

Abstract
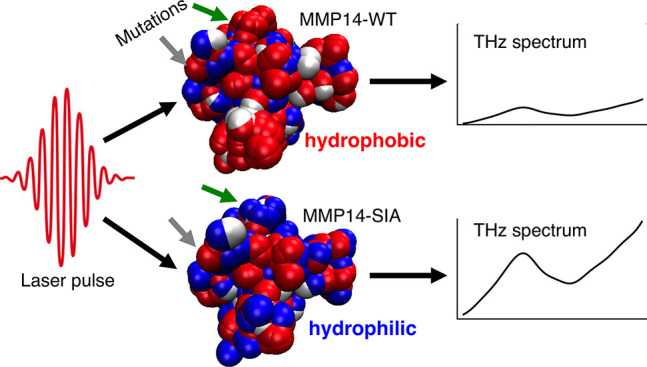
Although it is well-known that limited local mutations of enzymes, such as matrix metalloproteinases (MMPs), may change enzyme activity by orders of magnitude as well as its stability, the completely rational design of proteins is still challenging. These local changes alter the electrostatic potential and thus local electrostatic fields, which impacts the dynamics of water molecules close the protein surface. Here we show by a combined computational design, experimental, and molecular dynamics (MD) study that local mutations have not only a local but also a global effect on the solvent: In the specific case of the matrix metalloprotease MMP14, we found that the nature of local mutations, coupled with surface morphology, have the ability to influence large patches of the water hydrogen-bonding network at the protein surface, which is correlated with stability. The solvent contribution can be experimentally probed via terahertz (THz) spectroscopy, thus opening the door to the exciting perspective of rational protein design in which a systematic tuning of hydration water properties allows manipulation of protein stability and enzymatic activity.
Keywords: matrix metalloproteinase, THz spectroscopy, molecular dynamics, solvation science, local thermodynamics, rational design
Introduction
MMPs (matrix metalloproteinases) and ADAMs (a disintegrin and metalloproteinases) are a group of tightly regulated proteases which are of biological significance in the extracellular matrix (ECM) and consist of as many as 37 catalytically active proteins.1 They are multidomain Zn2+-dependent endopeptidases with an N-terminal prodomain (engaged in latency maintenance), a ∼130–260 residue globular catalytic domain, and additional functional and/or regulatory C-terminal domains. All proteolytically active MMPs/ADAMs share a HExxHxxGxxH/D + Zn2+ binding motif in their catalytic domain, in which the histidine residues coordinate the catalytic Zn2+,2,3 as well as a “Met turn” motif that forms a loop and is crucial for the structure of the catalytic cleft.4,5 Binding of a substrate to the catalytic site is coordinated by the structure of the catalytic cleft and by the so-called “S1’ pocket”. This pocket is hydrophobic in nature, variable in depth, and therefore one of the specificity determining factors among various family members.6
Each protease uniquely influences the properties of its substrate within the ECM and is associated with tissue-specific processes.3,7 MMP upregulation has been connected to several types of cancers as well as inflammatory diseases.8−10 As MMPs have a conserved catalytic site, the development of MMP inhibitors (MMPIs) that exhibit sufficient specificity and selectivity to target one member of the family is of fundamental interest.11 One solution is the development of protein engineered (PE) inhibitors, such as antibodies or TIMPs (tissue inhibitors of MMPs), that are based on MMP protein structures and target not only the catalytic site but nearby surface exposed residues which vary among family members.12 To date, some PE MMPIs have been successful in vitro or in vivo,13,14 indicating that there is potential to use such inhibitors as drugs. However, there remains much to be understood about the complex biochemical mechanism of their catalytic protein scaffolds. For instance, we still lack a clear understanding of the role of the solvent in determining their stability and selectivity toward their rich substrate portfolios.
In recent years, it has become more apparent that dynamical properties of proteins such as folding and substrate binding are influenced by their interaction with the solvent.15−19 Considering local thermodynamics (ΔG = ΔH – TΔS), some of the entropic contributions are governed by the solvent; for instance, the release of water from the protein surface into the bulk is entropically favorable.20,21 While the solvent contribution to ΔS is significant, predicting the local impact of solvent energy changes on the global thermodynamics remains a challenge, since protein surfaces are nonplanar and have a nonuniform distribution of polar and/or charged groups. Estimation of solvent free energy changes has been accomplished with the transfer model, where free energy changes in the peptide backbone and residue side chains have been used to predict the contribution of the solvent to folding/unfolding processes. Whereas the model is in general successful, a limitation of this model stems from the assumption of group additivity, which may not be relevant in instances in which electrostatics have an important effect, such as in the case of charge screening from salt solutes or clustering of charged groups on the protein surface.22,23 Recent theoretical investigations have concluded that the spatial arrangement of surface chemical and geometric features can tune local water properties in a surprisingly complex way.24−29 Here, we aim to explore “solvent tuning” and demonstrate that THz spectroscopy allows one to probe these subtle changes. Exchanging a hydrophobic group with a hydrophilic one in the middle of a hydrophobic patch has been shown to reduce the water density fluctuations, and hence local hydrophobicity in a very extended portion of the patch.30,31 Underlying this complexity is the competition in directional interactions (i.e., hydrogen bonds) with neighboring surface groups as well as with other water molecules.26 The subtle balance between water–surface interactions and water–water correlations can be tuned by varying specific parameters, such as the dimension of hydrophobic/hydrophilic patches and their spatial distribution,24,26,32 surface polarity and surface charge,33,34 surface morphology,32 and by adsorbed ions.28 Along with a suitable experimental probe, this is proposed to open the way for the design of enzymes with optimized dynamical and thermodynamical properties.25−28,32−34
Here we present the results of a combined study using terahertz (THz) spectroscopy, computational design, and classical molecular dynamics (MD) simulations to probe the change in hydration properties of the catalytic domains of human wild type MMP14 (MMP14-WT) and MMP9 (MMP9-WT) as well as the catalytic domain of the MMP14 protein which was computationally designed using the PROSS stability-design method (stabilized inactive mutant MMP14-SIA, see the Supporting Information (SI) for rational design details).35 These three MMP variants are structurally homologous but have different relative surface electrostatic potentials. Comparison of these three MMPs gives insight into the relationship of the surface properties to the THz response of the solvent, and in the specific case of MMP14 and its stabilized inactive mutant provides direct correlation of the THz response to the stability. Our results reveal significant differences in the THz response of MMP9, MMP14, and the MMP14 mutant, indicative of pronounced changes in the solvation shell. Notably, the changes in THz absorption increases with an increasingly positive surface electrostatic potential. These changes can be correlated to an increase in the hydrophilic nature of the protein surface, and in the specific case of MMP14-WT and MMP14-SIA the distribution of these hydrophilic groups in conjunction with the protein surface morphology act cooperatively to impact large patches of hydration water, revealing that surface mutations significantly impact the extended global, rather than local, solvation network. Taken together, these results demonstrate the possibility for a controlled solvent “tuning” for the stabilization of proteins. As part of a rational design of engineered enzymes and inhibitors, in future studies these protein hydration properties can be manipulated with a few ad hoc chosen mutations, thereby changing the stabilization and/or activity of proteins in a controlled way.
Results and Discussion
Water Mapping of Wild Type and Mutant MMPs via MD Simulations
In order to gain insight into the structure and dynamics of hydration water around MMPs, classical MD simulations were performed, where MMP14-WT, MMP9-WT, and MMP14-SIA were solvated in liquid water at room temperature (see the SI for simulation details). Briefly, MMP14-SIA comprises 17 mutations relative to the human wild type enzyme (Table S1) and exhibits an apparent melting temperature 16 °C greater than that of the wild type as well as a significantly greater heterologous expression levels in E. coli (see Figures S1 and S2 for characterization of the stabilized inactive mutant MMP14-SIA and the stabilized active mutant MMP14-SA). This stabilized design provided us with a unique opportunity to test the impact of polar surface mutations on a protein’s interaction with its hydration shell, since many stabilizing mutations increase surface polarity and charge.36 In the case of MMP14-SIA, 15 of the 17 mutations (all but A258Y and Y141F) are surface exposed, and of these five introduce new charges and one eliminates a charge (E248G). Thus, these designed mutations are likely to significantly alter the solvation properties of the enzyme.
There are pronounced structural differences in the inner part of the MMPs hydration layer (within 4.1 Å from the protein surface) with respect to bulk water, while in the outer hydration layer (up to 5.9 Å from MMP) an almost bulk-like environment is recovered (see Figure S3 and Table S2 in the SI). Water molecules within the inner hydration layer of all MMPs form less water–water hydrogen bonds (3.0 HBs/molecule) than in the bulk (3.3 HBs/molecule). For water molecules hydrating polar/hydrophilic MMP domains (bound water), this arises from the competition of water–water interactions and favorable H-bonding to the protein surface. The rest of hydration water molecules (unbound water, also referred to as wrap water) minimize the number of broken HBs by forming extended water–water HB structures parallel to the MMP surface (denoted as water-driven ordering).37 Similar extended two-dimensional water HB networks were observed before at planar hydrophobic interfaces and near model protein surfaces exposing both hydrophilic and hydrophobic patches.32−34,38 The degree of water-driven ordering is reduced in the proximity of polar surface regions due to preferential interactions with the MMP surface (denoted as MMP-driven ordering).34
In the following, “water-driven” is defined as water molecules that HB with other waters within the inner hydration layer, while “MMP-driven” refers to waters that form HBs with either the protein surface or waters in the outer hydration layer. To illustrate the balance between water-driven and MMP-driven orderings around hydrated MMPs, we quantify in Figure 1A–C both components in the inner hydration layer for bound and unbound water populations. We find that unbound water molecules (i.e., located above hydrophobic patches) are largely water-driven and on average form 2.0 HBs with other hydration water molecules (red in Figure 1A–C). In contrast, bound water (i.e., near hydrophilic patches) on average forms an almost equal water-driven and MMP-driven HBs. While the average properties of the bound/unbound hydration water populations are maintained between the MMP variants, their relative abundance is significantly modulated by the mutations. Figure 1D shows that the number of bound waters increases for MMP14-SIA with respect to the WT. This is almost exclusively due to water molecules interacting with amine, amide, and arginine groups, which are more abundant on the MMP14-SIA surface (for which 15 surface residues have been mutated relative to MMP14-WT; see the SI for exact mutations).
Figure 1.

Average number of HBs/molecule formed by bound and unbound water populations in the inner hydration layer dissected in terms of water-driven (red) and MMP-driven (blue) HB network components for MMP14-WT (A), MMP14-SIA (B), and MMP9-WT (C). The water-driven HBs account for HBs formed between water molecules within the inner hydration layer. The MMP-driven HBs are given by the sum of the water–MMP HBs and water–water HBs between water molecules of the inner and outer hydration layers. (D) Comparison between the average number of bound water molecules in the inner hydration layer of the two MMP14 mutants.
Evaluation of the fluctuations of interfacial water density can be used to map the local hydrophobicity/hydrophilicity, taking into account the environmental and topological context.24,30,31,39 Interfacial displacements and thus density fluctuations are typical for water in contact with hydrophobic surfaces but are largely suppressed for water at a hydrophilic surface.30,31,39 Density fluctuations can be quantified by computing the probability, P(N), to find N water molecules in a defined probing volume.30 We performed a group-specific, spatially resolved analysis of water density fluctuations around the MMP variants and introduce [⟨N2⟩ – ⟨N⟩2]/⟨N⟩ as the normalized average water number fluctuations in a spherical volume of 3.5 Å radius around each hydrophilic/-phobic group (e.g., −CH3, −CH2–, −OH, −NH2, etc.).31 Large/small water density fluctuations correspond to large/small average [⟨N2⟩ – ⟨N⟩2]/⟨N⟩ values.
The probability distribution, P (%), of the group specific [⟨N2⟩ – ⟨N⟩2]/⟨N⟩ values for MMP14-WT (red), -SIA (blue), and MMP9-WT (black) is illustrated in Figure 2A. All MMPs exhibit a bimodal distribution, with one maximum at low [⟨N2⟩ – ⟨N⟩2]/⟨N⟩ values (<0.4) and one maximum at high values (>0.7). We find that the probability of large water density fluctuations (>0.7) is increased for MMP14-WT, indicative of large hydrophobic patches, while hydrophilic surface patches with small fluctuations (<0.4) are more abundant at the MMP14-SIA surface. MMP9-WT shows an intermediate behavior, with values systematically in between the two MMP14s.
Figure 2.

(A) Probability distribution of the group specific water density fluctuations ([⟨N2⟩ – ⟨N⟩2]/⟨N⟩), calculated within spherical observation volumes around each surface group of the three MMP surfaces. The corresponding 3D spatially resolved maps are also shown for MMP14-WT (B), MMP9-WT (C), and MMP14-SIA (D). In the maps, each sphere identifies one hydrophilic/-phobic group at the MMP surface, while the color coding provides the magnitude of local water density fluctuations within a 3.5 Å radius from the group. The second color scale reports the associated free energy of wetting for the surface groups, ΔGwetting in kBT units (as deduced from eq 1). The green and gray arrows in panels (B) and (D) highlight two examples of surface patches containing mutated residues, L117K and G285S, respectively, which have been mutated from hydrophobic in MMP14-WT to hydrophilic in MMP-SIA.
To better visualize the hydrophilic/hydrophobic patches on MMPs surfaces, 3D spatially resolved maps of the group-specific water density fluctuations are reported in panels B–D of Figure 2. As can be seen, large hydrophobic patches (red areas) are formed on the MMP14-WT surface. The number of hydrophobic patches on the surface decreases in the order MMP14-WT > MMP9-WT > MMP14-SIA, in accordance with the expectation that the stabilized design would exhibit a more hydrophilic surface than the others. On the contrary, the larger number of bound waters interacting with polar groups of MMP14-SIA causes an increase in the areas occupied by hydrophilic patches (blue). The surprising result is that the mutation-induced changes in local hydrophobicity are not restricted to local spots, but rather extend over the whole MMP14 surface. Such changes in water density fluctuations around the various surface patches can be directly connected to the free energy associated with the hydration of the protein surface.
In particular, the probability that, due to density fluctuations, water evacuates the volume around an exposed surface group is directly related to the associated free energy cost via eq 1:39,40
| 1 |
where β = 1/kBT, ⟨N⟩ is the average number of water molecules in the chosen probe volume and ⟨(δN)2⟩v is the related mean-square fluctuation. βΔGdewetting can be seen as the free energy cost (in kBT units) to dewet the surface group exposed to the probing volume V, so that βΔGwetting = −βΔGdewetting is a local measure for the free energy of surface wetting. The group-specific, spatially resolved ΔGwetting values are reported in the 3D maps of panels B–D of Figure 2, as a second color-scale, showing how hydrophobic surface patches with increased density fluctuations (red areas) are less favorably wetted by water (βΔGwetting > −2.6kBT) than the hydrophilic ones (blue, for which βΔGwetting < −4.2kBT).
We find that local changes induced by mutations can cause an extended change from hydrophobic to hydrophilic. Overall, 15 surface residues were mutated (Table S1), but here we will focus on the specific impact of just two of these mutated residues, L117K and G285S: The green and gray arrows in Figure 2B/D indicate the positions of these mutations. In the case highlighted by the green arrow (L117K), a large hydrophobic patch (red in Figure 2B, with an area of about 4 nm2) is entirely converted into a hydrophilic one (blue in Figure 2D). The addition of a hydrophilic “spot” at the center of a hydrophobic patch is able to quench density fluctuations,30 and βΔGwetting decreases accordingly in the entire patch. This example shows how the L117K mutation does not contribute additively to βΔGwetting, but rather induces larger changes in the free energy that extend to the surrounding surface residues.
On the other hand, the gray arrow (G285S mutation) illustrates how introducing an isolated hydrophobic spot in the middle of a hydrophilic background has a negligible impact on density fluctuations, as illustrated by the predominant blue color persisting in the area near the gray arrow. In this case, the contribution of the mutated residue to the free energy is restricted to its own hydration (βΔGwetting changes color in the map only for the mutated spot) and is well described by an additive model where the global change induced in the MMP hydration free energy is simply the free energy difference to hydrate the residue before and after the mutation.
Together, the two discussed mutations show how the local topological context, i.e., the nature and disposition of the surrounding residues around a single mutation, plays a crucial role in determining the impact on hydration free energies. The fact that even a small number of specifically selected mutations, if strategically placed, can change the local hydrophilicity of a protein on a scale of several nanometers provides novel opportunities for protein engineering.
THz Spectroscopy Probes Significant Changes in the Solvation Dynamics for MMP14, MMP14-SIA Mutant, and MMP9
THz spectroscopy has been established as a sensitive method to probe changes in the solvation dynamics.17,41,42 Water has an extended hydrogen-bonding network in which hydrogen bonds break and reform on the order of sub-picoseconds. While infrared spectroscopy is sensitive the intramolecular vibrational modes of water, THz spectroscopy probes the low frequency, collective, intermolecular motions of the hydrogen-bonding network. In the frequency range up to 3 THz (100 cm–1), collective modes involving several water molecules are probed. Any change in the radial and angular intermolecular potential energy surface will affect the intermolecular water–water stretch, centered between 160 and 200 cm–1, and the hindered rotation of a single water molecule within the hydrogen network, centered between 350 and 700 cm–1, respectively.43 Coupling of water to the surface of a solute, such as a protein, introduces modifications to the water network that can extend several molecular layers from the solute surface. Water which has properties distinct from those of bulk water is defined as hydration shell water. THz spectroscopy, therefore, is sensitive to changes in the hydration shell and reports on spectral fingerprints corresponding to distinct hydration water populations (see the SI and ref (35) for more details). In previous studies, single point or multiple mutations lead to significant changes, which were probed by their distinct THz response.44,45
Figure 3 (left) shows the low frequency THz spectra measured by THz-TDS and FTIR of MMP14-WT as a function of protein concentration (see the SI for experimental details). The relative absorption, Δα = αsolution – αbuffer, is independent of the setup: both spectra fit together seamlessly. The spectra show an overall decrease of absorption in the entire frequency range compared to bulk. In Figure 3 (right), we compare the measured data for selected concentrations (solid lines) to the expected decrease in Δα due to volume exclusion of water by the protein (dashed lines). The volume exclusion was calculated assuming that the protein in solution has a spherical volume, determined from its radius (Rg = 24.7 Å for MMP14-WT; see Figure S4 in the SI), and displaces an equal volume of water from the solution. The expected decrease in the relative absorption is calculated via Δα(cprotein) = (Vprotein/Vtotal)αbuffer– αbuffer, with cprotein and Vprotein/Vtotal being the protein concentration and the relative volume of the proteins in the total volume, respectively. Neglecting density changes, we estimate that the observed decrease in the measured Δα relative to the pure volume exclusion Δα would correspond to a dynamical hydration shell extending 13 water layers (or a radius of 39 Å) from the protein surface. While low frequency modes of the HB network (<100 cm–1) extend over three hydration layers,43 and both theoretical and experimental studies have found the solvation shell to encompass water 40–50 Å from the solute surface,46,47 it is unlikely that this decrease can only be attributed to volume exclusion. We note that the decrease in Δα is frequency dependent and more pronounced in the frequency range between 250 and 550 cm–1, i.e., the frequency range of the water libration. Librations, i.e., hindered rotational motions of water molecules in the surrounding water cage, encompass distinct frequency regimes, where softer rotational motions (rocking librations) occur between 300 and 550 cm–1 and stiffer rotations (wagging librations) occur at higher frequencies (>550 cm–1).48 A blue shift of the librational band is observed in the case of a stiffening of the water network hydrating hydrophobic moieties.33 Therefore, we propose that stiffening of the water network due to protein–water interactions could contribute to the decreased absorption.
Figure 3.

Δα(ν) of MMP14-WT determined from THz-TDS and THz-FTIR with increasing protein concentration (left). The Δα(ν) of the lowest and highest concentration (right) greatly exceeds the expected decrease in Δα(ν) based on water volume exclusion. Error of the measurements are on average 0.6 cm–1 in the region 10–80 cm–1 and 5 cm–1 in the region 80–550 cm–1.
In Figure 4, we plot Δα of all MMP samples at 1 THz (left) and 2 THz (right) as a function of protein concentration as probed by THz time domain spectroscopy (see Figure S5 for full THz regime spectra). Also shown in Figure 4 as solid lines are the calculated Δα based solely on volume exclusion of water. The radius of gyration of MMP9-WT is Rg = 19.6 Å (see Figure S4 in SI). It was assumed that MMP14-SIA has the same radius as the WT. We find a clearly distinct concentration dependent Δα for MMP14-WT compared to MMP9-WT and MMP14-SIA mutant: MMP14-WT has a negative Δα, and the absolute magnitude of the decrease increases with an increase in protein concentration. The SIA form has positive values of Δα, which increases up to 40 μM; and beyond this point, Δα decreases but stays positive. MMP9-WT displays behavior intermediate of the two MMP14 variants, with slightly negative Δα values.
Figure 4.

Δα(ν) of MMP14 variants at 1 THz (left) and 2 THz (right) as a function of protein concentration. The solid line in each panel represents the expected decrease in Δα(ν) based on volume exclusion of water. Error bars are on average 0.6 cm–1.
Since the THz response is most sensitive to changes in hydration,41,17,42 it becomes clear that the site-specific mutations of MMP14 significantly alter the way in which water interacts with the protein surface. The mutations introduced in MMP14-SIA induce changes in the surface electrostatic potential: the charge of six surface exposed groups (L117K, Q120D, A139E, E248G, V270R, E286 K; Table S1) is altered, the majority of which result in a more positively charged surface. The change of a charge, from i.e., negative to positive, as well as surface charge density is known to influence the organization of water molecules within the interfacial regime.49−51 In a recent study, we could show that the THz absorption coefficient Δα provides a measure for local electrostatics of amino acids, with the positively charged state giving rise to an increased THz response.52 Considering the surface electrostatic potential of the three MMPs investigated here, the following trend of increasing positive potential is revealed: MMP14-WT < MMP9-WT < MMP14-SIA.11 We note that this trend corresponds surprisingly well with the observed THz absorption coefficients in which the THz response becomes more positive in sign for a more positively charged surface distribution. Based on the MD simulations, the increased THz absorption for more positively charged MMP surfaces is correlated with an increased hydrophilic nature.
Conclusions
In summary, experimental and theoretical characterization of MMP hydration reveals that changes in solvation may play a significant role in the stabilization of the designed variant. THz spectroscopy is able to probe these global solvation changes resulting from local mutations. We could correlate the dynamics of the extended HB network, as probed by the change in the THz absorption coefficient, and the surface electrostatic potential of MMP proteins. MD simulations explain changes in the hydrogen-bonding network by an increased population of bound water molecules at the protein surface. For MMP14, local mutations are shown to alter the hydrogen bond environment even for large patches, changing the local hydrophilicity. One key result from the MD simulations is that depending on the nature of the substituted residue and on the local surface topology and morphology, the effects of several mutations can propagate through the water HB network in the hydration layer and affect hydrophobicity and wetting on a scale of several nanometers. Moreover, depending on the local surface topology, the contribution of a muted residue to the free energy of protein hydration can be nonadditive, as shown here in the case of a hydrophilic residue introduced in a patch mostly composed by hydrophobic surface residues. In this scenario, the changes in hydration free energy cannot be quantitatively described by the existing models that rely on the assumption of group additivity since the effect of the local topological environment (distribution of neighboring amino acid residues) has to be explicitly considered together with the nature of the mutated residue and the eventual changes in the exposed surface area induced by the mutation.22 Our results provide insight into the understanding of such topological effects and suggest that tuning of protein hydration properties through point or “patch” mutations that take advantage of the surface topology could develop into a powerful tool for the rational design of proteins with increased stability and activity.
Experimental and Simulation Details
Design of the MMP14 Stabilized Mutant
To design a stable protein variant of MMP14 we applied a stability-design algorithm dubbed PROSS, a fully automated method implemented on a Web server (http://pross.weizmann.ac.il/bin/steps).35 The PROSS workflow comprises homology-based construction of a multiple sequence alignment to define at each position a set of amino acid options that frequently appear across the protein family natural diversity. Next, the target protein’s closest molecular structure has been used for Rosetta53 computational design simulations that identify a subset of mutations from the above amino acid options, each predicted to be independently stabilizing compared to the wild type sequence. Finally, Rosetta combinatorial sequence design is applied to scan all combinations of mutations from the above subset to identify optimal sequence variants, with substantially improved native-state energy.
To design MMP14 stabilized mutant, a human orthologue (PDB entry: 1BUV) served as an input in the PROSS run, and amino acid positions at the active site, defined as all positions within 5 Å from the binding interface (i.e., chain T) or from the metal ions, were held fixed during all simulation steps (see Table S1), yielding a small list of suggested mutants, out of which a variant bearing the highest amount of mutated residues was selected for manual inspection. In total, 14 mutations were suggested by PROSS, to be integrated into the sequence of human MMP14, based on the 1BUV structure (see Table S1, PROSS generated mutations). Following manual inspection of the aforementioned design and of the multiple sequence alignment, we decided to introduce three more mutations; Y141F, G285S and E286K. All three, in addition to the PROSS suggested S251N, match in sequence identity to the mouse orthologue of MMP14. Thus, the final design, referred to as MMP14-SA, comprised 17 or 15 mutations when compared to the human or mouse MMP14, respectively. MMP14-SA expression and thermostability were assayed and compared to that of mouse MMP14-WT, displaying a significant increase in protein expression and thermostability (Figure S1). Finally, in order to produce the catalytically inactive variant mutant (MMP14-SIA), we have introduced two additional mutations at the enzyme active site (H240E and E249H) of MMP14-SA.
MMP Protein Expression and Purification
The human catalytic domain (residues Y112–K292) of mouse MMP14-WT was prepared as previously described in the literature.54 Post expression the enzymes autodigest the hinge region, in vitro, following which the processed protein comprises residues Y112–S287.54,55 The mouse MMP14-WT and stabilized SIA and SA mutants catalytic domain (residues Y112-G288) were codon-optimized for expression in E. coli (Gen9, currently Ginkgo Boston). The three genes were cloned into the pET28-TevH expression vector with an N-terminal 6xHis tag followed by TEV cleavage site (https://pubmed.ncbi.nlm.nih.gov/18542865/) and was expressed in Bio-BL21(DE3) competent bacterial strains (Biolab, Ashkelon, Israel). Following expression overnight at 15 °C, the cells were harvested, washed, and lysed in an ice-cold lysis buffer (50 mM Tris pH8 at 4 °C, 0.5 M NaCl, 5 mM CaCl2, 5 mM imidazole, 10 μM MgCl2, 0.1% Brij35, lysozyme at 100 μg/mL and 2 pills of cOmplete protease inhibitor cocktail, Merck). The lysed culture was centrifuged for 1 h at 13 kRPM following supernatant collection and filtration (0.2 μm). The sample was loaded on a HisTrap (GE Healthcare) affinity column that was precalibrated with 50 mM Tris pH 8 (at 4 °C), 0.5 M NaCl, 5 mM CaCl2, and 10 mM imidazole. The protein was eluted with elution buffer of 50 mM Tris pH 8 (at 4 °C), 500 mM imidazole, 500 mM NaCl, 5 mM CaCl2, and 2.5 μM ZnCl2 in a single-step elution. Following affinity purification, the samples were submitted to size exclusion chromatography using the HiLoad 26/600 Superdex 75 pg column (GE Healthcare Life Sciences) in 25 mM Tris pH 8 (in 4 °C). Next, the protein was further submitted to ion exchange chromatography (IEC, MonoQ) that was washed with 25 mM Tris pH 8 (at 4 °C), following a gradient elution step with IEC elution buffer: 25 mM Tris pH 8 (at 4 °C) with 1 M of NaCl. The purity of all protein samples was evaluated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), concentrated in 50 mM Tris pH 8 (at 4 °C), 100 mM NaCl, 5 mM CaCl2, 2.5 μM ZnCl2 and 0.01% Brij35, aliquoted, and frozen in liquid nitrogen and stored at −80 °C until further use. The human MMP9-WT catalytic domain (residues 107–215, 391–443) was cloned into the pET28-TevH (https://pubmed.ncbi.nlm.nih.gov/18542865/) and also expressed in the E. coli BL21(DE3) strain. Cells were harvested, washed, lysed, and centrifuged to isolate the inclusion bodies. Inclusion bodies were solubilized through suspension in 8 M urea, 25 mM Tris, pH 8. The protein was purified using a HisTrap FF column (GE Healthcare, USA) and dialyzed slowly with a buffer of 6 M urea, 25 mM Tris, pH 8, and 25 mM NaCl at 4 °C. Further purification was done with a HiTrap Q HP column (GE Healthcare), and the protein was diluted to 0.1 mg/mL in 6 M urea, 25 mM Tris, pH 8, 250 mM NaCl. Refolding was achieved by dialysis with stepwise reduction of urea concentration. Final protein purification was done with a size exclusion column (HiLoad 26/600 Superdex 75 pg, GE Healthcare Life Sciences). The purity of all protein samples was checked with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Protein aliquots were frozen in liquid nitrogen and stored at −80 °C until further use.
Characterization of the wild type (WT), stabilized (SA), and stabilized inactive mutant (SIA) MMP14 samples (Figure S1) reveals increased thermal stability and overexpression of the stabilized mutants relative to the WT.
Simulation Details
Classical MD simulations were carried out using the GROMACS package.56 The CHARMM27 and TIP3P force fields were used for the MMPs and water, respectively.57,58 The adopted CHARMM27 model can lead to issues with protein secondary structure, however it was shown by a previous study that the combination of CHARMM27 and TIP3P reproduces with a satisfactory accuracy the structural, dynamical and vibrational properties of hydration water around proteins of the MMP family.58 It is also important to consider that the choice of the TIP3P water model affects the absolute values obtained for the number of H-bonds formed by water molecules, as well as the magnitude of density fluctuations discussed in the main text, which are known to depend on the chosen water model. These issues do not affect the conclusions of the present study, which are systematically based on a comparison and ranking of the 3 investigated proteins, and not on absolute values.
For all MMP14-WT, MMP9-WT and MMP14-SIA systems, the initial configuration in the simulations is systematically taken as the folded structure, obtained by optimizing at the chosen level of theory the configuration extracted from PDB: 1BUV. The folded configuration is maintained during the simulation time for all three MMPs. In the simulations, the mutations are allowed to perturb the global protein structure, which is unconstrained. After proper equilibration, including a first run in the NPT ensemble and a subsequent run in the NVT ensemble, 60 ns simulations were carried out in the NVT ensemble (298 K) and used for analysis. A time-step of 1 fs was employed and stretching motions involving H atoms were constrained. A cubic box with a length of 89 Å has been used for all simulations in combination with Three-Dimensional Periodic Boundary Conditions. The HBs are defined using the standard distance and angle criterion from Luzar,59 with O–X distance cutoff of 3.5 Å and H–O–X angle in the 0–30° range, where X = O for water–water HBs, and X can be either O or N for water-MMP HBs. Different criteria have been tested with H–O–X angle in the 0–40° range and O–X distance cutoff of 3.2 Å to ensure that our results are not biased by the chosen criterion. Water molecules are assigned to the inner/outer/bulk regions based on the distance from the MMP surface (see water-MMP radial pair distribution function in Figure S3). A water molecule in the inner hydration layer is further classified as bound water if it is located within 3.5 Å from MMP polar groups (i.e., H-bonding sites) and as unbound water otherwise.
Terahertz Spectroscopy
Terahertz (THz) absorption spectra were measured with a THz time domain spectrometer (THz-TDS, range 10–80 cm–1) and a FTIR spectrometer (80–600 cm–1). The THz-TDS is a custom-built spectrometer system that has been previously described,60 while the FTIR spectrometer is a commercially available spectrometer (Bruker Vertex 80 v) equipped with a helium cooled silicon bolometer detector. For the THz-TDS measurements, a demountable cell consisting of two z-cut quartz windows (4 mm thick) and a Teflon spacer (100 μm thick) was used. Spectra were measured 10 times with an integration time of 30 s per scan. In the case of the FTIR measurements, a similar demountable cell with diamond windows (0.5 mm thick) and a Kapton spacer (13 μm thick) was used. The spectra are an average of 128 scans with 1 cm–1 resolution. All samples were measured at least two times with each technique to ensure reproducibility. The sample cell temperature was kept constant at 20 °C with an external chiller, and the sample compartment was purged with nitrogen. The absorption coefficient (and refractive index if applicable) of the sample was determined from previously described methods.60,61 Error of THz-TDS and FTIR measurements are 0.6 and 5 cm–1, respectively.
Acknowledgments
This research was supported by ERC Advanced Grant 698437 and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC 2033 – 390677874 – RESOLV. Research in the Fleishman lab was supported by an Israel Science Foundation grant (1844/19) and by a charitable donation in the memory of Sam Switzer.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00155.
MMP14 stability design PROSS parameters; DSF stability analysis; Michaelis–Menten kinetic analysis of hMMP14-WT, mMMP14-WT, and MMP14-SA mutants on a standard MMP substrate; radial pair distribution functions (g(r)) calculated in between all MMP14-WT and MMP9-WT atoms and all water molecules oxygen atoms; average number of water–water HBs/molecule, q and t order parameters, and water dipole reorientation time (τμ, divided by the bulk reference, τμ(bulk)), for water molecules in the inner hydration layer, outer hydration layer, and bulk regions; hydrodynamic diameters of MMP14-WT and MMP9-WT in solution as a function of concentration measured by dynamic light scattering; THz-TDS absorption spectra of MMP14-WT, MMP14-SIA, and MMP9-WT (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Clark I.; Swingler T.; Sampieri C.; Edwards D. The Regulation of Matrix Metalloproteinases and Their Inhibitors. Int. J. Biochem. Cell Biol. 2008, 40 (6–7), 1362–1378. 10.1016/j.biocel.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Matthews B. W. Structural Basis of the Action of Thermolysin and Related Zinc Peptidases. Acc. Chem. Res. 1988, 21 (9), 333–340. 10.1021/ar00153a003. [DOI] [Google Scholar]
- Levin M.; Udi Y.; Solomonov I.; Sagi I. Next Generation Matrix Metalloproteinase Inhibitors — Novel Strategies Bring New Prospects. Biochim. Biophys. Acta, Mol. Cell Res. 2017, 1864 (11), 1927–1939. 10.1016/j.bbamcr.2017.06.009. [DOI] [PubMed] [Google Scholar]
- Bode W.; Gomis-Rüth F.-X.; Stöckler W. Astacins, Serralysins, Snake Venom and Matrix Metalloproteinases Exhibit Identical Zinc-Binding Environments (HEXXHXXGXXH and Met-Turn) and Topologies and Should Be Grouped into a Common Family, the ‘Metzincins.’. FEBS Lett. 1993, 331 (1–2), 134–140. 10.1016/0014-5793(93)80312-I. [DOI] [PubMed] [Google Scholar]
- Tallant C.; García-Castellanos R.; Baumann U.; Gomis-Rüth F. X. On the Relevance of the Met-Turn Methionine in Metzincins. J. Biol. Chem. 2010, 285 (18), 13951–13957. 10.1074/jbc.M109.083378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G.; Nagase H. Progress in Matrix Metalloproteinase Research. Mol. Aspects Med. 2008, 29 (5), 290–308. 10.1016/j.mam.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagi I.; Talmi-Frank D.; Arkadash V.; Papo N.; Mohan V. Matrix Metalloproteinase Protein Inhibitors: Highlighting a New Beginning for Metalloproteinases in Medicine. MNM 2016, 3, 31–47. 10.2147/MNM.S65143. [DOI] [Google Scholar]
- Zhang H.; Liu M.; Sun Y.; Lu J. MMP-14 Can Serve as a Prognostic Marker in Patients with Supraglottic Cancer. Eur. Arch Otorhinolaryngol 2009, 266 (9), 1427–1434. 10.1007/s00405-009-0943-6. [DOI] [PubMed] [Google Scholar]
- Sathyamoorthy T.; Tezera L. B.; Walker N. F.; Brilha S.; Saraiva L.; Mauri F. A.; Wilkinson R. J.; Friedland J. S.; Elkington P. T. Membrane Type 1 Matrix Metalloproteinase Regulates Monocyte Migration and Collagen Destruction in Tuberculosis. J. Immunol. 2015, 195 (3), 882–891. 10.4049/jimmunol.1403110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owyong M.; Chou J.; van den Bijgaart R. J.; Kong N.; Efe G.; Maynard C.; Talmi-Frank D.; Solomonov I.; Koopman C.; Hadler-Olsen E.; Headley M.; Lin C.; Wang C.-Y.; Sagi I.; Werb Z.; Plaks V. MMP9Modulates the Metastatic Cascade and Immune Landscape for Breast Cancer Anti-Metastatic Therapy. Life Sci. Alliance 2019, 2 (6), e201800226 10.26508/lsa.201800226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udi Y.; Fragai M.; Grossman M.; Mitternacht S.; Arad-Yellin R.; Calderone V.; Melikian M.; Toccafondi M.; Berezovsky I. N.; Luchinat C.; Sagi I. Unraveling Hidden Regulatory Sites in Structurally Homologous Metalloproteases. J. Mol. Biol. 2013, 425 (13), 2330–2346. 10.1016/j.jmb.2013.04.009. [DOI] [PubMed] [Google Scholar]
- Arkadash V.; Yosef G.; Shirian J.; Cohen I.; Horev Y.; Grossman M.; Sagi I.; Radisky E. S.; Shifman J. M.; Papo N. Development of High Affinity and High Specificity Inhibitors of Matrix Metalloproteinase 14 through Computational Design and Directed Evolution. J. Biol. Chem. 2017, 292 (8), 3481–3495. 10.1074/jbc.M116.756718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botkjaer K. A.; Kwok H. F.; Terp M. G.; Karatt-Vellatt A.; Santamaria S.; McCafferty J.; Andreasen P. A.; Itoh Y.; Ditzel H. J.; Murphy G. Development of a Specific Affinity-Matured Exosite Inhibitor to MT1-MMP That Efficiently Inhibits Tumor Cell Invasion in Vitro and Metastasis in Vivo.. Oncotarget 2016, 7 (13), 16773–16792. 10.18632/oncotarget.7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle A. G.; Cieplak P.; Nam D. H.; Shiryaev S. A.; Ge X.; Strongin A. Y. Selective Function-Blocking Monoclonal Human Antibody Highlights the Important Role of Membrane Type-1 Matrix Metalloproteinase (MT1-MMP) in Metastasis. Oncotarget 2017, 8 (2), 2781–2799. 10.18632/oncotarget.13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder H.; Fenimore P. W.; Chen G.; McMahon B. H. Protein Folding Is Slaved to Solvent Motions. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (42), 15469–15472. 10.1073/pnas.0607168103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball P. Water as an Active Constituent in Cell Biology. Chem. Rev. 2008, 108 (1), 74–108. 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- Grossman M.; Born B.; Heyden M.; Tworowski D.; Fields G. B.; Sagi I.; Havenith M. Correlated Structural Kinetics and Retarded Solvent Dynamics at the Metalloprotease Active Site. Nat. Struct. Mol. Biol. 2011, 18 (10), 1102–1108. 10.1038/nsmb.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur K.; Heisler I. A.; Meech S. R. Water Dynamics at Protein Interfaces: Ultrafast Optical Kerr Effect Study. J. Phys. Chem. A 2012, 116 (11), 2678–2685. 10.1021/jp2074539. [DOI] [PubMed] [Google Scholar]
- Pezzella M.; El Hage K.; Niesen M.; Shin S.; Willard A. P.; Meuwly M.; Karplus M. Water Dynamics Around Proteins: T-and R-States of Hemoglobin and Melittin. J. Phys. Chem. B 2020, 124, 6540. 10.1021/acs.jpcb.0c04320. [DOI] [PubMed] [Google Scholar]
- Lockett M. R.; Lange H.; Breiten B.; Heroux A.; Sherman W.; Rappoport D.; Yau P. O.; Snyder P. W.; Whitesides G. M. The Binding of Benzoarylsulfonamide Ligands to Human Carbonic Anhydrase Is Insensitive to Formal Fluorination of the Ligand. Angew. Chem. 2013, 125 (30), 7868–7871. 10.1002/ange.201301813. [DOI] [PubMed] [Google Scholar]
- Barnes R.; Sun S.; Fichou Y.; Dahlquist F. W.; Heyden M.; Han S. Spatially Heterogeneous Surface Water Diffusivity around Structured Protein Surfaces at Equilibrium. J. Am. Chem. Soc. 2017, 139 (49), 17890–17901. 10.1021/jacs.7b08606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton M.; Bolen D. W. Predicting the Energetics of Osmolyte-Induced Protein Folding/Unfolding. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (42), 15065–15068. 10.1073/pnas.0507053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton M.; Bolen D. W.. Application of the Transfer Model to Understand How Naturally Occurring Osmolytes Affect Protein Stability. In Methods in Enzymology; Elsevier, 2007; Vol. 428, pp 397–418. 10.1016/S0076-6879(07)28023-1 [DOI] [PubMed] [Google Scholar]
- Xi E.; Venkateshwaran V.; Li L.; Rego N.; Patel A. J.; Garde S. Hydrophobicity of Proteins and Nanostructured Solutes Is Governed by Topographical and Chemical Context. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (51), 13345–13350. 10.1073/pnas.1700092114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe J. I.; Shell M. S. Computational Discovery of Chemically Patterned Surfaces That Effect Unique Hydration Water Dynamics. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (32), 8093–8098. 10.1073/pnas.1807208115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe J.; Barry M.; DeStefano A.; Gokturk P. A.; Jiao S.; Robinson-Brown D.; Webber T.; Crumlin E. J.; Han S.; Shell M. S. Water Structure and Properties at Hydrophilic and Hydrophobic Surfaces. Annu. Rev. Chem. Biomol. Eng. 2020, 11 (1), 523–557. 10.1146/annurev-chembioeng-120919-114657. [DOI] [PubMed] [Google Scholar]
- Giovambattista N.; Rossky P. J.; Debenedetti P. G. Effect of Temperature on the Structure and Phase Behavior of Water Confined by Hydrophobic, Hydrophilic, and Heterogeneous Surfaces †. J. Phys. Chem. B 2009, 113 (42), 13723–13734. 10.1021/jp9018266. [DOI] [PubMed] [Google Scholar]
- Giovambattista N.; Debenedetti P. G.; Rossky P. J. Hydration Behavior under Confinement by Nanoscale Surfaces with Patterned Hydrophobicity and Hydrophilicity. J. Phys. Chem. C 2007, 111 (3), 1323–1332. 10.1021/jp065419b. [DOI] [Google Scholar]
- Tuladhar A.; Dewan S.; Pezzotti S.; Brigiano F. S.; Creazzo F.; Gaigeot M.-P.; Borguet E. Ions Tune Interfacial Water Structure and Modulate Hydrophobic Interactions at Silica Surfaces. J. Am. Chem. Soc. 2020, 142 (15), 6991–7000. 10.1021/jacs.9b13273. [DOI] [PubMed] [Google Scholar]
- Jamadagni S. N.; Godawat R.; Garde S. Hydrophobicity of Proteins and Interfaces: Insights from Density Fluctuations. Annu. Rev. Chem. Biomol. Eng. 2011, 2 (1), 147–171. 10.1146/annurev-chembioeng-061010-114156. [DOI] [PubMed] [Google Scholar]
- Sarupria S.; Garde S. Quantifying Water Density Fluctuations and Compressibility of Hydration Shells of Hydrophobic Solutes and Proteins. Phys. Rev. Lett. 2009, 103 (3), 037803. 10.1103/PhysRevLett.103.037803. [DOI] [PubMed] [Google Scholar]
- Cyran J. D.; Donovan M. A.; Vollmer D.; Siro Brigiano F.; Pezzotti S.; Galimberti D. R.; Gaigeot M.-P.; Bonn M.; Backus E. H. G. Molecular Hydrophobicity at a Macroscopically Hydrophilic Surface. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (5), 1520–1525. 10.1073/pnas.1819000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S.; Willard A. P. Characterizing Hydration Properties Based on the Orientational Structure of Interfacial Water Molecules. J. Chem. Theory Comput. 2018, 14 (2), 461–465. 10.1021/acs.jctc.7b00898. [DOI] [PubMed] [Google Scholar]
- Shin S.; Willard A. P. Water’s Interfacial Hydrogen Bonding Structure Reveals the Effective Strength of Surface-Water Interactions. J. Phys. Chem. B 2018, 122 (26), 6781–6789. 10.1021/acs.jpcb.8b02438. [DOI] [PubMed] [Google Scholar]
- Goldenzweig A.; Goldsmith M.; Hill S. E.; Gertman O.; Laurino P.; Ashani Y.; Dym O.; Unger T.; Albeck S.; Prilusky J.; Lieberman R. L.; Aharoni A.; Silman I.; Sussman J. L.; Tawfik D. S.; Fleishman S. J. Automated Structure-and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63 (2), 337–346. 10.1016/j.molcel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenzweig A.; Fleishman S. J. Principles of Protein Stability and Their Application in Computational Design. Annu. Rev. Biochem. 2018, 87 (1), 105–129. 10.1146/annurev-biochem-062917-012102. [DOI] [PubMed] [Google Scholar]
- Conti Nibali V.; Pezzotti S.; Sebastiani F.; Galimberti D. R.; Schwaab G.; Heyden M.; Gaigeot M.-P.; Havenith M. Wrapping up Hydrophobic Hydration-Locality Matters. J. Phys. Chem. Lett. 2020, 11, 4809. 10.1021/acs.jpclett.0c00846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzotti S.; Galimberti D. R.; Gaigeot M.-P. 2D H-Bond Network as the Topmost Skin to the Air-Water Interface. J. Phys. Chem. Lett. 2017, 8 (13), 3133–3141. 10.1021/acs.jpclett.7b01257. [DOI] [PubMed] [Google Scholar]
- Chandler D. Interfaces and the Driving Force of Hydrophobic Assembly. Nature 2005, 437 (7059), 640–647. 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- Garde S.; Hummer G.; García A. E.; Paulaitis M. E.; Pratt L. R. Origin of Entropy Convergence in Hydrophobic Hydration and Protein Folding. Phys. Rev. Lett. 1996, 77 (24), 4966–4968. 10.1103/PhysRevLett.77.4966. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus S.; Kim S. J.; Heyden M.; Yu X.; Heugen U.; Gruebele M.; Leitner D. M.; Havenith M. An Extended Dynamical Hydration Shell around Proteins. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (52), 20749–20752. 10.1073/pnas.0709207104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaab G.; Sebastiani F.; Havenith M. Ion Hydration and Ion Pairing as Probed by THz Spectroscopy. Angew. Chem., Int. Ed. 2019, 58 (10), 3000–3013. 10.1002/anie.201805261. [DOI] [PubMed] [Google Scholar]
- Heyden M.; Sun J.; Funkner S.; Mathias G.; Forbert H.; Havenith M.; Marx D. Dissecting the THz Spectrum of Liquid Water from First Principles via Correlations in Time and Space. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (27), 12068–12073. 10.1073/pnas.0914885107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding T.; Li R.; Zeitler J. A.; Huber T. L.; Gladden L. F.; Middelberg A. P. J.; Falconer R. J. Terahertz and Far Infrared Spectroscopy of Alanine-Rich Peptides Having Variable Ellipticity. Opt. Express 2010, 18 (26), 27431. 10.1364/OE.18.027431. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus S.; Meister K.; Prigozhin M. B.; DeVries A. L.; Havenith M.; Dzubiella J.; Gruebele M. Functional Importance of Short-Range Binding and Long-Range Solvent Interactions in Helical Antifreeze Peptides. Biophys. J. 2012, 103 (2), L20–L22. 10.1016/j.bpj.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyden M.; Tobias D. J.; Matyushov D. V. Terahertz Absorption of Dilute Aqueous Solutions. J. Chem. Phys. 2012, 137 (23), 235103. 10.1063/1.4772000. [DOI] [PubMed] [Google Scholar]
- Sushko O.; Dubrovka R.; Donnan R. S. Sub-Terahertz Spectroscopy Reveals That Proteins Influence the Properties of Water at Greater Distances than Previously Detected. J. Chem. Phys. 2015, 142 (5), 055101. 10.1063/1.4907271. [DOI] [PubMed] [Google Scholar]
- Novelli F.; Guchhait B.; Havenith M. Towards Intense THz Spectroscopy on Water: Characterization of Optical Rectification by GaP, OH1, and DSTMS at OPA Wavelengths. Materials 2020, 13 (6), 1311. 10.3390/ma13061311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W.; Seok S.; Kim D.; Tian C. S.; Shen Y. R. Sum-Frequency Spectroscopic Study of Langmuir Monolayers of Lipids Having Oppositely Charged Headgroups. Langmuir 2010, 26 (23), 18266–18272. 10.1021/la103129z. [DOI] [PubMed] [Google Scholar]
- Dreier L. B.; Nagata Y.; Lutz H.; Gonella G.; Hunger J.; Backus E. H. G.; Bonn M. Saturation of Charge-Induced Water Alignment at Model Membrane Surfaces. Sci. Adv. 2018, 4 (3), eaap7415 10.1126/sciadv.aap7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta C.; Mammetkuliyev M.; Benderskii A. V. Re-Orientation of Water Molecules in Response to Surface Charge at Surfactant Interfaces. J. Chem. Phys. 2019, 151 (3), 034703. 10.1063/1.5066597. [DOI] [PubMed] [Google Scholar]
- Sebastiani F.; Ma C. Y.; Funke S.; Baumer A.; Decka D.; Hoberg C.; Esser A.; Forbert H.; Schwaab G.; Marx D.; Havenith M. Probing Local Electrostatics of Glycine in Aqueous Solution by THz Spectroscopy. Angew. Chem., Int. Ed. 2021, 60, 3768. 10.1002/anie.202014133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead T. A.; et al. Optimization of Affinity, Specificity and Function of Designed Influenza Inhibitors Using Deep Sequencing. Nat. Biotechnol. 2012, 30 (6), 543. 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H.; Decaneto E.; Grossman M.; Havenith M.; Sagi I.; Lubitz W.; Knipp M. Crystallization and Preliminary X-Ray Crystallographic Analysis of the Catalytic Domain of Membrane Type 1 Matrix Metalloproteinase. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2014, 70 (2), 232–235. 10.1107/S2053230X13034857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaneto E.; Vasilevskaya T.; Kutin Y.; Ogata H.; Grossman M.; Sagi I.; Havenith M.; Lubitz W.; Thiel W.; Cox N. Solvent Water Interactions within the Active Site of the Membrane Type I Matrix Metalloproteinase. Phys. Chem. Chem. Phys. 2017, 19 (45), 30316–30331. 10.1039/C7CP05572B. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26 (16), 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Mackerell A. D.; Feig M.; Brooks C. L. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25 (11), 1400–1415. 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- Dielmann-Gessner J.; Grossman M.; Conti Nibali V.; Born B.; Solomonov I.; Fields G. B.; Havenith M.; Sagi I. Enzymatic Turnover of Macromolecules Generates Long-Lasting Protein-Water-Coupled Motions beyond Reaction Steady State. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (50), 17857–17862. 10.1073/pnas.1410144111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzar A. Resolving the Hydrogen Bond Dynamics Conundrum. J. Chem. Phys. 2000, 113 (23), 10663–10675. 10.1063/1.1320826. [DOI] [Google Scholar]
- Adams E. M.; Lampret O.; König B.; Happe T.; Havenith M. Solvent Dynamics Play a Decisive Role in the Complex Formation of Biologically Relevant Redox Proteins. Phys. Chem. Chem. Phys. 2020, 22, 7451. 10.1039/D0CP00267D. [DOI] [PubMed] [Google Scholar]
- Böhm F.; Schwaab G.; Havenith M. Mapping Hydration Water around Alcohol Chains by THz Calorimetry. Angew. Chem., Int. Ed. 2017, 56 (33), 9981–9985. 10.1002/anie.201612162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.