

Abstract
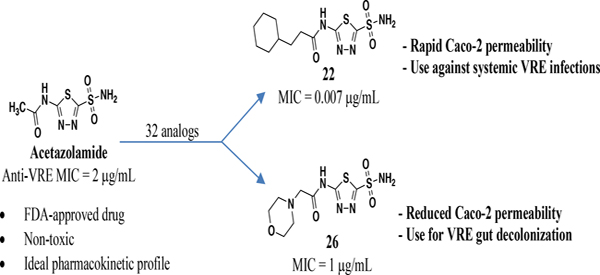
Vancomycin-resistant enterococci (VRE) are the second leading cause of hospital-acquired infections (HAIs) attributed to a drug-resistant bacterium in the United States and resistance to the frontline treatments is well documented. To combat VRE, we have repurposed the FDA-approved carbonic anhydrase drug acetazolamide to design potent anti-enterococcal agents. Through structure-activity relationship optimization we have arrived at two leads possessing improved potency against clinical VRE strains from MIC = 2 μg/mL (acetazolamide) to MIC = 0.007 μg/mL (22) and 1 μg/mL (26). Physicochemical properties were modified to design leads that either have high oral bioavailability to treat systemic infections or low intestinal permeability to treat VRE infections in the gastrointestinal tract. Our data suggests the intracellular target for the molecules are putative α-carbonic and γ-carbonic anhydrases and homology modeling and molecular dynamics simulations were performed. Together, this study presents potential anti-VRE therapeutic options to provide alternatives for problematic VRE infections.
Graphical Abstarct
INTRODUCTION
Vancomycin-resistant enterococci (VRE) are one of the leading causes of drug-resistant healthcare-associated infections (HAIs) in the United States, accounting for 35% of all enterococcus-related HAIs.1 In 2017, VRE infections were estimated at approximately 55,000 in the U.S. and led to a 10% mortality rate2. In addition to the increased rates of mortality there is an added economic burden as patients hospitalized with VRE infections endure prolonged hospital stays and significantly higher hospitalization costs.3 Moreover, the Centers for Disease Control and Prevention (CDC) categorizes VRE as a serious healthcare threat.2 The growing problem of VRE in the healthcare setting is exacerbated by the lack of effective treatments. Currently, the only two Food and Drug Administration (FDA)-approved therapeutic options for treatment of VRE are either linezolid or a combination of quinupristin and dalfopristin.4 Linezolid is marginally effective as the 30-day mortality rate for patients with VRE blood-stream infection is as high as 30%5 and it is also associated with toxicity such as myelosuppression, neuropathy and lactic acidosis.6 The combination therapy of quinupristin/dalfopristin is rarely used due to its high toxicity.7 Some studies have evaluated the efficacy of a linezolid/daptomycin combination against VRE infections,5 and while these show promise, the combination is not yet approved for VRE treatment. Further compounding the VRE problem is the rise of resistance to the FDA-approved therapies8,9 along with daptomycin10 and the fact that approximately 90% of vancomycin-resistant E. faecium clinical isolates are resistant to other antibiotics such as β-lactams.11 Given the increasing prevalence of VRE in healthcare settings and the rise of resistance to the current therapeutic options,12,13 there is a clear and urgent need for novel strategies to combat this infection.
Enterococcus is a common colonizer of the human gastrointestinal tract (GIT); however, the genus typically accounts for < 0.2% of the total human gut microbiota.14,15 Carriers of VRE can either present with symptoms of virulent infections or be asymptomatic. Asymptomatic colonization is the most problematic as it is often unnoticed in individuals admitted into healthcare settings, but is a strong predictor of HAI transmission and acquisition.16 Additionally, dysbiosis (imbalance of normal gut microbiota) caused by exposure of patients to antibiotics increases the density of VRE in the GIT allowing the pathogen to become a predominant species.17,18 This colonization event serves as the origination point for all other nosocomial infections caused by VRE and is a key step for bacteria to enter the bloodstream11,18,19. VRE colonization of the GIT is problematic for organ transplant patients20 and those in intensive care units21 and is linked to higher risk of death.22 More recently, the role of the GIT microbiome in stimulating inflammation, autoimmunity, and cancer immunity has been an emerging field of study.23 For example, perturbations of the microbiome may lead to increased Enterococcus load in the GIT, which in turn intensifies systemic autoimmune disorders such as graft-versus-host disease24 and systemic lupus erythematosus25. Thus, in addition to the need for new therapeutics to treat systemic VRE infection, decolonization strategies have been investigated as a promising approach for reduction of Enterococcus, including VRE, from the GIT before the pathogen can facilitate more problematic infections. However, there has been limited success to date with decolonization using antibiotics due to either low efficacy, poor patient tolerability, or further dysbiosis of the microbiome.26–28 Molecule absorption appears to be a large driver for efficacy as non-absorbable antibiotics outperformed absorbable antibiotics, thus a molecule that can be taken orally and not be systemically absorbed is preferred.26 Linezolid is highly absorbed when taken orally and when used alone is not effective at VRE decolonization for this reason.29,30
Our team has previously explored drug repurposing as an avenue to discover new anti-VRE agents.30–32 Repurposing FDA-approved drugs with well-characterized toxicology and pharmacology to find new applications is an attractive way to reduce time, cost, and risk associated with antimicrobial drug development.33–35 As part of our current efforts, we have identified FDA-approved carbonic anhydrase inhibitors (CAIs) (Figure 1) as a novel class of potent anti-enterococcal agents36 with MICs (minimum inhibitory concentrations) of 0.5 mg/mL, 4 mg/mL and 1 mg/mL for acetazolamide (AZM), methazolamide (MZM) and ethoxzolamide (EZM), respectively.
Figure 1.

FDA-approved carbonic anhydrase inhibitors with reported efficacy against VRE32.
AZM is currently approved as a mild diuretic for the treatment of glaucoma and congestive heart failure37 and is used chronically to treat epilepsy.38,39 Furthermore, AZM is listed on the World Health Organizations (WHO) list of essential medicines40 due to its safety profile, low cost, high efficacy, and high availability. Aside from the anti-enterococcal activity, AZM represents a particularly attractive starting point for optimization because it has an overall safe profile, as up to 1 gram dose/day can be given with no toxicity to humans,41 and possesses good pharmacokinetic properties with respect to rapid absorption, high renal clearance and no metabolites.42–46 Even though AZM has many positives as a starting point for optimization there are properties that could be improved. AZM is rapidly absorbed at doses up to 5–10 mg/kg but absorption is not efficient at higher doses47–49. This may be due to the fact that AZM would be classified as a low-permeable drug (apparent permeability coefficient, Papp, < 5.0 × 106 cm/s in Caco-2 assay) according to the Food and Drug Administration permeability classifications50. AZM may be comparable to linezolid in terms of antibacterial potency it lags well behind linezolid in terms of intestinal permeability. Thus, the scaffold could benefit not only from improved potency but also intestinal permeability for treatment of systemic infection. Conversely, for treatment and decolonization of GIT VRE infection, a parallel approach to develop a second lead with reduced permeability is also desirable.
Although AZM is a potent CAI against human carbonic anhydrases (double-digit nanomolar biochemical activity against various isoforms)42, it is safe for use in treating chronic diseases; thus, short-term treatment of acute bacterial infections would likely not cause adverse effects in humans. Moreover, CAIs in general, have garnered attention for their promise as antimicrobial agents51,52 as recent literature has characterized carbonic anhydrases in bacterial pathogens such as Helicobacter pylori,53–55 Burkholderia pseudomallei,56 Vibrio cholerae,57,58 and Neisseria gonorrhoeae59 to name a few. However, these studies all stop short of developing novel therapeutics to specifically target these pathogens and often do not even test CAIs for activity against the bacteria at all. With regards to Enterococcus spp. there are only two instances in which researchers noted the presence of putative genes encoding for carbonic anhydrases.60,61 Thus, even though carbonic anhydrases possess potential as antibacterial therapeutic targets they remain relatively underexplored for drug discovery purposes, especially against Enterococcus spp.
Based on its encouraging drug profile and anti-enterococcal properties, AZM shows considerable promise as a potential treatment for VRE. However, because VRE causes both systemic and GIT infections, it would be desirable to produce leads that have either: 1) both increased potency and permeability for systemic treatment, or 2) increased potency with reduced permeability to maintain drug concentration in the GIT as an effective decolonization strategy. Moreover, studies dedicated to explore the scaffold for antibiotic potential are necessary to fully validate carbonic anhydrases as novel antibacterial therapeutic targets. For these reasons we have undertaken a drug-repurposing and optimization study for AZM-based VRE inhibitors.
RESULTS
Chemistry
The majority of analogs were synthesized in two or three steps beginning with de-acylation of AZM using concentrated hydrochloric acid to form intermediate 1 (Scheme 1). This common intermediate served as the diversification point for analog modification on the newly exposed amine. The first set of analogs were synthesized by either coupling commercially available acyl chlorides, or first converting carboxylic acids to acyl chlorides using oxalyl chloride, with 1 to form a set of amide containing analogs (3 – 23). This set consisted of primarily linear, branched, or cyclic alkyl moieties as well as phenyl derivatives on the amide. The linker between the carbonyl of the amide and the pendant group varies in length from 0 – 2 methylenes. A second set of analogs containing heterocycles with either one or two methylene linkers were synthesized by first coupling 1 with either 2-chloroacetyl chloride to form intermediate 2a or 3-chloropropanoyl chloride to form 2b. These intermediates were then subjected to nucleophilic attack with various nitrogen containing cycloalkanes to arrive at derivatives 24 - 28. It should be noted that liquid-liquid extraction during work up often resulted in significant loss of the highly polar desired product into the aqueous phase. Multiple extractions were necessary to isolate the analogs. Thus, elimination of the liquid-liquid extraction and moving directly to concentration in vacuo followed by flash chromatography provided improved yields.
Scheme 1.

Synthetic route for analogs 3 – 28. Reagents and conditions: a) concentrated HCl (13 eq.), 95 °C, 24 hr, 74%; b) 1 (1.0 eq.), 2-chloroacetyl chloride (1.1 eq to yield 2a) or 3-chloropropanoyl chloride (1.1 eq to yield 2b), TEA (1.2 eq.), MeCN, 0 °C – rt 19 hr, 46 and 39%, respectively; c) R-COOH (1.1 eq.), oxalyl chloride (1.2 eq.), DMF (1 drop), DCM, 0 °C – rt, 2 hr, crude product carried into the next step; d). 1 (1.0 eq.), R2-COCl (from previous step or commercial source, 1.1 eq.), TEA (1.2 eq.), MeCN, 0 °C – rt, 14 hr, 4.5 – 71%; e) 2a or 2b (1.0 eq.), cyclic amine (2.0 eq.), TEA (2.0 eq.), anhydrous THF, 0 °C – rt, 20 hr, 82%.
Next, three analogs were synthesized to probe the role of the carbonyl for anti-VRE activity. These analogs were accessed via reductive-amination in which aldehydes were reacted with 1 to form the imine intermediate then immediately reduced using sodium cyanoborohydride to provide analogs 29 – 31 (Scheme 2).
Scheme 2.

Synthesis of analogs 29 - 31. Reagents and conditions: a) i. 1 (1.0 eq.), R-CHO (1.5 eq.), CH2Cl2, rt, 1 hr; ii. sodium cyanoborohydride (1.5 eq.), 0 °C – rt, 18 hr, 8 – 21%.
Finally, a control analog in which the sulfonamide was replaced with a methyl sulfone was synthesized from the commercially available 5-(methanesulfonyl)-1,3,4-thiadiazole-2-amine 32 (Scheme 3). This was then coupled with acetic anhydride to produce analog 33.
Scheme 3.

Synthesis of analog 33. Reagents and conditions: a) 32 (1.0 eq.), acetic anhydride (1.5 eq.), acetic acid, 60 °C, 1 hr, 42%.
Structure-activity relationship for anti-VRE activity
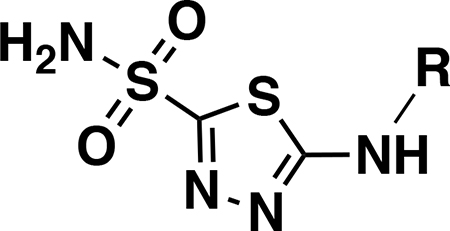
Analogs were initially tested versus the clinical multidrug-resistant VRE strain, E. faecium HM-965, to determine MICs in support of SAR around the AZM scaffold. The first set of derivatives probed the importance of lipophilic substituents on the amide of AZM (Table 1). Gradually increasing alkyl bulk directly next to the amide carbonyl was correlated to increased potency against VRE. For example, comparing AZM (methyl) to compounds 3 (ethyl), 4 (iso-propyl), and 5 (tert-butyl) provided an 8-fold increase in potency across the series of nearest neighbor analogs with the tert-butyl analog 5 displaying an MIC = 0.25 μg/mL compared to the parent AZM (MIC = 2 μg/mL). Inserting a methylene between the amide and the alkyl substituents represented by analogs 6 - 8 also provided a boost in potency. Additionally, the trend of increasing alkyl bulk correlating to VRE potency was maintained in this set of analogs. These SAR data points can be summarized by comparing both tert-butyl analog, 5 (MIC = 0.25 μg/mL) to 8 (MIC = 0.015 μg/mL), which contains a methylene linker.
Table 1.
Minimum inhibitory concentrations (MICs) for AZM-based analogs against E. faecium HM-965
 | |||||
|---|---|---|---|---|---|
| Compound | R | MICa | Compound | R | MICa |
| AZM |  |
2 | 17 |  |
0.25 |
| 3 | 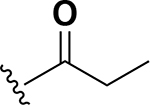 |
1 | 18 | 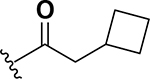 |
0.25 |
| 4 | 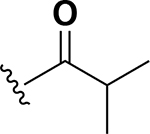 |
1 | 19 | 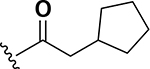 |
0.06 |
| 5 | 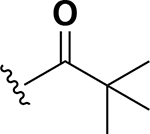 |
0.25 | 20 | 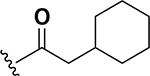 |
0.06 |
| 6 | 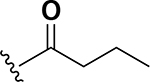 |
0.5 | 21 | 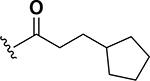 |
0.06 |
| 7 | 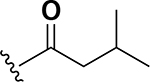 |
0.125 | 22 | 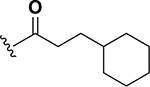 |
0.007 |
| 8 | 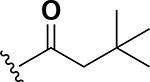 |
0.015 | 23 | 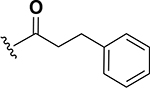 |
0.06 |
| 9 | 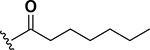 |
0.015 | 24 | 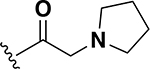 |
1 |
| 10 | 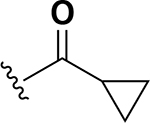 |
2 | 25 | 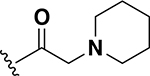 |
1 |
| 11 | 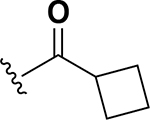 |
0.5 | 26 | 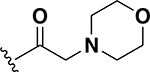 |
1 |
| 12 | 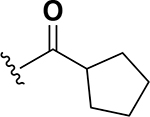 |
0.25 | 27 | 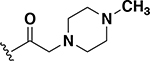 |
8 |
| 13 | 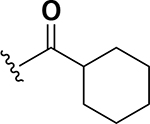 |
0.25 | 28 | 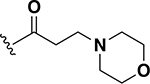 |
2 |
| 14 | 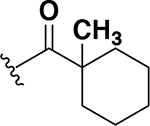 |
0.25 | 29 | 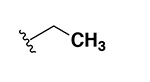 |
> 64 |
| 15 | 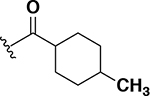 |
0.25 | 30 | 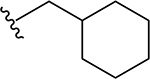 |
> 64 |
| 16 | 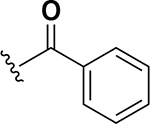 |
2 | 31 | 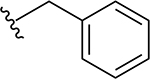 |
> 64 |
AZM = acetazolamide.
MIC values in μg/mL against E. faecium strain HM-965
The next set of analogs investigated the role of cyclic alkanes on VRE potency. As was observed with the branched alkyl derivatives, increased lipophilic bulk for the cyclic alkanes also led to improved anti-VRE potency. For example, as observed in the previous set of branched alkanes, potency again increased 8-fold across the series of analogs 10 (cyclopropyl), 11 (cyclobutyl), 12 (cyclopentyl), and 13 (cyclohexyl) with compound 13 displaying an MIC = 0.25 μg/mL (Table 1). Two additional cyclohexane-based derivatives with either a methyl at the 1-position (14) or 4-position (15) were equipotent to the cyclohexane alone. Inserting a methylene provided the same boost in potency as was observed with the branched alkane series described above. Analogs 17 – 20 extended the pendant cycloalkane by a single methylene linker and resulted in a 4- to 8-fold increase in anti-VRE activity with an MIC of 0.06 μg/mL for the cyclohexyl derivative 20. Taking this SAR trend one step further two analogs were synthesized that extended the pendant group with a two-methylene linker. No increase of potency was observed for the cyclopentane pendant group going from a one- to two-methylene linker (analogs 19 and 21, respectively). However, there was an 8-fold increase in potency observed for the cyclohexane pendant group moving from a single methylene (20, MIC = 0.06 μg/mL) to a two-methylene linker providing the most potent analog of the series against VRE (22, 0.007 μg/mL). Altogether, moving the cyclohexane from no linker to a two-methylene linker provided a 32-fold increase in anti-VRE activity.
The anti-VRE potency appeared to be enhanced by alkyl rings systems over aromatic pendent groups. Specifically, comparing the cyclohexane containing analog 13 to the phenyl derivative 16, the aromatic group resulted in 8-fold reduction of potency (0.25 μg/mL to 2 μg/mL, respectively) (Table 1). The same trend was observed with modifying the cyclohexane pendant group on analog 22 to a phenyl group in analog 23. These two nearest neighbor analog pairs illustrate the preference for saturated ring systems over the aromatic pendant groups for anti-VRE activity. Interestingly, while the pendant phenyl derivatives were less potent than their cyclohexane counterparts, the SAR trend of extending the linker from zero (16) to two-methylenes (23) proved robust as anti-VRE activity for the phenyl substituted analogs improved by 32-fold.
To this point only lipophilic alkyl and phenyl substitutions were made to the AZM. These modifications improved potency and are likely to increase permeability through the intestinal lining compared to AZM, making these analogs potential candidates for treating systemic VRE infections. However, the increased intestinal permeability may be detrimental toward decolonization of VRE from the gut. Thus, the next a set of analogs explored the incorporation of heteroatoms into the cycloalkane groups with the intent to derive analogs with increased polarity and decreased permeability that would be beneficial for gut decolonization. Across the set of five analogs, the insertion of heteroatoms was detrimental to anti-VRE activity. For example, the pyrrolidine (24) and piperidine (25) analogs each exhibited MICs of 1 μg/mL (Table 1). These values were 16-fold less potent than the cyclopentyl and cyclohexyl counterparts 19 and 20 (0.06 μg/mL each). The same was observed with the morpholine derivative 26. Activity was reduced even further with the methylpiperazine analog 27. Finally, the extension of the linker displayed relatively no effect on potency that was previously observed for the cyclohexyl derivatives (20 compared to 22). For example, derivatives to investigate extending the morpholine from a single methylene (26) to a two-methylene (28) linker was essentially equipotent (MIC = 2 μg/mL). This set of analogs indicates that the addition of heteroatoms in the pendant group reduces anti-VRE activity of the molecules compared to the alkyl derivative; however, they were still equipotent in activity to linezolid against VRE (Table 2).
Table 2.
MICs for selected analogs against a panel of VRE isolates
| VRE strains | Compounds/control drugs |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AZM | 5 | 7 | 8 | 9 | 12 | 13 | 17 | 19 | 20 | 21 | 22 | 23 | 26 | Van | Lin | |
| E. faecium HM-968 | 2 | 0.5 | 0.125 | 0.015 | 0.015 | 0.5 | 0.5 | 0.5 | 0.06 | 0.06 | 0.06 | 0.03 | 0.25 | 1 | 64 | 0.5 |
| E. faecium NR-28978 | 2 | 0.25 | 0.25 | 0.03 | 0.06 | 0.5 | 0.5 | 1 | 0.125 | 0.125 | 0.125 | 0.06 | 0.5 | 2 | 32 | 1 |
| E. faecium NR-31903 | 2 | 0.5 | 0.125 | 0.015 | 0.03 | 0.5 | 0.5 | 0.25 | 0.125 | 0.125 | 0.03 | 0.015 | 0.25 | 2 | >64 | 1 |
| E. faecium NR-32065 | 1 | 0.25 | 0.25 | 0.007 | 0.03 | 0.5 | 0.5 | 0.25 | 0.015 | 0.06 | 0.06 | 0.015 | 0.125 | 2 | 64 | 0.25 |
| E. faecium NR-31914 | 2 | 0.5 | 0.25 | 0.007 | 0.03 | 0.5 | 0.5 | 0.5 | 0.125 | 0.06 | 0.06 | 0.03 | 0.25 | 2 | 64 | 1 |
| E. faecium NR-31916 | 2 | 0.5 | 0.125 | 0.03 | 0.06 | 0.5 | 0.25 | 0.25 | 0.015 | 0.125 | 0.03 | 0.03 | 0.125 | 1 | >64 | 1 |
| E. faecium NR-32052 | 2 | 0.5 | 0.125 | 0.015 | 0.03 | 0.5 | 0.5 | 0.25 | 0.06 | 0.06 | 0.06 | 0.015 | 0.5 | 1 | 64 | 0.5 |
| E. faecium NR-31971 | 2 | 0.5 | 0.5 | 0.06 | 0.03 | 1 | 0.5 | 0.5 | 0.06 | 0.03 | 0.03 | 0.007 | 0.125 | 2 | 64 | 1 |
| E. faecium NR-31972 | 2 | 1 | 0.5 | 0.015 | 0.03 | 1 | 0.25 | 0.5 | 0.03 | 0.06 | 0.03 | 0.015 | 0.125 | 4 | >64 | 0.5 |
| E. faecalis HM-335 | 2 | 0.5 | 0.125 | 0.03 | 0.015 | 0.5 | 0.5 | 0.25 | 0.03 | 0.03 | 0.06 | 0.03 | 0.125 | 0.5 | >64 | 0.5 |
| E. faecalis HM-334 | 2 | 0.5 | 0.5 | 0.03 | 0.015 | 0.5 | 0.25 | 0.5 | 0.03 | 0.03 | 0.015 | 0.007 | 0.015 | 4 | 64 | 1 |
| MIC50 | 2 | 0.5 | 0.25 | 0.015 | 0.03 | 0.5 | 0.5 | 0.5 | 0.03 | 0.06 | 0.06 | 0.015 | 0.125 | 2 | 64 | 1 |
| MIC90 | 2 | 0.5 | 0.5 | 0.03 | 0.06 | 1 | 0.5 | 0.5 | 0.125 | 0.125 | 0.06 | 0.03 | 0.05 | 4 | >64 | 1 |
MIC50: minimum inhibitory concentration at which the compound/drug inhibited 50% of the tested strains, MIC90: minimum inhibitory concentration at which the compound/drug inhibited 90% of the tested strains. Lin = linezolid, Van = vancomycin. MIC values in μg/mL.
To conclude the SAR study for this series a limited set of analogs to explore modifications to the other parts of the AZM scaffold were tested. First the necessity of the sulfonamide was probed by testing the methylsulfone derivative 33. This substitution abolished the anti-VRE activity (Figure 2). Modifications to the thiadiazole core of AZM also led to loss of potency. Specifically, converting the thiadiazole to a thiazole central core with substitution of a carbon for nitrogen at the 4-position as seen in analog 34, also abrogated activity against VRE. The same was true for analog 35, which contained a central phenyl core in place of the thiadiazole. These analogs, combined with the previously reported activities for the FDA-approved CAIs36 shown in Figure 1 highlight the importance of the central thiadiazole core to the anti-VRE activity.
Figure 2.

Analogs 33 – 35 have MICs > 64 μg/mL against E. faecium HM-965.
Anti-VRE activity against additional VRE and VSE bacterial strains.
The activity of thirteen analogs (5, 7, 8, 9, 12, 13, 17, 19, 20, 21, 22, 23, 26) AZM, vancomycin and linezolid were tested against a panel of eleven VRE clinical isolates. As presented in Table 2, compounds 8 and 22 exhibited the most potent activity against the tested isolates inhibiting their growth at concentrations ranging from 0.007 to 0.06 μg/mL with MICs50 and MICs90 of 0.015 μg/mL and 0.03 μg/mL, respectively. These were followed closely by 9 and 19 (MIC50 of 0.03 μg/mL and MICs90 of 0.06 μg/mL and 0.125 μg/mL, respectively) then 20 and 21 (MIC50 of 0.06 μg/mL and MICs90 of 0.125 μg/mL and 0.06 μg/mL, respectively) while the rest ranged from MIC50 values of 0.125 – 2 μg/mL and MICs90 of 0.5 μg/mL and 4 μg/mL. Most compounds outperformed the drug of choice, linezolid, which exhibited an MIC50 and MIC90 values 1 μg/mL with range from 0.25 to 1 μg/mL. Finally, selected analogs along with linezolid and vancomycin were tested versus vancomycin-sensitive enterococci (VSE) strains (Table S1). These results indicate the molecules exhibit similar potencies between VRE and VSE and show no unexpected discrepancies between isolates.
Toxicity against human Caco-2 cell lines
Toxicity to host tissues is an important property that must be assessed for novel antimicrobial compounds in the drug discovery process. The ability of new compounds to exhibit their activity against the target microorganism without affecting the host (mammalian) tissues is highly important to confirm early in the drug discovery process. To determine if AZM analogs were toxic to eukaryotic cells, the viability of colon adenocarcinoma (Caco-2) cells, exposed to the most potent analogs, was assessed using the MTS assay. All the tested AZM analogs proved to be highly tolerable and non-toxic to mammalian cells at a concentration as high as 128 μg/mL (Figure 3, full data set Table S2). The 50% cytotoxic concentrations (CC50), the compounds’ concentration leading to 50% viability of the cells, for all the tested analogs was higher than 128 μg/mL. This concentration represents more than 8500-fold and 64-fold higher than the MIC50 values obtained for analogs 22 and 26 against clinical VRE isolates, respectively. We should note Caco-2 cells have high endogenous expression of human CA XIII62 and AZM binds to this isoform with a KI value of 16 nM.63
Figure 3.

Cell viability for Caco-2 cells dosed at 64 μg/mL (blue bars) and 128 μg/mL (red bars) for representative analogs.
Mechanism of action studies
During SAR optimization we began to investigate the potential mechanism of action for the AZM-based analogs. Recent literature described above has suggested carbonic anhydrases as potential therapeutic targets for antibacterial drug development. This, combined with the fact that AZM is a carbonic anhydrase inhibitor, led us to hypothesize that inhibition of a carbonic anhydrase might be the likely mechanism of action for this scaffold against VRE. Moreover, the SAR also pointed in this direction. For example, the sulfonamide moiety on AZM is necessary for chelation of a catalytic Zn2+ ion in the active site of carbonic anhydrases and removal of this functional group abrogates any binding to carbonic anhydrases. The same methylsulfone analog 33 was also inactive against VRE strains suggesting the role of chelation of a metal ion for anti-VRE activity. However, there is limited literature suggesting the presence of carbonic anhydrases in Enterococcus species and no reports of carbonic anhydrases characterized from this genus.
A common method to determine if an inhibitor is exhibiting cellular activity that is mediated through inhibition of a carbonic anhydrase is to test for activity under both normal culture conditions and in conditions with increased CO2 levels59,64. As CO2 is the natural substrate for carbonic anhydrases, the molecules will exhibit reduced activity in the CO2 conditions compared to normal conditions. This test was performed on five representative enterococcal strains with both AZM and 20 (Table 3). We observed that both AZM and 20 were inactive against these VRE strains when incubated in presence of 5% CO2 compared to incubation in the ambient air. The same was true for additional analogs against E. faecium ATCC 700221 under both conditions (Table S3). Linezolid was used as a control and, as expected, displayed no shift in potency between ambient air and 5% CO2 conditions suggesting the change in potency for AZM and derivatives is not an unintended effect of the increased CO2 but is from target engagement with carbonic anhydrase. These results, combined with the SAR data for the 33, suggest the intracellular target for these molecules is likely a carbonic anhydrase.
Table 3.
MICs of AZM and 20 under normal and CO2 conditions
| Strain | AZMa |
20a |
||
|---|---|---|---|---|
| normalb | CO2c | normalb | CO2c | |
| E. faecium NR-31971 | 2 | >64 | 0.03 | >64 |
| E. faecium NR-31972 | 2 | >64 | 0.06 | >64 |
| E. faecium HM-965 | 2 | >64 | 0.06 | >64 |
| E. faecalis HM-334 | 2 | >64 | 0.03 | >64 |
| E. faecium ATCC 700221 | 1 | >64 | 0.06 | >64 |
AZM = acetazolamide.
MIC values in μg/mL.
indicates standard conditions in ambient air.
indicates incubation in presence of 5% CO2.
Next, analogs 22, 26, AZM and linezolid were assessed in time-kill assays to determine whether the scaffolds exhibit bacteriostatic or bactericidal activity against VRE. All molecules were shown to be bacteriostatic versus E. faecium and significantly reduced the bacterial burden compared to the DMSO-treated controls (Figure 4). The control linezolid also displayed bacteriostatic activity consistent with the known mechanism for this antibiotic against Enterococcus strains65. It reduced the VRE burden more than the reduction observed with compounds 22 and 26 or AZM.
Figure 4.

Time-kill assay for analogs 22 and 26 along with AZM and linezolid compared to DMSO-treated controls over 24 hours at 37 °C. Limit of detection is 100 CFU/mL. The data were analyzed via two-way ANOVA with post-hoc Dunnett’s test for multiple comparisons. Asterisks (*) denote statistically significant different (p < 0.05) between treatments with either 22, 26, AZM or linezolid in comparison to DMSO-treated cells.
Enterococcus faecium genome encodes for both α- and γ-carbonic anhydrase classes
Only twice in previous literature have carbonic anhydrases been annotated in Enterococcus genomes. The first was in 1999 when Smith et al. compared genomes of various bacteria and archaea species and reported genes that were putative α- and γ-carbonic anhydrases (hereafter referred to as α-CA and γ-CA, respectively) in E. faecalis.60 In 2015, Cappasso et al. reported that E. faecalis only expressed γ-CA.61 However, there are no reports of carbonic anhydrases in genomes from E. faecium. Given the evidence that the inhibitors reported herein may be engaging a carbonic anhydrase as the intracellular target, we analyzed the genome of the E. faecium DO strain (TX0016/ATCC BAA-472) for putative carbonic anhydrase genes in Uniprot (https://www.uniprot.org). It was found that E. faecium DO encodes genes for both the α-CA and γ-CA.
The E. faecium α-CA (Efα-CA, Uniprot ID: Q3XYE8) is predicted to be 234 amino acids in length and shares high sequence homology with many predicted α-CAs across the Enterococcus genus. To determine the similarity of Efα-CA with characterized α-CAs from other genera, both bacterial and human, we performed a sequence alignment in Uniprot (Figure S1). H. pylori α-CA was the most similar to Efα-CA with the same sequence length of 234 amino acids, 23.9% identity and 37.2% similarity (Table S4). This equated to these two α-CAs sharing 61.1% sequence identity + similarity. The α-CA from N. gonorrhoeae was also very similar with a combined residue identity + similarity of 59.5%. Two human α-CAs, human CA 1 and CA 2, were around 50% sequence identity + similarity compared to Efα-CA.
The γ-CA class is much less characterized compared to α-CAs. Most of the structural and catalytic studies of γ-CAs has been for archaea species,66 however, there are recent reports characterizing the structures and enzymatic activity of bacterial γ-CAs.67,68 The E. faecium γ-CA (Efγ-CA, Uniprot accession: Q3XX77) is predicted to be 161 amino acids in length and shares high sequence homology with many predicted γ-carbonic anhydrases across the Enterococcus spp. To determine the similarity of Efγ-CA with γ-CAs characterized from other species we performed a sequence alignment in Uniprot (Figure S2). Of the organisms analyzed, Efγ-CA was most similar with the 165 amino acid γ-CA from C. difficile, sharing 77% of residues that were identical + similar (Table S5). The structure of C. difficile γ-CA has been deposited in the PDB (ID: 4MFG) from the Center for Structural Genomics of Infectious Disease (csgid.org) but has never been characterized for catalytic activity or published in any manuscript. Three other bacterial γ-CAs that have been previously reported in the literature (Escherichia coli,67 V. cholerae,57 and F. tularensis69) ranged from 62 – 65% identity + similarity to Efγ-CA and were generally larger proteins (179 – 184 amino acids). Finally, two thermostable microorganisms, Methanosarcina thermophila and Thermus thermophilus, displayed the lowest homology (approximately 50%) and were considerably larger (247 and 230 amino acids, respectively). Additionally, there are no ligand bound structures of γ-CA and only a few reports of inhibitory data with AZM. Thus, this class is understudied as a potential therapeutic target for treating bacterial infections.
Homology modeling of E. faecium carbonic anhydrases
Once the intracellular targets for the analogs were determined to likely be one or both of two previously uncharacterized carbonic anhydrases expressed in E. faecium, homology models were generated for each carbonic anhydrase. Efα-CA shared the highest sequence homology with the H. pylori α-CA; however, two critical residues in the active site differed between the species. The active site of H. pylori contains an alanine at position 192 and asparagine at position 108 (Figure S3A). E. faecium has a threonine in place of the alanine (position 180 in Efα-CA) and an aspartic acid in place of the asparagine (position 94 in Efα-CA, Figure S1). The Thr180 is particularly important because in species where this residue is conserved it forms contacts with the thiadiazole nitrogen atoms AZM. For example, in the AZM bound structure of human CA 2 (PDB: 3HS470), the corresponding Thr200 residue side chain forms a productive contact with the thiadiazole core (Figure S3B). To determine which would provide the higher quality homology model, we made two models for the Efα-CA: one using human CA 2 as the template and one using H. pylori α-CA using Prime71,72 (Schrödinger, LLC). The resulting Efα-CA homology models shared high structural similarity as the overlaid models aligned with the template structures. To assess the quality of the model we calculated a Schrödinger structural alignment score for each. Efα-CA based on human CA 2 had a score of 0.038 with a protein RMSD of 0.975 Å. Alternatively, the model based on the H. pylori α-CA template produced an alignment score of 0.006 and an RMSD value of 0.397 Å. According to the Prime software the lower the structural alignment score the better quality of the alignment for overlaid structures. A score of 0.7 or greater would indicate a poorly aligned structure. With this information we chose to move forward with the Efα-CA homology model based on H. pylori α-CA as the template (Figure 5A). The histidine residues that coordinate the Zn2+ and residues lining the active site are well conserved both in sequence and secondary structure with the exception of the Asn108 and Ala192 (Figure 5B). Both the high degree of sequence and structural homology between the template and the model provides confidence the homology model is an accurate prediction of the actual Efα-CA structure. Both a structural overlay (Figure S4) and surface representation for Efα-CA and H. pylori α-CA (Figure S5) are shown in supporting information.
Figure 5.

(A) Homology model of Efα-CA (gray ribbons) built with H. pylori α-CA crystal structure (PDB: 4YGF) as the template. Catalytic Zn2+ shown with dark grey sphere. (B) Zoom in of active site for Efα-CA model (light gray) overlaid with H. pylori α-CA crystal structure (magenta). Conserved residues and/or residues important to AZM binding shown in sticks. Amino acid numbers for H. pylori shown in labels. E. faecium amino acids that are different from H. pylori in parentheses. All images generated using PyMol.
Efγ-CA shared the highest sequence homology with C. difficile γ-CA, and since there are no ligand bound structures of carbonic anhydrase inhibitors with γ-CAs, we chose to use the C. difficile structure (PDB ID: 4MFG) as the template to build homology model of the Efγ-CAs. The Efγ-CA model exhibits the same trimeric catalytic unit found in this particular class of CA (Figure 6A and B) and shares high structural similarity to the C. difficile γ-CA with an alignment score of 0.015 (overlay of catalytic site histidine residues in Figure 6D). The surface representation (Figure 6C) indicates a deep groove below the catalytic Ni2+ ion that would be able to accept a ligand. The site was mapped using the software FTSite73 and was identified to be an acceptable ligand binding site. The groove is relatively long and deep providing an oval shaped site (Figure S6).
Figure 6.

Representations of Efγ-CA homology model. (A) Side view ribbon representation, Ni2+ (gray spheres); (B) Top view representation shows trimeric catalytic unit. (C) Side view surface representation, black arrow orients viewer to Ni2+. (D) Overlay of C. difficile γ-CA crystal structure (purple, PDB: 4MFG) and Efγ-CA homology model (gold). Overlaid active site histidine residues in sticks. Residue numbers correspond to of C. difficile γ-CA. H79 from one monomeric unit while H64’ and H84’ from adjacent monomer. Images generated using PyMol.
Both models appear to be of high quality compared to the reference crystal structures. In the case of the Efα-CA, it possesses the known carbonic anhydrase inhibitor binding site observed across the α-CA class. The Efγ-CA also has an observed ligand binding site mapped by FTSite. These models were selected to perform molecular dynamics simulations on prioritized ligands in order to better understand the observed SAR from the series.
Molecular dynamics simulation for analogs binding to E. faecium homology models
To try to better understand the SAR that was observed with this set of analogs, we performed molecular dynamics (MD) simulations using Desmond (D.E. Shaw Research, release 2020–1) via the Maestro interface using AZM, 22, and 26. The MDs were performed on both Efα-CA and Efγ-CA homology models and run on a 75 ns timescale. Beginning with Efα-CA, we first performed an MD simulation with AZM and compared the predicted pose of the same ligand with human CA 2 (PDB: 3HS4). In human CA 2, nitrogens of the AZM thiadiazole are positioned in proximity to Thr200 forming a hydrogen bond with the side-chain of the residue (Figure S3B). Thr199 formed side-chain mediated hydrogen bonds with sulfonamide while it is coordinated to the Zn2+ ion. The MD simulation reached convergence at about 10 ns and largely recapitulated these same interactions in the Efα-CA homology model (MD simulation report and video file in supporting information). It was predicted the side chain of Thr180 forms hydrogen bond contacts with 3-nitrogen of the thiadazole core (Figure 7A) as seen in the AZM-human CA 2 ligand bound structure. Additionally, the backbone amide carbonyl of Pro181 is predicted to from a productive interaction with proton on the amide of AZM. Both of these interactions are observed at > 60% of the 75 ns timescale. The water mediated interaction is predicted with Asn125, however, this appears to be weaker as the contact was only made 30% of the time.
Figure 7.

Ligand poses for AZM, 22 and 26 generated by MD simulation in the active site of the Efα-CA. Ligands, residues and waters important for ligand interactions shown as sticks. Polar hydrogens shown for clarity of proposed hydrogen bond interactions (yellow dashed lines). (A) Generated pose for AZM (green sticks) in the Efα-CA site. (B) Generated pose for 22 (cyan sticks). (C) Predicted binding pose for 26 (yellow sticks). (D) Overlaid ligands show the alternative positions adopted by analogs 22 and 26 compared to AZM. (E) Surface representation of Efα-CA with 22 (cyan sticks) and 26 (yellow sticks). The hydrophobic patch lined with Leu178, Pro181, and Pro182 shown as red surface. Polar patch lined by Glu67, Lys69, and Asn70 shown as blue surface. Images generated using PyMol.
Next an MD simulation was performed for 22 binding in the Efα-CA active site. This trajectory reached convergence at approximately 35 ns (MD simulation report and video file in supporting information). The ligand remained fairly rigid aside from the pendant cyclohexane moiety. Interestingly, contacts with the side chains of Thr179 and Thr180 were predicted to remain, albeit, with different atoms on the ligand (Figure 6B). The simulation predicts the thiadiazole core to be flipped in the active site relative to how AZM binds (Figure 6D) and the side-chain of Thr180 forms a hydrogen bond with the carbonyl of the amide on 22 while Thr179 forms hydrogen bonds with the sulfonamide similar to those observed with AZM and human CA 2. This may be a factor of the hydrophobic side chain migrating toward a hydrophobic patch on the side of the active site made up of Leu178, Pro181, and Pro182 (Figure 7E and S7). It is possible that for this hydrophobic interaction to take place with the low energy conformation of the ligand, that it must flip in the active site to accommodate this interaction. Indeed, it has previously been reported that the active site of CAs possess a hydrophobic patch and a polar patch, and this interaction predicted in the MD simulation is consistent with previously reported hydrophobic contacts of carbonic anhydrase inhibitors and CA active site.74,75 Additionally, reported structural data of other CAIs has shown the central aromatic core of the inhibitor can rotate in active site to take to access productive contacts with the surface of the CA.76 Regardless, the predicted interactions with Thr179 and Thr180 may be stronger interactions than those observed with AZM in the Efα-CA active site as they are predicted to be maintained for greater that 80% of the duration of the simulation. According to this simulation, it is possible this ligand, with the alternative orientation, forms stronger hydrogen bonds and additional hydrophobic interactions with the Efα-CA catalytic site.
The final MD simulation in the Efα-CA active site was performed with 26. This trajectory reached convergence quickly, within the first 10 ns (MD simulation report and video file in supporting information), and the thiadiazole core again adopted an alternative pose compared to that of AZM (Figure 6C and D). The ligand is predicted to form hydrogen bond contacts similar to those observed with 22 as Thr180 donated a hydrogen bond to the amide carbonyl of the ligand while Thr179 made the same contacts with the sulfonamide. However, the predicted strength of the Thr180 interaction with the amide carbonyl of 26 was reduced and predicted to interact approximately 50% of the time compared to 89% of the time with the carbonyl of 22. Alternatively, the Thr179 interaction was intact for almost the full duration of the simulation. Thus, there appears to be a trade-off in binding strength at each Threonine residue. The pendant morpholine group is lifted in the active site away from the hydrophobic patch and more in vicinity of a polar region of the protein active site lined by Glu67, Lys69, and Asn70 (Figure 7E and S8). However, there are no predicted contacts made by the morpholine moiety with these residues and in the video file (Analog 26 Efα-CA MD.mp4 in supplemental information) it shows the morpholine remained flexible and does not stay in any particular location in the active site. Contrast this with the video for 22 (Analog 22 EfaCA MD.mp4 in the supplemental) where the cyclohexane moiety is also relatively flexible and but it remains in a conformation that extends toward the hydrophobic patch for a longer duration of the simulation. This would indicate that no meaningful binding interaction are made or sustained. As a result of the morpholine lifting the ligand toward the polar region there was a new opportunity to form a water mediated hydrogen-bond with Asp94 and the proton on the amide nitrogen of 26. However, this interaction is only maintained 41% of the time and may be relatively weak compared to the hydrogen bonds directly to the ligand from residues.
Next MD simulations were performed for all three ligands into the newly generated Efγ-CA homology model. While these calculations may provide some insight into potential binding interactions, they should be interpreted with caution as there is no experimentally derived ligand bound structural data with γ-CA reported. Thus, we do not have a template to work from as we did with AZM to provide confidence in the simulations and these poses are speculative. Nonetheless, there is evidence reported in the literature that carbonic anhydrase inhibitors are capable of inhibiting γ-CAs,77,78 therefore, we know that the ligand can indeed bind to this potential target. However, at least in the case of the γ-CA from the oral pathogen Porphyromonas gingivalis, they tend to bind with weaker KI values in the upper nanomolar and low micromolar range compared to low nanomolar against human CA 2 for example.77 Regardless, MD simulations of the potential binding interactions may provide some insight into the observed SAR and future analog design.
Aside from the sulfonamide coordination with the Ni2+, in this case most of the proposed interactions took place via water mediated contacts with the catalytic site. Additionally, compared to the apo-Efγ-CA homology model, the active site in the MD simulated structures appear to be flexible as the cavity in which the ligand binds, opens up. The calculation indicated that helix α1 shifts approximately 3.3 Å upon binding (Figure S9A). The surface representations of the apo versus the predicted bound state for ligand 22 show the widening of the active site pocket to accommodate the ligand (Figures S9B and C). The widening may allow more access to the pocket for solvent water molecules leading to the aforementioned water mediated hydrogen bonds with the protein residues. For example, AZM is predicted to form relatively strong water mediated hydrogens bonds from the 3-nitrogen on the thiadiazole to Asp53 from the proton on the amide nitrogen to Glu109 on one monomer, while the amide carbonyl is predicted to form a water bridged hydrogen bond to Met85 on the other monomer lining the catalytic site (Figure 8A). These interactions all occur over 50% of the time during the simulation.
Figure 8.

Proposed binding poses of analogs from MD simulation with Efγ-CA. (A) Binding pose for AZM (green sticks). Residues associated with binding interactions are in sticks and labeled. Water molecules associated with binding interactions shown as red and white sticks. Predicted interactions shown as yellow dashed lines. Ni2+ ion shown as gray sphere. (B) Predicted binding pose for 22 (cyan sticks). (C) Predicted binding pose for 26 (yellow sticks). (D) Surface electrostatic map for Efγ-CA with ligands overlaid to show different in predicted binding modes. Blue surface indicates net positive charge and red surface indicates net negative charge. Images generated using PyMol.
Similar to what was observed in the Efα-CA binding for 22 and 26, these ligands also adopted alternative poses compared to AZM. Both were pulled toward one side of the site that displayed a weak positive surface charge (Figure 8D). The proposed interactions with this surface were made by the carbonyl oxygen on the amide of each ligand. In the case of 22 the ligand is a bit more extended in the active site with the carbonyl oxygen predicted to form a water mediated hydrogen bond with the backbone nitrogen of Ala103 for 54% of the simulation (Figure 8B). Analog 26 in predicted to make a water mediated contact with Gln45 but for only about 34% of the simulation indicating this interaction is slightly weaker (Figure 8C). Judging solely on the predicted strength of the interactions from the MD simulation, it would appear that AZM would be predicted to bind with the highest affinity followed by 22 and then 26. However, without experimental binding data, both structural and biochemical, it is difficult to make conclusions based on this model.
To summarize the MD simulations of ligands bound to both Efα-CA and Efγ-CA homology models were performed. In the case of Efα-CA the experimentally derived ligand bound structure for AZM to other α-CAs was mostly recapitulated and provided confidence in the simulation. In the calculations the central thiadiazole core of 22 and 26 appears to rotate in the active site compared to AZM. Further experimental evaluation is necessary to validate these proposed binding poses. The proposed poses for the ligands with Efγ-CA at this point are to be taken with a bit of caution as there is no structural information of an inhibitor bound to a γ-CA to use as a benchmark. Nonetheless, the data does provide some insight into the observed SAR that will be discussed below. All PDB files for the homology models and representative time points for ligands bound within the simulation, MD simulation report files, and .MP4 video files for each ligand binding simulation are provided in the supporting information.
In Vitro Solubility, Caco-2 Permeability, and Human Liver Microsome Stability
Compounds 22 and 26 were selected for in vitro ADME evaluation. We chose the most potent analog in 22, as well as 26 because it maintained appreciable activity and possesses physicochemical properties expected to reduce GIT permeability. It is our hypothesis that these compounds, exhibiting different physicochemical and potency profiles, would be effective against different forms of VRE infection, either systemic or gut colonization.
Aqueous solubility in phosphate buffered saline (PBS) at pH 7.4, Caco-2 permeability, and human liver microsome (HLM) stability were all determined for each analog (Table 4). 22 was measured to have a mean maximum solubility of 32.8 μM in PBS at pH 7.4 and 25 °C (Table 4), about 100-fold less soluble than AZM, which has a reported solubility of 3200 μM,45 although the AZM measurement was taken in water rather than PBS. The reduced solubility of 22 could be expected due to the addition of a highly lipophilic alkyl substituents; however, the peak solubility is still approximately 1500-fold greater than the MIC concentration against E. faecium HM-965 (32.8 μM converts to 10.4 μg/mL). Analog 26 displayed improved solubility compared to 22 with a mean maximum solubility of 126.6 μM. This maximum solubility is approximately 40-fold greater than the MIC for this analog against the same strain (126.6 μM converts to 39 μg/mL). Therefore, it is reasonable to believe the molecules should remain in solution at concentrations necessary to kill VRE in the respective compartments.
Table 4.
In Vitro ADME Profiles for AZM, 22, and 26
| Comp | LogPa | Solubility (μM)b | Papp A → Bc | Papp B → Ac | HLM % remainingd | Half-life (min) |
|---|---|---|---|---|---|---|
| AZM | −0.86 | 3200e | 0.19f | 0.77f | - | - |
| 22 | 1.49 | 32.8 | 6.5 | 14.1 | 62 | 74 |
| 26 | −1.54 | 126.6 | < 0.17g | <0.17g | 86 | 297 |
Calculated using pKCSM.71
Solubility in PBS at pH 7.4 at equilibrium at room temperature at 24 hr.
Bi-directional Caco-2 permeability with pH 6.5/7.4 for donor/receiver chambers, respectively. A = apical, B = basolateral. Papp is the apparent permeability coefficient (10−6 cm/sec).
HLM = human liver microsomes. Tested at 37 °C in 0.1 mg/mL HLMs. % remaining is percentage of comopound remaining in sample at 60 min time point and is average of 2 replicates.
Granero et al.45 measured in water at 25 °C.
Crowe et al.46 measured at pH 6.0/7.4 in donor/receiver chambers.
Detection was below limit of quantitation in receiver sample.
Permeability was also assessed for each molecule using the Caco-2 assay79, in both directions (apical➔basolateral and basolateral➔apical), was performed. To mimic the intestine-to-blood pH gradient, the donor samples were maintained at pH 6.5 (the pH of the lumen of the intestine) while the receiver sample was at pH 7.4 (blood pH). AZM has a previously reported Papp = 0.19 × 10−6 cm/s (A➔B) and 0.77 × 10−6 cm/s (B➔A) with a pH gradient of 6.0/7.4 (donor/receiver samples) (Table 4).46 The net transport for AZM is 0.24 (A➔B) and this was shown to be a result of efflux by P-glycoproteins (P-gp) as P-gp inhibitors increased the net transport up to 1.92 in the A➔B direction. Analog 22 exhibited an increase in Papp with a value of 6.5 × 10−6 cm/s in the A➔B direction and 14.1 × 10−6 cm/s in the B➔A direction resulting in a net transport 0.46 (A➔B). The increased rate of permeability and net transport can likely be attributed to the increase in LogP (calculated in PKCSM)80 for 22 compared to AZM making it more membrane permeable. Gratifyingly, the design of analog 26 managed to reduce the permeability almost completely as the concentration of molecule in the receiver chambers was below the limit of detection for the assay (83% of 26 was recovered from the sample chambers).
AZM is excreted through the kidneys and urine unmetabolized81,82. To assess metabolic stability of 22 and 26, the analogs were submitted for in vitro metabolism analysis in HLMs according to literature protocols83. The molecules did exhibit slow clearance within HLMs with 62% of 22 and 86% of 26 remaining after 60-minute incubation with estimated half-lives of 74 and 297 minutes, respectively. Additionally, according to permeability data, 26 would likely have little liver exposure in vivo as it would exhibit little intestinal absorption. These data indicate that the pharmacophore of analogs 22 and 26 would remain intact and in solution in the respective compartments in therapeutically relevant amounts to combat the VRE infection. Full tabular data for in vitro ADME experiments presented in Tables S6 – S8.
Evaluation of pharmacokinetics upon oral administration for analog 22
In preparation for future in vivo efficacy assays we chose to evaluate analog 22 in an in vivo pharmacokinetics assay to assess the oral bioavailability of this molecule in mice. Mice were dosed by oral gavage at 10 mg/kg and then plasma samples were collected at eight timepoints (10, 30, 60, 120, 240, 260, 480 and 1440 min post dose). The analog was shown to enter circulation rapidly and effectively with a plasma concentration of 184 ± 178 ng/mL at the 10 min time point and peaked at 30 min with a Cmax value of 456 ± 21 ng/mL (Figure 9, Table 5, and Table S9). The analogs had a measured area under the curve (AUCinf) value of 562 h*ng/mL. The time the molecule was in the plasma at a total drug concentration greater than the MIC90 (T>MIC90) is approximately 3 hrs, however, the concentration of unbound fraction is likely much less given that AZM is highly protein bound in plasma.43 The molecule is completely excreted at the 1440 min (24 hr) time point. This data would indicate that the lead molecule 22 is capable of entering the systemic circulation efficiently and would be a good a candidate for treating systemic forms of VRE infections.
Figure 9.

In vivo pharmacokinetic analysis of plasma concentrations for analog 22 in mice dosed orally. Seven time points shown, final time point at 1440 min was 0 ng/mL and not included on plot. Error bars indicate standard deviation at each time point (n = 3 per time point). Green dotted line represents the MIC concentration of 22 (0.007 μg/mL).
Table 5.
Experimental pharmacokinetic indices for analog 22
| Animal-Dose | Tmax (h)b | Cmax (ng/ml)c | AUCinf (h*ng/mL)d | AUC/D (h*kg*ng/mL/mg)e | MRT (h)f |
|---|---|---|---|---|---|
| PO-Mousea | 0.50 | 456 ± 21 | 562 | 56 | 1.35 |
Dosed by oral gavage to mice.
Tmax = time-point of maximum plasma concentration.
Cmax = maximum plasma concentration.
AUCinf = area under the curve extrapolated to timepoint infinity.
AUC/D = area under the curve divided by dose.
MRT = mean residence time.
DISCUSSION
Optimization of novel class of anti-VRE molecules based on the FDA approved CAI AZM was carried out. Data suggests the mechanism of action for this series may be inhibition of two previously uncharacterized carbonic anhydrases, an α-class and a γ-class, encoded for in the Enterococcus genome. The series exhibited tractable SAR trends with respect to the substitution on the amine portion of the 5-amino-1,3,4-thiadiazole-2-sulfonamide scaffold (Figure 10). A SAR trend that was observed across three matched molecular pairs (AZM and 29; 13 and 30; 16 and 31) was the removal of the amide carbonyl resulting in significant reduction or complete loss of anti-VRE potency. Secondly, it was observed that increasing 3-dimensional alkyl bulk off the amide yielded increased potency toward the pathogen. For example, the anti-VRE activity increased moving from methyl < ethyl = i-propyl < t-butyl for the series of AZM, 3, 4, and 5. It was more pronounced when the chain length was extended by one methylene with stepwise increases in potency moving from pendant methyl (3, MIC = 1 μg/mL), ethyl (6, 0.5 μg/mL), i-propyl (7, 0.125 μg/mL), and t-butyl (8, 0.015 μg/mL). The same could be observed when comparing cyclic alkyl moieties as the anti-VRE potency increased with larger ring size for the series of analogs 10 – 13 and 17 – 20.
Figure 10.

Summary of SAR for sulfonamide anti-VRE scaffold.
Extending the distance of the pendant group from the carbonyl also led to improved anti-VRE potency across several nearest neighbor analogs. One such example being the set of analogs moving the pendant cyclohexane moiety from directly next to the amide (13, MIC = 0.25 μg/mL), to a one-methylene (20, 0.06 μg/mL) and then to a two-methylene linker (22, 0.007 μg/mL). Interestingly, incorporation of polar atoms such as nitrogen or oxygen into the cycloalkane ring reduced activity and these molecules also did not adhere to previously observed SAR trends (analogs 24 – 28). Nonetheless, even though potency was reduced most of the analogs were equipotent to linezolid.
To round out SAR trends, three analogs were tested to explore the necessity of the sulfonamide and modifications to the central thiadazole-core. First, the sulfone containing analog 33, in which the sulfonamide nitrogen was replaced with a methyl group, exhibited a complete loss of activity consistent with SAR from this class of inhibitors and human carbonic anhydrase targets. The next derivative maintained the sulfonamide substituent while removing a nitrogen from the 4-position that flanks the sulfonamide in the central thiadiazole core to provide thiazole derivative (34). Surprisingly, this analog also displayed a complete loss of anti-VRE activity suggesting the nitrogen placement in the thiadiazole ring is equally as crucial for binding. This is further supported by analog 35, in which the thiadiazole core is replaced by a central phenyl core, also leading to a complete loss of activity. Taken together these analogs suggest the critical element for inhibition by this series of molecules must contain the sulfonamide with a nitrogen in the ring directly next to it (Figure 10).
In an attempt to better understand the observed SAR, homology models for the proposed intracellular targets Efα-CA and Efγ-CA were built and MD simulations were performed. We first attempted docking for all analogs to try to gain insight into molecule SAR but docking scores are not meant to provide comparative values for analogs.84 The more accurate method to evaluate binding is MD simulation; thus, given the heavy computational burden of these calculations, we carried them out for three molecules: AZM, 22, and 26 against each homology model. AZM served as a benchmark as there is sufficient ligand bound structural data to various α-CAs to evaluate the accuracy of the MD simulation. The binding mode was largely the same and formed similar contacts compared to the experimentally derived interactions for AZM providing confidence in the Efα-CA simulations. The atoms mainly involved in binding, aside from the normal sulfonamide interactions, according to the simulation were the 3-nitrogen within the thiadiazole ring and the nitrogen in the amide. These interactions may explain why the other FDA approved CAIs and analog 35 had lower efficacy against VRE as these were all more sterically hindered at the 3-nitrogen or lacked it completely. Surprisingly, the MD simulation did not predict any interactions with the amide carbonyl or the 4-nitrogen in the thiadiazole even though these atoms appear to be crucial for activity against VRE. We would have expected some interactions with these atoms given that removing led to losing the activity all together (see analogs 29 – 31 and 34). This discrepancy may be attributed to the fact that we are performing the MD simulations on a homology model and not on a known crystal structure. Another alternative explanation for the reduced efficacy of 34 and 35, is that lacking a 4-nitrogen in thiadiazole or substituting a phenyl ring in place of the thiadiazole, may be the modulated pKa of the sulfonamide nitrogen. It was shown recently that modulation of the pKa to make the sulfonamide more acidic improves the binding affinity and the on-rate for binding the human CA 2.76 AZM has a reported pKa of the sulfonamide of 7.285 and a predicted pKa of 7.3 using MarvinSketch (Version 18.28, ChemAxon, www.chemaxon.com). Analog 34, lacking the 4-nitrogen, has a predicted sulfonamide pKa increase to 8.43 and analog 35, with a central phenyl core, has a predicted sulfonamide pKa of 10.28. Moreover, other reports have estimated that the sulfonamide interaction accounts for up to 65% of the Gibbs free energy of binding to the CA in a series of fluorinated CAIs.86 While the analogs reported here are not directly comparable to the fluorinated derivatives it does underscore the importance of tuning the sulfonamide interaction with the metal. Thus, the combination of reduced interactions with the active site along with increasing the pKa of the sulfonamide may be two likely factors contributing to the loss of activity for these analogs.
Interestingly, the binding poses for 22 and 26 adopt alternative orientations that seem to accommodate for the increased bulk of the cyclohexyl and morpholine moieties. These orientations are predicted to still maintain the contact with Thr180, however, the hydrogen bond is now to the carbonyl on the amide rather than the 3-nitrogen in the thiadiazole. This binding pose would indicate increased importance of the carbonyl. Comparing a similar cyclohexane containing analog lacking the carbonyl and reducing the linker by a single methylene (30), the analog was again inactive against VRE suggesting this potential interaction may be real and is critical for inhibition. Compound 22 was the most potent of the three analogs tested experimentally and, according to the MD simulation calculation, this analog would also form the strongest interactions with the Efα-CA site. Moreover, the lipophilic cyclohexane tail is predicted to place itself along the well-known hydrophobic patch of the active site. Thus, the increased strength of the proposed hydrogen bonds combined with the added hydrophobic binding could partially explain the increased potency of 22 over the other analogs. The SAR trend of increasing length of the alkyl linker to improve potency also fits this binding pose as the lipophilic portions would be able to reach the hydrophobic patch more effectively without pulling the molecule away from the productive contacts with Thr180.
Finally, the difference in potency between 22 and 26 could also potentially be explained by where the morpholine tail on 26 resides during the simulation. This polar tail does not move toward the hydrophobic patch but rather is oriented toward a more polar region of the active site. However, even though it aligns itself with this region it does not form any new productive contacts. Furthermore, this pose may pull the amide carbonyl away from Thr180 in turn, reducing the strength of the hydrogen bond, as observed in the simulation. This pose could account for the reduced potency of all analogs that had heteroatoms inserted into the pendant cyclic group. Indeed, a new from study Glöckner et al. that investigated a congeneric series of alkyl substituents concluded that increasing alkyl length improves the kinetics on-rate (kon) of CAI ligands, via interaction with hydrophobic patch.87,88 Alternatively, addition of polar atoms into the alkyl chains reduced the kon. Combining this information with the sulfonamide interaction studies referenced above one could deduce that the hydrophobic tail of series presented herein could improve the kon to facilitate the sulfonamide interaction with the active site Zn2+ more efficiently. These more exhaustive thermodynamic and kinetic studies will be performed in the future to delineate the contributions of the substituents in out series toward Efα-CA binding.
The MD simulations for the Efγ-CA at this point are speculative and it is difficult to generate any meaningful conclusions with respect to observed SAR. However, they do provide predicted interactions that could be probed in the future.
This data combined with the fact that modification of the sulfonamide to the sulfone (33) abolishes VRE potency (previously seen in CAI SAR) and that the bacteria are no longer susceptible to the molecules in conditions with high CO2 strongly suggests the intracellular target for these molecules might be one or both of the carbonic anhydrases.
Even though the MD simulations for these three molecules does provide some insight into the predicted binding and observed SAR, there are still caveats to be kept in mind. First, potency toward the bacteria is not solely based on binding affinity to the targets, but also it is a cellular response that is heavily reliant on molecule permeability and access to the intracellular targets, an aspect that is much different and more difficult to predict in bacteria compared to human cells. Second, if these molecules are indeed inhibiting both classes of CA then the balance of polypharmacology would have an effect on the overall cellular potency. Third, we have yet to determine which class of CA is more essential to the survival of the bacteria. Questions regarding the expression levels of both the Efα-CA and Efγ-CA or the essentiality of the processes they regulate still need to be answered before we can determine if one or both are suitable drug targets. Also, does one CA compensate for the other making it beneficial to inhibit both? These items will be investigated in follow-up work.
Finally, in preparation of future in vivo work, analogs 22 and 26 were assessed for in vitro ADME properties including aqueous solubility in PBS buffer at pH 7.4, Caco-2 cell permeability, and HLM stability as well as in vivo oral bioavailability The analogs did exhibit reduced solubility compared to AZM; however, this could be expected given the addition of lipophilic substituents and they still maintained aqueous solubility well above the concentrations necessary for potency against the bacteria. The molecules also provided diverging profiles for Caco-2 permeability as 22 was sufficiently permeable, more than the reported values for AZM. This was also showed in the in vivo PK assay as 22 proved to be orally bioavailable and this data positions our team to assess various dose sizes and schedules for treatment of systemic VRE infections. Alternatively, analog 26 was designed to reduce cell permeability as a strategy to sequester the molecule in the human gut compartment for treatment of gut colonization. The permeability data suggests the strategy was successful as the molecule was not detected in the receiver samples. The increased permeability observed for 22 is likely a result of the addition of a highly lipophilic functionality that would allow for more efficient diffusion through a hydrophobic cellular membrane, although the molecule still appears to be a substrate for P-gp. On the other hand, the decreased permeability of 26 is likely a result of the addition of more polar heteroatoms into the alkyl ring, although, it is a bit surprising that these modifications reduced permeability to a degree that the molecule level was below the limit of detection in the receiver samples. For comparison, linezolid has a reported Papp (A ➜ B) = 18.1 × 10−6 cm/s and 31.2 × 10−6 cm/s (B ➜ A)89. This high permeability rate may be a contributing factor to the lack of efficacy for linezolid in VRE decolonization30.
Although AZM is reported to not be metabolized in vivo and is excreted unchanged through the urine, this attribute may not translate to 22 and 26 as these are new chemical entities so an assessment of molecules stability in HLMs was obtained. The molecules exhibited an overall stable profile in HLMs with 62% of 22 remaining and 86% of 26 remaining after 60-minute incubation. What this method does not account for is loss of molecule to non-specific binding to membranes of the microsomes; however, there may be some metabolic activity for 22 and less so for 26. There is no reported data of AZM in HLMs for comparison. Given the reduced permeability of 26, we are less concerned about any potential metabolic liability as this molecule would likely not make it to the liver in the first place. However, in vivo PK analysis would need to be performed to confirm the molecule does not enter the blood stream in appreciable amounts. Alternatively, even though 22 has an apparent half-life of approximately 74 minutes in HLMs, that could reduce the overall concentration of the active molecule in circulation after first pass metabolism.
Items that remain in question regarding this series that will be part of future investigation include modification to increase plasma half-life, and unbound drug concentration specifically. AZM is known to bind plasma protein at over 90%.43 While high plasma protein binding is typically an indicator of less free drug to access the desired target that is not that case for carbonic anhydrase inhibitors. AZM has better affinity for carbonic anhydrase than it does for human serum albumin and this pushes the equilibrium toward high concentrations at the target. For example, total drug concentration is much greater in red blood cells (RBCS) (>10-fold), where carbonic anhydrase 2 is located, than in plasma.90,91 Therefore, even though plasma protein binding is >90% for AZM there is less total drug in the plasma water than at the site of action and the equilibrium is always moving toward binding to carbonic anhydrase as a sink until sites are saturated. Given that the MIC90 for 22 is 0.030 μg/mL, which equates to 94.2 nM cellular potency, it is reasonable to assume that the affinity at the intracellular carbonic anhydrase target would be low nM range. Thus, even though it is unknown how effectively 22 binds to plasma proteins the molecule may behave similarly to AZM as far as distribution. Nonetheless, improving the plasma protein binding parameter will be a priority and further in vivo PK will be evaluated as part of future studies. Additional evaluation of potential metabolic by-products for 22 is also warranted to determine how much is excreted unchanged or to identify potential hot spots that may be susceptible to metabolism. Additional optimization of the lead 26 to improve upon the MIC90 while maintaining low GIT permeability is also a priority of future work. Finally, future in vivo studies are planned to assess the efficacy of these molecules in systemic and gut decolonization models of VRE.
CONCLUSION
In conclusion, we report a new class of potent anti-enterococcal agents based on the FDA-approved CAI, AZM. This scaffold has undergone SAR optimization to yield two potential leads for treatment of VRE infections. The most potent analog 22 was shown to have an MIC = 0.007 μg/mL versus clinical strains of VRE and an MIC90 value of 0.03 μg/mL. Alternatively, 26 had a measured MIC = 1 μg/mL and an MIC90 value of 4 μg/mL. Our data suggests the intracellular targets for these analogs might be uncharacterized Efα-CA and Efγ-CA. MD simulation studies indicate the analogs bind efficiently to both targets and provide insight into the observed SAR as well as potential future modifications. These molecules diverge in Caco-2 permeability where 22 exhibited increased permeability compared to AZM while 26 was effectively impermeable. Thus, it is our hypothesis that 22, with superior anti-VRE potency and permeability, will be effective against systemic VRE infections; while 26, with much reduced permeability will be particularly effective in VRE gut decolonization. Future studies are required and planned to test this hypothesis. To close, the lead molecules described represent promising, novel therapeutic treatment options for a wide variety of VRE infections and both possess potency and/or PK properties that are superior to the current standard treatment of VRE.
EXPERIMENTAL SECTION
Chemistry
General Methods.
1H and 13C NMR spectra were recorded on either a Bruker DRX500 (operating at 500 and 126 MHz) or Avance-III-800 spectrometer (operating at 800 and 200 MHz) in either CDCl3, MeOD-d4, or DMSO-d6 with or without the internal standard of TMS at 0.05% v/v. The chemical shifts (δ) reported as parts per million (ppm) and the coupling constants are reported as s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, m = multiplet. The purity of all final compounds was >95 % purity as assessed by HPLC according to current American Chemical Society guidelines for publication. Final compounds were analyzed on an Agilent 1200 series chromatograph. The chromatographic method utilized as ThermoScientific Hypersil GOLD C-18 or silica column. UV detection wavelength = 220/254 nm; flow-rate = 1.0 mL/min; mobile phases: 1) acetonitrile/water with 0.1% formic acid or 2) acetonitrile/water with 0.1% ammonium hydroxide. The mass spectrometer used is an Advion CMS-L Compact Mass Spectrometer with an ESI or an APCI source. Samples are submitted for analysis solubilized in 1:1 acetonitrile:water solution or using the atmospheric solids analysis probe (ASAP). Compounds were generally prepared according to scheme 1 and protocols are detailed below. The following analogs were purchased from commercial vendors, fully characterized to confirm identify and purity then used for biological evaluation: AZM (Alfa Aesar, Cat # L07562), EZM (Sigma Aldrich, Cat # 333328), 40 (Enamine, Cat # EN300–79477), and 41 (Combi-Blocks, Cat # A59503).
N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (acetazolamide, AZM).
AZM was purchased from a commercial vendor (Alfa Aesar, Cat # L07562) and characterized prior to biological evaluation. 1H NMR (500 MHz, DMSO-d6): δ 13.04 (s, 1H), 8.36 (s, 2H), 3.41 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 169.8, 164.6, 161.4, 22.7. APCI-MS−: m/z 220.9 [M - H]−. HPLC retention time: 7.935 min (mobile phase 1). HPLC Purity: 95.5%.
6-Ethoxybenzo[d]thiazole-2-sulfonamide (ethoxzolamide, EZM).
EZM was purchased from a commercial vendor (Sigma Aldrich, Cat # 333328) and characterized prior to biological evaluation. 1H NMR (500 MHz, DMSO-d6): δ 8.23 (s, 2H), 8.00 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 2.5 Hz, 1H), 7.20 (dd, J = 9.0, 2.5 Hz, 1H), 4.09 (q, J = 7.0 Hz, 2H), 1.34 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO-d6): δ 166.6, 158.3, 146.3, 137.9, 125.2, 118.1, 105.7, 64.2, 14.9. APCI-MS−: m/z 256.9 [M - H]−. HPLC retention time: 11.263 min (mobile phase 1). HPLC Purity: 98.3%.
5-Amino-1,3,4-thiadiazole-2-sulfonamide (1).
To a 100 mL roundbottom flask was added AZM (2.1 g, 9.3 mmol, 1 eq.) and concentrated HCl (10 mL, 120 mmol, 13 eq.) to form a white suspension. The reaction was refluxed at 95 °C and the white suspension became a colorless solution. After 24 h the reaction was cooled to rt and a white precipitate began to form. The reaction was removed from stirring and the white precipitate was allowed to settle. The excess HCl was decanted off and the remaining solid suspension containing as little HCl as possible was cooled in ice bath. This suspension was neutralized to pH 7 with 5.0 M aqueous NaOH solution. The white precipitate was filtered out by vacuum filtration to afford 2 as a white solid (1.23 g, 6.8 mmol, 74 %). 1H NMR (500 MHz, DMSO-d6): δ 8.02 (s, 2H), 7.77 (s, 2H). 13C NMR (126 MHz, DMSO-d6): δ 171.8, 158.5. APCI-MS+: m/z 180.9 [M + H]+. HPLC retention time: 2.174 min (mobile phase 2). HPLC Purity: 98.6%.
2-Chloro-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (2a).
To a vial was added the 2 (1.0 g, 5.6 mmol, 1 eq.) in anhydrous acetonitrile (25 mL) followed by addition of triethylamine (0.93 mL, 6.7 mmol, 1.2 eq.). The reaction was cooled to 0 °C then a solution of chloroacetyl chloride (0.49 mL, 6.1 mmol, 1.1 eq.) in acetonitrile (5 mL) was added over 15 minutes. The reaction was stirred at 0 °C for 6 hrs then allowed to warm to room temperature overnight. The solvent was removed in vacuo and the resulting crude material was treated with water (30 mL) and extracted with dichloromethane (3 × 40 mL). The organic layers were combined, washed with brine and dried over MgSO4. The suspension was filtered and the filtrate was concentrated. The crude product was purified by column chromatography (30 – 50% EtOAc:Hex) to afford intermediate 3a (0.66 g, 2.6 mmol, 46%) as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 13.37 (s, 1H), 8.33 (s, 2H), 4.48 (s, 2H). APCI-MS: m/z 256.9 [M + H]+.
3-Chloro-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)propanamide (2b).
Prepared according to same procedure as intermediate 3a using 2 (0.50 g, 2.8 mmol, 1 eq.), triethylamine (0.50 mL, 3.6 mmol, 1.3 eq.), and 3-chloropropanoyl chloride (0.46 g, 3.6 mmol, 1.3 eq.) to yield 2b (0.29 g, 1.1 mmol, 39%) as an off-white solid. 1H NMR (500 MHz, DMSO-d6): δ 13.15 (s, 1H), 8.30 (s, 2H), 3.88 (t, J = 6.3 Hz, 2H), 3.03 (t, J = 6.3 Hz, 2H). APCI-MS: m/z 270.8 [M + H]+.
General Procedure 1 for analogs 3 – 23. Described in detail for analog 3.
N-(5-Sulfamoyl-1,3,4-thiadiazol-2-yl)propionamide (3).92
To an argon flushed vial was added 1 (0.200 g, 1.1 mmol, 1 eq.) dissolved in 1 mL of acetonitrile, followed by triethylamine (187 μL, 0.27 mmol, 1.2 eq.) at 0 °C. Propionyl chloride (96 μL, 1.1 mmol, 1 eq.) was then diluted with acetonitrile (0.2 mL) and added drop-wise to the reaction mixture at 0 °C. The reaction mixture was stirred for 14 hours at room temperature. The reaction mixture was then worked up using DCM and washed with 2N HCl, followed by brine and was further purified by normal phase chromatography (0 – 100% ethyl acetate: hexane) to afford desired product 3 as a white solid (0.041 g, 0.17 mmol, 16%). 1H NMR (500 MHz, DMSO-d6): δ 12.98 (s, 1H), 8.34 (s, 2H), 2.56 – 2.52 (m, 2H), 1.11 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 173.3, 164.5, 161.5, 28.5, 9.1. ESI-MS: m/z 236.6 [M+H]+. HPLC retention time: 8.672 min (mobile phase 1). HPLC Purity: 98.2%.
N-(5-Sulfamoyl-1,3,4-thiadiazol-2-yl)isobutyramide (4).92
Prepared according to general procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), isobutyryl chloride (29 μL, 0.28 mmol, 1 eq.), and triethylamine (47 μL, 0.34 mmol, 1.2 eq.). Isolated as a white solid (0.049 g, 0.20 mmol, 71%). 1H NMR (500 MHz, DMSO-d6): δ 13.01 (s, 1H), 8.34 (s, 2H), 2.84–2.79 (m, 1H), 1.15 (d, J = 6.8 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 176.4, 164.7, 161.6, 34.3, 19.2. ESI-MS: m/z 250.6 [M+H]+. HPLC retention time: 9.553 min (mobile phase 1). HPLC Purity: 100%.
N-(5-Sulfamoyl-1,3,4-thiadiazol-2-yl)pivalamide (5).93
Prepared according to procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), pivaloyl chloride (34 μL, 0.28 mmol, 1 eq.), and triethylamine (47 mL, 0.34 mmol, 1.2 eq.). Isolated as a white solid (0.021 g, 0.080 mmol, 29%). 1H NMR (500 MHz, DMSO-d6): δ 12.75 (s, 1H), 8.33 (s, 2H), 1.27 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 173.5, 162.7, 159.2, 38.8, 28.5. APCI-MS: m/z 264.9 [M+H]+. HPLC retention time: 10.230 min (mobile phase 1). HPLC Purity: 99.2%
N-(5-Sulfamoyl-1,3,4-thiadiazol-2-yl)butyramide (6).92
Prepared according to procedure 1 using 1 (0.053 g, 0.29 mmol, 1 eq.), butyryl chloride (30 μL, 0.29 mmol, 1 eq.), and triethylamine (47 mL, 0.34 mmol, 1.2 eq.). Isolated as a white solid (0.009 g, 0.036 mmol, 17 %). 1H NMR (500 MHz, DMSO-d6): δ 12.98 (s, 1H), 8.29 (s, 2H), 2.48 (bs, 2H), 1.64–1.58 (m, 2H), 0.87 (t, J = 7.3 Hz). 13C (126 MHz, DMSO-d6: δ 172.5, 164.6, 161.4, 37.0, 18.2, 13.8. APCI-MS: m/z 251.0 [M+H]+ HPLC retention time: 9.465 min (mobile phase 1). HPLC Purity: 95.9%.
3-Methyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)butanamide (7).94
Prepared according to procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), isovaleryl chloride (34 μL, 0.28 mmol, 1 eq.), and triethylamine (47 mL, 0.34 mmol, 1.2 eq.). Isolated as a white solid (0.036 g, 0.14 mmol, 49 %). 1H NMR (500 MHz, DMSO-d6): δ 13.03 (s, 1H), 8.33 (s, 2H), 2.42 (d, J = 7.1 Hz, 2H), 2.11 (m, 1H), 0.93 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, DMSO-d6): δ 171.9, 164.6, 161.3, 44.0, 25.7, 22.5. APCI-MS: m/z 265.0 [M + H]+. HPLC retention time: 10.306 min (mobile phase 1). HPLC Purity: 100%.
3,3-Dimethyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)butanamide (8).
Prepared according to procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), 3,3-dimethylbutyryl chloride (39 μL, 0.28 mmol, 1 eq.), and triethylamine (47 μL, 0.34 mmol, 1.2 eq.). Isolated as a white solid (0.033g, 0.12 mmol, 43 %). 1H NMR (500 MHz, DMSO-d6): δ 12.98 (s, 1H), 8.32 (s, 2H), 2.42 (s, 2H), 1.01 (s, 9H). 13C NMR (126 MHz, DMSO-d6): δ 171.2, 164.5, 161.2, 48.1, 31.4, 29.7. APCI-MS: m/z 279.0 [M+H]+. HPLC retention time: 10.844 min (mobile phase 1). HPLC Purity: 100%.
N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)heptanamide (9).
Prepared according to procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), heptanoyl chloride (43 μL, 0.28 mmol, 1 eq.), and triethylamine (47 μL, 0.34 mmol, 1.2 eq.). Isolated as a white solid (0.028 g, 0.12 mmol, 35 %). 1H NMR (500 MHz, DMSO-d6): δ 13.00 (s, 1H), 8.33 (s, 2H), 1.65 – 1.57 (m, 2H), 1.34 – 1.20 (m, 8H), 1.86 (t, J = 7.1 Hz). 13C NMR (126 MHz, DMSO-d6): δ 172.4, 164.3, 161.1, 34.9, 30.9, 28.2, 24.4, 22.0, 14.0. APCI-MS: m/z 293.0 [M+H]+. HPLC retention time: 11.600 min (mobile phase 1). HPLC Purity: 99.2%.
N-(5-Sulfamoyl-1,3,4-thiadiazol-2-yl)cyclopropanecarboxamide (10).
Prepared according to procedure 1 using 1 (0.035 g, 0.19 mmol, 1 eq.), cyclopropanecarbonyl chloride (21 μL, 0.23 mmol, 1.2 eq.), and triethylamine (32 μL, 0.23 mmol, 1.2 eq.). Isolated as a white solid (0.010 g, 0.040 mmol, 21%). 1H NMR (500 MHz, DMSO-d6): δ 13.28 (s, 1H), 8.30 (s, 2H), 2.04 – 1.99 (m, 1H, CH), 1.03 – 0.97 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 173.2, 164.6, 161.5, 14.1, 9.6. APCI-MS: m/z 249.0 [M+H]+. HPLC retention time: 9.253 min (mobile phase 1). HPLC Purity: 100 %.
N-(5-Sulfamoyl-1,3,4-thiadiazol-2-yl)cyclobutanecarboxamide (11).
Prepared according to procedure 1 using 1 (0.040 g, 0.22 mmol, 1 eq.), cyclobutanecarbonyl chloride (26 μL, 0.27 mmol, 1.2 eq.), and triethylamine (37 μL, 0.27 mmol, 1.2 eq.). Isolated as a white solid (0.026 g, 0.057 mmol, 26%). 1H NMR (500 MHz, DMSO-d6): δ 12.88 (s, 1H), 8.29 (s, 2H), 3.43–3.36 (m, 1H), 2.25–2.18 (m, 2H), 2.17–2.14 (m, 2H), 1.98–1.89 (m, 1H), 1.83–1.76 (m, 1H). 13C NMR (126 MHz, DMSO-d6): δ 174.0, 164.6, 161.6, 38.5, 24.8, 18.0. ESI-MS: m/z 262.9 [M+H]+. HPLC retention time: 11.133 min (mobile phase 1). HPLC Purity: 100 %.
N-(5-Sulfamoyl-1,3,4-thiadiazol-2-yl)cyclopentanecarboxamide (12).
Prepared according to procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), cyclopentanecarbonyl chloride (34 μL, 0.28 mmol, 1.2 eq.), and triethylamine (46 μL, 0.33 mmol, 1.2 eq.). Isolated as a white solid (0.025 g, 0.089 mmol, 32%). 1H NMR (500 MHz, DMSO-d6): δ 12.98 (s, 1H), 8.29 (s, 2H), 2.99–2.93 (m, 1H), 1.91–1.84 (m, 2H), 1.73–1.67 (m, 2H), 1.64–1.60 (m, 2H), 1.57–1.50 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 175.6, 164.6, 161.6, 44.2, 30.1, 26.0. APCI-MS: m/z 277.1 [M + H]+. HPLC retention time: 10.498 min (mobile phase 1). HPLC Purity: 99.5%.
N-(5-Sulfamoyl-1,3,4-thiadiazol-2-yl)cyclohexanecarboxamide (13).
Prepared according to procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), cyclohexanecarbonyl chloride (34 μL, 0.28 mmol, 1.2 eq.), and triethylamine (46 μL, 0.33 mmol, 1.2 eq.). Isolated as a white solid (0.027 g, 0.094 mmol, 34%). 1H NMR (500 MHz, DMSO-d6): δ 12.97 (s, 1H), 8.33 (s, 2H), 2.62 – 2.54 (m, 1H), 1.86 (broad m, 2H), 1.74 (broad m, 2H), 1.46–1.37 (m, 2H), 1.33 – 1.12 (m, 4H). 13C NMR (126 MHz DMSO-d6): δ 175.3, 164.7, 161.5, 43.7, 28.9, 25.5, 25.3. APCI-MS: m/z 291.2 [M+H]+. HPLC retention time: 11.043 min (mobile phase 1). HPLC Purity: 95.2%.
1-methyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)cyclohexane-1-carboxamide (14).
Step 1:
To a vial was added the 1-methylcyclohexane-1-carboxylic acid (95 mg, 0.67 mmol, 1.1 eq.) in DCM (1 mL). This was cooled to 0 °C followed by addition of oxalyl chloride (0.40 mL, 0.80 mmol, 1.2 eq.) and DMF (1 drop). The reaction then allowed to warm to room temperature and stirred for 2 hrs. After 2 hrs the reaction was concentrated in vacuo and the crude acyl chloride was carried into step 2 uncharacterized.
Step 2:
Same as procedure 1 using 1 (0.12 g, 0.64 mmol, 1 eq.), crude acyl chloride (step 1), and triethylamine (100 μL, 0.70 mmol, 1.2 eq.). Isolated as a white solid (0.015 g, 0.050 mmol, 7.8%). 1H NMR (500 MHz, DMSO-d6): δ 12.68 (s, 1H), 8.28 (s, 2H), 2.09 – 2.04 (m, 2H), 1.49 – 1.26 (m, 8H), 1.19 (s, 3H). 13C NMR (200 MHz, DMSO-d6) δ 177.3, 164.8, 162.4, 43.4, 34.7, 25.5, 22.8. APCI-MS: m/z 305.1 [M+H]+. HPLC retention time: 11.416 min (mobile phase 1). HPLC Purity: 100 %.
4-methyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)cyclohexane-1-carboxamide (15).
Step 1:
To a vial was added the 4-methylcyclohexane-1-carboxylic acid (114 mg, 0.80 mmol, 1.4 eq.) in DCM (1 mL). This was cooled to 0 °C followed by addition of oxalyl chloride (0.40 mL, 0.80 mmol, 1.4 eq.) and DMF (1 drop). The reaction then allowed to warm to room temperature and stirred for 2 hrs. After 2 hrs the reaction was concentrated in vacuo and the crude acyl chloride was carried into step 2 uncharacterized.
Step 2:
Same as procedure 1 using 1 (0.10 g, 0.57 mmol, 1 eq.), crude acyl chloride (step 1), and triethylamine (100 μL, 0.70 mmol, 1.2 eq.). Isolated as a white solid (0.076 g, 0.25 mmol, 44%). 1H NMR (500 MHz, DMSO-d6) δ 12.95 (s, 1H), 8.29 (s, 2H), 2.45 (m, 1H), 1.80 (dd, 2H, J = 10, 5 Hz), 1.70 (dd, 2H, J = 10, 5 Hz), 1.40 (qd, J = 12.9, 3.3 Hz, 2H), 1.34 – 1.23 (m, 1H), 0.95 – 0.87 (qd, J = 12.9, 3.3 Hz, 2H), 0.84 (d, J = 6.5 Hz, 3H).13C NMR (200 MHz, DMSO-d6) δ 175.5, 164.7, 161.6, 43.0, 33.6, 31.8, 29.0, 21.6. APCI-MS: m/z 305.0 [M+H]+. HPLC retention time: 11.572 min (mobile phase 1). HPLC Purity: 96.8%.
N-(5-Sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide (16).93
Prepared according to procedure 1 using 1 (0.043 g, 0.24 mmol, 1 eq.), benzoyl chloride (34 μL, 0.28 mmol, 1.2 eq.), and triethylamine (40 μL, 0.28 mmol, 1.2 eq.). Isolated as a white solid (0.0035 g, 0.012 mmol, 5%). 1H NMR (500 MHz, DMSO-d6): δ 13.56 (s, 1H), 8.37 (s, 2H), 8.19 – 8.12 (m, 2H), 7.73 – 7.66 (m, 1H), 7.61–7.58 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 166.1, 165.0, 162.6, 133.7, 131.3, 129.1, 129.0. APCI-MS: m/z 284.9 [M+H]+. HPLC retention time: 10.288 min (mobile phase 1). HPLC Purity: 99.4%.
2-Cyclopropyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (17).
Step 1:
To a vial was added the 2-cyclopropyl acetic acid (31 mg, 0.31 mmol, 1.1 eq.) in DCM (1 mL). This was cooled to 0 °C followed by addition of oxalyl chloride (29 μL, 0.33 mmol, 1.2 eq.) and DMF (1 drop). The reaction then allowed to warm to room temperature and stirred for 1 hr. After 1 hr the reaction was concentrated in vacuo and the crude acyl chloride was carried into step 2 uncharacterized.
Step 2:
Same as procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), crude 2-cyclopropyl acetyl chloride (step 1), and triethylamine (46 μL, 0.33 mmol, 1.2 eq.). Isolated as a white solid (0.023 g, 0.088 mmol, 31%). 1H NMR (500 MHz, DMSO-d6): δ 12.95 (s, 1H), 8.29 (s, 2H), 2.39 (d, J = 7.1 Hz, 2H), 1.06–0.98 (m, 1H), 0.46 (q, J = 5.7 Hz, 2H), 0.17 (q, J = 4.9 Hz, 2H). 13C NMR (126 MHz, DMSO-d6): δ 172.2, 164.5, 161.6, 40.1, 7.4, 4.4. APCI-MS: m/z: 262.9 [M+H] +. HPLC retention time: 9.599 min (mobile phase 1). HPLC Purity: 99.1%.
2-Cyclobutyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (18).
Step 1:
To a vial was added the 2-cyclobutyl acetic acid (80 μL, 0.77 mmol, 1.4 eq.) in DCM (1 mL). This was cooled to 0 °C followed by addition of oxalyl chloride (0.40 mL, 0.85 mmol, 1.5 eq.) and DMF (1 drop). The reaction then allowed to warm to room temperature and stirred for 2 hrs. After 2 hrs the reaction was concentrated in vacuo and the crude acyl chloride was carried into step 2 uncharacterized.
Step 2:
Same as procedure 1 using 1 (0.10 g, 0.57 mmol, 1 eq.), crude 2-cyclobutyl acetyl chloride, and triethylamine (100 μL, 0.70 mmol, 1.2 eq.). Isolated as a white solid (0.033 g, 0.12 mmol, 42%). 1H NMR (500 MHz, DMSO-d6) δ 12.96 (s, 1H), 8.29 (s, 2H), 2.69 – 2.56 (m, 3H), 2.03 (m, 1H), 1.86 – 1.73 (m, 2H), 1.68 (q, J = 8.8 Hz, 2H). 13C NMR (200 MHz, DMSO-d6) δ 171.6, 164.7, 161.4, 42.3, 32.2, 27.6, 18.5. APCI-MS: m/z 277.0 [M+H]+. HPLC retention time: 10.490 min (mobile phase 1). HPLC Purity: 98.9 %.
2-Cyclopentyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (19).
Step 1:
To a vial was added the 2-cyclopentyl acetic acid (38 μL, 0.31 mmol, 1.1 eq.) in DCM (1 mL). This was cooled to 0 °C followed by addition of oxalyl chloride (29 μL, 0.33 mmol, 1.2 eq.) and DMF (1 drop). The reaction then allowed to warm to room temperature and stirred for 1 hr. After 1 hr the reaction was concentrated in vacuo and the crude acyl chloride was carried into step 2 uncharacterized.
Step 2:
Same as procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), crude 2-cyclopentyl acetyl chloride, and triethylamine (46 μL, 0.33 mmol, 1.2 eq.). Isolated as a white solid (0.035 g, 0.12 mmol, 43%). 1H NMR (500 MHz, DMSO-d6): δ 12.98 (s, 1H), 8.30 (s, 2H), 2.51 (d, J = 7.3 Hz, 2H), 2.25–2.18 (m, 1H), 1.75–1.69 (m, 2H), 1.61–1.54 (m, 2H), 1.52–1.45 (m, 2H), 1.17–1.10 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 172.2, 164.6, 161.3, 41.0, 36.5, 32.1, 24.8. APCI-MS: m/z 291.2 [M+H]+. HPLC retention time: 11.152 min (mobile phase 1). HPLC Purity: 100 %.
2-Cyclohexyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (20).
Prepared according to procedure 1 using 1 (0.050 g, 0.28 mmol, 1 eq.), 2-cyclohexylacetyl chloride (53 μL, 0.33 mmol, 1.2 eq.), and triethylamine (46 μL, 0.33 mmol, 1.2 eq.). Isolated as a white solid (0.033 g, 0.11 mmol, 39 %). 1H NMR (500 MHz, DMSO-d6): δ 13.00 (s, 1H), 8.32 (s, 2H), 2.42 (d, J = 7.1 Hz, 2H), 1.83–1.75 (m, 1H), 1.69 – 1.59 (m, 4H), 1.26–1.09 (m, 4H), 1.02–0.91 (m, 2H). 1H NMR (500 MHz, CD3OD-d4) δ 2.40 (d, J = 7.2 Hz, 2H), 1.90 – 1.82 (m, 1H), 1.76 – 1.69 (m, 4H), 1.68 – 1.63 (m, 1H), 1.33 – 1.25 (m, 2H), 1.23 – 1.16 (m, 1H), 1.08 – 0.99 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 171.8, 164.6, 42.8, 34.9, 32.7, 26.0, 25.8. 13C NMR (126 MHz, CD3OD-d4) δ 171.7, 164.9, 161.4, 42.6, 35.0, 32.5, 25.7, 25.6. APCI-MS: m/z 305.1 [M+H]+. HPLC retention time: 11.609 min (mobile phase 1). HPLC Purity: 100%.
3-Cyclopentyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)propanamide (21).
Prepared according to procedure 2 using 2 (0.050 g, 0.28 mmol, 1 eq.,), 3-cyclopentylpropanoyl chloride (51 μL, 0.33 mmol, 1.2 eq.), and triethylamine (46 μL, 0.33 mmol, 1.2 eq.). Isolated as a white solid (0.022 g, 0.076 mmol, 26 %). 1H NMR (500 MHz, DMSO-d6): δ 12.98 (s, 1H), 8.29 (s, 2H), 2.49 (t, J = 7.5 Hz, 2H), 1.72 – 1.67 (m, 3H), 1.63–1.54 (bm, 4H), 1.47 – 1.43 (m, 2H), 1.05 (bs, 2H). 13C NMR (126 MHz, DMSO-d6): δ 172.7, 164.6, 161.4, 39.4, 34.5, 32.3, 30.9, 25.0. APCI-MS: m/z 305.0 [M+H]+. HPLC retention time: 11.783 min (mobile phase 1). HPLC Purity: 99.2%.
3-Cyclohexyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)propanamide (22).
Step 1:
To a vial was added the 3-cyclohexylpropanoic acid (0.31 g, 2.0 mmol, 1.1 eq.) in DCM (5 mL). This was cooled to 0 °C followed by addition of oxalyl chloride (0.27 g, 2.1 mmol, 1.2 eq.) and DMF (1 drop). The reaction then allowed to warm to room temperature and stirred for 1 hr. After 1 hr the reaction was concentrated in vacuo and the crude 3-cyclohexylpropanyl chloride was carried into step 2 uncharacterized.
Prepared according to procedure 1 using 1 (0.32 g, 1.8 mmol, 1 eq.,), 3-cyclohexylpropanyl chloride (from step 1), and triethylamine (300 μL, 2.1 mmol, 1.2 eq.). Isolated as a white solid (0.18 g, 0.57 mmol, 32 %). 1H NMR (500 MHz, DMSO-d6): δ 13.00 (s, 1H), 8.33 (s, 2H), 2.54 (t, J = 7.7 Hz, 2H), 1.71 – 1.63 (m, 4H), 1.63 – 1.57 (m, 1H), 1.56 – 1.48 (m, 2H), 1.24 – 1.09 (m, 4H), 0.93 – 0.83 (m, 2H). 1H NMR (500 MHz, CD3OD-d4) δ 2.58 – 2.48 (m, 2H), 1.79 – 1.69 (m, 4H), 1.69 – 1.62 (m, 1H), 1.59 (q, J = 7.2 Hz, 2H), 1.33 – 1.12 (m, 4H), 0.98 – 0.87 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 172.8, 164.6, 161.4, 39.9, 32.8, 32.1, 26.4, 26.0. 13C NMR (126 MHz, CD3OD-d4) δ 172.8, 164.9, 161.6, 37.0, 32.6, 32.5, 31.9, 26.1, 25.8. APCI-MS: m/z 319.0 [M+H]+. HPLC retention time: 12.322 min (mobile phase 1). HPLC Purity: 100%.
3-Phenyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)propanamide (23).95
Prepared according to procedure 1 using 1 (0.025 g, 0.14 mmol, 1.0 eq.), 3-phenylpropanoyl chloride (0.023 mL, 0.15 mmol, 1.1 eq.), potassium carbonate (0.023 g, 0.17 mmol, 1.2 eq.). Isolated as a white solid (0.013 g, 0.042 mmol, 30%). 1H NMR (500 MHz, DMSO-d6): δ 13.02 (s, 1H), 8.29 (s, 2H), 7.30–7.12 (m, 5H), 2.91 (t, J = 7.6 Hz, 2H), 2.81 (t, J = 7.6 Hz, 2H). 13C NMR (126 MHz, DMSO-d6): δ 172.0, 164.5, 161.5, 140.8, 128.7, 128.6, 126.5, 37.0, 30.5. APCI-MS+: m/z 313.0 [M + H]+. HPLC retention time: 10.952 min (mobile phase 1). HPLC Purity: 98.2 %.
General Procedure 2 for analogs 24 - 28. Described in detail for analog 24.
2-(Pyrrolidin-1-yl)-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (24).
To a vial was added intermediate 2a (0.050 g, 0.19 mmol, 1 eq.) and anhydrous THF (3 mL) and cooled to 0 °C. This was followed by addition of triethylamine (0.054 mL, 0.39 mmol, 2.0 eq.) and pyrrolidine (0.021 g, 0.29 mmol, 1.5 eq.). The reaction stirred at 0 °C for 1 h then warmed to room temperature and stirred overnight. The reaction was then concentrated in vacuo and the crude product was adsorbed to Celite and purified by reverse-phase flash chromatography (5 – 100% acetonitrile:water) to afford 24 (0.013 g, 0.043 mmol, 22%) as an off-white solid. 1HNMR (500 MHz, DMSO-d6) δ 7.84 (s, 2H), 3.87 (s, 2H), 3.24–3.12 (m, 3H), 2.53–2.44 (m, 2H), 1.87 (t, J = 6.6 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 170.2, 169.7, 161.2, 58.8, 54.3, 23.1. APCI-MS: m/z 292.0 [M + H]+. HPLC retention time: 3.430 min (mobile phase 1). HPLC Purity: 96.8%.
2-(Piperidin-1-yl)-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (25).
Prepared according to general procedure 2 using 2a (0.050 g, 0.19 mmol, 1.0 eq.), triethylamine (0.050 mL, 0.39 mmol, 2.0 eq.) and piperidine (0.040 mL, 0.29 mmol, 2.0 eq.). Isolated as a white solid (0.049 g, 0.15 mmol, 81 %). 1H NMR (500 MHz, DMSO-d6): δ 7.96 (s, 2H), 3.66 (s, 2H), 2.92 (s, 4H), 1.69–1.62 (m, 4H), 1.44 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.7, 167.9, 162.0, 60.5, 53.5, 23.7, 22.4. APCI-MS: m/z 306.1 [M + H]+. HPLC retention time: 3.445 min (mobile phase 1). HPLC Purity: 95.3%.
2-(Morpholin-1-yl)-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (26).96
Prepared according to general procedure 2 using 2a (0.10 g, 0.39 mmol, 1 eq.), triethylamine (0.11 mL, 0.78 mmol, 2.0 eq.) and morpholine (0.068 g, 0.78 mmol, 2.0 eq.). Isolated as a white solid (0.056 g, 0.18 mmol, 47 %). 1H NMR (500 MHz, DMSO-d6): δ 3.56 – 3.53 (m, 4H), 3.19 (s, 2H), 2.46 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 173.0, 167.4, 160.9, 66.2, 62.9, 53.1. APCI-MS: m/z 308.1 [M+H]+. HPLC retention time: 3.441 min (mobile phase 1). HPLC Purity: 96.4%.
2-(4-Methylpiperazin-1-yl)-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)acetamide (27).96
Prepared according to general procedure 3 using 2a (0.05 g, 0.19 mmol, 1 eq.), triethylamine (0.050 mL, 0.39 mmol, 2.0 eq.) and 1-methylpiperazine (0.029 g, 0.29 mmol, 1.5 eq.). Isolated as a white solid (0.017 g, 0.052 mmol, 26 %). 1H NMR (500 MHz, DMSO-d6): δ 8.14 (s, 2H), 3.42 (s, 2H), 2.65 (brs, 4H), 2.52 (brs, 4H), 2.25 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 170.2, 163.8, 163.3, 60.3, 54.0, 51.9, 45.2. APCI-MS: m/z 321.1 [M+H]+. HPLC retention time: 3.358 min (mobile phase 1). HPLC Purity: 96.2%.
3-Morpholino-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)propanamide (28).
Prepared according to general procedure 3 using 2b (0.05 g, 0.18 mmol, 1.0 eq.), triethylamine (0.048 mL, 0.36 mmol, 2.0 eq.) and morpholine (0.032 g, 0.36 mmol, 2.0 eq.). Isolated as a white solid (0.049 g, 0.15 mmol, 82 %). 1H NMR (500 MHz, DMSO-d6): δ 8.28 (s, 2H), 3.54–3.46 (m, 4H), 2.71–2.61 (m, 4H), 2.36 (s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 171.7, 164.6, 161.5, 66.4, 53.8, 53.2, 32.8. APCI-MS: m/z 322.1 [M+H]+. HPLC retention time: 3.436 min (mobile phase 1). HPLC Purity: 99.3%.
General Procedure 3 for analogs 29 - 31. Described in detail for analog 29.
5-(ethylamino)-1,3,4-thiadiazole-2-sulfonamide (29).
Under argon atmosphere, acetic acid (0.02 ml) was added dropwise to the solution of 1 (0.11 g, 0.60 mmol) and acetaldehyde (40 μL, 0.90 mmol) in anhydrous CH2Cl2 (4 mL) and MeOH (4 mL) at room temperature. After 1 hour, sodium cyanoborohydride (0.057 g, 0.90 mmol) was added portionwise to the reaction mixture at 0 °C, then stirred at room temperature overnight. The reaction mixture was concentrated under vacuum and the crude product was purified by normal phase flash chromatography (0% MeOH/DCM ~10 % MeOH/DCM) to afford compound 29 (0.027 g, 0.13 mmol, 22 %) as white solid; 1H NMR (500 MHz, DMSO) δ 8.28 (s, 1H), 7.88 (s, 2H), 2.97 (q, J = 7.2 Hz, 2H), 1.01 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 172.2, 155.7, 38.2, 15.2. MS (APCI) m/z: 209.0 (M+1) +. HPLC retention time: 2.436 min (mobile phase 2). HPLC Purity: 96.8%.
5-((cyclohexylmethyl)amino)-1,3,4-thiadiazole-2-sulfonamide (30).
Prepared according to general procedure 3 using 1 (0.11 g, 0.60 mmol), cyclohexanecarbaldehyde (110 μl, 0.90 mmol) and sodium cyanoborohydride (0.057 g, 0.90 mmol) to afford compound 30 (0.023 g, 0.084 mmol, 14 %) as white solid. 1H NMR (500 MHz, DMSO) δ 8.36 (t, J = 5.8 Hz, 1H), 7.92 (s, 2H), 2.82 (t, J = 6.3 Hz, 2H), 1.64–1.71 (m, 4H), 1.58–1.63 (m, 1H), 1.44–1.36 (m, 1H), 1.20–1.09 (m, 3H), 0.91–0.80 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 172.1, 155.8, 49.3, 37.6, 30.3, 26.2, 25.6. MS (APCI) m/z: 277.0 (M+1) +. HPLC retention time: 11.295 min (mobile phase 1). HPLC Purity: 95.2%.
5-(benzylamino)-1,3,4-thiadiazole-2-sulfonamide (31).
Prepared according to general procedure 3 using 1 (0.11 g, 0.60 mmol), benzaldehyde (110 μl, 0.90 mmol) and sodium cyanoborohydride (0.057 g, 0.90 mmol) to afford compound 5 (0.013 g, yield 7.9% ) as white solid; 1H NMR (500 MHz, DMSO) δ 8.88 (s, 1H), 7.88 (s, 2H), 7.32–7.19 (m, 5H), 4.16 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 172.2, 155.7, 137.7, 128.7, 127.9, 127.6, 46.6. MS (APCI) m/z: 270.9 (M+1)+. HPLC retention time: 10.249 min (mobile phase 1). HPLC Purity: 95.5%.
N-(5-(Methylsulfonyl)-1,3,4-thiadiazol-2-yl)acetamide (33).
In a vial was added 32 (0.050 g, 1 eq., 0.28 mmol) followed by acetic anhydride (0.043 g, 1.5 eq., 0.42 mmol) in acetic acid (1 ml). The reaction mixture was stirred at 60 °C for 1 h. The solution was gradually cooled followed by the addition of water (10 ml). The suspension was cooled to 0 °C and the target compound was collected by filtration as a white solid (0.026 g, 0.12 mmol, 42%). 1H NMR (500 MHz, DMSO-d6): δ 13.26 (s, 1H), 3.52 (s, 3H), 2.27 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 170.0, 162.7, 162.6, 43.6, 22.7. HPLC retention time: 8.680 min (mobile phase 1). HPLC Purity: 99.8%.
N-(5-sulfamoylthiazol-2-yl)acetamide (34).
34 was purchased from a commercial vendor (Enamine, Cat # EN300–79477) and characterized prior to biological evaluation. 1H NMR (500 MHz, DMSO-d6): δ 12.45 (s, 1H), 7.81 (s, 1H), 7.68 (s, 2H), 2.19 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 169.3, 160.9, 140.4, 133.4, 22.4. APCI-MS+: m/z 222.0 [M + H]+. HPLC retention time: 7.874 min (mobile phase 1). HPLC Purity: 97.6%.
N-(4-sulfamoylphenyl)acetamide (35).
35 was purchased from a commercial vendor (Combi-Blocks, Cat # A59503) and characterized prior to biological evaluation. 1H NMR (500 MHz, DMSO-d6): δ 10.24 (s, 1H), 7.87 – 7.59 (m, 4H), 7.19 (s, 2H), 2.04 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 169.2, 142.5, 138.4, 127.0, 118.8, 24.4. APCI-MS: m/z 215.0 [M + H]+. HPLC retention time: 7.880 min (mobile phase 1). HPLC Purity: 97.5%.
Computational Modeling
Uniprot data mining and sequence alignments
To search for putative carbonic anhydrases in the genome of E. faecium we searched UniProt (https://www.uniprot.org)97 for the term “enterococcus faecium carbonic anhydrase”. We identified a carbonic anhydrase in the well-studied genome of E. faecium DO and chose to move forward with this particular species carbonic anhydrases. We found both an α- and γ-CA. We then used the “Align” tool in UniProt to align the newly found putative sequences from E. faecium with the sequences of previously reported α- and γ-CAs. The alignment files were loaded into ESPript 3.098 to generate the color coded sequence alignments.
Generation of homology models for Efα-CA and Efγ-CA
Homology models for Efα-CA and Efγ-CA were built based on the crystal structures of H. pylori (PDB ID 4YGF) and C. difficile (PDB ID 4MFG) retrieved from the Protein Data Bank,99 respectively. The sequences of the two proteins were collected from the Universal Protein Resource, UniProt. All sequence alignments were performed using the Prime STA sequence alignment method. Then, both homology models were built using Prime’s knowledge-based method in the Schrödinger suite (version 2020–1, Schrödinger, LLC: New York, NY, 2017), including all relevant ions and ligands.
Protein structure preparation and molecular docking
The Efα-CA and Efγ-CA homology models were then further preprocessed for docking using the Protein Preparation Wizard from the Schrödinger suite. Bond orders were assigned, hydrogen bonds were added, zero-order bonds to metals were formed, disulfide bonds were formed, and heteroatom states were generated between 7.0 ± 2.0 using Epik. H-bond assignments were performed at pH 7.4 using PROPKA. Then, all waters with fewer than 3 H-bonds to non-waters were removed. Finally, a restrained minimization using the OPLS3e forcefield was performed with heavy atoms converging to an RMSD of 0.3 Å.
To generate the docked structures of AZM, 22, and 26 with Efα-CA, Interactive Pose Prediction was used. A grid was generated from the workspace using AZM as the workspace reference ligand and the active site Zn2+ ion as an interaction constraint atom. After docking the reference ligand, 22 and 26 were docked. These structures were then used for the MD simulation. For Efγ-CA, a grid was generated using the Receptor Grid Generation application within Glide. The site was defined using the area denoted from FTMap (coordinates: X: 14.87, Y: −1.34, Z: 2.41) with the Ni2+ ion as an interaction constraint atom. AZM, 22, and 26 were prepared using LigPrep. Possible ionization states were generated at 7.0 ± 2.0 using Epik, metal binding states were included, and tautomers were generated.
Schrödinger’s Glide was used in extra precision (XP) to generate several poses per compound. Compounds were ranked based on the Glide-XP scoring function. The poses with the best Glide-XP score that also formed an interaction between the sulfonamide nitrogen and Ni2+ ion were visually inspected and selected for MD simulations.
Molecular dynamics simulation of analog binding
The top-scored docking poses were subjected to solvent-explicit, all-atom molecular dynamics simulation using the GPU-accelerated DESMOND software. TIP3P was selected as the solvent model, and the boundary conditions were defined as orthorhombic. The box size calculation method was defined as “buffer”, and the box was minimized. Ions and salts were excluded from within 5 Å of the ligand. Ions were added to neutralize each complex, and salt (NaCl) was added at a concentration of 0.15 M. Each molecular dynamic production run was carried out for 76 ns with 10 ps recording intervals. The NPT ensemble class was set to 300.0 K and the pressure was set to 1.01325 bar. All other options were kept at default values.
Biological Evaluation
Bacterial strains, media and chemicals
The enterococcal clinical isolates used in this study were obtained from Biodefense and Emerging Infections Research Resources Repository (BEI Resources) and the American Type Culture Collection (ATCC). All drugs used in this study were purchased from chemical vendors: linezolid (Chem-impex International, Wood Dale, IL), vancomycin hydrochloride (Gold Biotechnology, St. Louis, MO), acetazolamide (Sigma-Aldrich, St. Louis, MO). Trypticase soy broth (TSB), and trypticase soya agar (TSA) were purchased from Becton, Dickinson and Company (Cockeysville, MD). Phosphate buffered saline was purchased from Fisher Scientific (Waltham, MA). All synthesized compounds were prepared in a stock concentration of 10 mg/mL in DMSO.
Antibacterial activity of synthesized compounds against VRE strains
The minimum inhibitory concentrations (MICs) of the compounds and control drugs were determined using the broth microdilution method, according to guidelines outlined by the Clinical and Laboratory Standards Institute (CLSI100). Enterococci strains were grown aerobically overnight on trypticase soya agar plates at 37° C. Afterwards, a bacterial solution equivalent to 0.5 McFarland standard was prepared and diluted in trypticase soy broth (TSB) to achieve a bacterial concentration of about 5 × 105 CFU/mL and seeded in 96-well well plates. Compounds and control drugs were added and serially diluted along the plates. Plates were then, incubated aerobically at 37° C for 18–20 hours. MICs reported are the minimum concentration of the test agents that completely inhibited the visual growth of bacteria.
For the MIC results presented in Table 3 and Table S3, MICs were determined as described above for the results referred to as “normal” where the plates were incubated in the ambient air at 37 °C for 18 – 20 hours before recording MICs. For the “CO2” conditions, plates were incubated in presence of 5% CO2 at 37 °C for 18 – 20 hours before recording MICs.
Time-kill assay
A time kill analysis was conducted for acetazolamide, 22, 26, and the control antibiotic linezolid following a method described previously32. Briefly, an overnight culture of E. faecium ATCC 700221 was diluted in trypticase soy broth to reach a starting inoculum of 3.5 × 106 CFU/mL. Compounds and linezolid (all tested at 5× MIC, in triplicates), were incubated with the bacterial suspension for 24 hours. DMSO (the solvent for compounds and linezolid) served as a negative control. Samples were taken at 2h, 4h, 6h, 8h, 12h, and 24h post inoculation, diluted in PBS, and plated onto trypticase soya agar plates. Plates were incubated at 37 °C for 16 hours before counting colonies to determine viable CFU/mL.
Toxicity against human Caco-2 cell lines
To explore the tolerability of AZM analogs, the cytotoxicity profile of the most potent compounds was assessed against human colorectal adenocarcinoma (Caco-2) cells as described previously.101,102 Briefly, Caco-2 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), non-essential amino acids (1X), and penicillin-streptomycin at 37 °C with 5% CO2. The cells were incubated with different concentrations of AZM analogs in a 96-well plate at 37 °C with 5% CO2 in serum-free media for 2 hours. Control cells were treated with DMSO at a concentration equal to that in the compounds-treated samples. The assay reagent MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (Promega, Madison, WI, USA) was subsequently added and the plates were incubated for three hours. Absorbance readings (at OD490) were recorded using a kinetic microplate reader (Spectra MAX IX3, Molecular Device, CA, USA). The data are presented as percent cells viability after treatment with each compound relative to the viability in DMSO-treated control cells (average ± standard deviation). The 50% cytotoxic concentration (CC50) for each compound was determined and used to express the cytotoxicity pattern.
Aqueous Solubility
Assay were performed by Eurofins Panlabs (MO, USA) according to the following protocol from Lipinski et al.103 Aqueous solubility (μM) was determined by comparing the peak area of the principal peak in a calibration standard (200 μM) containing organic solvent (methanol/water, 60/40, v/v) with the peak area of the corresponding peak in a buffer sample. In addition, chromatographic purity (%) was defined as the peak area of the principal peak relative to the total integrated peak area in the HPLC chromatogram of the calibration standard.
Caco-2 permeability Assay
Assays and data analysis were performed by Eurofins Panlabs (MO, USA) according to the following protocol from Obach et al83. Calculations were carried out as follows:
Permeability
The apparent permeability coefficient (Papp) of the test compound was calculated using equation 1 below:
| Eq.1 |
where VR is the volume of the receiver chamber. CR,end is the concentration of the test compound in the receiver chamber at the end time point, Δt is the incubation time and A is the surface area of the cell monolayer. CD,mid is the calculated mid-point concentration of the test compound in the donor side, which is the mean value of the donor concentration at time 0 minute and the donor concentration at the end time point. CR,mid is the mid-point concentration of the test compound in the receiver side, which is one half of the receiver concentration at the end time point. Concentrations of the test compound were expressed as peak areas of the test compound.
Recovery of test compound from permeability assay
The recovery of the test compound was calculated as follows using equation 2:
| (2) |
where VD and VR are the volumes of the donor and receiver chambers, respectively. CD,end is the concentration of the test compound in the donor sample at the end time point. CR,end is the concentration of the test compound in the receiver sample at the end time point. CD0 is the concentration of the test compound in the donor sample at time zero. Concentrations of the test compound were expressed as peak areas of the test compound.
Human Liver Microsome Stability
Assays and data analysis were performed by Eurofins Panlabs (MO, USA) according to the protocol from Hidalgo et al.79
In vivo pharmacokinetics
Assays and data analysis were performed by Eurofins Panlabs (Taiwan). 24 mice (male, ICR strain) were dosed orally with 10 mg/kg 22 by oral gavage. Blood-plasma concentrations of molecules were analyzed at 8 time-points (10, 30, 60, 120, 240, 360, 480 and 1440 min). Each time point consisted of n = 3 mice. Samples were collected by cardiac puncture and 50 μL plasma was collected and mixed with 150 μL buffer containing 0.5 ng/μL of internal standard oxybutynin. Samples were diluted further with acetonitrile, vortexed and submitted for LC/MS/MS analysis. LC was run using an Agilent Poroshell 120 EC-C18 column 2.7μm (3.0 × 50mm) with Mobile Phase A: Acetonitrile/formic acid (0.2% v/v) and Mobile Phase B: water/formic acid (0.2% v/v). Samples were analyzed by multiple reaction monitoring and the plasma concentration was determined by comparison to a previously derived standard curve for the analog.
This study and the related standard operating procedures (SOPs) were reviewed and approved by Pharmacology Discovery Services Taiwan, Ltd. Institutional Animals Care and Use Committee (IACUC).
Supplementary Material
ACKNOWLEDGMENT
This work was funded by Purdue College of Pharmacy (D.P.F), Purdue Institute for Drug Discovery (D.P.F. and M.N.S), National Institute of Allergy and Infectious Diseases (NIAID) (1R01AI148523, M.N.S. and D.P.F.; 5R01AI134685, D.P.F.).
ABBREVIATIONS
- VRE
vancomycin-resistant enterococci
- SAR
Structure-activity relationship
- AZM
acetazolamide
- EZM
ethoxzolamide
- TLC
thin layer chromatography
- HPLC
high performance liquid chromatography
- MD
molecular dynamics
Footnotes
Supporting Information. The following items provided in the SI section include: 1) MICs of selected analogs against vancomycin-sensitive E. faecalis strains, 2) E. faecium ATCC 700221 reference strain in normal and CO2 conditions provided, 3) tabular Caco-2 cell viability, 4) in vitro ADMET data obtained from Eurofins Panlabs for 22 and 26, 5) tabular in vivo plasma concentrations for 22 upon oral administration, 6) homology model figures and files (.pdb format), 7) MD simulation reports, 8) structural files of representative MD timepoints (.pbd format), 9) videos of MD simulations for AZM, 22, 26 (.mp4 format), 10) NMR and MS spectra as well as HPLC chromatograms for each new analog synthesized, and 11) molecular formula strings for compounds are provided in .csv file. These files are available free of charge.
REFERENCES
- (1).Sievert DM; Ricks P; Edwards JR; Schneider A; Patel J; Srinivasan A; Kallen A; Limbago B; Fridkin S Antimicrobial-Resistant Pathogens Associated with Healthcare-Associated Infections Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect. Control Hosp. Epidemiol. 2013, 34, 1–14. [DOI] [PubMed] [Google Scholar]
- (2).Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2019; Atlanta, GA, 2019. [Google Scholar]
- (3).Stagliano D; Susi A; Adams D; Nyland C Abstracts of Papers, Open Forum Infectious Diseases Fall 2017, Oct 7, 201; Epidemiology, Associated Conditions, and Outcomes of Hospital Associated Vancomycin-Resistant Enterococcus Infections in the US Military Health Care System. Infectious Disease Society of America: Arlington, VA, 2017. [Google Scholar]
- (4).Rossolini GM; Arena F; Pecile P; Pollini S Update on the Antibiotic Resistance Crisis. Curr. Opin. Pharmacol. 2014, 18, 56–60. [DOI] [PubMed] [Google Scholar]
- (5).Britt NS; Potter EM; Patel N; Steed ME Effect of Continuous and Sequential Therapy among Veterans Receiving Daptomycin or Linezolid for Vancomycin-Resistant Enterococcus Faecium Bacteremia. Antimicrob. Agents Chemother. 2017, 61, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Watkins RR; Lemonovich TL; File TM Jr An Evidence-Based Review of Linezolid for the Treatment of Methicillin-Resistant Staphylococcus Aureus (MRSA): Place in Therapy. Core Evid. 2012, 7, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dhanda G; Sarkar P; Samaddar S; Haldar J Battle Against Vancomycin-Resistant Bacteria: Recent Developments in Chemical Strategies. J. Med. Chem. 2019, 62, 3184–3205. [DOI] [PubMed] [Google Scholar]
- (8).van Harten RM; Willems RJL; Martin NI; Hendrickx APA Multidrug-Resistant Enterococcal Infections: New Compounds, Novel Antimicrobial Therapies? Trends Microbiol. 2017, 25 (6), 467–479. [DOI] [PubMed] [Google Scholar]
- (9).Leavis HL; Bonten MJM; Willems RJL Identification of High-Risk Enterococcal Clonal Complexes: Global Dispersion and Antibiotic Resistance. Curr. Opin. Microbiol. 2006, 9 (5), 454–460. [DOI] [PubMed] [Google Scholar]
- (10).Munoz-Price LS; Lolans K; Quinn JP Emergence of Resistance to Daptomycin during Treatment of Vancomycin-Resistant Enterococcus Faecalis Infection. Clin. Infect. Dis. 2005, 41 (4), 565–566. [DOI] [PubMed] [Google Scholar]
- (11).Arias CA; Murray BE The Rise of the Enterococcus: Beyond Vancomycin Resistance. Nat. Rev. Microbiol. 2012, 10 (4), 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hayakawa K; Marchaim D; Pogue JM; Ho K; Parveen S; Nanjireddy P; Sunkara B; Singla M; Jagadeesh KK; Moshos JA Predictors and Outcomes of Linezolid-Resistant Vancomycin-Resistant Enterococcus: A Case-Case-Control Study. Am. J. Infect. Control 2012, 40, e261–e263. [DOI] [PubMed] [Google Scholar]
- (13).Dobbs TE; Patel M; Waites KB; Moser SA; Stamm AM; Hoesley CJ Nosocomial Spread of Enterococcus Faecium Resistant to Vancomycin and Linezolid in a Tertiary Care Medical Center. J. Clin. Microbiol. 2006, 44 (9), 3368–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Eckburg PB; Bik EM; Bernstein CN; Purdom E; Dethlefsen L; Sargent M; Gill SR; Nelson KE; Relman DA Diversity of the Human Intestinal Microbial Flora. Science (80-. ). 2005, 308 (5728), 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schloissnig S; Arumugam M; Sunagawa S; Mitreva M; Tap J; Zhu A; Waller A; Mende DR; Kultima JR; Martin J; Kota K; Sunyaev SR; Weinstock GM; Bork P Genomic Variation Landscape of the Human Gut Microbiome. Nature 2013, 493 (7430), 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ajao AO; Harris AD; Roghmann M; Johnson JK Systematic Review of Measurement and Adjustment for Colonization Pressure in Studies of Methicillin-Resistant Staphylococcus Aureus, Vancomycin-Resistant Enterococci, and Clostridium Difficile Acquisition. Infect. Control Hosp. Epidemiol. 2011, 3 (5), 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Donskey CJ; Chowdhry TK; Hecker MT; Hoyen CK; Hanrahan JA; Hujer AM; Hutton-Thomas RA; Whalen CC; Bonomo RA; Rice LB Effect of Antibiotic Therapy on the Density of Vancomycin-Resistant Enterococci in the Stool of Colonized Patients. N. Engl. J. Med. 2000, 343 (26), 1925–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ubeda C; Taur Y; Jenq RR; Equinda MJ; Son T; Samstein M; Viale A; Socci ND; Van Den Brink, M. R. M.; Kamboj M; Pamer EG Vancomycin-Resistant Enterococcus Domination of Intestinal Microbiota Is Enables by Antibiotic Treatment in Mice and Precedes Bloodstream Invasion in Humans. J. Clin. Invest. 2010, 120, 4332–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Brandl K; Plitas G; Mihu CN; Ubeda C; Jia T; Fleisher M; Schnabl B; DeMatteo RP; Pamer EG Vancomycin-Resistant Enterococci Exploit Antibiotic-Induced Innate Immune Deficits. Nature 2008, 455, 804–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Russell DL; Flood A; Zaroda TE; Acosta C; Riley MMS; Busuttil RW; Pegues DA Outcomes of Colonization with MRSA and VRE among Liver Transplant Candidates and Recipients. Am. J. Transplant. 2008, 8, 1737–1743. [DOI] [PubMed] [Google Scholar]
- (21).Wong MT; Kauffman CA; Standiford HC; Linden P; Fort G; Fuchs HJ; Porter SB; Wenzel RP; Group RVCS Effective Suppression of Vancomycin-Resistant Enterococcus Species in Asymptomatic Gastrointestinal Carriers by a Novel Glycolipodepsipeptide, Ramoplanin. Clin. Infect. Dis. 2001, 33, 1476–1482. [DOI] [PubMed] [Google Scholar]
- (22).Ziakas PD; Pliakos EE; Zervou FN; Knoll BM; Rice LB; Mylonakis E MRSA and VRE Colonization in Solid Organ Transplantation: A Meta-analysis of Published Studies. Am. J. Transplant. 2014, 14, 1887–1894. [DOI] [PubMed] [Google Scholar]
- (23).Zitvogel L; Kroemer G Immunostimulatory Gut Bacteria. Science 2019, 366, 1077–1078. [DOI] [PubMed] [Google Scholar]
- (24).Stein-Thoeringer CK; Nichols KB; Lazrak A; Docampo MD; Slingerland AE; Slingerland JB; Clurman AG; Armijo G; Gomes ALC; Shono Y; Staffas A; Burgos da Silva M; Devlin SM; Markey KA; Bajic D; Pinedo R; Tsakmaklis A; Littman ER; Pastore A; Taur Y; Monette S; Arcila ME; Pickard AJ; Maloy M; Wright RJ; Amoretti LA; Fontana E; Pham D; Jamal MA; Weber D; Sung AD; Hashimoto D; Scheid C; Xavier JB; Messina JA; Romero K; Lew M; Bush A; Bohannon L; Hayasaka K; Hasegawa Y; Verheschild MJGT; Cross JR; Ponce DM; Perales MA; Giralt SA; Jenq RR; Teshima T; Holler E; Chao NJ; Pamer EG; Peled JU; van den Brink MRM Lactose Drives Enterococcus Expansion to Promote Graft-versus-Host Disease. Science. 2019, 366, 1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Manfredo Vieira S; Hiltensperger M; Kumar V; Zegarra-Ruiz D; Dehner C; Khan N; Costa FRC; Tiniakou E; Greiling T; Ruff W; Barbieri A; Kriegel C; Mehta SS; Knight JR; Jain D; Goodman AL; Kriegel MA Translocation of a Gut Pathobiont Drives Autoimmunity in Mice and Humans. Science. 2018, 359, 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kauffman CA Therapeutic and Preventative Options for the Management of Vancomycin-Resistant Enterococcal Infections. J. Antimicrob. Chemother. 2003, 51, 23–30. [DOI] [PubMed] [Google Scholar]
- (27).Stiefel U; Pultz NJ; Helfand MS; Donskey CJ Efficacy of Oral Ramoplanin for Inhibition of Intestinal Colonization by Vancomycin-Resistant Enterococci in Mice. Antimicrob. Agents Chemother. 2004, 48, 2144–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Montecalvo MA; Horowitz H; Wormser GP; Seiter K; Carbonaro CA Effect of Novobiocin-Containing Antimicrobial Regimens on Infection and Colonization with Vancomycin-Resistant Enterococcus Faecium. Antimicrob. Agents Chemother. 1995, 39 (3), 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cheng VCC; Chen JHK; Tai JWM; Wong SCY; Poon RWS; Hung IFN; To KKW; Chan JFW; Ho P-L; Lo C-M Decolonization of Gastrointestinal Carriage of Vancomycin-Resistant Enterococcus Faecium: Case Series and Review of Literature. BMC Infect. Dis. 2014, 14, 514, DOI: 10.1186/1471-2334-14-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).AbdelKhalek A; Abutaleb NS; Mohammad H; Seleem MN Repurposing Ebselen for Decolonization of Vancomycin-Resistant Enterococci (VRE). PLoS One 2018, 13, e0199710, DOI: 10.1371/journal.pone.0199710.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).AbdelKhalek A; Abutaleb NS; Elmagarmid KA; Seleem MN Repurposing Auranofin as an Intestinal Decolonizing Agent for Vancomycin-Resistant Enterococci. Sci. Rep. 2018, 8, 8353, DOI: 10.1038/s41598-018-26674-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mohammad H; AbdelKhalek A; Abutaleb NS; Seleem MN Repurposing Niclosamide for Intestinal Decolonization of Vancomycin-Resistant Enterococci. Int. J. Antimicrob. Agents 2018, 51 (6), 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Brown D Antibiotic Resistance Breakers: Can Repurposed Drugs Fill the Antibiotic Discovery Void? Nat. Rev. Drug Discov. 2015, 14, 821–832. [DOI] [PubMed] [Google Scholar]
- (34).Oprea TI; Bauman JE; Bologa CG; Buranda T; Chigaev A; Edwards BS; Jarvik JW; Gresham HD; Haynes MK; Hjelle B; Hromas R; Hudson L; Mackenzie DA; Muller CY; Reed JC; Simons PC; Smagley Y; Strouse J; Surviladze Z; Thompson T; Ursu O; Waller A; Wandinger-Ness A; Winter SS; Wu Y; Young SM; Larson RS; Willman C; Sklar LA Drug Repurposing from an Academic Perspective. Drug Discov. Today. Ther. Strateg. 2011, 8, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ashburn TT; Thor KB Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [DOI] [PubMed] [Google Scholar]
- (36).Younis W; AbdelKhalek A; S Mayhoub A; N Seleem M In Vitro Screening of an FDA-Approved Library Against ESKAPE Pathogens. Curr. Pharm. Des. 2017, 23, 2147–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Van Berkel MA; Elefritz JL Evaluating Off-Label Uses of Acetazolamide. Bull. Am. Soc. Hosp. Pharm. 2018, 75, 524–531. [DOI] [PubMed] [Google Scholar]
- (38).Lim L; Foldvary N; Mascha E; Lee J Acetazolamide in Women with Catamenial Epilepsy. Epilepsia 2001, 42, 746–749. [DOI] [PubMed] [Google Scholar]
- (39).Patsalos PN; St. Louis EK The Epilepsy Prescriber’s Guide to Antiepileptic Drugs; Cambridge University Press, 2018; pp 1–10. [Google Scholar]
- (40).World Health Organization. Model List of Essential Medicines, 21st list, 2019, Geneva, Switzerland, 2019; DOI: 10.1016/S1473-3099(14)70780-7. [DOI] [Google Scholar]
- (41).Dollery CT; Boobis AR Therapeutic Drugs, 2nd Edition; Churchill Livingstone, London, United Kingdom, 1999, [Google Scholar]
- (42).Supuran CT Carbonic Anhydrases: Novel Therapeutic Applications for Inhibitors and Activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [DOI] [PubMed] [Google Scholar]
- (43).Gomolin H; Chapron J Elucidating Acetazolamide Renal Clearance. J. Clin. Pharmacol. 1992, 32, 1028–1032. [DOI] [PubMed] [Google Scholar]
- (44).Ritschel WA; Paulos C; Arancibia A; Agrawal MA; Wetzelsberger KM; Lücker PW Pharmacokinetics of Acetazolamide in Healthy Volunteers after Short-and Long-term Exposure to High Altitude. J. Clin. Pharmacol. 1998, 38, 533–539. [DOI] [PubMed] [Google Scholar]
- (45).Granero GE; Longhi MR; Becker C; Junginger HE; Kopp S; Midha KK; Shah VP Biowaiver Monographs for Immediate Release Solid Oral Dosage Forms : Acetazolamide. J. Pharm. Sci. 2008, 97, 3691–3699. [DOI] [PubMed] [Google Scholar]
- (46).Crowe A; Teoh Y; Crowe A; Teoh Y Limited P -Glycoprotein Mediated Efflux for Anti- Epileptic Drugs. J. Drug Target. 2008, 14 (5), 291–300. [DOI] [PubMed] [Google Scholar]
- (47).Woodbury DM Other Antiepileptic Drugs: Sulfonamide and Derivatives: Acetazolamide. Antiepileptic Drug 1982, 771–789. [Google Scholar]
- (48).Maren TH; Mayer E; Wadsworth BC Carbonic Anhydrase Inhibition. I. The Pharmacology of Diamox 2-Acetylamino-1, 3, 4-Thiadiazole-5-Sulfonamide. Bull. Johns Hopkins Hosp. 1954, 95, 199–243. [PubMed] [Google Scholar]
- (49).Reiss WG; Soles K Acetazolamide in the Treatment of Seizures. Ann. Pharmacother. 1996, 30, 514–519. [DOI] [PubMed] [Google Scholar]
- (50).Volpe DA; Faustino PJ; Ciavarella AB; Asafu-Adjaye EB; Ellison CD; Yu LX; Hussain AS Classification of Drug Permeability with a Caco-2 Cell Monolayer Assay. Clin. Res. Regul. Aff. 2007, 24, 39–47. [Google Scholar]
- (51).Supuran CT Bacterial Carbonic Anhydrases as Drug Targets: Toward Novel Antibiotics? Front. Pharmacol. 2011, 2, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Capasso C; Supuran CT Bacterial, Fungal and Protozoan Carbonic Anhydrases as Drug Targets. Expert Opin. Ther. Targets 2015, 19, 1689–1704. [DOI] [PubMed] [Google Scholar]
- (53).Modak JK; Liu YC; Supuran CT; Roujeinikova A Structure − Activity Relationship for Sulfonamide Inhibition of Helicobacter Pylori α - Carbonic Anhydrase. J. Med. Chem. 2016, 59, 11098–11109. [DOI] [PubMed] [Google Scholar]
- (54).Nishimori I; Minakuchi T; Morimoto K; Sano S; Onishi S; Takeuchi H; Vullo D Carbonic Anhydrase Inhibitors : DNA Cloning and Inhibition Studies of the r -Carbonic Anhydrase from Helicobacter Pylori, A New Target for Developing Sulfonamide and Sulfamate. J. Med. Chem. 2006, 49, 2117–2126. [DOI] [PubMed] [Google Scholar]
- (55).Takeuchi H; Supuran C; Onishi S; Nishimori I The Alpha and Beta Classes Carbonic Anhydrases from Helicobacter Pylori as Novel Drug Targets. Curr. Pharm. Des. 2008, 14, 622–630. [DOI] [PubMed] [Google Scholar]
- (56).Del S; Vullo D; Di P; Osman SM; Alothman Z; Donald WA; Supuran CT; Capasso C Sulfonamide Inhibition Profile of the c -Carbonic Anhydrase Identified in the Genome of the Pathogenic Bacterium Burkholderia Pseudomallei the Etiological Agent Responsible of Melioidosis. Bioorg. Med. Chem. Lett. 2017, 27, 490–495. [DOI] [PubMed] [Google Scholar]
- (57).Del Prete S; Vullo D; De Luca V; Carginale V; di Fonzo P; Osman SM; AlOthman Z; Supuran CT; Capasso C Anion Inhibition Profiles of α-, β- and γ-Carbonic Anhydrases from the Pathogenic Bacterium Vibrio Cholerae. Bioorganic Med. Chem. 2016, 24, 3413–3417. [DOI] [PubMed] [Google Scholar]
- (58).Del Prete S; De Luca V; Scozzafava A; Carginale V; Supuran CT; Capasso C Biochemical Properties of a New α-Carbonic Anhydrase from the Human Pathogenic Bacterium, Vibrio Cholerae. J. Enzyme Inhib. Med. Chem. 2014, 29, 23–27. [DOI] [PubMed] [Google Scholar]
- (59).Nafi BM; Miles RJ; Butler LO; Carter ND; Kelly C; Jeffery S Expression of Carbonic Anhydrase in Neisseriae and Other Heterotrophic Bacteria. J. Med. Microbiol. 1990, 32, 1–7. [DOI] [PubMed] [Google Scholar]
- (60).Smith KS; Jakubzick C; Whittam TS; Ferry JG Carbonic Anhydrase Is an Ancient Enzyme Widespread in Prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 15184–15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Capasso C; Supuran CT An Overview of the Alpha-, Beta- and Gamma-Carbonic Anhydrases from Bacteria: Can Bacterial Carbonic Anhydrases Shed New Light on Evolution of Bacteria? J. Enzyme Inhib. Med. Chem. 2015, 30, 325–332. [DOI] [PubMed] [Google Scholar]
- (62).Klijn C; Durinck S; Stawiski EW; Haverty PM; Jiang Z; Liu H; Degenhardt J; Mayba O; Gnad F; Liu J A Comprehensive Transcriptional Portrait of Human Cancer Cell Lines. Nat. Biotechnol. 2015, 33, 306–312. [DOI] [PubMed] [Google Scholar]
- (63).Di Fiore A; Monti SM; Hilvo M; Parkkila S; Romano V; Scaloni A; Pedone C; Scozzafava A; Supuran CT; De Simone G Crystal Structure of Human Carbonic Anhydrase XIII and Its Complex with the Inhibitor Acetazolamide. Proteins Struct. Funct. Bioinforma. 2009, 74 (1), 164–175. [DOI] [PubMed] [Google Scholar]
- (64).Sanders E; Maren TH Inhibition of Carbonic Anhydrase in Neisseria: Effects on Enzyme Activity and Growth. Mol. Pharmacol. 1967, 3, 204–215. [PubMed] [Google Scholar]
- (65).Bostic GD; Perri MB; Thal LA; Zervos MJ Comparative in Vitro and Bactericidal Activity of Oxazolidinone Antibiotics against Multidrug-Resistant Enterococci. Diagn. Microbiol. Infect. Dis. 1998, 30, 109–112. [DOI] [PubMed] [Google Scholar]
- (66).Kisker C; Schindelin H; Alber BE; Ferry JG; Rees DC A Left-hand Beta-helix Revealed by the Crystal Structure of a Carbonic Anhydrase from the Archaeon Methanosarcina Thermophila. EMBO J. 1996, 15, 2323–2330. [PMC free article] [PubMed] [Google Scholar]
- (67).Park HM; Park JH; Choi JW; Lee J; Kim BY; Jung CH; Kim JS Structures of the Gamma-Class Carbonic Anhydrase Homologue YrdA Suggest a Possible Allosteric Switch. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 920–926. [DOI] [PubMed] [Google Scholar]
- (68).Alber BE; Colangelo CM; Dong J; Stålhandske CMV; Baird TT; Tu C; Fierke CA; Silverman DN; Scott RA; Ferry JG Kinetic and Spectroscopic Characterization of the Gamma-Carbonic Anhydrase from the Methanoarchaeon Methanosarcina Thermophila. Biochemistry 1999, 38, 13119–13128. [DOI] [PubMed] [Google Scholar]
- (69).Del S; Vullo D; Osman SM; Alothman Z; Supuran CT; Capasso C Sulfonamide Inhibition Profiles of the b-Carbonic Anhydrase from the Pathogenic Bacterium Francisella Tularensis Responsible of the Febrile Illness Tularemia. Bioorg. Med. Chem. 2017, 25, 3555–3561. [DOI] [PubMed] [Google Scholar]
- (70).Sippel KH; Robbins AH; Domsic J; Genis C; Agbandje-Mckenna M; McKenna R High-Resolution Structure of Human Carbonic Anhydrase II Complexed with Acetazolamide Reveals Insights into Inhibitor Drug Design. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 992–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Jacobson MP; Pincus DL; Rapp CS; Day TJF; Honig B; Shaw DE; Friesner RA A Hierarchical Approach to All-atom Protein Loop Prediction. Proteins Struct. Funct. Bioinforma. 2004, 55, 351–367. [DOI] [PubMed] [Google Scholar]
- (72).Jacobson MP; Friesner RA; Xiang Z; Honig B On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [DOI] [PubMed] [Google Scholar]
- (73).Ngan CH; Hall DR; Zerbe B; Grove LE; Kozakov D; Vajda S FtSite: High Accuracy Detection of Ligand Binding Sites on Unbound Protein Structures. Bioinformatics 2012, 28, 286–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Jain A; Whitesides GM; Alexander RS; Christianson DW Identification of Two Hydrophobic Patches in the Active-Site Cavity of Human Carbonic Anhydrase II by Solution-Phase and Solid-State Studies and Their Use in the Development of Tight-Binding Inhibitors. J. Med. Chem. 1994, 37, 2100–2105. [DOI] [PubMed] [Google Scholar]
- (75).Tanpure RP; Ren B; Peat TS; Bornaghi LF; Vullo D; Supuran CT; Poulsen SA Carbonic Anhydrase Inhibitors with Dual-Tail Moieties to Match the Hydrophobic and Hydrophilic Halves of the Carbonic Anhydrase Active Site. J. Med. Chem. 2015, 58, 1494–1501. [DOI] [PubMed] [Google Scholar]
- (76).Glockner S; Ngo K; Wagner B; Heine A; Klebe G The Influence of Varying Fluorination Patterns on the Thermodynamics and Kinetics of Benzenesulfonamide Binding to Human Carbonic. Biomolecules 2020, 10, 509, DOI: 10.3390/biom10040509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Vullo D; Del Prete S; Osman SM; De Luca V; Scozzafava A; Alothman Z; Supuran CT; Capasso C Sulfonamide Inhibition Studies of the γ-Carbonic Anhydrase from the Oral Pathogen Porphyromonas Gingivalis. Bioorganic Med. Chem. Lett. 2014, 24, 240–244. [DOI] [PubMed] [Google Scholar]
- (78).Zimmerman S; Innocenti A; Casini A; Ferry JG; Scozzafava A; Supuran CT Carbonic Anhydrase Inhibitors. Inhibition of the Prokariotic Beta and Gamma-Class Enzymes from Archaea with Sulfonamides. Bioorg. Med. Chem. Lett. 2004, 14, 6001–6006. [DOI] [PubMed] [Google Scholar]
- (79).Hidalgo IJ; Raub TJ; Borchardt RT Characterization of the Human Colon Carcinoma Cell Line (Caco-2) as a Model System for Intestinal Epithelial Permeability. Gastroenterology 1989, 96, 736–749. [PubMed] [Google Scholar]
- (80).Pires DEV; Blundell TL; Ascher DB PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Yano I; Tanihara IH Pharmacokinetics and Pharmacodynamics of Acetazolamide in Patients with Transient Intraocular Pressure Elevation. Eur. J. Clin. Pharmacol. 1998, 54, 63–68. [DOI] [PubMed] [Google Scholar]
- (82).Wistrand PJ The Use of Carbonic Anhydrase Inhibitors in Ophthalmology And. Ann. New York Acad. Sci. 1984, 429, 609–619. [DOI] [PubMed] [Google Scholar]
- (83).Obach RS; Baxter JG; Liston TE; Silber BM; Jones BC; Macintyre F; Rance DJ; Wastall P The Prediction of Human Pharmacokinetic Parameters from Preclinical and in Vitro Metabolism Data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [PubMed] [Google Scholar]
- (84).Chen Y-C Beware of Docking! Trends Pharmacol. Sci. 2014, 36, 78–95. [DOI] [PubMed] [Google Scholar]
- (85).The Merck Index, 13th Edition. O’Neil MJ; Smith A; Heckelman PE, Eds. Merck & Co., Inc: Whitehouse Station, NJ, 1996, pp 11. [Google Scholar]
- (86).Krishnamurthy VM; Bohall BR; Kim C; Moustakas DT; Christianson DW; Whitesides GM Thermodynamic Parameters for the Association of Fluorinated Benzenesulfonamides with Bovine Carbonic Anhydrase II. Chem. Asian J. 2007, 2 (1), 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Glöckner S; Ngo K; Sager CP; Hüfner-Wulsdorf T; Heine A; Klebe G Conformational Changes in Alkyl Chains Determine the Thermodynamic and Kinetic Binding Profiles of Carbonic Anhydrase Inhibitors. ACS Chem. Biol. 2020, 15, 675–685. [DOI] [PubMed] [Google Scholar]
- (88).Gaspari R; Rechlin C; Heine A; Bottegoni G; Rocchia W; Schwarz D; Bomke J; Gerber H-D; Klebe G; Cavalli A Kinetic and Structural Insights into the Mechanism of Binding of Sulfonamides to Human Carbonic Anhydrase by Computational and Experimental Studies. J. Med. Chem. 2016, 59 (9), 4245–4256. [DOI] [PubMed] [Google Scholar]
- (89).Lakshminarayana SB; Huat TB; Ho PC; Manjunatha UH; Dartois V; Dick T; Rao SPS Comprehensive Physicochemical, Pharmacokinetic and Activity Profiling of Anti-TB Agents. J. Antimicrob. Chemother. 2015, 70, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Wallace SM; Riegelman S Uptake of Acetazolamide by Human Erythrocytes in Vitro. J. Pharm. Sci. 1977, 66, 729–731. [DOI] [PubMed] [Google Scholar]
- (91).Wallace SM; Shah VP; Riegelman S GLC Analysis of Acetazolamide in Blood, Plasma, and Saliva Following Oral Administration to Normal Subjects. J. Pharm. Sci. 1977, 66, 527–530. [DOI] [PubMed] [Google Scholar]
- (92).Schüttelkopf AW; Gros L; Blair DE; Frearson JA; van Aalten DMF; Gilbert IH Acetazolamide-Based Fungal Chitinase Inhibitors. Bioorg. Med. Chem. 2010, 18, 8334–8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Masereel B; Rolin S; Abbate F; Scozzafava A; Supuran CT Carbonic Anhydrase Inhibitors: Anticonvulsant Sulfonamides Incorporating Valproyl and Other Lipophilic Moieties. J. Med. Chem. 2002, 45, 312–320. [DOI] [PubMed] [Google Scholar]
- (94).Vaughan J J; Eichler J; Anderson G Heterocyclic Sulfonamides as Carbonic Anhydrase Inhibitors. 2-Acylamido-and 2-Sulfonamido-1, 3, 4-Thiadiazole-5-Sulfonamides. J. Org. Chem. 1956, 21, 700–701. [Google Scholar]
- (95).Arslan O; Küfrevioğlu ÖI; Nalbantoğlu B Synthesis and Investigation of Inhibition Effects of New Carbonic Anhydrase Inhibitors. Bioorg. Med. Chem. 1997, 5, 515–518. [DOI] [PubMed] [Google Scholar]
- (96).Turkmen H; Durgun M; Yilmaztekin S; Emul M; Innocenti A; Vullo D; Scozzafava A; Supuran CT Carbonic Anhydrase Inhibitors. Novel Sulfanilamide/Acetazolamide Derivatives Obtained by the Tail Approach and Their Interaction with the Cytosolic Isozymes I and II, and the Tumor-Associated Isozyme IX. Bioorg. Med. Chem. Lett. 2005, 15, 367–372. [DOI] [PubMed] [Google Scholar]
- (97).UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Robert X; Gouet P Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42, 320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Berman HM; Henrick K; Nakamura H Announcing the Worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980–981. [DOI] [PubMed] [Google Scholar]
- (100).Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard M7-A9, CLSI, Pennsylvania, PA, 2012. [Google Scholar]
- (101).Kotb A; Abutaleb NS; Seleem MA; Hagras M; Mohammad H; Bayoumi A; Ghiaty A; Seleem MN; Mayhoub AS Phenylthiazoles with Tert-Butyl Side Chain: Metabolically Stable with Anti-Biofilm Activity. Eur. J. Med. Chem. 2018, 151, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Hammad A; Abutaleb NS; Elsebaei MM; Norvil AB; Alswah M; Ali AO; Abdel-Aleem JA; Alattar A; Bayoumi SA; Gowher H From Phenylthiazoles to Phenylpyrazoles: Broadening the Antibacterial Spectrum toward Carbapenem-Resistant Bacteria. J. Med. Chem. 2019, 62, 7998–8010. [DOI] [PubMed] [Google Scholar]
- (103).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.