

Abstract
Methamphetamine (METH) is a globally abused, highly addictive stimulant. While investigations of the rewarding and motivational effects of METH have focused on neuronal actions, increasing evidence suggests that METH can also target microglia, the innate immune cells of the central nervous system, causing release of proinflammatory mediators and therefore amplifying the reward changes in the neuronal activity induced by METH. However, how METH induces neuroinflammatory responses within the central nervous system (CNS) is unknown. Herein, we provide direct evidence that METH creates neuroinflammation, at least in part, via the activation of the innate immune Toll-like receptor 4 (TLR4). Biophysical studies revealed that METH bound to MD-2, the key co-receptor of TLR4. Molecular dynamics simulations showed METH binding stabilized the active heterotetramer (TLR4/MD-2)2 conformation. Classic TLR4 antagonists LPS-RS and TAK-242 attenuated METH induced NF-κB activation of microglia, whereas added MD-2 protein boosted METH-induced NF-κB activation. Systemically administered METH (1 mg/kg) was found to specifically up-regulate expression of both CD11b (microglial activation marker) and the proinflammatory cytokine interleukin 6 (IL-6) mRNAs in the ventral tegmental area (VTA), but not in either the nucleus accumbens shell (NAc) or prefrontal cortex (PFC). Systemic administration of a non-opioid, blood-brain barrier permeable TLR4 antagonist (+)-naloxone inhibited METH-induced activation of microglia and IL-6 mRNA over-expression in VTA. METH was found to increase conditioned place preference (CPP) as well as extracellular dopamine concentrations in the NAc, with both effects suppressed by the non-opioid TLR4 antagonist (+)-naloxone. Furthermore, intra-VTA injection of LPS-RS or IL-6 neutralizing antibody suppressed METH-induced elevation of extracellular NAc dopamine. Taken together, this series of studies demonstrate that METH-induced neuroinflammation is, at least in part, mediated by TLR4-IL6 signaling within the VTA, which has the downstream effect of elevating dopamine in the NAc shell. These results provide a novel understanding of the neurobiological mechanisms underlying acute METH reward that includes a critical role for central immune signaling, and offers a new target for medication development for treating drug abuse.
Keywords: Toll-like receptor 4, myeloid differentiation protein 2, methamphetamine, microglia, (+)-naloxone
Graphical Abstract

INTRODUCTION
Methamphetamine (METH) is a globally abused, highly addictive stimulant. Its behavioral effects and high abuse potential arise from its ability to dramatically increase extracellular dopamine levels within the nucleus accumbens shell (NAc) 1, 2, an effect generally attributed to blockade and reversal of vesicular monoamine and dopamine transporters 3, 4. While investigations of the rewarding and motivational effects of METH have primarily focused on neuronal actions, increasing evidence suggests that METH can also target microglia, the innate immune cells of the central nervous system, resulting in neuroinflammation 5-7.
A role for central immune signaling in mediating the rewarding effects of other drugs of abuse has emerged in recent years. For example, the pattern-recognition receptor, Toll-like receptor 4 (TLR4) detects and responds to morphine 8-10 and cocaine 11, triggering a proinflammatory central immune signaling cascade. Historically, the study of TLR4 involvement in drug reward arose as a direct consequence of the discovery that morphine activated microglia in vivo and in vitro, resulting in the induction of proinflammatory products, including proinflammatory cytokines 9, 12, 13, which opposed the pain-relieving effects of opioids 12. Exploration of the mechanisms underlying morphine-induced microglial activation revealed that these effects of were not via classical, stereoselective opioid receptors but rather through activation of the non-stereoselective innate immune receptor, TLR4 9. Notably, unlike classical opioid receptors that can only bind (−)-isomers of antagonists like (−)-naloxone14, 15, TLR4 function is antagonized by both (+)- and (−)-naloxone 16, 17, leading to the use of (+)-naloxone as a blood-brain barrier permeable, TLR4 selective, non-opioid inhibitor of morphine’s effects on microglia 13.
Studies of opioids in the context of analgesia were soon extended to pursue the question of generalization. That is, work that followed explored: (a) whether TLR4 contributes to the rewarding effects of morphine and other opioids, and (b) whether TLR4 contributes to the actions of other drugs of abuse, as well. Through these lines of investigation, it was recognized that blockade of TLR4 suppresses morphine- and cocaine-induced proinflammatory signaling in vitro in both cell expression systems and isolated microglia 9, 11 , as well as in vivo 11, 13. Furthermore, TLR4 blockade suppresses classical neurochemical indices of drug reward, including morphine- and cocaine-induced increases in extracellular dopamine levels within the nucleus accumbens shell (NAc) 11, 13. At the level of behavior, blockade of TLR4 suppresses a variety of opioid- and cocaine-induced indices of drug reward, including conditioned place preference, drug self-administration, incubation of craving, and relapse/reinstatement of drug seeking 11, 13, 18. While studies of the neuroanatomical bases of TLR4 modulation of drug reward are still in their infancy, investigation of cocaine-TLR4 interactions revealed that cocaine-induced dopamine increases in NAc are dependent on interleukin-1β (IL-1β) signaling produced by TLR4 activation within the ventral tegmental area (VTA) 11, 18.
Collectively, the findings reviewed above led to the xenobiotic hypothesis of drug abuse 19, suggesting that TLR4 within the central nervous system (CNS) recognizes cocaine and morphine as “foreign”, and triggers central proinflammatory immune signaling. This is in keeping with the role of TLR4 as a detector of endogenous danger signals released by cellular stress and damage (danger associated molecular patterns [DAMPs]), microbes and other pathogens (MAMPs/PAMPs), and certain exogenous small molecules (xenobiotics; XAMPs) 19. While METH has been shown to induce microglial activation and proinflammatory responses within the CNS 5-7, how METH creates such neuroinflammation has remained a mystery. Were METH responded to as a xenobiotic and, more specifically as a XAMP acting at TLR4, this would predict that METH could exert its rewarding effects, in part, via microglial proinflammatory signaling events. Herein we provide direct evidence that: (1) METH creates its neuroinflammatory effects by targeting the lipopolysaccharide (LPS) binding pocket of the TLR4 accessory protein, myeloid differentiation protein 2 (MD-2); (2) METH-induced microglial activation is mediated by TLR4 signaling; and (3) METH-induced neuroinflammation is, at least in part, mediated by TLR4-IL6 signaling within the ventral tegmental area (VTA), which has the downstream effect of elevating dopamine in the NAc. These results demonstrate that METH-induced conditioned place preference (CPP; a behavioral reflection of drug reward) and increases in extracellular dopamine within the NAc (a neurochemical reflection of drug reward) are each mediated, at least in part, by TLR4 immune signaling.
RESULTS
METH binds to MD-2
MD-2 has been identified as a cell surface protein co-receptor required for TLR4 signaling, which is responsible for ligand recognition. To assess whether METH could be recognized by MD-2, biophysical binding of METH with MD-2 was performed. As shown in Fig. 1A, METH binding caused the quenching of MD-2 intrinsic fluorescence. By fitting the titration curve with a one site binding model, a dissociation constant (KD) of 6.7 ± 0.6 μM was obtained for METH interacting with MD-2. As a comparison, roxithromycin, a compound used as a negative control in MD-2 binding 20, showed negligible MD-2 fluorescence quenching, demonstrating the specific binding of METH to MD-2. Fig. 1B presents the lg(F0/F – 1) versus lg([METH]/μM) plot. A stoichiometry of 0.89 ± 0.08 and a KD of 7.1 ± 0.5 μM were obtained for the binding of METH to MD-2, which justifies the one-site binding model used for the nonlinear least square fit of the MD-2 quenching data (Fig. 1A). No apparent quenching of protein A intrinsic fluorescence was observed during the titration of METH into a protein A solution (Fig. 1C). The possibility that the observed MD-2-METH binding was due to protein A tag-METH interaction was excluded by testing the binding of METH to protein A. Bis-ANS, an MD-2 molecular probe, which has been shown to bind to the LPS binding pocket of MD-2 and its fluorescence intensity enhances when this MD-2 binding occurs 21. METH decreased Bis-ANS fluorescence in a concentration-dependent manner with Ki of 16.0 ± 3.7 μM (Fig. 1D), indicating that it competitively replaces Bis-ANS binding to the LPS binding pocket of MD-2 (Fig. 1D). Taken together, these data suggest that METH binds to the LPS binding pocket of MD-2.
Figure 1.
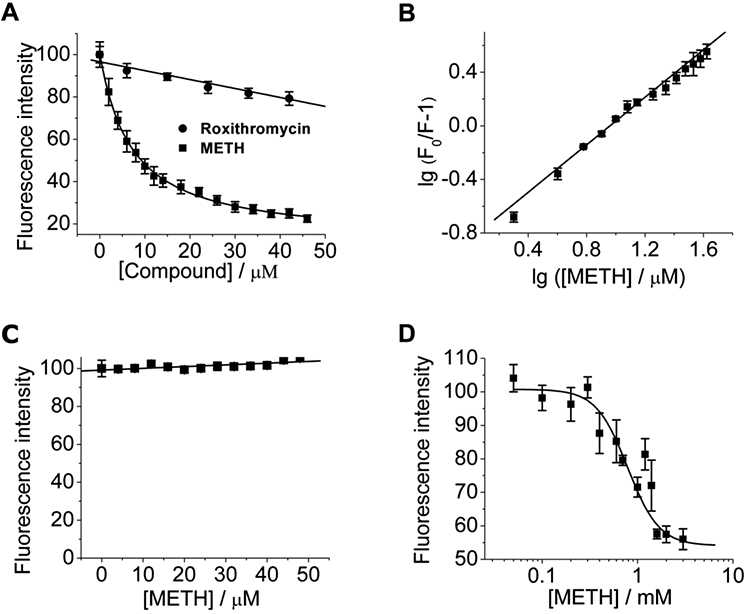
Biophysical characterizations of methamphetamine (METH) binding to MD-2. (A), Titration curves of MD-2 intrinsic fluorescence with the increasing compound concentrations. METH bound to MD-2 and caused the quenching of MD-2 intrinsic fluorescence, while roxithromycin, a compound used as a negative control, showed no apparent MD-2 binding activity. A dissociation constant (KD) of 6.7 ± 0.6 μM was obtained for METH-MD-2 interactions. (B), Re-plotting the data from (A) according to the equation: lg (F0/F – 1) = −lgKD + n × lg ([METH]), revealing a slope (n, stoichiometry) of 0.89 ± 0.08 and a KD = 7.6 ± 0.9 μM for the METH-MD-2 interactions. (C), Titration curve of protein A (negative control protein) intrinsic fluorescence with the increasing METH concentrations. 280 nm was used as the excitation, and emission at 308 nm (peak position) was plotted against the titrated METH concentration. METH failed to bind to protein A and showed negligible effect on the intrinsic fluorescence of protein A. (D), METH displaced Bis-ANS binding to MD-2. The fluorescence intensity of Bis-ANS increases upon MD-2 binding. METH caused the decrease of Bis-ANS fluorescence from the Bis-ANS/MD-2, indicating that METH replaced Bis-ANS binding to MD-2. Data fitting to a one-site competitive model gives a Ki of 16.0 ± 3.7 μM. Data were means ± SD; n = 3/group.
Molecular dynamics simulations of METH binding with TLR4/MD-2
To investigate how METH interacts with MD-2 and modulates TLR4 signaling, molecular dynamics simulations of METH interacting with the heterotetramer (TLR4/MD-2)2 were performed. METH was found to dock into the dimerization interface between the entrance of the MD-2 cavity and TLR4* that is from the adjacent copy of TLR4-MD-2. Specifically, METH overlapped with the R2 chain of Lipid A (Fig. S3), which agrees well with the biophysical binding results that METH binds to the LPS binding pocket of MD-2. During 20 ns molecular dynamics simulations, the root-mean square deviation (RMSD) value of the heterotetramer (TLR4/MD-2)2 (Fig. S4) stabilized. It should be noted that the active heterotetramer (TLR4/MD-2)2 structure (PDB ID: 3VQ2) was used for the molecular dynamics simulations. The ligand binding-induced MD-2 conformational changes were monitored by φ-ψ backbone dihedral angles in F126 loop, which are located in the core of the (TLR4/MD-2)/(TLR4*/MD-2*) oligomerization interface 22. The φ-ψ backbone dihedral angles in the F126 loop (G123, F126 and S127) underwent large transitions in the absence of METH, while the backbone dihedral angle of the F126 loop in MD-2 did not undergo apparent transitions in the presence of METH during the molecular dynamics simulations (Fig. 2). Fig. 3 shows the representative pose of METH binding to TLR4/MD-2 during the molecular dynamics simulations. The aromatic ring of METH was surrounded by the hydrophobic residues I80, I124, F126 and Y131 of MD-2. The METH side group N-methyl-2-azyl-propanyl interacted with F414 and S415 of TLR4* through hydrophobic and electrostatic interactions, respectively. These results showed that METH binding stabilized the active heterotetramer (TLR4/MD-2)2. It should be noted that TLR4/MD-2 is in a dynamic equilibrium in the cell membrane with populations of heterodimers and heterotetramers 23. METH binding stabilized the tetrameric form and could shift the equilibrium toward the activated state. Therefore, METH might activate TLR4 signaling and be a non-classic TLR4 agonist.
Figure 2.

The transitions of backbone dihedral angles of residues in the MD-2 F126 loop during the course of molecular dynamics simulations of active heterotetramer (TLR4/MD-2)2 (PDB ID: 3VQ2) in the absence or presence of METH. φ is defined as the dihedral angle formed by the atoms C(O)-N-C(α)-C(O) while ψ is defined by the atoms N-C(α)-C(O)-N.
Figure 3.

Representative pose of METH binding to TLR4/MD-2 within the 20 ns molecular dynamics simulations. METH was shown as green stick; MD-2 was shown as red cartoon; TLR4*, which is from the adjacent copy of TLR4-MD-2, was shown as cyan cartoon. Key residues of binding site in interacting with ligand were shown as cyan line model.
METH activates TLR4 signaling in BV-2 microglial cells
In order to test whether METH could activate TLR4 signaling, the effect of METH on TLR4 downstream NF-κB activity in BV-2 microglial cells was measured. TLR4 activation on microglia triggers an intracellular signaling cascade resulting in transcription factor NF-κB activation 24. As shown in Fig. 4A, METH induced NF-κB activation in a concentration-dependent manner. LPS-RS is a competitive LPS antagonist, which targets the LPS binding pocket of MD-2. TAK242 is a potent TLR4 antagonist, which binds selectively to Cys747 in the TIR domain of TLR4 and subsequently disrupts the ability of TLR4 to associate with TIRAP, therefore blocking TLR4 signaling 25. Both LPS-RS and TAK-242 were found to block METH induced NF-κB activation (Fig. 4B), which supports the hypothesis that METH-induced microglial activation was mediated by TLR4 signaling.
Figure 4.
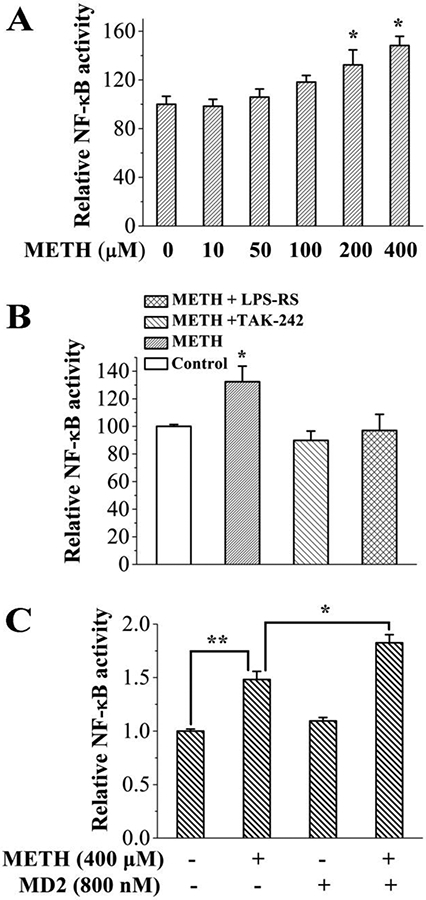
METH activates TLR4 signaling in microglial BV-2 cells. (A), METH induced NF-κB activation in a concentration dependent manner. *p<0.05 versus the untreated control group. (B), classic TLR4 antagonists LPS-RS (2 ng/mL) and TAK-242 (200 nM) blocked METH (200 μM)-induced NF-κB signaling. METH induced NF-κB signaling was significantly blunted by TAK-242 (p<0.05) and LPS-RS (p<0.05). The control group, METH+TAK-242, and METH+LPS-RS groups are not different from each other (p > 0.05). A One-way ANOVA was used for statistical analysis between different treatments. (C), MD-2 boosted METH induced NF-κB activation. A main effect of both METH (F(1, 10) = 102.3, p < 0.0001) and MD-2 (F(1, 10) = 19.3, p = 0.001) was observed, with the Bonferroni posthoc analysis revealing significant METH alone effects (p = 0.001), and further MD-2-enabled enhancements (p = 0.006) of NF-κB signaling over and above these. * p < 0.05; **, p < 0.01. BV-2 NF-κB reporter cells were treated with indicated concentration of METH, TAK-242, LPS-RS and/or MD-2 protein for 24 hours. NF-κB activity was measured by Dual-Glo Luciferase Assay System. All the data were represented by means ± SEMs; n = 3-4/group. A Two-way ANOVA was used for statistical analysis between different treatments.
MD-2 is a secreted protein. Thus, purified MD-2 was added into the cell culture to test whether MD-2 mediates METH induced TLR4 activation in microglial cells, As shown in Fig. 4C, the added MD-2 protein boosted METH induced NF-κB activation, which supports the direct role of MD-2 in METH induced TLR4 signaling activation in microglial cells. It should be noted that MD-2 is critical for TLR4 distribution in the cell 26. Knocking-down/out MD-2 would disrupt the distribution of TLR4 and hinder distribution of TLR4 to the plasma membrane 26. Therefore, a CRISPR/Cas9 or RNAi strategy was not used to investigate the role of MD-2 in METH induced microglial cell activation.
TLR4-IL6 signaling within VTA, at least in part, mediates METH induced neuroinflammation and elevation of extracellular NAc dopamine
As noted above, biophysical characterization and in vitro microglial cell studies indicate that METH binds to MD-2 and thereby activates TLR4 signaling. Previous findings with morphine 9 and cocaine 11 indicate that drug-induced TLR4 activation importantly contributes to the rewarding effects of these drugs, as reflected by conditioned place preference (CPP) and elevation of extracellular NAc dopamine. As shown in Fig. 5, METH produced robust increases in CPP (Fig. 5A) and extracellular NAc dopamine (Fig. 5B) compared to the saline control. As noted in the Introduction, (+)-Naloxone is a non-opioid TLR4 antagonist with good blood-brain-barrier (BBB) permeability 17 and fails to act as an opioid antagonist given the extreme stereoselectivity of classical opioid receptors only for (−)-isomers 14. Relevant to the present series of studies, the non-opioid TLR4 antagonist (+)-naloxone has been shown to suppress morphine 9 and cocaine 11 induced CPP and increase in extracellular dopamine within the NAc. Interestingly, (+)-naloxone (15 mg/kg) also inhibited METH-induced CPP (Fig. 5A) and increased extracellular NAc dopamine (Fig. 5B). 5 mg/kg of (+)-naloxone did not reliably block METH-induced dopamine increase within the NAc such that a dose-dependent effect was observed (Fig. 5C). It should be noted that (+)-naloxone treatment alone did not affect CPP or alter the basal dopamine level, suggesting that (+)-naloxone did not independently produce effects on dopamine signaling. We have previously established that (+)-naloxone treatment does not interfere with dopaminergic cell functioning, and that non-TLR4 stimulation of increased dopamine within the NAc is preserved in the presence of (+)-naloxone 11. These findings indicate that METH-induced increase of NAc dopamine is partially dependent on systemic TLR4 signaling.
Figure 5.

TLR4 antagonist (+)-naloxone inhibited METH induced conditioned place preference (A) and METH induced increase of NAc dopamine (B). (A), (+)-naloxone attenuated METH-induced conditioned place preference. A significant main effect of METH (1 mg/kg) was observed (F(1, 19) = 53.45, p < 0.0001) representative of the significantly increased conditioned place preference, compared to saline controls. A significant main effect (F(1, 19) = 8.08, p = 0.01) and interaction (F(1, 19) = 5.9, p = 0.03) were found for the attenuation of the METH-induced effect by (+)-naloxone (15 mg/kg). Posthoc analysis relieved a significant reduction in the METH conditioned place preference by (+)-naloxone (p = 0.007). ** p < 0.01; *** p < 0.001. Data are means ± SEMs; n = 5-6/group. Prior to drug treatment there were no differences in extracellular dopamine concentrations in the NAc shell across all groups (p > 0.05). Two-way ANOVA with Bonferroni corrected posthoc test was used for statistical analysis between different treatments. (B), (+)-naloxone attenuated METH-induced increase of extracellular dopamine within the NAc shell. METH (1 mg/kg) elevated extracellular dopamine in the NAc compared to saline controls. When testing the 2 x 2 effect of METH versus 15 mg/kg (+)-naloxone (and saline controls) a mixed effects ANOVA relieved a main effect of METH (F(1, 36) = 130.0, p < 0.0001) and time (F(8, 36) = 11.09, p < 0.0001). A main effect of (+)-naloxone treatment was observed (F(1, 36) = 5.6, p = 0.02) with a significant METH x (+)-naloxone x time effect reported (F(8, 18) = 3.5, p = 0.01). (C) The METH-induced effect was attenuated dose-dependently attenuated across the peak of the METH-dopamine response (40-80 mins) with a significant effect of treatment (F(4, 17) = 17.09, p < 0.0001) and a significant effect of (+)-naloxone to reduce this peak METH effect at 15 mg/kg p <0 0.02. It should be noted that 5 & 15 mg/kg (+)-naloxone + saline represent the combined two groups (5 mg/kg (+)-naloxone + saline as well as 15 mg/kg (+)-naloxone + saline). The extracellular dopamine concentration in the saline control group was 1.0 ± 0.3 nM. Data were means ± SEMs; n = 4/group.
In order to investigate the site-specificity within the CNS of METH-induced TLR4 signaling and examine whether TLR4 signaling is activated within the regions comprising the mesolimbic dopamine pathway, rats were administered a single intraperitoneal injection of METH and brains removed 30 min or 2 h later. Micropunches were collected from the PFC, NAc, and VTA; mRNA markers for proinflammatory factors were measured using quantitative RT-PCR for each region. mRNA was chosen for analysis as this approach allowed multiple genes to be probed from such small samples and provides evidence that the cells responded to METH exposure. mRNA for CD11b, a microglial activation marker, was upregulated in the VTA following METH administration (Fig. 6A) but was unaffected in either the NAc or the PFC. IL-1β mRNA was unaffected at all time-points in all three brain regions (Fig. 6B). However, mRNA for the proinflammatory cytokines tumor necrosis factor-α (TNF-α, Fig. 6C, p<0.01 at 30 min, p > 0.05 at 2 h) and interleukin-6 (IL-6, Fig. 6D, p < 0.05 at 30 min and 2 h) were upregulated in the VTA but not within the NAc or the PFC. These data indicate that systemically administered METH induces proinflammatory central immune signaling specifically within the VTA at the single low dose of METH (1 mg/kg) tested.
Figure 6.

A single systemic injection of 1 mg/kg METH upregulated mRNA markers of central immune activation within the VTA, but not within the PFC or NAc. (A), METH upregulated VTA mRNA expression of CD11b at both 30 min (*p < 0.05) and 2 hr (*p < 0.05) following treatment; however in the PFC and the NAc CD11b mRNA was not altered (p > 0.05). Two way ANOVA revealed a main effect of brain region (F(2,32)= 4.69, p = 0.016) and was followed by Bonferroni post-hoc tests. (B), A two-way ANOVA revealed no effect on IL-1β within the VTA, NAc, or PFC 30 min or 2 hrs following METH administration (F(4,28)= 1.53, p = 0.222). (C), METH produced an increase of TNF-α mRNA expression within the VTA at 30 min (**p < 0.01) which returned to baseline by 2 hrs (p > 0.05); there were no effects within the NAc or the PFC ( p> 0.05). Bonferroni post-hoc tests were preceded by two way ANOVA assessment of a main effect of time (F(2,31)= 6.15, p = 0.006) and brain region (F(2,31)= 3.70, p = 0.036). (D), A two way ANOVA revealed that METH produced an upregulation of IL-6 mRNA within the VTA (*p < 0.05) but not in the ventromedial PFC or the NAc (p>0.05); there was an effect of brain region (F(2,27) = 4.74, p=−0.0173). All the data are presented as means ± SEMs; n = 4-6/group.
In order to determine whether METH-induced central immune signaling within the VTA is TLR4-dependent in vivo, proinflammatory factor mRNA expression was measured in rats administered either METH (1 mg/kg) or vehicle (saline) with a pretreatment of (+)-naloxone (15 mg/kg, sub-cutaneous) or vehicle. Based on the overall pattern of results for VTA mRNA expression in Fig. 6, brains were removed 2 h later and micropunches of the VTA were collected and processed for RT-PCR. As shown in Fig. 7A, METH-induced upregulation of CD11b mRNA was inhibited by the non-opioid TLR4 antagonist (+)-naloxone. Similarly, METH-induced upregulation of IL-6 mRNA expression within the VTA was inhibited by (+)-naloxone administration (Fig. 7D). Additionally, neither METH nor (+)-naloxone administration alone produced any alterations in IL-1β (Fig. 7B) or TNF-α (Fig. 7C), as supported by the time course of IL-1β and TNF-α mRNA expression in Fig. 6. Together, these data indicate that TLR4-induced IL6 signaling within VTA, at least in part, mediates METH induced neuroinflammation.
Figure 7.
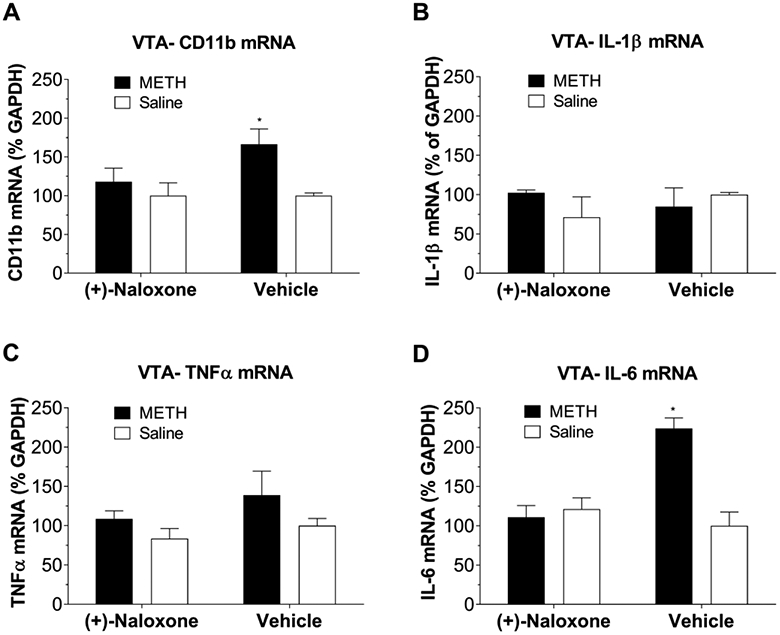
Systemic TLR4 antagonism via (+)-naloxone (15 mg/kg) prevented METH-induced up-regulation of mRNA for CD 11b and IL-6. Tissues were collected at 2 h after METH based on the overall pattern of VTA results in Figure 6. (A), METH administration upregulated mRNA expression for CD 11b within the VTA 2 h following administration (F(1,9)=6.06, p=0.034). METH + (+)-naloxone group was not different from saline control (p>0.05) or (+)-naloxone alone (p>0.05). (B), METH had no effect on mRNA expression of IL-1β within the VTA (interaction: F(1,9) = 0.016, p = 0.703). (C), METH also had no effect on mRNA expression of TNF-α (interaction: F(1,10) =0.11, p = 0.75) within the VTA two hours following METH injection. (D), Within the VTA, IL-6 is upregulated 2 h following METH and that (+)-naloxone suppressed this effect. Interaction: F(1,9)=19.27, p=0.001; Main effect of METH: F(1,9) = 13.74, p = 0.005; (+)-naloxone: F(1,9) = 8.99, p = 0.015. *p < 0.05. All the data were represented by means ± SEMs; n = 4-6/group. Two-way ANOVA followed by Bonferroni was used for statistical analysis.
In order to confirm that TLR4-IL6 signaling within VTA contributes to METH induced elevation of extracellular NAc dopamine, rats received an intra-VTA injection of the classic TLR4 antagonist LPS-RS (Fig. 8) or an anti-rat IL-6 neutralizing antibody (Fig. 9) followed 1 h later by an i.p. injection of METH. Timing of injections was based on pilot studies. As shown in Fig. 8, intra-VTA LPS-RS suppressed the NAc dopamine increase in rats receiving METH, while it had no effect on extracellular dopamine in the NAc of rats receiving systemic saline (instead of METH). Similarly, intra-VTA anti-IL-6 suppressed NAc dopamine levels in rats receiving METH, while it had no effect on extracellular dopamine levels in the NAc of rats receiving systemic saline. Taken together, the results across studies above showed TLR4-IL6 signaling within the VTA, at least in part, mediates METH induced neuroinflammation and elevation of extracellular NAc dopamine.
Figure 8.
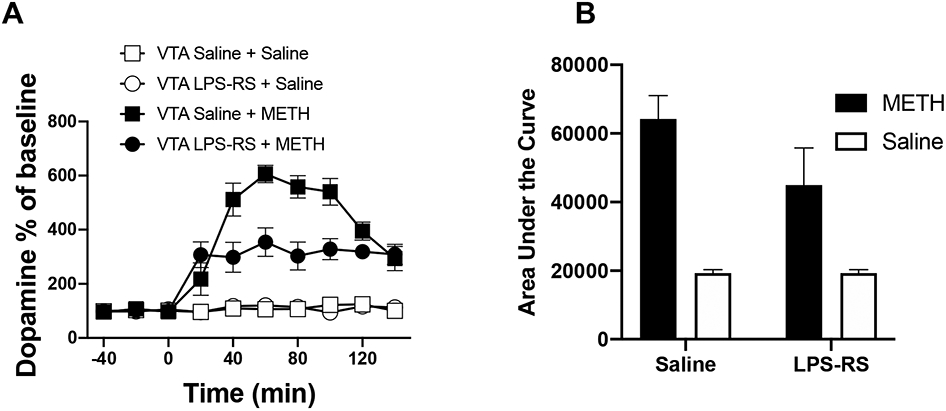
Classic TLR4 antagonist LPS-RS attenuated METH induced increase of NAc dopamine as measured by micro-dialysis (A) and the corresponding Area under Curve (AUC, B). Rats received an intra-VTA injection of either LPS-RS (5 μg in 1 μl) or equivolume saline, followed 1 h later by an i.p. injection of 1 mg/kg METH or equivolume saline. 1 mg/kg METH robustly increased extracellular dopamine levels in NAc. While intra-VTA anti-LPS-RS had no effect on extracellular dopamine levels in NAc of rats receiving systemic saline (instead of METH), it suppressed NAc dopamine levels in rats receiving METH. Mixed effects ANOVA revealed main effects of time (F(9, 153) = 32.15, p < 0.0001), METH (F(1, 153) = 474.7, p < 0.0001), LPS-RS (F(1, 153) = 26.46, p < 0.0001) and a time x METH x LPS-RS effect (F(9, 153) = 5.527, p < 0.0001). An integrated analysis of the area under the curve confirmed this finding with main effects of METH (F(1, 16) = 150.8, p < 0.0001), LPS-RS (F(1, 16) = 11.20, p = 0.004) and an interaction effect (F(1, 16) = 11.22, p = 0.004) observed using a 2 way ANOVA. Bonferroni corrected posthoc analysis found a significant LPS-RS reduction in the METH-induced AUC (p = 0.0013). Data were means ± SEMs; n = 5/group.
Figure 9.
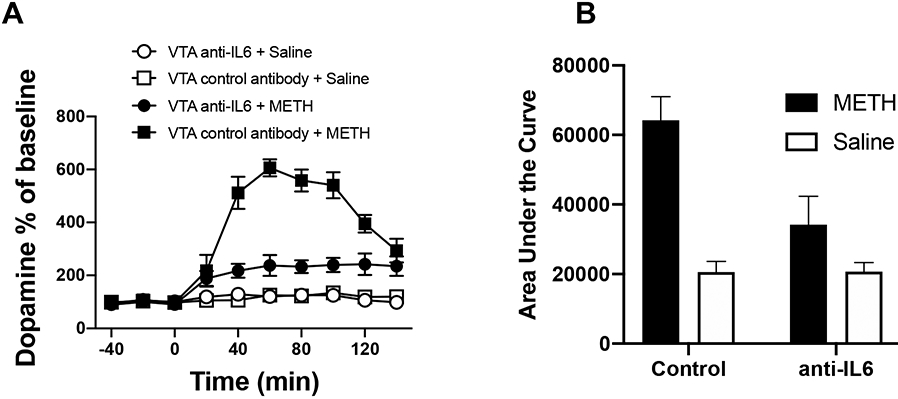
IL-6 neutralizing antibody attenuated METH induced increase of NAc dopamine as measured by micro-dialysis (A) and the corresponding Area under Curve (AUC, B). Rats were received an intra-VTA injection of an anti-rat IL-6 neutralizing antibody (0.2 μg in 1 μl), or control IgG (0.2 μg in 1μl), followed 1 h later by an i.p. injection of either 1 mg/kg METH or equivolume saline. 1 mg/kg METH robustly increased extracellular dopamine levels in NAc. While intra-VTA anti-IL-6 had no effect on extracellular dopamine levels in NAc of rats receiving systemic saline (instead of METH), it suppressed NAc dopamine levels in rats receiving METH (*** p < 0.001). Data were means ± SEMs; n = 4/group. A significant main effect of time (F(9, 40) = 30.7, p < 0.0001), METH (F(1, 40) = 407.9, p < 0.0001), anti-IL6 (F(1, 40) = 88.2, p < 0.0001) and a significant time x METH x anti-IL6 interaction term (F(9, 36) = 9.97, p < 0.0001) were obtained using mixed effects ANOVA. A two way ANOVA of the area under the curve data confirmed this finding with main effects of METH (F(1, 16) = 126.8, p < 0.0001), anti-IL6 (F(1, 16) = 34.5, p < 0.0001) and a significant interaction term (F(1, 16) = 35.2, p < 0.0001). Bonferroni corrected posthoc analysis found a significant anti-IL6 antibody reduction in the METH-induced AUC (p < 0.0001).
DISCUSSION
The data presented here indicate that METH binds to the LPS binding pocket of MD-2 and stabilizes the active heterotetramer (TLR4/MD-2)2 conformation. METH activates NF-κB signaling in microglia, an effect boosted by MD-2 protein and blocked by TLR4 antagonists LPS-RS and TAK-242. Furthermore, METH-induced TLR4 activation contributes to both CPP and increased extracellular levels of dopamine in the NAc shell. In addition, systemic METH administration upregulates mRNA expression for CD11b and IL-6, but not IL-1β, specifically in the VTA. The non-opioid TLR4 antagonist (+)-naloxone inhibits CD11b and IL-6 mRNA expression within VTA. This effect is independent of an action on opioid receptors given that opioid receptors are strictly stereoselective for (−)-isomers and fail to bind (+)-isomers, including (+)-naloxone14, 15. These data indicate that TLR4 signaling within the VTA, resulting in elevations in IL-6 gene activation, at least in part, mediates METH-induced neuroinflammation. Notably, either blocking TLR4 signaling by the classic TLR4 antagonist LPS-RS or blocking the effects of released IL-6 protein by anti-rat IL-6 neutralizing antibody within the VTA suppresses METH-induced elevations in extracellular NAc dopamine. These data indicate a direct role of VTA TLR4-IL6 signaling in regulating extracellular NAc dopamine levels and support that METH-induced elevations of IL6 mRNA likely result in IL6 protein as well. These findings also provide further support for the recently proposed xenobiotic hypothesis of drug abuse 9, 11, 19. This hypothesis suggests that: (i) in serving its immune-surveillance role, TLR4 together with its accessory proteins detect and respond to the presence of drugs of abuse, such as morphine, cocaine, and now METH, as foreign compounds and initiate proinflammatory immune signaling in response to the perceived threat, and (ii) drug-induced proinflammatory signaling paired with unique neuronal actions of each drug of abuse synergize to create their rewarding effects. It should be noted that pure TLR4 activation within the VTA by the classic TLR4 agonist LPS would induce a marginal dopamine increase in the NAc, compared to drugs of abuse 11. It is clear that TLR4 signaling comprises only part of the complexity of METH’s actions and hence TLR4 does not account for all of METH’s pharmacodynamic effects. METH can directly impact neuronal function creating a combined neural and immune response that, together, accounts for its in vivo actions. Hence, it is the combination of TLR4 activation and the presence of the drug of abuse with its own independent neuronal actions that culminates in modulating drug reward.
Previous reports have noted neurotoxic effects of METH, linking this to inflammatory responses by microglia and glutamatergic toxicity created by astrocytes 27. It is notable that the neurotoxic effects reported for METH are typically observed with much higher and repeated doses than the 1 mg/kg single dose of METH used in the present study. With much higher dosing of METH, neuroinflammatory responses in PFC and NAc have been observed28-30, effects not created by the single 1 mg/kg METH dose used here. The only prior report of METH-induced neuroinflammatory effects within the VTA also employed far higher METH doses than used here30. The aim of the present study was to explore the hypothesis that METH initiates central immune signaling via interaction with TLR4 and that the ability of METH to create conditioned place preference (CPP; a behavioral reflection of drug reward) and to increase NAc extracellular dopamine concentrations (a neurochemical reflection of drug reward) is, in part, dependent on TLR4-induced immune signaling. Based on past literature 31, 32 and pilot studies, a dose of 1 mg/kg of METH, which produced reliable CPP and produced robust dopamine elevation in the NAc (Fig. 5), was used in order to minimize the interference by METH neurotoxicity. Although there are reports of increased numbers and/or activation of microglial cells in the brains of human METH addicts 6 as well as microglial and central immune activation in animal models 33, 34, the mechanism(s) by which METH exerts such effects was unknown. The data in this study suggest that METH could signal via the TLR4-MD-2 complex to initiate proinflammatory signaling in at least microglia and perhaps in other TLR4-expressing cell types such as astrocytes, as well.
Notably, the data demonstrate a direct action of METH on MD-2, suggesting that METH itself can activate TLR4 rather than resulting indirectly via METH-induced release of DAMPs with agonist actions at TLR4. These data complement and extend the recent findings that METH upregulates TLR4 expression in microglial BV2 cells 35 and primary mouse astrocytes 36 and that siRNA knockdown of TLR4 suppressed METH-induced NF-κB activation and proinflammatory responses in both glial cell types 35, 36. The key role of the TLR4 accessory protein MD-2 is of considerable relevance. The brain regional heterogeneity and isolated cell type sensitivity to XAMP exposure has been linked not only to cell surface receptor expression but also to the extracellular environment and the requirement of the presence of accessory proteins 8, 37.
We have recently demonstrated that other drugs of abuse, including opioids 9 and cocaine 11 target MD-2 and activate TLR4 signaling. This TLR4 activation contributes to the rewarding effects of these drugs 9, 11. The TLR4 antagonist (+)-naloxone suppresses CPP and NAc dopamine increases in response to opioids and cocaine 9, 11. Herein, (+)-naloxone similarly suppresses METH-induced CPP and NAc dopamine release. Whether TLR4 blockade attenuates other behavioral indices of acute METH reward beyond suppressing METH-induced conditioned place preference remains to be investigated. What little is known to date is that while TLR4 blockade with chronically administered TLR4 antagonist (+)-naltrexone significantly suppressed incubated cue-induced opioid seeking, it failed to suppress incubated cue-induced METH seeking 38. (−)-Naltrexone, behaving similarly as the opioid inactive enantiomers (+)-naltrexone or (+)-naloxone in inhibiting TLR4 signaling 17 in addition to blocking opioid receptors, did not alter the reinforcing effects of intranasal METH 39. It will be important to explore a number of reward and motivational paradigms to define the behavioral consequences of METH-induced TLR4 activation. This is because (1) the mechanisms underlying incubation of craving for METH and opioids are distinct 40, (2) the neurocircuitry of rewarding effects of acute vs. chronic exposure to drugs of abuse are distinct 41, and (3) the observation in the present study of site-specific effects of METH on proinflammatory responses.
The findings across opioids 9, 13 , cocaine 11, and now METH suggest that TLR4 signaling may be an important mediator of drug-induced dopaminergic effects. Further, the data indicate that METH, like cocaine 11, 42 and opioids 9, 13, induces upregulation of proinflammatory markers, specifically within the VTA. Were METH administration to trigger the release of proinflammatory cytokines, such an effect might explain how TLR4 signaling influences dopaminergic functioning. Proinflammatory cytokines, released following TLR4 activation, can upregulate surface expression of AMPA and NMDA receptors, increase conductivity of NMDA receptors, increase spontaneous neurotransmitter release, and increase glutamate transmission, therefore exaggerating neuroexcitatory actions 19. However, many of these findings to date particularly focus on IL-1β and TNF-α. Although TNF-α mRNA upregulation is evident 30 min following METH administration, no alterations in IL-1β mRNA were detected at the time points tested. This is surprising considering the recent finding that intra-VTA IL-1β signaling, induced via TLR4, is required for cocaine to influence neuronal responses and drug seeking 11, 43. Although IL-6 was upregulated following METH administration, less is known about IL-6’s ability to influence neuronal functioning. Certain neuronal populations throughout the brain express IL-6 receptors 44 so it is reasonable to postulate that IL-6 could directly influence neuronal signaling, but how this signaling might be relevant to the mesolimbic dopamine pathway warrants further investigation.
It may be that the lack of effect on IL-1β partially explains the differences between the cocaine findings where TLR4 blockade abolishes cocaine-induced dopamine increases 11, and the METH data presented here where TLR4 attenuates, but does not block, METH-induced dopamine increases. This type of biased signaling induced by different TLR4 ligands is a hallmark of this type of complex receptor signaling cascade, evolutionarily allowing one receptor to trigger customized responses based on the cell, environment, and initiating trigger 17, 45. It is also important to note that METH has the ability to more dramatically affect dopamine concentrations than cocaine through METH actions on dopamine transporters, given that METH triggers reverse transport 46. While it may be that METH also activates dopaminergic signaling through TLR4-mediated proinflammatory mechanisms in the VTA that drive increased dopaminergic neuronal firing, METH mesolimbic dopaminergic effects, at least at the single low dose of METH tested, are likely less dependent on proinflammatory activation in the VTA given METH’s reversal of dopamine transporters 46.
A remaining question regards the cellular source of TLR4 through which METH induces neuroinflammation. TLR4 is predominately expressed on microglial cells within the CNS 47, therefore most of its effects have generally been attributed to microglial responses. However, astrocytes are also are immunocompetent and can participate in proinflammatory signaling 48. Although there is some controversy regarding TLR4 expression in astrocytes under basal conditions, cultured primary murine astrocytes have been shown to express low levels of TLR4 mRNA and demonstrate marked upregulation of TLR4 when exposed to TLR4 ligands 49. Astrocytes serve as important modulators of pre- and post-synaptic activity, synapse formation, function, plasticity, and elimination as well as regulation of glutamate transmission 50, in part via their role in the tripartite synapse 51. Importantly, astrocyte functioning can be closely tied to microglial activity; TLR4 signaling rapidly triggers a microglial proinflammatory response, subsequently activating astrocytes 52. When astrocytes shift from a basal state to an activated, proinflammatory state, they can release proinflammatory cytokines, D-serine (co-agonist of glutamate on NMDA receptors), and glutamate 52. Whether astrocytes importantly contribute to the effects reported here bear future investigation.
Conclusions
In summary, the findings indicate a novel mechanism by which METH initiates central immune activation; namely through TLR4-IL6 signaling. The data also point to the need for future research to clarify mechanistically how immune signaling contributes to elevations in extracellular dopamine levels in the NAc and the role of dopamine transporters in this process. Further, TLR4 signaling induced via METH appears to be initially selective to the VTA and contributes to METH-induced CPP and increases in extracellular NAc dopamine, correlates of drug reward. The present findings may have important implications for drug reward and merit further investigation as to how these mechanisms may contribute to the development of addiction and other pathological effects of METH. They also provide further support for the xenobiotic hypothesis of drug abuse. TLR4 may be a novel target for the development of pharmacotherapies to aid in the treatment of drug abuse and addiction. Additionally, the role of glial cells, TLR4, and proinflammatory mediators in dopamine cell functioning and toxicity have overarching implications for numerous diseases and/or neuropathic states effecting dopamine systems that could guide the development of pharmacotherapies aiming to treat these pathologies.
MATREIALS and METHODS
Subjects
Viral-free adult, male Sprague Dawley rats (275–350 g; Harlan) were pair-housed in standard Plexiglas cages with ad libitum food and water and maintained on a 12 h light/dark cycle (lights on 0700 h). Rats were allowed 1 week of acclimation before any procedures. Naïve animals were used for each study. All experimental procedures were approved by the Institutional Animal Care and Use Committee and conform to ARRIVE guidelines. All studies were performed blinded with respect to group assignment.
Drugs
METH (>98%) was purchased from Sigma-Aldrich (Cat. No., M8750; St. Louis, MO, USA). (+)-Naloxone was provided by Dr. Kenner Rice of the Drug Design and Synthesis Section, National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health (NIH) and no impurities were detected by TLC, NMR analysis, elemental analyses and optical rotation. Sources of all other compounds are as noted in appropriate methods sections below.
Titrations of MD-2 with METH
The insect expression human MD-2-pAcGP67A vector was provided by Dr Jie-Oh Lee (KAIST, Korea) 22 and high 5 insect cells were provided by Dr. Xuedong Liu (University of Colorado Boulder). MD-2 expression and purification were performed as described previously9. Briefly, baculovirus was prepared by co-transfection of SF-9 insect cells with the MD-2-pAcGP67A vector and bright linearized baculovirus DNA as described by the manufacturer’s protocol (BD Bioscience, San Diego, CA, USA). After 2-3 rounds of amplification, the MD-2 baculovirus suspension reached a titer of ~108/ml virus particles and was used to transfect high 5 insect cells to express MD-2. MD-2 was secreted into the medium. 3-4 days after transfection, the medium was harvested and subjected to IgG sepharose affinity purification. SDS-PAGE analysis showed that the purity of the prepared protein was >95%.
Fluorescence measurements were performed on a Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA, USA). All measurements were carried out under room temperature in a 2×10 mm quartz cell (Starna Cells, Atascadero, CA, USA). The wavelength of 280 nm was chosen to excite the Tyr and Trp residues in MD-2 fluorescence and emission of 300–450 nm was measured. 385 nm was chosen as the excitation wavelength of the extrinsic fluorescence probe Bis-ANS (Sigma Aldrich, St. Louis, MO) and emission at 420-550 nm was recorded. Appropriate baseline signals were subtracted from spectra obtained. Fluorescence was also corrected by the relation, Fcorr = Fobs anti-log (ODex + ODem/2) for the inner filter effect when necessary, where ODex and ODem are the optical densities at excitation and emission wavelengths, respectively.
For protein intrinsic fluorescence quenching assays, 0.5 μM of MD-2 was titrated with different concentrations of METH, and the fluorescence emission at 337 nm was plotted against METH concentration. The raw data was fitted by a non-linear least square method using the equation: F=0.5×(2×F0−FPL×(KD+[LT]+[PT] −((KD+[LT]+[PT]22−4×[LT]×[PT])0.5)), where [F], the observed fluorescence; F0, initial fluorescence of protein in the absence of ligand; FPL, adjustable parameter for protein–ligand complex molar fluorescence; KD, dissociation constant; [LT], total concentration of the ligand; [PT], total protein concentration. METH showed no fluorescence signal at the tested conditions. Roxithromycin, which has been reported to show no apparent binding to MD-2 20, served as a negative control compound. Protein A was used as a negative control protein.
For the competitive displacement assay, different concentrations of METH (0-5 mM) were titrated into MD-2 (0.5 μM) and Bis-ANS (0.5 μM) reaction mix. After establishing an overnight equilibrium at room temperature, the Bis-ANS fluorescence intensity was measured. The fluorescence intensity at 478 nm was plotted against METH concentration. Ki of METH was determined using the equation: Ki= Kapp/(1 + [Bis-ANS]/KD(Bis-ANS -MD-2)).
Molecular dynamics simulations of METH binding with TLR4/MD-2
The METH structure was optimized by Gaussian 09 software using B3LYP density functional method and 6-31G(d,p) basis set. Protein structure used for docking was extracted from the X-ray structure (PDB ID: 3VQ2). AutoDock Vina was performed to determine the docking poses of METH. The Iterated Local Search Globule Optimizer was applied to locate the most favorable binding site. TLR4/MD-2 was treated as a rigid body and semi-flexible docking was carried out. Optimal binding sites were searched in a box of 88 Å × 116 Å × 150 Å that covered the entire protein. Top 20 poses for METH were picked up using the scoring function of AutoDock Vina.
Molecular dynamics simulations were performed as described previously 53. NAMD 2.10 was used for the simulation of the best binding pose of METH with TLR4/MD-2 (2:2:2). The AMBER 03 force field was used for TLR4/MD-2. Atomic charges of METH were fitted by R.E.D. based on the quantum mechanics calculation. Other atomic parameters were treated with the general AMBER force field. All were solvated with TIP3P water molecules in a cubic box, with a minimum distance of 10 Å between the edge of the box and protein. Na+ and Cl− atoms were added to neutralize the system. Periodic boundary conditions were applied. The integration time step was set to 2 fs. All bonds involving hydrogen were constrained by the SHAKE algorithm. Particle-mesh Ewald method was used to calculate the long-range electrostatic interactions. Temperature was regulated using Langevin dynamics with the collision frequency of 5 ps−1. Pressure was scaled at 1 atm with the Nosé-Hoover Langevin piston method.
The solvated system was minimized using the conjugate gradient algorithm, following by heating gradually to 310 K. The volume of was adjusted under a constant number, pressure and temperature ensemble for 1 ns. Subsequently, eight independent 20 ns long NVT (constant Number of atoms, volume, and temperature) Molecular Dynamics simulations were performed for this system. The convergence of Molecular Dynamics simulations on the TLR4/MD-2 complexes was evaluated by the root-mean-square deviation (RMSD) of backbone atoms after superposition.
Dual luciferase NF-κB activity
The murine microglial BV-2 cell line was provided by Dr. Rona Giffard (Stanford University). NF-κB dual luciferase reporter microglia BV-2 cell line was constructed by Cignal Lenti NF-κB Reporter kit (SABiosciences, MD, USA) as described previously9. Firefly luciferase gene was placed under the control the NF-κB transcriptional response element and the constitutively expressing Renilla luciferase was placed under the control of the CMV promoter. The internal control Renilla luciferase overcomes technical variability and increases data reliability. NF-κB dual luciferase reporter BV-2 cells were cultured in DMEM medium supplemented with 10% FBS, penicillin (50 unit/mL), streptomycin (50 μg/mL) and puromycin (4 μg/mL). BV-2 reporter cells were seeded at a density of 1×104 cells/well in 96-well plates. After 24 h incubation, medium was changed to Opti-MEM medium supplemented with 0.5% FBS, penicillin (50 unit/mL), streptomycin (50 μg/mL) and 1% of non-essential amino acid and indicated concentration of METH and/or LPS-RS, TAK-242 (Invivogen; San Diego, California) or purified MD-2 protein; each treatment was run with four replicates 24 h later, NF-κB activity was analyzed by Dual-Glo Luciferase Assay System (Promega, Madison, MI, USA). It should be noted that TAK-242 is a potent TLR4 antagonist with an IC50 of 1.1-33 nM 54. 200 nM was chosen as the working concentration of TAK-242 herein. The ratio of Firefly luciferase activity to Renilla luciferase activity represents NF-κB activity. When data were analyzed, the NF-κB activity of the untreated media control group was set as 1.
Conditioned place preference (CPP)
CPP methods (Fig. S1) are in agreement with the detailed procedures of our prior studies 9, 11. On Day 1, rats were pre-exposed to the conditioning boxes and allowed to freely explore for 20 minutes. ANYmaze software was used to track the amount of time each rat spent in each compartment. Rats were randomly assigned to treatment groups and received either systemic (+)-naloxone (15 mg/kg) versus equivolume saline 10 min prior to METH (1 mg/kg) versus equivolume saline. Conditioning sessions were performed once daily for 45 min on Days 2-5 (total of 2 saline and 2 METH conditioning sessions) and were tested for place preference on Day 6 in a drug-free state.
In vivo microdialysis
In vivo microdialysis methods (Fig. S2) are in agreement with the detailed procedures of our prior studies 9, 11. Rats were placed in separate Plexiglas bowls with food and water ad libitum in the microdialysis testing room. Microdialysis guide cannulae were surgically implanted, aimed at the right or left NAc shell using stereotaxic coordinates relative to bregma: anterior/posterior = +1.7 mm; medial/lateral = +/−0.8 mm; relative to dura: dorsal/ventral = −5.6 mm, bite bar = 0 55, in a counterbalanced fashion. Microdialysis probes were inserted through each guide cannula and artificial CSF perfused through the probes using an infusion pump at a rate of 0.2 μL/min overnight. The next morning, the flow rate was increased to 1.5 μL/min for the duration of the experiment. Two h later, 3 baseline samples were collected and then drug treatments were administered. The sample collection tubes were changed every 20 min for a total of 4 h and stored at −80° C until high performance liquid chromatography analysis along with electrochemical detection, using a method previously described 9, 11.
For tissue collection and probe placement verification, rats were euthanized with intraperitoneal 65 mg/kg sodium pentobarbital before brain extraction and post-mortem fixation. Brains were cryostat sectioned and sections containing each rat’s cannula track were mounted on slides and stained with cresyl violet, cover-slipped, and viewed under a light microscope. To be included in data analysis, at least 75% of the probe had to be within the NAc shell.
In vivo drug administration
For METH, a dose of 1 mg/kg (intraperitoneal injection) was used, which was based on past literature 31, 32 and pilot studies.
For (+)-naloxone, 5 mg/kg and 15 mg/kg were used, which were based on the previous studies of (+)-naloxone inhibiting cocaine 11 and opioids 9 dependence and drug reward. It should be noted that (+)-naloxone has a short half life (t1/2, ~ 1.5 h) 56. Therefore, two successive 2.5 mg/kg or 7.5 mg/kg of (+)-naloxone subcutaneous injections with the interval of 15 min were employed to give more even in vivo drug concentration than one injection of 5 mg/kg or 15 mg/kg of (+)-naloxone.
For LPS-RS, a single intra-VTA injection of 5 μg in 1 μL was employed 10 min prior to a single, i.p. injection of 1 mg/kg METH, which was based on previous uses of LPS-RS inhibiting cocaine induced increases of NAc dopamine 11. For IL-6 antibody, a single intra-VTA injection of sheep anti-rat IL-6 neutralizing antibody (R&D, Cat. No., AF506; 0.2 μg in 1 μL) was used 1 h prior to a single, i.p. injection of 1 mg/kg METH.
Real-Time RT-PCR
After completion of the METH-course and (+)-naloxone/METH-course injections, rats were euthanized with i.p. 65 mg/kg sodium pentobarbital followed by transcardial perfusion with ice cold 0.9% heparinized saline. The brains were flash frozen in chilled isopentane, frozen on dry ice and stored at −80°C until the collection of tissue micropunches. Brains were cryostat sectioned (30 μm) at −20°C. The location of each region (VTA, NAc, PFC) was determined according to the Paxinos and Watson brain atlas 55. Circular micropunches of 0.25 cm in length were taken from each region on both hemispheres using blunt 18-gauge, stainless steel hypodermic tubing. Tissue micropunches were flash frozen in liquid nitrogen in 1.5 ml microcentrifuge tubes and stored at −80°C until mRNA extraction.
Total RNA was isolated from each tissue micro-punch by utilizing a standard method of phenol:chloroform 57. Detailed descriptions of RNA isolation, cDNA synthesis, PCR amplification protocols, and primer sequences are as previously published 58. PCR amplification of cDNA was performed using the QuantiTect SYBR Green PCR Kit (Qiagen, Valencia, CA). Formation of the PCR product was monitored in real time using the MyiQ Single-Color Real-Time PCR Detection System (BioRad, Hercules, CA). Relative gene expression was determined relative to the housekeeping gene GAPDH. It should be noted that the suitability of GAPDH as the reference gene was checked and a single dose of METH (1mg/kg) which did not alter GAPDH expression.
Statistics
All statistical tests were performed, and graphs created in GraphPad Prism Version 5. Data are presented as mean ± SD or mean ± SEM. Appropriate statistical analyses were chosen based on experimental design. The specific statistical analysis used is indicated in each figure caption for all studies. For all ANOVAs, Bonferroni post-hoc tests were used. As is standard, significance threshold was set to p < 0.05 for all analyses. Data collection and analyses were performed blinded to group assignment.
Supplementary Material
Acknowledgements
We thank the Network and Computing Center, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, National Supercomputer Center of China in Guangzhou and the Computing Center of Jilin Province for the supply of computational resources. We thank Dr Jie-Oh Lee (KAIST, Korea) for providing the insect expression human MD-2-pAcGP67A vector and Dr. Xuedong Liu (University of Colorado Boulder) for providing the high 5 insect cells.
Funding
This work was supported in part by grants from National Key Research and Development Program of China (2016YFC0800907), National Natural Science Foundation of China (21850410455, 21877106), the Open Funding of Beijing National Laboratory for Molecular Sciences, Australian Research Council Research Fellowship (DP110100297) and NIH grant DA033358. Drug synthesis was supported by the NIH Intramural Research Programs of the National Institute on Drug Abuse and the National Institute of Alcohol Abuse and Alcoholism.
Abbreviation
- METH
methamphetamine
- TLR4
Toll-like receptor 4
- NAc
nucleus accumbens shell
- VTA
ventral tegmental area
- PFC
prefrontal cortex
- IL-1β
interleukin-1β
- IL-6
interleukin 6
- CNS
central nervous system
- DAMPs
danger associated molecular patterns
- PAMPs
pathogens associated molecular patterns
- XAMPs
xenobiotics associated molecular patterns
- LPS
lipopolysaccharide
- MD-2
myeloid differentiation protein 2
- CPP
conditioned place preference
- RMSD
root-mean-square deviation
- BBB
blood-brain-barrier
Footnotes
The authors declare no competing financial interest.
Supporting Information
Graphic illustration of CPP testing; graphic illustration of microdialysis testing;overlap of the best binding pose of METH with classic TLR4 agonist Lipid A; Time evolution of the Cα RMSDfor TLR4/MD-2 interacting with METH.
References
- 1.Courtney KE, and Ray LA (2014) Methamphetamine: an update on epidemiology, pharmacology, clinical phenomenology, and treatment literature, Drug Alcohol Depend. 143, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vollm BA, de Araujo IE, Cowen PJ, Rolls ET, Kringelbach ML, Smith KA, Jezzard P, Heal RJ, and Matthews PM (2004) Methamphetamine activates reward circuitry in drug naive human subjects, Neuropsychopharmacology 29, 1715–1722. [DOI] [PubMed] [Google Scholar]
- 3.Chu PW, Seferian KS, Birdsall E, Truong JG, Riordan JA, Metcalf CS, Hanson GR, and Fleckenstein AE (2008) Differential regional effects of methamphetamine on dopamine transport, Eur. J. Pharmacol 590, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kokoshka JM, Vaughan RA, Hanson GR, and Fleckenstein AE (1998) Nature of methamphetamine-induced rapid and reversible changes in dopamine transporters, Eur. J. Pharmacol 361, 269–275. [DOI] [PubMed] [Google Scholar]
- 5.Fantegrossi WE, Ciullo JR, Wakabayashi KT, De La Garza R 2nd, Traynor JR, and Woods JH (2008) A comparison of the physiological, behavioral, neurochemical and microglial effects of methamphetamine and 3,4-methylenedioxymethamphetamine in the mouse, Neuroscience 151, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K, Iwata Y, Tsuchiya KJ, Suda S, Suzuki K, Kawai M, Takebayashi K, Yamamoto S, Matsuzaki H, Ueki T, Mori N, Gold MS, and Cadet JL (2008) Methamphetamine causes microglial activation in the brains of human abusers, J. Neurosci 28, 5756–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LaVoie MJ, Card JP, and Hastings TG (2004) Microglial activation precedes dopamine terminal pathology in methamphetamine-induced neurotoxicity, Exp. Neurol 187, 47–57. [DOI] [PubMed] [Google Scholar]
- 8.Hutchinson MR, Zhang YN, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, and Watkins LR (2010) Evidence that opioids may have toll-like receptor 4 and MD-2 effects, Brain Behav. Immun 24, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR, and Yin H (2012) Morphine activates neuroinflammation in a manner parallel to endotoxin, Proc. Natl. Acad. Sci. U S A 109, 6325–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang YX, Chu HC, Jiang YN, and Yuan L (2016) Morphine enhances IL-1 beta release through toll-like receptor 4-mediated endocytic pathway in microglia, Purinergic Signal. 12, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA, Pomrenze MB, Galer EL, Kopajtic TA, Li CM, Amat J, Larson G, Cooper DC, Huang Y, O'Neill CE, Yin H, Zahniser NR, Katz JL, Rice KC, Maier SF, Bachtell RK, and Watkins LR (2015) DAT isn't all that: cocaine reward and reinforcement require Toll-like receptor 4 signaling, Mol. Psychiatry 20, 1525–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, and Watkins LR (2007) Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward, ScientificWorldJournal 7, 98–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, van Steeg K, Kopajtic TA, Loram LC, Sfregola C, Galer E, Miles NE, Bland ST, Amat J, Rozeske RR, Maslanik T, Chapman TR, Strand KA, Fleshner M, Bachtell RK, Somogyi AA, Yin H, Katz JL, Rice KC, Maier SF, and Watkins LR (2012) Opioid activation of Toll-like receptor 4 contributes to drug reinforcement, J. Neurosci 32, 11187–11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacquet YF, Klee WA, Rice KC, Iijima I, and Minamikawa J (1977) Stereospecific and nonstereospecific effects of (+)- and (−)-morphine: evidence for a new class of receptors?, Science 198, 842–845. [DOI] [PubMed] [Google Scholar]
- 15.Marcoli M, Ricevuti G, Mazzone A, Pasotti D, Lecchini S, and Frigo GM (1989) A stereoselective blockade by naloxone of opioid and non-opioid-induced granulocyte activation, Int J Immunopharmacol 11, 57–61. [DOI] [PubMed] [Google Scholar]
- 16.Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, and Watkins LR (2008) Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4), Eur. J. Neurosci 28, 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Zhang Y, Peng Y, Hutchinson MR, Rice KC, Yin H, and Watkins LR (2016) Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4, Br. J. Pharmacol 173, 856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown KT, Levis SC, O'Neill CE, Northcutt AL, Fabisiak TJ, Watkins LR, and Bachtell RK (2018) Innate immune signaling in the ventral tegmental area contributes to drug-primed reinstatement of cocaine seeking, Brain Behav. Immun 67, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Cochran TA, Hutchinson MR, Yin H, and Watkins LR (2014) Drug addiction. In Microglia in Health and Disease pp 299–317, Springer. [Google Scholar]
- 20.Resman N, Gradisar H, Vasl J, Keber MM, Pristovsek P, and Jerala R (2008) FEBS Lett., Febs Letters 582, 3929–3934. [DOI] [PubMed] [Google Scholar]
- 21.Mancek-Keber M, and Jerala R (2006) Structural similarity between the hydrophobic fluorescent probe and lipid A as a ligand of MD-2, FASEB J 20, 1836–1842. [DOI] [PubMed] [Google Scholar]
- 22.Park BS, Song DH, Kim HM, Choi BS, Lee H, and Lee JO (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex, Nature 458, 1191–U1130. [DOI] [PubMed] [Google Scholar]
- 23.Latty SL, Sakai J, Hopkins L, Verstak B, Paramo T, Berglund NA, Cammarota E, Cicuta P, Gay NJ, Bond PJ, Klenerman D, and Bryant CE (2018) Activation of Toll-like receptors nucleates assembly of the MyDDosome signaling hub, Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeuchi O, and Akira S (2010) Pattern recognition receptors and inflammation, Cell 140, 805–820. [DOI] [PubMed] [Google Scholar]
- 25.Matsunaga N, Tsuchimori N, Matsumoto T, and Ii M (2011) TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules, Mol. Pharmacol 79, 34–41. [DOI] [PubMed] [Google Scholar]
- 26.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, and Miyake K (2002) Essential role of MD-2 in LPS responsiveness and TLR4 distribution, Nat. Immunol 3, 667–672. [DOI] [PubMed] [Google Scholar]
- 27.Kita T, Takeshima M, Wagner GC, Hozumi H, Miyazaki I, and Asanuma M (2008) New perspectives on the mechanism of methamphetamine-induced neurotoxicity, Nihon Shinkei Seishin Yakurigaku Zasshi 28, 49–61. [PubMed] [Google Scholar]
- 28.Wang B, Chen T, Xue L, Wang J, Jia Y, Li G, Ren H, Wu F, Wu M, and Chen Y (2019) Methamphetamine exacerbates neuroinflammatory response to lipopolysaccharide by activating dopamine D1-like receptors, Int. Immunopharmacol 73, 1–9. [DOI] [PubMed] [Google Scholar]
- 29.Nakajima A, Yamada K, Nagai T, Uchiyama T, Miyamoto Y, Mamiya T, He J, Nitta A, Mizuno M, Tran MH, Seto A, Yoshimura M, Kitaichi K, Hasegawa T, Saito K, Yamada Y, Seishima M, Sekikawa K, Kim HC, and Nabeshima T (2004) Role of tumor necrosis factor-alpha in methamphetamine-induced drug dependence and neurotoxicity, J. Neurosci 24, 2212–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frank MG, Adhikary S, Sobesky JL, Weber MD, Watkins LR, and Maier SF (2016) The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine, Brain Behav. Immun 51, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voigt RM, Mickiewicz AL, and Napier TC (2011) Repeated mirtazapine nullifies the maintenance of previously established methamphetamine-induced conditioned place preference in rats, Behav. Brain Res 225, 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berry JN, Neugebauer NM, and Bardo MT (2012) Reinstatement of methamphetamine conditioned place preference in nicotine-sensitized rats, Behav. Brain Res 235, 158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loftis JM, Choi D, Hoffman W, and Huckans MS (2011) Methamphetamine causes persistent immune dysregulation: a cross-species, translational report, Neurotox. Res 20, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goncalves J, Martins T, Ferreira R, Milhazes N, Borges F, Ribeiro CF, Malva JO, Macedo TR, and Silva AP (2008) Methamphetamine-induced early increase of IL-6 and TNF-alpha mRNA expression in the mouse brain, Ann. N. Y. Acad. Sci 1139, 103–111. [DOI] [PubMed] [Google Scholar]
- 35.Wan F, Zang S, Yu G, Xiao H, Wang J, and Tang J (2017) Ginkgolide B suppresses methamphetamine-induced microglial activation through TLR4-NF-κB signaling pathway in BV2 cells, Neurochem. Res 42, 2881–2891. [DOI] [PubMed] [Google Scholar]
- 36.Du SH, Qiao DF, Chen CX, Chen S, Liu C, Lin Z, Wang H, and Xie WB (2017) Toll-like receptor 4 mediates methamphetamine-induced neuroinflammation through Caspase-11 signaling pathway in astrocytes, Front. Mol. Neurosci 10, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eidson LN, and Murphy AZ (2013) Blockade of Toll-like receptor 4 attenuates morphine tolerance and facilitates the pain relieving properties of morphine, J. Neurosci 33, 15952–15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Theberge FR, Li X, Kambhampati S, Pickens CL, St Laurent R, Bossert JM, Baumann MH, Hutchinson MR, Rice KC, Watkins LR, and Shaham Y (2013) Effect of chronic delivery of the Toll-like receptor 4 antagonist (+)-naltrexone on incubation of heroin craving, Biol. Psychiatry 73, 729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoops WW, Pike E, Hays LR, Glaser PE, and Rush CR (2015) Naltrexone and bupropion, alone or combined, do not alter the reinforcing effects of intranasal methamphetamine, Pharmacol. Biochem. Behav 129, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Airavaara M, Pickens CL, Stern AL, Wihbey KA, Harvey BK, Bossert JM, Liu QR, Hoffer BJ, and Shaham Y (2011) Endogenous GDNF in ventral tegmental area and nucleus accumbens does not play a role in the incubation of heroin craving, Addict. Biol 16, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koob GF, and Volkow ND (2016) Neurobiology of addiction: a neurocircuitry analysis, Lancet Psychiatry 3, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cearley CN, Blindheim K, Sorg BA, Krueger JM, and Churchill L (2011) Acute cocaine increases interleukin-1beta mRNA and immunoreactive cells in the cortex and nucleus accumbens, Neurochem. Res 36, 686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hutchinson MR, and Watkins LR (2014) Why is neuroimmunopharmacology crucial for the future of addiction research?, Neuropharmacology 76 Pt B, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marz P, Otten U, and Rose-John S (1999) Neural activities of IL-6-type cytokines often depend on soluble cytokine receptors, Eur. J. Neurosci 11, 2995–3004. [DOI] [PubMed] [Google Scholar]
- 45.Zeuner MT, Kruger CL, Volk K, Bieback K, Cottrell GS, Heilemann M, and Widera D (2016) Biased signalling is an essential feature of TLR4 in glioma cells, Biochim. Biophys. Acta 1863, 3084–3095. [DOI] [PubMed] [Google Scholar]
- 46.Fleckenstein AE, Gibb JW, and Hanson GR (2000) Differential effects of stimulants on monoaminergic transporters: pharmacological consequences and implications for neurotoxicity, Eur. J. Pharmacol 406, 1–13. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, and Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex, J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong Y, and Benveniste EN (2001) Immune function of astrocytes, Glia 36, 180–190. [DOI] [PubMed] [Google Scholar]
- 49.Bowman CC, Rasley A, Tranguch SL, and Marriott I (2003) Cultured astrocytes express toll-like receptors for bacterial products, Glia 43, 281–291. [DOI] [PubMed] [Google Scholar]
- 50.Haydon PG, and Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling, Physiol. Rev 86, 1009–1031. [DOI] [PubMed] [Google Scholar]
- 51.Watkins LR, Hutchinson MR, Milligan ED, and Maier SF (2007) "Listening" and "talking" to neurons: implications of immune activation for pain control and increasing the efficacy of opioids, Brain Res. Rev 56, 148–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Narita M, Miyatake M, Shibasaki M, Tsuda M, Koizumi S, Narita M, Yajima Y, Inoue K, and Suzuki T (2005) Long-lasting change in brain dynamics induced by methamphetamine: enhancement of protein kinase C-dependent astrocytic response and behavioral sensitization, J. Neurochem 93, 1383–1392. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, Cui F, Chen H, Zhang T, Yang K, Wang Y, Jiang Z, Rice KC, Watkins LR, Hutchinson MR, Li Y, Peng Y, and Wang X (2018) Dissecting the Innate Immune Recognition of Opioid Inactive Isomer (+)-Naltrexone Derived Toll-like Receptor 4 (TLR4) Antagonists, J. Chem. Inf. Model 58, 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, Hazeki O, Kitazaki T, and Iizawa Y (2006) A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-Chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling, Mol. Pharmacol 69, 1288–1295. [DOI] [PubMed] [Google Scholar]
- 55.Paxinos G, and Watson C (1998) The rat brain in stereotaxic coordinates. 4th ed., 4 ed., Academic Press. [Google Scholar]
- 56.Lewis SS, Loram LC, Hutchinson MR, Li CM, Zhang Y, Maier SF, Huang Y, Rice KC, and Watkins LR (2012) (+)-Naloxone, an opioid-inactive toll-like receptor 4 signaling inhibitor, reverses multiple models of chronic neuropathic pain in rats, J. Pain 13, 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chomczynski P, and Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate phenol chloroform extraction, Anal. Biochem 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 58.Frank MG, Barrientos RA, Biedenkapp JC, Rudy JW, Watkins LR, and Maier SF (2006) mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging, Neurobiol. Aging 27, 717–722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.