

Abstract
Purpose of the review:
Coronary artery disease (CAD) is a common disease globally attributable to the interplay of complex genetic and lifestyle factors. Here, we review how genomic sequencing advances have broadened the fundamental understanding of the monogenic and polygenic contributions to CAD and how these insights can be utilized, in part by creating polygenic risk estimates, for improved disease risk stratification at the individual patient level.
Recent Findings:
Initial studies linking premature CAD with rare familial cases of elevated blood lipids highlighted high-risk monogenic contributions, predominantly presenting as familial hypercholesterolemia (FH). More commonly CAD genetic risk is a function of multiple, higher frequency variants each imparting lower magnitude of risk, which can be combined to form polygenic risk scores (PRS) conveying significant risk to individuals at the extremes. However, gaps remain in clinical validation of PRSs, most notably in non-European populations.
Summary:
With improved and more broadly utilized genomic sequencing technologies, the genetic underpinnings of coronary artery disease are being unraveled. As a result, polygenic risk estimation is poised to become a widely used and powerful tool in the clinical setting. While the use of PRSs to augment current clinical risk stratification for optimization of cardiovascular disease risk by lifestyle change or therapeutic targeting is promising, we await adequately powered, prospective studies, demonstrating the clinical utility of polygenic risk estimation in practice.
Keywords: heart disease, coronary artery disease, genetics, polygenic risk assessment
INTRODUCTION
The tools available to address and optimize preventative strategies and medical therapies for emergent and lifechanging cardiovascular events have greatly improved over time. Yet, while the number of deaths and burden of disease due to coronary artery disease (CAD) has decreased dramatically in the past few decades it remains the number one cause of death in the United States and worldwide[1]. The global prevalence of coronary heart disease (CHD) is estimated at 197.2 million individuals (20.1 million for US in 2018) and this disease accounted for 9.1 million annual deaths. In the US alone, 8.3% of men and 6.2% women are living with CHD with 720,000 individuals with new events and 335,000 individuals with recurrent events each year[1]. Gaps in preventative strategies exist not only in structuring care delivery across oftentimes disconnected healthcare and societal systems, but also in our identification and understanding of the most prescient signals that may act as early warning signs for acute events or indicate a higher lifetime (long-term) risk of disease for an individual for which appropriate actions should be taken.
Advances in data science have been vital to unveiling the factors related to early disease risk and prevention [2]. For CAD there has been great interest in understanding how genetics can augment existing risk stratification tools. Insights into the biologic pathways and intricate networks involved in the development and progression of CAD gained from the discovery of individual genes has been expertly reviewed in the past [3–5]. Here, we aim to review how genomic sequencing advances and modeling have improved our fundamental understanding of CAD and has set the stage for an exciting future in improved risk stratification aligned with the promise of personalized preventive medicine.
Coronary Artery Disease and Clinical Risk
Coronary artery disease (CAD) results from the complex interplay of a person’s individual environmental, lifestyle and genetic factors. CAD is the accumulation of atherosclerotic plaque within the arteries that supply the heart muscle with oxygen and nutrient rich blood that it requires for optimal sustained function. Symptoms of CAD manifest when the obstruction to blood flow created by a plaque exceeds the myocardial energy demands. This can occur incrementally over a period of time (months to years) eventually leading to chest pain (angina) or shortness of breath on exertion when that supply-demand mismatch threshold is achieved or can progress rapidly and unpredictably as in the case of unstable plaque rupture resulting in an acute heart attack.
Disruptions to multiple pathways of healthy cellular and physiologic homeostasis created by a person’s risk factors are responsible for initial plaque formation and propagation. Co-morbid disease risks such as elevated blood pressure, dyslipidemia, diabetes mellitus, chronic kidney disease and chronic inflammatory conditions such as rheumatoid arthritis and systemic lupus erythematosus are well established. Additionally, lifestyle practices including sedentary behavior, cigarette smoking, stress and unhealthy diet have also been implicated in the development of CAD. For patients, these modifiable risk factors have been included in an effort by the American Heart Association known as “Life’s Simple 7” to serve as a general guidepost for patients to achieve optimal cardiovascular health in the areas of smoking cessation, blood pressure control, lipid management, blood glucose control, healthy diet and body weight, and physical activity [1]. The tools that medical professionals utilize, mainly to determine the need or intensity of statin therapy, are typically more granular. These include, but are not limited to, the AHA/ACC Atherosclerotic Cardiovascular Disease (ASCVD) Pooled Cohort Equations, Framingham Risk Score – Cardiovascular Disease (FRS-CVD), and QRISK2 risk assessment tools which delineate 10-year risk for disease and adverse events based on inputs of age, sex, ethnicity, blood pressure, cholesterol levels, as well as smoking and diabetes status [6, 7]. While the QRISK2 assessment tool models for disease in 1st degree relatives < 60 years of age, the most used tools do not account for some of the most important factors – the genetic contribution to coronary disease. As such, in using the current risk stratification tools the individual genetic environments in which these clinical risk factors exist and which also likely influence are not considered, and the promise of individualized risk factor optimization is not fully actualized.
Heritability of CAD
The positive relationship between a family history for CAD has long been recognized through various epidemiologic studies and thus hinting at a genetic component and heritability for this disease separate [8]. Heritability can be thought of as the amount of variation in the disease phenotype resulting purely form genetic effects[9]. Twin studies have been helpful in the understanding of heritability as related to CAD. Results from the Swedish Twins Registry, following 21,004 monozygotic or dizygotic Swedish twins, clearly illustrated the link between genetics and premature CAD [10]. Here, monozygotic male twins had an odds ratio for death of 8.1, and 3.8 for dizygotic twins, if one twin died of CAD prior to reaching 55 years of age. The increased risk for death decreased as the age of the index case advanced in age. The Framingham Offspring Study, as part of the landmark Framingham Heart Study, further demonstrated that family history, beyond twins, as related to parental history of premature CAD is an independent and prospective risk factor for the future development of CAD [11]. In this study of 2,302 offspring of participants there was ~ 2 times the risk of adverse cardiovascular events (using multivariable adjustment including age) for the sons and daughters of parents who had experienced premature CAD. Adding family history to overall risk assessment was independent of existing lifestyle and clinical factors.
Soon the underpinnings of family history and our understanding of the molecular basis of CAD and influences of specific gene mutations and variants expanded. This occurred as our ability to probe the human genome progressed from classic Sanger sequencing, which is limited in scale and read length, to DNA microarrays, which enabled large-scale genome-wide association studies (GWAS), and finally to high-throughput next-generation sequencing [12]. Through these technological advances the link between our genes and biology ushered in a new era of genomic medicine for primary risk prediction, disease diagnosis and prognostication, and therapeutic targeting and discovery.
The influence of genetics on a person’s risk of developing a disease exists along a spectrum[9]. There are specific single-gene mutations that are highly penetrant that alter the function of the encoded product to such degree that disease is of the highest likelihood, such as in the case of LDLR mutations, encoding low-density lipoprotein receptor, in the development of familial hypercholesterolemia (FH). These are termed monogenic diseases as the disease is the result of just a single gene mutation. Monogenic diseases are typically rare. However, common complex diseases like coronary artery disease are most often the result of the cumulative effects of multiple gene variants each with a small individual effect but that in concert may exert higher genetic risk of disease. These are aptly named polygenic diseases. CAD has both monogenic and polygenic origins (Figure 1). In the case of polygenic CAD, it is thought that genetics explains in the range of 50% of an average individual’s overall risk of disease, with the other half attributed to lifestyle and environment, though significant gaps remain in the accounting for the entirety of the genetic contribution.
Figure 1. Basic overview of CAD Genetics.
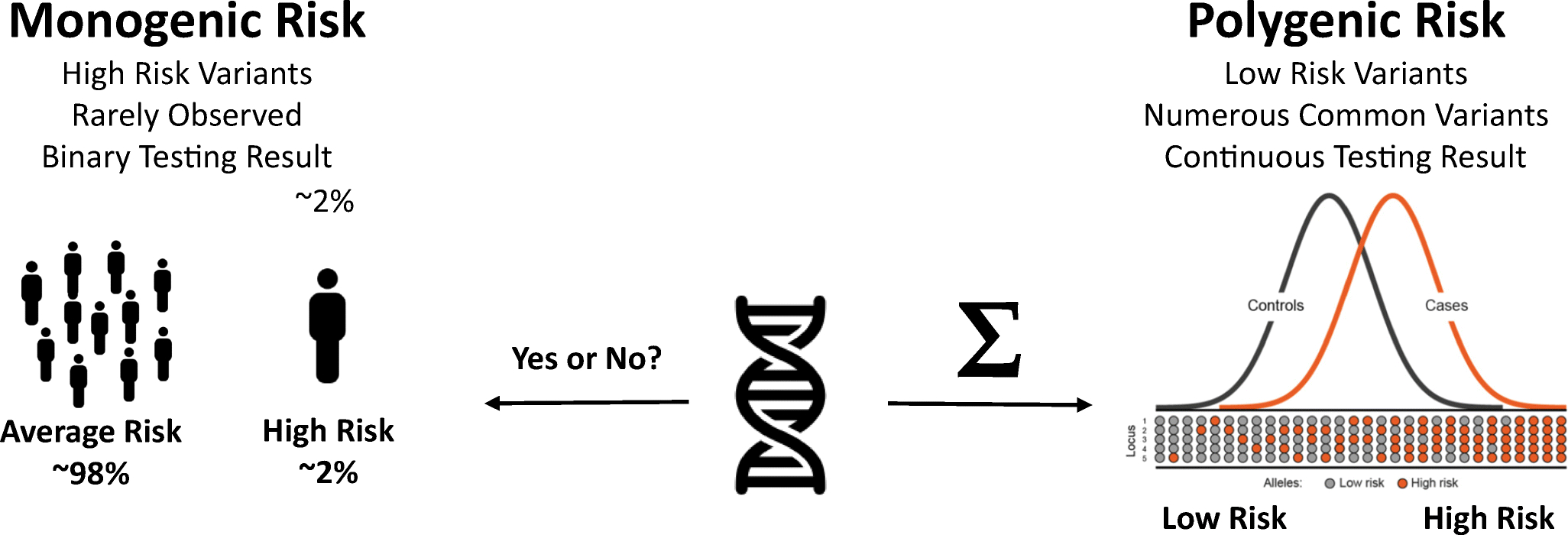
Genetic risk for CAD, and many common diseases, can be simplified into a monogenic (left) and polygenic (right) component. Monogenic risk variants tends to be rare, with small numbers of genetic mutations with large impacts on genetic risk. Polygenic risk variants tend to be more common, with more modest impacts on risk individually, and with cumulative effects that can be significant.
Familial Hypercholesterolemia and Monogenic Contributors to CAD
One of the most common genetic diseases in mankind, and the main monogenic driver of CAD, is familial hypercholesterolemia (FH) [13]. FH is typically an autosomal dominant disease that leads to massively elevated levels of low-density lipoprotein (LDL) cholesterol and results in increased risk of premature CAD and myocardial infarction (men < 55 years, women < 60 years). Strikingly, if left untreated 50% of men and 15% of women with FH will succumb to this disease in their early years[14]. Heterozygous FH – which results from carrying a defect in only one allele - is far more common (~1:250 persons) though presenting with a less severe phenotype, while homozygous FH is rare (1:150,000–300,000 persons) and can present dramatically with heart disease at very early ages [15, 16]. Patients were first diagnosed with FH based on laboratory values, family history and physical exam findings of cholesterol deposits in several tissues - tendon xanthomas, eyelid or skin xanthelasmata and arcus cornealis. It wasn’t until the LDL receptor (LDLR) from a patient with FH was sequenced in 1,985 showing a large deletion that the first insights into the molecular defects responsible for this disease were made known [17]. The disruption in receptor-mediated clearance as well as additional mechanisms leading to elevations in LDL cholesterol in FH have been elegantly reviewed elsewhere [18].
Known pathogenic mutations within LDLR now number over a thousand and account for the vast majority of FH cases (~95%). Shortly after the report of the LDLR truncation mutant, mutations in APOB effecting apolipoprotein-B100 and leading to defective binding of LDL to LDLR and elevated cholesterol in six unrelated individuals were described [19]. There are currently over 30 individual mutations within APOB identified in FH though account for roughly 5% of the disease [20]. Gain-of-function mutations in proprotein convertase subtilisin/kexin type 9 (PCSK9) responsible for FH were later identified from positional cloning in 23 French families [21]. Mutations in PCSK9 however are rare and account for less than 1% of FH. The discovery of PCSK9’s role in cholesterol metabolism through well-designed family studies illustrating both gain-of-function and loss-of-function variants and subsequently leading to an entirely new class of hypolipidemic agents has been heralded as a pivotal moment in translational medicine – moving from bedside to benchtop [22]. Several other genes have been implicated in monogenic cholesterol disorders culminating in premature CAD including LDLRAP1 (LDL receptor adapter protein), APOE (apolipoprotein E), STAP1 (signal-transducing adapter protein 1), ABCG5 (sterolin 1), and ABCG8 (sterolin 2)[23–26]. The identification of causal mutations in individuals with suspected FH is critical for downstream cascade testing of family members as well as for determination of the timing and intensity of therapeutics[13, 27]. As sequencing efforts continue in large, multi-ethnic populations additional causal mutations in other genes are expected and multiple genes (polygenic hypercholesterolemia) are estimated to account for ~15% of this disease [28].
Given the profound increased risk for adverse cardiovascular outcomes for individuals with FH and the beneficial impact of start lipid-lowering therapy in these patients, identifying affected individuals and cascade testing of relatives is increasingly important[29]. Unfortunately, if relaying solely on clinical factors, many cases of FH go undetected and undiagnosed. A study of 50,726 patients with linked electronic health records and exome sequencing data found that only 24% of carriers for FH mutations would have been flagged for as FH based on current clinical criteria for probable of definite diagnosis [30]. Implementing standardized genetic testing in affected individuals and their family members as was done in an Estonian population has been shown to increase the diagnosis of FH and successfully guide them to initiating appropriate treatment[31]. As such, a recent Expert Consensus Panel recommended genetic testing for casual mutations in LDLR, APOB and PCSK9 in individuals with known or probable FH with subsequent cascade testing of family members[32].
The Polygenic Nature of Coronary Artery Disease
Unlike monogenic Mendelian diseases, the majority of CAD within the population is influenced by polygenic inheritance attributed to numerous common and rare variants with small effects throughout the genome [5]. As DNA sequencing technologies and analytic platforms improved, and in-turn became more cost-effective and scalable, research efforts using genomics broadened greatly and expanded in size. This enabled the proliferation of GWAS [33]. GWAS is an experimental design that is aimed at identifying relationships between traits or diseases with DNA variants and whose power to do so improves with increasing sample size. The first GWAS in CAD was reported simultaneously by three independent groups in 2007 and identified a region on chromosome 9p21 associated with CAD [34–36]. Taken together these three reports included over 11,000 individuals as cases with CAD and 37,000 individuals as controls without heart disease for discovery and validation. Despite these impressive numbers, the population studied was predominantly of European ancestry. Interestingly, the variants at 9p21 are found in a region that does not encode for a specific protein product or regulatory region and set off a hunt for what biologic role the variant could play to increase the risk of CAD by ~50% for persons with 2 copies of the risk block. While the definitive role of 9p21 in CAD is controversial, this risk region is known to act in vascular smooth muscle cells via increased expression of the long non-coding RNA ANRIL leading to altered cell adhesion, contraction and proliferation associated with CAD and other 9p21 associated disease phenotypes [37].
Based on the clinically observed phenotypic relationships between dyslipidemias and CAD, the next wave of studies assessed for genetic associations not only to CAD but also to lipid traits using a variety of approaches. Several GWAS increasingly identified and validated the significance of a growing number of variants including SORT1, LPA, MRAS, PHACTR1 and novel variants within genes already known to be involved in the disease process such as LDLR and PCSK9 [38–43]. Interestingly, results from a mendelian randomization study illustrated a disconnect between genetic mechanisms that increase circulating HDL and any protective effect for myocardial infarction a presage for the lack of benefit observed in forthcoming clinical trials focused on therapies raising HDL [44]. However, several studies used whole-genome or exome sequencing to identify rare and low-frequency variants effecting non-LDL pathways that highlight the role of triglycerides in the development of CAD[45–49]. Though despite these well-designed studies in select populations the portion of disease potentially explained by the collection of risk variants remained low and each newly discovered variant only imparting small risk increase over that seen in control populations.
Understanding the strength in GWAS and ability to identify variants of interest with genome-wide significance comes in part with larger sample sizes and increasing diversity, new consortiums were formed to combine their data sets into meta-analysis. From these efforts came nearly 20 additional novel associated variants with CAD plus independent validation of previously associated variants [50, 51]. In 2013 taking this even further with the inclusion of 63,746 CAD cases and 130,681 controls, the largest CAD GWAS meta-analysis performed up to that time reported an additional 15 loci associated with CAD (bringing total number of observed loci associated with CAD to 46) [52]. Given that less than half of these variants (17 of 46) were known to be involved in pathways related to blood pressure and blood lipids, these data further supported the impact of genomics on CAD risk even outside of traditional clinical risk factors and helped broaden the understanding of the complex biology of CAD. The success of these early meta-GWAS in CAD risk variant discovery and validation and the entry of new datasets, namely the UK Biobank, soon grew the number of CAD associated variants to more than 160 independent loci [53–57]. Most recently, combining an additional 25,892 CAD cases and 142,336 controls from a Japanese population in a trans-ancestry meta-analysis of existing CAD cohorts from CARDIoGRAMplusC4D and UK Biobank, an additional 40 new loci associated with CAD were identified[58]. Though, the addition of these 40 new loci only increased the known heritability of CAD by 1.12%.
Development of Polygenic Risk Scores for CAD
In an effort to utilize the combined disease risk associations of the newly appreciated genetic variants discovered from GWAS and other studies discussed above, the use of polygenic risk scores for CAD (CAD PRS) became increasingly popular (Table 1). A PRS is created by the weighted sum of detected risk loci and aims to quantify an individuals’ underlying genetic predisposition to disease [9, 59]. PRSs have been developed and shown clinical utility for various diseases such as atrial fibrillation, diabetes mellitus, breast cancer and prostate cancer[60, 61]. The CAD PRS emerged as a promising genetic CAD metric with personal and clinical utility especially given the shortcomings of traditional risk assessment tools that do not account or any genetic contribution to disease [62, 63]. Initiated by early efforts of CAD GWASs, geneticists derived the prototype of CAD PRSs with dozens of common variants, explaining ~10% of heritability overall [51, 52]. Initial CAD PRSs utilized limited variants and the work focused on specific populations, mainly of European ancestry, but despite only modest gains in risk prediction, the association of CAD PRS with adverse cardiovascular events showed promise[64, 65]. Despite the moderate statistical power due to limited cohort size and the use of a genotyping array targeting pre-selected loci, the re-analysis of statin prevention trials illustrated the value of CAD-PRSs with 27 or 57 SNPs as an orthogonal predictor capable to stratify high-risk group with greater benefit from initiation of statin therapy and adherence to a favorable lifestyle practices [66–69].
Table 1:
Description of leading studies used to develop and validate various CAD PRS models
| Derivation | Target | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Discovery GWAS | Training/Testing | Validation | ||||||||||||
| Method | Description | Cohort | Ncase | Ncontrol | PRS (NSNP) | Cohort | Ncase | Ncontrol | Ref | Cohort | Ncase or Ntreatment | Ncontrol or Nplacebo | Performance | Ref |
| P+T | pruning and p-value thresholding | CARDIoGRAMplusC4D | 22,333 | 64,762 | PRSMega (27) | community case: MDCS 1st prevention: JUPITER, ASCOT-LLA 2nd prevention: CARE, PROVE-IT |
3,477 | 44,944 | 67 | FOURIER | 7,163 | 7,135 | 6 | |
| ODYSSEY | 9,462 | 9,462 | 72 | |||||||||||
| CARDIoGRAMplusC4D | 22,333 | 64,762 | PRSTada (50) | Malmö Diet and Cancer study (MDCS) | 2,213 | 21,382 | 69 | ARIC | 1,230 | 6,584 | HR: 1.75 (1.46–2.10) | 66 | ||
| WGHS | 971 | 20,251 | HR: 1.94 (1.58–2.39) | |||||||||||
| MDCS | 2,903 | 19,486 | HR: 1.98 (1.76–2.23) | |||||||||||
| eMERGE Phase III (European) | 5,887 | 39,758 | OR: 1.28 (1.25–1.32) HR: 1.20 (1.15–1.25) |
73 | ||||||||||
| eMERGE Phase III (African) | 527 | 7,070 | OR: 1.05 (0.98–1.14) HR: 1.05 (0.94–1.17) |
|||||||||||
| eMERGE Phase III (Hispanic) | 299 | 2,194 | OR: 1.20 (1.05–1.35) HR: 1.13 (0.93–1.36) |
|||||||||||
| CADIoGRAMplusC4D | 63,746 | 130,681 | PRSNataragan (57) | WOSCOPS | 604 | 4,288 | 68 | ODYSSEY | 9,462 | 9,462 | 72 | |||
| JUPITER | 108 | 8603 | ||||||||||||
| ASCOT-LLA | 4070 | 149 | ||||||||||||
| CARDIA | 1154 | |||||||||||||
| BioImage | 4392 | |||||||||||||
| LDpred | Bayesian PRS method adjusting marginal SNP effect size with LD reference | CADIoGRAMplusC4D | 63,746 | 130,681 | PRSKhera (6,630,150) | UK Biobank Phase II | 8,676 | 260,302 | 61 | MHI Biobank Phase 1 (IcWGS) | 974 | 976 | CAD Prevalence OR: 1.64 (1.48–1.81) | 77 |
| WHI Biobank Phase 2 (array) | 2,492 | 817 | CAD Prevalence OR: 1.55 (1.38–1.73) | |||||||||||
| CARTaGENE | 173 | 5,589 | CAD Prevalence OR: 1.69 (1.44–1.99) | |||||||||||
| MDCS | 4,122 | 24,434 | HR: 1.45 (1.40–1.49) | 75 | ||||||||||
| eMERGE Phase III (European) | 5,887 | 39,758 | OR: 1.66 (1.62–1.71) HR: 1.50 (1.43–1.56) |
73 | ||||||||||
| eMERGE Phase III (African) | 527 | 7,070 | OR: 1.30 (1.21–1.41) HR: 1.19 (1.07–1.33) |
|||||||||||
| eMERGE Phase III (Hispanic) | 299 | 2,194 | OR: 1.42 (1.25–1.61) HR: 1.16 (0.96–1.41) |
|||||||||||
| Meta-analysis | reciprocal two-stage sequential discovery and replication design with independent SNPs | CADIoGRAMplusC4D | 88,192 | 162,544 | 161 | UK Biobank | 34,541 | 261,984 | 57 | |||||
| UK Biobank | 34,541 | 261,984 | ||||||||||||
| Meta-analysis | meta-score based on 3 previous scores | CARDIoGRAMplusC4D | 63,746 | 130,681 | PRSInouye (1,745,179) | UK Biobank | 22,242 | 460,387 | 70 | MHI Biobank Phase 1 (IcWGS) | 974 | 976 | CAD Prevalence OR: 1.74 (1.57–1.93) | 77 |
| WHI Biobank Phase 2 (array) | 2,492 | 817 | CAD Prevalence OR: 1.60 (1.43–1.80) | |||||||||||
| WTCCC-CAD | 1,926 | 2,938 | CARTaGENE | 173 | 5,589 | CAD Prevalence OR: 1.75 (1.49–2.05) | ||||||||
| eMERGE Phase III (European) | 5,887 | 39,758 | OR: 1.73 (1.68–1.78) HR: 1.53 (1.46–1.60) |
73 | ||||||||||
| MIGEN-Harps | 488 | 531 | eMERGE Phase III (African) | 527 | 7,070 | OR: 1.40 (1.30–1.52) HR: 1.27 (1.13–1.43) |
||||||||
| eMERGE Phase III (Hispanic) | 299 | 2,194 | OR: 1.93 (1.67–2.22) HR: 1.53 (1.23–1.90) |
|||||||||||
| Trans-ancestry meta-GWAS | fixed-effect and random-effect meta-analyses | CARDIoGRAMplusC4D | 60,801 | 123,504 | PRSKoyama (75,028) | Independent Japanese cohort | 1,827 | 9,172 | 58 | |||||
| UK Biobank | 34,541 | 261,984 | ||||||||||||
| Biobank Japan | 25,892 | 142,336 | ||||||||||||
| AnnoPred | Bayesian PRS framework leveraging functional annotation | UK Biobank | 4,746 | 88,182 | PRSYe (2,994,054) | UK Biobank | 3,467 | 172,771 | 107 | |||||
| SCT | stacked clumping and thresholding | UK Biobank | 7,912 | 121,941 | PRSBolli (300,238) | UK Biobank | 15,433 | 262,900 | 108 | |||||
With the ever-increasing efficiency of DNA sequencers, genotyping technology, and statistical genetics techniques, as well as investment in national biobanks of genotypic and phenotypic information, the development of more complex genetic risk models is now possible. The development of many of these large (100,000+ individuals) datasets and prediction models rely upon imputation, which allows for the inference of whole genome genotypes for common genetic variation from an initially sparse genotyping assay. While these statistical techniques are mature and accurate, they can lead to bias in application across disparate genetic ancestry. With this in mind, Inouye et al. developed a genomic risk score (metaGRSCAD) by meta-analysis to model variant effect sizes (with 1.7M variants), improving to explain up to 26% of CAD heritability [70]. Alternatively, Khera et al. utilized a Bayesian framework to comprise a genome-wide polygenic score (GPSCAD) with 6.6M variants, estimating variant effect sizes with linkage disequilibrium (LD) adjustment [61]. As a complement to conventional risk factors, both PRSs were validated to predict the fold change of disease susceptibility, stratify individuals with different trajectories of risk, and inform tailored therapeutic intervention in independent datasets or clinical trials [71–77]. Although not unexpected given the multiple shared biologic pathways across diseases, the use of CAD PRSs even extends outside of strict coronary disease events alone as there are independent associations with peripheral arterial disease, aortic aneurysm, stroke and carotid artery disease as well [78, 79].
The Clinical Utility of Polygenic Risk Scores
While the ability to stratify future disease risk on a population level based on CAD PRS has been well established with several groups illustrating improved risk stratification and net reclassification in large datasets, there does remain a healthy debate about if it is time and how best to integrate a CAD PRS into clinical practice[80–85]. Accordingly, current guidelines for the prevention of cardiovascular disease from professional societies in the US and Europe lack the inclusion of CAD PRS [70]. This could be due to the absence of convincing outcome data from large-scale, multi-ethnic, prospective studies incorporating CAD PRS into risk optimization and therapeutic guidance strategies. When used prospectively in small sized, limited scope studies, CAD PRS may lead to increased use of statin therapy but inconclusive improvements in diet and exercise[86, 87]. Though, as has been recognized in other studies delivering genomic information to patients, the knowledge of genetic risk did not increase personal health anxieties. Rather than instilling fear of disease in a patient, CAD PRS has the opportunity to provide a patient with an important orthogonal risk enhancer, similar to the role of coronary artery calcium (CAC), to motivate and empower healthy lifestyle and optimal medical therapies. We have seen that individuals with high CAD PRS achieve the largest risk reduction on statin therapy for both primary and secondary prevention compared to persons with low CAD PRS risk, significantly lowering the number needed to treat, and lifestyle remains an incredibly vital component of lowering event risk over time[66, 67]. The relative risk reduction with statin therapy in low versus high CAD PRS individuals was 34% vs 50% for primary prevention and 3% vs 47% for secondary prevention[67]. Individuals are not destined for disease just because they have a high CAD PRS.
Apart from somatic mutations that accumulate with age and environment and are known to drive certain cancer risks and sometimes risk for CAD, our genetic risk for CAD is largely set from birth. Unlike traditional clinical risk stratification tools based on age and chronic conditions such as hypertension and metabolic disease that may not manifest until mid or late life, a person will know their CAD PRS from the first day of life[75]. Even in the absence of traditional risk factors a lifetime risk of CAD can be established early with the use of CAD PRS. Incorporating this concept, recent work suggested that CAD PRS could be used in concert with widely used CAC testing to help establish the optimal time of first CAC screening based on tiers of CAD PRS[88, 89]. Additionally, a CAD PRS appears to act as a potentiometer for monogenic CVD either dialing up or dialing down the combined impact for disease[90].
Mirroring the improvements on CAD relative risk reduction that high CAD PRS has with statin therapy, recent analysis of landmark secondary prevention trials, ODYSSEY and FOURIER, with PCSK9 inhibitor therapy illustrated that these therapies had the greatest benefit for individuals with elevated genetic risk [72, 76]. In these studies, the relative risk reduction for clinical events in low versus high CAD PRS groups was 13% vs 31% (ODYSSEY) and 13% vs 37% (FOURIER). This is on top of high intensity statin therapy for which 70% of patients in FOURIER and 90% of patients in ODYSSEY were already taking.
Though lacking prospective validation, in this era of higher cost for novel therapies CAD PRS may prove key to matching the right patients with the most cost-effective therapies to achieve the greatest individualized benefit. While the promise of incorporating CAD PRS into clinical pathways is strong, the impact of CAD PRS is not fully understood as it does appear to have variation across sex, certain risk factors such as cigarette smoking, and most importantly, across ethnicities[91–94].
The Performance of Polygenic Risk Scores Across Ethnicities
Recent studies urged a significant attenuation of cross-population prediction accuracy to hamper the utility of CAD PRSs [73, 93]. Most of the studies predominantly conducted with European-decent population and heavily underrepresented the global demographic diversity or human evolutionary history [95]. The current Eurocentric sampling is inadequate to discover the disparate genetic architectures, differentiated LD patterns and non-genetic factors with gene-by-environment (GxE) interactions among the populations [73, 94, 96]. Recent studies have reported foreseeable improvement by trans-ethnic meta-analysis[58]. Growing efforts were also promoted to diversify the exploratory samples in genomic research with harmonized phenotypic definition and case ascertainment [97, 98]. To leverage current large-scale datasets and understanding of genetic studies, efforts attempted to bridge this gap by functional fine-mapping, the goal of which was to identify causal variants shared across populations [99, 100]. A recent study demonstrated that regulatory annotation partitioning can maintain the portability of PRS models from Europeans applying towards East Asians [101]. However, all current polygenic prediction method only allowed for input from one to multiple homogeneous subpopulations. Future work is needed for admixed population with higher genome complexity. Promising direction could extend to incorporate ancestry-specific effect size estimation and local ancestry adjustment [102, 103].
In addition, increased adoption and connectivity of personal electronic health records (EHRs) can expedite genomic discovery and personalized medicine implementation[97]. Wearable sensors and devices also enable the impending collection of comprehensive exogenous factors to quantify individual envirotypes [104]. Recent perspective has outlined the potentials for developing and integrating risk predictions with PRSs and biobank-linked EHR data [105]. Few cohort studies demonstrated the potential improvement by delineating the multiplicative interaction of modifiable risk factors with CAD PRSs [91, 106–108]. High-level risk stratification considering environmental factors and GxE association would potentially further disentangle the bias between different populations.
CONCLUSIONS
It is without a doubt that the rapid advances in genomic medicine have now enabled a more complete assessment of cardiovascular risk that had previously remained unaccounted for. By using readily available, increasingly affordable and clinically meaningful genetic assessment tools, clinicians now can better inform patients about their own baseline genetic risks in order to create personalized and strategic risk optimization strategies. As our genetic knowledge and analytical techniques expand, these risk optimization strategies will be further tailored to each individual. Our group has developed the MyGeneRank platform (https://mygenerank.scripps.edu/) specifically to assist an individual in determining their CAD PRS using their smartphone device which can be determined in minutes and shared with their clinical care team. PRSs will continue to improve as genomic research expands to include a better representation of ethnicities and populations under-represented in current biomedical research. In addition, the call for more standardized reporting, data sharing, especially metadata, within the scientific community is essential to improve accuracy in the real-world and the ability to translate findings into the clinical setting[109]. Studies investigating the role of CAD in diverse populations must be conducted and shared openly to ensure that our tools to intervene early to prevent clinical disease are accessible to all.
ACKNOWLEDGMENTS
EDM and AT are supported by UL1TR002550 from NCATS/NIH to The Scripps Research Institute. AT acknowledges additional support from NHGRI/NIH HG010881.
ABBREVIATIONS
- CAD
coronary artery disease
- EHR
electronic health record
- FH
familial hypercholesterolemia
- GWAS
genome wide association study
- LD
linkage disequilibrium
- LDL
low density lipoprotein
- LDLR
low density lipoprotein receptor
- PRS
polygenic risk score
Footnotes
Conflict of Interest: Evan D. Muse and Ali Torkamani are co-founders of GeneXwell. In addition, Dr. Torkamani has a patent pending on polygenic risk score generation and adjustment. Mr. Chen has nothing to disclose.
COMPLIANCE WITH ETHICAL STANDARDS
Human and Animal Rights and Informed Consent: This article does not contain any studies with human or animal subjects performed by any of the authors.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
REFERENCES:
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021. doi: 10.1161/CIR.0000000000000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topol EJ. High-performance medicine: the convergence of human and artificial intelligence. Nat Med. 2019;25(1):44–56. doi: 10.1038/s41591-018-0300-7. [DOI] [PubMed] [Google Scholar]
- 3.Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18(6):331–44. doi: 10.1038/nrg.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018;114(9):1241–57. doi: 10.1093/cvr/cvy084. [DOI] [PubMed] [Google Scholar]
- 5.Musunuru K, Kathiresan S. Genetics of Common, Complex Coronary Artery Disease. Cell. 2019;177(1):132–45. doi: 10.1016/j.cell.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Hippisley-Cox J, Coupland C, Vinogradova Y, Robson J, Minhas R, Sheikh A et al. Predicting cardiovascular risk in England and Wales: prospective derivation and validation of QRISK2. BMJ. 2008;336(7659):1475–82. doi: 10.1136/bmj.39609.449676.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;74(10):e177–e232. doi: 10.1016/j.jacc.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ten Kate LP, Boman H, Daiger SP, Motulsky AG. Familial aggregation of coronary heart disease and its relation to known genetic risk factors. Am J Cardiol. 1982;50(5):945–53. doi: 10.1016/0002-9149(82)90400-3. [DOI] [PubMed] [Google Scholar]
- 9.Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. 2018;19(9):581–90. doi: 10.1038/s41576-018-0018-x. [DOI] [PubMed] [Google Scholar]
- 10.Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330(15):1041–6. doi: 10.1056/NEJM199404143301503. [DOI] [PubMed] [Google Scholar]
- 11.Lloyd-Jones DM, Nam BH, D’Agostino RB Sr., Levy D, Murabito JM, Wang TJ et al. Parental cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults: a prospective study of parents and offspring. JAMA. 2004;291(18):2204–11. doi: 10.1001/jama.291.18.2204. [DOI] [PubMed] [Google Scholar]
- 12.Churko JM, Mantalas GL, Snyder MP, Wu JC. Overview of high throughput sequencing technologies to elucidate molecular pathways in cardiovascular diseases. Circ Res. 2013;112(12):1613–23. doi: 10.1161/CIRCRESAHA.113.300939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478–90a. doi: 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henderson R, O’Kane M, McGilligan V, Watterson S. The genetics and screening of familial hypercholesterolaemia. J Biomed Sci. 2016;23:39. doi: 10.1186/s12929-016-0256-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akioyamen LE, Genest J, Shan SD, Reel RL, Albaum JM, Chu A et al. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open. 2017;7(9):e016461. doi: 10.1136/bmjopen-2017-016461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur Heart J. 2016;37(17):1384–94. doi: 10.1093/eurheartj/ehw028. [DOI] [PubMed] [Google Scholar]
- 17.Lehrman MA, Schneider WJ, Sudhof TC, Brown MS, Goldstein JL, Russell DW. Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science. 1985;227(4683):140–6. doi: 10.1126/science.3155573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4(4):214–25. doi: 10.1038/ncpcardio0836. [DOI] [PubMed] [Google Scholar]
- 19.Soria LF, Ludwig EH, Clarke HR, Vega GL, Grundy SM, McCarthy BJ. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc Natl Acad Sci U S A. 1989;86(2):587–91. doi: 10.1073/pnas.86.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers. 2017;3:17093. doi: 10.1038/nrdp.2017.93. [DOI] [PubMed] [Google Scholar]
- 21.Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–6. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 22.Sabatine MS. PCSK9 inhibitors: clinical evidence and implementation. Nat Rev Cardiol. 2019;16(3):155–65. doi: 10.1038/s41569-018-0107-8. [DOI] [PubMed] [Google Scholar]
- 23.Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292(5520):1394–8. doi: 10.1126/science.1060458. [DOI] [PubMed] [Google Scholar]
- 24.Awan Z, Choi HY, Stitziel N, Ruel I, Bamimore MA, Husa R et al. APOE p.Leu167del mutation in familial hypercholesterolemia. Atherosclerosis. 2013;231(2):218–22. doi: 10.1016/j.atherosclerosis.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Fouchier SW, Dallinga-Thie GM, Meijers JC, Zelcer N, Kastelein JJ, Defesche JC et al. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ Res. 2014;115(6):552–5. doi: 10.1161/CIRCRESAHA.115.304660. [DOI] [PubMed] [Google Scholar]
- 26.Rios J, Stein E, Shendure J, Hobbs HH, Cohen JC. Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum Mol Genet. 2010;19(22):4313–8. doi: 10.1093/hmg/ddq352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, Parikh VN et al. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ Genom Precis Med. 2020;13(4):e000067. doi: 10.1161/HCG.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 28.Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. The Lancet. 2013;381(9874):1293–301. doi: 10.1016/s0140-6736(12)62127-8. [DOI] [PubMed] [Google Scholar]
- 29.Besseling J, Hovingh GK, Huijgen R, Kastelein JJP, Hutten BA. Statins in Familial Hypercholesterolemia: Consequences for Coronary Artery Disease and All-Cause Mortality. J Am Coll Cardiol. 2016;68(3):252–60. doi: 10.1016/j.jacc.2016.04.054. [DOI] [PubMed] [Google Scholar]
- 30.Abul-Husn NS, Manickam K, Jones LK, Wright EA, Hartzel DN, Gonzaga-Jauregui C et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science. 2016;354(6319). doi: 10.1126/science.aaf7000. [DOI] [PubMed] [Google Scholar]
- 31.Alver M, Palover M, Saar A, Lall K, Zekavat SM, Tonisson N et al. Recall by genotype and cascade screening for familial hypercholesterolemia in a population-based biobank from Estonia. Genet Med. 2019;21(5):1173–80. doi: 10.1038/s41436-018-0311-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sturm AC, Knowles JW, Gidding SS, Ahmad ZS, Ahmed CD, Ballantyne CM et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2018;72(6):662–80. doi: 10.1016/j.jacc.2018.05.044. [DOI] [PubMed] [Google Scholar]
- 33.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet. 2017;101(1):5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316(5830):1491–3. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 35.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316(5830):1488–91. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357(5):443–53. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo Sardo V, Chubukov P, Ferguson W, Kumar A, Teng EL, Duran M et al. Unveiling the Role of the Most Impactful Cardiovascular Risk Locus through Haplotype Editing. Cell. 2018;175(7):1796–810 e20. doi: 10.1016/j.cell.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358(12):1240–9. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- 39.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40(2):161–9. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–28. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 41.Erdmann J, Grosshennig A, Braund PS, Konig IR, Hengstenberg C, Hall AS et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41(3):280–2. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myocardial Infarction Genetics C, Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41(3):334–41. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466(7307):714–9. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. The Lancet. 2012;380(9841):572–80. doi: 10.1016/s0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102–6. doi: 10.1038/nature13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45(11):1345–52. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helgadottir A, Gretarsdottir S, Thorleifsson G, Hjartarson E, Sigurdsson A, Magnusdottir A et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet. 2016;48(6):634–9. doi: 10.1038/ng.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khera AV, Won HH, Peloso GM, O’Dushlaine C, Liu D, Stitziel NO et al. Association of Rare and Common Variation in the Lipoprotein Lipase Gene With Coronary Artery Disease. JAMA. 2017;317(9):937–46. doi: 10.1001/jama.2017.0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–83. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coronary Artery Disease Genetics C. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43(4):339–44. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 51.Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43(4):333–8. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Consortium CAD, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121–30. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Howson JMM, Zhao W, Barnes DR, Ho WK, Young R, Paul DS et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat Genet. 2017;49(7):1113–9. doi: 10.1038/ng.3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49(9):1392–7. doi: 10.1038/ng.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017;49(9):1385–91. doi: 10.1038/ng.3913. [DOI] [PubMed] [Google Scholar]
- 57.van der Harst P, Verweij N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ Res. 2018;122(3):433–43. doi: 10.1161/CIRCRESAHA.117.312086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koyama S, Ito K, Terao C, Akiyama M, Horikoshi M, Momozawa Y et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet. 2020;52(11):1169–77. doi: 10.1038/s41588-020-0705-3.* This study highlights the importance of ethnicity in PRS derivation when translating findings into non-European populations. One of the largest trans-ethnic meta-analysis to date for discovery of novel loci associated with CAD.
- 59.Chatterjee N, Shi J, Garcia-Closas M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat Rev Genet. 2016;17(7):392–406. doi: 10.1038/nrg.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen H, Liu X, Brendler CB, Ankerst DP, Leach RJ, Goodman PJ et al. Adding genetic risk score to family history identifies twice as many high-risk men for prostate cancer: Results from the prostate cancer prevention trial. Prostate. 2016;76(12):1120–9. doi: 10.1002/pros.23200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–24. doi: 10.1038/s41588-018-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DeFilippis AP, Young R, Carrubba CJ, McEvoy JW, Budoff MJ, Blumenthal RS et al. An analysis of calibration and discrimination among multiple cardiovascular risk scores in a modern multiethnic cohort. Ann Intern Med. 2015;162(4):266–75. doi: 10.7326/M14-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rana JS, Tabada GH, Solomon MD, Lo JC, Jaffe MG, Sung SH et al. Accuracy of the Atherosclerotic Cardiovascular Risk Equation in a Large Contemporary, Multiethnic Population. J Am Coll Cardiol. 2016;67(18):2118–30. doi: 10.1016/j.jacc.2016.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thanassoulis G, Peloso GM, Pencina MJ, Hoffmann U, Fox CS, Cupples LA et al. A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: the Framingham Heart Study. Circ Cardiovasc Genet. 2012;5(1):113–21. doi: 10.1161/CIRCGENETICS.111.961342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tikkanen E, Havulinna AS, Palotie A, Salomaa V, Ripatti S. Genetic risk prediction and a 2-stage risk screening strategy for coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33(9):2261–6. doi: 10.1161/ATVBAHA.112.301120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR et al. Genetic Risk, Adherence to a Healthy Lifestyle, and Coronary Disease. N Engl J Med. 2016;375(24):2349–58. doi: 10.1056/NEJMoa1605086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield MJ, Devlin JJ et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. The Lancet. 2015;385(9984):2264–71. doi: 10.1016/s0140-6736(14)61730-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U, Mehran R et al. Polygenic Risk Score Identifies Subgroup With Higher Burden of Atherosclerosis and Greater Relative Benefit From Statin Therapy in the Primary Prevention Setting. Circulation. 2017;135(22):2091–101. doi: 10.1161/CIRCULATIONAHA.116.024436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tada H, Melander O, Louie JZ, Catanese JJ, Rowland CM, Devlin JJ et al. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J. 2016;37(6):561–7. doi: 10.1093/eurheartj/ehv462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, Dudbridge F et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J Am Coll Cardiol. 2018;72(16):1883–93. doi: 10.1016/j.jacc.2018.07.079.* The findings presented here integrate large genomic datasets to show the benefit of polygenic risk scoring for risk stratification above and beyond traditional risk factors.
- 71.Aragam KG, Dobbyn A, Judy R, Chaffin M, Chaudhary K, Hindy G et al. Limitations of Contemporary Guidelines for Managing Patients at High Genetic Risk of Coronary Artery Disease. J Am Coll Cardiol. 2020;75(22):2769–80. doi: 10.1016/j.jacc.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Damask A, Steg PG, Schwartz GG, Szarek M, Hagstrom E, Badimon L et al. Patients With High Genome-Wide Polygenic Risk Scores for Coronary Artery Disease May Receive Greater Clinical Benefit From Alirocumab Treatment in the ODYSSEY OUTCOMES Trial. Circulation. 2020;141(8):624–36. doi: 10.1161/CIRCULATIONAHA.119.044434.* The findings presented here with PSCK9 inhibitor therapy extend the statin findings from Mega et al 2015 showing the utility of CAD PRS to identify patients who receive the greatest benefit from therapy.
- 73.Dikilitas O, Schaid DJ, Kosel ML, Carroll RJ, Chute CG, Denny JA et al. Predictive Utility of Polygenic Risk Scores for Coronary Heart Disease in Three Major Racial and Ethnic Groups. Am J Hum Genet. 2020;106(5):707–16. doi: 10.1016/j.ajhg.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Emdin CA, Bhatnagar P, Wang M, Pillai SG, Li L, Qian HR et al. Genome-Wide Polygenic Score and Cardiovascular Outcomes With Evacetrapib in Patients With High-Risk Vascular Disease: A Nested Case-Control Study. Circ Genom Precis Med. 2020;13(1):e002767. doi: 10.1161/CIRCGEN.119.002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hindy G, Aragam KG, Ng K, Chaffin M, Lotta LA, Baras A et al. Genome-Wide Polygenic Score, Clinical Risk Factors, and Long-Term Trajectories of Coronary Artery Disease. Arterioscler Thromb Vasc Biol. 2020;40(11):2738–46. doi: 10.1161/ATVBAHA.120.314856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marston NA, Kamanu FK, Nordio F, Gurmu Y, Roselli C, Sever PS et al. Predicting Benefit From Evolocumab Therapy in Patients With Atherosclerotic Disease Using a Genetic Risk Score: Results From the FOURIER Trial. Circulation. 2020;141(8):616–23. doi: 10.1161/CIRCULATIONAHA.119.043805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wunnemann F, Sin Lo K, Langford-Avelar A, Busseuil D, Dube MP, Tardif JC et al. Validation of Genome-Wide Polygenic Risk Scores for Coronary Artery Disease in French Canadians. Circ Genom Precis Med. 2019;12(6):e002481. doi: 10.1161/CIRCGEN.119.002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ntalla I, Kanoni S, Zeng L, Giannakopoulou O, Danesh J, Watkins H et al. Genetic Risk Score for Coronary Disease Identifies Predispositions to Cardiovascular and Noncardiovascular Diseases. J Am Coll Cardiol. 2019;73(23):2932–42. doi: 10.1016/j.jacc.2019.03.512. [DOI] [PubMed] [Google Scholar]
- 79.Timmerman N, de Kleijn DPV, de Borst GJ, den Ruijter HM, Asselbergs FW, Pasterkamp G et al. Family history and polygenic risk of cardiovascular disease: Independent factors associated with secondary cardiovascular events in patients undergoing carotid endarterectomy. Atherosclerosis. 2020;307:121–9. doi: 10.1016/j.atherosclerosis.2020.04.013. [DOI] [PubMed] [Google Scholar]
- 80.Abraham G, Havulinna AS, Bhalala OG, Byars SG, De Livera AM, Yetukuri L et al. Genomic prediction of coronary heart disease. Eur Heart J. 2016;37(43):3267–78. doi: 10.1093/eurheartj/ehw450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Elliott J, Bodinier B, Bond TA, Chadeau-Hyam M, Evangelou E, Moons KGM et al. Predictive Accuracy of a Polygenic Risk Score-Enhanced Prediction Model vs a Clinical Risk Score for Coronary Artery Disease. JAMA. 2020;323(7):636–45. doi: 10.1001/jama.2019.22241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mars N, Koskela JT, Ripatti P, Kiiskinen TTJ, Havulinna AS, Lindbohm JV et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat Med. 2020;26(4):549–57. doi: 10.1038/s41591-020-0800-0. [DOI] [PubMed] [Google Scholar]
- 83.Mosley JD, Gupta DK, Tan J, Yao J, Wells QS, Shaffer CM et al. Predictive Accuracy of a Polygenic Risk Score Compared With a Clinical Risk Score for Incident Coronary Heart Disease. JAMA. 2020;323(7):627–35. doi: 10.1001/jama.2019.21782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Riveros-Mckay F, Weale ME, Moore R, Selzam S, Krapohl E, Sivley RM et al. An Integrated Polygenic Tool Substantially Enhances Coronary Artery Disease Prediction. Circ Genom Precis Med. 2021. doi: 10.1161/CIRCGEN.120.003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jarmul J, Pletcher MJ, Hassmiller Lich K, Wheeler SB, Weinberger M, Avery CL et al. Cardiovascular Genetic Risk Testing for Targeting Statin Therapy in the Primary Prevention of Atherosclerotic Cardiovascular Disease: A Cost-Effectiveness Analysis. Circ Cardiovasc Qual Outcomes. 2018;11(4):e004171. doi: 10.1161/CIRCOUTCOMES.117.004171. [DOI] [PubMed] [Google Scholar]
- 86.Knowles JW, Zarafshar S, Pavlovic A, Goldstein BA, Tsai S, Li J et al. Impact of a Genetic Risk Score for Coronary Artery Disease on Reducing Cardiovascular Risk: A Pilot Randomized Controlled Study. Front Cardiovasc Med. 2017;4:53. doi: 10.3389/fcvm.2017.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kullo IJ, Jouni H, Austin EE, Brown SA, Kruisselbrink TM, Isseh IN et al. Incorporating a Genetic Risk Score Into Coronary Heart Disease Risk Estimates: Effect on Low-Density Lipoprotein Cholesterol Levels (the MI-GENES Clinical Trial). Circulation. 2016;133(12):1181–8. doi: 10.1161/CIRCULATIONAHA.115.020109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Severance LM, Carter H, Contijoch FJ, McVeigh ER. Targeted Coronary Artery Calcium Screening in High-Risk Younger Individuals Using Consumer Genetic Screening Results. JACC Cardiovasc Imaging. 2021. doi: 10.1016/j.jcmg.2020.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Severance LM, Contijoch FJ, Carter H, Fan CC, Seibert TM, Dale AM et al. Using a genetic risk score to calculate the optimal age for an individual to undergo coronary artery calcium screening. J Cardiovasc Comput Tomogr. 2019;13(4):203–10. doi: 10.1016/j.jcct.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fahed AC, Wang M, Homburger JR, Patel AP, Bick AG, Neben CL et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun. 2020;11(1):3635. doi: 10.1038/s41467-020-17374-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hindy G, Wiberg F, Almgren P, Melander O, Orho-Melander M. Polygenic Risk Score for Coronary Heart Disease Modifies the Elevated Risk by Cigarette Smoking for Disease Incidence. Circ Genom Precis Med. 2018;11(1):e001856. doi: 10.1161/CIRCGEN.117.001856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang Y, Hui Q, Gwinn M, Hu YJ, Quyyumi AA, Vaccarino V et al. Sexual Differences in Genetic Predisposition of Coronary Artery Disease. Circ Genom Precis Med. 2020. doi: 10.1161/CIRCGEN.120.003147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51(4):584–91. doi: 10.1038/s41588-019-0379-x.* An important perspective addresssing the Eurocentric bias in current genomic studies calling for improved diversity in future studies.
- 94.Gola D, Erdmann J, Lall K, Magi R, Muller-Myhsok B, Schunkert H et al. Population Bias in Polygenic Risk Prediction Models for Coronary Artery Disease. Circ Genom Precis Med. 2020;13(6):e002932. doi: 10.1161/CIRCGEN.120.002932. [DOI] [PubMed] [Google Scholar]
- 95.Skoglund P, Mathieson I. Ancient Genomics of Modern Humans: The First Decade. Annu Rev Genomics Hum Genet. 2018;19:381–404. doi: 10.1146/annurev-genom-083117-021749. [DOI] [PubMed] [Google Scholar]
- 96.Wang M, Menon R, Mishra S, Patel AP, Chaffin M, Tanneeru D et al. Validation of a Genome-Wide Polygenic Score for Coronary Artery Disease in South Asians. J Am Coll Cardiol. 2020;76(6):703–14. doi: 10.1016/j.jacc.2020.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abul-Husn NS, Kenny EE. Personalized Medicine and the Power of Electronic Health Records. Cell. 2019;177(1):58–69. doi: 10.1016/j.cell.2019.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590(7845):290–9. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet. 2018;19(8):491–504. doi: 10.1038/s41576-018-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weissbrod O, Hormozdiari F, Benner C, Cui R, Ulirsch J, Gazal S et al. Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat Genet. 2020;52(12):1355–63. doi: 10.1038/s41588-020-00735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Amariuta T, Ishigaki K, Sugishita H, Ohta T, Koido M, Dey KK et al. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat Genet. 2020;52(12):1346–54. doi: 10.1038/s41588-020-00740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Atkinson EG, Maihofer AX, Kanai M, Martin AR, Karczewski KJ, Santoro ML et al. Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat Genet. 2021;53(2):195–204. doi: 10.1038/s41588-020-00766-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maples BK, Gravel S, Kenny EE, Bustamante CD. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am J Hum Genet. 2013;93(2):278–88. doi: 10.1016/j.ajhg.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Engelhard MM, Oliver JA, McClernon FJ. Digital envirotyping: quantifying environmental determinants of health and behavior. NPJ Digit Med. 2020;3:36. doi: 10.1038/s41746-020-0245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li R, Chen Y, Ritchie MD, Moore JH. Electronic health records and polygenic risk scores for predicting disease risk. Nat Rev Genet. 2020;21(8):493–502. doi: 10.1038/s41576-020-0224-1. [DOI] [PubMed] [Google Scholar]
- 106.Rao DC, Sung YJ, Winkler TW, Schwander K, Borecki I, Cupples LA et al. Multiancestry Study of Gene-Lifestyle Interactions for Cardiovascular Traits in 610 475 Individuals From 124 Cohorts: Design and Rationale. Circ Cardiovasc Genet. 2017;10(3). doi: 10.1161/CIRCGENETICS.116.001649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ye Y, Chen X, Han J, Jiang W, Natarajan P, Zhao H. Interactions Between Enhanced Polygenic Risk Scores and Lifestyle for Cardiovascular Disease, Diabetes, and Lipid Levels. Circ Genom Precis Med. 2021;14(1):e003128. doi: 10.1161/CIRCGEN.120.003128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bolli A, Di Domenico P, Pastorino R, Busby GB, Botta G. Risk of Coronary Artery Disease Conferred by Low-Density Lipoprotein Cholesterol Depends on Polygenic Background. Circulation. 2021. doi: 10.1161/CIRCULATIONAHA.120.051843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wand H, Lambert SA, Tamburro C, Iacocca MA, O’Sullivan JW, Sillari C et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature. 2021;591(7849):211–9. doi: 10.1038/s41586-021-03243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]