

Abstract
Adult cardiomyocytes are postmitotic cells that undergo very limited cell division. Thus, cardiomyocyte death as occurs during myocardial infarction has very detrimental consequences for the heart. Mitochondria have emerged as an important regulator of cardiovascular health and disease. Mitochondria are well established as bioenergetic hubs for generating ATP but have also been shown to regulate cell death pathways. Indeed many of the same signals used to regulate metabolism and ATP production, such as calcium and reactive oxygen species, are also key regulators of mitochondrial cell death pathways. It is widely hypothesized that an increase in calcium and reactive oxygen species activate a large conductance channel in the inner mitochondrial membrane known as the PTP (permeability transition pore) and that opening of this pore leads to necroptosis, a regulated form of necrotic cell death. Strategies to reduce PTP opening either by inhibition of PTP or inhibiting the rise in mitochondrial calcium or reactive oxygen species that activate PTP have been proposed. A major limitation of inhibiting the PTP is the lack of knowledge about the identity of the protein(s) that form the PTP and how they are activated by calcium and reactive oxygen species. This review will critically evaluate the candidates for the pore-forming unit of the PTP and discuss recent data suggesting that assumption that the PTP is formed by a single molecular identity may need to be reconsidered.
Keywords: calcium, cell death, mitochondria, permeability, reactive oxygen species
CELL DEATH VIA THE MITOCHONDRIAL PERMEABILITY TRANSITION PORE
The mitochondrial PTP (permeability transition pore) is a large conductance channel in the inner mitochondrial membrane that is thought to play an important role in cell death.1 The PTP was first described by Haworth and Hunter,2 who showed that addition of high levels of calcium to bovine myocardial mitochondria induced a nonspecific increase in permeability of the inner mitochondrial membrane. They further showed that the calcium-induced permeability was readily reversible if calcium was chelated. Although PTP was initially written off as a laboratory phenomenon with minimal clinical consequence, additional study showed that PTP played an important role in initiating cell death in many diseases.1,3,4
It has been shown that opening of the PTP collapses the mitochondrial membrane potential and allows equilibration of water and solutes <1.5 kD.2,5 Classically, when PTP is induced in media containing solutes <1.5 kD, the mitochondria swell due to an influx of water. PTP opening can be induced in mitochondria depleted of endogenous energy substrate and in the presence of uncoupler, showing that the formation of PTP is not energy dependent.2,6 In addition to the classic, sustained high-conductance PTP opening, it has also been shown that transient low conductance activation of PTP can occur.7,8 Inhibition of transient opening of the PTP has been reported to inhibit preconditioning.9 It has been suggested that transient opening of the PTP might be a release valve to reduce high levels of matrix calcium.10
Initial studies demonstrated that calcium is a potent activator of the PTP. Subsequently, it was demonstrated that Mg++, ADP, H+, and NADH increased the Km of calcium-induced PTP formation such that higher levels of calcium are needed to induce PTP opening. Reactive oxygen species (ROS) were also shown to activate PTP opening.11 Beyond these endogenous modulators, many pharmacological inhibitors and potentiators of the PTP have been identified, such as the ANT (adenine nucleotide translocase) inhibitor, bongkrekic acid, and the ANT activator atractyloside.12 These 2 modulators are proposed to work at the ANT by locking the carrier protein in different confirmations.13 It was shown that any drug that stabilized the conformation of ANT in which it faces the cytosol (the c conformation) enhanced PTP opening, whereas stabilization of the ANT in the matrix (m) conformation inhibits PTP opening.3 CsA (cyclosporin A) was originally shown by Fournier et al14 to block mitochondrial calcium efflux and allow mitochondria to accumulate large amounts of calcium. Crompton et al15 then showed that ability of mitochondria to retain calcium in the presence of CsA was due to CsA inhibition of the PTP. Using a photoactive CsA derivative, CypD (cyclophilin D), a mitochondrial peptidyl-prolyl cis-trans isomerase was shown to be the target of CsA.16 Genetic deletion studies have confirmed that CypD is an activator of PTP opening.17 CsA has been demonstrated to reduce cell death in many studies of ischemia-reperfusion injury.18,19 CsA is also known to bind calcineurin and cause immunomodulation20; however, CypD inhibitors that do not bind to calcineurin, such as Debio 025 and NIM811 (N-methyl-4-isoleucine cyclosporin), have been shown to block PTP,21,22 suggesting that inhibition of calcineurin is not required. In addition, CsA blocks ischemic preconditioning; this effect has been attributed to blocking transient opening of PTP during the preconditioning period.9 Additional work will be needed to determine the details and physiological/pathological implications of transient PTP opening.23,24
PTP opening has been proposed as the key driver of ischemia/reperfusion (I/R) injury.3,5,25 Many of the endogenous potentiators of the PTP, including calcium and ROS, increase during I/R injury (Figure 1).26 Inhibitors of the PTP have been shown to reduce infarct size following I/R. CsA, for example, has been shown to reduce infarct size following I/R, even if the drug infusion was started after the onset of ischemia.27 It has been shown that acidic pH reduces cell death in I/R,28,29 and this has been attributed to inhibition of the PTP by acid pH during ischemia.30–32 Low ischemic pH inhibits PTP during ischemia such that PTP opening only occurs during reperfusion when pH is restored.28 This is suggested to provide a window of opportunity to administer PTP inhibitors at the start of reperfusion. Of note, inhibition of PTP by low pH only occurs in de-energized mitochondria.33 If mitochondria are energized, low pH has been shown to stimulate PTP opening.33 Thus, it appears that low pH will only inhibit PTP if the mitochondria are de-energized. Inhibition of the PTP and cardioprotection have been demonstrated in cells, isolated hearts,18 and in vivo models.34 In a phase II trial, the administration of CsA during acute myocardial infarction decreased infarct size as assessed by magnetic resonance imaging,35 although a subsequent phase III trial showed no benefit to CsA treatment.36 Potential reasons why CsA has been beneficial in animal studies yet failed to show benefit in clinical trials have been discussed elsewhere.37 It is worth pointing out that although CsA is the most commonly used inhibitor of the pore, it works through binding CypD, which is only a regulator of the pore. Therefore, it is possible that the stimulus for myocardial infarction present in the phase III trial is overcoming the inhibition that is conferred through inhibiting CypD.
Figure 1. Calcium and reactive oxygen species (ROS) lead to activation of the mitochondrial PTP (permeability transition pore) and initiation of cell death.

A, At the end of ischemia and the first few seconds of reperfusion, an increase in mitochondrial calcium can occur via MCU (mitochondrial calcium uniporter) which can activate PTP. There is also a buildup of succinate which upon conversion to fumarate by complex II, generates large levels of QH2 (ubiquinol) which drives reverse electron transport (RET) of complex I leading to ROS production. As shown in B, inhibition of PTP,. inhibition of MCU and inhibition of complex II (or complex I) are potential mechanisms to reduce PTP opening and reduce cell death. CypD indicates cyclophilin D. Illustration credit: Ben Smith.
Beyond I/R injury, the PTP is thought to play a role in the necrotic cell death that is present in chronic heart failure, potentially driven by calcium mishandling by the cardiac myocytes in the disease. However, a role for PTP opening in heart failure is not clear, and furthermore, mice lacking CypD had increased susceptibility to heart failure.10 Outside the field of cardiology, there is evidence that the mitochondrial PTP is involved in neuronal plasticity,38 in ischemia in other organs such as kidney, as well as muscular dystrophies.39,40 Although activation of PTP is involved in cell death in most if not all tissues and in many diseases, this review is focused on cardiomyocytes and ischemia and reperfusion.
Approaches to inhibit PTP have been hampered by the lack of detailed knowledge of the molecular identity of the PTP. Over the years, there have been many hypotheses regarding the molecular identity of the PTP. The gold standard approach for confirming the molecular identity of the putative PTP candidates has relied on the ability of genetic manipulation of a gene or a gene family to abolish CsA-sensitive PTP opening. This approach has failed to confirm most, if not all, of the candidates. Here, we describe the major competing hypotheses and provide historical context to summarize the current understanding of the molecular identity of the PTP. We will also highlight contemporary studies that provide some evidence that the assumption that the PTP is formed by a single molecular identity may need to be reconsidered.
ANT/VDAC/CypD
Formation of the Theory
An early proposal suggested that the PTP was formed at contact sites between the inner and outer mitochondrial membranes. Research by the Brdiczka lab showed that the ANT was the primary regulator of contact sites.41 The formation of these contacts was related to the ADP level in the cell. Further work with a CypD affinity matrix, comprised of a glutathione S-transferase/cyclophilin D fusion protein bound to glutathione agarose, pulled down both VDAC (voltage-dependent anion carrier) and ANT, and, when these were reconstituted into phosphatidylcholine liposomes, they retained characteristics of the PTP.42,43 It was shown that CypD bound to ANT in a CsA-dependent manner. Furthermore, reconstituted and purified ANT (from Neurospora crassa) was found to form nonspecific 300 to 600 pico-siemen (pS) channels, further supporting its presumed role in PTP44,45; however, patch clamping of the megachannel which was proposed to be the PTP, resulted in 1.3 nano-siemen (nS) channels. Many proteins can form nonspecific channels when reconstituted into liposomes or bilayers, and therefore, the size of the conductance needs to be considered. It is also important to verify the findings in intact mitochondria. It was proposed that under physiological conditions VDAC and ANT form a complex at contact sites, and under pathological conditions, such as I/R injury, this complex could deform into the PTP. The involvement of ANT in the PTP was readily reconcilable with the fact that ADP, atractyloside, and bongkrekic acid were known modulators of the PTP, and all are known to bind to ANT.46,47 Other studies showed that Bax (BCL2-associated X) could regulate PTP opening in conjunction with ANT.48 In this study, Bax was shown to co-immunoprecipitate with ANT, and ectopic expression of Bax induced cell death, but not without the presence of ANT, and both ANT and Bax were necessary for atractyloside to induce channel formation in artificial membranes.48 Thus, at the turn of the millennium, it was generally hypothesized that ANT/VDAC formed the basic unit of the PTP, and recruitment of additional proteins, such as CypD, Bax, hexokinase, or translocator protein modulated the activation of the PTP.4 The ANT/VDAC/CypD model is depicted in Figure 2.
Figure 2. ANT (adenine nucleotide transporter)/VDAC (voltage-dependent anion carrier) hypothesis.

CypD indicates cyclophilin D; and ROS, reactive oxygen species. Illustration credit: Ben Smith.
Mounting Evidence Against the Theory
Studies showing that CsA-sensitive mitochondrial swelling was still present when components of the ANT/VDAC model were genetically deleted led to a decline in acceptance of this hypothesis. In 2004, in a study by Kokoszka et al,49 liver mitochondria lacking ANT1 and ANT2 mitochondria were challenged with both uncoupler and calcium. It was shown that a CsA-sensitive PTP occurred in response to uncoupler in both wild-type (WT) and knockout strains. However, PTP opening in the ANT knockout required an order of magnitude more calcium than in WT. These data were interpreted as showing that ANT was a modulator of the calcium sensitivity of the PTP instead of the pore-forming species itself.
Genetic knockouts of VDAC were also tested independently in 2 laboratories. Previous studies showed that mammals possess 3 highly conserved isoforms of the VDAC gene, and although all had initially been considered as possibilities for involvement in the PTP,50 VDAC1 had emerged as the leading candidate in the early 2000s (primarily due to its association with a putative PTP inhibitor Ro 68–3400).51 In 2006, the Bernardi lab showed that the characteristics of PTP were indistinguishable between VDAC1−/− and WT mitochondria and that the putative inhibitor Ro 68–3400 was still able to inhibit PTP, suggesting that VDAC is not the target for PTP inhibition by Ro 68–3400.52 Soon after, in 2007, it was shown that in mitochondria lacking VDAC1 and VDAC3, PTP was preserved.53 Experiments were also done in embryonic fibroblasts, which showed that VDAC2 null mitochondria displayed intact PTP formation (this was necessary because VDAC2−/− is embryonic lethal). These data suggest that the VDACs as a family were dispensable for PTP formation.53 Furthermore, in a later study, it was shown that PTP formation, as measured by mitochondria or mitoplast swelling or calcium uptake, could occur in mitoplasts, which are devoid of an outer mitochondrial membrane.54
Phosphate Carrier and a Restructured Theory
Leung et al55 later showed that the antibody to ANT also recognized the phosphate carrier (PiC), which led to consideration of a role for the PiC in the PTP. It was noted that the PiC binds to CypD in a CsA dependent manner. This led to the hypothesis that the PiC undergoes a CypD-mediated conformational change in response to high levels of calcium and is the pore-forming component of the PTP; in this model, ANT modulates the PiC conformational change.55
Although the proposed PiC model did reconcile some data, this model was inconsistent with other data in the literature. For example, although a study in the late 1990s utilizing patch-clamp analysis found that the PiC could form a channel in response to calcium, the characteristics were more similar to the inner mitochondrial anion channel rather than the PTP.56 Furthermore, additional studies found that cardiac-specific genetic deletion of the PiC did not block PTP opening but did lead to greater calcium uptake capacity, attenuation of I/R injury, and protected isolated cells from calcium-related overload–induced death.57,58 A similar study showed that both overexpression and underexpression of the PiC did not affect PTP formation.58 It was concluded that the PiC was not essential to PTP opening but likely a key modulator. Interpretation of these data is somewhat complicated because altering the level of the PiC will change the phosphate level and thus the ADP/ATP level in the mitochondria, which is a known modulator of the pore. Furthermore, a complete knockout (KO) of the PiC would drastically lower the level of inorganic phosphate in the cell, which could interfere with the accumulation and buffering of calcium, an activator of the PTP. Taken together, the data suggest that the PiC is not the pore-forming component of the PTP. Currently, the PiC is thought to provide a regulatory role in PTP formation, primarily through its regulation of inorganic phosphate levels.59
Misfolded Proteins
Formation of the Theory
He and Lemasters60 suggested that PTP, in some instances, becomes unregulated or unable to be inhibited by CsA or Mg2+. Various PTP inducers were used at multiple concentrations and PTP opening, CypD membrane localization, and PTP diameter were measured. The data showed 2 populations of pores, a population that was regulated by CsA and Mg2+, and another that was not. Their findings showed that, in general, stronger induction of the PTP results in unregulated PTP formation and less intense induction of the PTP results in regulated PTP formation.
He and Lemasters60 proposed that the PTP is formed by an aggregate of amphipathic membrane proteins that are regulated by chaperone proteins. They hypothesized that the exposure to oxidative stress caused misfolding of native proteins, which would bring hydrophilic regions capable of forming pores into close proximity with the lipid bilayer. After misfolding occurred, chaperone proteins such as CypD would bind to the misfolded proteins. This binding was hypothesized to be an attempt of CypD to restore the misfolded proteins to their native state. The further addition of calcium to the CypD-chaperoneprotein cluster would result in opening of a regulated pore, and CsA would antagonize this CypD-mediated opening. When the total number of the pore complexes outnumbered the available chaperone proteins, the pores would become unregulated by CsA and Mg2+. The authors noted that ANT would be frequently involved in this complex, as it is the most abundant inner mitochondrial membrane protein but would not be a necessary component of the pore.
Critical Evaluation and Rebuttal
The misfolded proteins model was proposed in the early 2000s and has not been refuted or supported by any direct evidence. However, the concept that the PTP is a heterogeneous group of denatured proteins is not easily reconcilable with the data showing that PTP regulation is modulated by matrix pH, membrane potential, and adenine nucleotides.61,62
Complex V: The F1F0-ATPase
Structure and Function of the F1F0-ATPase
The F1F0-ATPase, known as complex V in the electron transport chain, consists of 2 distinct domains. The F1 domain is located in the mitochondrial matrix, and the F0 domain is embedded in the inner mitochondrial membrane.63 The F1 moiety has 3 copies of the α and β-subunits, and 1 γ, δ, and ε subunit and comprises the catalytic portion of the enzyme. The F0 portion of the enzyme is comprised of the c-ring (which has 8 c-subunits in bovine tissue64) and the peripheral stalk, which contains 1 copy of the b, d, F6, and OSCP (oligomycin sensitive-conferring protein) subunits. The peripheral stalk serves to transmit the mechanical force from the c-ring rotor to the catalytic region to provide the requisite energy to produce ATP from ADP and Pi (inorganic phosphate). The F1F0-ATPase forms dimers that are organized into rows.65 Two distinct theories have been developed recently implicating different subunits of the F1F0-ATPase as the inner membrane pore-forming unit of the PTP.
Dimer Theory
In 2009, Giorgio et al66 reported that CypD could bind to the F1F0-ATPase OSCP (oligomycin sensitive conferring protein) subunit. Furthermore, it was noted that CypD binding was reversed by the addition of CsA and that CypD modulated the catalytic activity of the ATPase. Chinopoulos et al67 also reported that CypD modulated the activity of the F1 F0 -ATPase. Giorgio et al68 later showed that ATP synthase dimers reconstituted in lipid bilayers could be induced to form channels with conductance similar to that of the PTP. This pore formation could not be duplicated when ATP synthase monomers were placed in the membrane. This information, combined with the observation that many modulators of PTP were known regulators of ATP synthase activity (such as magnesium, adenine nucleotides, Pi, and membrane potential), led to the hypothesis PTP formed at the junction of 2 F1F0-ATP synthase proteins.
To test this hypothesis, the Bernardi lab took the approach of mutating residues of the F1F0-ATP synthase that could potentially confer the calcium and pH sensitivity of the PTP. Mutation of Thr 163 to Ser in the β-subunits of the F1F0-ATP synthase in Rhodospirillum rubrum maintained the ability of the F1F0-ATP synthase to hydrolyze MgATP, but this mutation was not able to hydrolyze CaATP.69 These findings have been interpreted to suggest that Thr 163 is essential for calcium binding. Of note, with WT β-subunit, F1F0-ATP synthase hydrolysis of MgATP is coupled to the generation of a proton gradient; however, hydrolysis of CaATP does not generate a proton gradient.69 This suggests that the F1 conformational state is different depending on calcium versus magnesium binding and that the conformational state with calcium binding is not able to couple to proton translocation. These findings would be consistent with the hypothesis that the replacement of magnesium with calcium in the F1F0-ATP synthase is the inciting event that precipitates a conformational change in the functional protein dimer to the PTP.70 Giorgio et al71 recently tested the role of Thr 163 (the bovine equivalent to Thr159) in regulating calcium-activated PTP and found that T159S mutations exhibited a decrease in calcium sensitivity to PTP opening.
Additional support for the F1F0-ATP synthase dimer model was provided by studies showing that mutation of H112 of the OSCP subunit abolishes the inhibition of the PTP by low pH, which occurs in the absence of substrates. The mutants had similar F1F0-ATP synthase activity under physiological pH as well as acidic conditions as compared to WT, indicating that the failure of acidic pH to protect was not likely due to baseline differences in energy stores of the mutants.72 These findings support the hypothesis that the F1F0-ATP synthase mediates PTP formation. A representative model of the dimer theory is shown in Figure 3.
Figure 3. F1F0-ATPase dimer hypothesis.

In normal mitochondria, the F1F0ATPase is thought to self-assembly into a ribbon dimer structure. Bernardi et al61,70 have proposed that PTP (permeability transition pore) channel formation could occur at a monomer-monomer interface following a calcium-dependant and reactive oxygen species–dependent change in conformation. Illustration credit: Ben Smith.
Concerns Raised About This Model
A challenge to the F1F0-ATP synthase dimer theory came from the Walker lab in studies in which several subunits of the peripheral stalk (the b subunit and OSCP) were genetically deleted in haploid cells,73 and CsA-sensitive PTP formation still could be initiated by addition of calcium. These data were interpreted as showing that if the F1F0-ATP synthase is the pore, the site of CypD interaction with the pore is not at the OSCP or b subunit. The lack of CypD binding at these deleted sites directly challenged the ATP synthase dimer model, which is partially predicated on the OSCPCypD interaction. Bernardi et al have suggested that the PTP channels formed in these cells were smaller than the normal PTP, based upon the slower rate of swelling in the knockouts.74 Bernardi noted that in all of the experiments deleting subunits of the F1F0-ATP synthase that the deletions may be leading to the development of a vestigial ATP synthase, which could have formed a small, but functional PTP; this could account for the slower rate of swelling in the peripheral stalk knockouts in the Walker experiments.74
The Walker lab published a recent article showing that a CsA-sensitive permeability is still present following deletion of 5 proteins (subunits e, f, g, the 6.8kD proteolipid, and DAPIT [diabetes-associated protein in insulin-sensitive tissues]) associated with the lateral stalk.75 They show that individual deletion of subunits e, f, g, and the 6.8kD proteolipid all block the formation of dimers. They also show that these mutants generate a membrane potential and transport calcium into the mitochondria. These dimerization-null ATP synthase mutants challenge the dimer model as a PTP-like activity persists in the absence of dimers, suggesting that ATP synthase dimerization is not required for this PTP-like activity. As mentioned above, Bernardi has raised questions about the size of this PTP channel.
C-Ring Theory
In 2011, Alavian et al76 reported that Bcl-xL (B-cell lymphoma extra-large) binds to ATP synthase at the β-subunit and modulates ATP synthase activity. This was based on prior data showing that Bcl and Bcl-xL could localize to the inner mitochondrial membrane and are involved in cell death.77 These data led to the investigation of the F1F0-ATP synthase for specific subunits that were capable of forming pores similar to the PTP. In 2014, Alavian et al78 reported that purified c-subunits formed a voltage-sensitive channel when reconstituted into liposomes. Additionally, they reported that persistently high calcium caused detachment of the c-ring from the F1 subunit. Finally, depletion of the c-subunit decreased PTP formation, and exogenous addition of the β-subunit increased the probability of PTP closure. In this theory, when the conditions for PTP are met, the F1 portion of the F1F0-ATP synthase releases from the F0 portion and the previously inhibited channel pore in the middle of the c-subunit allows passage of solutes. Under physiological conditions, the F1 subunit, particularly the β-subunit, inhibits conduction across the pore. Figure 4 illustrates the c-subunit theory.
Figure 4. C-ring hypothesis.
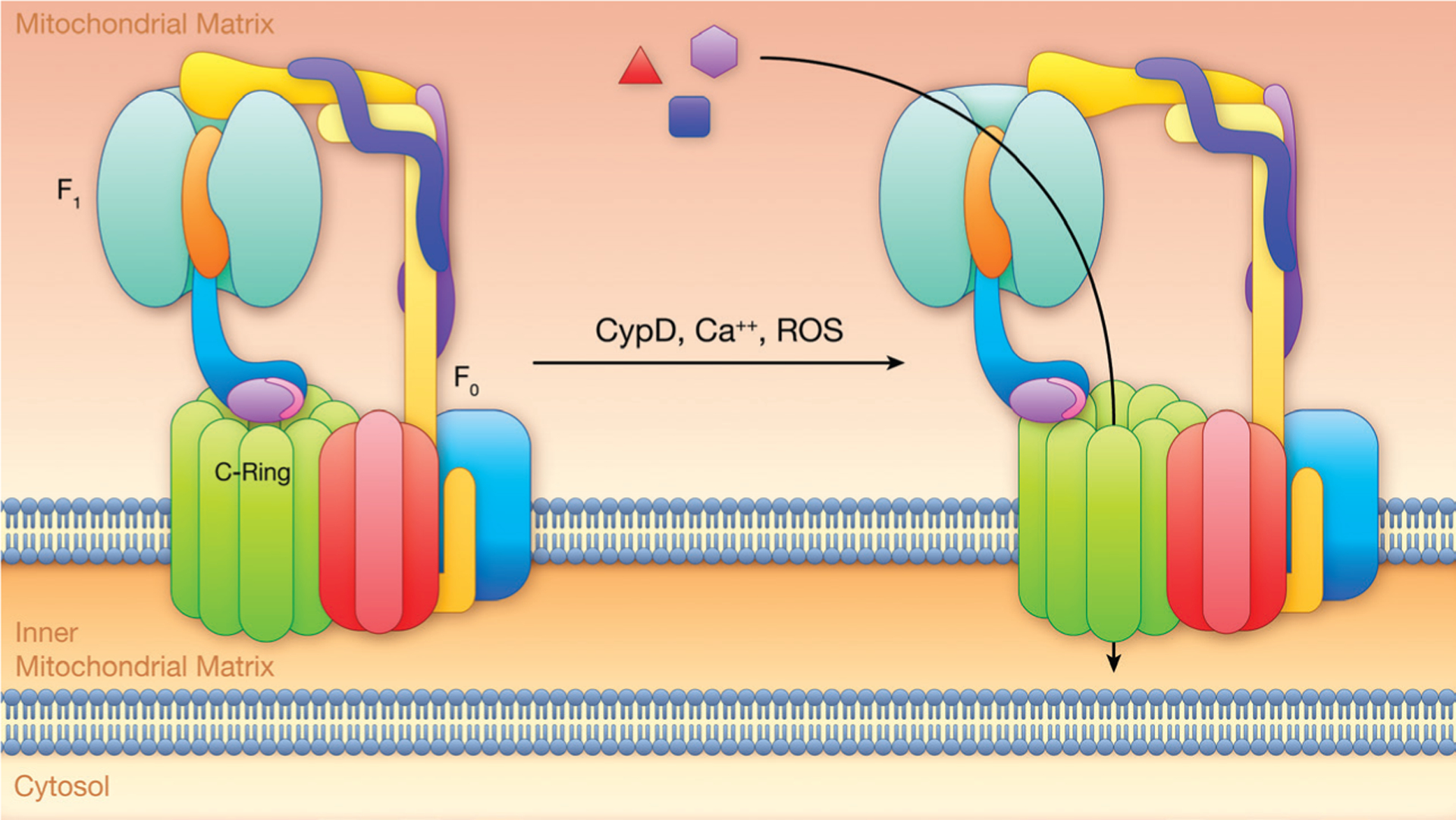
Calcium, reactive oxygen species (ROS), and cellular stress signaling activation of the mitochondrial permeability transition pore and initiation of cell death through a potential pore in the c-ring. In this theory, the F1F0-ATPase would likely have to had cleared a lipid plug from the interior of the c-ring. CypD indicates cyclophilin D; and OSCP, oligomycin sensitive-conferring protein. Illustration credit: Ben Smith.
Concerns Raised With This Model
The Walker laboratory produced deletions of the 3 genes that contribute to the c-subunit in haploid cells and found that a CsA-sensitive PTP persisted in this model.79 Beyond the genetic studies performed on the c-subunit, some questions have been raised about the plausibility of the mechanism regarding the c-ring hypothesis. The internal surface of the c-ring is thought to be lipophilic, and therefore, would be unlikely to conduct solutes. Questions have been raised as to how the lipid plug that usually occupies the center of the c-ring is dislodged during PTP activation.79,80 Furthermore, atomistic simulations did not support hydration of the c-ring to form a pore.81 Finally, it is important to consider whether or not the dislodging of the lipid plug would be readily reversible, which would be necessary if the PTP activates transiently as suggested.80
There are inherent difficulties in testing any hypothesis regarding the F1F0-ATP synthase, as the loss of the protein will drastically change the ATP production of the cell, resulting in altered matrix ATP/ADP (which are known modulators of the PTP). Furthermore, genetic deletion in mammals may not be possible, as deletions of this enzyme (or even subunits) may be embryonic lethal. To this end, the models that have been used to test the dimer and c-ring hypotheses have so far been limited to cells, mitochondria, or lipid bilayers. Another approach being used is to make point mutations in key subunits that would be consistent with the known properties of the PTP.
Recent Studies Suggesting a New Understand of PTP
The Possibility of Multiple Pores
Although distinct potential molecular identities (ANT, PiC, F1F0-ATP synthase dimers, F1F0-ATP synthase c-ring) have been proposed to be the inner membrane component of the PTP, all have been called in to question because a CsA-sensitive permeability remained following genetic deletion of the associated genes. This has led investigators to propose mechanisms that have multiple pore-forming components.
A recent article on the deletion of the c-subunit of the F1F0 ATP synthase showed that although CsA-sensitive PTP could be induced, the resultant pore had a much lower conductance than WT PTP.82 Neginskaya et al82 using the same haploid cells lacking the c-subunit as in the Walker study performed patch-clamp analysis of these cells and showed that the conductance of the pore was ≈300 pS, which is lower than the 1.3 nS channel typically associated with the PTP. They also showed that a 300 pS channel that persists in the cells lacking the c-subunit was inhibited by bongkrekic acid, an inhibitor of ANT. It was also noted that ANT has been reported to form a 300 pS channel. The authors concluded that the classic PTP was not formed in these knockout mitochondria, that mitochondrial permeability increase may be caused by another channel that shares the characteristic CsA sensitivity noted for the PTP. Additionally, the authors concluded that the lack of the c-subunit may enhance PTP formation through other proteins, such as ANT, which was supported by showing that the alternative channel was sensitive to the ANT inhibitor, bongkrekic acid.
Consistent with this concept, Karch et al83 showed that the quadruple deletion of ANT1, ANT2, ANT4, and CypD results in the loss of PTP. The ANT family of proteins are a pore-forming species that could be consistent with the PTP. The PTP could still be observed in the triple ANT knockout, in which CsA could then completely inhibit PTP formation. Importantly, patch clamping in the triple ANT KO showed that the permeability increase was not due to a classical PTP. The authors suggest that these data are reconcilable with 2 distinct molecular identities of the PTP.
The hypothesis that both ANT and the F1F0-ATPsynthase can form permeability pores might suggest that the ATP synthasome or perhaps the assembly or disassembly of the synthasome might be important in induction of the PTP. Beutner et al84 provide data showing that dimers of the F1F0-ATPsynthase, the ANT, and the PiC can assemble into a synthasome complex. They further show that CypD is needed for the disassembly of the synthasome into its components. This hypothesis would suggest that the ATP synthasome is assembled under high work conditions and that disassembly of the synthasome is needed to generate the PTP. This hypothesis also provides a physiological role for CypD. One might ask what conditions favor disassembly of the synthasome. Conditions of low work (low ADP) and high substrate or high calcium would enhance ΔΨ and lead to generation of ROS. It is plausible that this would trigger disassembly of the synthasome modulated by CypD.
Taken together, these data would be consistent with CypD promoting disassembly of the synthasome to F1F0- ATPsynthase dimers, which forms the 1.5 nS channel; in this theory, CsA would inhibit the formation of the PTP by inhibiting the disassembly of the synthasome. If subunits of the F1F0-ATPsynthase are deleted, this alters the assembly of the synthasome and can lead to the generation of other smaller channels that can be inhibited by CsA. Additional studies will be needed to fully elucidate the role of the F1F0-ATPsynthase and other components of the synthasome in generating the PTP.
REGULATION BY CALCIUM
Another strategy for reducing the PTP is to reduce mitochondrial calcium, as calcium is a well-established activator of PTP.85 Calcium has long been known to enter the mitochondria via an electrophoretic pathway using the mitochondrial membrane potential as the driving force. In 2011, 2 groups independently identified the pore-forming protein responsible for mitochondrial calcium uptake: a protein previously identified as CCD109A (coiled-coil domain containing 109A), which has been renamed MCU (mitochondrial calcium uniporter).86,87 It was shown that this protein is part of a large complex responsible for mitochondrial calcium entry (Figure 5). This complex is currently proposed to be a tetramer of MCU88–90 along with several EF hand proteins known as mitochondrial calcium uptake protein (MICU) 1, 2, or 3 (MICU1, MICU2 and MICU3) and essential MCU regulator (EMRE) also known as single-pass membrane protein with aspartate rich tail (SMDT1).91 EMRE is a single-pass membrane protein associated with the uniporter complex, which appears to essential for calcium uptake in metazoans.92 In the yeast Saccharomyces cerevisiae, which lacks MCU, EMRE must be coexpressed with human MCU to reconstitute mitochondrial calcium uniporter activity.93 However, MCU from fungi that lack EMRE can transport calcium in the absence of EMRE.93 Wang et al94 performed cryo-EM (cryogenic electron microscopy) studies of human MCU and EMRE to better understand why metazoan MCU requires EMRE. They compared the structure of HsMCU (human MCU) with and without EMRE and showed that the juxtamembrane loop of HsMCU blocks the calcium channel and that EMRE forms a stent to move the juxtamembrane loop and open the channel (Figure 5).
Figure 5. MCU (mitochondrial calcium uniporter) complex: A shows the MCU complex in the open conformation and B shows it in the closed conformation.
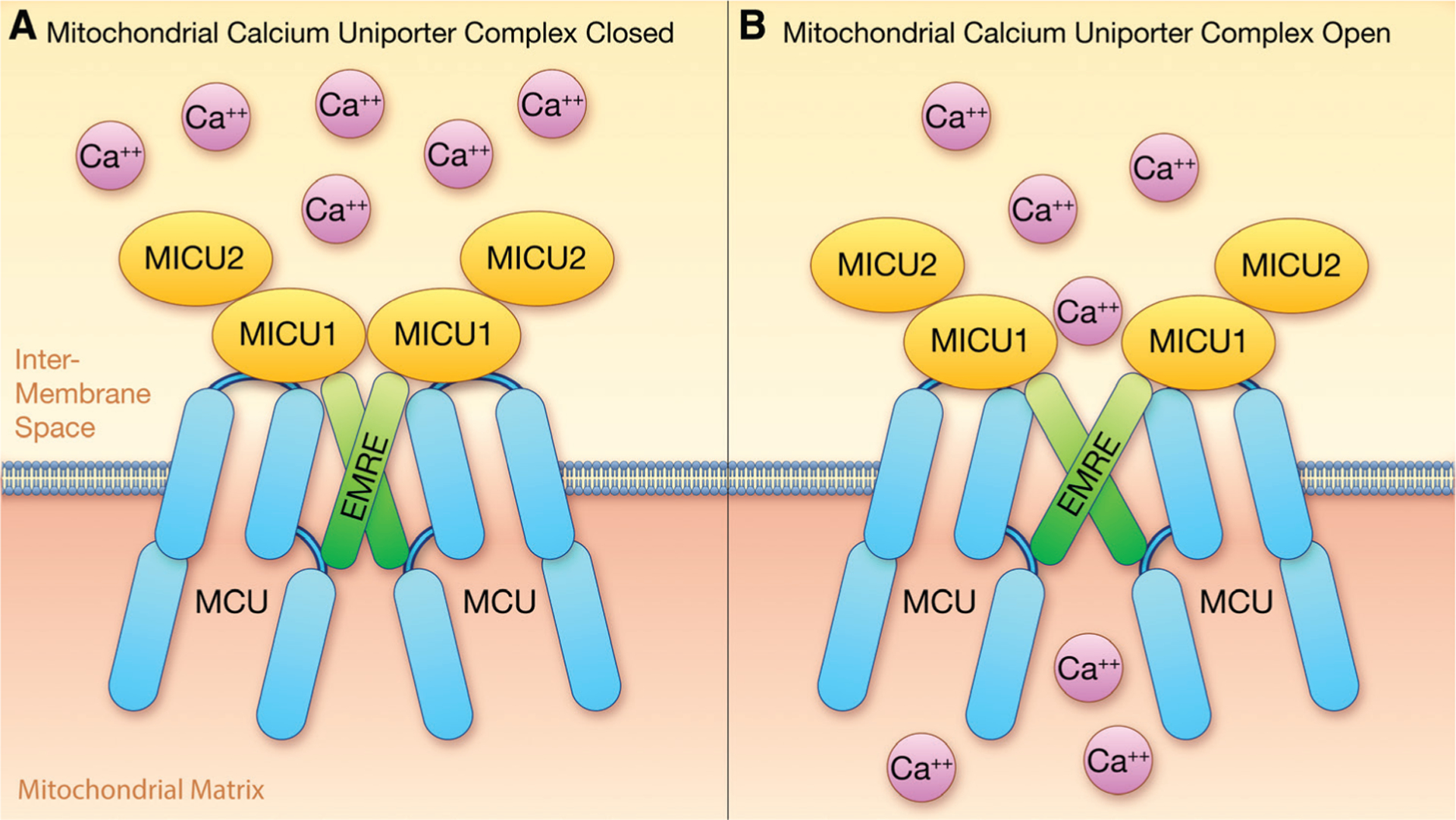
EMRE indicates essential MCU regulator; and MICU, mitochondrial calcium uptake protein. Illustration credit: Ben Smith.
MICU1, 2, and 3 are EF hand proteins that bind calcium and regulate MCU activity. MICU1 binds to EMRE and MCU and has been proposed to reduce MCU mediated calcium uptake at low calcium levels, but increase calcium uptake at high calcium levels.95–97 MICU1 has been shown to interact with the DIME sequence of MCU, and this interaction is proposed to facilitate the role of MICU1 as a gatekeeper to reduce calcium entry at low extramitochondrial calcium levels.98,99 Liu et al95 found that loss of MICU1 led to a perinatal lethality rate of ≈85%. However, with age, the MICU1−/− mice seemed to improve in phenotype as well as regain close-to-normal mitochondrial calcium homeostasis, concurrent with a decrease in EMRE protein expression. Liu et al95 also found that crossing the MICU1-KO with an EMRE+/− mouse rescued the perinatal lethality. Interestingly, Tufi et al100 have suggested that MICU1 has a function in addition to regulating MCU. They show in flies that loss of MICU1 is lethal and it is not rescued by loss of MCU or loss of EMRE; loss of MCU or EMRE alone are not lethal and therefore if the only function of MICU1 is to regulate MCU it would be expected that deleting MCU would rescue the lethality of loss of MICU1. Interestingly, Gottschalk et al101 report that MICU1 localizes with MICOS (mitochondrial contact site and cristae organizing system). Additional studies will be needed to sort out the role of MICU1.
As MICU1 binds directly to EMRE and MCU, MICU1 can regulate MCU in the absence of MICU2. In contrast, MICU2 binds to MICU1, and therefore, MICU2 cannot regulate MCU in the absence of MICU1. The affinity of MICU2 for calcium is lower than MICU1 and so depending on the ratio of MICU1 to MICU2 in the complex the calcium sensitivity of the MCU can be regulated.102 MCUb, which has high homology to MCU, has been shown to inhibit calcium uptake when it is incorporated into the tetramer. The DIME sequence is similar in MCU and MCUb. Thus the mechanism by which MCUb inhibits calcium uptake is not fully elucidated, but a recent study showed that MCUb can alter the MCU complex stoichiometry which could alter mitochondrial calcium uptake.103
In addition to calcium uptake mechanisms, matrix calcium is also regulated by mitochondrial calcium efflux. Studies in the 1970 showed that when the MCU was inhibited with ruthenium red, a mitochondrial calcium efflux occurred, which was activated by extramitochondrial sodium.104 The protein responsible, a NCLX (sodium, calcium, lithium exchanger), was identified (slc8B1; solute carrier family 8, member B1) in 2010 and shown to mediate sodium-dependent mitochondrial calcium efflux.105 A proton-calcium exchanger has also been proposed to play a role in mitochondrial calcium efflux. LETM1 (leucine zipper EF-hand containing transmembrane protein 1), an EF-hand protein, was identified by a genome-wide small interfering RNA (RNAi) screen as a mitochondrial calcium proton antiporter.106 However, LETM has also been suggested to mediate potassium-proton exchange.107 Additional studies will be needed to sort out the role of LETM in mitochondrial calcium homeostasis. Studies in which both NCLX and LETM were expressed in HeLa cells showed that NCLX overexpression enhanced mitochondrial calcium efflux, but LETM overexpression did not.108 Because mitochondrial calcium efflux is sodium dependent, a change in cytosolic sodium can alter mitochondrial calcium. It has been suggested that an increase in cytosolic sodium, as occurs during heart failure, leads to a decrease mitochondrial calcium, and this decrease in mitochondrial calcium might impair energetics and by altering NAD(P)H might alter antioxidant balance.109,110
MCU, Mitochondrial Calcium, and Heart Disease
An increase in mitochondrial calcium has been shown to occur during ischemia and reperfusion, and this increase in calcium is proposed to activate the PTP.26 Thus inhibition of MCU during ischemia has been proposed as means to reduce cell death in I/R (Figure 1). Studies performed in a mouse model in which MCU was deleted in the germline did not show protection in a perfused heart model of I/R.111 Mitochondria from these MCU-KO hearts did not take up calcium and did not exhibit PTP opening; however, when the hearts were subjected to global ischemia and reperfusion, they showed similar infarct size to the WT hearts. In another study, mice expressing a cardiac-specific dominant-negative MCU were also not protected from I/R injury.112 The MCU pore-forming sequence was mutated and overexpressed in the heart using the alphamyosin heavy chain promoter, which turns on at birth. Because MCU exists as a tetramer, the mutated MCU acts as a dominant-negative. Cardiac mitochondria from mice overexpressing this dominant-negative MCU did not take up calcium and did not exhibit PTP opening.112 However, similar to the mice with germline deletion of MCU, these hearts did not show a reduction in infarct size when they were subject to an in vivo model of I/R. In contrast to these studies in which MCU was knocked out at birth or before, studies in which MCU is deleted in adult heart show cardioprotection. The Elrod lab and the Molkentin lab developed a mouse with a tamoxifen-inducible deletion of MCU from the heart.113,114 The mouse was studied independently in the 2 labs, and both found that when MCU is deleted in the adult hearts, there is a reduction in infarct size in an in vivo model of I/R. Taken together, the data suggest that if MCU is deleted before birth, the heart is not protected from I/R, whereas deletion of MCU in the adult heart results in cardioprotection.
Although the precise reason for the difference is not clear, the data are consistent with the hypothesis that deletion of MCU before birth results in an adaptation in the heart which alters I/R death. Interestingly, CsA protects in WT but not in germline MCU-KO hearts.111 It is known that with strong induction of PTP, CsA is no longer able to inhibit PTP formation. Therefore, the lack of protection by CsA in the germline MCU-KO might be due to a more robust activation of the PTP. Another possibility is that in the germline MCU-KO hearts, a new PTP independent mode of death now predominates, and this may be the case in the germline MCU-KO hearts. However, inhibition of the RIP (receptor-interacting protein) kinase necroptosis pathway using necrostatin or genetic ablation of RIP3 was protective in the WT heart but not in the germline MCU-KO hearts.115 Thus, if germline MCU-KO hearts upregulate another cell death pathway, it is not the RIP1/3 pathway. It is possible that some adaptation occurs in the mice in which MCU is deleted before birth, and this adaptation allows activation of PTP independent of calcium activation and CypD (since it is not inhibited by CsA). This adaptation could be clinically relevant as CsA did not protect in a recent clinical trial.36
As discussed, calcium uptake into mitochondria can also be regulated by altering levels of the regulators of the MCU complex, and if alterations in these regulators occur, they could alter the susceptibility to PTP initiated death. A loss of function mutation in MICU1 has been reported in human patients and is associated with ataxia which has been attributed to mitochondrial calcium overload.116 Additional mutations in MICU1 have also been identified which are also consistent with mitochondrial calcium overload.117 An increase in MICU1 and NCXL has been reported in failing human hearts, presumably consistent with compensation to reduce mitochondrial calcium overload.118 MICU2 has been shown to be increased at the transcriptional level in both mice and humans with cardiovascular disease.119 Mice with deletion of MICU2 had evidence of mild diastolic dysfunction. Patients with a null mutation in MICU2 have been reported to have altered mitochondrial calcium regulation and a severe neurodevelopmental disorder.120 Paillard et al102 demonstrated that the stoichiometry of MICU1 and MICU2 vary among different tissues and that the ratio of MICU1/MCU is regulated by the availability of MICU1. At low levels of MICU1/MCU, there is a decreased threshold for calcium entry into the mitochondria. Furthermore, overexpression of MICU1 in the heart leads to contractile dysfunction. This raises the interesting speculation that alterations in the MICU1/MICU2 might occur with age or disease and alter susceptibility to calcium overload and PTP opening.
MCUb levels vary among different tissues and can be regulated in disease state.121 A recent study by Lambert et al103 shows that MCUb insertion into the MCU complex displaces MCU and thereby alters MICU1/2 association with the complex because MCUb does not directly bind these gatekeepers and thereby alters the calcium sensitivity of MCU. They further show an increased incorporation of MCUb into the complex during I/R, which would be expected to decrease calcium entry into the mitochondria. In addition, they showed that transgenic MCUb overexpression was protective from I/R and displayed reduced mitochondrial swelling in isolated cardiac mitochondria.
NCLX has been shown to be increased in failing human hearts.118 Cardiac-specific overexpression of NCLX reduced mitochondrial calcium loading and I/R death. In mice, in which NCLX was acutely deleted in the heart, 87% of the mice died within 14 days. The death was attributed to mitochondrial calcium overload and was partially rescued by inhibiting the PTP by crossing the NCLX-KO mice with CypD-KO mice.
REGULATION BY ROS
ROS is another well-established activator of the PTP, which is known to increase during I/R and heart failure and is, therefore, another potential target to reduce cardiomyocyte death (Figure 1). Could strategies be undertaken on reperfusion to reduce ROS generation? A number of studies tested the hypothesis that treatment with antioxidants would reduce I/R injury, and the majority of these studies failed to show protection.122–125 Furthermore, clinical trials in the 1990s, testing whether superoxide dismutase would be beneficial in patients undergoing angioplasty following acute myocardial ischemia, failed to show a beneficial effect.126,127 However, one limitation of these early studies is that the antioxidants were not targeted to the mitochondria. This prompted the development of mitochondrial-targeted antioxidants such as mitochondrial-targeted ubiquinol (MitoQ).128,129 MitoQ has shown great promise in reducing both I/R injury128 and heart failure.130 MitoQ also has been tested in a patient study for Parkinson disease but showed no improvement over placebo.131 A small human trial for hepatitis C did show a reduction in markers of liver damage in the MitoQ-treated patients.132 It should be mentioned that ROS also has signaling roles in the heart and that prolonged treatment with drugs such as MitoQ could interfere with beneficial ROS signaling. For example, it has been shown that antioxidants can block cardioprotective signaling.133
Another potential strategy to reduce ROS would be to block its production; this would be analogous to inhibiting calcium uptake into the mitochondria. One limitation of this strategy is that there appear to be many sources of ROS generation during I/R, including the electron transport chain, reverse electron transport at complex I fueled by an increase in succinate during ischemia, monoamine oxidase, and NADPH oxidases. It may be difficult to inhibit all the relevant sources of ROS. However, there has been promising data suggesting that inhibition of reverse electron transport (eg, by lowering levels of succinate) reduces infarct size following I/R.134–136 An increase in succinate is a common finding during ischemia, and there are data suggesting that on reperfusion the accumulated succinate can drive reverse electron transport of complex I generating ROS (Figure 1). Although there are several mechanisms that can produce succinate in the mitochondria, Chouchani et al134,135 have suggested that succinate accumulates during ischemia due reverse operation of complex II. It is proposed that QH2 (ubiquinol) is oxidized by SDH (succinate dehydrogenase; complex II) with fumarate as an electron acceptor resulting in the generation of succinate.134 Others have suggested that succinate accumulation during ischemia results from canonical Krebs cycle activity.137 Inhibition of SDH with malonate has been shown to reduce levels of succinate and reduce infarct size. However, some issues have been raised about the extent of reverse electron transport at complex I during reperfusion.138 Future studies will be needed to work out the details of this mechanism.
SUMMARY AND FUTURE DIRECTION
PTP-mediated cell death is well documented in I/R injury, and strategies to reduce PTP in I/R have generally been showed to be cardioprotective. However, CsA a classic desensitizer of the PTP did not show protection in a recent clinical trial. CsA inhibits by binding CypD, and it is known that with strong stimuli PTP can become independent of CypD. This has led to the drive to identify inhibitors that directly inhibit the PTP pore-forming unit. This effort is limited by the lack of knowledge of the molecular identity of the PTP. As discussed in this review, it is possible that multiple pores or PTPs can form and this might account for the lack of protection in the clinical trial. Thus, a major goal of future studies is to identify the PTP(s) to allow rational development of inhibitors.
Although CypD is known to activate the PTP, the mechanism responsible is unclear. Similarly, although it is well accepted that an increase in calcium and ROS are triggers of PTP and cell death the mechanisms by which they activate the PTP are unclear. Until we know the identity of the PTP, it will be difficult to determine how calcium and ROS activate PTP opening.
In summary, in spite of considerable effort and studies, the precise mechanism by which an increase in calcium and ROS lead to cardiomyocyte death is still largely unknown. Just as the identification of MCU lead to an explosion of our understanding of mitochondrial calcium transport and regulation, the identification of the PTP is needed to spur our understanding of PTP and its role in cell death.
Sources of Funding
This work was supported by intramural funding from the National Heart Lung and Blood Institute at the National Institutes of Health (ZIA HL002066 and ZIA HL006059) and Foundation Leducq (6CVD04). T.M.B. was supported by the NIH Medical Research Scholars Program a public-private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, the American Association for Dental Research, the Colgate-Palmolive Company, and other private donors.
Nonstandard Abbreviations and Acronyms
- ANT
adenine nucleotide transporter
- CsA
cyclosporin A
- CypD
cyclophilin D
- EMRE
essential MCU regulator
- LETM1
leucine zipper-EF-hand containing transmembrane protein 1
- MCU
mitochondrial calcium uniporter
- MICOS
mitochondrial contact site and cristae organizing system
- MICU
mitochondrial calcium uptake protein
- NCLX
sodium-calcium-lithium exchanger
- OSCP
oligomycin sensitive-conferring protein
- PiC
phosphate carrier
- PTP
permeability transition pore
- RIP
receptor-interacting protein
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SMDT1
single-pass membrane protein with aspartate rich tail
- VDAC
voltage-dependent anion carrier
Footnotes
Disclosures
None.
REFERENCES
- 1.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion–a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9 [DOI] [PubMed] [Google Scholar]
- 2.Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1979;195:460–467. doi: 10.1016/0003-9861(79)90372-2 [DOI] [PubMed] [Google Scholar]
- 3.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787:1402–1415. doi: 10.1016/j.bbabio.2008.12.017 [DOI] [PubMed] [Google Scholar]
- 4.Crompton M The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341 (pt 2):233–249. [PMC free article] [PubMed] [Google Scholar]
- 5.Di Lisa F, Bernardi P. A CaPful of mechanisms regulating the mitochondrial permeability transition. J Mol Cell Cardiol. 2009;46:775–780. doi: 10.1016/j.yjmcc.2009.03.006 [DOI] [PubMed] [Google Scholar]
- 6.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0 [DOI] [PubMed] [Google Scholar]
- 7.Ichas F, Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim Biophys Acta. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4 [DOI] [PubMed] [Google Scholar]
- 8.Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725–734. doi: 10.1016/S0006-3495(99)77239-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hausenloy D, Wynne A, Duchen M, Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109:1714–1717. doi: 10.1161/01.CIR.0000126294.81407.7D [DOI] [PubMed] [Google Scholar]
- 10.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680–3687. doi: 10.1172/JCI43171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Assaly R, de Tassigny Ad, Paradis S, Jacquin S, Berdeaux A, Morin D. Oxidative stress, mitochondrial permeability transition pore opening and cell death during hypoxia-reoxygenation in adult cardiomyocytes. Eur J Pharmacol. 2012;675:6–14. doi: 10.1016/j.ejphar.2011.11.036 [DOI] [PubMed] [Google Scholar]
- 12.Block MR, Lauguin GJ, Vignais PV. Atractyloside and bongkrekic acid sites in the mitochondrial ADP/ATP carrier protein. An appraisal of their unicity by chemical modifications. FEBS Lett. 1981;131:213–218. doi: 10.1016/0014-5793(81)80370-5 [DOI] [PubMed] [Google Scholar]
- 13.Rottenberg H, Marbach M. Regulation of Ca2+ transport in brain mitochondria. II. The mechanism of the adenine nucleotides enhancement of Ca2+ uptake and retention. Biochim Biophys Acta. 1990;1016:87–98. doi: 10.1016/0005-2728(90)90010-2 [DOI] [PubMed] [Google Scholar]
- 14.Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr. 1987;19:297–303. doi: 10.1007/bf00762419 [DOI] [PubMed] [Google Scholar]
- 15.Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 16.Tanveer A, Virji S, Andreeva L, Totty NF, Hsuan JJ, Ward JM, Crompton M. Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur J Biochem. 1996;238:166–172. doi: 10.1111/j.1432-1033.1996.0166q.x [DOI] [PubMed] [Google Scholar]
- 17.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434 [DOI] [PubMed] [Google Scholar]
- 18.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162 [DOI] [PubMed] [Google Scholar]
- 19.Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. J Mol Cell Cardiol. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k [DOI] [PubMed] [Google Scholar]
- 20.Azzi JR, Sayegh MH and Mallat SG. Calcineurin inhibitors: 40 years later, can’t live without …. J Immunol. 2013;191:5785–5791. [DOI] [PubMed] [Google Scholar]
- 21.Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22 [DOI] [PubMed] [Google Scholar]
- 22.Tiepolo T, Angelin A, Palma E, Sabatelli P, Merlini L, Nicolosi L, Finetti F, Braghetta P, Vuagniaux G, Dumont JM, et al. The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1−/− myopathic mice. Br J Pharmacol. 2009;157:1045–1052. doi: 10.1111/j.1476-5381.2009.00316.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyman L, Coleman AK, Zhao G, Wescott AP, Joca HC, Greiser BM, Karbowski M, Ward CW, Lederer WJ. Dynamics of the mitochondrial permeability transition pore: Transient and permanent opening events. Arch Biochem Biophys. 2019;666:31–39. doi: 10.1016/j.abb.2019.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu X, Kwong JQ, Molkentin JD, Bers DM. Individual cardiac mitochondria undergo rare transient permeability transition pore openings. Circ Res. 2016;118:834–841. doi: 10.1161/CIRCRESAHA.115.308093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz Longacre L, Kloner RA, Arai AE, Baines CP, Bolli R, Braunwald E, Downey J, Gibbons RJ, Gottlieb RA, Heusch G, et al. ; National Heart, Lung, and Blood Institute, National Institutes of Health. New horizons in cardioprotection: recommendations from the 2010 national heart, lung, and blood institute workshop. Circulation. 2011;124:1172–1179. doi: 10.1161/CIRCULATIONAHA.111.032698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy E, Steenbergen C. What makes the mitochondria a killer? Can we condition them to be less destructive? Biochim Biophys Acta. 2011;1813:1302–1308. doi: 10.1016/j.bbamcr.2010.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinbrenner C, Liu GS, Downey JM, Cohen MV. Cyclosporine A limits myocardial infarct size even when administered after onset of ischemia. Cardiovasc Res. 1998;38:676–684. doi: 10.1016/s0008-6363(98)00064-9 [DOI] [PubMed] [Google Scholar]
- 28.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bond JM, Chacon E, Herman B, Lemasters JJ. Intracellular pH and Ca2+ homeostasis in the pH paradox of reperfusion injury to neonatal rat cardiac myocytes. Am J Physiol. 1993;265:C129–C137. doi: 10.1152/ajpcell.1993.265.1.C129 [DOI] [PubMed] [Google Scholar]
- 30.Halestrap AP. Calcium-dependent opening of a non-specific pore in the mitochondrial inner membrane is inhibited at pH values below 7. Implications for the protective effect of low pH against chemical and hypoxic damage. Biochemical J. 1991;278:715–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabó I, Zoratti M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem. 1992;267:2934–2939. [PubMed] [Google Scholar]
- 32.Szabó I, Bernardi P, Zoratti M. Modulation of the mitochondrial megachannel by divalent cations and protons. J Biol Chem. 1992;267:2940–2946. [PubMed] [Google Scholar]
- 33.Kristian T, Bernardi P, Siesjö BK. Acidosis promotes the permeability transition in energized mitochondria: implications for reperfusion injury. J Neurotrauma. 2001;18:1059–1074. doi: 10.1089/08977150152693755 [DOI] [PubMed] [Google Scholar]
- 34.Skyschally A, Schulz R, Heusch G. Cyclosporine A at reperfusion reduces infarct size in pigs. Cardiovasc Drugs Ther. 2010;24:85–87. doi: 10.1007/s10557-010-6219-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142 [DOI] [PubMed] [Google Scholar]
- 36.Cung TT, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, Bonnefoy-Cudraz E, Guérin P, Elbaz M, Delarche N, et al. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med. 2015;373:1021–1031. doi: 10.1056/NEJMoa1505489 [DOI] [PubMed] [Google Scholar]
- 37.Bernardi P, Di Lisa F. Cyclosporine before PCI in acute myocardial infarction. N Engl J Med. 2016;374:88–90. doi: 10.1056/NEJMc1514192. [DOI] [PubMed] [Google Scholar]
- 38.Stockburger C, Miano D, Pallas T, Friedland K, Müller WE. Enhanced neuroplasticity by the metabolic enhancer piracetam associated with improved mitochondrial dynamics and altered permeability transition pore function. Neural Plast. 2016;2016:8075903. doi: 10.1155/2016/8075903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ascah A, Khairallah M, Daussin F, Bourcier-Lucas C, Godin R, Allen BG, Petrof BJ, Des Rosiers C, Burelle Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am J Physiol Heart Circ Physiol. 2011;300:H144–H153. doi: 10.1152/ajpheart.00522.2010 [DOI] [PubMed] [Google Scholar]
- 40.Angelin A, Bonaldo P, Bernardi P. Altered threshold of the mitochondrial permeability transition pore in Ullrich congenital muscular dystrophy. Biochim Biophys Acta. 2008;1777:893–896. doi: 10.1016/j.bbabio.2008.03.026 [DOI] [PubMed] [Google Scholar]
- 41.Bücheler K, Adams V, Brdiczka D. Localization of the ATP/ADP translocator in the inner membrane and regulation of contact sites between mitochondrial envelope membranes by ADP. A study on freeze-fractured isolated liver mitochondria. Biochim Biophys Acta. 1991;1056:233–242. doi: 10.1016/s0005-2728(05)80054-4 [DOI] [PubMed] [Google Scholar]
- 42.Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x [DOI] [PubMed] [Google Scholar]
- 43.Beutner G, Ruck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 1996;396:189–195. doi: 10.1016/0014-5793(96)01092-7 [DOI] [PubMed] [Google Scholar]
- 44.Woodfield K, Rück A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336 (pt 2):287–290. doi: 10.1042/bj3360287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry. 1996;35:8483–8488. doi: 10.1021/bi960833v [DOI] [PubMed] [Google Scholar]
- 46.Henderson PJ, Lardy HA. Bongkrekic acid. An inhibitor of the adenine nucleotide translocase of mitochondria. J Biol Chem. 1970;245:1319–1326. [PubMed] [Google Scholar]
- 47.Novgorodov SA, Gudz TI, Jung DW, Brierley GP. The nonspecific inner membrane pore of liver mitochondria: modulation of cyclosporin sensitivity by ADP at carboxyatractyloside-sensitive and insensitive sites. Biochem Biophys Res Commun. 1991;180:33–38. doi: 10.1016/s0006-291x(05)81250-1 [DOI] [PubMed] [Google Scholar]
- 48.Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HL, Prévost MC, Xie Z, Matsuyama S, Reed JC, et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027 [DOI] [PubMed] [Google Scholar]
- 49.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blachly-Dyson E, Forte M. VDAC channels. IUBMB Life. 2001;52:113–118. doi: 10.1080/15216540152845902 [DOI] [PubMed] [Google Scholar]
- 51.Cesura AM, Pinard E, Schubenel R, Goetschy V, Friedlein A, Langen H, Polcic P, Forte MA, Bernardi P, Kemp JA. The voltage-dependent anion channel is the target for a new class of inhibitors of the mitochondrial permeability transition pore. J Biol Chem. 2003;278:49812–49818. doi: 10.1074/jbc.M304748200 [DOI] [PubMed] [Google Scholar]
- 52.Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1(−/−) mitochondria. Biochim Biophys Acta. 2006;1757:590–595. doi: 10.1016/j.bbabio.2006.02.007 [DOI] [PubMed] [Google Scholar]
- 53.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sileikyte J, Petronilli V, Zulian A, Dabbeni-Sala F, Tognon G, Nikolov P, Bernardi P, Ricchelli F. Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor). J Biol Chem. 2011;286:1046–1053. doi: 10.1074/jbc.M110.172486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herick K, Krämer R, Lühring H. Patch clamp investigation into the phosphate carrier from Saccharomyces cerevisiae mitochondria. Biochim Biophys Acta. 1997;1321:207–220. doi: 10.1016/s0005-2728(97)00050-9 [DOI] [PubMed] [Google Scholar]
- 57.Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, Huang T, Molkentin JD. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014;21:1209–1217. doi: 10.1038/cdd.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutiérrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J Mol Cell Cardiol. 2014;72:316–325. doi: 10.1016/j.yjmcc.2014.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015;21:206–214. doi: 10.1016/j.cmet.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2 [DOI] [PubMed] [Google Scholar]
- 61.Bernardi P The mitochondrial permeability transition pore: a mystery solved? Front Physiol. 2013;4:95. doi: 10.3389/fphys.2013.00095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baines CP. The molecular composition of the mitochondrial permeability transition pore. J Mol Cell Cardiol. 2009;46:850–857. doi: 10.1016/j.yjmcc.2009.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walker JE. The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans. 2013;41:1–16. doi: 10.1042/BST20110773 [DOI] [PubMed] [Google Scholar]
- 64.Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci U S A. 2010;107:16823–16827. doi: 10.1073/pnas.1011099107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blum TB, Hahn A, Meier T, Davies KM, Kühlbrandt W. Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows. Proc Natl Acad Sci U S A. 2019;116:4250–4255. doi: 10.1073/pnas.1816556116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem. 2009;284:33982–33988. doi: 10.1074/jbc.M109.020115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chinopoulos C, Konràd C, Kiss G, Metelkin E, Töröcsik B, Zhang SF, Starkov AA. Modulation of F0F1-ATP synthase activity by cyclophilin D regulates matrix adenine nucleotide levels. FEBS J. 2011;278:1112–1125. doi: 10.1111/j.1742-4658.2011.08026.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nathanson L, Gromet-Elhanan Z. Mutations in the beta-subunit Thr(159) and Glu(184) of the Rhodospirillum rubrum F(0)F(1) ATP synthase reveal differences in ligands for the coupled Mg(2+)- and decoupled Ca(2+)-dependent F(0)F(1) activities. J Biol Chem. 2000;275:901–905. doi: 10.1074/jbc.275.2.901 [DOI] [PubMed] [Google Scholar]
- 70.Bernardi P, Di Lisa F, Fogolari F, Lippe G. From ATP to PTP and back: a dual function for the mitochondrial ATP synthase. Circ Res. 2015;116:1850–1862. doi: 10.1161/CIRCRESAHA.115.306557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giorgio V, Burchell V, Schiavone M, Bassot C, Minervini G, Petronilli V, Argenton F, Forte M, Tosatto S, Lippe G, et al. Ca2+ binding to F-ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017;18:1065–1076. doi: 10.15252/embr.201643354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Antoniel M, Jones K, Antonucci S, Spolaore B, Fogolari F, Petronilli V, Giorgio V, Carraro M, Di Lisa F, Forte M, et al. The unique histidine in OSCP subunit of F-ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep. 2018;19:257–268. doi: 10.15252/embr.201744705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He J, Carroll J, Ding S, Fearnley IM, Walker JE. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci U S A. 2017;114:9086–9091. doi: 10.1073/pnas.1711201114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bernardi P Why F-ATP synthase remains a strong candidate as the mitochondrial permeability transition pore. Front Physiol. 2018;9:1543. doi: 10.3389/fphys.2018.01543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carroll J, He J, Ding S, Fearnley IM, Walker JE. Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc Natl Acad Sci U S A. 2019;116:12816–12821. doi: 10.1073/pnas.1904005116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hockenbery D, Nuñez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0 [DOI] [PubMed] [Google Scholar]
- 78.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci U S A. 2017;114:3409–3414. doi: 10.1073/pnas.1702357114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gerle C On the structural possibility of pore-forming mitochondrial FoF1 ATP synthase. Biochim Biophys Acta. 2016;1857:1191–1196. doi: 10.1016/j.bbabio.2016.03.008 [DOI] [PubMed] [Google Scholar]
- 81.Zhou W, Marinelli F, Nief C and Faraldo-Gómez JD. Atomistic simulations indicate the c-subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. eLife. 2017;6. doi: 10.7554/eLife.23781. [DOI] [Google Scholar]
- 82.Neginskaya MA, Solesio ME, Berezhnaya EV, Amodeo GF, Mnatsakanyan N, Jonas EA, Pavlov EV. ATP Synthase C-Subunit-deficient mitochondria have a small cyclosporine A-Sensitive channel, but lack the permeability transition pore. Cell Rep. 2019;26:11–17.e2. doi: 10.1016/j.celrep.2018.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, Molkentin JD. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci Adv. 2019;5:eaaw4597. doi: 10.1126/sciadv.aaw4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beutner G, Alanzalon RE, Porter GA Jr. Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci Rep. 2017;7:14488. doi: 10.1038/s41598-017-14795-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- 86.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nguyen NX, Armache JP, Lee C, Yang Y, Zeng W, Mootha VK, Cheng Y, Bai XC, Jiang Y. Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nature. 2018;559:570–574. doi: 10.1038/s41586-018-0333-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baradaran R, Wang C, Siliciano AF, Long SB. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature. 2018;559:580–584. doi: 10.1038/s41586-018-0331-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoo J, Wu M, Yin Y, Herzik MA Jr, Lander GC, Lee SY. Cryo-EM structure of a mitochondrial calcium uniporter. Science. 2018;361:506–511. doi: 10.1126/science.aar4056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kamer KJ, Mootha VK. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol. 2015;16:545–553. doi: 10.1038/nrm4039 [DOI] [PubMed] [Google Scholar]
- 92.Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–1382. doi: 10.1126/science.1242993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kovács-Bogdán E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, Huber RJ, Myre MA, Blower MD, Mootha VK. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci U S A. 2014;111:8985–8990. doi: 10.1073/pnas.1400514111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y, Nguyen NX, She J, Zeng W, Yang Y, Bai XC, Jiang Y. Structural Mechanism of EMRE-Dependent gating of the human mitochondrial calcium uniporter. Cell. 2019;177:1252–1261.e13. doi: 10.1016/j.cell.2019.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu JC, Liu J, Holmström KM, Menazza S, Parks RJ, Fergusson MM, Yu ZX, Springer DA, Halsey C, Liu C, et al. MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 2016;16:1561–1573. doi: 10.1016/j.celrep.2016.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kamer KJ, Grabarek Z, Mootha VK. High-affinity cooperative Ca2+ binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 2017;18:1397–1411. doi: 10.15252/embr.201643748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, Rubin E, Csordás G, Seifert EL, Hoek JB, et al. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat Commun. 2016;7:10955. doi: 10.1038/ncomms10955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Paillard M, Csordás G, Huang KT, Várnai P, Joseph SK, Hajnóczky G. MICU1 Interacts with the D-Ring of the MCU Pore to Control Its Ca2+ flux and sensitivity to Ru360. Mol Cell. 2018;72:778–785.e3. doi: 10.1016/j.molcel.2018.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Phillips CB, Tsai CW, Tsai MF. The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. Elife. 2019;8. doi: 10.7554/eLife.41112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tufi R, Gleeson TP, von Stockum S, Hewitt VL, Lee JJ, Terriente-Felix A, Sanchez-Martinez A, Ziviani E, Whitworth AJ. Comprehensive genetic characterization of mitochondrial Ca2+ uniporter components reveals their different physiological requirements in vivo. Cell Rep. 2019;27:1541–1550.e5. doi: 10.1016/j.celrep.2019.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gottschalk B, Klec C, Leitinger G, Bernhart E, Rost R, Bischof H, Madreiter-Sokolowski CT, Radulović S, Eroglu E, Sattler W, et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat Commun. 2019;10:3732. doi: 10.1038/s41467-019-11692-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Paillard M, Csordás G, Szanda G, Golenár T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spät A, et al. Tissue-Specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 2017;18:2291–2300. doi: 10.1016/j.celrep.2017.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW. MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation. 2019;140:1720–1733. doi: 10.1161/CIRCULATIONAHA.118.037968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Crompton M, Künzi M, Carafoli E. The calcium-induced and sodiuminduced effluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier. Eur J Biochem. 1977;79:549–558. doi: 10.1111/j.1432-1033.1977.tb11839.x [DOI] [PubMed] [Google Scholar]
- 105.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107:436–441. doi: 10.1073/pnas.0908099107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–147. doi: 10.1126/science.1175145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nowikovsky K, Froschauer EM, Zsurka G, Samaj J, Reipert S, Kolisek M, Wiesenberger G, Schweyen RJ. The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf-Hirschhorn syndrome. J Biol Chem. 2004;279:30307–30315. doi: 10.1074/jbc.M403607200 [DOI] [PubMed] [Google Scholar]
- 108.De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J Biol Chem. 2014;289:20377–20385. doi: 10.1074/jbc.M113.540898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu T, O’Rourke B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J Bioenerg Biomembr. 2009;41:127–132. doi: 10.1007/s10863-009-9216-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A. 2015;112:9129–9134. doi: 10.1073/pnas.1504705112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34. doi: 10.1016/j.celrep.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22. doi: 10.1016/j.celrep.2015.06.00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Parks RJ, Menazza S, Holmström KM, Amanakis G, Fergusson M, Ma H, Aponte AM, Bernardi P, Finkel T, Murphy E. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc Res. 2019;115:385–394. doi: 10.1093/cvr/cvy218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, et al. ; UK10K Consortium. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet. 2014;46:188–193. doi: 10.1038/ng.2851 [DOI] [PubMed] [Google Scholar]
- 117.Lewis-Smith D, Kamer KJ, Griffin H, Childs AM, Pysden K, Titov D, Duff J, Pyle A, Taylor RW, Yu-Wai-Man P, et al. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol Genet. 2016;2:e59. doi: 10.1212/NXG.0000000000000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature. 2017;545:93–97. doi: 10.1038/nature22082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bick AG, Wakimoto H, Kamer KJ, Sancak Y, Goldberger O, Axelsson A, DeLaughter DM, Gorham JM, Mootha VK, Seidman JG, et al. Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc Natl Acad Sci U S A. 2017;114:E9096–E9104. doi: 10.1073/pnas.1711303114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shamseldin HE, Alasmari A, Salih MA, Samman MM, Mian SA, Alshidi T, Ibrahim N, Hashem M, Faqeih E, Al-Mohanna F, et al. A null mutation in MICU2 causes abnormal mitochondrial calcium homeostasis and a severe neurodevelopmental disorder. Brain. 2017;140:2806–2813. doi: 10.1093/brain/awx237 [DOI] [PubMed] [Google Scholar]
- 121.Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32:2362–2376. doi: 10.1038/emboj.2013.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gallagher KP, Buda AJ, Pace D, Gerren RA, Shlafer M. Failure of superoxide dismutase and catalase to alter size of infarction in conscious dogs after 3 hours of occlusion followed by reperfusion. Circulation. 1986;73:1065–1076. doi: 10.1161/01.cir.73.5.1065 [DOI] [PubMed] [Google Scholar]
- 123.Nejima J, Knight DR, Fallon JT, Uemura N, Manders WT, Canfield DR, Cohen MV, Vatner SF. Superoxide dismutase reduces reperfusion arrhythmias but fails to salvage regional function or myocardium at risk in conscious dogs. Circulation. 1989;79:143–153. doi: 10.1161/01.cir.79.1.143 [DOI] [PubMed] [Google Scholar]
- 124.Ooiwa H, Stanley A, Felaneous-Bylund AC, Wilborn W, Downey JM. Superoxide dismutase conjugated to polyethylene glycol fails to limit myocardial infarct size after 30 min ischemia followed by 72 h of reperfusion in the rabbit. J Mol Cell Cardiol. 1991;23:119–125. doi: 10.1016/0022-2828(91)90099-8 [DOI] [PubMed] [Google Scholar]
- 125.Uraizee A, Reimer KA, Murry CE, Jennings RB. Failure of superoxide dismutase to limit size of myocardial infarction after 40 minutes of ischemia and 4 days of reperfusion in dogs. Circulation. 1987;75:1237–1248. doi: 10.1161/01.cir.75.6.1237 [DOI] [PubMed] [Google Scholar]
- 126.Murohara Y, Yui Y, Hattori R, Kawai C. Effects of superoxide dismutase on reperfusion arrhythmias and left ventricular function in patients undergoing thrombolysis for anterior wall acute myocardial infarction. Am J Cardiol. 1991;67:765–767. doi: 10.1016/0002-9149(91)90538-v [DOI] [PubMed] [Google Scholar]
- 127.Flaherty JT, Pitt B, Gruber JW, Heuser RR, Rothbaum DA, Burwell LR, George BS, Kereiakes DJ, Deitchman D, Gustafson N. Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation. 1994;89:1982–1991. doi: 10.1161/01.cir.89.5.1982 [DOI] [PubMed] [Google Scholar]
- 128.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com [DOI] [PubMed] [Google Scholar]
- 129.Murphy MP. Understanding and preventing mitochondrial oxidative damage. Biochem Soc Trans. 2016;44:1219–1226. doi: 10.1042/BST20160108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ribeiro Junior RF, Dabkowski ER, Shekar KC, O Connell KA, Hecker PA, Murphy MP. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic Biol Med. 2018;117:18–29. doi: 10.1016/j.freeradbiomed.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM; Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148 [DOI] [PubMed] [Google Scholar]
- 132.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x [DOI] [PubMed] [Google Scholar]
- 133.Forbes RA, Steenbergen C, Murphy E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ Res. 2001;88:802–809. doi: 10.1161/hh0801.089342 [DOI] [PubMed] [Google Scholar]
- 134.Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016;23:254–263. doi: 10.1016/j.cmet.2015.12.009 [DOI] [PubMed] [Google Scholar]
- 135.Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pell VR, Chouchani ET, Murphy MP, Brookes PS, Krieg T. Moving forwards by blocking back-flow: the yin and yang of MI therapy. Circ Res. 2016;118:898–906. doi: 10.1161/CIRCRESAHA.115.306569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang J, Wang YT, Miller JH, Day MM, Munger JC, Brookes PS. Accumulation of succinate in cardiac ischemia primarily occurs via canonical krebs cycle activity. Cell Rep. 2018;23:2617–2628. doi: 10.1016/j.celrep.2018.04.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Andrienko TN, Pasdois P, Pereira GC, Ovens MJ, Halestrap AP. The role of succinate and ROS in reperfusion injury - A critical appraisal. J Mol Cell Cardiol. 2017;110:1–14. doi: 10.1016/j.yjmcc.2017.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]