

Abstract
Over the last several hundred years, donkeys have adapted to high-altitude conditions on the Tibetan Plateau. Interestingly, the kiang, a closely related equid species, also inhabits this region. Previous reports have demonstrated the importance of specific genes and adaptive introgression in divergent lineages for adaptation to hypoxic conditions on the Tibetan Plateau. Here, we assessed whether donkeys and kiangs adapted to the Tibetan Plateau via the same or different biological pathways and whether adaptive introgression has occurred. We assembled a de novo genome from a kiang individual and analyzed the genomes of five kiangs and 93 donkeys (including 24 from the Tibetan Plateau). Our analyses suggested the existence of a strong hard selective sweep at the EPAS1 locus in kiangs. In Tibetan donkeys, however, another gene, i.e., EGLN1, was likely involved in their adaptation to high altitude. In addition, admixture analysis found no evidence for interspecific gene flow between kiangs and Tibetan donkeys. Our findings indicate that despite the short evolutionary time scale since the arrival of donkeys on the Tibetan Plateau, as well as the existence of a closely related species already adapted to hypoxia, Tibetan donkeys did not acquire adaptation via admixture but instead evolved adaptations via a different biological pathway.
Keywords: Kiang, Donkey, High altitude, Adaptation, Selection
INTRODUCTION
Domestic donkeys have been used as draft animals by humans for over 5 000 years (Beja-Pereira et al., 2004). Despite the highly restricted distribution of their wild progenitor, the arid-adapted African wild ass (Beja-Pereira et al., 2004; Ma et al., 2020), donkeys demonstrate a propensity to adapt to a wide range of environments, including high-altitude habitats on the Tibetan Plateau.
The genetic mechanisms underlying high-altitude adaptation have been studied extensively in multiple mammalian species, including dog, yak, chiru, human, and many other animals (Beall et al., 2010; Foll et al., 2014; Ge et al., 2013; Gou et al., 2014; Huerta-Sánchez et al., 2014; Lorenzo et al., 2014; Qiu et al., 2012; Qu et al., 2013; Simonson et al., 2010; Wang et al., 2014; Wang et al., 2016). Various studies have demonstrated the importance of the endothelial PAS domain-containing protein 1 (EPAS1) gene, also known as hypoxia-inducible factor-2-alpha (HIF-2α), which exhibits activity under low oxygen conditions (Beall et al., 2010; Huerta-Sánchez et al., 2014; Miao et al., 2017; vonHoldt et al., 2017). Specifically, these studies show that both dogs and humans from Tibet obtained the EPAS1 allele, which is necessary for their adaptation to high-altitude conditions, via hybridization with closely related lineages that were already adapted to the Tibetan Plateau (Huerta-Sánchez et al., 2014; Miao et al., 2017; vonHoldt et al., 2017).
Interestingly, kiangs, which belong to a lineage that shared a common ancestor with donkeys ~1.47–1.75 million years ago (Jónsson et al., 2014), also inhabit the Tibetan Plateau. The close geographic proximity of these two closely related species suggests the possibility that, as for dogs (Gou et al., 2014), cattle (Wu et al., 2018), and humans (Huerta-Sánchez et al., 2014), adaptive admixture may have facilitated the adaptation of donkeys to low-oxygen conditions. This scenario is likely given the propensity of equid species to interbreed, including kiangs and donkeys, despite their large karyotypic differences (2ndonkey=62, 2nkiang=52) (Jónsson et al., 2014). Alternatively, the kiang and Tibetan donkey may have acquired their high-altitude adaptations independently, potentially via the same or different biological pathways. To test these hypotheses, we de novo assembled the genome of a kiang individual and analyzed the genomes of 93 domestic donkeys (24 from Tibetan Plateau, 28 from Chinese lowland, eight from Iran, 26 from Africa, and seven from Middle Asia) and five kiangs.
MATERIALS AND METHODS
De novo assembly of kiang genome
A blood sample of a male kiang was collected from Beijing Zoo in 2015. We de novo assembled its genome via a whole-genome shotgun approach. DNA was isolated from blood tissue using standard cetyltrimethylammonium bromide (CTAB) extraction and libraries were prepared following the protocols provided by Illumina. Multiple paired-end and mate-pair libraries were constructed with variable fragment lengths ranging from 220 bp to 17 kb (Supplementary Table S1). All libraries were sequenced through the Illumina HiSeq 2000 & 2500 sequencing platform. In total, 400.92 Gb of raw reads (~174× coverage of the kiang genome) with an average read length of 126 bp were generated for genome assembly. Using these data, the de novo genome was assembled by ALLPATHS-LG (Gnerre et al., 2011).
Positively selected genes (PSGs) based on dN/dS ratio (non-synonymous substitutions per non-synonymous site (dN) to synonymous substitutions per synonymous site (dS))
Human, donkey, horse, pig, and rhino genome sequences were downloaded from the Ensembl database. In consideration of alternative splicing variants, the longest transcripts were selected to represent genes. First, we performed all-to-all BLASTp analysis with an e-value cutoff of 1e−5. To weigh the similarity between gene pairs, we assigned an H-score (BLAST bit score) ranging from 0 to 100, calculated by score (G1G2)/max (score (G1G1), score (G2G2)). Next, we built a hierarchy graph by hcluster_sg (Li et al., 2006), requiring the minimum edge (score) to be greater than 5 and the minimum edge density to be larger than 0.34 to form a cluster. Gene family clustering ceased immediately once there was more than one out-group gene.
We used MUSCLE (Edgar, 2004) and MAFFT (Katoh et al., 2002) software for multiple sequence alignments to identify gene families. After that, the protein alignments were back translated to nucleotide alignments to build a phylogenetic tree with TreeBeST (http://treesoft.sourceforge.net/treebest.shtml), which uses a built-in algorithm to construct the best tree reconciled with a species tree and roots the tree by minimizing the number of duplications and losses. Using gene trees, the pairwise relationships (orthologous and within-species paralogous genes) can be inferred.
Multiple sequence alignments of the one-to-one orthologous genes were performed using PRANK (Löytynoja & Goldman, 2008). After alignment and trimming, we identified 5 778 high-confidence one-to-one orthologous genes in the kiang, human, donkey, horse, pig, and rhino genomes. The branch site model in the Codeml program in the PAML package (Yang, 2007) was used to detect PSGs in the kiang lineage, with 164 PSGs thus identified.
Expression profile analysis of PSGs in kiang using human expression data
As it is difficult to obtain expression data for kiangs, we used publicly available human expression data to examine the expression patterns of genes that are positively selected in kiangs. Analysis was performed as described in our previous study (Li et al., 2013). Human gene expression data (Human U133A Gene Atlas) from 84 tissues or cells were downloaded from BioGPS (Wu et al., 2016) (http://biogps.org/#goto=welcome) with the GEO code GSE1133. To avoid bias expression in different tissues, the expression levels of PSGs were normalized by dividing each tissue value by the average whole-genome expression level. Only the top 10 tissues/cell lines are presented.
Genome re-sequencing
Tissues for DNA extraction were stored in alcohol at −80 °C. Genomic DNA was prepared by standard phenol-chloroform extraction. Sequence libraries were constructed according to the Illumina library preparation pipeline and sequenced using the Hiseq 2500 platform. The genomes of five kiangs and 75 domestic donkeys (24 from Tibetan Plateau, 13 from Chinese plains, eight from Iran, 23 from Africa, and seven from Middle Asia) were re-sequenced in this study (genomes of 18 domestic donkeys were provided by Wang et al. (2020) (Supplementary Table S9). Zebra data were downloaded from a previously published study as an outgroup (Jónsson & Schubert, 2014).
Read mapping and variant calling
Before alignment, reads were trimmed based on their quality scores using the quality trimming program Btrim (Kong, 2011). Quality-filtered reads were mapped to our kiang de novo reference using the alignment algorithm BWA-MEM (Pavlidis et al., 2013). Single nucleotide polymorphisms (SNPs) were detected using the Genome Analysis Toolkit (GATK) (McKenna et al., 2010). Duplicate read pairs were first identified using the Picard tools (http://picard.sourceforge.net/). We applied hard filters according to GATK guidance, with the following criteria used to filter raw SNPs: QD<2.0, FS>60.0, MQ<40.0, HaplotypeScore>13.0, MappingQualityRankSum<–12.5, ReadPosRankSum<–8.0, -cluster 3 -window 10. All SNPs were annotated using the ANNOVAR program (Wang et al., 2010).
Population structure analysis
To infer the population relationships among different domesticated donkey populations, population structure was deduced using ADMIXTURE, a tool for maximum-likelihood (ML) estimation of individual ancestries from multi locus SNP genotype datasets (Alexander et al., 2009), with different K values from 2 to 5.
Detection of selective sweep
We calculated the genome-wide distribution of population fixation statistics FST and nucleotide diversity θπ with a window size of 50 kb and a step size of 25 kb. Putative selection targets were extracted with the top 5% of log ratios for both θπ and FST. Our approach was to identify genomic regions with high differentiation between Chinese plain donkeys (n=28) and Tibetan donkeys (n=24). The locus-specific branch length (LSBL) of Tibetan donkeys was calculated by pairwise FST distances with dTP, dTF, and dPF (P represents Chinese plain donkeys, F represents foreign donkeys, T represents Tibetan donkeys), where LSBLTibetan=(dTP+dTF–dPF)/2 (Shriver et al., 2004).
To detect whether a selective sweep (a beneficial allele that recently reached fixation due to strong positive natural selection) has occurred in the kiang population, we calculated nucleotide diversity around exonic substitutions with a non-overlapping window size of 10 kb using vcftools v0.1.11 (Danecek et al., 2011).
SweeD analysis
The SweeD v4.0.0 program (Pavlidis et al., 2013) was used to detect selective sweeps for the three populations (i.e., kiangs, Tibetan donkeys, and plain donkeys) using a 10 kb non-overlapping window. This program implements the composite-likelihood ratio (CLR) statistic, which identifies regions with significant deviations from the neutral site frequency spectrum (SFS).
Coalescent simulation
To determinate the threshold for detection of outlier windows, we conducted coalescent simulations using the msms v3.2rc program (Ewing & Hermisson, 2010) based on demographic parameters derived from the best-fitting model inferred by δaδi (Gutenkunst et al., 2009) (Supplementary Table S21). For neutrality, only intergenic SNPs with more than 40-fold coverage at the population-level and minor allele frequencies (MAF)>0.01 were considered. Fixed sites in the kiang population were considered as ancestor alleles. A total of 15 divergence models were considered among the three populations, i.e., Chinese plain, Tibetan, and Foreign plain donkeys (Nigeria, Kenya, Egypt, Iran, and Kyrgyzstan). The model with the maximum log-likelihood value was chosen as the best one. We simulated genotypes corresponding to a 50–100 kb region with the same sample size as the real data 10 000 times according to the estimation from the best model. We converted the .ms format files into .vcf format by a custom Perl script. We calculated theFST, LSBL, and log π-ratio using the same pipeline as mentioned above for these sequences. The statistical significance between the simulated and observed data was measured using the randtest function in the ade4 R package. The recombination rate used here was 1 cM/Mb, and the mutation rate and generation time were 7.242×10−9 per site per generation and eight years, respectively (McVean et al., 2004; Orlando et al., 2013). The commands used for running the msms software were as follows: For Chinese plain, Tibetan, and Foreign plain donkey: java -jar msms3.2rc-b163.jar -ms 186 10000 -N 10000 -I 3 82 56 48 -t 14.484 -r 400 50000 -n 1 0.9474 -n 2 1.0707 -n 3 1.0904 -m 1 2 2.2049 -m 2 1 2.1552 -m 2 3 1.9153 -m 3 2 2.0253 -g 1 0.928 -g 2 0.898 -g 3 0.769 -ej 0.00195 3 2 -en 0.001953 2 0.8279 -ej 0.00897 2 1 -en 0.00897 1 0.5171 -threads 10. For kiang: java -jar msms3.2rc-b163.jar -ms 12 10000 -t 107.2 -r 400 100000 -threads 10.
Analysis of genetic introgression
We inferred gene flow among the different donkey (Kyrgyzstan, Nigeria, Kenya, Egypt, Iran, Tibet, and Chinese plain) and kiang populations, with zebra as the outgroup species, based on maximum-likelihood (ML) implemented in TreeMix. The command was "-i input -noss -m migration events –root zebra -o output", and migration events from 1 to 4 were gradually added to the ML tree. Genetic introgression events were also detected using the D-statistic (ABBA-BABA test) in ADMIXTOOLS (Patterson et al., 2012). We calculated the fd statistic, a modified version of the D-statistic described in Martin et al. (2015), using sliding window analysis with 50 kb windows.
Gene enrichment analysis
Gene Ontology (GO) enrichment analyses were performed using the DAVID program (https://david.ncifcrf.gov/).
RESULTS
Kiang genome assembly
We first de novo assembled the kiang genome using ~400 Gb of data sequenced by the Illumina Hiseq 2000 & 2500 platform from multiple paired-end and mate-pair libraries constructed with varying length fragments (220 bp to 17 kb). The scaffold and contig N50 sizes of the draft genome were 17 Mb and 264 kb, respectively ( Figure 1A; Supplementary Text, Figures S1, S2 and Tables S1–S3). We assessed the completeness of our assembly by aligning the protein-coding genes of the horse to the kiang genome using BLAT software (Kent, 2002). We retrieved 22 308 of 22 632 horse coding sequences (>98%) in the kiang assembly, indicating a gene region completeness of over 98.00% (Supplementary Table S4). This completeness was also supported by a high BUSCO (Benchmarking Universal Single-Copy Orthologs) score (Simão et al., 2015) of >96%, which indicated that our assembly contained the vast majority of near-universal single-copy orthologs (Supplementary Table S5). The gene model sets predicted by multiple methods were integrated using GLEAN to form a comprehensive and non-redundant gene set. After filtering short genes (<150 bp), we identified a total of 27 178 protein-coding genes with an average gene length of ~17 204 bp and a mean exon length of ~157 bp (Supplementary Tables S6, S7). Approximately 760 Mb of repeat sequences were identified by RepeatMasker, accounting for ~32% of our assembly (Supplementary Table S8).
Figure 1.

Genome evolution in kiangs
A: Distribution of structural variants compared with horse genome. Tracks (outside to inside) show chromosomes, a: indel density, b: insertion density, c: deletion density, d: translocation density, e: gene density, f: repeat density. Density of indels, insertions, deletions, and translocations, was calculated from a 1 Mb non-overlapping sliding window, and 500 kb non-overlapping sliding window for gene density and repeat density of the horse. B: Expression analysis of REGs based on human expression data. Analysis was performed as described previously (Li et al., 2013). Human gene expression data (Human U133A Gene Atlas) in 84 tissues or cells were downloaded from BioGPS (Wu et al., 2016) (http://biogps.org/#goto=welcome). Relative expression level of REGs in each tissue was calculated by mean expression value of REGs in tissue divided by average whole-genome expression value. Only top 10 tissues/cell lines are presented. Species tree of six mammals was used to detect positively selected genes in kiang lineage (as foreground lineage) by branch site model in PAML. C: McDonald-Kreitman (MK) test identified several genes related to immunity, DNA damage, energy metabolism, and angiogenesis under positive selection in kiang lineage.
Rare genetic introgression between kiangs and Tibetan donkeys
To assess the possibility of introgression between kiangs and Tibetan donkeys, we analyzed the genomes of five kiangs and 93 domestic donkeys (24 from Tibetan Plateau, 28 from Chinese lowland, eight from Iran, 26 from Africa, and seven from Middle Asia), including the 80 genomes generated in this study (Figure 2A; Supplementary Table S9), with a median depth of 7.50× and coverage of 96.79% of the assembled genome. We mapped the re-sequenced reads to the draft kiang genome for polymorphism calling for analysis of population genetics. After mapping the sequenced reads to the kiang reference genome, we called a total of 22 056 186 SNPs, including 81 592 non-synonymous and 68 064 synonymous SNPs, using the GATK pipeline (Supplementary Tables S10, S11 and Figures S3–S5).
Figure 2.
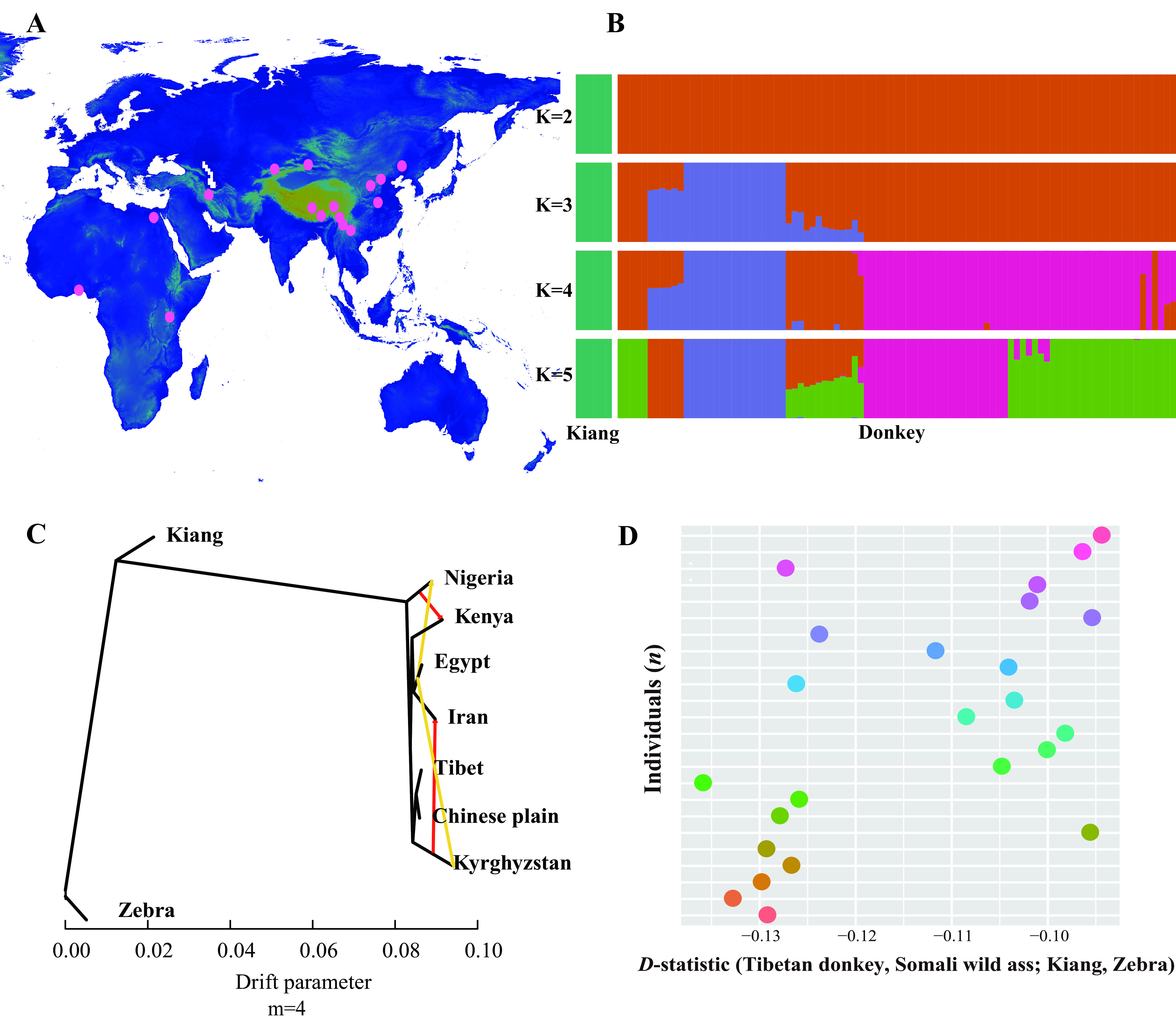
Population genetics analysis of kiangs and domestic donkeys
A: Geographical location of domestic donkeys with re-sequenced genomes. Blue through light green indicate low to high altitude. B: Population structure analysis by Admixture with K from 2 to 5. C, D: No genetic introgression between kiang and Tibetan donkey was revealed by D-statistic and TreeMix.
ADMIXTURE analysis separated kiangs from Tibetan donkeys without any admixture signals (Figure 2B). TreeMix analyses did not detect a migration edge between the Tibetan donkeys and kiangs, further suggesting that introgression between these lineages did not occur (Figure 2C; Supplementary Figure S6). Considering potential introgression between the Asian wild ass and domestic donkey (Jónsson et al., 2014), we calculated the D-statistic (ABBA-BABA test) of ADMIXTOOLS in the form (Tibetan donkey, Somali wild ass; Kiang, Zebra), which yielded a D-value<0 (|Z|>3;Figure 2D; Supplementary Figure S7 and Table S12). This pattern suggested gene flow between the Somali wild ass and kiang or between the Tibetan donkey and zebra. Additional analyses using the fd statistic (Martin et al., 2015) did not identify any gene flow signals between kiangs and Tibetan donkeys (Supplementary Table S13).
We then computed the fd statistics in non-overlapping 50 kb sliding windows across the Tibetan donkey genome to further assess whether undetected low-level gene flow (i.e., below the detection threshold of ADMIXTURE and D-statistics) could have left a localized footprint in the genome. The level of divergence (dxy) between the kiang and Tibetan donkey in the top 1% of fd regions, was, on average, slightly higher (0.3337) than in the rest of the genome (0.3105). This pattern did not support genetic introgression. Furthermore, we manually checked the windows with the top four highest fd values. The phylogenetic tree suggested a potential genetic introgression signature in these segments from Tibetan donkeys to kiangs (Supplementary Figure S8), although it may also be attributable to incomplete lineage sorting. Therefore, these results suggest rare genetic introgression between kiangs and Tibetan donkeys, although we cannot absolutely exclude introgression at some small regions.
Genomic substitutions underlying kiang evolution
The lack of admixture between kiangs and Tibetan donkeys indicates that these species acquired their adaptation to high altitude independently. To assess whether these processes of adaptation involved similar pathways, we used the dN/dS ratio to identify rapidly evolving genes (REGs) in the kiang genome. After identifying 5 778 high-confidence one-to-one orthologous genes among the kiang, human, donkey, horse, pig, and rhino genomes, we used the branch site model in the Codeml program of PAML (Yang, 2007) to detect genes under positive selection in the kiang lineage. This analysis yielded 164 protein-coding REGs with elevated dN/dS ratios in the lineage leading to kiangs (P<0.05) (Supplementary Table S14) (Zhang et al., 2005).
We then used the BioGPS dataset (Wu et al., 2016), which contains expression data from 84 human tissues/cell types, to characterize the function of the REGs, as described in our previous study (Li et al., 2013). The REGs displayed high expression levels in cell lines and tissues related to the immune system, thus supporting the function of some REGs in immunity (Figure 1B). The rapid evolution of immune genes has been commonly reported in different mammals and is likely due to an evolutionary “arms race” with pathogens (Kosiol et al., 2008). Additional gene enrichment analysis did not identify any significantly enriched terms but indicated that eight REGs were involved in the pathway “regulation of growth”, and four REGs (EP300, P2RX3, CREBBP, and ALDH2) were involved in the pathway “response to oxygen levels” (Supplementary Table S15).
We then examined gene interactions among REGs using the BioGRID database (Stark et al., 2006) (https://thebiogrid.org/). We found frequent gene-gene interactions among the REGs. Interestingly, many of these interactions involved EP300 as a hub gene, which showed the second highest number of interactions with other genes (Supplementary Figure S9). EP300 has been identified as a co-activator of HIF1α and plays a role in the stimulation of hypoxia-induced genes such as VEGF (Zhang et al., 2013). However, as EP300 has many other functions, future studies are necessary to identity the functional consequences of rapid EP300 evolution.
False-positive branch site tests can be high due to many confounding factors, like multi-nucleotide mutations (Venkat et al., 2018). Therefore, we further leveraged our re-sequencing data to identify fixed amino acid substitutions in the kiang lineage using the McDonald-Kreitman (MK) test in the PopGenome package (Pfeifer et al., 2014). This analysis identified a total of 30 genes under positive selection in the kiang lineage, including genes related to immunity, DNA damage, energy metabolism, and angiogenesis (Figure 1C; Supplementary Table S16, P<0.05). None of these genes, however, overlapped with the REGs identified by PAML, likely due to the different statistical principles used. PAML assumes that amino acid differences are fixed. This assumption, however, is likely to be violated when comparing closely related lineages such as kiangs and donkeys.
Interestingly, the MK test detected some genes involved in vascular development, an important component for hypoxia adaptation. For example, the TEK gene encodes the TEK receptor tyrosine kinase, a receptor that binds to the ligand angiopoietin-1 and mediates a signaling pathway during embryonic vascular development (Puri et al., 1999). NOTCH1 encodes the notch receptor 1 in the notch signaling pathway, a key pathway for angiogenesis (Limbourg et al., 2005).
Hard selective sweep in EPAS1 in kiangs
To detect positive-selection signals in the kiang population, we explored population genetics including nucleotide diversity (in 10 kb windows) and CLR of a sweep model using the SweeD program (Pavlidis et al., 2013). We identified a total of 248 genes in the top 1% of CLR values and 1 141 genes in windows that showed the lowest 1% of nucleotide diversity. A total of 34 genes were found to overlap between these analyses (Supplementary Table S17). Demographic history simulation also indicated that these genes evolved under positive selection compared to the null demographic model (P<0.01). However, no GO category was significantly enriched in this set of 34 genes. The functional consequences of these candidate PSGs were unclear, and thus require future validation and study. In addition to the high-altitude environment, there may be other forces driving the rapid evolution of these genes.
The adaptive evolution of EPAS1 is tightly coupled to hypoxia adaptation in Tibetan people and animals (Beall et al., 2010; Gou et al., 2014; Huerta-Sánchez et al., 2014; Lorenzo et al., 2014; Simonson et al., 2010; Wang et al., 2014). Here, simulation of demographic history supported signatures of selective sweep across EPAS1 in the kiang population with significantly lower nucleotide diversity and higher CLR values (P<0.01). By comparing population re-sequencing data from donkeys and kiangs, at theEPAS1 locus, we found a non-synonymous substitution in the kiang population (Figure 3). However, using the same methodology, we found no evidence of positive selection at EPAS1 in the Tibetan donkey and no evidence that it was affected by adaptive admixture from the kiang (see following section).
Figure 3.
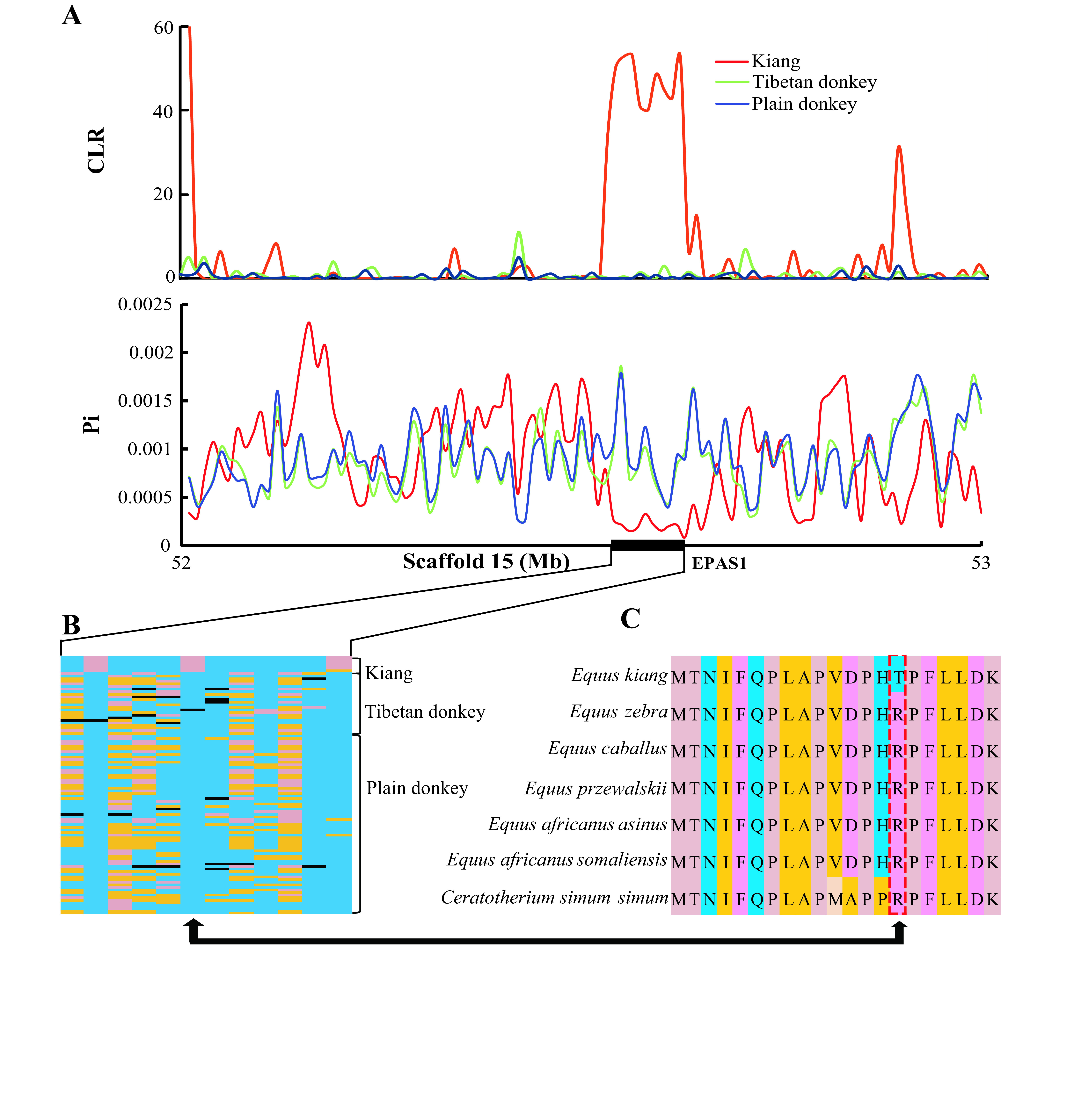
Hard selective sweep in EPAS1 in kiangs
A: Composite-likelihood ratio (CLR) detected by SweeD and nucleotide diversity levels around EPAS1 gene in different populations, including kiang, Tibetan donkey, and plain donkey. Results indicate that this gene likely experienced a hard selective sweep in kiangs. B: Haplotype of nucleotide mutations in EPAS1 showing high level of divergence between kiangs and domestic donkeys. Pink, yellow, blue, and black indicate genotypes of Homozygous variant, Heterozygote, Homozygous reference, and No call, respectively. C: Partial EPAS1 amino acid sequences among different species.
The signature of selection in EPAS1 corroborates the hard selective sweep, in which a beneficial allele has recently reached fixation due to strong positive natural selection. We further evaluated the hard selective sweep mode of adaptation at the genome-wide scale in the kiang. A hard selective sweep will deepen diversity around those changes most likely to have functional consequences (i.e., amino acid substitutions) (Enard et al., 2014). As described in previous research investigating the patterns of hard selective sweeps in humans (Hernandez et al., 2011), we explored diversity levels across non-synonymous and synonymous mutations fixed in the kiang population (Figure 4) Consistent with the finding in the human population (Hernandez et al., 2011), the diversity around the non-synonymous mutations was similar to that around the synonymous mutations (Figure 4). This indicates that genome-wide hard selective sweeps may be rare in kiangs, as reported in humans (Hernandez et al., 2011).
Figure 4.
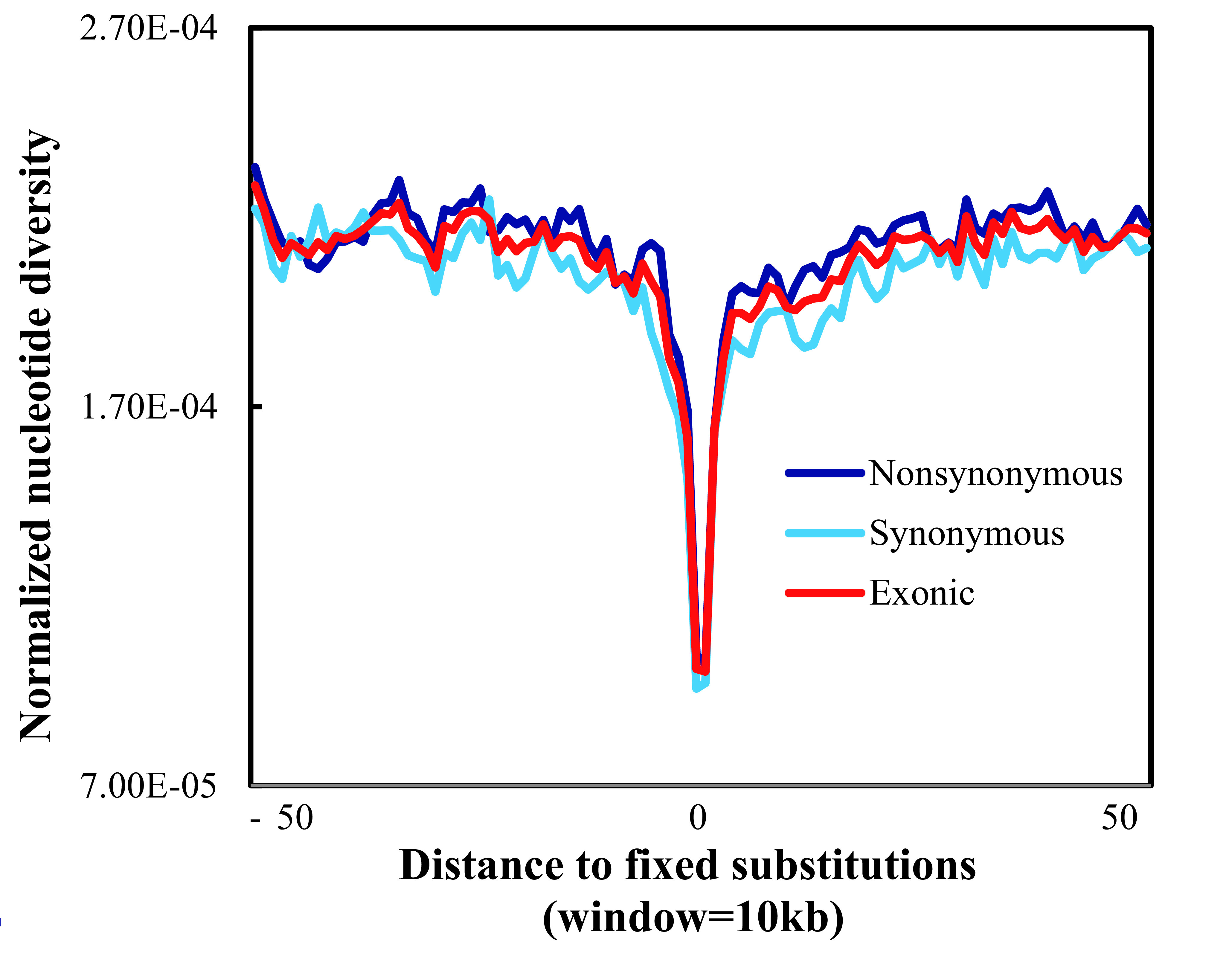
Rare hard selective sweep in kiangs at genome-wide scale
Normalized nucleotide diversity was calculated as nucleotide diversity level in kiang population divided by donkey-kiang divergence around fixed substitutions using a non-overlapping window size of 10 kb.
Evidence for selective sweep at EGLN1 in Tibetan donkeys
To investigate the potential genetic mechanism underlying high-altitude adaptation in Tibetan domestic donkeys, we performed population genetics analyses on the genomes of 93 donkeys. The phylogenetic tree and ancestry estimate analysis by ADMIXTURE (Supplementary Figures S10, S11) indicated that Tibetan donkeys are a genetically homogeneous subpopulation that diverged from the other six populations of donkeys (Kyrgyzstan, Nigeria, Kenya, Egypt, Iran, and lowland China) sequenced in this study. The pattern of population variation also supports the out-of-Africa theory for the domestic donkey, with a higher level of genetic diversity (Supplementary Table S18), private variants (Supplementary Figure S12), and a higher decay rate of linkage disequilibrium (LD) (Supplementary Figure S13).
To investigate natural selection in the Tibetan donkey, we first computed the FST (Akey et al., 2002) between Tibetan and Chinese plain donkeys across their genomes. Here, we found that the genic region exhibited a significantly higher FST value than the intergenic region (Figure 5A, P<2.2e-16). In addition, we divided SNPs into different classes according to theFST value (e.g., 0–0.1, 0.1–0.2, 0.2–0.3, 0.3–0.4, 0.4–0.5, >0.5), and found that population differentiation was more pronounced at non-synonymous SNPs than other types of SNPs ( Figure 5B, P=0.003 by chi-square test; Supplementary Figure S14). A pattern of excess genic SNPs with high FST values (>0.4) between Tibetan domestic donkeys and lowland donkeys was found when we constrained the analyses to SNPs presenting similar minor allele frequencies (Figure 5C; Supplementary Figure S14). This suggests that positive natural selection has, at least partly, driven population differentiation between Tibetan and lowland donkeys.
Figure 5.
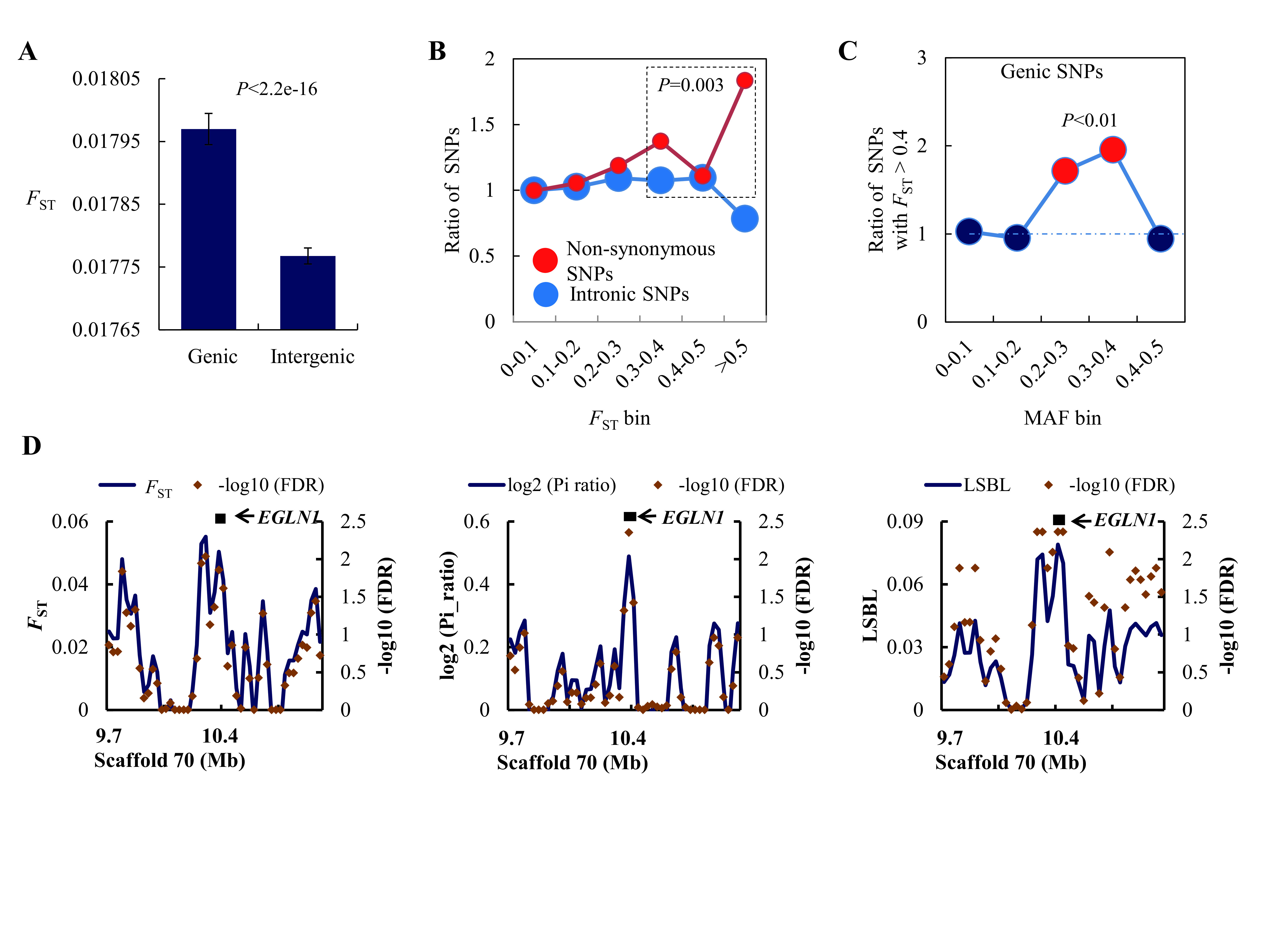
Evidence of high-altitude adaptation in Tibetan domestic donkeys
A: By comparing the genomes of Tibetan donkey populations and others, the genic region exhibited significantly higher FST values than the intergenic region. Statistical significance was calculated by Mann-Whitney U test. B: Population differentiation was more pronounced in non-synonymous SNPs than other types of SNPs. Statistical significance was calculated by chi-square test. C: A pattern of excess genic SNPs with high FST values (>0.4) between Tibetan domestic donkeys and lowland donkeys was found when constraining analyses to SNPs presenting similar minor allele frequencies (MAF). Statistical significance was calculated by chi-square test. D: Landscape ofFST, Pi (nucleotide diversity), and LSBL values corroborates strong positive selection on EGLN1 gene. –log10 transformed FDR P-values are presented.
To further explore the genetic mechanisms underlying high-altitude adaptation, we identified PSGs in the Tibetan donkey lineage by computing the FST, LSBL, and nucleotide diversity ratio (Δπ) between Tibetan and Chinese plain donkeys using sliding windows across the donkey genomes (Figure 5D; Supplementary Figure S15). These summary statistics were compared to simulated ones based on a neutral demographic model inferred by δaδi (Gutenkunst et al., 2009). A total of 158 candidate genes were identified by all three methods (FDR-corrected P<0.01) (Supplementary Figure S16 and Table S19). However, no gene category was found to be significantly enriched. We also manually checked the candidate PSGs detected by each method. One specific candidate was particularly noted: i.e.,EGLN1. This gene displayed a significantly higher LSBL (FDR-corrected P=0.0044), significantly lower nucleotide diversity (FDR-corrected P=0.0043), and borderline significant FST (FDR-corrected P=0.014) (Figure 5D). The EGLN1 gene, which encodes for HIF prolyl 4-hydroxylase 2 (PHD2), is a key gene for hypoxia adaptation in Tibetans, alongside EPAS1 (Bigham et al., 2010; Lorenzo et al., 2014; Peng et al., 2011; Simonson et al., 2010; Xiang et al., 2013; Xu et al., 2011). Therefore, our results indicate that Tibetan donkeys did not acquire their ability to withstand high altitude via adaptive introgression or through mutations of the EPAS1 gene, suggesting that kiangs and Tibetan donkeys acquired adaptations independently and through different biological pathways.
Potential independent adaptation to high altitude between kiangs and Tibetan domestic donkeys
Although EPAS1 and EGLN1 do not appear to have evolved in parallel in kiangs and Tibetan donkeys, it is possible that their parallel adaptation to high altitudes involved other genes. To test this hypothesis, we aligned sequencing reads from kiangs and donkeys to the horse reference genome (outgroup) and ran SweeD using these alignments. This allowed us to limit any issue arising from reference bias and identify candidate PSGs in both kiangs and Tibetan domestic donkeys (Figure 6). Among the 2 243 10 kb windows (top 1%) under potential positive selection, only 11 windows (0.49%) distributed on different chromosomes were shared between the two populations, covering 22 protein-coding genes (hypergeometric P=8.08e-11), none of which were related to high-altitude adaption (Supplementary Table S20). Thus, our results suggest that no parallel adaptation to high altitude occurred between these two closely related species.
Figure 6.
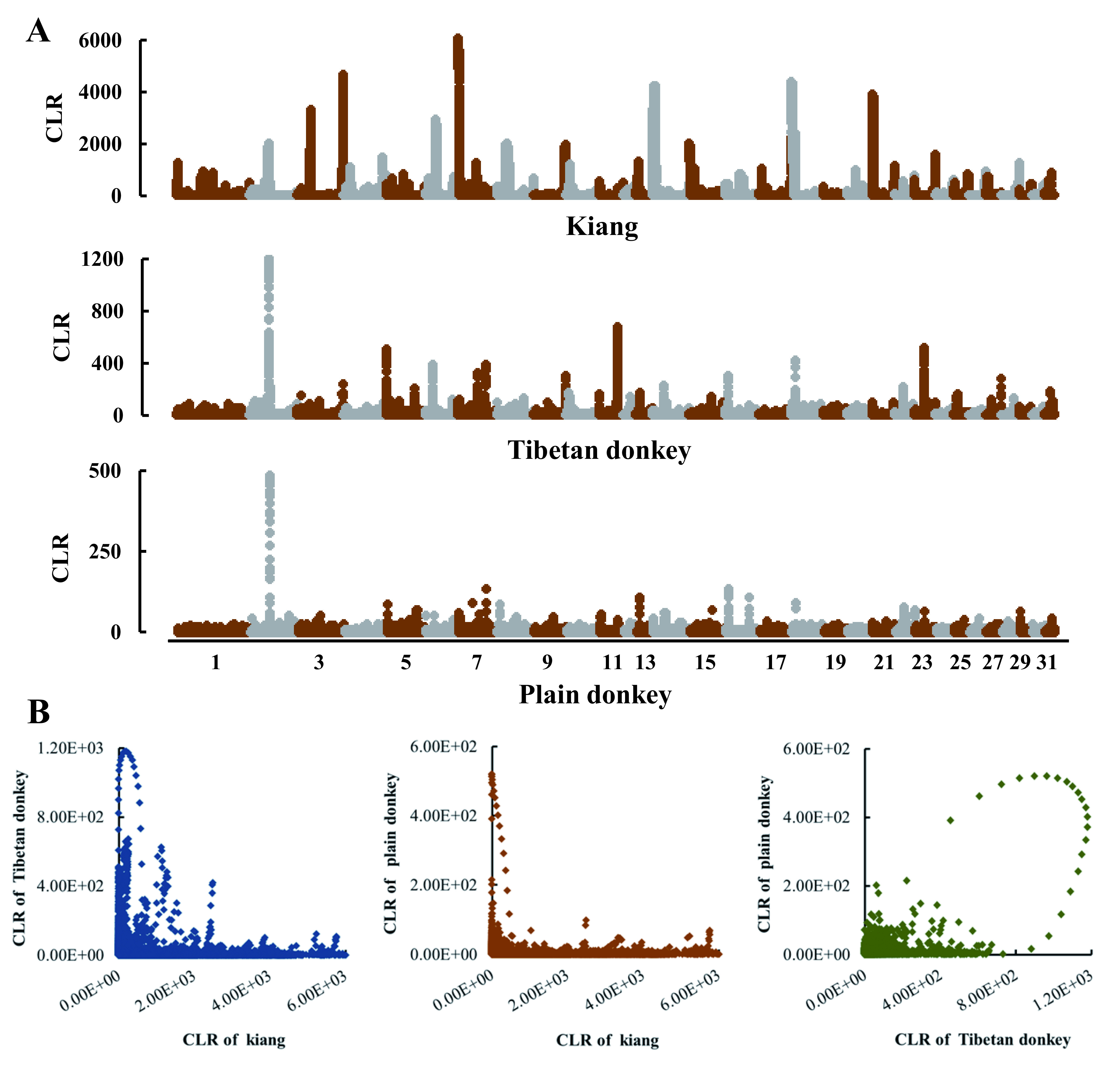
Selective sweep analysis by SweeD in kiang, Tibetan donkey, and plain donkey populations (A) and rare high CLR regions overlapped in the three populations(B)
DISCUSSION
The extreme environment of plateau regions can lead to hypoxia in animals, representing a considerable challenge for life, particularly for introduced livestock. In the present study, we assembled a draft de novo genome of the kiang and performed large-scale re-sequencing of kiang and domestic donkey genomes. Our findings demonstrated that kiangs and Tibetan donkeys have utilized different genes (EPAS1 and EGLN1, respectively) to adapt to the low-oxygen conditions associated with living at high altitudes. Interesting, both EPAS1 and EGLN1 are the two most important genes for high-altitude adaptation in Tibetans and other plateau animals (Beall et al., 2010; Foll et al., 2014; Ge et al., 2013; Gou et al., 2014; Huerta-Sánchez et al., 2014; Lorenzo et al., 2014; Qiu et al., 2012; Qu et al., 2013; Simonson et al., 2010; Wang et al., 2014; Wang et al., 2016). This suggests that the number of potential biological pathways involved in high-altitude adaptation in mammals may be limited.
While EPAS1 is a clear candidate for adaptation to high altitudes in kiangs, other genes not detected in our analyses may also be involved. This is likely to be the case given the small sample size (n=5) of kiang genomes available for this study. Future study based on additional samples will help to clarify the population structure and demographic history of kiangs, as well as identify signatures of positive natural selection.
Our findings indicate that Tibetan donkeys did not acquire their ability to withstand high altitudes via adaptive introgression with kiangs. Although hybrids between kiangs and horses, donkeys, and wild asses have been reported in captivity (Gray, 1972; Hay, 1859; Kinloch, 1869), e.g., a male kiang-donkey hybrid was born in London Zoological Gardens in 1920 (Flower, 1929), no evidence exists that kiang hybrids can reproduce. Rare genetic introgression between kiangs and Tibetan donkeys may also be due to limited encounters given the short time that donkeys have been living on the Tibetan Plateau. Given their biological similarities, however, the adaptive variants in both EGLN1 and EPAS1 described here could provide markers for breeding more resilient donkeys in other high-altitude regions of the world.
DATA AVAILABILITY
All sequences reported in this study have been deposited in the Genome Sequence Archive database (http://gsa.big.ac.cn/) under Accession ID (CRA001222).
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
D.D.W. and Y.P.Z. designed and led the project, D.D.W., L.Z., and L.A.F.F. prepared the manuscript. L.Z., H.Q.L., X.L.T., and C.M.J. performed data analysis, C.F.W., X.G., S.W., M.S.W., M.C.W., X.L.L, J.L.H., and H.K.Z. performed part of the data analysis, H.C., A.E., A.C.A., R.A.M.A., O.O., S.C.O., O.J.S., M.G.F, S.C.O., B.A., and J.K.L. performed some sampling. All authors read and approved the final version of the manuscript.
Funding Statement
This work was supported by the National Natural Science Foundation of China (31621062), Strategic Priority Research Program of the Chinese Academy of Sciences (XDA2004010302), and Second Tibetan Plateau Scientific Expedition and Research (STEP) Program (2019QZKK05010703). D.D.W. was supported by the National Natural Science Foundation of China (91731304, 31822048), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13020600), Qinghai Department of Science and Technology Major Project, and State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, Yunnan University (2018KF001). Sampling of this work was also supported by the Animal Branch of the Germplasm Bank of Wild Species, Chinese Academy of Sciences (Large Research Infrastructure Funding)
Contributor Information
Chang-Fa Wang, Email: wangcf1967@163.com.
Ya-Ping Zhang, Email: zhangyp@mail.kiz.ac.cn.
Laurent A. F. Frantz, Email: laurent.frantz@qmul.ac.uk.
Dong-Dong Wu, Email: wudongdong@mail.kiz.ac.cn.
References
- 1.Akey JM, Zhang G, Zhang K, Jin L, Shriver MD Interrogating a high-density SNP map for signatures of natural selection. Genome Research. 2002;12(12):1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander DH, Novembre J, Lange K Fast model-based estimation of ancestry in unrelated individuals. Genome Research. 2009;19(9):1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beall CM, Cavalleri GL, Deng LB, Elston RC, Gao Y, Knight J, et al Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders . Proceedings of the National Academy of Sciences of the United States of America. 2010;107(25):11459–11464. doi: 10.1073/pnas.1002443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beja-Pereira A, England PR, Ferrand N, Jordan S, Bakhiet AO, Abdalla MA, et al African origins of the domestic donkey. Science. 2004;304(5678):1781. doi: 10.1126/science.1096008. [DOI] [PubMed] [Google Scholar]
- 5.Bigham A, Bauchet M, Pinto D, Mao XY, Akey JM, Mei R, et al Identifying signatures of natural selection in tibetan and andean populations using dense genome scan data. PLoS Genetics. 2010;6(9):e1001116. doi: 10.1371/journal.pgen.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Enard D, Messer PW, Petrov DA Genome-wide signals of positive selection in human evolution. Genome Research. 2014;24(6):885–895. doi: 10.1101/gr.164822.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ewing G, Hermisson J MSMS: a coalescent simulation program including recombination, demographic structure and selection at a single locus . Bioinformatics. 2010;26(16):2064–2065. doi: 10.1093/bioinformatics/btq322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flower SS List of the vertebrated animals exhibited in the Gardens of the Zoological Society of London, 1828–1927 centenary edition in 3 volumesVol 1: mammals . Nature. 1929;124(3135):836. [Google Scholar]
- 11.Foll M, Gaggiotti OE, Daub JT, Vatsiou A, Excoffier L Widespread signals of convergent adaptation to high altitude in Asia and America. The American Journal of Human Genetics. 2014;95(4):394–407. doi: 10.1016/j.ajhg.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge RL, Cai QL, Shen YY, San A, Ma L, Zhang Y, et al Draft genome sequence of the Tibetan antelope. Nature Communications. 2013;4:1858. doi: 10.1038/ncomms2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, et al High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(4):1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gou X, Wang Z, Li N, Qiu F, Xu Z, Yan DW, et al Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Research. 2014;24(8):1308–1315. doi: 10.1101/gr.171876.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray AP. 1972. Mammalian Hybrids. 2nd ed. Farnham Royal, Slough, United Kingdom: Commonwealth Agricultural Bureaux.
- 16.Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genetics. 2009;5(10):e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hay WE Notes on the kiang of Thibet (E. kiang) . Proceedings of the Zoological Society of London. 1859;27:353–357. [Google Scholar]
- 18.Hernandez RD, Kelley JL, Elyashiv E, Melton SC, Auton A, McVean G, et al Classic selective sweeps were rare in recent human evolution. Science. 2011;331(6019):920–924. doi: 10.1126/science.1198878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huerta-Sánchez E, Jin X, As an, Bianba Z, Peter BM, Vinckenbosch N, et al Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature. 2014;512(7513):194–197. doi: 10.1038/nature13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jónsson H, Schubert M, Seguin-Orlando A, Ginolhac A, Petersen L, Fumagalli M, et al Speciation with gene flow in equids despite extensive chromosomal plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(52):18655–18660. doi: 10.1073/pnas.1412627111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katoh K, Misawa K, Kuma KI, Miyata T MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research. 2002;30(14):3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kent WJ BLAT—the BLAST-like alignment tool. Genome Research. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinloch AAA. 1869. Large Game Shooting in Thibet and the North West. Oxford: Harrison.
- 24.Kong Y Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics. 2011;98(2):152–153. doi: 10.1016/j.ygeno.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Kosiol C, Vinař T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, et al Patterns of positive selection in six mammalian genomes. PLoS Genetics. 2008;4(8):e1000144. doi: 10.1371/journal.pgen.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Coghlan A, Ruan J, Coin LJ, Hériché JK, Osmotherly L, et al TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Research. 2006;34(suppl_1):D572–D580. doi: 10.1093/nar/gkj118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Vonholdt BM, Reynolds A, Boyko AR, Wayne RK, Wu DD, et al Artificial selection on brain-expressed genes during the domestication of dog. Molecular Biology and Evolution. 2013;30(8):1867–1876. doi: 10.1093/molbev/mst088. [DOI] [PubMed] [Google Scholar]
- 28.Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111(14):1826–1832. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenzo FR, Huff C, Myllymäki M, Olenchock B, Swierczek S, Tashi T, et al A genetic mechanism for Tibetan high-altitude adaptation. Nature Genetics. 2014;46(9):951–956. doi: 10.1038/ng.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Löytynoja A, Goldman N Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science. 2008;320(5883):1632–1635. doi: 10.1126/science.1158395. [DOI] [PubMed] [Google Scholar]
- 31.Ma XY, Ning T, Adeola AC, Li J, Esmailizadeh A, Lichoti JK, et al Potential dual expansion of domesticated donkeys revealed by worldwide analysis on mitochondrial sequences. Zoological Research. 2020;41(1):51–60. doi: 10.24272/j.issn.2095-8137.2020.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin SH, Davey JW, Jiggins CD Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Molecular Biology and Evolution. 2015;32(1):244–257. doi: 10.1093/molbev/msu269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McVean GAT, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304(5670):581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- 35.Miao BP, Wang Z, Li YX Genomic analysis reveals hypoxia adaptation in the Tibetan Mastiff by introgression of the gray wolf from the Tibetan Plateau. Molecular Biology and Evolution. 2017;34(3):734–743. doi: 10.1093/molbev/msw274. [DOI] [PubMed] [Google Scholar]
- 36.Orlando L, Ginolhac A, Zhang GJ, Froese D, Albrechtsen A, Stiller M, et al Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature. 2013;499(7456):74–78. doi: 10.1038/nature12323. [DOI] [PubMed] [Google Scholar]
- 37.Patterson N, Moorjani P, Luo YT, Mallick S, Rohland N, Zhan YP, et al Ancient admixture in human history. Genetics. 2012;192(3):1065–1093. doi: 10.1534/genetics.112.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pavlidis P, Živković D, Stamatakis A, Alachiotis N SweeD: likelihood-based detection of selective sweeps in thousands of genomes. Molecular Biology and Evolution. 2013;30(9):2224–2234. doi: 10.1093/molbev/mst112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng Y, Yang ZH, Zhang H, Cui CY, Qi XB, Luo XJ, et al Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Molecular Biology and Evolution. 2011;28(2):1075–1081. doi: 10.1093/molbev/msq290. [DOI] [PubMed] [Google Scholar]
- 40.Pfeifer B, Wittelsbürger U, Ramos-Onsins SE, Lercher MJ PopGenome: an efficient swiss army knife for population genomic analyses in R. Molecular Biology and Evolution. 2014;31(7):1929–1936. doi: 10.1093/molbev/msu136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puri MC, Partanen J, Rossant J, Bernstein A Interaction of the TEK and TIE receptor tyrosine kinases during cardiovascular development. Development. 1999;126(20):4569–4580. doi: 10.1242/dev.126.20.4569. [DOI] [PubMed] [Google Scholar]
- 42.Qiu Q, Zhang GJ, Ma T, Qian WB, Wang JY, Ye ZQ, et al The yak genome and adaptation to life at high altitude. Nature Genetics. 2012;44(8):946–949. doi: 10.1038/ng.2343. [DOI] [PubMed] [Google Scholar]
- 43.Qu YH, Zhao HW, Han NJ, Zhou GY, Song G, Gao B, et al Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau. Nature Communications. 2013;4:2071. doi: 10.1038/ncomms3071. [DOI] [PubMed] [Google Scholar]
- 44.Shriver MD, Kennedy GC, Parra EJ, Lawson HA, Sonpar V, Huang J, et al The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Human Genomics. 2004;1(4):274. doi: 10.1186/1479-7364-1-4-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 46.Simonson TS, Yang YZ, Huff CD, Yun HX, Qin G, Witherspoon DJ, et al Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329(5987):72–75. doi: 10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- 47.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M BioGRID: a general repository for interaction datasets. Nucleic Acids Research. 2006;34(suppl 1):D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venkat A, Hahn MW, Thornton JW Multinucleotide mutations cause false inferences of lineage-specific positive selection. Nature Ecology & Evolution. 2018;2(8):1280–1288. doi: 10.1038/s41559-018-0584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.vonHoldt B, Fan ZX, Ortega-Del Vecchyo D, Wayne RK EPAS1 variants in high altitude Tibetan wolves were selectively introgressed into highland dogs . PeerJ. 2017;5:e3522. doi: 10.7717/peerj.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang C, Li H, Guo Y, Huang J, Sun Y, Min J, Wang J, Fang X, Zhao Z, Wang S, et al Donkey genomes provide new insights into domestication and selection for coat color. Nature Communications. 2020;11(1):6014. doi: 10.1038/s41467-020-19813-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang GD, Fan RX, Zhai WW, Liu F, Wang L, Zhong L, et al Genetic convergence in the adaptation of dogs and humans to the high-altitude environment of the Tibetan Plateau. Genome Biology and Evolution. 2014;6(8):2122–2128. doi: 10.1093/gbe/evu162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang K, Li MY, Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang MS, Yang HC, Otecko NO, Wu DD, Zhang YP Olfactory genes in Tibetan wild boar. Nature Genetics. 2016;48(9):972–973. doi: 10.1038/ng.3631. [DOI] [PubMed] [Google Scholar]
- 54.Wu CL, Jin XF, Tsueng G, Afrasiabi C, Su AI BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Research. 2016;44(D1):D313–D316. doi: 10.1093/nar/gkv1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu DD, Ding XD, Wang S, Wójcik JM, Zhang Y, Tokarska M, et al Pervasive introgression facilitated domestication and adaptation in the Bos species complex . Nature Ecology & Evolution. 2018;2(7):1139–1145. doi: 10.1038/s41559-018-0562-y. [DOI] [PubMed] [Google Scholar]
- 56.Xiang K, Ouzhuluobu, Peng Y, Yang ZH, Zhang XM, Cui CY, et al Identification of a Tibetan-specific mutation in the hypoxic gene EGLN1 and its contribution to high-altitude adaptation . Molecular Biology and Evolution. 2013;30(8):1889–1898. doi: 10.1093/molbev/mst090. [DOI] [PubMed] [Google Scholar]
- 57.Xu SH, Li SL, Yang YJ, Tan JZ, Lou HY, Jin WF, et al A genome-wide search for signals of high-altitude Adaptation in Tibetans. Molecular Biology and Evolution. 2011;28(2):1003–1011. doi: 10.1093/molbev/msq277. [DOI] [PubMed] [Google Scholar]
- 58.Yang ZH PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 59.Zhang B, Day DS, Ho JW, Song LY, Cao JJ, Christodoulou D, et al A dynamic H3K27ac signature identifies VEGFA-stimulated endothelial enhancers and requires EP300 activity. Genome Research. 2013;23(6):917–927. doi: 10.1101/gr.149674.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang JZ, Nielsen R, Yang ZH Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Molecular Biology and Evolution. 2005;22(12):2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data to this article can be found online.
Data Availability Statement
All sequences reported in this study have been deposited in the Genome Sequence Archive database (http://gsa.big.ac.cn/) under Accession ID (CRA001222).