

Abstract
Epidemiologic studies often rely on questionnaire data, exposure measurement tools, and/or biomarkers to identify risk factors and the underlying carcinogenic processes. An emerging and promising complementary approach to investigate cancer etiology is the study of somatic “mutational signatures” that endogenous and exogenous processes imprint on the cellular genome. These signatures can be identified from a complex web of somatic mutations thanks to advances in DNA sequencing technology and analytical algorithms. This approach is at the core of the Sherlock-Lung study (2018–ongoing), a retrospective case-only study of over 2,000 lung cancers in never-smokers (LCINS), using different patterns of mutations observed within LCINS tumors to trace back possible exposures or endogenous processes. Whole genome and transcriptome sequencing, genome-wide methylation, microbiome, and other analyses are integrated with data from histological and radiological imaging, lifestyle, demographic characteristics, environmental and occupational exposures, and medical records to classify LCINS into subtypes that could reveal distinct risk factors. To date, we have received samples and data from 1,370 LCINS cases from 17 study sites worldwide and whole-genome sequencing has been completed on 1,257 samples. Here, we present the Sherlock-Lung study design and analytical strategy, also illustrating some empirical challenges and the potential for this approach in future epidemiologic studies.
Keywords: genomic analyses, histology, lung cancer, mutational signatures, never-smokers, radiological imaging
Abbreviations
- CT
computed tomography
- FFPE
formalin-fixed paraffin-embedded
- H&E
hematoxylin and eosin
- LCINS
lung cancer in never-smokers
- GWAS
genome-wide association studies
- QC
quality control
- SBS
single-base substitution
- WGS
whole-genome sequencing
Lung cancer in never-smokers (LCINS) accounts for 10%–25% of lung cancer cases (1) and ranks among the most common causes of cancer mortality (2, 3). Compared with former and current smokers with lung cancer, the predominant histology observed in never-smokers is adenocarcinoma (4). Geographic variability in lung cancer risk among never-smokers is also observed (5, 6), likely due to regional differences in lifestyle and in environmental and occupational exposures. Some established environmental risk factors associated with LCINS include exposure to secondhand smoke (5, 7–10), radon (11–15), outdoor (16, 17) and indoor (18–21) air pollution, and asbestos (22, 23), which have been reviewed extensively (Table 1). Other risk factors include history of respiratory diseases, such as tuberculosis (24–26), pneumonia (24, 25, 27), and asthma (27–29). However, most LCINS cases have no known risk factors, highlighting the critical need for etiological studies.
Table 1.
A Summary Table of Established Risk Factors for Lung Cancer in Never-Smokers
| Exposure | Study Types | Exposure Assessment | Summary of Findings/Public Health Implications | Selected References |
|---|---|---|---|---|
| Secondhand tobacco smoke | Meta-analysis, pooled analysis, review | Questionnaire data, in-person interviews | Increased risk of LCINS for spousal secondhand smoke exposure as well as workplace exposure has been consistently observed regardless of study design, geographic region, etc. Associations between childhood exposures to secondhand smoke and LCINS have generally been weaker and not as consistent. Secondhand smoke exposure is an important risk factor in all geographical regions. | (6–10) |
| Outdoor pollution | Cohort, meta-analysis | Quantitative estimates of residential exposure to outdoor pollutants (e.g., inverse distance-weighted interpolation methods geocoded to baseline residential address, fixed site monitors, land use regression) | Different types of outdoor pollutants have been studied, including ozone, PM, and nitrogen oxides. Most consistently reported association with LCINS has been with PM2.5. | (16, 17, 93–96) |
| Indoor pollution | Case-control, cohort, meta-analysis, pooled analysis | Questionnaire data, in-person interviews on household air pollution | Different sources of indoor pollution have been studied. These include indoor cooking oil, coal fuel, waste and dung combustion, and biomass fuel, among others. The association between indoor pollution and LCINS has consistently been stronger among women, which is likely due to women having greater exposure to fuel combustion products at home than men. Approximately half of the world’s population is still exposed to indoor air pollution from domestic cooking and/or heating with solid fuels. | (18–21, 97–100) |
| Asbestos | Meta-analysis, pooled analysis | Questionnaire data, in-person interview of occupational exposure history, environmental monitoring/quantitative measurements of fibers linked to job title | Association between asbestos exposure and LCINS observed, generally stronger among men than women likely due to differences in exposure levels. Although most dangerous asbestos types are no longer used, other siliceous fibers and chrysotile are still incorporated into building projects in developing nations. | (22, 23, 101) |
| Radon | Meta-analysis, pooled analysis, review | Occupational exposure, residential exposure | Increased risk of LCINS is consistently seen in miners; level of association less clear with residential exposure. Population attributable fraction for residential radon exposure is higher in Europe. | (6, 11–13, 15, 102–104) |
| Pneumonia | Meta-analysis, pooled analysis | Self-reported history of pneumonia | Increased risk of LCINS observed with pneumonia. Substantial public health interest due to the large population diagnosed with pneumonia. | (24, 25, 27) |
| Tuberculosis | Meta-analysis, pooled analysis, review | Self-reported history of tuberculosis | Previous diagnosis of tuberculosis has been found to have an independent effect on LCINS. Although the incidence of tuberculosis is low in North America, it is common in low- and middle-income countries and affects millions; therefore, the possible association with lung cancer risk is of public health importance. | (6, 24–27) |
| Asthma | Case-control, meta-analysis, pooled analysis | Self-reported history of asthma, physician-diagnosed asthma | Some studies have reported an association between asthma and LCINS, but association is unclear and published literature is mixed. | (27–29, 105, 106) |
Abbreviations: LCINS, lung cancers in never-smokers; PM, particulate matter.
Throughout life, somatic cells acquire mutations, many of which occur well before the development of cancer (30). Advances in sequencing technologies combined with the development of novel computational methods can decipher the characteristic patterns of somatic mutations, termed “mutational signatures,” imprinted by the activities of endogenous and exogenous mutational processes (31). Distinct mutational signatures can now be identified from the thousands of somatic substitutions, insertions/deletions, copy number alterations, and structural rearrangements (32) observed in cancerous or normal somatic genomes (30–32), using the sequence context of each alteration (33, 34). Mutational signatures can reveal established exposures (e.g., tobacco smoking in lung cancer (35), ultraviolet light in skin cancer (36), or alcohol (37) or aflatoxin (38) exposure in liver cancer). Mutational signatures can also identify failure of known endogenous processes (e.g., defective DNA mismatch repair (39), errors in homologous recombination repair pathways (40), or loss of both polymerase proofreading and mismatch repair function (41)). Similar “marks” are likely imprinted on the genomes of LCINS.
The objective of the Sherlock-Lung study is to identify mutational signatures and relate them to past exogenous and endogenous processes by analyzing the cancer genome of 2,000 ethnically diverse LCINS cases identified through previous research efforts. The analytical approach used in this retrospective case-only study design is novel in that the different patterns of mutations observed within tumors can be used to infer prior probable exposures, some occurring years before diagnosis, even in the absence of exposure data. The starting point is the identification of mutational signatures in tumor samples and linking them to potential exogenous exposures and endogenous biological processes in external databases (42). When information on environmental and lifestyle exposures is available for the cases, we can also estimate the magnitude of etiological heterogeneity by relating exposure data to tumor subtypes in case-only analyses (43). In contrast, more traditional analytical approaches typically analyze mutational data and relate it to exposures reported by the cases. Moreover, the approach we use here allows for the identification of potential new or unexpected risk factors for LCINS, as it happened, for example, with the identification of the plant-derived aristolochic acid, through its specific mutational patterns, as a risk factor for a subset of hepatocellular carcinoma (44) and clear cell renal cell carcinoma (45). We acknowledge that the case-only design does not allow estimation of relative risks for the association between specific exposures and the risk of developing LCINS, but the identification of potential heterogeneity of exposure–tumor mutation associations will lay the foundation for future cohort or case-control studies to obtain such relative risks. Notably, the analysis of genomic data can also lead to a greater understanding of the endogenous mutational mechanisms (e.g., deficient DNA repair (40) or apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC)-related mutations (46)) triggering or facilitating clonal outgrowth in the presence or absence of exogenous exposures.
Briefly, the 2 primary aims of the Sherlock-Lung study are to: 1) identify exogenous exposures and endogenous processes involved in LCINS through the analysis of mutational signatures and other molecular characteristics, and 2) develop an integrated molecular, histological, and radiological classification of LCINS (Figure 1). We also discuss secondary aims with their attendant challenges and opportunities.
Figure 1.
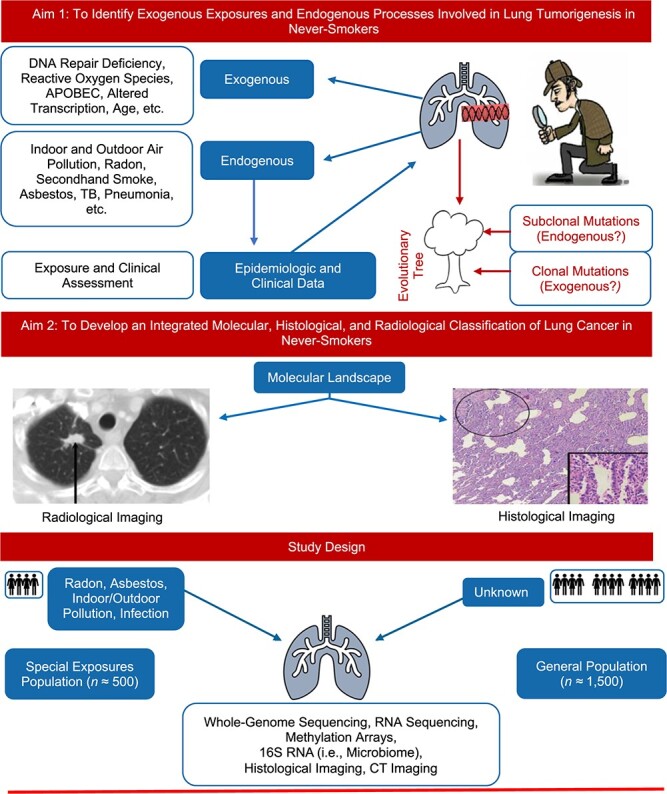
Aims of the Sherlock-Lung study, worldwide, 2018–ongoing.
METHODS
Study design
Sherlock-Lung will include 2,000 LCINS patients with treatment-naive fresh frozen tumor specimens and a source of germline DNA, of any histological type, but primarily adenocarcinoma, of all stages, ages, and sexes. Surgical samples will mostly come from stages I–IIIA (resectable lesions), while biopsies will also include more advanced cases. LCINS whose diagnosis was based only on imaging evaluation are excluded. A subgroup of patients (n ~ 500) will be sought who have known history of high exposure to established lung cancer risk factors (“special exposures populations”), while the remaining cases will be patients with no known exposures to established risk factors (“general populations”) (47) (Figure 1).
Study population and sample/data collection.
Retrospective collection of data and biospecimens from 2,000 LCINS cases with fresh frozen tumor specimens requires contacting many potential sources. Accordingly, we have established contact with institutions identified through publications, conferences, extensive web searches, and personal relationships; recruitment has been pursued by follow-up e-mail and phone/video calls. Sample collection began in 2019 and to date, LCINS cases have been drawn from tissue biobanks, hospital case series, population-based or hospital-based case-control studies, and clinical trials with fresh frozen lung tumor and a source of germline DNA samples. To capture a variety of exposures and genetic background across geographical regions, the study plan is to recruit at least 100 cases each from Asia, Africa, Central and South America, and the Middle East, in addition to those of European descent. Of note, germline data from this multiethnic population will increase the diversity of existing large-scale genome-wide association studies (GWAS) (>78% of participants in published GWAS are of European ancestry (48), and approximately 71.8% of these samples are collected from the United States, the United Kingdom, and Iceland (49)). Based on the power calculations from the “Mutographs of Cancer” whole-genome sequencing (WGS) data, 100 samples should be sufficient to detect 20% enrichment of mutations associated with a given exposure, whereas 2,000 samples can detect a 5% enrichment (50).
Subject prioritization.
Prioritization of subject selection is based on tumor sample requirements, exposure to specific risk factors, and availability/quality of data on exposure assessment, pathology, and imaging. The optimal sample requirements are listed in Web Figure 1 (available at https://doi.org/10.1093/aje/kwaa234). Overall, samples from cases with documented high exposures to established lung carcinogens (special exposures populations) have the highest priority as they have the most potential to identify a mutational signature associated with a distinct exogenous risk factor. Given the rarity and importance of these samples, we are willing to extend inclusion criteria and collect formalin-fixed paraffin-embedded (FFPE) samples for validation and pertinent analyses if frozen biospecimens are not available (e.g., for cases with high exposure to wood and coal smoke from Colombia, Table 2).
Table 2.
Lung Cancer Samples From Never-Smoker Patients Identified During the Initial 20 Months of Sherlock-Lung, Worldwide, 2018–Ongoing
| Region | Cases With Data and Samples Received | Pending Cases a | Potential Additional Cases b | |||
|---|---|---|---|---|---|---|
| General | Special Exposures | General | Special Exposures | General | Special Exposures | |
| North America | 353 | 0 | 55 | 180 | 138 | 0 |
| Europe | 424 | 0 | 149 | 47 | 9 | 125 |
| Asia | 434 | 28 | 300 | 200 | 200 | 90 |
| Central/South America | 0 | 131c | 108 | 19 | 477 | 213 |
| Africa | 0 | 0 | 80 | 70 | 100 | 30 |
| Middle East | 0 | 0 | 72 | 0 | 40 | 0 |
| Australia | 0 | 0 | 0 | 5 | 0 | 40 |
| Caribbean | 0 | 0 | 50 | 0 | 30 | 0 |
| All regions | 1,211 | 159 | 814 | 521 | 994 | 498 |
a Written commitments to provide data/samples has been received from the institutions.
b Institutions have expressed interest in participating in Sherlock-Lung and efforts are underway to establish collaborations.
c All samples received to date are fresh frozen specimens with the exception of 106 formalin-fixed paraffin-embedded samples from South America.
For cases without clear exposures (general populations), the minimum requirement for enrollment includes availability of data on environment or residence, lifestyle, demographics, histology, and >1 fresh frozen tissue sample paired with a source of germline DNA (i.e., whole blood, buffy coat, normal lung tissue, saliva, or buccal cells) per subject. Additional criteria for prioritization, in order of importance, include:
Ethnic diversity and geographic region.
Availability of multiple normal tissue samples (n = 4) to study the lifetime accumulation of mutations and presence of mutations in cancer driver genes.
Multiple hematoxylin and eosin (H&E) slides for histological classification.
Lung computed tomography (CT)-scan imaging data for radiological classification.
Availability of FFPE tissue blocks from the tumor center and periphery for tumor microenvironment analysis.
Availability of multiple tumor tissue samples (n = 4) for clonal evolution analysis.
Availability of plasma samples for circulating tumor DNA analysis.
Collection of exposure data.
We rely on 3 data sources to collect data on exposures to the major risk factors, including secondhand tobacco smoke, asbestos, radon, indoor and outdoor air pollution, and previous lung/respiratory tract diseases: 1) self-reported household, occupational, and lifestyle exposures; 2) medical records; 3) residential data (location of longest residence), which we plan to link to geogenic mapping and satellite data. The documented exposures must have occurred at least a decade prior to cancer diagnosis and must have lasted, cumulatively, for at least 1 year. Some case series will have extensive data on these risk factors while other case series will have limited data, depending on the approach used to recruit cases.
Code of ethics.
Because the National Cancer Institute is only receiving deidentified samples and data from collaborating centers, has no direct contact or interaction with study subjects, and does not use or generate identifiable private information, Sherlock-Lung has been determined to constitute “Not Human Subject Research (NHSR)” based on the Federal Common Rule (45 CFR 46; https://www.ecfr.gov/). Contributing cases are required to confirm collection under a local IRB-approved study.
Laboratory and analytical methods
Aim 1: Characterize the genomic landscape of LCINS and relationship with exposures and endogenous processes.
LCINS will be characterized using WGS, whole transcriptome, and genome-wide methylation analyses to describe the mutational burden of single nucleotide variants (SNVs), mutational signatures, alterations in major cancer driver genes and in lineage-specific surfactant genes, somatic copy number alterations (SCNAs), structural variants (SVs), germline variation, methylation patterns, gene expression, telomere length, presence of new tumor epitopes, viral sequences, lung microbiota, and additional genomic changes in LCINS (Table 3). Moreover, we plan to analyze germline genetic variants from lung GWAS (51–54) or polygenic risk scores in relation to quantifiable tumor genomic alterations.
Table 3.
Summary of the Experimental Plans to Characterize Lung Cancers in Sherlock-Lung, Worldwide, 2018–Ongoing
| Experimental Plan | No. of Samples/Imaging Per Subject | Subjects |
|---|---|---|
| Aim 1: Characterize the genomic landscape of LCINS and relationship with exposures and endogenous processes | ||
| Whole genome sequencing | T coverage 80× = 1, Bcoverage 40× = 1 | ~2000 |
| RNA sequencing + methylation array | T = 1, N = 1 | ~1,000 |
| Cancer-driver gene targeted sequencing or WES + SNP arraya | N coverage 1,000× = 4, Bcoverage 400× = 1 or Ncoverage 200× = 4, Bcoverage 100x = 1 | ~500 |
| 16S ribosomal RNA | T = 1, N = 1 | ~2000 |
| Aim 2: Develop an integrated molecular, histological, and radiological classification of LCINS. | ||
| H&E slides | Up to 6 tumor blocks per tumor | ~2000 |
| Lung CT–scan imaging | Diagnostic imaging (and before or after diagnosis if available) | ~2000 |
Abbreviations: B, number of blood samples; CT, computed tomography; LCINS, lung cancers in never-smokers; N, number of normal lung tissue samples; SNP, single-nucleotide polymorphism; T, number of lung tumor samples; WES, whole-exome sequencing.
a Ongoing pilot study in normal lung tissue samples.
Sample processing and quality control.
Samples from collaborating sites are received at a central laboratory for staging and preparation. Our central laboratory requires a minimum of 2 tissue samples of 40 mg each to extract DNA and RNA for genomic analyses. If collaborating sites send lung tissue specimens, these are first sent to a facility for validation of pathology and nucleotide extraction (DNA and RNA) with standard quality control (QC) metrics (Web Appendix 1). When already-extracted tumor DNA samples are provided, confirmatory quantification and QC are performed at a central laboratory following similar procedures. QC-approved DNA samples are then sent to a genomic center for whole genome sequencing, while methylation, microbiome, and RNA sequencing analyses are conducted at the central laboratory.
Whole-genome sequencing.
To date, studies of the genomic landscape of lung cancer have included mostly smokers and relied largely on targeted sequencing or whole-exome sequencing (55–60). However, the larger number of somatic mutations in whole-genome sequences, of which approximately 98% is not covered by whole-exome sequencing, provides increased power for signature decomposition. Moreover, WGS can better reveal structural rearrangements, detailed copy number profile, noncoding mutations, and other genomic changes not captured by whole-exome sequencing. Sherlock-Lung tumor tissue samples will be analyzed by WGS with an average coverage of 80×, using blood samples or other germline DNA sources (coverage of approximately 40×) as reference. For high-priority cases with uncertain sample quality, we will conduct deeper tumor sequencing (coverage of approximately 120×–200×) to increase the probability of detecting low-allele-fraction or subclonal mutations.
Using current bioinformatic tools, separation of clonal from subclonal mutations within each tumor enables inference of evolutionary trajectories of the mutational processes in tumors. In previous analyses of lung adenocarcinomas in smokers, we found that mutations assigned to the tobacco smoking signature (single-base substitution (SBS) 4) were predominantly clonal and therefore involved in tumor initiation (57). We will extend this analysis to normal tissue adjacent to the tumor tissue to improve our knowledge of early clonal expansion of cancer-driver mutations that are widely present in clinically normal tissues albeit in lower cell fractions (61–63). A pilot study is underway in normal tissue to identify the optimal approach to detect genomic changes in a small fraction of cells (Table 3).
RNA sequencing.
RNA sequencing analysis of tumor/normal tissue pairs is being carried out in approximately 1,000 cases, using 2 × 150 base pairs paired-end sequencing with a target depth of 100 million reads, to identify transcribed alterations and assess gene expression, gene splicing mutations, fusions, and tumor immune microenvironment.
Whole-genome DNA methylation analysis.
In contrast to stable genetic events, epigenetic states are reversible and responsive to environmental stressors (64). Understanding the epigenetic landscape that is specific to LCINS will help dissect the nongenetic factors contributing to the tumorigenesis through interaction with gene expression regulation and mutagenesis. To address this question and describe global DNA methylation patterns, their impact on gene expression, the presence of 5′—C—phosphate—G—3′ (CpG) island methylator phenotype (65), and methylation features associated with mutational signatures, DNA methylation analysis in tumor/normal tissue pairs from approximately 1,000 cases is being analyzed using Infinium EPIC arrays (approximately 850,000 probes; Illumina, Inc., San Diego, California).
Gene expression and methylation data will also be used to deconvolute the tumor microenvironment and its constituents (e.g., infiltrating immune cell populations) (66–68), in conjunction with H&E and immunofluorescent analyses. These data will also enable assessment of sample purity based on copy number alteration and single-nucleotide variant allele frequency data (69).
Microbiome analysis.
Recent studies have characterized the microbiota in both normal and tumor lung tissue. We observed that the proportion of microbiota species in normal lung tissue can be distinct from other organs and associated with environmental exposures related to lung cancer risk, including air pollution (70). Another study found a distinct lung microbiome in patients with lung cancer and particular genomic changes (71). It was also shown that local microbiota promote inflammation associated with lung adenocarcinoma via interaction with immune cells (72). The lung cancer microbiome was also characterized in The Cancer Genome Atlas (TCGA) data using a custom data analysis pipeline (73). In Sherlock-Lung, we will characterize the taxonomic and functional profiles of lung microbiota using 16S rRNA data to investigate the contribution to risk and progression of LCINS.
Aim 2: Develop an integrated molecular, histological, and radiological classification of LCINS.
Histological classification.
Integrated histological and molecular studies, largely based on smokers, have suggested that lung adenocarcinomas could be further classified into subtypes by combining histological features with genomics, transcriptomic, and epigenomic changes (74). Different histological subtypes (e.g., lepidic, acinar, papillary) suggest distinct biological pathways with implications for distinct etiological risk factors and clinical outcomes. In Sherlock-Lung, we are collecting, digitally scanning, annotating, and examining H&E slides from all available tissue blocks (up to 6 per tumor) to evaluate the tumor histological landscape. The original pathology report is collected for diagnostic review.
Radiological classification.
The widespread use of CT scans for lung cancer screening has resulted in a dramatic increase in the number of indeterminate ground-glass opacities (75), many with multifocal components, which makes judgment on the extent and benefit of surgical resection challenging. Collection of CT-scan images with reports enables integration of radiological imaging, including ground-glass opacities, with histological and molecular features. Histological and CT images are being archived for future studies, including application of deep learning/artificial intelligence–based algorithms to evaluate diagnostic and prognostic features.
Secondary aims
We plan further data collection and analyses (Table 4), including mining electronic medical records (e.g., the Clinical Practice Research Datalink, https://www.cprd.com/), to explore novel associations between LCINS and medical conditions or chronic medication use that can help inform genomic analyses and interpretation; lineage phylogenetic analysis (76–79) to reconstruct the tumor evolution using multiple tumor and normal tissue samples, possibly including single cell approaches; the analysis of density, colocalization, and spatial architectures of cells in the tumor microenvironment, to investigate cancer immunoediting (80, 81) using H&E-stained slides in conjunction with multiplex immunofluorescence staining of specific markers; the analysis of circulating tumor DNA (82) using low pass WGS; in-silico (46, 83, 84) and experimental (85, 86) validation of mutational signatures; and development of novel analytical approaches for the integrative analyses.
Table 4.
Secondary Aims, Sherlock-Lung, Worldwide, 2018–Ongoing
| Objective | Experimental Plans |
|---|---|
| Electronic medical records | Analysis of medical conditions and long-term medication use in cases of LCINS |
| Tumor microenvironment | Quantification and spatial analysis of immune, endothelial, stromal cells |
| H&E | |
| Multiplex immunofluorescent markers | |
| Digital imaging and spatial analysis using HALO imaging platforma | |
| Liquid biopsy | Circulating tumor DNA (ctDNA) |
| Deep target sequencing of driver genes + low-pass WGS | |
| Comparative analysis of WGS in T/ctDNA | |
| Clonal evolution | Multiregion tumor and normal tissue samples |
| Phylogenetic analysis | |
| Single cell analysis | |
| Laboratory validation | Organoid/CRISPR/engineered cell lines |
| Mutational signatures and other genomic changes | |
| Development of algorithms | Novel approaches for mutational signature analyses |
| Integrated analysis of -omics data and radiological and pathological imaging |
Abbreviations: CRISPR, clustered regularly interspaced short palindromic repeats; H&E, hematoxylin and eosin; LCINS, lung cancers in never-smokers; WGS, whole-genome sequencing.
a HALO image analysis platform (Indica labs, Albuquerque, New Mexico, USA).
Data sharing
The genomic data and digital imaging database from this study will be made available in accordance with National Institutes of Health policy through the National Cancer Institute’s Genomic Data Commons.
RESULTS
As of June 2, 2020, we have received samples and data from 1,370 LCINS cases from 17 collaborating institutions in North America, Europe, Asia, and Central and South America (Table 2). Of these, 159 cases have known high levels of special exposures (28 from Asia and 131 from Central and South America). The disproportionately low number of cases with high exposures to known risk factors underscores the challenge of collecting high-quality frozen specimens from cases with documented exposures at least a decade prior to cancer diagnosis, especially given that these high-level of exposures are primarily seen in cases from low-and middle-income countries.
We have received written commitment from institutions to provide additional fresh frozen and/or FFPE samples from 1,560 cases. In our experience, selected centers provided less than 50% of the promised frozen samples, and QC-related exclusions further decreased the number of samples available for WGS. Thus, if needed, we could collect more samples from 1,342 cases from other potential collaborators (Table 4). In addition to the geographical regions already represented, we anticipate samples from other regions, including Africa and the Middle East. We are prioritizing collection of samples from the regions outside Europe and the United States, especially those with documented exposures to known LCINS risk factors.
Frozen specimens collected from the 1,370 LCINS cases identified to date include 1,017 tumor and 820 nontumor lung-tissue specimens and 644 blood DNA samples. Out of a total of 1,837 lung tissue specimens received, 1,798 were shipped to the laboratory for DNA extraction. Of these, 210 failed tissue QC or pathology review (11.7%). The quality varied by region; Asia had the lowest proportion of tissues to fail QC (3.8%), followed by North America (11.6%) and Europe (14.1%). Samples from Central and Southern America are still under review. We found that if the lung tissue specimens passed QC or pathology review, extracted DNA was of high quality (94.9% of the extracted DNA passed QC for sequencing). Similarly, when collaborating sites sent existing extracted DNA, the quality was excellent, with 94.4% passing QC and only 8 samples out of 1,257 failing sequencing (Table 5).
Table 5.
Sample Collection, Processing, and Sequencing Attempts According to Geographical Region for Cases of Lung Cancer in Never-Smokers With Data/Samples Received as of June 2, 2020, Sherlock-Lung, Worldwide, 2018–Ongoing
| Frozen Lung Tissue | DNA Samples Received | WGS Attempted and Completed a | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor (n = 1,017) | Nontumor Lung (n = 820) | Tumor (n = 385) | Nontumor Lung (n = 153) | Germline DNA (n = 644) | Tumor (n = 447) | Normal Lung (n = 196) | Germline DNA (n = 614) | ||||||||||
| Region (No. of Sites) | No of LCINS Subject (n = 1,370) | No. | % Extracted DNA Passed QC | No. | % Extracted DNA Passed QC | No. | % Passed DNA QC | No. | % Passed DNA QC | No. | % Passed DNA QC | No. | % Passed Sequencing | No. | % Passed Sequencing | No. | % Passed Sequencing |
| North America (8) | 353 | 139 | 68.9 | 41 | 70.7 | 262 | 83.6 | 90 | –b | 336 | 95.8 | 207 | 98.1 | 12 | 100 | 312 | 98.7 |
| Europe (4) | 424 | 387 | 68.9 | 340 | 66.2 | 123 | 98.4 | 63 | 100 | 303 | 100 | 126 | 100 | 70 | 100 | 302 | 100 |
| Asia (2) | 462c | 434 | 89.2 | 434 | 88.0 | 114 | 100 | 114 | 100 | ||||||||
| Central/South America (3) | 131d | 57 | –b | 5 | –b | 5 | –b | ||||||||||
Abbreviations: LCINS, lung cancers in never-smokers; QC, quality control; WGS, whole-genome sequencing.
a Of total sequencing attempted and completed across all sample types (n = 1,257), 8 samples failed.
b Data under review.
c Twenty-eight out of 462 cases have known special exposures.
d All 131 cases have known special exposures.
Notably, analysis of mutational signatures was important for excluding samples that were erroneously included in the study. For example, 2 samples were dominated by signature SBS7, attributed to ultraviolet exposure. Upon repeat review by 3 different pathologists, it was determined that the samples were not from primary lung tumors but from metastases of skin squamous cell carcinomas. Moreover, we identified 1 sample dominated by SBS4, attributed to tobacco smoking, and verified that the case was a current smoker, erroneously reported as a never-smoker. We also found 1 sample with a high level of signature SBS31 attributed to platinum-based chemotherapy. Upon retrieval of clinical data from previous hospitalizations, we found that this subject had a prior tumor treated with platinum and bevacizumab.
DISCUSSION
Here we have presented the framework of a large integrative study that uses mutational signatures and other genomic features to complement questionnaire and exposure assessment approaches to identify potential factors contributing to lung tumorigenesis in never-smokers. In tumorigenesis, endogenous factors, such as developmental and differentiation programs, or exogenous factors, such as mutagenic exposures, pathogens, and inflammation (32), can leave a “signature” on both tumor and adjacent nontumor tissue, which can be captured by the analysis of the mutations within trinucleotide or pentanucleotide context. To date, this approach has identified over 50 different mutational signatures across cancer types, many of which have mapped to one or more environmental or endogenous events (31, 32, 35). Moreover, mutational signatures can have potential clinical value as predictors of therapeutic response in cancer (87).
The second primary objective is to categorize LCINS based on molecular and clinical characteristics. According to The Cancer Genome Atlas analyses, lung adenocarcinomas (mostly from smokers) can be further classified into subtypes using a combination of histological features and transcriptomic, epigenomic, and genomic changes (74). Our study of nonsmoking cases might reveal pathways in adenocarcinomas (or other histological types) previously hidden by the strong effect of tobacco smoking, possibly allowing further tumor classification, which in turn might have treatment implications. The Cancer Genome Atlas has estimated that at least 1 in 10 cancer cases across cancer types might be classified and treated differently using a molecular taxonomy instead of current histopathology-based classification (88).
The use of WGS for the analysis of mutational signatures and tumor subclassification has several challenges and limitations, which extend beyond LCINS. Based on initial sample collection (Tables 2 and 5) and the analysis of mutational signatures in these samples, we have learned a series of lessons (Figure 2).
Figure 2.

Lessons learned from the Sherlock-Lung study, worldwide, 2018–ongoing.
Collecting frozen specimens can be challenging. Despite technological advancements, WGS analysis currently requires unfragmented DNA from fresh frozen tissue specimens. As noted above, we aim to gather samples from diverse racial/ethnic groups and geographical locations across 6 continents to ensure diversity of exposures and ancestry. However, in regions that lack resources and proximity to hospitals, have limited cancer screening programs, or often incur misdiagnoses (e.g., lung cancer can be initially misdiagnosed as tuberculosis), cancer diagnoses are often delayed, when surgery is a treatment option for a very small percentage of such cases. In the absence of surgical specimens, tumor biopsies have been collected. Although biopsies can be obtained from advanced tumors, balancing the stage distribution, which is skewed towards early stage in surgical cases, the materials have been either too small or necrotic. Additionally, only a subset of hospitals had the infrastructure to rapidly freeze, maintain, and ship adequate samples.
Collection of samples from high-income countries, although expected to be more feasible, is daunting. For example, 3 well-established institutions in North America initially identified 494 cases, but only 215 samples could be retrieved. Moreover, there has been variability in the provision of adequate samples—some centers effectively provided frozen tissue, others had extracted DNA, while others could provide FFPE blocks, tissue sections (with often different section numbers and thickness), H&E slides, or digitally scanned images, necessitating great flexibility from the central laboratory. Also, we encountered delays due to country-specific restrictions/policies on data and biospecimens sharing; accordingly, we developed a solution to conduct tumor profiling analyses locally with careful validation of comparable platforms, procedures, and QC, as in our central laboratory.
Collection of samples from individuals with high exposure to known risk factors for lung cancer remains a major challenge. Exposures to these risk factors is most relevant decades before the cancer diagnosis, when they potentially had an impact on tumor initiation. These exposures have been high in the past for many countries, including those where current exposure levels are low because of stringent occupational exposure limits and public health interventions (e.g., asbestos exposure). However, tumor samples from lung cancer cases with previous high levels of exposures, even drawn from occupational cohorts, exist only as FFPE archived tissue blocks, which currently limit their utility for genomic analyses. Sample collection in low- and middle-income countries, where exposures are still high, might not have had exposures in the most important etiological time window for lung cancer and might experience challenges described above. As an alternative approach to identify exposure-specific mutational signatures, we will utilize known experimental mutational signatures of environmental or microbial mutagens generated by exposing pluripotent stem cells, cell lines, or organoids to different dosages of mutagens (38, 85, 89, 90). We will test these mutagen signatures in our samples and estimate the proportion of mutations that can be explained by them. These analyses could shed light on potential exposures associated with the mutations, which will require further epidemiologic and experimental validation.
Many algorithms have been proposed to identify mutational signatures from a composite of genomic changes. However, they often lack consensus on analysis and result interpretation, their parameters can vary across tissue or cancer types (91), supervised fitting of signatures can lead to false results, and no recognized gold standard exists (92). Although these issues are less likely to be important for signatures with distinct patterns (e.g., APOBEC-related signatures SBS2 and SBS13), others, particularly the so called “flat” signatures (e.g., SBS3, SBS5, or SBS40) characterized by similar mutation distributions across trinucleotide contexts, cannot be always robustly separated. Moreover, there are several mutational signatures identified across cancer types whose origin is still unknown (32). The analytical plan envisions the use of multiple algorithms, the pentanucleotide context, and the verification of mutation enrichment and distributions to confirm signatures.
In conclusion, Sherlock-Lung is predicated on the examination of the genomic tumor landscape with careful clinical and exposure assessment to enable investigation of LCINS etiology. We have described the study design and objectives as well as opportunities and challenges of this integrative approach. With the reduction in cost for sequencing technologies and the progress of analytical tools, similar studies across different cancer types could become a viable approach for future epidemiologic investigation of risk factors for distinct cancers.
Supplementary Material
ACKNOWLEDGMENTS
Author affiliations: Integrative Tumor Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Rockville, Maryland, United States (Maria Teresa Landi, Naoise C. Synnott, Tongwu Zhang, Wei Zhao, Michael Kebede, Jian Sang, Laura Mendoza, Marwil Pacheco, Mustapha Abubakar, Montserrat Garcia-Closas); Cancer Prevention Fellowship Program, Division of Cancer Prevention, National Cancer Institute, Rockville, Maryland, United States (Naoise C. Synnott); Westat, Inc., Rockville, Maryland, United States (Jennifer Rosenbaum); Biostatistics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Rockville, Maryland, United States (Bin Zhu, Jianxin Shi); Laboratory of Translational Genomics, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Maryland, United States (Jiyeon Choi); Cancer Genomics Research Laboratory, Frederick National Laboratory for Cancer Research, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Rockville, Maryland, United States (Belynda Hicks); Occupational and Environmental Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Rockville, Maryland, United States (Neil E. Caporaso, Nathaniel Rothman, Qing Lan); Genome Integrity and Structural Biology Laboratory, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina, United States (Dmitry A. Gordenin); Big Data Institute, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom (David C. Wedge); Manchester Cancer Research Centre, The University of Manchester, Manchester, United Kingdom (David C. Wedge); Department of Cellular and Molecular Medicine, Department of Bioengineering, Moores Cancer Center, University of California, San Diego, California, United States (Ludmil B. Alexandrov); and Office of the Director, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Rockville, Maryland, United States (Montserrat Garcia-Closas, Stephen J. Chanock).
This work was supported by the Intramural Research Program of the National Cancer Institute, Division of Cancer Epidemiology and Genetics, National Institutes of Health. Part of this work was also supported by the National Institutes of Health Intramural Research Program Project Z1AES103266 (to D.A.G).
We thank Drs. Amy Berrington, Ludmila Prokunina, and Laufey Amundadottir, Division of Cancer Epidemiology and Genetics, National Cancer Institute, for their critical review of the study design; Drs. Rena Jones, Neil Caporaso, Melissa Friesen, and Debra Silverman (Division of Cancer Epidemiology and Genetics) for their help in designing the collection instrument for epidemiologic data; Drs. Christine Ambrosone (Roswell Park Cancer Center); Chris Amos (Baylor University); Oscar Arrieta (Instituto Nacional de Cancerologia, Mexico); Yohan Bossé (Laval University); Paul Brennan (International Agency for Research on Cancer); David Christiani (Harvard University); Dario Consonni (University of Milan, Italy); Paul Hofman (University of Nice, France); Tobias Peikert and Brian Bartholmai (Mayo Clinic); Chao Agnes Hsiung (National Health Research Institutes, Zhunan, Taiwan); Geoffrey Liu (University of Toronto, Canada); Bonnie Rothberg (Yale University); Matthew Schabath (Moffitt Cancer Center); Hongbing Shen (Nanjing Medical University, China); and Maria Wong (University of Hong Kong) for their help in assembling the first collection of specimens and data; Drs. Naoko Ishibe and Susan Viet (Westat) for the literature review on risk factors for LCINS; Drs. Mary Olanich, Yelena Golubeva, and Petra Lenz (Cancer Genomics Research Laboratory, Frederick National Laboratory for Cancer Research, Division of Cancer Epidemiology and Genetics), and Maire Duggan (University of Calgary, Canada) for the development of standard operating procedures for sample collection and histological imaging; and Drs. Nuria Lopez-Bigas (European Bioinformatics Institute, Barcelona) and Hannah Carter (University of California San Diego) for the useful discussions on analytical approaches. We also thank the members of the Sherlock-Lung Advisory Board, Drs. Matthew Meyerson (Broad Institute), John Samet (University of Colorado), Margaret Spitz (Baylor College of Medicine), Ronald Summers (National Institutes of Health Clinical Center), Michael Thun (American Cancer Society), and William Travis (Memorial Sloan Kettering Cancer Center) for their support and positive feedback throughout the study.
Conflict of interest: none declared.
REFERENCES
- 1. Couraud S, Zalcman G, Milleron B, et al. Lung cancer in never smokers—a review. Eur J Cancer. 2012;48(9):1299–1311. [DOI] [PubMed] [Google Scholar]
- 2. Samet JM, Avila-Tang E, Boffetta P, et al. Lung cancer in never smokers: clinical epidemiology and environmental risk factors. Clin Cancer Res. 2009;15(18):5626–5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cho J, Choi SM, Lee J, et al. Proportion and clinical features of never-smokers with non-small cell lung cancer. Chin J Cancer. 2017;36(1):e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Subramanian J, Govindan R. Lung cancer in never smokers: a review. J Clin Oncol. 2007;25(5):561–570. [DOI] [PubMed] [Google Scholar]
- 5. Thun MJ, Hannan LM, Adams-Campbell LL, et al. Lung cancer occurrence in never-smokers: an analysis of 13 cohorts and 22 cancer registry studies. PLoS Med. 2008;5(9):e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sisti J, Boffetta P. What proportion of lung cancer in never-smokers can be attributed to known risk factors? Int J Cancer. 2012;131(2):265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Office on Smoking and Health . The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. http://www.ncbi.nlm.nih.gov/books/NBK44324/. Accessed September 22, 2020. [PubMed]
- 8. Brennan P, Buffler PA, Reynolds P, et al. Secondhand smoke exposure in adulthood and risk of lung cancer among never smokers: a pooled analysis of two large studies. Int J Cancer. 2004;109(1):125–131. [DOI] [PubMed] [Google Scholar]
- 9. Kim AS, Ko HJ, Kwon JH, et al. Exposure to secondhand smoke and risk of cancer in never smokers: a meta-analysis of epidemiologic studies. Int J Environ Res Public Health. 2018;15(9):1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim CH, Lee YC, Hung RJ, et al. Exposure to secondhand tobacco smoke and lung cancer by histological type: a pooled analysis of the International Lung Cancer Consortium (ILCCO). Int J Cancer. 2014;135(8):1918–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Darby S, Hill D, Deo H, et al. Residential radon and lung cancer—detailed results of a collaborative analysis of individual data on 7148 persons with lung cancer and 14,208 persons without lung cancer from 13 epidemiologic studies in Europe. Scand J Work Environ Health. 2006;32(suppl 1):1–83. [PubMed] [Google Scholar]
- 12. Krewski D, Lubin JH, Zielinski JM, et al. Residential radon and risk of lung cancer: a combined analysis of 7 North American case-control studies. Epidemiology. 2005;16(2):137–145. [DOI] [PubMed] [Google Scholar]
- 13. Lorenzo-Gonzalez M, Ruano-Ravina A, Torres-Duran M, et al. Lung cancer and residential radon in never-smokers: a pooling study in the northwest of Spain. Environ Res. 2019;172:713–718. [DOI] [PubMed] [Google Scholar]
- 14. Lubin JH, Boice JD Jr, Edling C, et al. Lung cancer in radon-exposed miners and estimation of risk from indoor exposure. J Natl Cancer Inst. 1995;87(11):817–827. [DOI] [PubMed] [Google Scholar]
- 15. Torres-Durán M, Barros-Dios JM, Fernández-Villar A, et al. Residential radon and lung cancer in never smokers. A systematic review. Cancer Lett. 2014;345(1):21–26. [DOI] [PubMed] [Google Scholar]
- 16. Hamra GB, Guha N, Cohen A, et al. Outdoor particulate matter exposure and lung cancer: a systematic review and meta-analysis. Environ Health Perspect. 2014;122(9):906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang WS, Zhao H, Wang X, et al. An evidence-based assessment for the association between long-term exposure to outdoor air pollution and the risk of lung cancer. Eur J Cancer Prev. 2016;25(3):163–172. [DOI] [PubMed] [Google Scholar]
- 18. Hosgood HD 3rd, Boffetta P, Greenland S, et al. In-home coal and wood use and lung cancer risk: a pooled analysis of the International Lung Cancer Consortium. Environ Health Perspect. 2010;118(12):1743–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kurmi OP, Arya PH, Lam KB, et al. Lung cancer risk and solid fuel smoke exposure: a systematic review and meta-analysis. Eur Respir J. 2012;40(5):1228–1237. [DOI] [PubMed] [Google Scholar]
- 20. Zhao Y, Wang S, Aunan K, et al. Air pollution and lung cancer risks in China—a meta-analysis. Sci Total Environ. 2006;366(2–3):500–513. [DOI] [PubMed] [Google Scholar]
- 21. Vermeulen R, Downward GS, Zhang J, et al. Constituents of household air pollution and risk of lung cancer among never-smoking women in Xuanwei and Fuyuan, China. Environ Health Perspect. 2019;127(9):97001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ngamwong Y, Tangamornsuksan W, Lohitnavy O, et al. Additive synergism between asbestos and smoking in lung cancer risk: a systematic review and meta-analysis. PLoS One. 2015;10(8):e0135798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olsson AC, Vermeulen R, Schüz J, et al. Exposure-response analyses of asbestos and lung cancer subtypes in a pooled analysis of case-control studies. Epidemiology. 2017;28(2):288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brenner DR, Boffetta P, Duell EJ, et al. Previous lung diseases and lung cancer risk: a pooled analysis from the International Lung Cancer Consortium. Am J Epidemiol. 2012;176(7):573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brenner DR, McLaughlin JR, Hung RJ. Previous lung diseases and lung cancer risk: a systematic review and meta-analysis. PLoS One. 2011;6(3):e17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang HY, Li XL, Yu XS, et al. Facts and fiction of the relationship between preexisting tuberculosis and lung cancer risk: a systematic review. Int J Cancer. 2009;125(12):2936–2944. [DOI] [PubMed] [Google Scholar]
- 27. Denholm R, Schüz J, Straif K, et al. Is previous respiratory disease a risk factor for lung cancer? Am J Respir Crit Care Med. 2014;190(5):549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gardner LD, Loffredo CA, Langenberg P, et al. Associations between history of chronic lung disease and non-small cell lung carcinoma in Maryland: variations by sex and race. Ann Epidemiol. 2018;28(8):543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qu YL, Liu J, Zhang LX, et al. Asthma and the risk of lung cancer: a meta-analysis. Oncotarget. 2017;8(7):11614–11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev. 2014;24(100):52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steele CD, Tarabichi M, Oukrif D, et al. Undifferentiated sarcomas develop through distinct evolutionary pathways. Cancer Cell. 2019;35(3):441–456.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Macintyre G, Goranova TE, De Silva D, et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat Genet. 2018;50(9):1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alexandrov LB, Ju YS, Haase K, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354(6312):618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Zeeland AA, Vreeswijk MP, de Gruijl FR, et al. Transcription-coupled repair: impact on UV-induced mutagenesis in cultured rodent cells and mouse skin tumors. Mutat Res. 2005;577(1–2):170–178. [DOI] [PubMed] [Google Scholar]
- 37. Letouzé E, Shinde J, Renault V, et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat Commun. 2017;8(1):1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang MN, Yu W, Teoh WW, et al. Genome-scale mutational signatures of aflatoxin in cells, mice, and human tumors. Genome Res. 2017;27(9):1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meier B, Volkova NV, Hong Y, et al. Mutational signatures of DNA mismatch repair deficiency in C. elegans and human cancers. Genome Res. 2018;28(5):666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Polak P, Kim J, Braunstein LZ, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet. 2017;49(10):1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haradhvala NJ, Kim J, Maruvka YE, et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat Commun. 2018;9(1):1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Phillips DH. Mutational spectra and mutational signatures: insights into cancer aetiology and mechanisms of DNA damage and repair. DNA Repair (Amst). 2018;71:6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Begg CB, Zhang ZF. Statistical analysis of molecular epidemiology studies employing case-series. Cancer Epidemiol Biomarkers Prev. 1994;3(2):173–175. [PubMed] [Google Scholar]
- 44. Poon SL, Pang ST, McPherson JR, et al. Genome-wide mutational signatures of aristolochic acid and its application as a screening tool. Sci Transl Med. 2013;5(197):e101. [DOI] [PubMed] [Google Scholar]
- 45. Scelo G, Riazalhosseini Y, Greger L, et al. Variation in genomic landscape of clear cell renal cell carcinoma across Europe. Nat Commun. 2014;5:5135. [DOI] [PubMed] [Google Scholar]
- 46. Chan K, Roberts SA, Klimczak LJ, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47(9):1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Landi MT, Zhang T, Garcia-Closas M, et al. Sherlock-Lung: tracing lung cancer mutational processes in never smokers [abstract]. Cancer Res. 2019;79(13 suppl):Abstract SY26-02. [Google Scholar]
- 48. Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. 2016;538(7624):161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Peterson RE, Kuchenbaecker K, Walters RK, et al. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell. 2019;179(3):589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cancer Research UK . The Mutographs Project. https://www.mutographs.org. Accessed October 7, 2019.
- 51. Landi MT, Chatterjee N, Yu K, et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am J Hum Genet. 2009;85(5):679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McKay JD, Hung RJ, Han Y, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49(7):1126–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lan Q, Hsiung CA, Matsuo K, et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat Genet. 2012;44(12):1330–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seow WJ, Matsuo K, Hsiung CA, et al. Association between GWAS-identified lung adenocarcinoma susceptibility loci and EGFR mutations in never-smoking Asian women, and comparison with findings from Western populations. Hum Mol Genet. 2017;26(2):454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48(6):607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shi J, Hua X, Zhu B, et al. Somatic genomics and clinical features of lung adenocarcinoma: a retrospective study. PLoS Med. 2016;13(12):e1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376(22):2109–2121. [DOI] [PubMed] [Google Scholar]
- 59. Luo W, Tian P, Wang Y, et al. Characteristics of genomic alterations of lung adenocarcinoma in young never-smokers. Int J Cancer. 2018;143(7):1696–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee JJ, Park S, Park H, et al. Tracing oncogene rearrangements in the mutational history of lung adenocarcinoma. Cell. 2019;177(7):1842–1857.e21. [DOI] [PubMed] [Google Scholar]
- 61. Risques RA, Kennedy SR. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 2018;14(1):e1007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Martincorena I. Somatic mutation and clonal expansions in human tissues. Genome Med. 2019;11(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yoshida K, Gowers KHC, Lee-Six H, et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature. 2020;578(7794):266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mazor T, Pankov A, Johnson BE, et al. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell. 2015;28(3):307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4(12):988–993. [DOI] [PubMed] [Google Scholar]
- 66. Chakravarthy A, Furness A, Joshi K, et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun. 2018;9(1):3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen B, Khodadoust MS, Liu CL, et al. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol. 2018;1711:243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173(2):291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zheng X, Zhang N, Wu HJ, et al. Estimating and accounting for tumor purity in the analysis of DNA methylation data from cancer studies. Genome Biol. 2017;18(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yu G, Gail MH, Consonni D, et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 2016;17(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Greathouse KL, White JR, Vargas AJ, et al. Interaction between the microbiome and TP53 in human lung cancer. Genome Biol. 2018;19(1):123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jin C, Lagoudas GK, Zhao C, et al. Commensal microbiota promote lung cancer development via gammadelta T cells. Cell. 2019;176(5):998–1013.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Maddi A, Sabharwal A, Violante T, et al. The microbiome and lung cancer. J Thorac Dis. 2019;11(1):280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang Y, Fu F, Chen H Management of Ground-Glass Opacities in the Lung cancer Spectrum. Ann Thorac Surg. 2020;110(6):1796–1804. [DOI] [PubMed] [Google Scholar]
- 76. de Bruin EC, McGranahan N, Mitter R, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346(6206):251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jamal-Hanjani M, Hackshaw A, Ngai Y, et al. Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol. 2014;12(7):e1001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Negrao MV, Quek K, Zhang J, et al. TRACERx: tracking tumor evolution to impact the course of lung cancer. J Thorac Cardiovasc Surg. 2018;155(3):1199–1202. [DOI] [PubMed] [Google Scholar]
- 79. Turajlic S, Xu H, Litchfield K, et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell. 2018;173(3):595–610.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. [DOI] [PubMed] [Google Scholar]
- 81. Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci. 2013;1284(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yeh P, Hunter T, Sinha D, et al. Circulating tumour DNA reflects treatment response and clonal evolution in chronic lymphocytic leukaemia. Nat Commun. 2017;8:14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Saini N, Roberts SA, Klimczak LJ, et al. The impact of environmental and endogenous damage on somatic mutation load in human skin fibroblasts. PLoS Genet. 2016;12(10):e1006385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Saini N, Sterling JF, Sakofsky CJ, et al. Mutation signatures specific to DNA alkylating agents in yeast and cancers. Nucleic Acids Res. 2020;48(7):3692–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kucab JE, Zou X, Morganella S, et al. A compendium of mutational signatures of environmental agents. Cell. 2019;177(4):821–836.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Petljak M, Alexandrov LB, Brammeld JS, et al. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell. 2019;176(6):1282–1294.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Davies H, Glodzik D, Morganella S, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23(4):517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hoadley KA, Yau C, Wolf DM, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158(4):929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Boot A, Huang MN, Ng AWT, et al. In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Res. 2018;28(5):654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Drost J, van Boxtel R, Blokzijl F, et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science. 2017;358(6360):234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Degasperi A, Amarante TD, Czarnecki J, et al. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat Cancers. 2020;1(2):249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Koh G, Zou X, Nik-Zainal S. Mutational signatures: experimental design and analytical framework. Genome Biol. 2020;21(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gharibvand L, Shavlik D, Ghamsary M, et al. The association between ambient fine particulate air pollution and lung cancer incidence: results from the AHSMOG-2 study. Environ Health Perspect. 2017;125(3):378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Huang F, Pan B, Wu J, et al. Relationship between exposure to PM2.5 and lung cancer incidence and mortality: a meta-analysis. Oncotarget. 2017;8(26):43322–43331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cui P, Huang Y, Han J, et al. Ambient particulate matter and lung cancer incidence and mortality: a meta-analysis of prospective studies. Eur J Public Health. 2015;25(2):324–329. [DOI] [PubMed] [Google Scholar]
- 96. Raaschou-Nielsen O, Andersen ZJ, Beelen R, et al. Air pollution and lung cancer incidence in 17 European cohorts: prospective analyses from the European Study of Cohorts for Air Pollution Effects (ESCAPE). Lancet Oncol. 2013;14(9):813–822. [DOI] [PubMed] [Google Scholar]
- 97. Kim C, Gao YT, Xiang YB, et al. Home kitchen ventilation, cooking fuels, and lung cancer risk in a prospective cohort of never smoking women in Shanghai, China. Int J Cancer. 2015;136(3):632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Raspanti GA, Hashibe M, Siwakoti B, et al. Household air pollution and lung cancer risk among never-smokers in Nepal. Environ Res. 2016;147:141–145. [DOI] [PubMed] [Google Scholar]
- 99. Barone-Adesi F, Chapman RS, Silverman DT, et al. Risk of lung cancer associated with domestic use of coal in Xuanwei, China: retrospective cohort study. BMJ. 2012;345:e5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhang Y, Chen K, Zhang H. Meta-analysis of risk factors on lung cancer in non-smoking Chinese female. Zhonghua Liu Xing Bing Xue Za Zhi. 2001;22(2):119–121. [PubMed] [Google Scholar]
- 101. Berry G, Liddell FDK. The interaction of asbestos and smoking in lung cancer: a modified measure of effect. Ann Occup Hyg. 2004;48(5):459–462. [DOI] [PubMed] [Google Scholar]
- 102. Oh SS, Koh S, Kang H, et al. Radon exposure and lung cancer: risk in nonsmokers among cohort studies. Ann Occup Environ Med. 2016;28:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhang ZL, Sun J, Dong JY, et al. Residential radon and lung cancer risk: an updated meta- analysis of case-control studies. Asian Pac J Cancer Prev. 2012;13(6):2459–2465. [DOI] [PubMed] [Google Scholar]
- 104. Neuberger JS, Gesell TF. Residential radon exposure and lung cancer: risk in nonsmokers. Health Phys. 2002;83(1):1–18. [DOI] [PubMed] [Google Scholar]
- 105. Rosenberger A, Bickeboller H, McCormack V, et al. Asthma and lung cancer risk: a systematic investigation by the international lung cancer consortium. Carcinogenesis. 2012;33(3):587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Santillan AA, Camargo CA Jr, Colditz GA. A meta-analysis of asthma and risk of lung cancer (United States). Cancer Causes Control. 2003;14(4):327–334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.