loss and intermediate‐loss energies. A pseudo‐colour overlay of these electron energy loss images on the osmium TEM yields a colour‐EM image of the cells. (B) Chemical structures of DAB‐lanthanide metal chelates; first generation, Ln‐DAB2 and second generation, Ln2‐DAB
2.2. Samples
Three sets of samples were prepared of three different cellular targets, namely using mitochondrial matrix‐directed APEX2, histone H2BNnucleosome and EdU‐DNA. Also, each of these three samples were labelled with DAB conjugated to three different lanthanide chelates: cerium, lanthanum and neodymium, respectively. The cerium and lanthanide DAB chelates were of second generation (Ln2‐DAB) and the neodymium DAB chelate was of the first generation (Ln‐DAB2).
Finally, a fourth set of samples were prepared, that served as a control, mitochondrial matrix‐directed APEX2 labelled with unmodified DAB, that is, DAB without any lanthanide chelate bound to it. For detailed description of the preparation protocols for each of the sample, please refer to the supplementary section.
2.2.1. Specimen processing postsecondary fixation
Postfixative was removed from cells and were rinsed with 0.1 M sodium cacodylate buffer pH 7.4 (5×1 minutes) on ice. Cells were washed with ddH2O at room temperature (5 × 1 minutes) followed by an ice‐cold graded dehydration ethanol series of 20%, 50%, 70%, 90%, 100% (anhydrous) for 1 minute each and 2×100% (anhydrous) at room temperature for 1 minute each. Cells were infiltrated with one part Durcupan ACM epoxy resin (44610, Sigma‐Aldrich) to one part anhydrous ethanol for 30 minutes, 3 times with 100% Durcupan resin for 1 hours each, a final change of Durcupan resin and immediately placed in a vacuum oven at 60°C for 48 hours to harden. Cells were identified, cut out by jewel saw and mounted on dummy blocks with Krazy glue. Coverslips were removed and 100 nm thick specimen sections were created with a Leica Ultracut UCT ultramicrotome and Diatome Ultra 45°, 4 mm wet diamond knife. Sections were picked up with 50 mesh gilder copper grids (G50, Ted Pella, Inc) and carbon coated on both sides with a Cressington 208 Carbon Coater for 15 second at 3.4 volts.
2.3. Electron microscopy
Electron microscopy was performed with a JEOL JEM‐3200EF transmission electron microscope, equipped with a LaB6 source operating at 200 KV. The microscope is fitted with an in‐column Omega filter after the intermediate lenses and before the projector lenses. The Cs and Cc aberration coefficients of the objective lens are 3.2 and 3.0 mm, respectively. Conventional TEM and EFTEM images were collected using a condenser aperture of size 120 μm, an objective aperture of size 30 μm and entrance aperture of size 120 μm was used. Additionally, a selective area aperture of size 50 μm was used for acquisition of electron energy‐loss spectra. The spectrometer energy resolution is 2 eV, measured as the FWHM of the zero‐loss peak. Before the acquisition of elemental maps, the sample was preirradiated with a low beam dose of ∼3.5×104 e–/nm2 for about 20 minutes to stabilise the sample and to reduce contamination.46
The conventional TEM images and electron energy loss spectra were acquired on an Ultrascan 4000 CCD from Gatan (Pleasanton, CA, USA). All the elemental maps, irrespective of whether it is a high‐loss or an intermediate‐loss map, were acquired on a DE‐12 camera, which is a DDD from Direct Electron LP (San Diego, CA, USA). The maps presented in this paper were acquired either by the traditional 3‐window method3, 47 or by the EFTEM Spectrum Imaging (EFTEM SI) technique.48, 49, 50, 51 For the 3‐window method, to mitigate the effects of sample drift, instead of acquiring a single preedge 1, preedge 2 and postedge image for long exposures, a series of images of shorter durations for each of the preedge and the postedge were acquired. These images were drift corrected and merged to form a single image preedge or postedge image as the case may be. Also, instead of sequentially collecting the entire series of images at one energy window before proceeding to the next energy window, we acquired a set of images successively through all the energy windows and then acquire the next set through all the energy windows and so on. This interlaced acquisition reduces detrimental effects due to sample shrinkage/warping and high‐tension instabilities over time has been previously described in more detail.23 The acquisition of both the 3‐window method and EFTEM SI was accomplished by writing a macro in Serial EM for the control DE‐12 detector.52 The high‐loss EFTEM images were aligned using the Template Matching plug‐in in ImageJ53 and the intermediate‐loss EFTEM images were aligned using the filters and alignment routines of Digital Micrograph (Gatan, Inc). The elemental maps for the 3‐window method was computed using the EFTEM‐TomoJ plug‐in of ImageJ54 and the elemental maps for the EFTEM SI were computed using the Digital Micrograph (Gatan, Inc). The EFTEM images were dark current subtracted only and not gain normalised to avoid the fixed pattern noise arising due to uncertainties in flat‐fielding.30 All the elemental maps presented in this paper were acquired at the full resolution of the DE‐12 detector without any binning applied to them. The SNR of the elemental map was obtained by dividing the signal by the standard deviation of the background intensity.55, 56 The signal was calculated by subtracting the mean intensity of regions that contained the lanthanide from the mean intensity of regions that represented the background. Please see the supplementary section in this paper for the detailed TEM and spectrometer acquisition parameters for each of the datasets presented.
3. RESULTS AND DISCUSSION
The method for colour EM that we developed depends upon the sequential oxidative deposition of Ln‐DAB2 (Figure 1), consisting of a single lanthanide ion, Ln3+ bound to the chelate (DTPA) conjugated to one of the amino groups of two diaminobenzidine molecules23 by two of its carboxylate groups. To deposit more lanthanide per oxidised DAB, we synthesised Ln2‐DAB (Figure 1B), by a multistep synthesis (Figure S1) from 3,3’‐dinitrobenzidine by bromination to the 5,5’‐dihalo derivative, followed by Heck reaction with ethyl acrylate. BOC‐protection of the amino groups was necessary to avoid their intramolecular cyclisation with the orthoacrylic esters. Catalytic hydrogenation of acrylic and nitro groups and saponification of the ethyl ester gave the resulting diacid that was coupled to N‐aminoethyl‐EDTA via an intermediate bis‐NHS ester to give the BOC‐protected DAB derivative conjugated to two EDTA chelates. Acid deprotection and titration with Ce3+ or Ln3+ gave the final product, Ln2‐DAB, in which 2 lanthanide ions are bound per DAB and would therefore deposit fourfold more Ln3+ per oxidised DAB than our first‐generation chelate, Ln‐DAB2.
HEK293T tissue culture cells transiently transfected with the genetically encoded peroxidase; APEX2 were fixed with 2% glutaraldehyde and incubated with Ce2‐DAB and H2O2 for 5 minutes until a faint darkening of the cells was visible by bright field microscopy. Followed by postfixation with ruthenium tetroxide, RuO4, and then dehydrated, infiltrated, embedded and sectioned for TEM.57 Previously, the EFTEM elemental maps for the lanthanide/s were obtained at their high‐loss M4,5 edge, which were then overlaid in pseudo‐colour onto a conventional electron micrograph to create the so‐called colour EM (see Figure 1).23
The main drawback of obtaining elemental maps at the M4,5 high‐loss region for the lanthanides is that the total intensity is very low and requires long exposures at relatively high dose and dose rates.30 The background intensity varies as a function of Energy Loss E, in the so‐called power‐law form I = AE – r, with values for parameters A and r being dependent on the experimental conditions.3 The overall signal intensity drops exponentially with energy loss, reaching negligible levels for energy loss above 2000 eV, and this is the practical limit of the technique.2 For example, it has been shown that only 5×10−5 fraction of the total incident electron dose contributed to the oxygen K edge (onset at 532 eV) for a sample of 40 nm amorphous ice.44
It has been suggested that for lanthanides, the N4,5 edge occurring at much lower energy‐loss can also be potentially used for elemental mapping.45 Figures 2(A) and (B) show the raw spectra of the high‐loss M4,5 edge region (onset at 883 eV) and the intermediate‐loss N4,5 edge region (onset at 110 eV) for cerium, respectively. The N4,5 edge spectrum was acquired at a fraction of the dose of the M4,5 edge spectrum. The two distinct white lines of the M4,5 edge prominently stand out from the background in Figure 2(A), in contrast to the N4,5 edge which is barely discernible from the background as a small bump in Figure 2(B) (see where the arrow points). In Figure 2(D), after the background subtraction, the saw tooth shape of the N4,5 edge is apparently evident.
FIGURE 2.
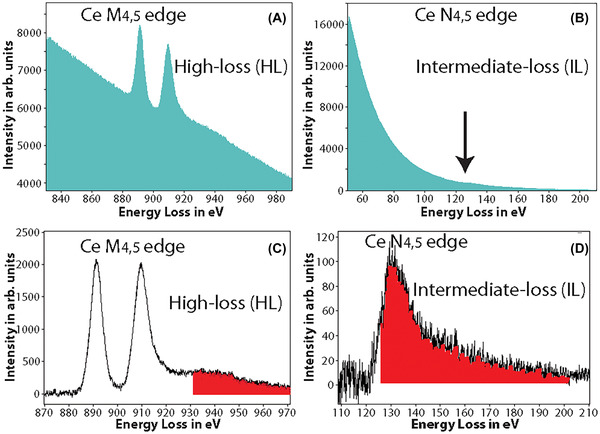
(A) The raw spectrum acquired at the high‐loss M4,5 edge of Ce, the two white line peaks with onset ∼883 eV is clearly visible. (B) The raw spectrum acquired at the intermediate‐loss N4,5 edge of Ce, edge is barely visible as a small bump in the spectrum (see arrow direction). (C) The background subtracted spectrum of (A), the M4,5 edge of Ce has a high signal‐above‐background ratio (SBR). The red‐shaded region shows the Ce extended energy‐loss fine structure (EXELFS) bleed into the Pr edge. (D) The background subtracted spectrum of (B), the N4,5 edge of Ce is now clearly visible, though with a much lower SBR. The red‐shaded region shows the part of the Ce edge that bleeds into the Pr edge
As outlined above, the total intensity and the SNR are significantly better at the intermediate‐loss region than at the high‐loss region. However, for edge visibility and detection, in addition to a good SNR, it is important to have a good signal‐above‐background ratio (SBR).3 Therefore, if the concentration of the lanthanide metal is low, then in spite of the good SNR, the SBR of the N4,5 edge can be so low that it cannot be distinguished from the background, and obtaining an elemental map at this edge will not be feasible. Also, it should be emphasised that, unlike M4,5 which is distinct and well separated for the lanthanides, the N4,5 edge has significant overlap from edge of adjacent lanthanides. The instance, the N4,5 edge of Ce (onset at 110 eV) will be difficult to distinguish from N4,5 edge of Pr (onset at 113 eV); for a more detailed discussion on this, please see the supplementary section ‘Criteria for edge selection in EFTEM elemental mapping’. However, this should not limit one to obtain EFTEM elemental maps of lanthanides at N4,5 edge, when there is no overlapping edge from other lanthanides or endogenous elements in the sample.
Figure 3(A) shows the conventional TEM image of mitochondrial matrix‐APEX2 labelled with Ce2‐DAB (second generation), Figures 3(B) and 3B’) show the Ce spectra of high‐loss M4,5 edge and the intermediate‐loss N4,5 edge obtained on this region. A high‐loss and an intermediate‐loss elemental maps were acquired on the sample at the same dose rate and total dose (total dose for the high‐loss acquisition was ∼3% higher than the intermediate‐loss acquisition). Figures 3(C)–(C’’) and 3(D)–(D’’) show the aligned and summed preedges and postedge of the high‐loss and intermediate‐loss acquisition, respectively. It is very clear comparing these two sets of images that the preedges and the postedge of the intermediate‐loss show superior SNR and detail compared to the high‐loss, as is expected. For the intermediate loss, the mitochondria in the postedge image (Figure 3D’’) looks sharper than the preedge images (Figures 3D and D’) because the mitochondrial matrix is loaded with Ce, and there is an increase in signal in this region relative to the background. However, it is for the high loss that the differences between the postedge image (Figure 3C’’) and the preedge images (Figures 3C and 3C’) are most striking. The preedge images at high‐loss are very blurry and noisy in comparison to the postedge. The reason for this striking difference at the high‐loss is the much higher SBR of the M4,5 edge in comparison to the N4,5 edge. The SBR measured from the spectra for the signal and background integrated for a 30 eV window is 1.0 and 0.3 for the high‐loss and the intermediate‐loss region, respectively.
FIGURE 3.
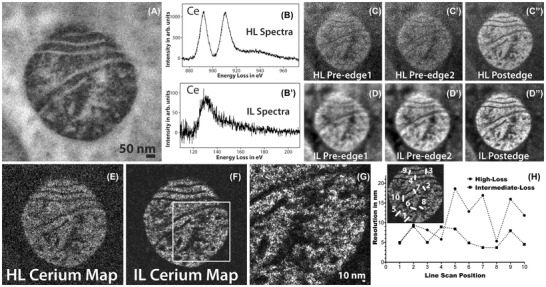
(A) Conventional TEM image of mitochondrial matrix‐APEX2 labelled with Ce2‐DAB (second generation). (B) The background subtracted high‐loss spectrum acquired on the sample showing the M4,5 edge of Ce. (B’) The background subtracted intermediate‐loss spectrum acquired on the sample showing the N4,5 edge of Ce. (C), (C’), (C’’) The preedge 1, preedge 2 and postedge acquired for the high‐loss M4,5 cerium edge at 815 eV, 855 eV and 899 eV, respectively. Each energy‐plane is a sum of 3 images acquired at an exposure of 100 sec/image. (D), (D’), (D’’) The preedge 1, preedge 2 and postedge acquired for the intermediate‐loss N4,5 cerium edge at 75 eV, 98 eV and 138 eV, respectively. Each energy‐plane is a sum of 29 images acquired at an exposure of 10 seconds/image. The dose and dose rate were nearly the same for both the high‐loss and intermediate‐loss acquisitions. (E) The high‐loss Ce elemental map. (F) The intermediate‐loss Ce elemental map. (G) Magnified view of the intermediate‐loss Ce elemental map. (H) The relative resolution comparison between the high‐loss and the intermediate‐loss Ce elemental map, measured by the width of the intensity between 25% and 75% of the peak, for 10 different regions taken exactly the same position of the sample for both the maps (see the inset). To enhance image display, a Gaussian blur of radius 1 was applied to the conventional image and Gaussian blur of radius 2 was applied to the EFTEM images and maps
Figures 3(E) and (F) show the elemental maps computed by the 3‐window method for the high‐loss and the intermediate‐loss, respectively. Both of which were acquired as a series with shorter exposure/energy plane. The intermediate‐loss was acquired at an exposure of 10 seconds/image; the high‐loss required much longer exposures of 100 seconds/image to be able to accumulate enough signal. The complete details of the acquisition can be found in the supplementary section. The intermediate‐loss elemental map is considerably sharper with a higher SNR; the SNR for the intermediate‐loss map was 7.4 and for the high‐loss map was 1.8, when calculated on the same region of the respective maps. It has been suggested that in a plot of the intensity distribution across an edge in an image, the measure of the width of the intensity between 0.25 and 0.75 of its peak value can be appropriated as the resolution.58 The edge width is proportional to the beam broadening due to the point spread function (PSF) of the imaging system. The region between the matrix and the cristae or the exterior background in the elemental maps (Figures 3E and F) serves as an edge for the measurement of the achieved resolution in the intermediate‐loss and high‐loss map. It should be cautioned that unlike greyscale conventional TEM/STEM images, where the intensity on either side of the edge is reasonably uniform, the pixel intensity of elemental maps vary significantly from one pixel to another because the intensity is proportional to the concentration of the element in that pixel. Therefore, measuring resolution from the edge step profile for elemental maps can be misleading. However, if the edge step profile is taken on precisely the same region of the image for both the high‐loss and the intermediate‐loss maps, then this can be used as a measure of relative degradation of resolution between the two maps. We obtained edge profiles at 10 precise same locations of the sample on both the maps; width (nm) of the intensity between 0.25 and 0.75 of its peak value are shown in Figure 3(E).
The resolution from this calculation for the high‐loss and the low‐loss maps are 11.0 ± 3.0 and 6.1 ± 2.0 nm, respectively. We would like to emphasise that this only implies that the resolution of the map acquired at the intermediate‐loss region is ∼1.8 times better than the map acquired at the high‐loss region and does not make any claim that these values are the absolute achievable resolution for the maps acquired at the respective energy‐loss region.
As demonstrated above, both the SNR and the resolution of the elemental map are somewhat better at intermediate‐loss than at high‐loss. The correctness of the power law background subtraction at the intermediate‐loss is not well known. However, there can be some speculation that the contrast that is seen in the intermediate‐loss elemental map may be density contrast of the heavily stained region containing cerium + ruthenium (secondary fixative), erroneously being interpreted as a cerium elemental map. On the other hand, for the high‐loss 3‐window method (energy‐loss > 200 eV), the accuracy of the power‐law background subtraction, and the optimisation of the position and width of the energy‐loss EFTEM images are well established.14 Therefore, there needs to be additional validation that the contrast that is seen in the intermediate‐loss Ce map is indeed an elemental contrast. To test this hypothesis, a control sample was made by labelling mitochondrial matrix‐APEX2 with unmodified DAB (i.e. with no bound lanthanide). The intensity of the mitochondrial matrix staining with DAB was comparable to that of the Ce2‐DAB sample, but containing only ruthenium and no cerium. If an intermediate‐loss map is acquired on the control sample with the same experimental settings as that of the mitochondrial matrix‐APEX2 labelled with Ce2‐DAB (Figure 3F), and if the map shows positive contrast for the matrix region of the mitochondria, then it can be concluded that contrast seen in Figure 3(F) is not a true Ce elemental contrast. Figures 4(A)–(D) show a high‐loss and intermediate‐loss map acquired using the 3‐window method on the control sample, with the same spectrometer settings as in Figure 3 (see the supplementary section for details). The mitochondrial matrix in the high‐loss map (Figure 4C) shows a faint positive contrast indicating an under subtraction of the background, and in the intermediate‐loss map (Figure 4D), it shows a negative contrast due to over subtraction of the background. Also, in Figure 4(D), although the mitochondrial matrix shows negative contrast, the spherical stained structures show positive contrast (see where the arrows point). Therefore, it can be concluded that in this case, mitochondrial matrix showing a negative contrast and not a positive contrast (i.e. density contrast) would have led to a partial suppression of the Ce elemental signal, if the sample did contain Ce (control sample does not contain Ce). Figures 4(E)–(H) illustrate another example of high‐loss and intermediate‐loss elemental maps acquired on the control sample by the 3‐window method; in this case, the intermediate‐loss map was acquired at a slightly different spectrometer settings than the previous example in Figure 4(D). The intermediate‐loss map was acquired for slit width of 15 eV instead of 20 eV, and the preedge 1, preedge 2 and postedge were acquired at 84 eV, 102 eV and 135 eV, respectively. In the previous example (Figure 4D), the preedge 1, preedge 2 and postedge were acquired at 75 eV, 98 eV and 138 eV, respectively. The high‐loss map (Figure 4G) shows a faint positive contrast as in the previous example (Figure 4C); however, the intermediate‐loss map (Figure 4H) now shows a positive contrast instead of a negative contrast for the mitochondria. It can be deduced from Figures 4(D) and (H) that the intermediate‐loss elemental maps are more susceptible to inconsistencies in background subtraction even for small changes in the slit width or position. The lower SBR of the intermediate‐loss N4,5 edge is the primary reason for this contrast inversion.
FIGURE 4.
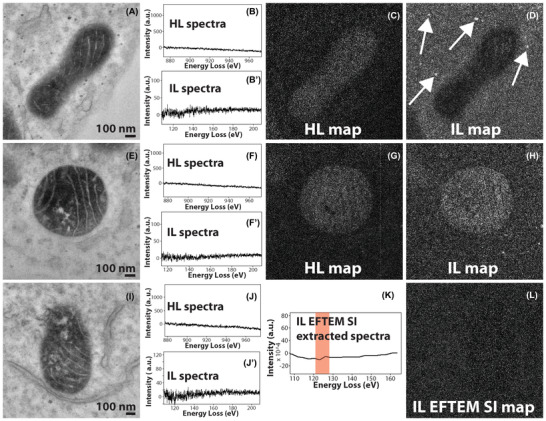
(A) Conventional TEM image of the control sample of mitochondrial matrix‐APEX2 labelled with plain DAB (no Ce or any other lanthanides). (B), (B’) The background subtracted high‐loss and intermediate‐loss spectrum acquired on the mitochondrial matrix region of (A), confirming the absence of Ce in the sample. (C) The high‐loss elemental map computed by the 3‐window method, for preedge 1, preedge 2 and postedge obtained for a slit width of 30 eV at 815 eV, 855 eV and 899 eV, respectively. The image shows a positive contrast for the matrix region, implying an under subtraction of the background. (D) The intermediate‐loss elemental map computed by the 3‐window method, for preedge 1, preedge 2 and postedge obtained for a slit width of 20 eV at 75 eV, 98 eV and 138 eV, respectively. The image shows a negative contrast for the matrix region, implying an over subtraction of the background. However, the spherical stained structures show a positive contrast (see the arrows), implying inconsistencies in background subtraction. (E) Conventional TEM image of a different region of the same control sample. (F), (F’) The background subtracted high‐loss and intermediate‐loss spectrum acquired on the mitochondrial matrix region of (D), confirming the absence of Ce in the sample. (G) The high‐loss elemental map computed by the 3‐window method, for preedge 1, preedge 2 and postedge obtained for a slit width of 30 eV at 815 eV, 855 eV and 899 eV, respectively. The image shows a positive contrast for the matrix region, implying an under subtraction of the background. (H) The intermediate‐loss elemental map computed by the 3‐window method, for preedge 1, preedge 2 and postedge obtained for a slit width of 15 eV at 84 eV, 102 eV and 135 eV, respectively. The image shows a positive contrast for the matrix region, implying an under subtraction of the background. (I) Conventional TEM image of a different region of the same control sample. (J), (J’) The background subtracted high‐loss and intermediate‐loss spectrum acquired on the mitochondrial matrix region of (G), confirming the absence of Ce in the sample. (K) The virtual intermediate‐loss spectrum extracted from the EFTEM SI image‐series, corroborating the results in (H’). (L) The intermediate‐loss elemental map computed by the EFTEM SI method, for an acquisition acquired from 90 to 150 eV, for a slit width of 8 eV and an energy step of 3 eV. The signal was integrated from the region represented by the red box in (K). The image shows a no contrast and only noise for the matrix region, implying a correct subtraction of the background. To enhance image display, a Gaussian blur of radius 1 was applied to the conventional images and Gaussian blur of radius 2 was applied to the EFTEM maps
From the discussion presented above, it is evident that the best technique to acquire an elemental map at the intermediate‐loss region is the EFTEM Spectrum Imaging (EFTEM SI).32, 50, 51, 59 This technique involves acquiring a series of EFTEM images with a narrow slit, and with the spectrometer continuously stepping through energies (or energy‐loss), from the region representing the edge to the region representing the background (prior to the edge onset). The spectrometer is stepped in such a way that there is a slight energy overlap between successive acquisitions. From the thus acquired EFTEM SI image series, a spectrum can be virtually extracted using the SI picker tool in Digital Micrograph (Gatan, Inc). Since this extracted spectrum directly correlates to the image series, the user has complete flexibility to choose which region (i.e. which subset of images) contributes to the background and which region contributes to the elemental signal. Additionally, the user can compensate for any chemical shifts or spectrum energy shifts postacquisition.
Figures 4(I)–(L) show the EFTEM SI intermediate‐loss acquisition on the same control sample shown in the previous two examples (Figures 4A–H). Figures 4(J) and J’ are the high‐loss and intermediate‐loss background subtracted spectra that confirm that there is no Ce in the sample. Figure 4(K) is the background subtracted virtual spectrum extracted from the EFTEM SI image series, also confirming absence of Ce. Figure 4(L) displays the Ce intermediate‐loss elemental map that was computed from the energy‐loss region indicated by the red box in Figure 4(K), that is, the region that would have contained the N4,5 edge of Ce (the control does not contain Ce). This map shows neither a positive or a negative contrast at the mitochondrial matrix region and is only noise, clearly authenticating the accuracy of background extrapolation of the EFTEM SI technique over the 3‐window method.
Figure 5 illustrates some examples of EFTEM SI intermediate‐loss elemental maps of three different cellular targets, namely using mitochondrial matrix‐directed APEX2, histone H2B‐Nucleosome and EdU‐DNA labelled with three different lanthanides: cerium, lanthanum and neodymium, respectively. This demonstrates the important capability advanced by this method to provide chemical identity of a specific genetically introduced probe providing a base for lanthanide‐based identification by analytical EM in a background of context‐highlighting electron density free of the chemical label.
FIGURE 5.
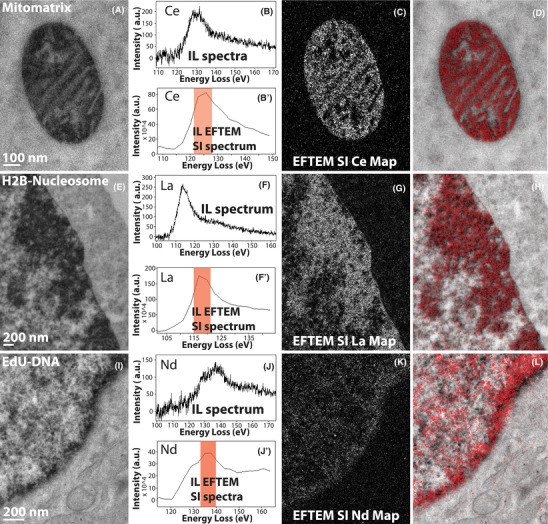
(A) Conventional TEM image of mitochondrial matrix‐APEX2 labelled with Ce2‐DAB (second generation). (B) The background subtracted intermediate‐loss spectrum acquired on the mitochondrial matrix region of (A), confirming the presence of Ce in the sample. (B’) The virtual intermediate‐loss spectrum extracted from the EFTEM SI image‐series, corroborating the results in (B). (C) The intermediate‐loss Ce elemental map computed by the EFTEM SI, the region (or images) contributing to the signal is enclosed in the red box in (B’). (D) Single colour EM. (E) Conventional TEM image of MiniSOG‐H2B labelled with La2‐DAB (second generation). (F) The background subtracted intermediate‐loss spectrum acquired on the nucleus of (d), confirming the presence of La in the sample. (F’) The virtual intermediate‐loss spectrum extracted from the EFTEM SI image‐series, corroborating the results in (E). (G) The intermediate‐loss La elemental map computed by the EFTEM SI, the region (or images) contributing to the signal is enclosed in the red box in (F’). (H) Single colour EM. (I) Conventional TEM image of DNA incubated with EdU and clicked with Fe‐TAML‐azide for oxidation of Nd‐DAB2 (first‐generation lanthanide DAB). (J) The background subtracted intermediate‐loss spectrum acquired on the nucleus of (G), confirming the presence of Nd in the sample. (J’) The virtual intermediate‐loss spectrum extracted from the EFTEM SI image‐series, corroborating the results in (H). (K) The intermediate‐loss Nd elemental map computed by the EFTEM SI, the region (or images) contributing to the signal is enclosed in the red box in (J’). (L) Single colour EM. To enhance image display, a Gaussian blur of radius 1 was applied to the conventional image and Gaussian blur of radius 2 was applied to the EFTEM images and maps
Figures 5(A)–(D) demonstrate the acquisition of EFTEM SI intermediate‐loss cerium elemental map on the mitochondrial matrix‐APEX2 labelled with Ce2‐DAB. Figure 5(B) shows the real background subtracted intermediate‐loss spectrum acquired on the mitochondrial region of (A), showing presence of Ce. In Figure 5(B’), the virtual spectrum extracted from the EFTEM SI image‐series, and subsequently background subtracted can be seen. Though the extracted spectrum of Figure 5(B’) is of a lower energy resolution than the actual spectra of Figure 5(B), the two closely match on the general shape and profile of the N4,5 edge. Computing the elemental map with the aid of the spectrum in Figure 5(B’), with the flexibility to choose parameters postacquisition, gives undisputable confidence in the correctness of the final result. Figure 5(C) shows the Ce elemental map, computed from the energy‐loss region indicated by the red box and Figure 5(D) displays the colour EM image of the pseudo‐coloured elemental map overlaid over the conventional TEM image. The colour EM image was generated by an ImageJ plugin that we developed, and this algorithm has been described elsewhere.23
The main drawback of acquiring elemental maps by the EFTEM SI method over the 3‐window method is that it necessitates the use of a narrow slit width and with an energy overlap between successive images, thereby requiring a far higher electron dose. For example, the EFTEM SI intermediate‐loss elemental map acquisition in Figure 5(C), required 2.6 times the dose of the 3‐window acquisition in Figure 3(F), for an elemental map with substantially lower SNR. The SNR of the EFTEM SI elemental map in Figure 5(C) is 1.6 in comparison to the SNR of 7.4 for the 3‐window method in Figure 3(F). However, the EFTEM SI acquisition in this case was oversampled, and considerable reduction in dose can be achieved by making subtle changes in the acquisition parameters. To illustrate, the EFTEM SI intermediate‐loss elemental map in Figure 5(C) was acquired between an energy‐loss of 90 eV to 150 eV, with a slit width of 8 eV and an energy step of 3 eV, resulting in a total of 21 images. The region that used for background subtraction was 98.6–107.1 eV and that for the Ce signal was 121.4–127.1 eV. The regions from 90 to 98.5 eV and 121.2 to 150 eV did not contribute in any way for the computation of the elemental map. Therefore, an acquisition from ∼94 to 134 eV should be sufficient and is wide enough to compensate for any chemical shifts (−2 eV, +7 eV). An EFTEM SI acquisition with a 5 eV energy step instead of 3 eV would reduce the total number of images in the EFTEM SI stack from 21 to 9, reducing the total dose by ∼60%. This would then make the dose required for the EFTEM SI acquisition comparable to the dose required for the 3‐window method.
Figures 5(E)–(F) illustrate the EFTEM SI intermediate‐loss lanthanum elemental map acquisition on the MiniSOG‐H2B labelled with La2‐DAB. The actual spectrum (Figure 5F) and the spectrum extracted from the EFTEM SI stack (Figure 5F’) show excellent correlation in shape and profile. The lanthanum elemental map shows good signal with a clean background in Figure 5(G), and Figure 5(H) shows the corresponding colour overlay. Figures 5(I)–(L) show the EFTEM SI intermediate‐loss neodymium elemental map acquired on the EdU‐DNA labelled with Nd‐DAB2. The actual spectrum and the extracted EFTEM SI spectrum show good correlation similar to the previous two examples, but the signal in the neodymium elemental map (Figure 5K) is faint. The reason for the poor neodymium signal is that, unlike the previous two examples, this sample was labelled with first‐generation lanthanide DAB rather than the second generation. Therefore, in this case, there is four times less metal (i.e. Nd) per DAB, with corresponding lower signal amplification, and therefore the signal‐above‐background (SBR) is low. To accommodate for the low SBR, three sets of EFTEM SI stack were acquired on the sample, subsequently aligned and added to create the summed EFTEM SI stack to improve the SNR.51 Therefore, even for cases where the concentration of the lanthanide is low, EFTEM SI method of acquiring intermediate‐loss elemental maps is still possible.
The main limitation of working at the N4,5 edge instead of the M4,5 edge for the lanthanides is that these have very broad edge profiles, extending well into the edge profiles of many adjacent lanthanides (see Figure 2). Therefore, from the perspective of colour EM, extending it from single colour to multicolour, where the spectral signal from two or more lanthanides each labelling a distinct macromolecule/protein can be unambiguously detected and separated,23 would be difficult. The immediately ensuing question is what is the degree and scale of the spectral overlap at the N4,5 edge region, if the two (or more) lanthanides are chosen such that they are well separated in atomic number, like La (Z = 57) and Yb (Z = 70). In such a scenario will it be possible to do multicolour EM at the intermediate‐loss region? This premise can be tested easily by recording an EFTEM SI acquisition of the H2B‐Nucleosome sample labelled with La2‐DAB, starting from the La N4,5 edge (onset at 99 eV) and continuing through the Yb N4,5 edge (onset at 185 eV). Subsequent computation of the elemental map at the La N4,5 edge will show the genuine La signal in the sample, while computation of the elemental map at the Yb N4,5 edge will show the bleed‐through of the La signal into the Yb N4,5 edge.
Figure 6(A) shows the conventional TEM image of H2B‐Nucleosome sample labelled with La2DAB. An EFTEM SI series was acquired on this region from 77 eV to 212 eV, with an energy slit width of 8 eV and energy step of 3 eV. After the background was fit and extrapolated, the signal was integrated for an energy‐width of ∼6eV, at different energy‐loss regions representing the edge of La (115.2–121 eV), Ce (121–126.9 eV), Nd (132.8–138.6 eV), Gd (150.4–156.2 eV) and Yb (194.4–200.3 eV). The La EFTEM SI map shown in Figure 6(B) displays strong La signal at the H2B region of the nucleus as expected. The Ce EFTEM SI map (Figure 6C) of the same region indicates that there is significant bleed‐through of the La signal into the Ce edge, but it gets fainter at the Nd edge (Figure 6D) and is almost negligible at the Gd edge (Figure 6E) and Yb edge (Figure 6F). The multiple linear least‐squares (MLLS) fitting is a powerful technique in EELS/EFTEM to separate overlapping edges by precisely mapping out the spectral shape resulting from fitting the spectra extracted from the EFTEM SI series to a reference spectra.60 Figures 6(G) and (H) show the MLLS fit algorithm of the EFTEM SI series to a previously acquired spectra of La and Ce, respectively. The bleed‐through of the La signal into the Ce intermediate‐loss region in Figure 6(C) is completely removed by applying the MLLS technique. This clearly shows that signal from adjacent lanthanides can indeed be effectively separated at the intermediate‐loss region by using advanced computational techniques like MLLS. The panels 6g and 6h were processed after a bin 4 to speed up the computation and the MLLS subroutine of Digital Micrograph (Gatan, Inc) was used to process the datasets. Although this paper does not provide any results of multicolour EM, the discussion above gives some confidence that it can be done in the intermediate‐loss region. We are currently in the process of chemically synthesising DAB chelates of lanthanides, other than the three listed in this manuscript (La, Ce and Nd) such as Gd and Yb, and will explore multicolour EM at the intermediate‐loss region in a future manuscript.
FIGURE 6.

(A) Conventional TEM image of H2B‐Nucleosome sample labelled with La2DAB. (B) La EFTEM SI map, with strong La signal as expected. (C) Ce EFTEM SI map of the same sample indicates that there is significant bleed‐through of the La signal into the Ce edge, that gets fainter at the Nd edge (D) and is almost negligible in the Gd edge (E) and Yb edge (F). La (G) and Ce (H) maps computed with the multiple linear least‐squares (MLLS) fitting to La and Ce spectra acquired separately. The MLLS subroutine of Digital Micrograph (Gatan, Inc) is a linear fitting algorithm that can be applied to discriminate overlapping edges. With MLLS fitting, the bleed‐through of the La signal into the Ce edge can be effectively removed (H). The MLLS fit was applied to images after binning by 4 (G, H); all other panels are unbinned images
4. CONCLUSION
As an extension of the technique of colour EM, we have expanded both the labelling chemistry of the lanthanide‐chelated DABs and the different strategies of targeting specific subcellular structures/macromolecules. The chemical synthesis of the novel second‐generation DAB‐lanthanide chelates has been demonstrated. These second‐generation DAB‐lanthanides bind two chelated lanthanide ions per DAB molecule, instead of two DAB molecules chelated to a single lanthanide ion as in the first‐generation lanthanide‐DABs. Therefore, it provides for a 4× increase in the number of lanthanide atoms for every oxidised DAB molecule, resulting in significant signal amplification in the EELS core‐loss signal. Additionally, we introduced the idea of using the N4,5 edge (intermediate‐loss) instead of the M4,5 edge (high‐loss) for the acquisition and computation of the lanthanide elemental maps. The substantially greater total characteristic signal due to the much higher inelastic scattering cross‐sections at the intermediate‐loss region provides a compelling reason for this strategy. For the same acquisition parameters, the intermediate‐loss map provides ∼4× increase in signal‐to‐noise and ∼2× increase in resolution, in comparison to the high‐loss map. However, due to the much lower signal‐above‐background at the intermediate‐loss region, the intermediate‐loss elemental maps of the lanthanides are more prone to artefacts due to the inconsistencies in the background subtraction. Most of these artefacts and issues in the lanthanide intermediate‐loss elemental map can be eliminated, if the maps are acquired using the more sophisticated EFTEM Spectrum Imaging (EFTEM SI) instead of the 3‐window method. The intermediate‐loss EFTEM SI elemental maps were used to generate colour EM images of mitochondrial matrix‐directed APEX2 labelled with second‐generation Ce2‐DAB, histone H2B‐Nucleosome labelled with second generation La2‐DAB and EdU‐DNA labelled with first‐generation Nd‐DAB2. All the examples demonstrated in the paper are of single‐colour EM only. The main limitation of using N4,5 edge or intermediate‐loss map for the lanthanides is the broader edge profile includes large overlapping regions with adjacent lanthanide edge profiles and hinders extension of this technique to enable multicolour EM, where 2 or more lanthanide chelated DABs are simultaneously used to label different subcellular targets. However, if there is sufficiently large difference in Z between the lanthanides, for example, lanthanum and gadolinium, or cerium and ytterbium, then multicolour EM should be potentially possible in the intermediate‐loss region and will be explored in the future.
4.1. Criteria for edge selection in EFTEM elemental mapping
An element can have several ionisation edges, some being major edges and others being minor edges. For example, cerium (Ce) has major edges at onset of 20 eV (O2,3), 110 eV (N4,5), 883 eV (M5) and 901 eV (M4); minor edges at onset of 207 eV (N2,3), 290 eV (N1), 1185 eV (M3), 1273 eV (M2) and 1435 eV (M1).45 The edge onset energy can vary by about (−2 eV, +7 eV) due to bonding effects called chemical shifts, in addition to the spread due to chromatic aberration of the imaging system, which can range from few electronvolts to tens of microelectronvolts depending on the instrument.2 The edge that is selected for elemental mapping/quantification needs to fulfil certain requirements. First, the edge should be a major edge in the range ∼100–2000 eV,3 the optimum EELS energy region being within 1000 eV.1 Second, the edge should have a distinct shape, for example, a sawtooth or peaked at threshold like white‐line for easy identification.3 Finally, the edge location (in eV) should be distinct with no overlapping edge due to other adjacent elements in the periodic table.
For Ce, as described above there are potentially four major edges; however the first major edge O2,3 at 20 eV does not satisfy any of the three conditions. However, using complex instrumentation like monochromators fitted to a state‐of‐the‐art TEM and with the aid of mathematical tools such as nonlinear multivariate statistical analysis, ability to differentiate material compositions based on subtle differences in edge <50 eV has been shown.61, 62
The N4,5 edge (see Figures 2B and D) satisfies the first two conditions: it is a major peak > 100eV and has a sharp and distinct sawtooth shape. However, the N4,5 edge for Ce is at 110 eV, and for praseodymium (Pr) it is at 113 eV, and due to the chemical shifts and the chromatic aberrations of the imaging system, unambiguous identification of the element solely based on this edge is not possible. The last two of these edges, the M5 and the M4 edge are very close to each other (see Figures 2A and C), and for the purpose of elemental mapping, they are generally considered together as the M4,5 edge. The M4,5 edge for Ce has the so called white lines shape, that starts at ∼883 eV and extends to ∼920 eV. This edge is sufficiently separately from the M4,5 edge of Pr, which starts only at ∼931 eV is the preferred choice for elemental mapping/quantification. In fact, we have previously shown a multicolour EM of two astrocyte processes contacting the same synapse, with one astrocyte being labelled with Cerium‐conjugated DAB and the other with Praseodymium conjugated DAB.23 It should be pointed out that, although the Cerium M4,5 edge is explicitly separated from the Praseodymium, the extended energy‐loss fine structure (EXELFS) of Ce bleeds into the Pr edge and beyond (see the area shaded in red in Figure 2C); this spectral bleed‐through has to be mathematically subtracted when maps of adjacent elements in the periodic table are computed.23
Alternatively, if there is already a priori information on the elemental composition of the sample, and the primary purpose of using EELS/EFTEM is not element identification but localisation or distribution of a particular element in the sample, then the criteria for the edge selection for the particular element can be slightly relaxed. For example, if a single cellular protein/organelle in cells was labelled by the deposition of only one lanthanide conjugated to diaminobenzidine, and the localisation of this protein/organelle is to be visualised by acquiring an EFTEM elemental map of the specific lanthanide metal, in a so‐called single colour EM. In such a scenario, the elemental map for the lanthanide can be potentially acquired on the intermediate‐loss region of the N4,5 edge instead of the high‐loss region of the M4,5 edge, provided that there are no overlapping edge from any of the endogenous elements in the sample.
4.2. Synthesis of Ln2‐DAB
2,2’‐Dibromo‐6,6’‐dinitrobenzidine, 2: 2,2’‐dinitrobenzidine (2.7 g, 10 mmol) was suspended in glacial acetic acid at 100°C with stirring, cooled to room temperature and bromine (1.13 mL, 22 mmol) in glacial acetic acid (10 mL) was added dropwise. After 30 minutes at room temperature, LC‐MS showed reaction was incomplete, so an additional 0.5 mL of bromine was added. After 30 minutes, water (100 mL) was added and the crude product collected by filtration, air‐dried and boiled in 95% EtOH (100 ml), cooled and refiltered to give the product 2 as an orange powder (2.34 g, 54%). ES‐MS (m/z) [M]+, [M+ H]+ for C12H9Br2N4O4, 430.9, 432.9. Found, 431.0, 433.0.
Diethyl 2,2’‐dinitrobenzidine‐2,2’‐bis‐propenoate, 3: Compound 2 (1.1 g, 2.5 mmol), ethyl acrylate (2. 5 mL, 23.5 mmol), triethylamine (1 mL, 7.1 mmol). Tri(o‐tolyl) phosphine (30 mg, 0.1 mmol) and palladium acetate (10 mg, 0.045 mmol) suspended in dry DMF were heated with stirring at 80°C for 3 hours and filtered hot through a glass sinter. After evaporation to dryness, the solid was suspended in 95% EtOH (50 mL) and filtered to yield the product as a brick‐red solid (1.17 g, 99%). ES‐MS (m/z) [M]+, [M+ H]+ for C22H23N4O8, 471.1. Found, 471.2.
Diethyl 2,2’‐dinitro‐N,N’‐tetra(t‐butyloxycarbonyl)benzidine‐2,2’‐bis‐propenoate, 4: Compound 3 (1.0 g, 2.1 mmol) was heated at 50°C in dry DMF (40 mL) with di‐t‐butyl carbonate (2.3 g, 10. 6 mmol) and DMAP (50 mg, 0.41 mmol) for 3 hours. The reaction mix was evaporated and the product purified by silica gel column chromatography by eluting with 10–25% ethyl acetate‐hexane to give an oil (1.69, 92%). ES‐MS (m/z) [M]+, [M+ Na]+ for C42H54N4NaO16, 893.3. Found, 893.4.
2,2’‐Diamino‐N,N’‐tetra(t‐butyloxycarbonyl)benzidine‐2,2’‐bis‐propanoic acid, 5: Compound 4 (1.69 g, 1.93 mmol) was hydrogenated in ethyl acetate‐ethanol (1:1) with Pd/C for 6 days at rt and pressure. After filtration and evaporation to dryness, the residue was dissolved in dioxane (10 mL) and methanol (10 mL) followed by aqueous 1M‐ NaOH (3 mL) was then added. After overnight reaction under N2, the organic solvents were removed by evaporation. Water and additional NaOH was then added until all solid was dissolved. After LC‐MS indicated complete saponification, glacial HOAc was added dropwise to give a precipitate that was collected by filtration after chilling in ice. The product was dried over P2O5 in vacuum overnight to give the desired product as an off‐white solid (1.40 g, 96%). ES‐MS (m/z) [M]+, [M+ H]+ for C38H55N4O12, 759.4. Found, 759.4. LC‐MS showed 20% of the material has lost one BOC group during saponification. Tri‐BOC, ES‐MS (m/z) [M]+, [M+ H]+ for C33H47N4O10, 659.3. Found, 659.3.
2,2’‐Diamino‐N,N’‐tetra(t‐butyloxycarbonyl)benzidine‐2,2’‐bis‐(propanoic succinimidyl ester), 6: Compound 5 (1.40 g, ∼2 mmol), N‐hydroxysuccinimide (0.58 g, 5 mmol) and N‐ethyl‐N′‐(3‐dimethylaminopropyl)carbodiimide (EDC; 0. 96 g, 5 mmol) were dissolved in dry DMF and kept overnight at room temperature. The desired products (tetra and tri‐Boc) were purified by prep RP‐HPLC using a water‐acetonitrile‐0.05% trifluoroacetic acid gradient and lyophilisation of the immediately frozen fractions. Yield of off‐white powder, 1.14 g (60%). Tetra‐Boc, ES‐MS (m/z) [M]+, [M+ H]+ for C46H61N6O16, 953.4. Found, 953.4. Tri‐Boc, ES‐MS (m/z) [M]+, [M+ H]+ for C41H53N6O14, 953.4. Found, 953.4.
N‐(N‐2‐aminoethylacetamido)‐ethanediamine‐N,N’,N’‐triacetic acid, 7. EDTA free acid (161 mg, 0.55 mmol) was heated in dry DMSO (4 mL) under nitrogen with a heat gun until dissolved. After cooling to room temperature, EDC (106 mg, 0.55 mmol) was added with stirring. After 30 minutes, N‐hydroxysuccinimide (63 mg, 0.55 mmol) was added followed by N‐Boc ethylenediamine (174 μL, 1.1 mmol) after an additional 30 minutes. The reaction was kept overnight, evaporated to dryness and purified by prep RP‐HPLC eluting with a gradient of water‐acetonitrile‐0.05% trifluoroacetic acid. Yield, 120 mg (27%) white solid. ES‐MS (m/z) [M]+, [M+ H]+ for C17H31N4O9, 435.2. Found, 435.3. The BOC group was removed by dissolving the solid in trifluoroacetic acid for 30 minutes, evaporation and lyophilisation of the resulting oil from 50% water‐acetonitrile‐0.05% trifluoroacetic acid.
Compound 8; 6 (130 mg, 0.14 mmol) was added to a solution of 7 (150 mg of BOC derivative cleaved as above, 0.35 mmol) in DMSO (1 mL) with triethylamine (0.24 mL, 1.75 mmol) at room temperature. After 24 hours, acetic acid (0.25 mL) was added and the product separated by prep RP‐HPLC eluting with a water‐acetonitrile‐0.05% trifluoroacetic acid gradient. Lyophilisation gave a white solid, 165 mg (84%). ES‐MS (m/z) [M]+, [M+ H]+ for C62H95N12O24, 1391.7. Found, 1391.8. BOC groups were removed by treating with TFA for 15 minutes at rt followed by immediate evaporation. The desired product was purified by prep RP‐HPLC eluting with a water‐acetonitrile‐0.05% trifluoroacetic acid gradient. Lyophilisation gave a white solid, 80 mg. ES‐MS (m/z) [M]+, [M+ H]+ for C42H63N12O16, 991.4. Found, 992.0. Titration with solutions of CeCl3 using arsenazo III pH 7.2 as endpoint indicator in 100 mM MOPS buffer gave 60% purity by weight indicating 6 TFA molecules are present in the resulting product.
Ln2‐DAB, 9: To a 2 mg/mL (1.2 mM) solution of 8 in 0.1 M sodium cacodylate pH 7.4, 25 μL LnCl3 (where Ln = La, Ce, Pr, Nd; 100 mM solutions in water, except Nd was a 100 mM solution in 0.1 N HCl) were added per mL and the pH adjusted to pH 7.4 with 1 M‐NaOH.
4.3. Sample preparation
4.3.1. Mitochondrial matrix‐APEX2 labelled with Ce2‐DAB (second‐generation lanthanide DAB) and DAB
HEK293T cells were cultured on imaging plates containing poly‐d‐lysine coated glass bottom No. 0 coverslips (P35GC‐0‐14C, MatTek Corporation). Cells were transiently transfected with Mitochondrial matrix‐APEX2 fusion using Lipofectamine 3000 (Life Technologies). APEX2 was fused to C‐ terminal fusion of mito matrix.5 After 16 hours of transfection, cells were fixed with 2% glutaraldehyde (18426, Ted Pella Incorporated) in 0.1 M sodium cacodylate buffer, pH 7.4 (18851, Ted Pella Incorporated) containing 1 mM CaCl2 for 5 minutes at 37°C and then on ice for 55 minutes. Fixative was removed and cells were rinsed with 0.1 M sodium cacodylate buffer pH 7.4 (5×1 minutes) on ice. On a set of plates, an enzymatic reaction with 1.2 mM Ce2‐DAB with 5 mM H2O2 (from 30% stock) in 0.1 M sodium cacodylate buffer, pH 7.4 containing 1 mM CaCl2 buffer solution at pH 7.4 for 5 minutes was completed. On a separate control plate of transfected cells was an enzymatic reaction of 2.5 mM DAB with 5 mM H2O2 in 0.1 M sodium cacodylate buffer, pH 7.4 containing 1 mM CaCl2 buffer solution at pH 7.4 for 5 minutes. After reactions, all plates of cells were rinsed with 0.1 M sodium cacodylate buffer pH 7.4 (5×1 minutes) on ice and then were posted fixed with 0.100% ruthenium tetroxide (20700‐05, Electron Microscopy Sciences) containing 2 mM CaCl2 and in 0.1 M sodium cacodylate buffer, pH 7.4 for 20 minutes.
4.3.2. MiniSOG‐H2B labelled with La2‐DAB (second‐generation lanthanide DAB)
HEK293T cells were cultured on imaging plates containing poly‐d‐lysine coated glass bottom No. 0 coverslips (P35GC‐0‐14C, MatTek Corporation). Cells were transiently transfected with miniSOG‐H2B fusion using Lipofectamine 3000 (Life Technologies). MiniSOG was fused to N‐terminus of H2B. After 16 hours of transfection, cells were fixed with 2% glutaraldehyde (18426, Ted Pella Incorporated) in 0.1 M sodium cacodylate buffer, pH 7.4 (18851, Ted Pella Incorporated) containing 1 mM CaCl2 for 5 minutes at 37°C and then on ice for 55 minutes. Fixative was removed, cells were rinsed with 0.1 M sodium cacodylate buffer pH 7.4 (5×1min) on ice and treated for 30 minutes in blocking buffer consisting of 50 mM glycine, 10 mM KCN and 5 mM aminotriazole to reduce nonspecific back‐ground reaction of diaminobenzidine (DAB). Preequilibration with second‐generation La2‐DAB was carried out in 0.1 M sodium cacodylate buffer, pH 7.4 containing 1 mM CaCl2 buffer solution at pH 7.4 that was added to the plates for 30 minutes following filtration with a 0.22 μm Millex 33 mm PES sterile filter (SLGSR33RS, Sigma‐Aldrich) at room temperature. Photooxidation was performed by blowing medical grade oxygen over the La2‐DAB solution and cells were illuminated using a standard FITC filter set (EX470/40, DM510, BA520) with intense light from a 150W xenon lamp. Illumination was stopped as soon as a light brown precipitate occurred after 8–10 minutes. After reaction, the La2‐DAB solution was removed and cells were rinsed with 0.1 M sodium cacodylate buffer pH 7.4 (5×1 minutes) on ice and then were posted fixed with 1% osmium tetroxide (19150, Electron Microscopy Sciences) containing 0.8% potassium ferrocyanide, 2 mM CaCl2 and in 0.1 M sodium cacodylate buffer, pH 7.4 for 30 minutes.
4.3.3. DNA labelled with EdU and clicked with Fe‐TAML‐azide for oxidation of Nd‐DAB2 (first‐generation lanthanide DAB)
Small epithelial airway cells (SEAC) were cultured on imaging plates containing poly‐d‐lysine coated glass bottom No. 0 coverslips (P35GC‐0‐14C, MatTek Corporation). SEAC were incubated with EdU (1149‐100, Click Chemistry Tools) the night before for 12 hours. The cells were fixed with 2% glutaraldehyde (18426, Ted Pella Incorporated) in 0.1 M sodium cacodylate buffer, pH 7.4 (18851, Ted Pella Incorporated) containing 1 mM CaCl2 for 5 minutes at 37°C and then on ice for 55 minutes. Fixative was removed and cells were washed with 0.1 M sodium cacodylate buffer pH 7.4 (5×2 minutes) on ice, with 1× PBS (2×2 minutes) at room temperature and rinsed two times quickly with 1%BSA‐1XPBS by using a 0.22 μm Millex 33 mm PES sterile filter (SLGSR33RS, Sigma‐Aldrich) at room temperature. Cells were click reacted with 1 mL solution containing and added in the order listed, 900 μL click buffer (50 mM HEPES pH 7.6 100 mM NaCl, 0.1% saponin), 10 μL CuSO4 (100 mM), 1 μL Fe‐TAML‐azide (14 mM stock, giving 28 μM, Mackay et al, manuscript in preparation) and 50 μL of freshly made sodium ascorbate (100 mM) and kept covered at room temperature for 60 minutes with protection from light. During the halfway point of the click reaction, an additional 50 μL of freshly made sodium ascorbate solution (100 mM) was added to the incubation solution. Cells were then washed with filtered 1%BSA‐1XPBS (2×2 minutes) at room temperature, 1× PBS (2×2 minutes) at room temperature, 50 mM Bicine‐100mM NaCl buffer at pH 8.3 (2×2 minutes) and reacted with 2.5 mM Nd2‐DAB in 50 mM Bicine‐100 mM NaCl buffer solution with 40 mM H2O2 (from 30%) at pH 8.3 for 15 minutes. Cells were then washed with filtered 1%BSA‐1XPBS (2×1 minutes) at room temperature, 1× PBS (2×1 minutes) at room temperature, 50 mM Bicine‐100 mM NaCl buffer at pH 8.3 (2×2 minutes), rinsed with 0.1 M sodium cacodylate buffer pH 7.4 (5×1 minutes) on ice and then were posted fixed with 1% osmium tetroxide (19150, Electron Microscopy Sciences) containing 0.8% potassium ferrocyanide, 2 mM CaCl2 and in 0.1 M sodium cacodylate buffer, pH 7.4 for 30 minutes.
4.4. EFTEM acquisition parameters
For the high‐loss EFTEM images of the mitochondrial matrix‐APEX2 labelled with Ce2‐DAB by the 3‐window method (see Figure 3E), a series of three images with a 100 second exposure/image was acquired by the DE‐12 detector at a frame rate of 0.04 fps and a magnification of 10 kX (pixel size 0.5 nm) for each energy window. The preedge 1, preedge 2 and postedge were acquired for a slit width of 30 eV at energy shifts of 815 eV, 855 eV and 899 eV, respectively. The EFTEM images were acquired at a dose rate of ∼1.2×10−4 PA/nm2 and the total dose for the acquisition was ∼6.8×105 e–/nm2.
For the intermediate‐loss EFTEM images of the mitochondrial matrix‐APEX2 labelled with Ce2‐DAB by the 3‐window method (see Figure 3F), a series of 29 images with a 10‐second exposure/image was acquired by the DE‐12 detector at a frame rate of 2 fps and a magnification of 10 kX (pixel size 0.5 nm) for each energy window. The preedge 1, preedge 2 and postedge were acquired for a slit width of 20 eV at energy shifts of 75 eV, 98 eV and 138 eV, respectively. The EFTEM images were acquired at a dose rate of ∼1.2×10−4 PA/nm2 and the total dose for the acquisition was ∼6.6×105 e–/nm2.
For the high‐loss EFTEM images of the control sample of mitochondrial matrix‐APEX2 labelled with plain DAB by the 3‐window method (see Figure 4C), a series of five images with a 100‐second exposure/image was acquired by the DE‐12 detector at a frame rate of 0.04 fps and a magnification of 10 kX (pixel size 0.5 nm) for each energy window. The preedge 1, preedge 2 and postedge were acquired for a slit width of 30 eV at energy shifts of 815 eV, 855 eV and 899 eV, respectively. The EFTEM images were acquired at a dose rate of ∼1.2×10−4 PA/nm2 and the total dose for the acquisition was ∼1.1×106 e–/nm2.
For the intermediate‐loss EFTEM images of the control sample of mitochondrial matrix‐APEX2 labelled with plain DAB by the 3‐window method (see Figure 4D), a series of 30 images with a 10‐second exposure/image was acquired by the DE‐12 detector at a frame rate of 2 fps and a magnification of 10 kX (pixel size 0.5 nm) for each energy window. The preedge 1, preedge 2 and postedge were acquired for a slit width of 20 eV at energy shifts of 75 eV, 98 eV and 138 eV, respectively. The EFTEM images were acquired at a dose rate of ∼1.2×10−4 PA/nm2 and the total dose for the acquisition was ∼6.8×105 e–/nm2.
For the high‐loss EFTEM images of the control sample of mitochondrial matrix‐APEX2 labelled with plain DAB by the 3‐window method (see Figure 4G), a series of five images with a 100‐second exposure/image was acquired by the DE‐12 detector at a frame rate of 0.04 fps and a magnification of 10 kX (pixel size 0.5 nm) for each energy window. The preedge 1, preedge 2 and postedge were acquired for a slit width of 30 eV at energy shifts of 815 eV, 855 eV and 899 eV, respectively. The EFTEM images were acquired at a dose rate of ∼1.2×10−4 PA/nm2 and the total dose for the acquisition was ∼1.1×106 e–/nm2.
For the intermediate‐loss EFTEM images of the control sample of mitochondrial matrix‐APEX2 labelled with plain DAB by the 3‐window method (see Figure 4H), a series of 30 images with a 10‐second exposure/image was acquired by the DE‐12 detector at a frame rate of 2 fps and a magnification of 10 kX (pixel size 0.5 nm) for each energy window. The preedge 1, preedge 2 and postedge were acquired for a slit width of 15 eV at energy shifts of 84 eV, 102 eV and 135 eV, respectively. The EFTEM images were acquired at a dose rate of ∼1.2×10−4 PA/nm2 and the total dose for the acquisition was ∼6.8×105 e–/nm2.
For the intermediate‐loss EFTEM images of the control sample of mitochondrial matrix‐APEX2 labelled with DAB by the EFTEM SI method (see Figure 4L), the stack was acquired by the DE‐12 detector from energy‐loss of 165 eV to 87 eV, with a slit width of 8 eV and energy step of 3 eV. The stack was acquired at a magnification of 12 kX (pixel size 0.4 nm), frame rate of 0.1 fps and exposure of 60 seconds per individual energy plane. The EFTEM images were acquired at a dose rate of ∼1.8×10−4 PA/nm2 and the total dose for the acquisition was ∼1.9×106 e–/nm2.
For the intermediate‐loss EFTEM images of the mitochondrial matrix‐APEX2 labelled with Ce2‐DAB by the EFTEM SI method (see Figure 5C), the stack was acquired by the DE‐12 detector from energy‐loss of 150–90 eV, with a slit width of 8 eV and energy step of 3 eV. The stack was acquired at a magnification of 12 kX (pixel size 0.4 nm), frame rate of 0.1 fps and exposure of 60 seconds per individual energy plane. The EFTEM images were acquired at a dose rate of ∼1.8×10−4 PA/nm2 and the total dose for the acquisition was ∼1.5×106 e–/nm2.
For the intermediate‐loss EFTEM images of the MiniSOG‐H2B labelled with La2‐DAB by the EFTEM SI method (see Figure 5G), the stack was acquired by the DE‐12 detector from energy‐loss of 78–144 eV, with a slit width of 8 eV and energy step of 3 eV. The stack was acquired at a magnification of 10 kX (pixel size 0.5 nm), frame rate of 0.1 fps and exposure of 60 seconds per individual energy plane. The EFTEM images were acquired at a dose rate of ∼2.6×10−4 PA/nm2 and the total dose for the acquisition was ∼2.2×106 e–/nm2.
For the intermediate‐loss EFTEM images of the EdU‐DNA labelled with Nd‐DAB2 by the EFTEM SI method (see Figure 5K), the stack was acquired by the DE‐12 detector from energy‐loss of 96 eV to 165 eV, with a slit width of 8 eV and energy step of 3 eV. The stack was acquired at a magnification of 10 kX (pixel size 0.5 nm), frame rate of 0.1 fps and exposure of 60 seconds per individual energy plane. The EFTEM images were acquired at a dose rate of ∼2.6×10−4 PA/nm2 and the total dose for the acquisition was ∼2.3×106 e–/nm2.
For the intermediate‐loss EFTEM images of the MiniSOG‐H2B labelled with La2‐DAB by the EFTEM SI method (see Figure 6B), the stack was acquired by the DE‐12 detector from energy‐loss of 77–212 eV, with a slit width of 8 eV and energy step of 3 eV. The stack was acquired at a magnification of 6 kX (pixel size 0.8 nm), frame rate of 0.1 fps and exposure of 60 seconds per individual energy plane.
Supporting information
FIGURE S1. Scheme of the synthesis of Ln2‐DAB. Reagents, solvents, conditions, (A) Br2, acetic acid, rt; (B) ethyl acrylate, Pd(OAc)2, (o‐tolyl)3phosphine, NEt3, DMF, 80°C; (C) BOC anhydride, DMAP, DMF, 80°C; (D) 1 atm H2, Pd‐C, EtOAc‐EtOH, rt; (E) NaOH, dioxane‐MeOH, rt; (F) HOSu, EDC, DMF, rt; (G) eda‐EDTA, NEt3, DMSO, rt; (H) TFA, rt; (I) LnCl3, pH 7 buffer, rt.
ACKNOWLEDGEMENTS
We would like to thank Rob Bilhorn and Benjamin Bammes of Direct Electron for help with the DE‐12 detector. We would like to thank David Mastronarde of University of Colorado Boulder for help with SerialEM. This work was supported by NIH grants R01GM086197 and R24GM137200.
Ramachandra R, Mackey MR, Hu J, et al. Elemental mapping of labelled biological specimens at intermediate energy loss in an energy‐filtered TEM acquired using a direct detection device. Journal of Microscopy. 2021;283:127–144. 10.1111/jmi.13014
Mark H. Ellisman and Stephen R. Adams are cosenior authors.
Contributor Information
Mark H. Ellisman, Email: mellisman@ucsd.edu.
Stephen R. Adams, Email: sadams@health.ucsd.edu.
REFERENCES
- 1.Knoll, M., & Ruska, E. (1932). The electron microscope. vol 78, p 318. Zeitschrift für Physik, 79(9–10), 699–699. [Google Scholar]
- 2.Carter, C. B., & Williams, D. B. (2009). Transmission electron microscopy. New York: Springer. [Google Scholar]
- 3.Egerton, R. F. (1996). Electron energy‐loss spectroscopy. New York: Plenum Press. [Google Scholar]
- 4.Goldstein, J. I., Newbury, D. E., Echlin, P., Joy, D. C., Lyman, C. E., Lifshin, E., … Michael, J. R. (2003). Scanning electron microscopy and microanalysis. New York: Springer. [Google Scholar]
- 5.Muller, D. A., Kourkoutis, L. F., Murfitt, M., Song, J. H., Hwang, H. Y., Silcox, J., … Krivanek, O. L. (2008). Atomic‐scale chemical imaging of composition and bonding by aberration‐corrected microscopy. Science, 319(5866), 1073–1076. [DOI] [PubMed] [Google Scholar]
- 6.Muller, D. A. (2009). Structure and bonding at the atomic scale by scanning transmission electron microscopy. Nature Materials, 8(4), 263–270. [DOI] [PubMed] [Google Scholar]
- 7.Varela, M., Lupini, A. R., van Benthem, K., Borisevich, A. Y., Chisholm, M. F., Shibata, N., … Pennycook, S. J. (2005). Materials characterization in the aberration‐corrected scanning transmission electron microscope. Annual Review of Materials Research, 35, 539–569. [Google Scholar]
- 8.Aronova, M. A., Kim, Y. C., Harmon, R., Sousa, A. A., Zhang, G., & Leapman, R. D. (2008). Three‐dimensional elemental mapping of phosphorus by quantitative electron spectroscopic tomography (QuEST) (Reprinted from J. Struct. Biol, vol 160, pg 35‐48, 2007). Journal of Structural Biology, 161(3), 322–335. [DOI] [PubMed] [Google Scholar]
- 9.Aronova, M. A., Kim, Y. C., Pivovarova, N. B., Andrews, S. B., & Leapman, R. D. (2009). Quantitative EFTEM mapping of near physiological calcium concentrations in biological specimens. Ultramicroscopy, 109(3), 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aronova, M. A., & Leapman, R. D. (2012). Development of electron energy‐loss spectroscopy in the biological sciences. MRS Bulletin, 37(1), 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Somlyo, A. P., & Shuman, H. (1982). Electron‐probe and electron‐energy loss analysis in biology. Ultramicroscopy, 8(1–2), 219–233. [DOI] [PubMed] [Google Scholar]
- 12.Egerton, R. F. (2012). Tem‐eels: A personal perspective. Ultramicroscopy, 119, 24–32. [DOI] [PubMed] [Google Scholar]
- 13.Grogger, W., Varela, M., Ristau, R., Schaffer, B., Hofer, F., & Krishnan, K. M. (2005). Energy‐filtering transmission electron microscopy on the nanometer length scale. Journal of Electron Spectroscopy, 143(2–3), 139–147. [Google Scholar]
- 14.Hofer, F., Grogger, W., Kothleitner, G., & Warbichler, P. (1997). Quantitative analysis of EFTEM elemental distribution images. Ultramicroscopy, 67(1–4), 83–103. [Google Scholar]
- 15.Lozano‐Perez, S., Bernal, V. D., & Nicholls, R. J. (2009). Achieving sub‐nanometre particle mapping with energy‐filtered TEM. Ultramicroscopy, 109(10), 1217–1228. [DOI] [PubMed] [Google Scholar]
- 16.Verbeeck, J., Van Dyck, D., & Van Tendeloo, G. (2004). Energy‐filtered transmission electron microscopy: An overview. Spectrochimica Acta Part B, 59(10–11), 1529–1534. [Google Scholar]
- 17.Leapman, R. D. (2017). Application of EELS and EFTEM to the life sciences enabled by the contributions of Ondrej Krivanek. Ultramicroscopy, 180, 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosman, M., & Keast, V. J. (2008). Optimizing EELS acquisition. Ultramicroscopy, 108(9), 837–846. [DOI] [PubMed] [Google Scholar]
- 19.Goping, G., Pollard, H. B., Srivastava, M., & Leapman, R. (2003). Mapping protein expression in mouse pancreatic islets by immunolabeling and electron energy loss spectrum‐imaging. Microscopy Research and Technique, 61(5), 448–456. [DOI] [PubMed] [Google Scholar]
- 20.Hunt, J. A., & Williams, D. B. (1991). Electron energy‐loss spectrum‐imaging. Ultramicroscopy, 38(1), 47–73. [Google Scholar]
- 21.Kothleitner, G., & Hofer, F. (2003). Elemental occurrence maps: A starting point for quantitative EELS spectrum image processing. Ultramicroscopy, 96(3–4), 491–508. [DOI] [PubMed] [Google Scholar]
- 22.Leapman, R. D. (2003). Detecting single atoms of calcium and iron in biological structures by electron energy‐loss spectrum‐imaging. Journal of Microscopy – Oxford, 210, 5–15. [DOI] [PubMed] [Google Scholar]
- 23.Adams, S. R., Mackey, M. R., Ramachandra, R., Palida Lemieux, S. F., Steinbach, P., Bushong, E. A., … Tsien, R. Y. (2016). Multicolor electron microscopy for simultaneous visualization of multiple molecular species. Cell Chemical Biology, 23(11), 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pirozzi, N. M., Hoogenboom, J. P., & Giepmans, B. N. G. (2018). ColorEM: Analytical electron microscopy for element‐guided identification and imaging of the building blocks of life. Histochemistry and Cell Biology, 150(5), 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scotuzzi, M., Kuipers, J., Wensveen, D. I., de Boer, P., Hagen, K. W., Hoogenboom, J. P., & Giepmans, B. N. G. (2017). Multi‐color electron microscopy by element‐guided identification of cells, organelles and molecules. Scientific Reports – UK, 7, 45970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boassa, D., Lemieux, S. P., Lev‐Ram, V., Hu, J., Xiong, Q., Phan, S., … Ngo, J. T. (2019). Split‐miniSOG for spatially detecting intracellular protein‐protein interactions by correlated light and electron microscopy. Cell Chemical Biology, 26(10), 1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sastri, M., Darshi, M., Mackey, M., Ramachandra, R., Ju, S., Phan, S., … Perkins, G. A. (2017). Sub‐mitochondrial localization of the genetic‐tagged mitochondrial intermembrane space‐bridging components Mic19, Mic60 and Sam50. Journal of Cell Science, 130(19), 3248‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heil, T., & Kohl, H. (2010). Optimization of EFTEM image acquisition by using elastically filtered images for drift correction. Ultramicroscopy, 110(7), 745–750. [DOI] [PubMed] [Google Scholar]
- 29.Aoyama, K., Matsumoto, R., & Komatsu, Y. (2002). How to make mapping images of biological specimens – Data collection and image processing. Journal of Electron Microscopy, 51(4), 257–263. [DOI] [PubMed] [Google Scholar]
- 30.Ramachandra, R., Bouwer, J. C., Mackey, M. R., Bushong, E., Peltier, S. T., Xuong, N. H., & Ellisman, M. H. (2014). Improving signal to noise in labeled biological specimens using energy‐filtered TEM of sections with a drift correction strategy and a direct detection device. Microscopy and Microanalysis, 20(3), 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaffer, B., Grogger, W., & Kothleitner, G. (2004). Automated spatial drift correction for EFTEM image series. Ultramicroscopy, 102(1), 27–36. [DOI] [PubMed] [Google Scholar]
- 32.Terada, S., Aoyama, T., Yano, F., & Mitsui, Y. (2001). Time‐resolved acquisition technique for elemental mapping by energy‐filtering TEM. Journal of Electron Microscopy, 50(2), 83–87. [DOI] [PubMed] [Google Scholar]
- 33.Egerton, R. F. (1984). Parallel‐recording systems for electron‐energy loss spectroscopy (Eels). Journal of Electron Microscopy Technique, 1(1), 37–52. [Google Scholar]
- 34.Faruqi, A. R., Cattermole, D. M., & Raeburn, C. (2003). Direct electron detection methods in electron microscopy. Nuclear Instruments and Methods A, 513(1–2), 317–321. [Google Scholar]
- 35.Xuong, N. H., Milazzo, A. C., Leblanc, P., Duttweiler, F., Bouwer, J. C., Peltier, S. T., … Matis, H. S. (2004). First use of a high‐sensitivity active pixel sensor array as a detector for electron microscopy. Proceedings of the SPIE, 5301, 242. [DOI] [PubMed] [Google Scholar]
- 36.Milazzo, A. C., Leblanc, P., Duttweiler, F., Jin, L., Bouwer, J. C., Peltier, S., … Xuong, N. H. (2005). Active pixel sensor array as a detector for electron microscopy. Ultramicroscopy, 104(2), 152–159. [DOI] [PubMed] [Google Scholar]
- 37.Faruqi, A. R., Henderson, R., Pryddetch, M., Allport, P., & Evans, A. (2005). Direct single electron detection with a CMOS detector for electron microscopy. Nuclear Instruments and Methods A, 546(1–2), 170–175. [Google Scholar]
- 38.Xuong, N. H., Jin, L., Kleinfelder, S., Li, S. D., Leblanc, P., Duttweiler, F., … Ellisman, M. (2007). Future directions for camera systems in electron microscopy. Methods in Cell Biology, 79, 721–739. [DOI] [PubMed] [Google Scholar]
- 39.Jin, L., Milazzo, A. C., Kleinfelder, S., Li, S. D., Leblanc, P., Duttweiler, F., … Xuong, N. H. (2008). Applications of direct detection device in transmission electron microscopy. Journal of Structural Biology, 161(3), 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milazzo, A. C., Lanman, J., Bouwer, J. C., Jin, L., Peltier, S. T., Johnson, J. E., … Ellisman, M. H. (2009). Advanced detector development for electron microscopy enables new insight into the study of the virus life cycle in cells and Alzheimer's disease. Microscopy and Microanalysis, 15, 8–9. [Google Scholar]
- 41.Milazzo, A. C., Moldovan, G., Lanman, J., Jin, L. A., Bouwer, J. C., Klienfelder, S., … Xuong, N. H. (2010). Characterization of a direct detection device imaging camera for transmission electron microscopy. Ultramicroscopy, 110(7), 741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baek, D., Zachman, M., Goodge, B., Lu, D., Hikita, Y., Hwang, H. Y., & Kourkoutis, L. F. Direct electron detection for atomic‐resolution EELS mapping at cryogenic temperature. Microscopy and Microanalysis, 24(Suppl 21). 454–455. [Google Scholar]
- 43.Hart, J. L., Lang, A. C., Leff, A. C., Longo, P., Trevor, C., Twesten, R. D., & Taheri M. L. (2017). Direct detection electron energy‐loss spectroscopy: A method to push the limits of resolution and sensitivity. Scientific Reports, 7(1). 10.1038/s41598-017-07709-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maigné, A., Wolf, M. (2018). Low‐dose electron energy‐loss spectroscopy using electron counting direct detectors. Microscopy, 67(suppl_1), i86–i97. 10.1093/jmicro/dfx088 [DOI] [PubMed] [Google Scholar]
- 45.Ahn, C. C., & Krivanek, O. L. (1983). EELS atlas. Warrendale, PA: Gatan. [Google Scholar]
- 46.Egerton, R. F., Li, P., & Malac, M. (2004). Radiation damage in the TEM and SEM. Micron (Oxford, England: 1993), 35(6), 399–409. [DOI] [PubMed] [Google Scholar]
- 47.Berger, A., & Kohl, H. (1993). Optimum imaging parameters for elemental mapping in an energy filtering transmission electron‐microscope. Optik, 92(4), 175–193. [Google Scholar]
- 48.Kortje, K. H. (1994). Image‐Eels – Simultaneous recording of multiple electron‐energy‐loss spectra from series of electron spectroscopic images. Journal of Microscopy, 174, 149–159. [Google Scholar]
- 49.Lavergne, J. L., Martin, J. M., & Belin, M. (1992). Interactive electron‐energy‐loss elemental mapping by the imaging‐spectrum method. Microscopy and Microanalysis, 3(6), 517–528. [Google Scholar]
- 50.Schaffer, B., Kothleitner, G., & Grogger, W. (2006). EFTEM spectrum imaging at high‐energy resolution. Ultramicroscopy, 106(11–12), 1129–1138. [DOI] [PubMed] [Google Scholar]
- 51.Watanabe, M., & Allen, F. I. (2012). The SmartEFTEM‐SI method: Development of a new spectrum‐imaging acquisition scheme for quantitative mapping by energy‐filtering transmission electron microscopy. Ultramicroscopy, 113, 106–119. [Google Scholar]
- 52.Mastronarde, D. N. (2005). Automated electron microscope tomography using robust prediction of specimen movements. Journal of Structural Biology, 152(1), 36–51. [DOI] [PubMed] [Google Scholar]
- 53.Tseng, Q. Z., Wang, I., Duchemin‐Pelletier, E., Azioune, A., Carpi, N., Gao, J., … Balland, M. (2011). A new micropatterning method of soft substrates reveals that different tumorigenic signals can promote or reduce cell contraction levels. Lab on A Chip, 11(13), 2231–2240. [DOI] [PubMed] [Google Scholar]
- 54.Messaoudi, C., Aschman, N., Cunha, M., Oikawa, T., Sorzano, C. O. S., & Marco, S. (2013). Three‐dimensional chemical mapping by EFTEM‐TomoJ including improvement of SNR by PCA and ART reconstruction of volume by noise suppression. Microscopy and Microanalysis, 19(6), 1669–1677. [DOI] [PubMed] [Google Scholar]
- 55.He, Z. L., & Zhou, J. Z. (2008). Empirical evaluation of a new method for calculating signal‐to‐noise ratio for microarray data analysis. Applied and Environmental Microbiology, 74(10), 2957–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waters, J. C. (2009). Accuracy and precision in quantitative fluorescence microscopy. Journal of Cell Biology, 185(7), 1135–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hayat, M. A. (1981). Fixation for electron microscopy. New York: Academic Press., [Google Scholar]
- 58.Reimer, D. L., & Kohl, H. (2008). Transmission electron microscopy. 5, New York: Springer., [Google Scholar]
- 59.Schaffer, B., Grogger, W., Kothleitner, G., & Hofer, F. (2008). Application of high‐resolution EFTEM SI in an AEM. Analytical and Bioanalytical Chemistry, 390(6), 1439–1445. [DOI] [PubMed] [Google Scholar]
- 60.LEAPMAN, R. D., & SWYT, C. R. (1988). Separation of overlapping core edges in electron energy loss spectra by multiple‐least‐squares fitting. Ultramicroscopy, 26, 393–403. [DOI] [PubMed] [Google Scholar]
- 61.Pfannmoller, M., Flugge, H., Benner, G., Wacker, I., Sommer, C., Hanselmann, M., … Schroder, R. R. (2011). Visualizing a homogeneous blend in bulk heterojunction polymer solar cells by analytical electron microscopy. Nano Letters, 11(8), 3099–3107. [DOI] [PubMed] [Google Scholar]
- 62.Pfannmoller, M., Flugge, H., Benner, G., Wacker, I., Kowalsky, W., & Schroder, R. R. (2012). Visualizing photovoltaic nanostructures with high‐resolution analytical electron microscopy reveals material phases in bulk heterojunctions. Synthetic Metals, 161(23–24), 2526–2533. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Scheme of the synthesis of Ln2‐DAB. Reagents, solvents, conditions, (A) Br2, acetic acid, rt; (B) ethyl acrylate, Pd(OAc)2, (o‐tolyl)3phosphine, NEt3, DMF, 80°C; (C) BOC anhydride, DMAP, DMF, 80°C; (D) 1 atm H2, Pd‐C, EtOAc‐EtOH, rt; (E) NaOH, dioxane‐MeOH, rt; (F) HOSu, EDC, DMF, rt; (G) eda‐EDTA, NEt3, DMSO, rt; (H) TFA, rt; (I) LnCl3, pH 7 buffer, rt.