

Abstract
Pathologic roles of interleukin (IL)-2, IL-9, and IL-15, have been implicated in multiple T-cell malignancies and autoimmune diseases. BNZ-1 is a selective and simultaneous inhibitor of IL-2, IL-9, and IL-15, which targets the common gamma chain signaling receptor subunit. In this first-in-human study, 18 healthy adults (n = 3/cohort) received an intravenous dose of 0.2, 0.4, 0.8, 1.6, 3.2, or 6.4 mg/kg infused over ≤5 minutes on day 1 and were followed for 30 days for safety and pharmacokinetic/pharmacodynamic sample collection. No dose-limiting toxicities, infusion reactions, or serious or severe treatment-emergent adverse events were observed. Headache was the only treatment-emergent adverse event in >1 subject (n = 3). Peak and total BNZ-1 exposure was generally dose proportional, with a terminal elimination half-life of ~5 days. Pharmacodynamic effects of BNZ-1 on regulatory T cells (Tregs, IL-2), natural killer (NK) cells (IL-15) and CD8 central memory T cells (Tcm, IL-15) were measured by flow cytometry and used to demonstrate target engagement. For Tregs, 0.2 mg/kg was an inactive dose, while a maximum ~50% to 60% decrease from baseline was observed on day 4 after doses of 0.4 to 1.6 mg/kg, and higher doses produced an 80% to 93% decrease from baseline on day 15. Similar pharmacodynamic trends were observed for natural killer cells and CD8 Tcm, although decreases in CD8 Tcm were more prolonged. These subpopulations returned to/toward baseline by day 31. T cells (total, CD4, and CD8), B cells, and monocytes were unchanged throughout. These preliminary results suggest that BNZ-1 safely and selectively inhibits IL-2 and IL-15, which results in robust, reversible immunomodulation.
Keywords: common gamma cytokines, interleukin-2, Interleukin-15, natural killer cells, regulatory T cells
The common gamma chain (γ c) family of cytokines (interleukin [IL]-2, IL-4, IL-7, IL-9, IL-15, IL-21) is an important family of cytokines that controls major immune responses and has overlapping roles in T- and B-lymphocyte development and function.1 IL-2 is primarily produced by CD4 T cells in response to antigenic stimulation but is also produced, in lower amounts, by other immune cells (CD8 T cells, natural killer [NK] cells, activated dendritic cell, and mast cells) and is crucial for the maintenance of regulatory T cells (Tregs) and drives both CD4 and CD8 T cells to become terminally differentiated effector T cells during an immune response.1,2 IL-15 shares structural and receptor signaling subunits with IL-2, which create multiple functional redundancies.3 Both induce the proliferation and the cytolytic activity of NK cells and CD8 T cells, and the proliferation and differentiation of stimulated B cells. IL-15 is also critical for the homeostasis of memory phenotype and antigen-specific memory CD8 T cells and is best characterized for its role in maintaining memory pools of CD8 T cells. Aberrant IL-2 and IL-15 activity contribute to or pathologically drive many different human diseases including T-cell malignancies,4,5 graft-versus-host disease,6 and autoimmune diseases (eg, alopecia areata).7,8 IL-9 is another γ c cytokine that is produced by subsets of activated T-helper cells (Th2, Th9, and Th17 cells), which induces the activation of airway and intestinal epithelial cells, B cells, eosinophils, and mast cells.9–11 IL-9 has been identified as a promoter of oncogenesis in hematologic malignancies and as a regulator of allergic and autoimmune diseases.9,12 Binding of each cytokine to its private receptor subunit recruits the γ c molecule to the complex, enabling the heterodimerization of their cytoplasmic domains with activation of the Janus family tyrosine kinase, JAK1 (in association with the private chain) and JAK3 (in association with γ c), which phosphorylate signal transducer and activator of transcription (STAT) proteins STAT3 and STAT5, respectively.1,13 In addition to the JAK-STAT pathway, γ c cytokines act through the phosphatidylinositol 3′-kinase/protein kinase B and extracellular signal-regulated kinase/mitogen-activated protein kinase intracellular signaling pathways.2
Approaches to blocking disease-driving γ c cytokines in the clinic have utilized monoclonal antibodies targeted against a single cytokine in the γ c family (IL-1514) or JAK inhibitors,15 which partially inhibit signaling of all six γ c cytokines in this family, since they target only the JAK/STAT pathway and do not block phosphatidylinositol 3′-kinase/protein kinase B and extracellular signal-regulated kinase/mitogen-activated protein kinase pathways. A potentially more effective and safer targeted therapeutic approach may be achieved through a selective and simultaneous inhibition of multiple cytokines in the γ c family, first proposed by our previous study.16 Recent understanding of the structural biology of cytokines and their receptors has made it possible to design peptides that target the cytokine-receptor interface and selectively and simultaneously block multiple γ c cytokines. These technological advances led to the discovery and development of BNZ-1, a PEGylated, 24-amino acid peptide that binds to the γ c subunit of γ c cytokine receptors. Intriguingly, BNZ-1 selectively blocks IL-2, IL-9, and IL-15 signaling, while not affecting IL-4, IL-7, or IL-21, as shown in our previous publication (in which BNZ-1 is referred to as BNZ132-1).16 However, inhibition of IL-9 was achieved at concentrations 10 times higher (5 nM) than those that induced completed inhibition of IL-2 and IL-15 (0.5 nM). Therefore, it is likely that IL-9 blockade may be detectable only in patients with elevated IL-9 signaling where partial inhibition may produce a phacodynamic effect. BNZ-1 has been shown to block the temporal expansion of regulatory T cells in mice exposed to continued presence of human IL-2 and completely protected mice from an IL-15–mediated experimental leukemia model.16 Thus, BNZ-1 is a proven in vivo inhibitor of IL-2 and IL-15 action. BNZ-1 has also been recently tested in a murine model of graft-versus-host disease that occurs in immunod-eficient NSG mice engrafted with human peripheral blood mononuclear cells (PBMCs), which results in a life-threatening graft-versus-host disease accompanied by significant elevations in IL-6 and interferon-γ and complete loss of fur that are all reversed by BNZ-1. Investigational new drug–enabling safety studies have been completed that, along with the preclinical proof-of-concept studies, have demonstrated that BNZ-1 safely blocks the pathologic activity of IL-2 and IL-15 in humans, which supports the initial clinical testing of BNZ-1.
The purpose of this first-in-human study was to characterize the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of a range of single intravenenous (IV) doses of BNZ-1 in healthy adults.
Methods
This was a single-site, open-label, ascending, single-dose study of BNZ-1 in healthy adults. The study (clinicaltrials.gov NCT03046459) was conducted in compliance with applicable regulatory requirements, the Declaration of Helsinki, the International Conference on Harmonisation Guidance on Good Clinical Practice, and the study site’s Institutional Review Board. Prior to initiation of any study-specific procedures, informed consent was obtained for all subjects.
This was the first study of BNZ-1 conducted in humans. Therefore, selection of starting dose was made using the US Food and Drug Administration (FDA) guidance for maximum recommended starting doses in healthy volunteers, using FDA-guided conversion of no-observed-adverse-effect level of 50 mg/kg/dose in a mouse model and a safety factor of 10.17 Based on this method, the chosen starting dose of BNZ-1 was 0.4 mg/kg, with a 2× step-up for subsequent dose cohorts (Figure 1). Following evaluation of PD data from the 0.4 mg/kg cohort, which showed a significant PD effect, a lower-dose cohort of 0.2 mg/kg was added in order to characterize the low end of the exposure-response relationship (eg, identify a no effect level). As such, 6 sequential cohorts with a total of 18 adult subject were enrolled (Cohort 1: 0.4 mg/kg; Cohort 2: 0.8 mg/kg; Cohort 3: 1.6 mg/kg; Cohort 4: 3.2 mg/kg; Cohort 5: 6.4 mg/kg; Cohort 6: 0.2 mg/kg). BNZ-1 was administered as a single slow IV bolus over a maximum of 5 minutes. The dose administered was dependent upon the subject’s weight and cohort assignment. Dose escalation proceeded if the prior dose was considered safe and well tolerated, as defined by a review of all available safety data through day 15 by the Safety Review Committee (composed of the study principal investigator, the medical monitor, and the sponsor medical representative). The total enrollment of the study ultimately depended on whether any participant developed dose-limiting toxicity requiring cohort expansion by 3. The minimum number of subjects needed to complete the study was 18.
Figure 1.
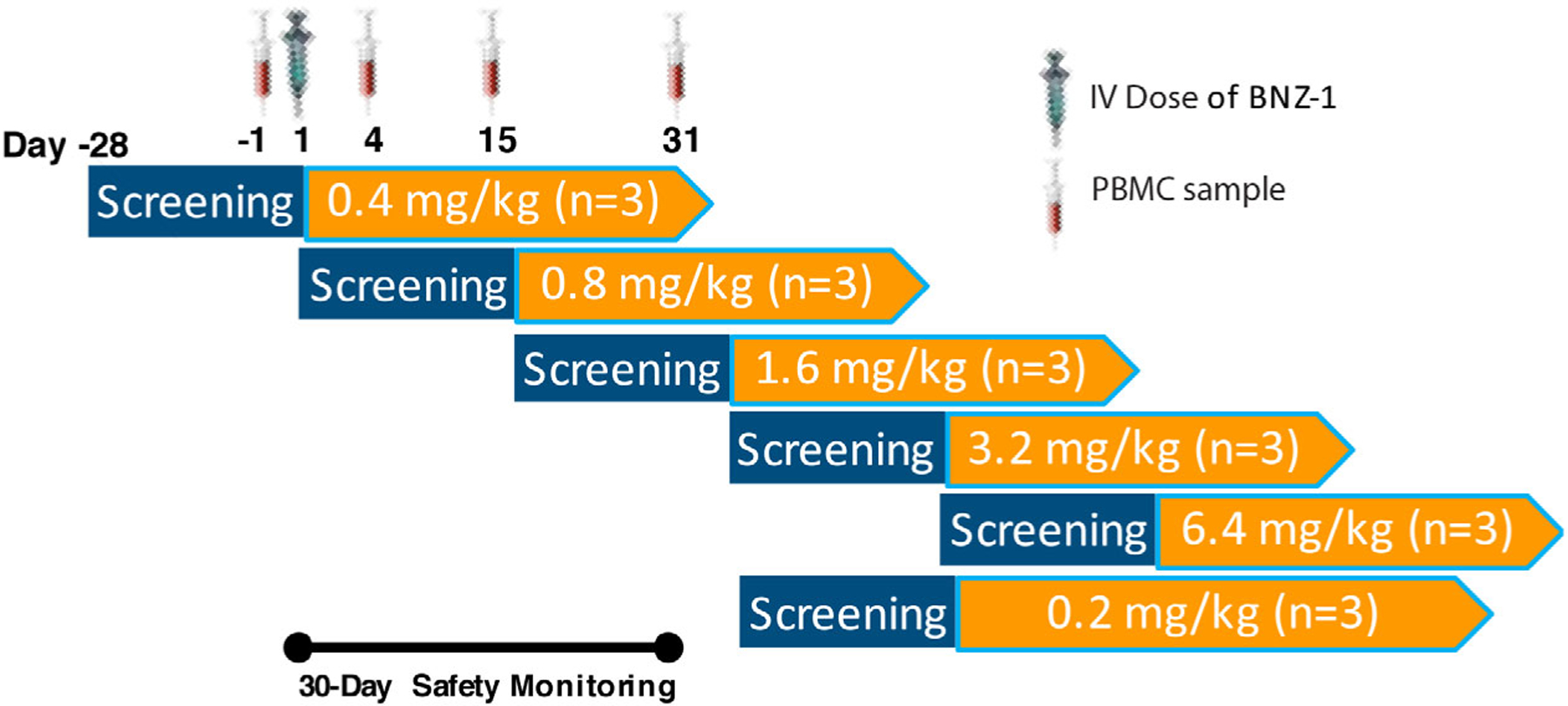
The first-in-human study included a 4-week screening period, a single IV dose of BNZ-1 administered on day 1, and a 30-day safety monitoring and sample collection period. PBMC samples for flow cytometry were collected before dosing and 3, 14, and 30 days after dosing. The starting dose of 0.4 mg/kg was estimated to be a no-effect dose based on preclinical data. However, 0.4 mg/kg was shown to have significant pharmacodynamic activity, so a lower dose of 0.2 mg/kg was added during the study to try and identify a no-effect dose. IV, intravenous; PBMC, peripheral blood mononuclear cell.
Key inclusion criteria required that subjects were healthy adults between 18 and 50 years of age, inclusive, and willing and able to provide informed consent. Female subjects needed to be nonpregnant and non-lactating and on an effective method of birth control for 3 months before and for the duration of the study. No use of prescription or over-the-counter drugs, including herbals, was allowed from 7 days before study drug administration through study completion except for oral contraceptives. All subjects had to be up to date on their immunizations per current Centers for Disease Control and Prevention/Department of Health and Human Services recommended immunizations for adults, as demonstrated by immunization records or antibody titers.
Key exclusion criteria were as follows: (1) clinically significant liver or kidney disease; (2) history of malignancy in the past 5 years; (3) recent systemic infection in the past month; (4) having received other investigational products or therapy in the past 30 days before enrollment; (5) clinically significant abnormal clinical laboratory results at baseline; and (6) having received a live vaccine immunization within 30 days before the study drug.
After informed consent was obtained and within a period of 30 days before dosing, all screening tests required to establish eligibility were completed. All subjects returned on day −1 and were admitted to the inpatient unit, at which time entry procedures and baseline clinical evaluation and laboratory blood work before dosing were performed. Subjects were considered enrolled once the investigator had confirmed they met all eligibility criteria on day 1.
On day 1, patients were dosed with BNZ-1, and blood samples (approximately 6 mL) for quantification of plasma levels of BNZ-1 were collected before dosing, immediately after dosing, and at 0.08, 0.5, 1, 2, 4, 8, and 24 hours after dosing. Subjects were discharged from the inpatient unit after clinical evaluation and laboratory blood work on day 2 and returned to the site for outpatient visits on days 4, 6, 8, 15, 22, and 31. At these time points, subjects were assessed for adverse events, had a physical examination, and had blood collections for clinical safety labs, PK, and PD. The final clinical evaluation and blood collection occurred on day 31.
Study Outcome Measures
The primary outcome measure was safety, defined by the incidence, severity, and relationship of treatment-emergent adverse events (TEAEs), and clinically significant changes on clinical labs (hematology, serum biochemistry, liver enzymes), vital signs, and physical examination, and the incidence of antidrug antibodies. A TEAE was defined as an adverse effect that started or worsened after the start of the study drug IV infusion. All TEAEs occurring during this clinical trial were coded using the Medical Dictionary for Regulatory Activities, Version 19.1, tabulated by system organ class, summarized by severity (grade 1 = mild, grade 2 = moderate, grade 3 = severe, grade 4 = life-threatening or disabling, and grade 5 = death) and relatedness to study drug. Secondary outcome measures included the PK of BNZ-1, which was measured in the plasma using a validated enzyme-linked immunosorbent assay (ELISA), as determined by the following PK parameters: maximum observed plasma concentration (Cmax), area under the plasma concentration–time curve (AUC) from time 0 to time of last measurable concentration (AUC0-t) and to infinity (AUC0-inf), time to Cmax, clearance, volume of distribution, apparent terminal elimination rate constant, and terminal elimination half-life (t1/2). PK parameters were calculated using WinNonLin, Version 6.3 (Certara, Princeton, New Jersey). The other secondary outcome measure was the PD of BNZ-1 as determined by flow cytometric analysis of whole blood samples collected on days 4, 15, and 31, to determine the mean percent change from baseline for total T cells, CD4 T cells, CD8 T cells, Tregs, NK cells, CD8 central memory T cells (Tcm), B cells, and monocytes. The Tregs (IL-2), NK cells (IL-15), and CD8 Tcm (IL-15) were considered relevant PD biomarkers for demonstrating target engagement and exposure-response for BNZ-1, while the other populations were considered in the context of specificity and safety. In addition, the maximum observed percent change from baseline at any time after dosing was calculated only for Tregs, NK cells, and CD8 Tcm.
ELISA and Flow Cytometry Methods
Sandwich ELISA for the Detection of BNZ-1 in Human Plasma.
Microlon 96-well polystyrene plates (Greiner Bio-One, Monroe, North Carolina) were coated with 100 μL of 10 μg/mL goat polyclonal antibody specific for BNZ-1 (Genscript, Piscataway, New Jersey) diluted in phosphate-buffered saline (PBS). Plates were covered with polyester Whatman adhesive sealer (GE Healthcare, Chicago, Illinois) and incubated overnight at 4°C. The following day, coating solution was aspirated, and wells were washed 4 times with Quantikine Wash Buffer 1 (R&D Systems, Minneapolis, Minnesota) using the BioTek 405 TS Microplate washer (BioTek, Winooski, Vermont). Plates were blocked with 300 μL of PBS containing 0.05% Tween-20 (MilliporeSigma, St. Louis, Missouri) and 1× casein (10× Casein Solution; Vector Labs, Burlingame, California) for 2 hours in the dark at 27°C. Reference standards and patient samples were cleared by centrifugation at 5000 × g for 5 minutes and diluted 40-fold in ELISA buffer, and 150 μL of each sample was added to the plate in duplicate, covered, and incubated at 27°C for 2 hours in the dark.
After aspiration and washing, 150 μL of 0.2 μg/mL of biotin-conjugated anti-PEG antibody (Detection Antibody Solution, Genscript) diluted in 5% mouse serum was added to each well. Plates were covered and incubated at 27°C for 2 hours in the dark. Wells were aspirated and washed as above, and 100 μL of Streptavidin-HRP Solution (R&D Systems), diluted 1:200 in 1× Reagent Diluent Concentrate 2, was added. Plates were covered and incubated at 27°C for 30 minutes in the dark. Wells were aspirated and washed as above, and 100 μL of Substrate Reagent (R&D Systems) was added. Plates were covered and incubated at 27°C for 12 minutes in the dark; 50μL of Stop Solution (R&D Systems) was added to each well, and the optical density at 450 nm was measured on a TECAN Genios microplate reader (Tecan Systems, San Jose, California). Data were analyzed with a 4PL regression function using the software R (R Foundation for Statistical Computing, Vienna, Austria).
Flow Cytometry Methods.
Two million PBMCs were washed twice with 1 mL flow staining solution (0.5% fetal bovine serum/PBS) at 500 × g for 1 minute using a microcentrifuge. The pellet was incubated with 20 μL of antibody mixture (concentration of each antibody was preoptimized) in the flow staining solution. Extracellular staining was performed by incubating the cells with antibodies for 45 minutes on ice, followed by 2 washes with 1 mL of flow staining solution. The cell-surface antibodies were cross-linked by incubating the stained cells in 0.5% paraformaldehyde in PBS for 30 minutes on ice. For intracellular staining, surface-stained cells (without paraformaldehyde fixation) were washed twice in 1 mL of serum-free PBS, incubated with 0.5 mL of fixation buffer (from the Foxp3 staining kit, eBIO-SCIENCE; Thermo Fisher Scientific, Waltham, Massachusetts) on ice for 30 minutes. The fixed cells were incubated with 0.5 mL of permeabilization buffer (from the Foxp3 staining kit) 10 minutes at room temperature and incubated with 20 μL of antibody mixture for 20 minutes at room temperature. The stained cells were washed twice in 1 mL of serum-free PBS and analyzed by FACSAria II polychromatic fluorescence cell analyzer/sorter (BD Biosciences). Publication-grade flow cytometry results were generated by reanalyzing the FCS files from the FACSAria analyzer using FlowJo software (FlowJo LLC, Ashland, Oregon).
Statistical Analysis Plan
The sample size of 18 (n = 3/cohort) was driven by clinical considerations to balance the need to minimize exposure of subjects to the study drug and the need to provide adequate safety, tolerability, and PK/PD information at each dose level. All subjects receiving any amount of BNZ-1 were included in the safety analysis. The PK, PD, and immunogenicity populations consisted of all subjects who received BNZ-1 and had at least 1 postdose PK, PD, or immunogenicity sample collected. Descriptive statistics of the PK and PD parameters were generated using SAS Version 9.3 (SAS Institute, Cary, North Carolina). Summary statistics, including sample size (n), arithmetic mean, standard deviation, coefficient of variation, standard error of the mean, median, minimum, and maximum were calculated for all nominal concentration time points.
Results
Subject and Baseline Demographics
A total of 18 adult subjects were enrolled and completed the study between November 2016 and May 2017 at 1 clinical site in the United States. Because no subject experienced a dose-limiting toxicity in any of the cohorts, no additional subjects were enrolled in the study as per protocol. The 0.2-mg/kg cohort was added after the PD results from the 0.4-mg/kg dose cohort demonstrated significant activity of BNZ-1 and was the fourth cohort dosed. The doses administered in this study ranged from 12.5 mg to 805 mg. Table 1 summarizes baseline demographics for all subjects. A total of 11 (61%) male and 7 (39%) female subjects, with a mean age of 36.4 years (range, 23–49 years), participated in the trial. Subjects were predominantly white (78%) and Hispanic (56%).
Table 1.
Subject Baseline Demographics
| Demographic | Category | Overall (n = 18) |
|---|---|---|
| Sex | Female | 11 (61%) |
| Male | 7 (39%) | |
| Race | American Indian/Alaska Native | 1 (6%) |
| Black or African American | 2 (11%) | |
| Native Hawaiian/Pacific Islander | 1 (6%) | |
| White | 14 (78%) | |
| Ethnicity | Hispanic or Latino | 10 (56%) |
| Not Hispanic or Latino | 8 (44%) | |
| Age (y) | Mean | 36.4 |
| SD | 7.47 | |
| Minimum | 23 | |
| Median | 36.5 | |
| Maximum | 49 | |
| Weight (kg) | Mean | 81.2 |
| SD | 21.0 | |
| Minimum | 57.2 | |
| Median | 74.2 | |
| Maximum | 126.6 | |
| BMI (kg/m2) | Mean | 29.54 |
| SD | 6.59 | |
| Minimum | 21.90 | |
| Median | 27.86 | |
| Maximum | 42.05 |
BMI, body mass index; SD, standard deviation.
Safety
There were no deaths, dose-limiting toxicities, serious or severe TEAEs, or TEAEs leading to discontinuation of the study drug or from the trial. Eleven of the 18 (61%) subjects experienced a total of 25 TEAEs, most of which were of mild severity (grade 1: 80%; Table 2). Thirteen adverse effects were considered possibly related to the study drug, and 12 were considered unlikely to be related. The only TEAE occurring in >1 subject each was headache (n = 3). No relationship between dose and frequency of TEAEs was observed. No clinically significant laboratory abnormalities of hematology, liver enzymes, metabolic panel, or coagulation were observed. In addition, no clinically significant changes were noted for subject vital signs or electrocardiograms. All blood samples analyzed across all subjects throughout the study period were negative for the presence of anti-BNZ-1 antibodies, although interfering levels of BNZ-1 were in the samples from the top 3 dose cohorts and cannot be considered conclusive.
Table 2.
Treatment-Emergent Adverse Events (TEAEs) During Study: Overall and Common
| BNZ-1 Dose Cohort (n = 3/Cohort) | |||||||
|---|---|---|---|---|---|---|---|
| 0.2 mg/kg | 0.4 mg/kg | 0.8 mg/kg | 1.6 mg/kg | 3.2 mg/kg | 6.4 mg/kg | Overall | |
| Total TEAEs observed | 6 | 10 | 2 | 1 | 3 | 3 | 25 |
| TEAE by severity | |||||||
| Mild | 6 | 6 | 2 | 1 | 3 | 2 | 20 (80%) |
| Moderate | 0 | 4 | 0 | 0 | 0 | 1 | 5 (20%) |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Common TEAEs (> 1 subject overall) | |||||||
| Headache | 1 (33%) | 1 (33%) | 0 | 1 (33%) | 0 | 0 | 3 (17%) |
Pharmacokinetics of BNZ-1
Mean plasma BNZ-1 concentration-time profiles following a single-dose IV administration of 0.2, 0.4, 0.8, 1.6, 3.2, or 6.4 mg/kg in cohorts of 6 subjects each were analyzed on a semilog scale (Figure 2). Overall, exposure as determined by Cmax increased with escalating doses. Cmax values ranged between 9.91 and 386 μg/mL and were reached 5 minutes after the end of drug infusion for the 0.2-, 0.4-, and 3.2-mg/kg doses, and 2 hours following the end of infusion for the 0.8-, 1.6-, and 6.4-mg/kg doses (Table 3). In general, after reaching peak concentration, there was an initial decline due to distribution, followed by a slower elimination phase consistent with clearance. The extent (AUC0-t) and total extent (AUC0-inf) also increased in a predominantly proportional manner with increasing dose, with values ranging from 934 to 45,900 μg · h/mL for AUC0-t and 1280 to 46 700 μg · h/mL for AUC0-inf. The mean apparent elimination t1/2 was between 107 and 130 hours across all dose levels. Mean clearance values ranged from 10.8 to 13.5 mL/h, and steady-state volume of distribution ranged from 1660 to 2020 mL across all dose levels studied.
Figure 2.
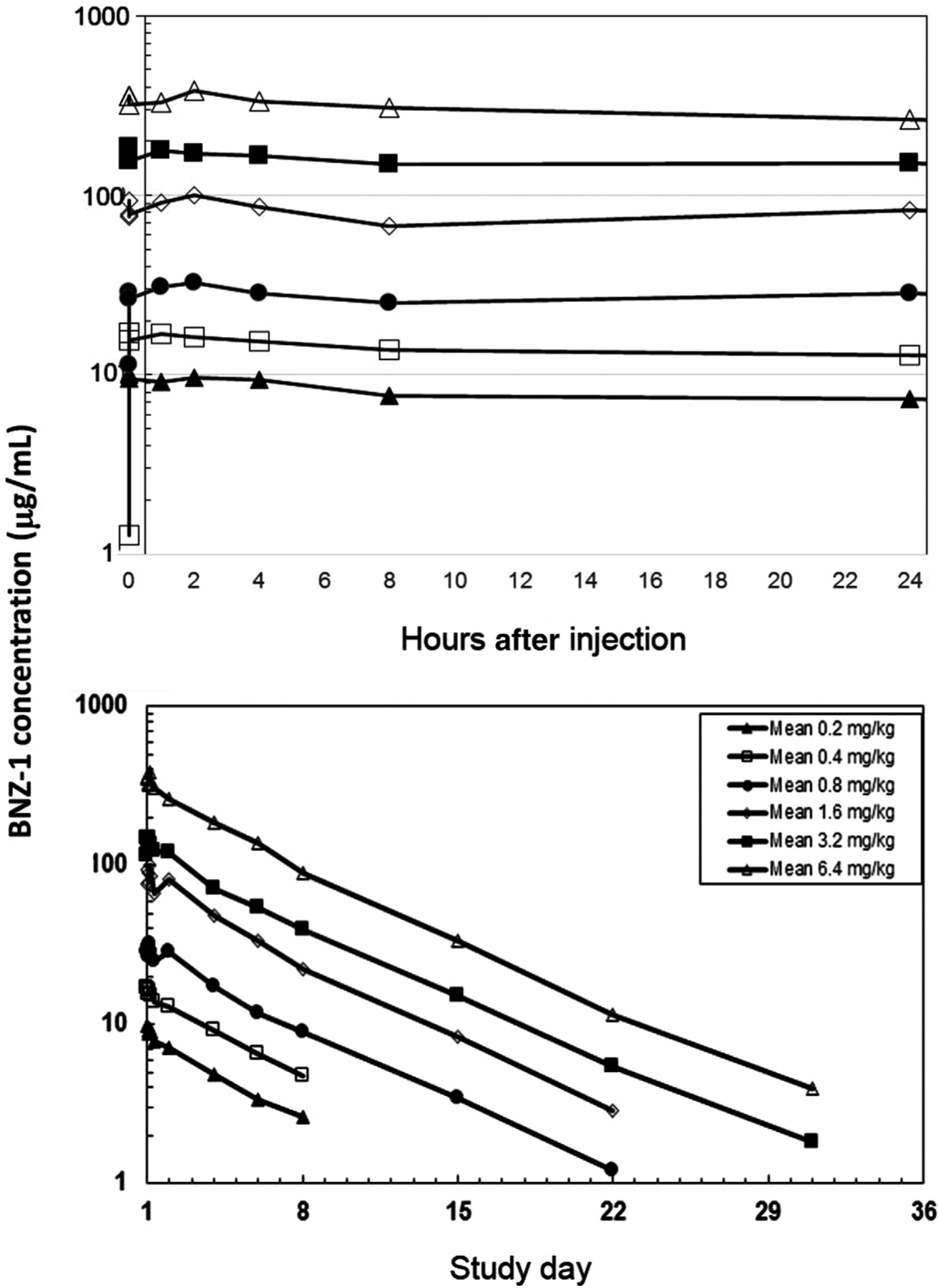
A single IV dose of BNZ-1 was administered over ≤5 minutes starting at time = 0, day 1. PK samples were collected at 0.08, 0.5, 1, 2, 4, 8, and 24 hours after dosing, and on days 4, 6, 8, 15, 22, and 31. The plot shows the average plasma concentration at each time point (n = 3). The upper figure represents the distribution phase (up to 24 hours), and the lower figure shows the elimination phase (days 2–36). IV, intravenous; PK, pharmacokinetic.
Table 3.
Plasma BNZ-1 Pharmacokinetic Parameters Following a Single Intravenous Infusion
| Pharmacokinetic Parameters | BNZ-1 Dose Group | |||||
|---|---|---|---|---|---|---|
| 0.2 mg/kg (n = 3) | 0.4 mg/kg (n = 3) | 0.8 mg/kg (n = 3) | 1.6 mg/kg (n = 3) | 3.2 mg/kg (n = 3) | 6.4 mg/kg (n = 3) | |
| AUC0-t (μg • h/mL) | 934 (35.7) | 1780 (42.0) | 4140 (22.4) | 1170 (13.1) | 19 600 (2.9) | 45 900 (21.6) |
| AUC0-inf (μg • h/mL) | 1280 (31.0) | 2260 (29.4) | 4450 (19.1) | 12 200 (14.1) | 20 100 (2.4) | 46 700 (21.4) |
| AUC%extrap | 26.3 ± 12.0 | 21.1 ± 9.38 | 6.90 ± 3.32 | 3.87 ± 1.47 | 2.44 ± 0.613 | 1.62 ± 0.870 |
| Cmax (μg/mL) | 9.91 (22.9) | 17.6 (6.5) | 31.4 (30.9) | 101 (4.3) | 153 (17.8) | 386 (43.0) |
| tmax (h) | 0.115 (0.100, 0.140) | 0.105 (0.104, 1.02) | 2.02 (2.02, 2.06) | 2.02 (1.88, 2.08) | 0.173 (0.165, 2.11) | 0.173 (0.166, 2.08) |
| Kel (1/h) | 0.00658 ± 0.000960 | 0.00663 ± 0.00196 | 0.00599 ± 0.000542 | 0.00622 ± 0.000990 | 0.00557 ± 0.00148 | 0.00539 ± 0.000780 |
| t1/2 (h) | 107 ± 15.5 | 112 ± 39.0 | 116 ± 10.9 | 113 ± 16.9 | 130 ± 31.7 | 130 ± 17.4 |
| CL (mL/h) | 11.9 ± 2.92 | 13.3 ± 3.78 | 13.4 ± 1.86 | 12.3 ± 3.65 | 10.8 ± 2.47 | 13.5 ± 2.86 |
| Vd (mL) | 1760 ± 309 | 1950 ± 290 | 2020 ± 179 | 1710 ± 474 | 1660 ± 135 | 2020 ± 95.5 |
AUC0-inf, area under the plasma concentration–time curve from time 0 to time of last measurable concentration; AUC0-t, area under the plasma concentration–time curve from time 0 to time of last measurable concentration; AUC%extrap, percentages of area under curve extrapolated to infinity; CL, clearance; Cmax, maximum observed plasma concentration; Kel, apparent terminal elimination rate constant; t1/2, terminal elimination half-life; tmax, time to maximum observed plasma concentration; Vd, volume of distribution.
AUCs and Cmax values are presented as geometric mean and geometric coefficient of variation.
Tmax values are presented as median (minimum, maximum). AUC%extrap, Kel, t1/2, CL, and Vd are presented as mean ± standard deviation.
Pharmacodynamic Effects
The PD activity of BNZ-1 was characterized using flow cytometry of PBMCs obtained before dosing and at multiple times after dosing to calculate the change from baseline for regulatory T cells (Tregs; IL-2 effect), NK cells (IL-15 effect), and CD8 Tcm (IL-15 effect), while other major leukocyte populations (CD4 and CD8 T cells, B cells, and monocytes) were analyzed primarily to demonstrate the selective PD effects of BNZ-1 (Figure 3).
Figure 3.
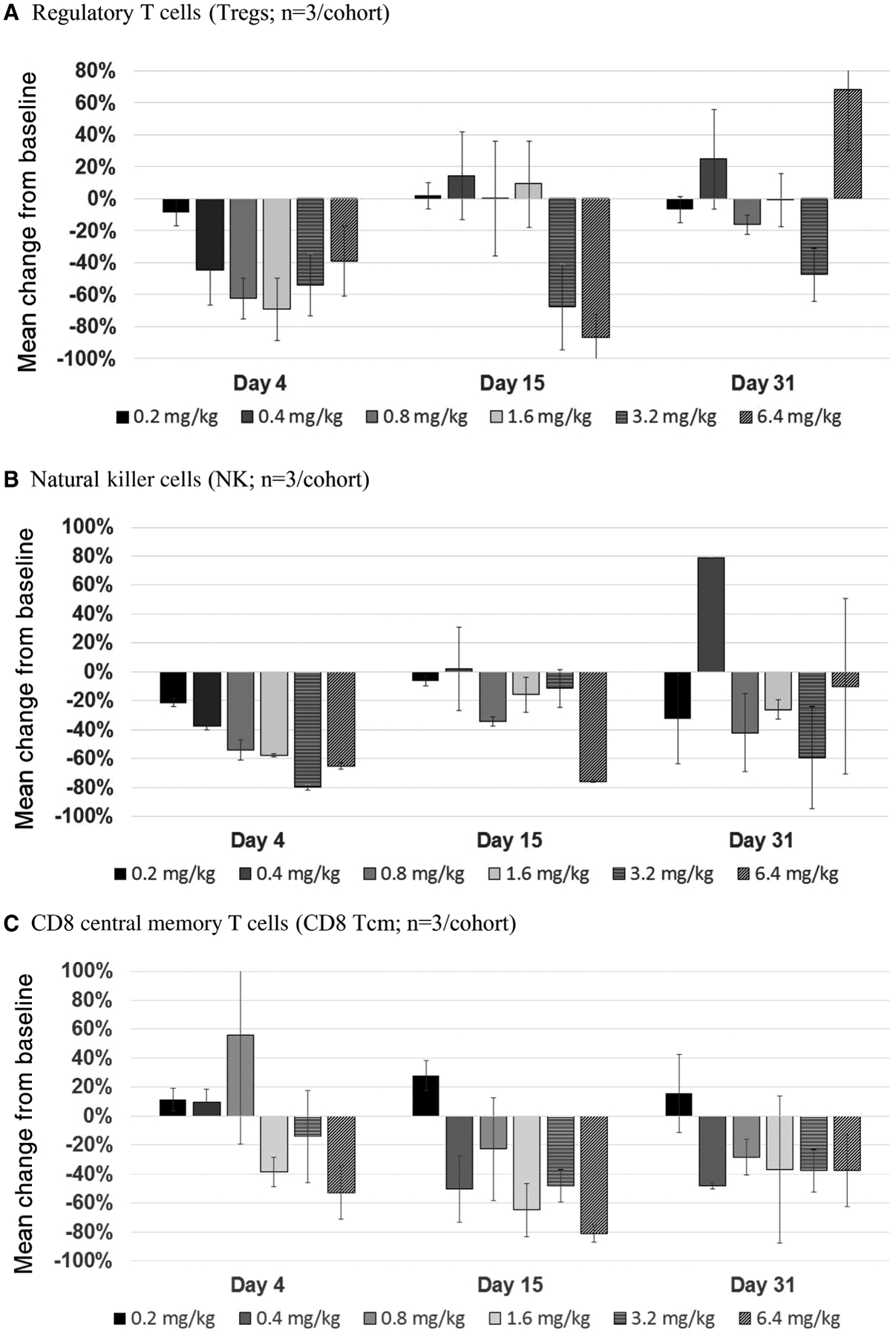
Whole blood samples for isolation and staining of PBMCs were collected before dosing on day −1 and after dosing on days 4, 15, and 31 and used to measure Tregs (A), NK cells (B), and CD8 central memory T cells (C). Samples were shipped overnight to the lab, stained the next day, and analyzed by FACSAria II polychromatic fluorescence cell analyzer. The graphs show the average percent change from baseline, calculated using the following equation: % Change = (% postdose – % predose) ÷ % predose. Each bar represents n = 2–3, and standard deviation values are shown. PBMCs, peripheral blood mononuclear cells.
Following a single IV dose of BNZ-1, a reduction from baseline in Tregs was observed by day 4 across doses (Figure 3A) that ranged from 8% to 69%. The reduction on day 4 increased with dose except for the 3.2- and 6.4-mg/kg doses, which had a delayed maximal PD effect. The maximum effect, as determined by the greatest postadministration reduction in Treg counts, was observed at day 4 for all dose cohorts except for the 3.2- and 6.4-mg/kg cohorts, where by day 15 (4 subjects) or 31 (2 subjects) Treg counts had reached a mean reduction of 69% and 93% from baseline, respectively. By day 15, Tregs in circulation returned to baseline for all dose levels apart from the 3.2- and 6.4-mg/kg doses, where circulating Treg counts continued to be reduced out to day 31 and day 15, respectively.
Following a single IV dose of BNZ-1, a reduction from baseline in NK cells was observed across all doses by day 4 (Figure 3B) that ranged from 22% to 65% and generally increased with dose. By day 15, NK cells in circulation returned to near baseline or baseline levels for all dose levels except for the 1.6 and 6.4 mg/kg doses where circulating NK counts remained below baseline on day 31. The maximum effect, as determined by greatest postadministration reduction in NK cells, was observed at day 4 for all dose cohorts except for the 6.4-mg/kg cohort where the percent change from baseline at any time after dosing occurred on day 15 and reached a mean reduction of 76% from baseline. NK cells returned to baseline by day 31, except for the 1.6- and 6.4-mg/kg doses, which remained below baseline on day 31.
In addition to NK cells and Tregs, the following major leukocyte populations were measured as part of the immunophenotyping flow cytometry panels: CD4 and CD8 T cells, B cells, and monocytes. In addition to the major leukocyte populations, analyses of subsets of CD8 T cells (naïve, stem memory, central memory, effector memory, and terminal effector) were performed, with a focus on the central memory CD8 T-cell subset (CD8 Tcm), which is known to be regulated by IL-15. The flow cytometry results demonstrated that CD8 Tcm was the only subset of CD8 T cells in which a dose/exposure-related reduction from baseline was observed. All doses of BNZ-1 produced a drop in CD8 Tcm from baseline by day 4 that generally increased by day 15, except the lowest dose (0.2 mg/kg), which had no measurable effect at any time point (Figure 3C). The mean maximum observed effect was an 81% decrease from baseline on day 15 in the 6.4-mg/kg cohort, followed by a 65% decrease in the 1.6-mg/kg cohort. Most cohorts returned to baseline by day 31. No effects of BNZ-1 were observed on the other evaluated cell populations (total, CD4, and CD8 T cells; B cells; and monocytes; data not shown).
Discussion
A mechanistic rationale exists for developing a multi-cytokine inhibitor targeting the γ c family due to their roles, sometimes redundant, in the pathogenesis of oncologic and immunologic diseases. Alopecia areata and T-cell leukemia and lymphoma are examples of human diseases linked to the concurrent dysregulation of IL-2, IL-15, and/or IL-9.7,8,18–25 BNZ-1 was rationally engineered to inhibit IL-2 and IL-15 signaling through the shared γ c receptor16 and was subsequently found to also inhibit IL-9, albeit with a >10-fold lower potency.16 Preclinical murine leukemia tumor studies using the IL-15 transgenic mice we developed16,23 and in vivo cytokine challenge studies16 have demonstrated the ability of BNZ-1 to block the pathogenic effects of IL-2 and IL-15 in vivo.16
To our knowledge, this was the first study of BNZ-1 in humans designed to characterize the preliminary safety of BNZ-1 and the PK and PD of a range of single IV doses of BNZ-1. The PK of BNZ-1 was well characterized in this study, and consistent with its IV route of administration, exposure was generally proportional across the wide range of doses tested (13–805 mg). The mean apparent elimination t1/2 of ~5 days and clearance of BNZ-1 did not differ significantly across the range of tested doses, which suggests that there may be little difference between single- and multiple-dose PK parameters of BNZ-1 across the range of doses planned for the multiple-dose studies (0.25 mg/kg to 4 mg/kg). The PK profile of BNZ-1 is consistent with the PK profiles of other PEGylated peptides and supports a once-weekly dosing regimen at lower doses and possibly an every-other-week dosing regimen of BNZ-1 at higher doses, which is being tested in a multiple-ascending-dose study in healthy subjects.
The PD effects of BNZ-1 in our study demonstrated that both IL-2 and IL-15 signaling were selectively antagonized, with the lowest dose having little effect, while the highest doses produced ~80% or greater decrease of Tregs, NK cells, and CD8 Tcm from baseline. The low doses/exposures produced less of a PD effect with a shorter duration, while high doses/exposures of BNZ-1 produced robust PD effects that lasted for up to 30 days after dosing. This demonstrates the opportunity to select doses to titrate the desired extent and duration of effect of BNZ-1. Equally important to the demonstrated PD potency to reduce Tregs, NK cells, and CD8 Tcm, BNZ-1 lacked a PD effect on all other major leukocyte subsets evaluated using flow cytometry (total T cells, CD4 T cells, CD8 T cells, B cells, and monocytes). This is consistent with the current understanding of the known roles for IL-2 and IL-15 on these populations of cells and suggests that a single dose of BNZ-1 effectively blocks the signaling of these 2 cytokines in an exposure-dependent manner. We were unable to determine if BNZ-1 inhibits the function of IL-9 in this study because of the previously discussed lower potency and the enrollement of healthy volunteers where we were measuring cytokine functions of the resting immune system. However, the safety results from this study demonstrated that high doses of BNZ-1 were generally well tolerated, suggesting that IL-9 inhibition by BNZ-1 may be possible in patients with an overactivity of IL-9 as part of their disease pathobiology. The inhibition of IL-2 and IL-15 function predictably decreased the number of of NK, CD8 Tcm, and Treg cells. The robust ability of BNZ-1 to decrease Tregs suggests that BNZ-1 may have potential in combination or stand-alone therapy for malignancies where reducing the number of Tregs has been proposed as a mechanism of enhancing antitumor immunity, which includes solid tumors (eg, melanoma, pancreatic cancer) and hematologic malignancies (eg, lymphoma).26–28 A clinical study of BNZ-1 in patients with refractory cutaneous T-cell lymphoma is currently ongoing (NCT03239392).
As predicted from preclinical experience, the PD effects of BNZ-1 were transient with cell population counts returning to or toward baseline for all cohorts by day 31, demonstrating the reversibility of BNZ-1 that coincided with the clearance of BNZ-1 from the blood of test subjects. This reversibility plus the lack of PD effect on the other major leukocytes further support the preclinical characterization of BNZ-1 as a potent, selective immunomodulator, which may lead to a potentially better safety profile as compared to other broad-acting immunosuppressants that might be targeting the same γ c family of cytokines.
In terms of safety, despite the observed potent PD effects, BNZ-1 was generally well tolerated, as no subject experienced a dose-limiting toxicity, infusion reaction, or other significant TEAEs, which allowed the study to be completed with the minimal predefined number of subjects. Infusion reactions to immune-targeted therapies is common due to cytokine release syndrome,29 which was not observed in preclinical nonhuman primate studies and not expected because BNZ-1 is a cytokine antagonist. However, cytokine release syndrome may be relevant only for cancer patients where an effect on malignant cells may trigger this event. Overall, no relationship between dose and incidence of TEAEs was observed. No subjects experienced any clinically significant effects on laboratory hematology panels (eg, no lymphopenia or leukopenia), which further supports BNZ-1 as a selective immunomodulator. The safety results from this study support the continued clinical development of BNZ-1. However, given that this is a single-dose study, no conclusions about long-term safety can be made until additional ongoing multiple-dose studies evaluating the longer-term safety of BNZ-1 are completed.
The PK and PD results suggest that single IV doses of BNZ-1 achieve exposures that are highly active across a wide range, enabling potential therapeutic dosing regimens using weekly or every-other-week dosing, which will be explored in future clinical studies.
Conclusion
The results of this first-in-human study suggest that a range of single IV doses of BNZ-1 were well tolerated with a good safety profile. The exposure-response results demonstrated that BNZ-1 produces selective and temporary decreases of Tregs, NK cells, and CD8 Tcm that are related to the inhibition of IL-2 and/or IL-15 signaling across a range of exposures that can be safely achieved in healthy subjects. These results supported the initiation of a phase 1/2 dose-ranging study to characterize the preliminary safety and efficacy of BNZ-1 in patients with large granular lymphocyte leukemia or refractory cutaneous T-cell lymphoma (NCT03239392) in which the dysregulation of IL-15 and/or IL-2 has been implicated. In support of this proposal, we have recently reported that leukemic cells from patients with CD8-type large granular lymphocyte leukemia respond to IL-2 and IL-15 ex vivo and that BNZ-1 effectively blocks the proliferation, activation, and viability of these leukemic cells.30
Acknowledgments
The authors thank the subjects for their participation in this study and the Celerion clinical site staff and project team, including Dr. Danielle Armas (study principal investigator). The authors also thank Samorn Biosciences for their assistance with preparation of the original draft of the manuscript.
Footnotes
Conflicts of Interest
PAF, ND, AB, LAM, WJK, AR, NA, and YT own stock or received stock options from Bioniz Therapeutics. JCZ, XW, and TAW declare no conflicts of interest.
Data Accessibility
The authors declare that all data presented in this work will be publicly shared. For this purpose, the reader should contact the corresponding author, Nazli Azimi, at nazli@bioniz.com.
References
- 1.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9(7):480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: biology, design and application. Trends Immunol. 2015;36(12):763–777. [DOI] [PubMed] [Google Scholar]
- 3.Waldmann TA. The biology of IL-15: implications for cancer therapy and the treatment of autoimmune disorders. J Investig Dermatol Symp Proc. 2013;16(1):S28–S30. [DOI] [PubMed] [Google Scholar]
- 4.Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood. 2017;129(9):1082–1094. [DOI] [PubMed] [Google Scholar]
- 5.Mishra A, Sullivan L, Caligiuri MA. Molecular pathways: interleukin-15 signaling in health and in cancer. Clin Cancer Res. 2014;20(8):2044–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hechinger AK, Smith BA, Flynn R, et al. Therapeutic activity of multiple common gamma-chain cytokine inhibition in acute and chronic GVHD. Blood. 2015;125(3):570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xing L, Dai Z, Jabbari A, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. 2014;20(9):1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suarez-Farinas M, Ungar B, Noda S, et al. Alopecia areata profiling shows TH1, TH2, and IL-23 cytokine activation without parallel TH17/TH22 skewing. J Allergy Clin Immunol. 2015;136(5):1277–1287. [DOI] [PubMed] [Google Scholar]
- 9.Deng Y, Wang Z, Chang C, Lu L, Lau CS, Lu Q. Th9 cells and IL-9 in autoimmune disorders: Pathogenesis and therapeutic potentials. Hum Immunol. 2017;78(2):120–128. [DOI] [PubMed] [Google Scholar]
- 10.Goswami R, Kaplan MH. A brief history of IL-9. J Immunol. 2011;186(6):3283–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noelle RJ, Nowak EC. Cellular sources and immune functions of interleukin-9. Nat Rev Immunol. 2010;10(10):683–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagato T, Kobayashi H, Kishibe K, et al. Expression of interleukin-9 in nasal natural killer/T-cell lymphoma cell lines and patients. Clin Cancer Res. 2005;11(23):8250–8257. [DOI] [PubMed] [Google Scholar]
- 13.Waldmann TA, Chen J. Disorders of the JAK/STAT pathway in T cell lymphoma pathogenesis: implications for immunotherapy. Annu Rev Immunol. 2017;35:533–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris JC, Janik JE, White JD, et al. Preclinical and phase I clinical trial of blockade of IL-15 using Mikbeta1 monoclonal antibody in T cell large granular lymphocyte leukemia. Proc Natl Acad Sci U S A. 2006;103(2):401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackay-Wiggan J, Jabbari A, Nguyen N, et al. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight. 2016;1(15):e89790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nata T, Basheer A, Cocchi F, et al. Targeting the binding interface on a shared receptor subunit of a cytokine family enables the inhibition of multiple member cytokines with selectable target spectrum. J Biol Chem. 2015;290(37):22338–22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.US Food and Drug Administration. FDA Guidance for Industry: Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. 2005. https://www.fda.gov/media/72309/download. Accessed August 22, 2019.
- 18.Zambello R, Facco M, Trentin L, et al. Interleukin-15 triggers the proliferation and cytotoxicity of granular lymphocytes in patients with lymphoproliferative disease of granular lymphocytes. Blood. 1997;89(1):201–211. [PubMed] [Google Scholar]
- 19.Shimbara A, Christodoulopoulos P, Soussi-Gounni A, et al. IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J Allergy Clin Immunol. 2000;105(1 Pt 1):108–115. [DOI] [PubMed] [Google Scholar]
- 20.Meresse B, Chen Z, Ciszewski C, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21(3):357–366. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Nourbakhsh B, Ciric B, Zhang GX, Rostami A. Neutralization of IL-9 ameliorates experimental autoimmune encephalomyelitis by decreasing the effector T cell population. J Immunol. 2010;185(7):4095–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nowak EC, Weaver CT, Turner H, et al. IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med. 2009;206(8):1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato N, Sabzevari H, Fu S, et al. Development of an IL-15-autocrine CD8 T-cell leukemia in IL-15-transgenic mice requires the cis expression of IL-15Ralpha. Blood. 2011;117(15):4032–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jabri B, Abadie V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat Rev Immunol. 2015;15(12):771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oh CK, Raible D, Geba GP, Molfino NA. Biology of the interleukin-9 pathway and its therapeutic potential for the treatment of asthma. Inflamm Allergy Drug Targets. 2011;10(3):180–186. [DOI] [PubMed] [Google Scholar]
- 26.Berger CL, Tigelaar R, Cohen J, et al. Cutaneous T-cell lymphoma: malignant proliferation of T-regulatory cells. Blood. 2005;105(4):1640–1647. [DOI] [PubMed] [Google Scholar]
- 27.Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. 2006;108(3):804–811. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang TT, Yang J, Zhang Y, et al. IL-2 and IL-15 blockade by BNZ-1, an inhibitor of selective gamma-chain cytokines, decreases leukemic T-cell viability. Leukemia. 2019;33(5):1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]