

Abstract
Biased agonists, which selectively stimulate certain signaling pathways controlled by a G protein-coupled receptor (GPCR), hold great promise as drugs that maximize efficacy while minimizing dangerous side effects. Biased agonists of the μ-opioid receptor (μOR) are of particular interest as a means to achieve analgesia through G protein signaling without dose-limiting side effects such as respiratory depression and constipation. Rational structure-based design of biased agonists remains highly challenging, however, because the ligand-mediated interactions that are key to activation of each signaling pathway remain unclear. We identify several compounds for which the R- and S-enantiomers have distinct bias profiles at the μOR. These compounds serve as excellent comparative tools to study bias because the identical physicochemical properties of enantiomer pairs ensure that differences in bias profiles are due to differences in interactions with the μOR binding pocket. Atomic-level simulations of compounds at μOR indicate that R- and S-enantiomers adopt different poses that form distinct interactions with the binding pocket. A handful of specific interactions with highly conserved binding pocket residues appear to be responsible for substantial differences in arrestin recruitment between enantiomers. Our results offer guidance for rational design of biased agonists at μOR and possibly at related GPCRs.
Introduction
The severity of the ongoing opioid crisis highlights the need for the development of safer drugs to treat chronic pain effectively.1 The activation of the μ-opioid receptor (μOR) by opioids such as morphine or fentanyl can lead to powerful analgesic effects but also to a number of adverse dose-limiting side effects including constipation, tolerance, dependence, and respiratory depression that can lead to death in overdoses.2,3 Recent work has suggested that while the analgesic properties of these drugs result from μOR-mediated G protein signaling, certain undesired side effects, including potentially lethal respiratory depression, arise from μOR-mediated β-arrestin (β-arr) signaling.4−7 These results suggest that an ideal analgesic would act as an agonist for μOR while selectively stimulating G protein signaling over β-arrestin signaling. The search for such “functionally selective” or “biased” ligands has become the focus of a great deal of ongoing work not only at μOR7−16 but also at many other G protein-coupled receptors (GPCRs), where therapeutic benefits come from activation of certain receptor signaling pathways and undesired side effects from other signaling pathways mediated by the same receptor.17−20
To date, a number of biased μOR ligands have been described in the literature that are reported to have an improved pharmacological profile over currently marketed opioids. Herkinorin, a derivative of the naturally occurring psychotropic salvinorin A, was shown to act as an agonist of the G protein signaling pathway at μOR without promoting β-arrestin recruitment.15,16 Similarly, a derivative of the naturally occurring compound mitragynine has been shown to activate μOR with minimal β-arrestin recruitment.8 Preclinical studies of TRV-130 (oliceridine) showed some degree of bias toward G protein signaling,13,14 but more recent work found no significant bias for this ligand;21 the Food and Drug Administration (FDA) recently approved it but only for intravenous use in controlled clinical settings, noting that the risk of respiratory depression persisted.22 Virtual screening at μOR yielded compound PZM21, which demonstrated exceptional selectivity for μOR over other opioid receptors and an apparent bias for G protein signaling,9 although more recent work failed to replicate this bias profile.23 Finally, a series of G protein-biased μOR ligands were discovered and extensively characterized by Schmid and co-workers.7 These publications focus on ligands with a variety of chemical scaffolds, suggesting promising inroads toward a safer, more effective treatment of pain.
Despite the progress made in the discovery of biased μOR ligands, the structural mechanism by which these compounds achieve bias remains elusive. To decipher this mechanism and relate it to ligand structure to facilitate rational design, one first requires knowledge of how such biased ligands bind to their target.17,24 The relatively small number of well-characterized biased μOR ligands, and the high chemical diversity of these compounds, make it difficult to identify which ligand–receptor interactions lead to activation of one signaling pathway over another. For example, herkinorin lacks a positively charged group that is characteristic of many μOR agonists. Even attributing changes in the signaling profile between closely related compound analogues to differing receptor interactions is not always possible, as minor changes to chemical substituents can have significant effects not only on receptor interactions but also on physicochemical properties that affect assay output, such as water solubility.
To circumvent these difficulties, we identify and study enantiomer pairs (i.e., pairs of compounds that represent mirror images of one another but are not superimposable) with distinct bias profiles at the μOR. Enantiomers share physical and chemical properties but present different binding interfaces to the receptor. If a pair of enantiomers have different bias profiles, one can relate these differences directly to differences in the interactions the enantiomers form with the receptor. In particular, analysis of the signaling profiles is not confounded by factors such as solubility or spatial volumes, which can all influence performance and readout in assays.
Here, we describe four enantiomer pairs of μOR-agonist ligands (the purified R- and S-enantiomers of four compound analogues), where changing the stereochemistry of a single chiral center consistently results in functionally distinct bias profiles, which are reproduced for all analogues. For each pair, the R- and S-enantiomers achieve similar full response (Emax) in cyclic adenosine monophosphate (cAMP) production (a measure of G protein signaling), while the S-enantiomer achieves much higher levels of arrestin recruitment than the R-enantiomer.
Molecular dynamics (MD) simulations indicate that the R- and S-enantiomers of each pair adopt distinct poses when bound to μOR. These poses for each pair share several receptor interactions but display key differences that provide a possible explanation for their disparate signaling profiles. Importantly, the receptor interactions for a given enantiomer (R- or S-) were reproduced for different analogues. The distinct interactions observed in simulation for enantiomers with distinct bias profiles allow for the development of a pharmacophore for biased ligand design at μOR. These findings hold substantial promise as a guide in the continuing development of next-generation biased opioid analgesics and also have important implications for the design of biased agonists for other GPCR targets with similar structures.
Results and Discussion
Discovery of Biased μOR-Activating Ligand Enantiomeric Pairs
Internal screening efforts were run on a Pfizer proprietary compound library to identify novel biased agonists of μOR with potential as improved analgesics. Compounds were screened in G protein and β-arrestin (with coexpression of GRK2) mode assays, using the known unbiased μOR agonist [d-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO) as a reference compound (see Materials and Methods). From this effort, racemic compound 1 (Figure 1A; cAMP EC50 = 1.4 nM [Emax = 104%], β-arrestin EC50 = 72 nM [Emax = 97%]) was identified as an attractive starting point for further optimization. As compound 1 is a racemic mixture, the separate enantiomers were obtained and characterized. Interestingly, the two enantiomers (compounds 1R and 1S) showed different signaling profiles. Compound 1S had a similar profile to the racemic compound 1, while compound 1R showed a more pronounced Emax bias profile (see Table 1 for all assay measurements).
Figure 1.

Enantiomers give rise to different bias profiles. (A) Compound 1 (racemic mixture, left) was identified in an initial Pfizer screen as a hit for further optimization. Purification of the individual enantiomers revealed distinct bias profiles, as characterized by the measurement of G protein-mediated signaling (cAMP production) and β-arrestin recruitment. (B) Enantiomers of four piperazine analogues were purified. Activity curves for compounds 2S (C) and 2R (D) show that the S-enantiomer showed higher arrestin recruitment at saturating conditions than the R-enantiomer, whereas both enantiomers reached a comparable Emax in G protein activity. The same is true for the other three pairs of enantiomers (Table 1 and Figure S1). Standard errors are shown for all experimentally determined data points. GRK2 was coexpressed in all β-arrestin recruitment assays.
Table 1. Bias Profiles of the Presented Enantiomersa.

cAMP and β-arr Emax relative to DAMGO (ref (44)). GRK2 was coexpressed in all β-arrestin recruitment assays. Both 2R and 2S have an analogue substituent with the chirality shown.
The cationic (charged piperazine base) and phenol moieties of these compounds are common to many opioid ligands, including morphinans. To examine whether the different signaling profiles observed for 1R and 1S are driven by the substituted urea vector or the branched aromatic system attached to the chiral carbon, compound analogues replacing the thiophene with different hydrophobic groups were synthesized and tested, confirming that the R- and S-enantiomers of multiple analogues generally reproduce the signaling profile signatures of 1R and 1S (see Table 1 and Figures 1 and S1). These compounds suggest that the stereochemistry of the branched aromatic moiety is the driver for the different observed bias profiles, presumably due to differences in ligand–receptor interactions between enantiomers. For this reason, a rigorous comparative computational modeling study of the complexes formed by several compounds with μOR was undertaken.
MD Simulations Reveal that R- and S-Enantiomers Form Distinct Receptor Interactions
To ascertain how the binding poses and ligand–receptor interactions differ between the two enantiomers within each pair, we first docked compounds 2S and 2R to an experimentally determined active-state μOR structure. These enantiomers docked in very similar poses that form receptor interactions commonly observed in cocrystal structures of opioid receptors:25−29 a salt bridge between the positively charged ligand ammonium group and D3.32 (we use the Ballesteros–Weinstein residue numbering,30,31 where the number before the period indicates the transmembrane helix (TM) in which a residue is found), a hydrogen-bonding group oriented toward H6.52, a π-stack interaction between the phenyl group and Y7.43, and placement of the hydrophobic tail between transmembrane helices 2 and 3 (TM2 and TM3) (Figure 2).
Figure 2.

MD simulations reveal distinct poses for different ligand enantiomers. Simulations of pairs of enantiomers (e.g., 2S and 2R shown) were initiated from docked poses. The S- and R-enantiomers formed similar interactions in their initial poses (left), including a π-stack with Y7.43 and a salt bridge with D3.32. During simulations, 2S maintained all of the initial interactions while also forming a water-mediated interaction with H6.52 (top right). By contrast, 2R underwent a reorientation, breaking the initial π-stack with Y7.43 and instead reorienting toward Y3.33 in a potential T−π stack interaction, while it retained the water-mediated interaction with H6.52 (bottom right). This reorientation also significantly weakens the direct salt bridge interaction with D3.32 for 2R. An overlay of the final poses (right) highlights the difference in the orientation of the phenyl group attached to the chiral center.
To account for the effects of receptor motion, solvation, and internal ligand conformational strain, we then performed MD simulations of these enantiomers in complex with the μOR, starting from the docked poses. For each enantiomer, we performed three simulations, each ∼1 μs in length.
Throughout the simulations, the S-enantiomer maintains all of the original interactions, including π–π interaction with Y7.43 on TM7 (Figure 3B) and the salt bridge with D3.32 (Figure 3D), while also forming a water-mediated interaction with H6.52 (Figure 2 and Table S1). The R-enantiomer, however, quickly reorients its phenyl group to a distinct pose to relieve ligand conformational strain. This reorientation shifts the phenyl group attached to the chiral center away from TM7, such that it forms a T−π interaction with Y3.33 on TM3 (Figures 2 and 3C). This shift of the phenyl group significantly weakens the salt bridge with D3.32 (Figure 3D). The R-enantiomer retains some interactions in common with the S-enantiomer, including a water-mediated interaction with H6.52 and placement of the hydrophobic tail (the substituents that distinguish compound analogues) between TM2 and TM3 (Figure 2). In both enantiomers, the urea group forms no stable direct interactions with the receptor, although it occasionally forms short-lived water-mediated interactions with Q2.60.
Figure 3.

In simulation, R- and S-enantiomers form different stable interactions with the receptor. (A) The poses of 2S (left) and 2R (right) adopted in MD simulations. In 2R, the phenyl group attached to the chiral center breaks an initial interaction with Y7.43 (maintained in 2S) to instead engage Y3.33. Panels (B)–(D) quantify ligand–receptor interactions by showing interatomic distances throughout each simulation. (B) Distance between the 4-position of the phenyl ring attached to the chiral center and the ζ carbon of Y7.43. (C) Distance between the 4-position of the phenyl ring attached to the chiral center and the γ carbon of Y3.33. (D) Shortest distance between the ligand cationic nitrogen and either side-chain oxygen of D3.32.
To validate that these enantiomer-specific interactions could be generalized, we carried out similar simulations of a second pair of enantiomers, 1S and 1R, which differ in the hydrophobic tail that interacts with TM2/TM3. These ligands docked in very similar poses to 2S and 2R, forming many of the same interactions. In simulation, the 1S enantiomer maintained its initial pose, apart from a shift in the position of the hydrophobic tail that differs between analogues. The 1R enantiomer underwent a similar reorientation to that observed for 2R, adopting a final pose nearly identical to that of 2R (Figures S2 and S3).
The consistency among ligand pairs allows a key conclusion to be made based on both the pharmacological and simulation data. The S-enantiomers, which are observed to have a higher Emax for β-arrestin recruitment, display a strongly favorable salt bridge with D3.32 and a concurrent π–π interaction with Y7.43. In contrast, the R-enantiomers, which have lower maximal β-arrestin recruitment activity, display a weaker salt bridge to D3.32, as well as engagement with Y3.33 instead of Y7.43. In other words, the ligands undergoing stabilizing interactions with Y7.43 and D3.32 favor strong arrestin recruitment. The ligands for which these interactions are attenuated can activate G protein signaling to a similar extent without favoring β-arrestin recruitment as strongly.
Development of Biased Signaling Pharmacophore for μOR
The broadly reproducible results for the R- and S-enantiomers across compounds 1–4 allow for correlation of experimentally observed bias profiles with enantiomer-specific interactions. Using this information, we developed a pharmacophore for biased signaling at μOR (Figure 4). If two enantiomers reach the same Emax in a given signaling pathway, the interactions they share in common are likely sufficient to activate that pathway. In the case of each of these enantiomer pairs, both compounds reach the same Emax in the G protein signaling pathway and share several key interactions. Conversely, if one enantiomer cannot activate a given pathway to the same extent as the other (i.e., does not reach the same Emax), this suggests that it lacks critical interactions for activation of that pathway. This is the case for the β-arrestin recruitment pathway, which the R-enantiomer cannot activate to the same extent as the S-enantiomer.
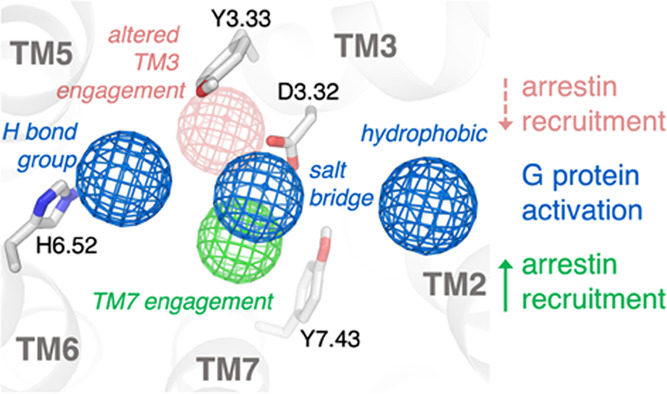
Figure 4.
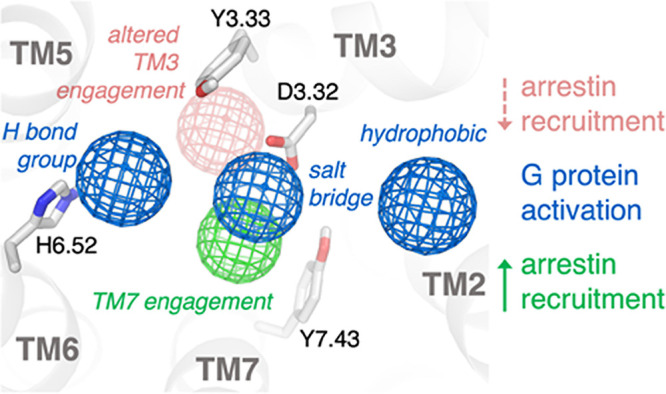
Pharmacophore for biased ligand design at the μ-opioid receptor. Through analysis of the conserved interactions observed in simulation across the enantiomer pairs presented here, a pharmacophore for biased ligand design at μOR can be determined. Ligands that interact strongly with Y7.43 and D3.32 have higher β-arrestin activity, while those that instead engage with Y3.33 and concomitantly weaken interaction with D3.32 have lower β-arrestin activity. All ligands studied activate G protein signaling to a similar extent, and all form an interaction with D3.32, place a hydrogen-bonding group toward TM6 to form water-mediated interactions with H6.52, and place a hydrophobic group in contact with TM2 and/or TM3.
Across ligands, each enantiomer reaches comparable Emax in G protein signaling, which suggests that their shared receptor interactions, though different in relative strength, are enough to activate that pathway to the same degree. Simulations of two pairs of R- and S-enantiomers studied here identify three key shared interactions between the different enantiomers (Figures 2 and S2): a salt bridge interaction with D3.32, a water-mediated hydrogen bond with H6.52, and placement of a hydrophobic group in contact with TM2 and/or TM3.
Conversely, the observation that one enantiomer elicits a weaker response than the other at a given signaling pathway suggests that the former enantiomer lacks some of the requisite interactions to activate that pathway fully. For the compounds we studied, the S-enantiomers, with higher β-arrestin activity, engage Y7.43 in a stabilizing interaction and have a strong direct salt bridge to D3.32. In contrast, the R-enantiomers, with lower β-arrestin activity, engage Y3.33 in a stabilizing interaction and have weakened interaction with D3.32 as a result, suggesting that either Y7.43 engagement or a strong salt bridge to D3.32 are required for full activation of the β-arrestin pathway by these ligands.
While the extent of signaling pathway activation (Emax) is most instructive in describing the differences between R- and S-enantiomers, a discussion of the overall compound bias must also include EC50, the concentration at which half-maximal signaling activation occurs. The relative potency at which the R- and S-enantiomers activate either the G protein or β-arrestin signaling pathway may be explained by how well each enantiomer binds to the lowest-energy binding pocket conformation presented by the receptor when coupled to either a G protein or a β-arrestin.
The S-enantiomer appears ideally suited to fit the binding pocket presented by the receptor in its G protein-coupled state, whose binding pocket conformation matches that from which our simulations were initiated.32 In contrast, the R-enantiomer conformation does not easily fit in this manner. Instead, the R-enantiomer quickly rearranges in simulation, requiring concomitant conformational changes in the receptor’s binding pocket. Specifically, Y3.33 adapts its position to accommodate the phenyl group of the R-enantiomer. This required conformational change in the receptor apparently decreases the potency of the R-enantiomer relative to the S-enantiomer as measured for G protein signaling. No structure of a μOR–arrestin complex state is available, but our experimental measurements suggest that both enantiomers bind more weakly to the arrestin-coupled state, with a larger decrease in potency for the S-enantiomer than for the R-enantiomer.
The ligand–receptor interactions formed by the ligands we studied are similar to those of other μOR agonists that stimulate G protein signaling. The G protein-biased ligands from Schmid and co-workers7 are analogous to the ligands described herein and can easily be placed to make the same interactions that we have correlated with G protein signaling activation. Similarly, recent structural studies reveal that DAMGO also makes these key interactions.32 In addition, PZM21 has been predicted to make each of these three key interactions.9
Comparing interactions predicted to lead to β-arrestin activation by the S-enantiomers we studied with interactions formed by other μOR agonists again reveals strong support for this pharmacophore. PZM21, which is believed to be G protein-biased, is not predicted to make any direct interaction with TM7.9 The ligands from Schmid and co-workers7 all lack a functional group that could interact with TM7 in a similar manner to the S-enantiomers in this study, suggesting one potential explanation for the lack of β-arrestin activity in their ligands. DAMGO, a balanced agonist that leads to full β-arrestin activation, has been observed to make a direct interaction with Y7.43, identical to what our pharmacophore predicts would be needed to stimulate β-arrestin recruitment.
While this pharmacophore appears to fit many ligands, one cocrystallized ligand, the balanced agonist BU72, suggests that there is still more at play. BU72 displays interactions with D3.32 and H6.52 and orients a hydrophobic group toward TM2 and TM3, which fits our pharmacophore for activation of the G protein signaling pathway. However, BU72 is not observed to make any direct interaction with TM7, which we predict to be important for β-arrestin signaling. It is possible that the strength of BU72’s interaction with D3.32 and the overall fit of its morphinan scaffold to the μOR active site are strong enough that stabilizing interactions with Y7.43 are not required for BU72 to stimulate β-arrestin signaling. It is unlikely that engagement with Y7.43 is the only interaction that drives β-arrestin recruitment, and it is possible that different interactions could reproduce the same effect. BU72’s full β-arrestin recruitment suggests that while our pharmacophore appears to generalize across many biased ligands and provides useful information for future drug design, it probably does not account for all of the possible ways in which β-arrestin signaling can be achieved at μOR.
We note that our experiments used an amplified assay (cAMP concentration) as a measure of G protein signaling and an unamplified assay (β-arrestin recruitment) as a measure of β-arrestin signaling. Comparison of data from amplified and unamplified assays can confound the quantification of ligand bias.33 To ensure a quantifiable response for arrestin signaling, we coexpressed GRK2 in all β-arrestin recruitment assays.34
The differences we observed in ligand–receptor interactions between enantiomers might lead to distinct bias profiles in more than one way. Different ligand–receptor interactions might favor different receptor conformations with different preferences for arrestin binding relative to G protein binding. Different interactions might also lead to differences in binding kinetics, affecting the residence time for each enantiomer at μOR. Several studies have shown that ligand residence time plays an important role in biased signaling35 and that β-arrestin signaling occurs at longer time scales than G protein signaling.36,37 The more strongly stabilizing interactions for the S-enantiomer here relative to the R-enantiomer could result in a longer residence time at the receptor and as a result more β-arrestin recruitment.
Our simulations do not determine whether different conformations of the intracellular side of the receptor are favored by each ligand and, if so, what those conformations are. To prevent the receptor from transitioning spontaneously toward its inactive state during simulation,38,39 and to ensure that different conformational changes observed in the binding pocket were due to differences in the bound ligands rather than to the allosteric effects of conformational changes on the intracellular side of the receptor, we restrained the intracellular surface of the receptor to its crystallographic active-state conformation, which appears to match that observed in the lower-resolution structure of the μOR in complex with its cognate G protein. Such restraints do not preclude local conformational changes in the binding pocket; indeed, such changes have been observed in previous studies that used similar restraints.40,41 We cannot rule out the possibility that the use of restraints may have introduced artifacts in our results, although the fact that the pharmacophore model suggested by our simulations agrees with structural and pharmacological observations on a variety of other ligands suggests that our conclusions are likely robust to any such artifacts.
In addition, we did not perform simulations of μOR in complex with β-arrestin because no structure of a μOR–arrestin complex is available. Structures of other GPCRs in complex with arrestins and G proteins suggest that conformational changes in the receptor will be subtle,42,43 but we cannot rule out the possibility that ligand–receptor interactions might differ substantially in the presence of arrestin.
Conclusions
As the opioid crisis shows no signs of abating, the need for safer analgesics is pressing. One of the most promising routes to achieving this would be biased agonism of μOR, a target with extremely strong clinical validation for the treatment of pain. While a number of putative μOR biased compounds have been identified, structure-led optimization has been hindered by a lack of understanding of the critical interactions that enable biased signaling. Here, we have demonstrated the importance of ligand engagement with residues on both TM3 and TM7 in determining β-arrestin recruitment activity through the pharmacological characterization and structural modeling of enantiomer pairs of multiple ligand analogues. These analyses, as well as comparisons to other biased and balanced μOR ligands, allowed for the development of a biased signaling pharmacophore that highlights key interactions required for G protein signaling and β-arrestin recruitment activation.
We propose that, in combination with consideration of ligand residence times, this pharmacophore could provide a valuable guide in the future design of biased ligands for μOR, facilitating the design of safer, more effective, opioid-based analgesics. Given similarities in sequence and structure across the GPCR family, which includes the targets of nearly a third of all drugs, our results may also prove useful in guiding the design of biased ligands for other GPCRs.
Materials and Methods
Cell Culture
Experimental methods have been published previously44 but are included here for clarity. PathHunter OPRM1 β-arrestin U2OS cells (U2OS-μ) were purchased from DiscoveRx (Birmingham, UK). Cells were grown in the modified Eagle’s medium, containing 2 mM of GlutaMAX, 10% fetal calf serum (FCS), 500 μg·mL–1 of geneticin, and 250 μg·mL–1 of hygromycin B at 37 °C and 5% CO2 in a humidified incubator. Cells were seeded in flasks at 2.2 × 104 cells·cm–2 and passaged every 3 days.
CHOK1 cells expressing μ-opioid receptors (CHO-μ) were grown in Dulbecco’s modified Eagle’s medium (DMEM)/F12, containing 2 mM of GlutaMAX, 10% FCS, and 300 μg·mL–1 of zeocin at 37 °C and 5% CO2 in a humidified incubator. Cells were seeded in flasks at 2.2 × 103 cells·cm–2 and passaged every 3 days. For assay use, cells were harvested and cryopreserved at 5 × 106 cells·mL–1 in 90% FCS and 10% dimethyl sulfoxide (DMSO) and stored in vapor-phase liquid nitrogen.
Membrane Preparation
U2OS-μ were grown in 225 cm2 flasks until 90% confluent, detached with TrypLE, and centrifuged for 5 min at 1000g. Cells were resuspended in ice-cold buffer (20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), 1 mM MgCl2). All subsequent steps were performed at 4 °C. The cell suspension was homogenized using a T25 Ultra Turrax homogenizer (IKA, Staufen, Germany) with three 10 s bursts. The cell homogenate was centrifuged for 30 min at 1000g, and the supernatant was collected and centrifuged at 55 000g for 45 min before resuspending in a buffer. Protein concentration was determined using the Bradford assay with bovine serum albumin (BSA) as a standard. Aliquots were stored at −80 °C.
Compound Synthesis
The general route to the piperazines is shown in Scheme 1. The racemic Ellman sulfimine (prepared from benzaldehyde) was treated with 3-methoxybenzyl Grignard to provide the racemic sulfimine 2, which could be deprotected using HCl in dioxane to provide the racemic amine 3. The piperazine was prepared by condensation with the dichloro reagent 4. To avoid the use of this mustard compound, we did try to protect the amine as a tosyl amide. We found that this reagent did not cleanly form the piperazine. The benzyl reagent 4 required heating in dimethylformamide (DMF) to affect the transformation. Debenzylation followed by urea formation utilizing phenylchloroformate and (3-thienylmethyl)amine provided 7. Demethylation with BBr3 and separation of enantiomers by chromatography gave the desired analogues 1S and 1R.
Scheme 1.
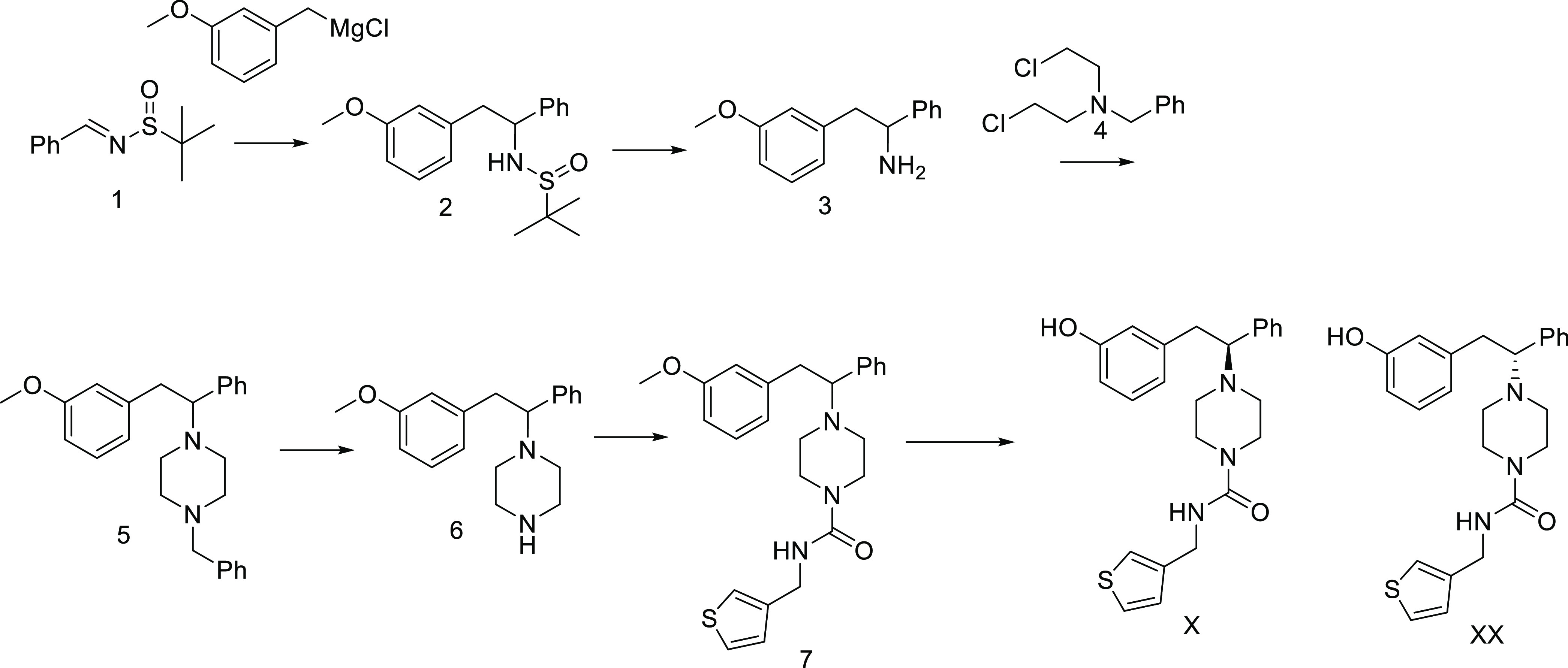
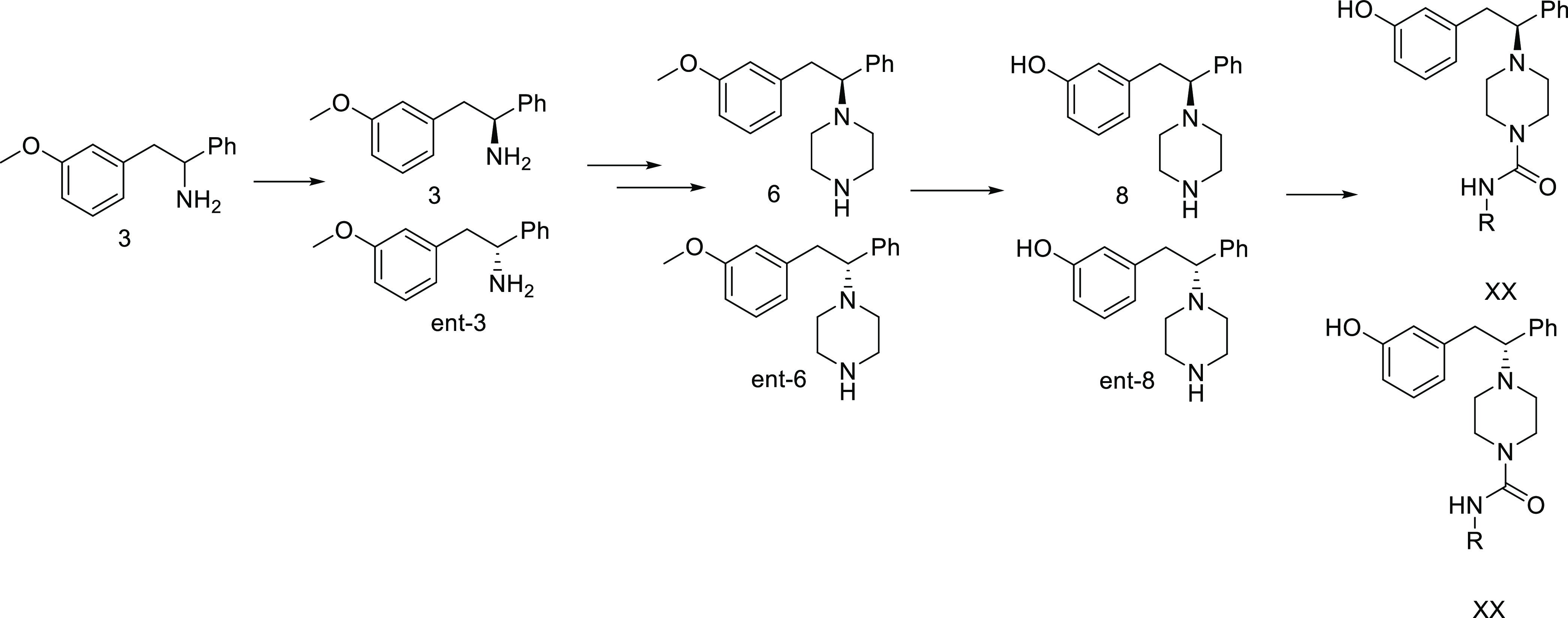
Attempts to use the nonracemic Ellman sulfimine lead to mixtures of diastereomers of 2. These could be separated using chromatography. However, a simpler method to separate the enantiomers on scale was to perform a classic resolution on 3 using (−)-dibenzoyltartaric acid in EtOH. Absolute stereochemistry was assigned by comparison of rotation to literature values. With gram quantities of each enantiomer in hand, we proceeded to develop a parallel synthesis of these analogues. To this end, we prepared 6 as shown in Scheme 2 from each enantiomer. At this stage, we demethylated using dodecanethiol and NaOH in DMSO to prepare 8 and ent-8 as separate enantiomers. In parallel, these were treated with disuccinimidyl carbonate in acetonitrile with diisopropylethylamine, followed by a variety of amines. The crude reactions were purified by high-performance liquid chromatography (HPLC) to provide the desired analogues.
Scheme 2.
Compound Preparation
Compounds were serially diluted in DMSO to produce 11-point, half-log concentration–response curves in 384-well acoustic qualified polypropylene plates (LabCyte, Sunnyvale, CA). Assay plates were prepared as required via the acoustic transfer of appropriate volumes from the serialized compound plate using an ECHO550 (LabCyte). Pfizer standard 145 and DMSO were also added to the plates as hundred % effect (HPE) and vehicle, respectively.
cAMP Homogeneous Time-Resolved Fluorescence (HTRF) Assay
Four microliters of assay buffer (Hanks’ balanced salt solution (HBSS) with calcium, magnesium, and 20 mM HEPES) containing 1.6 μM NKH477 was added to 384-well black HiBase plates (Greiner, Stonehouse, UK) containing 20 nL per well compound. Cryopreserved CHO-μ were thawed, centrifuged at 1000g, and resuspended to 2.5 × 105 cells·mL–1 in assay buffer containing 0.5 mM IBMX. The suspension was incubated for 30 min at room temperature prior to dispensing 4 μL per well into the assay plates. Plates were incubated at 37 °C for 30 min before the addition of cAMP Femto 2 HTRF reagents (Cisbio, Codolet, France) according to the manufacturer’s instructions. The plates were read after 1 h incubation at room temperature using an Envision reader (PerkinElmer).
β-Arrestin2 Recruitment Assay
U2OS-μ cells from the culture were harvested and resuspended at 3.75 × 105 cells·mL–1. The cells were seeded into 384-well CellStar plates (Greiner) at 7500 cells per well and incubated at 37 °C and 5% CO2 in a humidified incubator overnight. For the assay, 100 nL per well compounds were dispensed onto the cells using an ECHO550 (LabCyte). The plates were briefly centrifuged and incubated for 2 h at 37 °C. The assay was terminated by the addition of PathHunter detection reagent (DiscoveRx) according to the manufacturer’s instructions. The plates were read after 1 h incubation at room temperature using an Envision reader.
For the coexpression of GRK2, a modified baculovirus system was used to express GRK2 from a CMV promoter in mammalian cells (BacMam Life Technologies, CA). U2OS-μ were resuspended at 3.75 × 105 cells·mL–1 and supplemented with GRK2 Bacmam to achieve the required multiplicity of infection (MOI) prior to seeding into plates and assaying as described above.
Ligand Docking
Prior to docking, all ligands were prepared using the standard ligand prep option in LigPrep (Schrödinger). Glide SP (Schrödinger) was used to generate docked poses in an active-state structure of μOR (PDB: 5C1M). The docking grid was centered on the orthosteric binding pocket, defined as the centroid of the BU72 cocrystallized ligand in the crystal structure (PDB code 5C1M). All ligand poses within a score of 100 were kept after the initial Glide screen, and the best 400 poses were kept for energy minimization (distance-dependent dielectric constant of 2.0 with a maximum of 100 minimization steps) using the OPLS_2005 force field. Ligand sampling was flexible, nitrogen inversions and ring conformations (within an energy window of 2.5 kcal·mol–1) were both sampled, and Epik state penalties were added to docking scores. Crystallographic water molecules were retained for docking. Poses resulting from docking were then visually inspected and compared to existing crystal structures of the MOR in complex with agonists. For each ligand, we selected a pose that shared the following properties with the cocrystallized ligand: a salt bridge between the ligand cationic nitrogen and D3.32, and high overlap of the ligand phenol group (particularly the hydroxyl) with the analogous group of BU72 in the crystal structure 5C1M. Selected poses are shown in Figure S4. After performing the MD simulations described below, we confirmed that both the salt bridge and the water-mediated receptor interactions formed by the phenol group remained stable.
Molecular Dynamics Simulation System Setup
Simulations of μOR with ligands were initiated from the docked poses generated at an active-state structure of μOR (PDB: 5C1M),29 as described in the Ligand Docking section. We retained crystallographic waters, except when preparing the “no crystallographic waters” control simulation described below. We removed all other non-receptor molecules, including the cocrystallized nanobodies. Prime (Schrödinger) was used to model missing side chains and loops, and neutral acetyl and methylamide groups were added to cap protein termini. In all simulations, all titratable residues remained in their dominant protonation state at pH 7.0. H6.52 was represented with hydrogen on the epsilon nitrogen, except when preparing the “H6.52 HID” control simulation described below.
The prepared protein structures were aligned on the transmembrane helices to the “Orientation of Proteins in Membrane” (OPM)46 structure of PDB 5C1M. The prepared systems were then hydrated using the Dowser plugin in Visual Molecular Dynamics (VMD), with any new waters that overlapped with the retained crystallographic waters removed. The aligned systems were then inserted into a pre-equilibrated palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayer using in-house simulation preparation software.47 Sodium and chloride ions were added to neutralize each system for a final concentration of 150 mM. Dimensions of the membrane bilayer and surrounding water were chosen to maintain at least a 35 Å buffer between proteins in the x–y plane of the membrane and 20 Å in the membrane-normal z direction, when using periodic boundary conditions. The final systems, which varied in number of atoms and size, are listed in Table S2.
We performed two sets of control simulations with 2S bound to probe the effects of (1) the inclusion of crystallographic waters and (2) the protonation state of H6.52 (Table S2). These were set up exactly as described above, except that (1) in the “no crystallographic waters” control condition, we removed all crystallographic waters at the beginning of the system setup process and (2) in the “H6.52 HID” control condition, we represented H6.52 with a hydrogen on the delta nitrogen rather than the epsilon nitrogen.
The “no crystallographic waters” simulations reproduced the results of our non-control simulations with 2S bound. In each of the three “no crystallographic waters” simulations, waters from the extracellular solvent entered into the binding pocket. A water-mediated interaction between the ligand and H6.52 spontaneously reformed within 100 ns and then remained stable (Table S1).
In the “H6.52 HID” simulations, no water-mediated interaction formed between H6.52 and the phenol group, and neither the phenol specifically nor the ligand overall adopted any stable pose. Our other simulations, in which H6.52 is protonated on the epsilon nitrogen, are more consistent with 5C1M and other experimentally determined high-resolution structures of opioid receptors (PDB entries 4DKL, 4DHJ, and 6PT2) that show a water-mediated interaction between H6.52 and a ligand phenol group.
Simulation Protocols
The CHARMM36 parameter set was used for proteins, lipids, and salt ions.48−52 The CHARMM TIP3P model was employed for water. Parameters for all ligands were generated using the CHARMM general force field (CGenFF) using the ParamChem server.53−56 An inspection of ligand parameter penalties assigned by CGenFF was performed to confirm that the parameters assigned high penalties (i.e., those deemed least certain by CGenFF) were reasonable and that any errors in these parameters would not interfere substantially with conclusions drawn from simulation (see the Ligand Parameters section in the Supporting Information).
Three independent simulations were performed for each condition listed in Table S2. We collected a total of 12 μs of simulation trajectory across all conditions. For each simulation, the prepared receptor was overlaid with the experimentally determined structure of the β2-adrenergic receptor bound to Gs (PDB entry 3SN6).57 All μOR residues that were found to be within 5 Å of Gs following the overlay had a 5 kcal·mol–1·Å–2 harmonic restraint placed on their nonhydrogen atoms to ensure the μOR would remain in its active state throughout simulation, in the absence of the Gs protein.
Simulations were performed using the CUDA-enabled version of PMEMD in Amber16 on one to two graphical processing units (GPUs).58 Each system underwent a similar equilibration and minimization procedure. Systems were heated in the NVT ensemble from 0 to 100 K over 12.5 ps and then from 100 to 310 K over 125 ps with 10 kcal·mol–1·Å–2 harmonic restraints on all nonhydrogen lipid and protein atoms. Systems were then equilibrated in the NPT ensemble at 1 bar, with a starting 5 kcal·mol–1·Å–2 harmonic restraint placed on all heavy protein atoms and reduced in a stepwise fashion by 1 kcal·mol–1·Å–2 every 2 ns for a total of 10 ns and then by 0.1 kcal·mol–1·Å–2 every 2 ns for an additional 20 ns. Production simulations were carried out in the NPT ensemble at 310 K and 1 bar using a Langevin thermostat for temperature coupling and a Monte Carlo barostat for pressure coupling. All simulations employed a time step of 4 fs with hydrogen mass repartitioning.59 All bond lengths involving hydrogen atoms were constrained by SHAKE. Nonbonded interactions were cut off at 9.0 Å, while long-range electrostatic interactions were calculated using the particle mesh Ewald (PME) with an Ewald coefficient of approximately 0.31 Å and an interpolation order of 4. The fast Fourier transform (FFT) grid size was chosen such that the width of a single grid cell was approximately 1 Å. Trajectory snapshots were saved every 200 ps.
Analysis Protocols for Molecular Dynamics Simulations
The AmberTools15 CPPTRAJ package was used to reimage and center all resulting trajectories.60 Simulations were visualized and analyzed using VMD.61 Time traces from individual simulations, such as those displayed in Figure 3, were smoothed using a moving average with a window size of 50 ns and visualized using the PyPlot package from Matplotlib.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grant R01GM127359 (R.O.D.), Pfizer Inc. (R.O.D.), NIH Biomedical Informatics Training Grant T15-LM007033-33 (S.A.H.), and the European Molecular Biology Organization Long-Term Fellowship ALTF 544-2019 (D.A.).
Glossary
Abbreviations
- GPCR
G protein-coupled receptor
- μOR
μ-opioid receptor
- MD
molecular dynamics
- TM
transmembrane helix
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.1c00585.
Activity curves for the identified biased μOR-activating ligand enantiomer pairs (Figure S1); simulations of a second enantiomer pair match the results observed in the initial pair (Figure S2); chiral benzyl in the 1S/R pair behaves similarly to the 2S/R pair (Figure S3); docked ligand poses selected as starting points for MD simulations (Figure S4); frequency of water-mediated interaction between the ligand phenol group and H6.52 in all simulations (Table S1); system size and trajectory lengths for all included MD simulations (Table S2); and parameters for compounds 1R, 1S, 2R, and 2S (PDF)
Author Present Address
∇ Sosei Heptares, Steinmetz Building, Granta Park, Cambridge CB21 6DG, U.K
Author Present Address
# Discovery Sciences, R&D, AstraZeneca, Cambridge SK10 4TG, U.K.
Author Present Address
⊥ Computational and Structural Chemistry, Merck & Co., South San Francisco, California 94080, United States
Author Contributions
○ B.K. and S.A.H. contributed equally to this work.
The authors declare no competing financial interest.
Notes
Ligand force field parameters are included in the Supporting Information. MD trajectories and data are available at https://doi.org/10.5281/zenodo.4796912. The authors used the following software under license to Stanford University: Maestro and Glide 2017 (system prep), Amber16 (simulations), VMD 1.9.2 (visualization and analysis), Prism 9 (data analysis and figure generation), Chemdraw 16.0.1.4 (figure generation), and PyMol 1.8.6.2 (rendering).
Supplementary Material
References
- Volkow N. D.; Collins F. S. The Role of Science in Addressing the Opioid Crisis. N. Engl. J. Med. 2017, 377, 391–394. 10.1056/NEJMsr1706626. [DOI] [PubMed] [Google Scholar]
- Matthes H. W. D.; Maldonado R.; Simonin F.; Valverde O.; Slowe S.; Kitchen I.; Befort K.; Dierich A.; LeMeur M.; Dolle P.; Tzavara E.; Hanoune J.; Roques B. P.; Kieffer B. L. Loss of Morphine-Induced Analgesia, Reward Effect and Withdrawal Symptoms in Mice Lacking the Mu-Opioid-Receptor Gene. Nature 1996, 383, 819–823. 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Boom M.; Niesters M.; Sarton E.; Aarts L.; Smith T. W.; Dahan A. Non-Analgesic Effects of Opioids: Opioid-Induced Respiratory Depression. Curr. Pharm. Des. 2012, 18, 5994–6004. 10.2174/138161212803582469. [DOI] [PubMed] [Google Scholar]
- Bohn L. M.; Lefkowitz R. J.; Gainetdinov R. R.; Peppel K.; Caron M. G.; Lin F. T. Enhanced Morphine Analgesia in Mice Lacking Beta-Arrestin 2. Science 1999, 286, 2495–2498. 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bohn L. M.; Gainetdinov R. R.; Lin F. T.; Lefkowitz R. J.; Caron M. G. Mu-Opioid Receptor Desensitization by Beta-Arrestin-2 Determines Morphine Tolerance but Not Dependence. Nature 2000, 408, 720–723. 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Raehal K. M.; Walker J. K. L.; Bohn L. M. Morphine Side Effects in Beta-Arrestin 2 Knockout Mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- Schmid C. L.; Kennedy N. M.; Ross N. C.; Lovell K. M.; Yue Z. Z.; Morgenweck J.; Cameron M. D.; Bannister T. D.; Bohn L. M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165. 10.1016/j.cell.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruegel A. C.; Gassaway M. M.; Kapoor A.; Varadi A.; Majumdar S.; Filizola M.; Javitch J. A.; Sames D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J. Am. Chem. Soc. 2016, 138, 6754–6764. 10.1021/jacs.6b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Lin H.; Aryal D. K.; McCorvy J. D.; Dengler D.; Corder G.; Levit A.; Kling R. C.; Bernat V.; Hubner H.; Huang X. P.; Sassano M. F.; Giguere P. M.; Lober S.; Duan D.; Scherrer G.; Kobilka B. K.; Gmeiner P.; Roth B. L.; Shoichet B. K. Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature 2016, 537, 185. 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soergel D. G.; Subach R. A.; Burnham N.; Lark M. W.; James I. E.; Sadler B. M.; Skobieranda F.; Violin J. D.; Webster L. R. Biased Agonism of the Mu-Opioid Receptor by Trv130 Increases Analgesia and Reduces on-Target Adverse Effects Versus Morphine: A Randomized, Double-Blind, Placebo-Controlled, Crossover Study in Healthy Volunteers. Pain 2014, 155, 1829–1835. 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- Viscusi E. R.; Webster L.; Kuss M.; Daniels S.; Bolognese J. A.; Zuckerman S.; Soergel D. G.; Subach R. A.; Cook E.; Skobieranda F. A Randomized, Phase 2 Study Investigating TRV130, a Biased Ligand of the Mu-Opioid Receptor, for the Intravenous Treatment of Acute Pain. Pain 2016, 157, 264–272. 10.1097/j.pain.0000000000000363. [DOI] [PubMed] [Google Scholar]
- Soergel D. G.; Subach R. A.; Sadler B.; Connell J.; Marion A. S.; Cowan C. L.; Violin J. D.; Lark M. W. First Clinical Experience with Trv130: Pharmacokinetics and Pharmacodynamics in Healthy Volunteers. J. Clin. Pharmacol. 2014, 54, 351–357. 10.1002/jcph.207. [DOI] [PubMed] [Google Scholar]
- Chen X. T.; Pitis P.; Liu G. D.; Yuan C.; Gotchev D.; Cowan C. L.; Rominger D. H.; Koblish M.; DeWire S. M.; Crombie A. L.; Violin J. D.; Yamashita D. S. Structure Activity Relationships and Discovery of a G Protein Biased Mu Opioid Receptor Ligand, [(3-Methoxythiophen-2-Yl)Methyl]A2[(9r)-9-(Pyridin-2-Y1)-6-Oxaspiro-[4.5]Clecan-9-Yl]Ethylpamine (Trv130), for the Treatment of Acute Severe Pain. J. Med. Chem. 2013, 56, 8019–8031. 10.1021/jm4010829. [DOI] [PubMed] [Google Scholar]
- DeWire S. M.; Yamashita D. S.; Rominger D. H.; Liu G. D.; Cowan C. L.; Graczyk T. M.; Chen X. T.; Pitis P. M.; Gotchev D.; Yuan C.; Koblish M.; Lark M. W.; Violin J. D. A G Protein-Biased Ligand at the Mu-Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphines. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- Groer C. E.; Tidgewell K.; Moyer R. A.; Harding W. W.; Rothman R. B.; Prisinzano T. E.; Bohn L. M. An Opioid Agonist That Does Not Induce Mu-Opioid Receptor - Arrestin Interactions or Receptor Internalization. Mol. Pharmacol. 2007, 71, 549–557. 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb K.; Tidgewell K.; Simpson D. S.; Bohn L. M.; Prisinzano T. E. Antinociceptive Effects of Herkinorin, a Mop Receptor Agonist Derived from Salvinorin a in the Formalin Test in Rats: New Concepts in Mu Opioid Receptor Pharmacology: From a Symposium on New Concepts in Mu-Opioid Pharmacology. Drug Alcohol Depend. 2012, 121, 181–188. 10.1016/j.drugalcdep.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A. K.; Singh G.; Ghosh E. Emerging Structural Insights into Biased GPCR Signaling. Trends Biochem. Sci. 2014, 39, 594–602. 10.1016/j.tibs.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Pupo A. S.; Duarte D. A.; Lima V.; Teixeira L. B.; Parreiras-e-Silva L. T.; Costa-Neto C. M. Recent Updates on Gpcr Biased Agonism. Pharmacol. Res. 2016, 112, 49–57. 10.1016/j.phrs.2016.01.031. [DOI] [PubMed] [Google Scholar]
- Zhou X. E.; Melcher K.; Xu H. E. Understanding the GPCR Biased Signaling through G Protein and Arrestin Complex Structures. Curr. Opin. Struct. Biol. 2017, 45, 150–159. 10.1016/j.sbi.2017.05.004. [DOI] [PubMed] [Google Scholar]
- Violin J. D.; Crombie A. L.; Soergel D. G.; Lark M. W. Biased Ligands at G-Protein-Coupled Receptors: Promise and Progress. Trends Pharmacol. Sci. 2014, 35, 308–316. 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Vasudevan L.; Vandeputte M.; Deventer M.; Wouters E.; Cannaert A.; Stove C. P. Assessment of Structure-Activity Relationships and Biased Agonism at the Mu Opioid Receptor of Novel Synthetic Opioids Using a Novel, Stable Bio-Assay Platform. Biochem. Pharmacol. 2020, 113910 10.1016/j.bcp.2020.113910. [DOI] [PubMed] [Google Scholar]
- FDA. FDA Approves New Opioid for Intravenous Use in Hospitals, Other Controlled Clinical Settings. Https://Www.Fda.Gov/News-Events/Press-Announcements/Fda-Approves-New-Opioid-Intravenous-Use-Hospitals-Other-Controlled-Clinical-Settings. https://www.fda.gov/news-events/press-announcements/fda-approves-new-opioid-intravenous-use-hospitals-other-controlled-clinical-settings (accessed Sept 30, 2020).
- Hill R.; Disney A.; Conibear A.; Sutcliffe K.; Dewey W.; Husbands S.; Bailey C.; Kelly E.; Henderson G. The Novel Mu-Opioid Receptor Agonist PZM21 Depresses Respiration and Induces Tolerance to Antinociception. Br. J. Pharmacol. 2018, 175, 2653–2661. 10.1111/bph.14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisler J. W.; Xiao K. H.; Thomsen A. R. B.; Lefkowitz R. J. Recent Developments in Biased Agonism. Curr. Opin. Cell Biol. 2014, 27, 18–24. 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G.; Giguere P. M.; Katritch V.; Huang X. P.; Thompson A. A.; Cherezov V.; Roth B. L.; Stevens R. C. Molecular Control of Delta-Opioid Receptor Signalling. Nature 2014, 506, 191–196. 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal Structure of the Micro-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S.; Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Weis W. I.; Kobilka B. K. Structure of the Delta-Opioid Receptor Bound to Naltrindole. Nature 2012, 485, 400–404. 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Wacker D.; Mileni M.; Katritch V.; Han G. W.; Vardy E.; Liu W.; Thompson A. A.; Huang X. P.; Carroll F. I.; Mascarella S. W.; Westkaemper R. B.; Mosier P. D.; Roth B. L.; Cherezov V.; Stevens R. C. Structure of the Human Kappa-Opioid Receptor in Complex with JDTIC. Nature 2012, 485, 327–332. 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W. J.; Manglik A.; Venkatakrishnan A. J.; Laeremans T.; Feinberg E. N.; Sanborn A. L.; Kato H. E.; Livingston K. E.; Thorsen T. S.; Kling R. C.; Granier S.; Gmeiner P.; Husbands S. M.; Traynor J. R.; Weis W. I.; Steyaert J.; Dror R. O.; Kobilka B. K. Structural Insights into Mu-Opioid Receptor Activation. Nature 2015, 524, 315. 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H.. [19] Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. Methods in Neurosciences; Elsevier, 1995; Vol. 25, pp 366–428. [Google Scholar]
- Pándy-Szekeres G.; Munk C.; Tsonkov T. M.; Mordalski S.; Harpsoe K.; Hauser A. S.; Bojarski A. J.; Gloriam D. E. GPCRdb in 2018: Adding GPCR Structure Models and Ligands. Nucleic Acids Res. 2018, 46, D440–D446. 10.1093/nar/gkx1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A.; Hu H.; Maeda S.; Zhang Y.; Qu Q.; Paggi J. M.; Latorraca N. R.; Hilger D.; Dawson R.; Matile H.; Schertler G. F. X.; Granier S.; Weis W. I.; Dror R. O.; Manglik A.; Skiniotis G.; Kobilka B. K. Structure of the Micro-Opioid Receptor-Gi Protein Complex. Nature 2018, 558, 547–552. 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conibear A. E.; Kelly E. A Biased View of Mu-Opioid Receptors?. Mol. Pharmacol. 2019, 96, 542–549. 10.1124/mol.119.115956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis A.; Kliewer A.; Kelly E.; Henderson G.; Christie M. J.; Schulz S.; Canals M. Critical Assessment of G Protein-Biased Agonism at the Mu-Opioid Receptor. Trends Pharmacol. Sci. 2020, 41, 947–959. 10.1016/j.tips.2020.09.009. [DOI] [PubMed] [Google Scholar]
- Herenbrink C. K.; Sykes D. A.; Donthamsetti P.; Canals M.; Coudrat T.; Shonberg J.; Scammells P. J.; Capuano B.; Sexton P. M.; Charlton S. J.; Javitch J. A.; Christopoulos A.; Lane J. R. The Role of Kinetic Context in Apparent Biased Agonism at Gpcrs. Nat. Commun. 2016, 7, 10842 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy S. K.; Drake M. T.; Nelson C. D.; Houtz D. A.; Xiao K. H.; Madabushi S.; Reiter E.; Premont R. T.; Lichtarge O.; Lefkowitz R. J. β-Arrestin-dependent Protein-Independent Erk1/2 Activation by the Beta 2 Adrenergic Receptor. J. Biol. Chem. 2006, 281, 1261–1273. 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Ahn S. K.; Shenoy S. K.; Wei H. J.; Lefkowitz R. J. Differential Kinetic and Spatial Patterns of Beta-Arrestin and G Protein-Mediated Erk Activation by the Angiotensin Ii Receptor. J. Biol. Chem. 2004, 279, 35518–35525. 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Dror R. O.; Arlow D. H.; Maragakis P.; Mildorf T. J.; Pan A. C.; Xu H.; Borhani D. W.; Shaw D. E. Activation Mechanism of the Beta2-Adrenergic Receptor. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 18684–18689. 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R.; Zou Y.; Dror R. O.; Mildorf T. J.; Arlow D. H.; Manglik A.; Pan A. C.; Liu C. W.; Fung J. J.; Bokoch M. P.; Thian F. S.; Kobilka T. S.; Shaw D. E.; Mueller L.; Prosser R. S.; Kobilka B. K. The Dynamic Process of Beta(2)-Adrenergic Receptor Activation. Cell 2013, 152, 532–542. 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy J. D.; Butler K. V.; Kelly B.; Rechsteiner K.; Karpiak J.; Betz R. M.; Kormos B. L.; Shoichet B. K.; Dror R. O.; Jin J.; Roth B. L. Structure-Inspired Design of Beta-Arrestin-Biased Ligands for Aminergic Gpcrs. Nat. Chem. Biol. 2018, 14, 126–134. 10.1038/nchembio.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth S. A.; Kelly B.; Valant C.; Michaelis J. A.; Mastromihalis O.; Thompson G.; Venkatakrishnan A. J.; Hertig S.; Scammells P. J.; Sexton P. M.; Felder C. C.; Christopoulos A.; Dror R. O. Cryptic Pocket Formation Underlies Allosteric Modulator Selectivity at Muscarinic GPCRs. Nat. Commun. 2019, 10, 3289 10.1038/s41467-019-11062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Masureel M.; Qu Q.; Janetzko J.; Inoue A.; Kato H. E.; Robertson M. J.; Nguyen K. C.; Glenn J. S.; Skiniotis G.; Kobilka B. K. Structure of the Neurotensin Receptor 1 in Complex with Beta-Arrestin 1. Nature 2020, 579, 303–308. 10.1038/s41586-020-1953-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus D. P.; Hu H.; Robertson M. J.; Kleinhenz A. L. W.; Wingler L. M.; Capel W. D.; Latorraca N. R.; Lefkowitz R. J.; Skiniotis G. Structure of the M2 Muscarinic Receptor-Beta-Arrestin Complex in a Lipid Nanodisc. Nature 2020, 579, 297–302. 10.1038/s41586-020-1954-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winpenny D.; Clark M.; Cawkill D. Biased Ligand Quantification in Drug Discovery: From Theory to High Throughput Screening to Identify New Biased Mu Opioid Receptor Agonists. Br. J. Pharmacol. 2016, 173, 1393–1403. 10.1111/bph.13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickolls S. A.; Humphreys S.; Clark M.; McMurray G. Co-Expression of GRK2 Reveals a Novel Conformational State of the Micro-Opioid Receptor. PLoS One 2013, 8, e83691 10.1371/journal.pone.0083691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize M. A.; Lomize A. L.; Pogozheva I. D.; Mosberg H. I. OPM: Orientations of Proteins in Membranes Database. Bioinformatics 2006, 22, 623–625. 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Betz R. M. Dabble, 2.7.3, 2017. (http://doi.org/10.5281/zenodo.836914).
- Best R. B.; Mittal J.; Feig M.; MacKerell A. D. Inclusion of Many-Body Effects in the Additive Charmm Protein Cmap Potential Results in Enhanced Cooperativity of Alpha-Helix and Beta-Hairpin Formation. Biophys. J. 2012, 103, 1045–1051. 10.1016/j.bpj.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E. M.; Mittal J.; Feig M.; MacKerell A. D. Optimization of the Additive Charmm All-Atom Protein Force Field Targeting Improved Sampling of the Backbone Phi, Psi and Side-Chain Chi(1) and Chi(2) Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. W.; Zhao Y. Q.; Wang Y. J.; Huang J. F. Molecular Dynamics Simulations and Statistical Coupling Analysis Reveal Functional Coevolution Network of Oncogenic Mutations in the Cdkn2a-Cdk6 Complex. FEBS Lett. 2013, 587, 136–141. 10.1016/j.febslet.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. Update of the Charmm All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. Automation of the CHARMM General Force Field (CGenFF) Ii: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. Automation of the Charmm General Force Field (Cgenff) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; MacKerell A. D. Charmm General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Shen N.; Polani N. K.; Fan Y.; Ghosh J.; Herath C.; Marru S.; Pierce M.; Pamidighantam S. V.; Sheetz M.; MacKerell A. D.. Paramchem Force Field Parametrization Engine: Initial Guess Generation and Dihedral Parameter Optimization. Abstracts of Papers of the American Chemical Society; American Chemical Society, 2012; Vol. 244. [Google Scholar]
- Rasmussen S. G.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; Mathiesen J. M.; Shah S. T.; Lyons J. A.; Caffrey M.; Gellman S. H.; Steyaert J.; Skiniotis G.; Weis W. I.; Sunahara R. K.; Kobilka B. K. Crystal Structure of the Beta2 Adrenergic Receptor-Gs Protein Complex. Nature 2011, 477, 549–555. 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Gotz A. W.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with Amber on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- Hopkins C. W.; Le Grand S.; Walker R. C.; Roitberg A. E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. 10.1021/ct5010406. [DOI] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. Ptraj and Cpptraj: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.