

Abstract
Oleanolic acid (OA) is a natural cosmeceutical compound with various skin beneficial activities including inhibitory effect on hyaluronidase but the anti-hyaluronidase activity and mechanisms of action of its synthetic analogues remain unclear. Herein, a series of OA derivatives were synthesised and evaluated for their inhibitory effects on hyaluronidase. Compared to OA, an induction of fluorinated (6c) and chlorinated (6g) indole moieties led to enhanced anti-hyaluronidase activity (IC50 = 80.3 vs. 9.97 and 9.57 µg/mL, respectively). Furthermore, spectroscopic and computational studies revealed that 6c and 6g can bind to hyaluronidase protein and alter its secondary structure leading to reduced enzyme activity. In addition, OA indole derivatives showed feasible skin permeability in a slightly acidic environment (pH = 6.5) and 6c exerted skin protective effect by reducing cellular reactive oxygen species in human skin keratinocytes. Findings from the current study support that OA indole derivatives are potential cosmeceuticals with anti-hyaluronidase activity.
Keywords: Oleanolic acid, indole, hyaluronidase, skin permeability, skin protection
Graphical Abstract

Highlights
A series of novel oleanolic acid (OA) derivatives are synthesised
OA indole derivatives 6a-6g show potent inhibition on hyaluronidase activity
6c and 6g interacts with hyaluronidase protein to reduce enzyme activity
6c and 6g are skin permeable at pH 6.5 and 6c shows antioxidant effect in human skin keratinocytes
Introduction
Oleanolic acid (OA) is a pentacyclic triterpenoid that is ubiquitous in the plant kingdom. It is a bioactive component of numerous botanical extracts that are commonly used as cosmeceutical ingredients in various skincare products. The skin beneficial effects of OA are supported by several biological studies showing that it may exert promising anti-inflammatory, antioxidant, and anti-skin-ageing activities1–3. It also has been reported that OA’s anti-skin-ageing effects may be attributed to its inhibitory effect on several skin wrinkle formation related enzymes such as hyaluronidase (HAase)4, which is a key enzyme for the metabolism of hyaluronic acid (also known as hyaluronan; HA). HAase catalyses the reaction of degradation of HA to form monosaccharides by cleaving its glycosidic bonds5. HAases can be categorised into different types by their origins and mechanism of action. For instance, mammalian produced HAases, also termed as the endo-β-N-acetylhexosaminidases, can break down the β-1,4-glycosidic linkages of HA to form tetrasaccharides, whereas HAases produced by worms (i.e. leech) are endo-β-D-glucuronidases, which catalyses the cleavage of the β-1,3-glycosidic bonds to release pentasaccharides and hexasaccharides6. In addition, the types of HAases, including acidic ones (activated in acidic environment; pH of 3 to 4) and neutral ones (biologically functional at pH of 5 to 8), are varied by their catalytic activity in different pH environment6. HA is one of the major components that comprise the skin extracellular matrix (ECM) system, which is a three-dimensional network with crucial physiological functions such as maintaining the skin structure and retaining water molecules7. Published dermatological studies suggest that abnormal hyperactivity of HAase may lead to exacerbated degradation of skin epidermal HA, which is a histochemical hallmark for aged skin8. The HA-HAase homeostasis is a crucial factor that mediates a variety of skin ageing related physiological events including ECM structural integrity, wrinkle formation, and skin moisture retention9,10. Thus, HAase inhibitors are considered as promising cosmeceuticals with potential anti-skin-ageing effects, thereby attracted considerable amount of research interest11–13.
Several biochemical based assays have been developed to evaluate the inhibitory effects of small molecules on the activity of HAase. Viscosimetry assays measuring the ‘half viscosity time’ or ‘half time of the substrate’ were first used to evaluate HAase activity14. More advanced analytical methods, such as a spectrophotometric HAase assay measuring the undegraded HAase substrate15, were developed to accurately assess HAase activity. Moreover, the development of microplate based enzymatic HAase assays enabled the measurement of HAase activity at a larger scale, which led to the discovery of numerous HAase inhibitors16,17. Among these inhibitors, phytochemicals from medicinal plants, including polyphenols, terpenes (including monoterpenes, diterpenes, sesquiterpenes, and triterpenes) and their glycosides, showed promising anti-HAase effects with different mechanisms of action6,18,19. Notably, pentacyclic triterpenoids, such as OA and ursolic acid (UA), have been identified as active HAase inhibitors and are often used as the positive controls in anti-HAase enzymatic assays2,4. In addition, a series of pentacyclic triterpenoid analogues with an UA skeleton were chemically synthesised and a quantitative structure activity relationship study showed that the modifications (e.g. acetylation and methylation) of UA can result in enhanced inhibitory effects on HAase20. Moreover, augmented anti-HAase activity of a group of carboxamide derivatives via the induction of indole moieties has been reported21. However, to date, it is unclear whether enhanced anti-HAase activity can be achieved by the induction of indole groups to the OA skeleton.
In light of our laboratory’s research interest to study the potential skin beneficial effects of HAase inhibitors, herein, we aimed to: 1) synthesise a series of OA derivatives with an indole moiety and evaluate their inhibition against bovine testicular HAase; 2) study the leading compounds’ mechanisms of inhibition using biophysical and computational methods; 3) evaluate OA indole derivatives’ cosmeceutical properties including skin permeability and antioxidant activity in human skin cells.
Materials and methods
Chemicals and reagents
Hydrogen peroxide (H2O2) solution, dimethyl sulfoxide (DMSO), bovine serum albumin (BSA), HAase from bovine testes (type IV-S; CAS number: 37326–33-3), and hyaluronic acid from rooster comb (mw from 1000000 to 4000000 Da; CAS Number: 9067–32-7) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Oleanolic acid (OA), penylhydrazine hydrochloride, aldehydes, selectflour, and potassium hydroxide were obtained from Macklin Co., Ltd. (Shanghai, China; purity > 98%). All other solvents used in this study were purchased from Aladdin Chemical (Shanghai, China). The Jones reagent was prepared with a method as previously reported22.
Spectrometer
1H NMR and 13 C NMR spectra were recorded on a Bruker DPX-500 MHz spectrometer (Bruker Biospin, Rheinstetten, German) with chemical shifts (d) given in parts per million (ppm) with trimethylsilane (TMS) as an internal standard. High-resolution mass spectra (HRMS) were obtained on a Thermo Scientific Q Exactive UHMR (ultra-high mass range) hybrid quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific; Waltham, MA, USA).
General procedure for the preparation of OA analogues
Synthesis of oleanolic acid analogue 2
OA (500 mg, 1.1 mmol) was added into acetone (20 mL) and stirred at 0 °C until it was completely dissolved and then the Jones reagent (5 mL) was slowly added into the solution and stirred until the colour of the mixture turned yellow for 5 min. The reaction was monitored by the thin layer chromatography (TLC) and acetone in the reaction mixture was removed under reduced pressure after the reaction completed. The remaining reactants were dissolved in ethyl acetate and washed with deionised water, and then dried over anhydrous magnesium sulphate. Next, the crude product was purified on a silica gel column with petroleum ether/ethyl acetate (v/v = 12:1, Rf = 0.3) to obtain the intermediate analogue 2 with a yield of 80.6% (white solid). 1H NMR (500 MHz, CDCl3) δ 5.23 (t, J = 3.5 Hz, 1H), 2.77 (dd, J = 13.7, 4.1 Hz, 1H), 2.53–2.43 (m, 1H), 2.34–2.26 (m, 1H), 1.96–1.87 (m, 2H), 1.87–1.78 (m, 2H), 1.75–1.61 (m, 3H), 1.60–1.49 (m, 5H), 1.43–1.39 (m, 3H), 1.30–1.23 (m, 4H), 1.19–1.13 (m, 2H), 1.08 (s, 3H), 1.01 (s, 3H), 0.98 (s, 3H), 0.96 (s, 3H), 0.86 (s, 3H), 0.84 (s, 3H), 0.74 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 217.75, 183.75, 143.65, 122.40, 55.31, 47.45, 46.89, 46.58, 45.82, 41.73, 41.04, 39.27, 39.10, 36.81, 34.15, 33.80, 33.06, 32.40, 32.16, 30.68, 27.68, 26.45, 25.84, 23.56, 23.49, 22.91, 21.45, 19.55, 17.01, 15.02. HRMS (ESI-MS) m/z: [M + Na]+ calcd for C30H46NaO3: 477.3339; found: 477.3342.
Synthesis of oleanolic acid analogue 3
OA (500 mg, 1.1 mmol) and selectfluor (tetrafluoroborate, 3.3 mmol) were dissolved in the mixed solution of dioxane (4 mL) and nitromethane (6 mL), and stirred at 80 °C for 4 h monitored by the TLC. The reaction mixture was concentrated under reduced pressure and then extracted with ethyl acetate. The mixture was then washed with deionised water and dried with anhydrous magnesium sulphate. Next, the crude product was purified on a silica gel column with petroleum ether/ethyl acetate (v/v = 8:1, Rf=0.3) to obtain the intermediate analogue 3 as white solid with a yield of 50.3%. 1H NMR (500 MHz, CDCl3) δ 4.58 (dt, J = 46.6, 2.6 Hz, 1H), 3.24 (dd, J = 11.6, 4.6 Hz, 1H), 2.20–2.07 (m, 2H), 2.03–1.91 (m, 2H), 1.91–1.81 (m, 2H), 1.77–1.70 (m, 2H), 1.70–1.63 (m, 3H), 1.63–1.52 (m, 5H), 1.51–1.35 (m, 3H), 1.35–1.26 (m, 3H), 1.25 (s, 3H), 1.21 (dd, J = 14.1, 6.5 Hz, 1H), 1.13 (s, 3H), 1.01 (d, J = 2.6 Hz, 6H), 0.92 (s, 3H), 0.89 (s, 3H), 0.79 (s, 3H), 0.78–0.73 (m, 1H). 13 C NMR (151 MHz, CDCl3) δ 179.44, 97.37, 96.01,88.04, 78.74, 55.00, 50.87, 44.67, 44.35, 41.93, 41.71f, 38.89, 38.54, 36.41, 34.09, 33.52, 33.22, 31.52, 27.99, 27.47, 27.36, 27.18, 25.73, 23.80, 21.07, 18.32, 18.03, 17.69, 16.07, 15.38. HRMS (ESI-MS) m/z: [M + Na]+ calcd for C30H47FNaO3: 497.3401; found: 497.3405.
Synthesis of oleanolic acid analogues 4
Analogue 3 (500 mg, 1.1 mmol) in acetone (20 mL) was dissolved and stirred at 0 °C followed by adding the Jones reagent (5 mL) and stirring until the mixture turned yellow for 5 min and monitored by the TLC. The reaction mixture was then removed under reduced pressure and the remainder was dissolved in ethyl acetate and washed with deionised water. After drying the extract over anhydrous magnesium sulphate, it was purified on a silica gel column with petroleum ether/ethyl acetate (v/v = 12:1, Rf=0.3) to obtain the intermediate 4 as white solid with a yield of 69.6%. 1H NMR (500 MHz, CDCl3) δ 4.60 (dt, J = 46.6, 2.6 Hz, 1H), 2.59–2.50 (m, 1H), 2.47 (tt, J = 11.8, 3.6 Hz, 1H), 2.30–2.14 (m, 1H), 2.14–2.09 (m, 1H), 2.09–2.01 (m, 1H), 2.00–1.91 (m, 3H), 1.91–1.79 (m, 2H), 1.78–1.72 (m, 1H), 1.72–1.62 (m, 4H), 1.62–1.53 (m, 2H), 1.53–1.47 (m, 2H), 1.45–1.37 (m, 2H), 1.36–1.31 (m, 2H), 1.27 (s, 3H), 1.24 (dd, J = 9.3, 4.5 Hz, 1H), 1.18 (s, 2H), 1.12 (s, 3H), 1.06 (s, 3H), 1.04–0.99 (m, 6H), 0.92 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 179.44, 96.69, 88.04, 78.74, 55.00, 50.87, 44.67, 44.35, 41.93, 41.71, 41.70, 38.89, 38.54, 36.41, 34.09, 33.52, 33.22, 31.52, 27.99, 27.47, 27.36, 27.18, 25.73, 23.80, 21.07, 18.32, 18.01, 17.82, 16.07, 15.38. HRMS (ESI-MS) m/z: [M + Na]+ calcd for C30H45FNaO3: 495.3245; found: 495.3245.
Synthesis of oleanolic acid analogues 5a-5c
Analogue 4 (0.2 mmol) and substituted phenylhydrazines (0.4 mmol) were dissolved in acetic acid with refluxing at 110 °C overnight. The reaction was monitored by TLC and the reaction mixture was concentrated under reduced pressure. The remainder was dissolved in ethyl acetate and the organic layer was washed with deionised water, and then dried over anhydrous magnesium sulphate. The crude product was purified on a silica gel column with petroleum ether/ethyl acetate (v/v = 20:1–36:1) to obtain intermediate analogues 5a-5c.
Analogue 5a: followed by aforementioned general procedures, 5a was prepared via the Fischer indole synthesis of 4 with 4-(trifluoromethoxy) phenylhydrazine hydrochloride and purified with petroleum ether/ethyl acetate (v/v = 20:1, Rf = 0.50); light yellow solid; yield: 53.2%. 1H NMR (500 MHz, CDCl3) δ 4.58 (dt, J = 46.6, 2.6 Hz, 1H), 3.24 (dd, J = 11.6, 4.6 Hz, 1H), 2.20–2.07 (m, 2H), 2.03–1.91 (m, 2H), 1.91–1.81 (m, 2H), 1.77–1.70 (m, 2H), 1.70–1.63 (m, 3H), 1.63–1.52 (m, 5H), 1.51–1.35 (m, 3H), 1.35–1.26 (m, 3H), 1.25 (s, 3H), 1.21 (dd, J = 14.1, 6.5 Hz, 1H), 1.13 (s, 3H), 1.01 (d, J = 2.6 Hz, 6H), 0.92 (s, 3H), 0.89 (s, 3H), 0.79 (s, 3H), 0.78–0.73 (m, 1H). 13 C NMR (151 MHz, CDCl3) δ 179.47, 142.89, 142.63, 134.41, 128.40, 120.85, 114.93, 110.78, 110.69, 107.13, 96.57, 88.13, 52.91, 51.05, 44.44, 43.43, 41.91, 38.69, 37.59, 36.94, 34.30, 34.14, 33.23, 32.82, 31.57, 30.86, 29.73, 27.62, 27.40, 26.12, 23.78, 23.18, 21.13, 18.58, 17.82, 17.77, 16.40. HRMS (ESI-MS) m/z: [M + H]+ calcd for C37H48F4NO3: 630.3565; found: 630.3567.
Analogue 5b: followed by aforementioned general procedures, 5b was prepared via the Fischer indole synthesis of 4 with 4-fluorophenylhydrazine hydrochloride and purified with petroleum ether/ethyl acetate (v/v = 20:1, Rf = 0.35); light yellow solid; yield: 64.3%. 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 7.23 (d, J = 7.8 Hz, 1H), 7.00 (td, J = 7.9, 4.8 Hz, 1H), 6.87 (ddd, J = 11.3, 7.8, 0.6 Hz, 1H), 4.69 (dt, J = 46.8, 2.4 Hz, 1H), 2.89 (t, J = 14.6 Hz, 1H), 2.25 (d, J = 15.2 Hz, 1H), 2.23–2.09 (m, 3H), 2.09–1.97 (m, 2H), 1.97–1.88 (m, 2H), 1.80 (dd, J = 13.0, 1.7 Hz, 1H), 1.72–1.64 (m, 5H), 1.49–1.40 (m, 3H), 1.36 (s, 3H), 1.33–1.31 (m, 4H), 1.28 (s, 2H), 1.26 (s, 3H), 1.24 (s, 3H), 1.03 (s, 3H), 0.96 (s, 3H), 0.94 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 179.44, 149.30, 141.58, 131.91, 124.01, 119.23, 113.82, 107.40, 106.25, 96.63, 88.11, 52.96, 51.05, 44.44, 43.44, 41.95, 41.85, 38.71, 37.61, 37.20, 34.29, 34.13, 33.24, 32.84, 31.57, 30.89, 29.73, 27.62, 27.40, 26.12, 23.79, 23.20, 21.13, 18.58, 17.82, 17.81, 16.39. HRMS (ESI-MS) m/z: [M + H]+ calcd for C36H48F2NO2: 564.3648; found: 564.3651.
Analogue 5c: followed by aforementioned general procedures, 5c was prepared via the Fischer indole synthesis of 4 with 2-chlorophenylhydrazine hydrochloride and purified with petroleum ether/ethyl acetate (v/v = 32:1, Rf = 0.49); light yellow solid; yield: 57.3%. 1H NMR (500 MHz, CDCl3) δ 7.93 (s, 1H), 7.37 (d, J = 7.7 Hz, 1H), 7.14 (dd, J = 7.6, 0.8 Hz, 1H), 7.03 (t, J = 7.7 Hz, 1H), 4.69 (dt, J = 46.3, 2.4 Hz, 1H), 2.86 (d, J = 14.9 Hz, 1H), 2.25 (d, J = 15.0 Hz, 1H), 2.23–2.14 (m, 2H), 2.14–2.04 (m, 2H), 2.04–1.88 (m, 3H), 1.83–1.77 (m, 1H), 1.72–1.61 (m, 5H), 1.49–1.39 (m, 3H), 1.38–1.34 (m, 4H), 1.32 (s, 3H), 1.30–1.26 (m, 5H), 1.24 (s, 3H), 1.03 (s, 3H), 0.96 (s, 3H), 0.94 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 179.55, 141.69, 133.38, 129.78, 120.65, 119.94, 116.76, 116.08, 107.86, 96.75, 88.22, 53.08, 51.17, 44.55, 43.55, 42.06, 41.99, 38.83, 37.70, 37.29, 34.41, 34.25, 33.35, 32.95, 31.69, 30.99, 27.73, 27.51, 26.24, 23.90, 23.29, 21.24, 18.70, 17.935, 17.930, 16.49. HRMS (ESI-MS) m/z: [M + H]+ calcd for C36H48FClNO2: 580.3352; found: 580.3354.
Synthesis of oleanolic acid analogues 6a-6g
The indole ring substituted analogues 6a-6g were synthesised by employing substituted phenylhydrazine in the Fischer indolization. Analogue 2 (0.2 mmol) and substituted phenylhydrazines (0.4 mmol) were dissolved in acetic acid with refluxing at 110 °C overnight. The reaction was monitored by the TLC. After the reaction completed, acetic acid in the reaction mixture was removed under reduced pressure followed by dissolving the remainder in ethyl acetate. The organic layer was washed with deionised water and dried over anhydrous magnesium sulphate. The crude product was separated by preparative silica gel chromatography in petroleum ether/ethyl acetate (7:1 to 12:1).
Analogue 6a: followed by aforementioned general procedures, 6a was prepared via the Fischer indole synthesis of 2 with 2-nitrophenylhydrazine hydrochloride and purified with petroleum ether/ethyl acetate (v/v = 8:1, Rf = 0.33); light red solid; yield: 30.2%. 1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 2.2 Hz, 1H), 8.25 (s, 1H), 8.03 (dd, J = 8.9, 2.3 Hz, 1H), 7.29 (d, J = 8.9 Hz, 1H), 5.41 (t, J = 3.6 Hz, 1H), 2.90 (dd, J = 13.0, 3.9 Hz, 1H), 2.80 (d, J = 15.1 Hz, 1H), 2.22 (d, J = 15.2 Hz, 1H), 2.14–2.08 (m, 2H), 2.01 (td, J = 13.6, 4.1 Hz, 1H), 1.82 (ddd, J = 26.9, 12.3, 5.9 Hz, 3H), 1.71–1.59 (m, 2H), 1.63–1.40 (m, 4H), 1.39–1.32 (m, 1H), 1.32 (s, 3H), 1.30–1.21 (m, 3H), 1.19 (d, J = 2.3 Hz, 6H), 1.18–1.13 (m, 1H), 0.96 (s, 3H), 0.94 (s, 3H), 0.92 (s, 3H), 0.86 (s, 3H), 0.92–0.81 (m, 1H). 13 C NMR (151 MHz, CDCl3) δ 184.25, 144.38, 143.38, 141.22, 139.40, 127.82, 122.73, 116.97, 115.34, 110.13, 109.51, 53.03, 46.67, 46.37, 45.80, 41.80, 41.12, 39.43, 38.11, 36.40, 34.12, 33.85, 33.10, 32.48, 32.06, 30.73, 30.70, 27.74, 25.82, 23.57, 23.41, 23.13, 22.92, 19.25, 16.82, 15.55. HRMS spectrum of (compound 6a). HRMS (ESI-MS) m/z: [M-H]- calcd for C36H47N2O4: 571.3541; found: 571.3514.
Analogue 6b: followed by aforementioned general procedures, 6b was prepared via the Fischer indole synthesis of 2 with 2-fluorophenylhydrazine and purified with petroleum ether/ethyl acetate (v/v = 7:1, Rf = 0.35); light yellow solid; yield: 59.1%. 1H NMR (500 MHz, CDCl3) δ 7.88 (s, 1H), 7.19 (d, J = 7.8 Hz, 1H), 6.97 (td, J = 7.8, 4.6 Hz, 1H), 6.83 (dd, J = 11.2, 7.8 Hz, 1H), 5.57–5.29 (m, 1H), 2.94–2.85 (m, 1H), 2.76 (d, J = 14.9 Hz, 1H), 2.20 (d, J = 15.1 Hz, 1H), 2.14–2.08 (m, 2H), 2.01 (td, J = 13.6, 4.0 Hz, 1H), 1.88–1.73 (m, 3H), 1.71–1.48 (m, 5H), 1.46–1.32 (m, 3H), 1.31 (s, 3H), 1.29–1.20 (m, 3H), 1.19 (s, 6H), 1.16–1.11 (m, 1H), 0.96 (d, J = 2.2 Hz, 6H), 0.92 (s, 3H), 0.86 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 184.30, 150.29, 143.42, 141.75, 132.04, 124.04, 122.83, 119.17, 113.80, 107.82, 106.20, 53.14, 46.70, 46.39, 45.87, 41.82, 41.13, 39.43, 38.15, 36.87, 34.07, 33.85, 33.10, 32.48, 32.13, 30.96, 30.72, 27.76, 25.82, 23.59, 23.45, 23.32, 22.96, 19.25, 16.90, 15.54. HRMS (ESI-MS) m/z: [M-H]- calcd for C36H47FNO2: 544.3596; found: 544.3573.
Analogue 6c: followed by aforementioned general procedures, 6c was prepared via the Fischer indole synthesis of 2 with 2,4-difluorophenylhydrazine hydrochloride and purified with petroleum ether/ethyl acetate (v/v = 8:1, Rf = 0.35); white solid; yield: 53.9%. 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 10.7 Hz, 1H), 6.89 (dd, J = 9.0, 2.2 Hz, 1H), 6.67 (ddd, J = 11.4, 9.5, 2.2 Hz, 1H), 5.55–5.30 (m, 1H), 2.90 (dd, J = 14.0, 4.6 Hz, 1H), 2.69 (d, J = 15.0 Hz, 1H), 2.18 (d, J = 15.2 Hz, 1H), 2.14–2.07 (m, 2H), 2.03 (td, J = 13.6, 4.1 Hz, 1H), 1.81 (ddt, J = 24.6, 17.7, 10.4 Hz, 3H), 1.72–1.49 (m, 6H), 1.46–1.35 (m, 3H), 1.33 (d, J = 6.7 Hz, 3H), 1.30–1.22 (m, 3H), 1.21 (d, J = 3.3 Hz, 6H), 0.99–0.91 (m, 9H), 0.88 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 184.71, 157.58, 155.71, 149.09, 147.14, 143.43, 130.87, 122.76, 120.51, 108.18, 98.88, 95.89, 53.07, 46.71, 46.37, 45.86, 41.81, 41.10, 39.42, 38.14, 36.74, 34.18, 33.86, 33.11, 32.49, 32.10, 30.89, 30.72, 27.76, 25.81, 23.59, 23.42, 23.32, 22.93, 19.24, 16.89, 15.52. HRMS (ESI-MS) m/z: [M-H]- calcd for C36H46F2NO2: 562.3502; found: 562.3478.
Analogue 6d: followed by aforementioned general procedures, analogue 6d was prepared via the Fischer indole synthesis of 2 with 2,4-dimethylphenylhydrazine hydrochloride and purified with petroleum ether/ethyl acetate (v/v = 12:1, Rf = 0.30); white solid; yield: 71.1%. 1H NMR (500 MHz, CDCl3) δ 7.45 (s, 1H), 7.08 (s, 1H), 6.77 (s, 1H), 5.40 (d, J = 3.6 Hz, 1H), 2.89 (dd, J = 14.1, 4.5 Hz, 1H), 2.73 (d, J = 14.9 Hz, 1H), 2.43 (d, J = 16.0 Hz, 6H), 2.17 (d, J = 14.9 Hz, 1H), 2.14–2.06 (m, 2H), 2.01 (td, J = 13.5, 3.8 Hz, 1H), 1.87–1.72 (m, 3H), 1.72–1.45 (m, 5H), 1.45–1.36 (m, 3H), 1.28 (d, J = 16.2 Hz, 7H), 1.18 (d, J = 7.6 Hz, 6H), 1.01–0.90 (m, 9H), 0.86 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 184.00, 143.35, 140.75, 133.83, 128.42, 128.05, 123.36, 122.97, 119.13, 115.46, 107.01, 53.27, 46.69, 46.41, 45.88, 41.83, 41.14, 39.43, 38.11, 36.91, 34.03, 33.87, 33.11, 32.51, 32.18, 31.03, 30.73, 27.77, 25.84, 23.59, 23.46, 23.37, 22.98, 21.41, 19.30, 16.91, 16.71, 15.51. HRMS (ESI-MS) m/z: [M-H]- calcd for C38H52NO2: 554.4004; found: 544.3978.
Analogue 6e: followed by aforementioned general procedures, 6e was prepared via the Fischer indole synthesis of 2 with 4-bromophenylhydrazine hydrochloride and purified with petroleum ether/ethyl acetate (v/v = 8:1, Rf = 0.35); white solid; yield: 40.8%. 1H NMR (500 MHz, CDCl3) δ 7.76 (s, 1H), 7.54 (d, J = 1.9 Hz, 1H), 7.18 (dd, J = 8.5, 1.8 Hz, 1H), 5.40 (t, J = 3.6 Hz, 1H), 2.96–2.83 (m, 1H), 2.70 (d, J = 14.9 Hz, 1H), 2.15 (d, J = 15.0 Hz, 1H), 2.13–2.04 (m, 2H), 2.01 (td, J = 13.6, 2.9 Hz, 1H), 1.88–1.67 (m, 3H), 1.68–1.46 (m, 5H), 1.44–1.34 (m, 2H), 1.30–1.21 (m, 9H), 1.18 (d, J = 9.4 Hz, 6H), 0.95 (s, 3H), 0.92 (s, 6H), 0.85 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 184.37, 143.27, 141.53, 134.52, 129.33, 123.17, 122.62, 119.97, 117.01, 108.14, 104.02, 52.95, 46.52, 46.20, 45.68, 41.63, 40.91, 39.24, 37.93, 36.69, 33.90, 33.67, 32.94, 32.31, 31.93, 30.76, 30.55, 27.57, 25.65, 23.42, 23.26, 23.14, 22.76, 19.06, 16.74, 15.35. HRMS (ESI-MS) m/z: [M-H]- calcd for C36H47BrNO2: 604.2796; found: 604.2770.
Analogue 6f: followed by aforementioned general procedures, 6f was prepared via the Fischer indole synthesis of 2 with 2-bromophenylhydrazine hydrochloride and purified with petroleum ether/ethyl acetate (v/v = 8:1, Rf = 0.35); white solid; yield: 47.7%. 1H NMR (500 MHz, CDCl3) δ 7.83 (s, 1H), 7.38 (d, J = 7.7 Hz, 1H), 6.97 (t, J = 7.7 Hz, 1H), 5.42 (t, J = 3.7 Hz, 1H), 2.90 (dd, J = 13.8, 4.7 Hz, 1H), 2.76 (d, J = 15.0 Hz, 1H), 2.22 (d, J = 15.1 Hz, 1H), 2.16–2.10 (m, 2H), 2.06–1.98 (m, 1H), 1.90–1.74 (m, 3H), 1.72–1.50 (m, 6H), 1.43 (dt, J = 11.4, 2.4 Hz, 2H), 1.34 (s, 3H), 1.28 (t, J = 3.6 Hz, 2H), 1.26–1.16 (m, 9H), 0.97 (d, J = 3.3 Hz, 6H), 0.94 (s, 3H), 0.88 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 184.55, 143.45, 141.71, 134.70, 129.51, 123.35, 122.80, 120.15, 117.19, 108.32, 104.21, 53.13, 46.70, 46.38, 45.86, 41.81, 41.09, 39.42, 38.11, 36.87, 34.08, 33.85, 33.12, 32.49, 32.11, 30.95, 30.73, 27.75, 25.83, 23.60, 23.45, 23.32, 22.94, 19.24, 16.92, 15.54. HRMS (ESI-MS) m/z: [M-H]- calcd for C36H47BrNO2: 604.2796; found: 604.2772.
Analogue 6g: followed by aforementioned general procedures, 6g was prepared via the Fischer indole synthesis of 2 with 4-chlorophenylhydrazine hydrochloride and purified with petroleum ether/ethyl acetate (v/v = 7:1, Rf = 0.30); white solid; yield: 42.3%. 1H NMR (500 MHz, CDCl3) δ 7.86 (s, 1H), 7.32 (d, J = 7.8 Hz, 1H), 7.11 (dd, J = 7.6, 1.0 Hz, 1H), 6.99 (t, J = 7.7 Hz, 1H), 5.39 (t, J = 3.7 Hz, 1H), 2.88 (dd, J = 13.9, 4.6 Hz, 1H), 2.75 (d, J = 15.0 Hz, 1H), 2.20 (d, J = 15.1 Hz, 1H), 2.15–2.07 (m, 2H), 2.01 (td, J = 13.6, 4.0 Hz, 1H), 1.87–1.75 (m, 3H), 1.68–1.47 (m, 6H), 1.45–1.39 (m, 2H), 1.32 (s, 3H), 1.26–1.25 (m, 1H), 1.24–1.12 (m, 9H), 0.95 (d, J = 2.8 Hz, 6H), 0.92 (s, 3H), 0.86 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 183.86, 143.45, 141.76, 133.25, 129.76, 122.80, 120.42, 119.74, 116.60, 115.95, 108.16, 53.15, 46.67, 46.38, 45.88, 41.83, 41.14, 39.43, 38.12, 36.84, 34.08, 33.85, 33.10, 32.48, 32.13, 30.95, 30.72, 27.76, 25.81, 23.59, 23.45, 23.31, 22.97, 19.25, 16.91, 15.53. HRMS (ESI-MS) m/z: [M-H]- calcd for C36H47ClNO2: 560.3301; found: 560.3276.
Synthesis of oleanolic acid analogues 7a-7c
The pyridine ring substituted analogues 7a-7c were synthesised by employing the o-amino benzaldehyde and o-amino pyridylaldehyde in the Claisen Schmidt condensation. Analogue 2 (0.2 mmol) and o-amino benzaldehyde or o-amino pyridylaldehyde (0.4 mmol) were dissolved in saturated potassium hydroxide alcohol solution with refluxing at 90 °C for 5 h monitored by the TLC. After the reaction completed, alcohol in the reaction mixture was removed under reduced pressure followed by dissolving the remainder in ethyl acetate. The organic layer was washed with deionised water, and dried over anhydrous magnesium sulphate. The crude product was separated by preparative silica gel chromatography in petroleum ether/ethyl acetate (v/v 6:1 to 8:1).
Analogue 7a: followed by aforementioned general procedures, 7a was prepared via the Claisen Schmidt condensation of 2 with m-amino benzaldehyde and purified with petroleum ether/ethyl acetate (v/v = 6:1, Rf = 0.44); white solid; yield: 80.3%. 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 7.6 Hz, 1H), 7.75–7.64 (m, 2H), 7.59 (t, J = 7.3 Hz, 1H), 7.41 (t, J = 7.2 Hz, 1H), 7.36 (d, J = 4.3 Hz, 4H), 7.34–7.29 (m, 1H), 5.40 (t, J = 3.4 Hz, 1H), 5.10 (q, J = 12.6 Hz, 2H), 3.03–2.89 (m, 2H), 2.54 (d, J = 15.4 Hz, 1H), 2.05–1.98 (m, 2H), 1.77–1.68 (m, 5H), 1.67–1.62 (m, 2H), 1.61–1.48 (m, 5H), 1.45–1.41 (m, 5H), 1.41–1.35 (m, 2H), 1.24–1.21 (m, 2H), 1.20 (s, 3H), 1.16–1.12 (m, 1H), 0.95 (s, 3H), 0.92 (s, 3H), 0.86 (s, 3H), 0.72 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 177.48, 166.06, 147.40, 143.79, 136.44, 135.27, 128.90, 128.68, 128.45, 128.45, 128.06, 128.06, 128.01, 127.97,500 0.92, 126.55, 125.39, 122.50, 66.00, 53.95, 46.88, 46.02, 45.90, 45.59, 42.00, 41.64, 40.25, 39.26, 36.24, 33.93, 33.14, 32.42, 32.39, 32.30, 30.75, 27.66, 25.74, 25.35, 23.66, 23.52, 23.15, 20.49, 16.71, 15.04. HRMS (ESI-MS) m/z: [M + H]+ calcd for C44H56NO2: 630.43056; found: 630.42859.
Analogue 7b: followed by aforementioned general procedures, 7b was prepared via the Claisen Schmidt condensation of 2 with 2-amino-3-pyridinecarboxaldehyde and purified with petroleum ether/ethyl acetate (v/v = 8:1, Rf = 0.44); white solid; yield: 66.0%. 1H NMR (600 MHz, CDCl3) δ 9.01 (dd, J = 4.2, 1.9 Hz, 1H), 8.07 (dd, J = 8.1, 1.9 Hz, 1H), 7.75 (s, 1H), 7.38 (dd, J = 8.1, 4.2 Hz, 1H), 7.35 (d, J = 4.4 Hz, 3H), 7.33–7.29 (m, 1H), 5.39 (t, J = 3.4 Hz, 1H), 5.09 (q, J = 12.6 Hz, 2H), 3.00–2.93 (m, 2H), 2.55 (d, J = 15.5 Hz, 1H), 2.12 (s, 1H), 2.06–1.96 (m, 3H), 1.78–1.63 (m, 6H), 1.62–1.53 (m, 3H), 1.53–1.50 (m, 2H), 1.49 (s, 3H), 1.47 (s, 3H), 1.42–1.32 (m, 2H), 1.28–1.23 (m, 1H), 1.19 (s, 3H), 1.14 (dd, J = 10.5, 3.0 Hz, 1H), 0.94 (s, 3H), 0.91 (s, 3H), 0.86 (s, 3H), 0.71 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 177.45, 170.37, 155.01, 152.20, 143.89, 136.42, 136.24, 136.13, 130.41, 128.45, 128.45, 128.06, 128.06, 127.97, 122.33, 122.14, 121.23, 66.01, 53.68, 46.86, 45.90, 45.72, 45.56, 42.00, 41.64, 40.77, 39.25, 36.23, 33.91, 33.14, 32.39, 32.36, 32.24, 30.75, 27.64, 25.73, 25.38, 23.65, 23.51, 23.13, 20.46, 16.68, 15.08. HRMS (ESI-MS) m/z: [M + H]+ calcd for C43H55N2O2: 631.42581; found: 631.42365.
Analogue 7c: followed by aforementioned general procedures, 7c was prepared via the Claisen Schmidt condensation of 2 with 4-amino-3-pyridinecarboxaldehyde and purified with petroleum ether/ethyl acetate (v/v = 8:1, Rf = 0.4); white solid; yield: 56.0%. 1H NMR (500 MHz, CDCl3) δ 9.12 (s, 1H), 8.62 (d, J = 5.9 Hz, 1H), 7.83 (s, 1H), 7.81 (d, J = 5.9 Hz, 1H), 7.36 (s, 2H), 7.35 (s, 2H), 7.34–7.29 (m, 1H), 5.39 (t, J = 3.5 Hz, 1H), 5.09 (q, J = 12.6 Hz, 2H), 3.03–2.91 (m, 2H), 2.56 (d, J = 15.5 Hz, 1H), 2.09–2.05 (m, 1H), 2.04–1.97 (m, 2H), 1.78–1.72 (m, 2H), 1.72–1.61 (m, 5H), 1.60–1.47 (m, 5H), 1.43 (s, 3H), 1.42 (s, 3H), 1.41–1.34 (m, 2H), 1.19 (s, 3H), 1.16–1.10 (m, 1H), 0.94 (s, 3H), 0.92 (s, 3H), 0.85 (s, 3H), 0.71 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 177.45, 171.86, 151.59, 149.40, 145.20, 143.85, 136.42, 134.81, 130.79, 128.45, 128.45, 128.05, 128.05, 127.97, 122.63, 122.32, 121.78, 66.00, 53.75, 46.86, 46.05, 45.89, 45.53, 41.99, 41.62, 40.91, 39.24, 36.15, 33.91, 33.13, 32.38, 32.36, 32.22, 30.75, 27.64, 25.73, 25.39, 23.65, 23.50, 23.13, 20.47, 16.69, 15.07. HRMS (ESI-MS) m/z: [M + H]+ calcd for C43H55N2O2: 631.42581; found: 631.42407.
Synthesis of oleanolic acid analogues 8a-8c
The analogues 8a-8c were synthesised by employing substituted 2-nitrobenzaldehyde in the Claisen Schmidt Condensation. Analogue 2 (0.2 mmol) and 2-nitrobenzaldehyde (0.4 mmol) were dissolved in saturated potassium hydroxide alcohol solution at room temperature for 10 h monitored by the TLC. After the reaction completed, alcohol in the reaction mixture was removed under reduced pressure followed by dissolving the remainder in ethyl acetate. The organic layer was washed with deionised water, and dried over anhydrous magnesium sulphate and. The crude product was separated by preparative silica gel chromatography in petroleum ether/ethyl acetate (v/v 6:1 to 8:1).
Analogue 8a: followed by aforementioned general procedures, 8a was prepared via the Claisen Schmidt condensation of 2 with 5-hydroxy-2-nitrobenzaldehydeand purified with petroleum ether/ethyl acetate (v/v = 8:1, Rf = 0.3); White solid; Yield: 52.5%. 1H NMR (500 MHz, CDCl3) δ 8.15 (d, J = 9.1 Hz, 1H), 7.63 (s, 1H), 7.36–7.29 (m, 5H), 6.92 (dd, J = 9.1, 2.5 Hz, 1H), 6.63 (d, J = 2.4 Hz, 1H), 5.20 (t, J = 3.3 Hz, 1H), 5.13–5.03 (m, 2H), 2.86 (dd, J = 13.7, 3.9 Hz, 1H), 2.62 (d, J = 15.5 Hz, 1H), 2.02–1.87 (m, 3H), 1.69 (ddt, J = 31.3, 27.5, 8.4 Hz, 6H), 1.60–1.53 (m, 3H), 1.49–1.39 (m, 4H), 1.36–1.31 (m, 2H), 1.20 (s, 3H), 1.16 (s, 3H), 1.11 (d, J = 5.8 Hz, 3H), 1.11–1.05 (m, 2H), 0.89 (s, 3H), 0.87 (s, 3H), 0.84 (s, 3H), 0.61 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 209.33, 178.17, 160.96, 143.67, 140.49, 136.11, 136.01, 134.96, 134.89, 128.51, 128.51, 128.14, 128.09, 128.05, 127.67, 122.10, 116.87, 115.92, 66.37, 53.68, 46.94, 46.25, 45.79, 45.46, 42.98, 42.02, 41.68, 39.24, 36.88, 33.84, 33.07, 32.30, 32.04, 30.71, 28.66, 27.58, 25.69, 23.64, 23.36, 23.06, 22.85, 20.06, 16.58, 15.22. HRMS (ESI-MS) m/z: [M-H]- calcd for C44H54NO6: 692.39566; found: 692.39282.
Analogue 8 b: followed by aforementioned general procedures, 8 b was prepared via the Claisen Schmidt condensation of 2 with 5-methoxy-2-nitrobenzaldehydeand purified with petroleum ether/ethyl acetate (v/v = 8:1, Rf = 0.32); white solid; yield: 59.0%. 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 9.2 Hz, 1H), 7.63 (d, J = 2.1 Hz, 1H), 7.35–7.28 (m, 5H), 6.93 (dd, J = 9.2, 2.7 Hz, 1H), 6.68 (dd, J = 2.9, 0.6 Hz, 1H), 5.24 (t, J = 3.5 Hz, 1H), 5.10–5.01 (m, 2H), 3.90 (s, 3H), 2.88 (dd, J = 13.8, 4.1 Hz, 1H), 2.59 (d, J = 15.6 Hz, 1H), 2.00–1.89 (m, 2H), 1.83–1.76 (m, 1H), 1.73–1.64 (m, 4H), 1.61–1.54 (m, 4H), 1.49–1.39 (m, 4H), 1.35–1.28 (m, 3H), 1.19 (s, 3H), 1.15 (s, 3H), 1.12 (s, 3H), 1.09–1.06 (m, 1H), 0.90 (s, 3H), 0.88 (s, 3H), 0.86 (s, 3H), 0.62 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 207.58, 177.37, 163.08, 143.97, 141.04, 136.35, 135.30, 135.14, 134.83, 128.44, 128.44, 128.05, 128.05, 127.98, 127.65, 121.98, 115.90, 113.29, 66.01, 56.05, 53.59, 46.81, 45.98, 45.89, 45.29, 42.82, 41.98, 41.58, 39.22, 36.78, 33.84, 33.08, 32.29, 31.99, 30.69, 28.85, 27.59, 25.65, 23.62, 23.45, 23.08, 22.76, 20.11, 16.56, 15.19. HRMS (ESI-MS) m/z: [M + Na]+ calcd for C45H57NO6Na: 730.40781; found: 730.40540.
Analogue 8c: followed by aforementioned general procedures, 8c was prepared via the Claisen Schmidt Condensation of 2 with 4-acetamidobenzaldehyde and purified with petroleum ether/ethyl acetate (v/v = 6:1, Rf = 0.38); white solid; yield: 34.0%. 1H NMR (500 MHz, CDCl3) δ 7.55 (d, J = 8.4 Hz, 2H), 7.47 (s, 1H), 7.39 (d, J = 8.5 Hz, 2H), 7.36–7.32 (m, 4H), 7.32–7.28 (m, 1H), 5.34 (t, J = 3.4 Hz, 1H), 5.07 (dd, J = 31.4, 12.5 Hz, 2H), 2.98 (d, J = 16.3 Hz, 1H), 2.94 (dd, J = 13.8, 3.8 Hz, 1H), 2.26 (dd, J = 16.2, 2.1 Hz, 1H), 2.20 (s, 3H), 2.03–1.86 (m, 3H), 1.75–1.68 (m, 4H), 1.64–1.55 (m, 3H), 1.49 (dd, J = 17.8, 11.4 Hz, 3H), 1.44–1.30 (m, 4H), 1.23–1.20 (m, 2H), 1.19 (s, 3H), 1.16 (s, 3H), 1.13 (s, 3H), 0.93 (s, 3H), 0.91 (s, 3H), 0.82 (s, 3H), 0.65 (s, 3H). 13 C NMR (151 MHz, CDCl3) δ 207.87, 177.46, 168.32, 143.81, 138.12, 136.86, 136.36, 132.96, 131.86, 131.43, 131.43, 128.44, 128.44, 128.06, 128.06, 127.98, 122.31, 119.34, 119.34, 66.02, 52.93, 46.87, 45.88, 45.42, 45.12, 44.28, 42.03, 41.66, 39.22, 36.21, 33.89, 33.11, 32.34, 31.93, 30.74, 29.79, 27.61, 25.66, 24.74, 23.70, 23.65, 23.13, 22.66, 20.36, 16.53, 15.28. HRMS (ESI-MS) m/z: [M + Na]+ calcd for C46H59NO4Na: 712.43363; found: 712.43085.
Sample preparation
Given that OA and its derivatives are poorly dissolved in phosphate buffer solution (PBS), we first dissolved test samples in methyl sulfoxide (DMSO) as stock solutions at a concentration of 4 mg/mL. The DMSO stock solution of each sample was diluted with PBS to reach to desired concentrations and the DMSO content in the assay reaction mixture was less than 1%, which did not induce significant inhibition of the enzyme activity (inhibition rate less than 3%).
Hyaluronidase inhibition assay
Inhibitory effect on bovine testicular HAase activity was evaluated using a reported method with minor modifications23. It is noted that long HA chains and HAase may form of non-active complexes at low ionic condition, which can impede the catalytic activity of HAase and disrupt the HAase inhibition assay24. This interference can be avoided by adding a positively charged protein to balance the ionic strength of the reaction system and recover the activity of HAase24. Therefore, bovine serum albumin (BSA; 0.01%) was added to the reaction buffer (PBS; 20 mM) in acidic condition (pH 3.75), which provides an optimal electrostatic environment for the enzyme reaction. Test samples (5 μL; concentration ranging from 2.5 to 40 μg/mL) were pre-incubated with HAase protein (7.5 U/mL; 95 μL) in the BSA solution at 37 °C for 10 min. Then hyaluronic acid (HA; 100 μL) was added to the pre-incubated mixture at 37 °C for 45 min. HA (undegraded) was precipitated by adding an acidic BSA solution (1 mL; pH = 3.75) containing 0.1% BSA in sodium acetate (24 mM) and acetic acid (79 mM) for 10 min. The absorbance was then measured at a wavelength of 600 nm using a microplate reader (SpectraMax M2, Molecular Devices Corp., operated by SoftmaxPro v.4.6 software, Sunnyvale, CA, USA). Each sample was tested at least in three replicates. The inhibition rate was calculated using following formula: Inhibition% = 1 – [ODHA – ODsample]/[ODHA – ODHAase] × 100%. The IC50 value of each sample was determined by analysing its inhibition rates at different concentrations with a nonlinear regression algorithm by GraphPad Prism.
Lineweaver–Burk plot
The kinetic parameters for HAase inhibition of 6c and 6g were obtained using a reported method with minor modifications25. Briefly, four concentrations of 6c and 6g near the IC50 value were selected for the inhibition assay. The HAase inhibition activity of 6c and 6g was measured at a series of concentrations of HA solutions (0.125–2.0 µM and 0.2–1.0 µM, respectively). The inhibition type of test compounds was determined by the Lineweaver-Burk plots.
Circular dichroism (CD) measurement
The secondary structure of HAase protein was monitored by circular dichroism analysis with a Jasco J-720 spectropolarimeter (Jasco, Tokyo, Japan) using the following parameters: wavelength: 190 − 250 nm; bandwidth: 1 nm; path length of quartz cell: 1 mm26. HAase protein (0.2 mg/mL) was incubated with test samples at two concentrations (10 and 20 μg/mL) for 10 min prior to the CD measurements.
Molecular docking
Auto Dock 4.2 software was used for the molecular docking study provided by the Rhode Island IDeA Network of Biomedical Research Excellence (RI-INBRE)27. The three-dimensional rigid structure of HAase was obtained from the protein data bank (PDB ID: 2PE4). The structures of OA derivatives were generated by Chemdraw 3 D (PerkinElmer Inc.; Waltham, MA, USA). All water molecules were removed and essential hydrogen atoms as well as Kollman charges were added into the HAase protein, followed by the computation of the Gasteiger charges. The amino acid residues of Gln18 and Arg20 were employed as the active sites for docking simulation to study the bind mode and interaction force. The size of the grid box was defined as 80 Å × 80 Å × 80 Å in the x, y, and z dimension with a grid spacing of 0.375 Å. The docking simulations were conducted with the Lamarckian genetic algorithm and the conformation with the lowest free binding energy was selected for analysis.
Skin permeability
A computational method, SwissADME, was used to predict the skin permeability of OA and its derivatives using a previously reported method28. Briefly, the chemical structures of OA and its derivatives were imported by using their simplified molecular input line entry specification (SMILES) file generated by ChemDraw (PerkinElmer Inc.; Waltham, MA, USA). The SwissADME algorithm (http://www.swissadme.ch/) was used to obtain skin penetration related parameters including Log S(ESOL), Log S(Ali), and Log Kp. Next, a biochemical based assay, namely, the parallel artificial membrane permeability assay (PAMAPA), was performed to measure the skin permeabilities of OA and its derivatives using a reported method with minor modifications28. The PAMPA assay kit (including Prisma HT buffers, membrane plate, reference standards) was purchased from Pion, Inc. (Billerica, MA, USA) and the assay was performed according to the manufacturer’s instructions. Test compounds and reference standards (i.e. warfarin, piroxicam, and verapamil) were prepared in DMSO stock solution (10 mM). The Prisma HT buffers were prepared and adjusted to pH at 6.5 and 7.4. In the deep-well plate, the Prisma HT buffer (1 mL) was added and mixed thoroughly and then the diluted sample (50 µM; 180 µL) was added to the donor (bottom) plate in the Pion sandwich apparatus and the Prisma HT buffer (200 µL) was added to the acceptor (top plate). The sandwich apparatus was then incubated and stirred at room temperature for 24 h. The UV absorbance of the donor and acceptor plates were recorded using a plate reader with the PION PAMPA analysis software to obtain the -LogPe values of the samples.
Cell culture and viability
Human keratinocytes (HaCaT) cells were purchased from Sigma Aldrich Co. (St. Louis, MO, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies; Gaithersburg, MD, USA) with 10% fetal bovine serum (Gibico; Grand Island, NY, USA) at 37 °C in the presence of 5% CO2. The cell viability of HaCaT treated with or without OA and its derivatives was measured by the XTT assay according to manufacturer’s instructions (XTT Cell proliferation Kit II; Roche, USA). Briefly, HaCaT cells were seeded in 96-well plates at 5 × 103 cells per well and allowed to attach for 12 h. Then OA and its derivatives (100 µL; 10 µg/mL) were incubated with cells for 24 h, followed by adding the XTT reagent (50 µL; 0.3 mg/mL) in each well and incubated for 4 h. The absorbance of each well was measured at wavelengths of 450 nm using a SpectraMax M2 plate reader (Molecular Devices; Sunnyvale, CA, USA).
Cellular reactive oxygen species (ROS)
HaCaT cells were seeded in 96-well plates at 6 × 103 cells per well and allowed to attach for 12 h. Then the cells were incubated with test samples (10 µg/mL) for 24 h. Next, the cell culture medium was removed and fresh phosphate buffer solution (PBS; 100 µL) containing a fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFDA; 20 µM) was added to the cells and incubated for 20 min. Then the cells were treated with H2O2 (100 µL; 200 µM) for 1 h followed by measuring the fluorescence intensity of each well with excitation and emission wavelengths of 485 and 525 nm, respectively, using a SpectraMax M2 plate reader29.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA). Data were expressed as the mean value ± standard deviation (SD). The significance of difference was analysed using a one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls multiple comparison test (SNK).
Results and discussion
Synthesis of oleanolic acid analogues
A series of OA derivatives were synthesised via structural modifications at OA’s C-2, C-3, C-28, C-12 and C-13 positions (Scheme 1). OA was first fluorinated with selectfluor (tetrafluoroborate) to obtain the intermediate analogue 3. Another intermediate analogue 4 was obtained by modifying compound 3 with the Jones reagent. The target compounds 5a-5c were prepared through condensation of compound 4 with various substituted phenylhydrazine and purified by column chromatography. Analogue 2 was synthesised by modifying OA at the C-3 position, which was oxidised to carbonyl with the Jones reagent. In addition, compounds 6a-6g were synthesised by employing a variety of substituted phenylhydrazine to compound 2 using the Fischer indolization approach. Similarly, target compounds 7a-7c and 8a-8c were synthesised through the condensation of compound 2 with various aldehydes via the Claisen Schmidt condensation. The structures of OA analogues were confirmed by their spectroscopic data including 1H NMR, 13 C NMR and HRMS.
Scheme 1.

Synthesis routes of OA analogues 2–4, 5a-5c, 6a-6g, 7a-7c and 8a-8c. Reagents and conditions: (a) selectflour, dioxane, nitromethane, 80 °C, 4 h; (b) the Jones reagent, acetone, at 0 °C; (c) RPhNHNH2, CF3COOH, acetic acid, 115 °C, overnight; (d) BnCl, K2CO3, r.t., overnight; (e) R-CHO (o-amino benzaldehyde or o-amino pyridylaldehyde), KOH, ethanol, refluxed, 5 h; (f) R-CHO, KOH, ethanol, r.t., overnight.
OA derivatives inhibit the activity of HAase enzyme
The inhibitory effects of compounds 2-8c against HAase enzyme activity were evaluated. The inhibitory effect of OA analogues on HAase was first evaluated at a threshold concentration of 40 µg/mL. OA (1) showed a moderate inhibitory activity with an inhibition rate of 59.3% and the intermediates 2–4 only had weak inhibitory effects with inhibition rates less than 37.3% (Table 1). Among the OA derivatives, analogues 6a, 6b, 6c, 6d, 6e, 6f, and 6g bearing indole moieties showed the most potent anti-HAase effects with an inhibition rate of 100.9, 100.2, 86.3, 93.5, and 98.1%, respectively. Analogues 5a-5c only showed weak inhibitory effect (18.7–37.6% inhibition, respectively). Similarly, the anti-HAase activities of OA derivatives 7a-7c and 8a-8c (22.6–32.5% and 20.2–25.2% inhibition rate, respectively) were weaker than analogues 6. Next, the inhibitory effects of OA derivatives with promising enzyme inhibition activity (6a-6g) were further evaluated by obtaining their IC50 values. OA derivatives 6a-6g had IC50 values ranging from 9.5–16.2 µg/mL (or 16.7–28.9 µM) (Figure 1).
Table 1.
Inhibitory effect of 1 (OA) and its chemical synthesised derivatives 2–8 (at 40 μg/mL) on the enzyme activity of HAase.
| Compound | MW (g/mol) | Functional group | Inhibition (%) | |
|---|---|---|---|---|
| 1 | 456.7 | / | 59.3 ± 2.4 | |
| 2 | 454.7 | / | 15.9 ± 4.3 | |
| 3 | 474.7 | / | 12.7 ± 3.9 | |
| 4 | 472.7 | / | 37.3 ± 1.4 | |
| 5a | 629.8 | R1= | 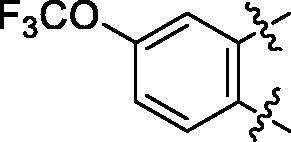 |
18.7 ± 3.8 |
| 5b | 563.8 | R1= | 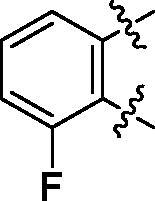 |
25.6 ± 3.8 |
| 5c | 580.2 | R1= | 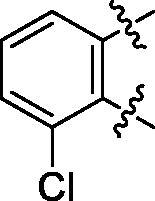 |
37.6 ± 4.7 |
| 6a | 572.8 | R2= | 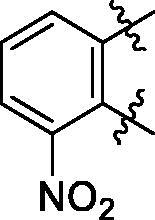 |
100.9 ± 4.3 |
| 6b | 545.8 | R2= | 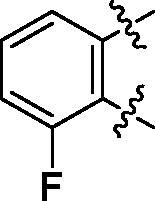 |
100.4 ± 2.4 |
| 6c | 563.8 | R2= | 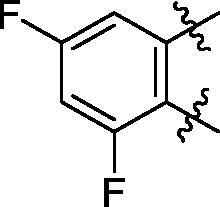 |
86.3 ± 3.3 |
| 6d | 555.8 | R2= | 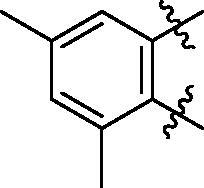 |
93.5 ± 3.9 |
| 6e | 606.7 | R2= | 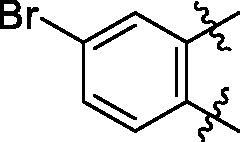 |
83.7 ± 3.3 |
| 6f | 606.7 | R2= | 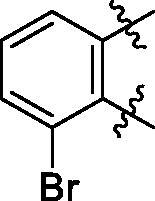 |
100.6 ± 3.6 |
| 6g | 562.2 | R2= | 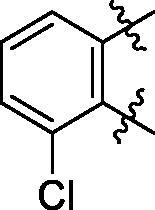 |
98.1 ± 0.4 |
| 7a | 629.9 | R3= | 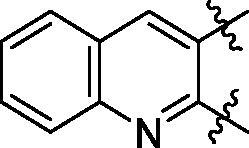 |
27.9 ± 4.0 |
| 7b | 630.9 | R3= | 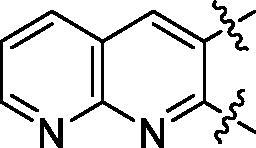 |
21.3 ± 3.9 |
| 7c | 630.9 | R3= | 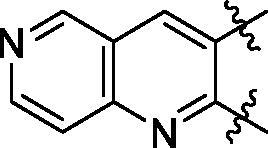 |
32.5 ± 1.7 |
| 8a | 693.9 | R4= | 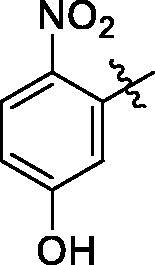 |
20.2 ± 2.6 |
| 8b | 708.0 | R4= | 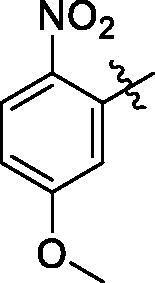 |
31.8 ± 0.6 |
| 8c | 690.0 | R4= | 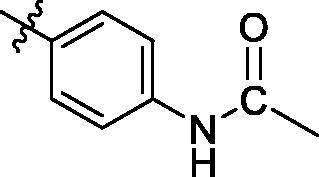 |
25.2 ± 4.4 |
Figure 1.

Inhibitory effects of leading compounds 6a-6g on the activity of bovine testicular HAase enzyme activity. The inhibition rates (%) of each compound were measured at five concentrations (1, 5, 10, 20, and 40 µg/mL) and an inhibition curve was constructed to calculate the IC50 value of each test compound using nonlinear regression.
Some preliminary structure and activity relationship (SAR) can be observed based on the anti-HAase activity of OA analogues bearing different functional groups at 40 µg/mL. For instance, decarboxylation at C-28 position (analogues 3–5; inhibition rate of 12.7–37.6%) did not increase the anti-HAase activity as compared to OA indole derivatives with a carboxyl group at the C-28 position (analogues 6; inhibition rate of 86.3–100.6%). In addition, OA derivatives with a carboxyl group and an indole group (analogues 6; inhibition rate: 86.3–100.6%) showed superior inhibitory effects as compared to carbonylated OA derivatives with functional groups including pyridine ring (analogues 7; inhibition rate: 22.6–30.1%) or benzaldehyde ring (analogues 8; inhibition rate: 20.2–31.8%). However, there is a lack of convincing evidence to support the conclusion whether the induction of halide groups on the OA indole skeleton (6b, 6c, 6e, 6f, and 6g; inhibition rate: 83.7–100.6%) have superior anti-HAase activity as compared to the analogues with the same skeleton but bearing non-halide moieties 6a and 6d (inhibition rate: 93.5–100.9%). Further SAR studies are required to elucidate the impact of indole moieties on the OA derivatives’ anti-HAase effects. We further evaluated the mechanisms of inhibition of two leading compounds 6c and 6g.
OA indole derivatives 6c and 6g interact with HAase protein
We next studied OA indole derivatives 6c’ and 6g’s mechanisms of inhibition on HAase with spectroscopic assays and computational method. First, the Lineweaver-Burk plot of 6c or 6g at various concentrations in the presence of substrate solutions (HA) at several concentrations was constructed, which showed straight lines on the x-axis at different points (Figure 2). This suggests that 6c and 6g had mixed type of competitive and non-competitive inhibitions on HAase. Second, the effects of HAase inhibitors (6c and 6g) on the secondary structure of the enzyme protein were evaluated by the CD spectra. CD is a sensitive tool for determining the secondary structure of proteins and can be used to monitor protein’s conformational changes induced by binding interactions with small molecules30. The CD spectrum of intact HAase protein yielded two typical far-UV CD signals at 205 and 224 nm, which are in response to the peptide bond absorption, representing typical α-helical structures of the enzyme protein (Figure 3). Moreover, changes in the near UV region (320–260 nm) of CD spectra indicted that binding interactions occurred between 6c or 6g and the aromatic amino acid side chains of the enzyme protein, indicating the changes in protein’s tertiary structure. Quantitatively, co-incubation of HAase protein and 6c or 6g showed a reduction of the α-helix content from 35.4% to 26.1 and 29.8%, respectively. In addition, the random coil content increased from 39.3% to 42.5 and 44.7%, respectively. This suggests that HAase inhibitors disturbed the hydrogen force in the secondary structure of HAase resulting in an impaired enzyme activity. This observation is in agreement with several reported studies using CD technique to show that natural products based HAase inhibitors (for e.g. flavonoids including baicalin, liquiritin, and rutin) can bind to HAase protein and alter its secondary structures, thereby leading to undermined HAase activity31,32.
Figure 2.

Enzyme reaction kinetics of 6c (A) and 6g (B) were measured by constructing the Lineweaver-Burk plots. Four concentrations (5–40 µg/mL) were co-incubated with HAase in the presence of substrate solutions (HA) at concentrations of 0.125–2.0 µM and 0.2–1.0 µM, respectively.
Figure 3.

Effects of 6c (A) and 6g (B) on the secondary structure of HAase protein characterised by far-UV CD spectra measurements.
Next, the interactions between HAase protein and OA analogues were studied by molecular docking, which predicted that OA indole derivatives including 6c and 6g were able to bind to HAase protein (Figure 4(A,B)). Further binding analysis of two leading HAase inhibitors 6c and 6g showed that they interacted with enzyme protein’s amino acid residues including Arg20, Gln18, Phi22, Arg191 and Ala192 (Figure 4(C,D)). In addition, 6c and 6g formed several molecular forces including hydrogen bond and alkyl-pi bond to stabilise the formation of the ligand-protein complex, which also contributed to the reduced enzyme activity. Furthermore, the free binding energy to HAase protein and the binding constant of compounds 6c, 6d, 6e, 6f, and 6g were predicted as −9.61, −8.93, −8.15, −9.03, and −8.85 kcal/mol, and 7.85, 13.45, 15.12, 10.44, and 14.77 µM, respectively. The predicted binding parameters was in agreement with data obtained from the enzyme inhibition assay. For instance, 6c, as one of the most active HAase inhibitor among all of the OA analogues, had the lowest free binding energy (−9.61 kcal/mol) and binding constant (7.85 µM). Data from biochemical (enzyme kinetic), biophysical (CD), and computational assays support that the protein-ligand interaction contributed to the inhibitory effect of OA derivatives on HAase enzyme activity. Apart from binding to HAase protein’s catalytic or allosteric site and altering the secondary structure of enzyme protein, other types of inhibition are involved in HAase inhibitors’ mechanisms of action. For instance, studies have shown that several factors including pH conditions and ionic strength are critical for HAase’s activity and some glycoproteins HAase inhibitors (also termed as inter-α-inhibitors) showed magnesium dependent inhibition on HAase at specific pH environment33. However, further studies are warranted to confirm whether OA derivatives’ inhibition HAase was mediated by aforementioned mechanism.
Figure 4.

Interactions between HAase protein and OA derivatives predicted by molecule docking. OA derivatives’ potential binding pocket on HAase protein (A); Enlarged view of the binding site of leading compounds 6c and 6 g on HAase protein (B); The interactions between HAase protein’s amino acid residues and compound 6c (C) and 6 g (D).
Several factors should be accounted for the development of HAase inhibitors based cosmeceuticals for their potential skin-ageing applications. For example, HAase inhibitors may need to penetrate the skin barrier and reach to specific skin layers (i.e. basement membrane and interstitial matrix) to exert desired biological effects. Therefore, it is critical to characterise the physico-chemical properties, such as the skin permeability, of OA derived HAase inhibitors.
Permeability of OA and its derivatives
The skin permeability of OA and its derivatives was assessed by in silico and in vitro models. First, the skin permeability coefficient (Log Kp; cm/s) of OA and its derivatives (6a-6g) was predicted by the SwissADME algorithm based on their physico-chemical properties including molecular weight, lipophilicity, and Log Po/w. The more negative the coefficient value is, the less possible for the compound to penetrate the skin barrier. The SwissADME method predicted that OA and its derivatives have feasible skin permeability with Log Kp values of −3.77 and −3.21 to −2.46 cm/s, respectively (Figure 5(A)). Notably, structural modified OA derivatives showed superior skin permeability as compared to OA. This observation was further supported by OA derivatives’ apparent permeability (Log Pe) value obtained from the PAMPA assay at pH conditions of 6.5 and 7.4. Compounds 6a-6g had -Log Pe values of of 5.13, 4.86, 4.88, 4.98, 5.03, 5.09, and 4.87, respectively, in a slightly acidic condition (pH = 6.5; Figure 5(B)). The PAMPA assay reference standards including warfarin, piroxicam, and verapamil, respectively representing low, medium, and high skin permeability, had a -Log Pe value of 5.75, 5.44, and 5.04, respectively, at pH = 6.5. This suggest that the skin permeability of several OA derivatives including 6b, 6c, and 6g was enhanced at pH = 6.5 as compared to OA (4.86–4.88 v.s. 5.04), which supported the prediction of the skin permeability of OA derivatives in the SwissADME model. However, OA’ and its derivatives’ (6a-6g) -Log Pe value significantly increased to 5.3 and 6.46, 6.37, 6.12, 5.73, 5.61, 5.69, and 5.24, respectively, when the pH condition adjusted to 7.4 (Figure 5(C)), suggesting that the skin permeability of OA derivatives may be undermined in a slightly basic environment.
Figure 5.

Skin permeability of OA and its derivatives (6a-6g) predicted by computational SwissADME method and obtained the PAMPA assay. Skin permeability coefficient predicted by the SwissADME (A). Skin permeability (apparent permeability; -Log Pe value) obtained by the PAMPA assay at pH = 6.5 (B) and 7.4 (C). Statistical significant difference was considered as a (p < 0.05) when compared to the reference compound warfarin; b (p < 0.05) when compared to the reference compound piroxicam; c (p < 0.05) when compared to the reference compound verapamil.
Data obtained from the PAMPA assays provided useful insights for the further development of OA indole derivatives as cosmeceuticals. For instance, OA derivatives are more skin permeable in a slightly acidic condition (pH = 6.5). This might be favourable in healthy skin conditions given that the skin’s surface and uppermost layers (for e.g. stratum corneum) are acidic34. In addition, the acidic pH is preferrable for a variety of cosmetic products, such as cleansers and toners (pH 4.5–6), serums (pH 4–6), vitamin C (ascorbic acid) products (pH 2.6–3.2) and retinol products (pH 4.0–6.6)35. However, there is a limitation in our current study. The skin permeabilities of the OA derivatives were assessed using computational method (the SwissADME prediction) and membrane based in vitro model (the PAMPA assay). Results obtained from these in silico and in vitro assays may not accurately reflect OA derivatives’ skin permeability. Skin is the largest organ with perhaps the most complex biological functions in the body. One of the major physiological purposes of skin is to protect the body from external substances including pathogen, UV radiation, and free radicals36,37. This protection is enabled by the involvement of numerous skin cells including dermis (which is consisting of fibroblasts, sweat glands, dermal adipose cells, mast cells, and infiltrating leucocytes) and epidermis (which is constituted by keratinocytes, melanocytes, Langerhans cells, and Merkel cells)38,39. Altogether, these cells form four distinct layers including the stratum basale, stratum spinosum, stratum granulosum, and stratum corneum, which determines to the rigidity of skin permeability39,40. Therefore, further evaluations of the skin permeability of OA derivatives using in vivo models are warranted.
OA derivative 6c reduces H2O2-induced cellular reactive oxygen species (ROS) in HaCaT cells
The potential skin beneficial effects of OA and its derivatives were evaluated by cellular assays. First, cytotoxicity of OA and its derivatives was evaluated in human keratinocytes HaCaT cells. Treatment with OA and its derivatives 6a-6g at a concentration of 10 µg/mL resulted in a cell viability of 96.68 and 94.13–104.55%, respectively, suggesting that these compounds are non-toxic to HaCaT cells at the tested concentration (Figure 6(A)). In addition, the skin protective effect of OA derivatives was evaluated against H2O2-induced oxidative stress by assessing the level of ROS generated in HaCaT cells. The ROS level in H2O2-stimulated cells was elevated by 13-fold as compared to the control group. Treatment with compound 6c (10 µg/mL) showed antioxidant effect on HaCaT cells by decreasing the ROS level by 23.6% as compared to the control group (H2O2-exposed cells) (Figure 6(B)). Other OA analogues did not significantly ameliorate H2O2-induced oxidative stress.
Figure 6.

Effects of OA and its derivatives on the cell viability of human keratinocyte HaCaT cells (A) and their antioxidant effect against H2O2-induced production of ROS in HaCaT cells (B). Data are presented as means ± SD from experiments with three replicates. Statistical significant difference was considered as a (p < 0.01) when compared to the model (H2O2-exposed) group. n.s. = not significant.
Apart from the anti-HAase activity, OA also shows inhibitory effects on the activity of skin-ageing related enzymes including elastase41 and collagenase42, which may contribute to the overall anti-skin-ageing activity of OA. Similar to the anti-HAase activity, it is possible that the OA indole derivatives may exert enhanced anti-elastase and anti-collagenase effects but further enzymatic assays are warranted to confirm this. In addition, it was reported that OA inhibits particulate matter induced release of matrix metalloproteinase 1 (as known as interstitial collagenase) in dermal fibroblasts3. The current study also showed that OA inhibits particulate matter induced skin ageing which results in the release of pro-inflammatory cytokines including tumour necrosis factor-alpha and interleukin 6. Thus, OA’s anti-inflammatory effects may also have contributed to the overall skin protective effects against skin ageing. Moreover, an in vivo study using a mouse model showed the skin protective of OA against toxin (12-O-teradecanoyl-phorbol-13-acetate) induced skin carcinogenesis via the inhibition of aberrant gene expressions43. Therefore, further studies on the evaluations of OA indole derivatives’ skin beneficial effects using in vivo models are warranted.
Conclusion
In summary, a series of OA analogues were structurally modified by the induction of indole moieties via the Fischer indole synthesis. Several OA indole derivatives bearing halogen atom(s) showed superior anti-HAase activity as compared to the parent compound OA. The mechanistic studies using biochemical, biophysical, and computational assays suggest that the inhibitory effects of OA derivatives on HAase may be associated with their binding capacity to HAase protein. Moreover, OA derivatives showed feasible skin permeability from in silico (SwissADME) and in vitro (PAMPA) assays. Further biological evaluations of potential skin protective effects of OA derived HAase inhibitors in human keratinocytes showed that OA analogue with enhanced skin permeability also exerted cellular antioxidant effects. Findings from the current study provide useful insight on the development of OA based HAase inhibitors as cosmeceutical agents but further investigations are warranted to assess their anti-skin-ageing effects using in vivo models.
Acknowledgements
Spectrometric data were acquired from instruments located at the University of Rhode Island in the RI-INBRE core facility supported by Grant P20GM103430 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). H.H., Z.L., and P.W. were supported by National Natural Science Foundation of China (No. 81803390), Guangdong Basic and Applied Basic Research Foundation (No. 2021A1515010221), Special Fund Project of Science and Technology Innovation Strategy of Guangdong Province 2018 and 2020 [No. Jiangke(2018)352 and Jiangke(2020)182], the project of Jiangmen city social welfare innovation platform construction (No. 2018090103460022105), Department of Education of Guangdong Province (No. 2020KZDZX1202, 2018KTSCX236, 2017KSYS010 and 2016KCXTD005), Jiangmen Program for Innovative Research Team (No. 2018630100180019806), and the Youth Foundation of Wuyi University (No. 2017td01). S.N. was supported by funding from China Scholarship Council (201808210448).
Disclosure statement
The authors declare no competing interests.
References
- 1.Sultana N, Ata A.. Oleanolic acid and related derivatives as medicinally important compounds. J Enzyme Inhibition Med Chem 2008;23:739–56. [DOI] [PubMed] [Google Scholar]
- 2.Liu J. Oleanolic acid and ursolic acid: research perspectives. J Ethnopharmacol. 2005;100:92–4. [DOI] [PubMed] [Google Scholar]
- 3.Kim YJ, Lee JE, Jang HS, et al. Oleanolic acid protects the skin from particulate matter-induced aging. Biomol Therapeutics 2021;29:220–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nema NK, Maity N, Sarkar B, Mukherjee PK.. Cucumis sativus fruit-potential antioxidant, anti-hyaluronidase, and anti-elastase agent. Arch Dermatol Res 2011;303:247–52. [DOI] [PubMed] [Google Scholar]
- 5.Jung H. Hyaluronidase: an overview of its properties, applications, and side effects. Arch Plastic Surg 2020;47:297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Girish K, Kemparaju K, Nagaraju S, Vishwanath B.. Hyaluronidase inhibitors: a biological and therapeutic perspective. Curr Med Chem 2009;16:2261–88. [DOI] [PubMed] [Google Scholar]
- 7.Papakonstantinou E, Roth M, Karakiulakis G.. Hyaluronic acid: a key molecule in skin aging. Dermato Endocrinol 2012;4:253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stern R, Maibach HI.. Hyaluronan in skin: aspects of aging and its pharmacologic modulation. Clin Dermatol. 2008;26:106–22. [DOI] [PubMed] [Google Scholar]
- 9.Manuskiatti W, Maibach HI.. Hyaluronic acid and skin: wound healing and aging. Inter J Dermatol 1996;35:539–44. [DOI] [PubMed] [Google Scholar]
- 10.Gupta RC, Lall R, Srivastava A, Sinha A.. Hyaluronic acid: Molecular mechanisms and therapeutic trajectory. Front Vet Sci 2019;6:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garg C, Khurana P, Garg M.. Molecular mechanisms of skin photoaging and plant inhibitors. Inter J Green Pharm 2017;11:S217–S232. [Google Scholar]
- 12.Liyanaarachchi GD, Samarasekera JKRR, Mahanama KRR, Hemalal KDP.. Tyrosinase, elastase, hyaluronidase, inhibitory and antioxidant activity of Sri Lankan medicinal plants for novel cosmeceuticals. Industrial Crops Products 2018;111:597–605. [Google Scholar]
- 13.Hetta MH. Hyaluronidase inhibitors as skin rejuvenating agents from natural source. Inter J Phytocosmetics Natural Ingred 2020;7:4. http://www.ijpni.org. [Google Scholar]
- 14.Swyer GI, Emmens CW.. A modified method for the viscosimetric assay of hyaluronidase. Biochem J 1947;41:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benchetrit LC, Pahuja SL, Gray ED, Edstrom RD.. A sensitive method for the assay of hyaluronidase activity. Anal Biochem 1977;79:431–437. [DOI] [PubMed] [Google Scholar]
- 16.Tung JS, Mark GE, Hollis GF.. A microplate assay for hyaluronidase and hyaluronidase inhibitors. Anal Biochem 1994;223:149–152. [DOI] [PubMed] [Google Scholar]
- 17.Frost GI, Stern R.. A microtiter-based assay for hyaluronidase activity not requiring specialized reagents. Anal Biochem 1997;251:263–269. [DOI] [PubMed] [Google Scholar]
- 18.Scotti L, Kumar Singla R, Mitsugu Ishiki H, et al. Recent advancement in natural hyaluronidase inhibitors. Curr Top Med Chem 2016;16:2525–31. [DOI] [PubMed] [Google Scholar]
- 19.Mio K, Stern R.. Inhibitors of the hyaluronidases. Matrix Biol 2002;21:31–37. [DOI] [PubMed] [Google Scholar]
- 20.Abdullah NH, Thomas NF, Sivasothy Y, et al. Hyaluronidase inhibitory activity of pentacylic triterpenoids from Prismatomeris tetrandra (Roxb.) K. schum: Isolation, synthesis and QSAR Study. Inter J Mol Sci 2016;17:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olgen S, Kaessler A, Nebioğlu D, Jose J.. New potent indole derivatives as hyaluronidase inhibitors. Chem Biol Drug Design 2007;70:547–551. [DOI] [PubMed] [Google Scholar]
- 22.Wu P, He H, Ma H, et al. Oleanolic acid indole derivatives as novel α-glucosidase inhibitors: Synthesis, biological evaluation, and mechanistic analysis. Bioorg Chem 2021; 107:104580. [DOI] [PubMed] [Google Scholar]
- 23.Chaiyana W, Chansakaow S, Intasai N, Kiattisin K, et al. Chemical constituents, antioxidant, anti-MMPs, and anti-hyaluronidase activities of Thunbergia laurifolia lindl. Leaf extracts for skin aging and skin damage prevention. Molecules 2020;25:1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lenormand H, Deschrevel B, Vincent JC.. pH effects on the hyaluronan hydrolysis catalysed by hyaluronidase in the presence of proteins: Part I. Dual aspect of the pH-dependence. Matrix Biol 2010;29:330–337. [DOI] [PubMed] [Google Scholar]
- 25.Ma H, Wang L, Niesen DB, et al. Structure activity related, mechanistic, and modeling studies of gallotannins containing a glucitol-core and α-glucosidase. RSC Adv. 2015;5:107904–107915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma H, Liu W, Frost L, et al. The hydrolyzable gallotannin, penta-O-galloyl-β-d-glucopyranoside, inhibits the formation of advanced glycation endproducts by protecting protein structure. Mol BioSyst 2015;11:1338–1347. [DOI] [PubMed] [Google Scholar]
- 27.Liu C, Ma H, Slitt AL, Seeram NP.. Inhibitory effect of cannabidiol on the activation of NLRP3 inflammasome is associated with its modulation of the P2X7 receptor in human monocytes. J Natural Products 2020;83:2025–2029. [DOI] [PubMed] [Google Scholar]
- 28.Liu C, Xu Y, Kirk RD, et al. Inhibitory effects of skin permeable glucitol-core containing gallotannins from red maple leaves on elastase and their protective effects on human keratinocytes. J Funct Foods 2020;75:104208. [Google Scholar]
- 29.Li H, Dasilva NA, Liu W, et al. Thymocid®, a standardized black cumin (Nigella sativa) seed extract, modulates collagen cross-linking, collagenase and elastase activities, and melanogenesis in murine B16F10 melanoma cells. Nutrients 2020;12:2146–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zsila F, Bikadi Z, Fitos I, Simonyi M.. Probing protein binding sites by circular dichroism spectroscopy. Curr Drug Disc Technol 2004;1:133–153. [DOI] [PubMed] [Google Scholar]
- 31.Li X, Liu H, Yang Z, et al. Study on the interaction of hyaluronidase with certain flavonoids. J Mol Struc 2021;1241:130686. [Google Scholar]
- 32.Li X, Xu R, Cheng Z, et al. Comparative study on the interaction between flavonoids with different core structures and hyaluronidase. Spectrochimica Acta Part A 2021; 262:120079. [DOI] [PubMed] [Google Scholar]
- 33.Mio K, Carrette O, Maibach HI, Stern R.. Evidence that the serum inhibitor of hyaluronidase may be a member of the inter-alpha-inhibitor family. J Biol Chem 2000;275:32413–32421. [DOI] [PubMed] [Google Scholar]
- 34.Proksch E. pH in nature, humans and skin. J Dermatol 2018;45:1044–1052. [DOI] [PubMed] [Google Scholar]
- 35.Abels C, Angelova-Fischer I.. Skin care products: age-appropriate cosmetics. Curr Probl Dermatol 2018;54:173–182. [DOI] [PubMed] [Google Scholar]
- 36.Khavkin J, Ellis DAF.. Aging skin: Histology, physiology, and pathology. Facial Plastic Surg Clinics North Am 2011;19:229–234. [DOI] [PubMed] [Google Scholar]
- 37.Voegeli R, Gierschendorf J, Summers B, Rawlings AV.. Facial skin mapping: from single point bio-instrumental evaluation to continuous visualization of skin hydration, barrier function, skin surface pH, and sebum in different ethnic skin types. Inter J Cosmetic Sci 2019;41:411–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Proksch E, Brandner JM, Jensen JM.. The skin: an indispensable barrier. Exp Dermatol 2008;17:1063–1072. [DOI] [PubMed] [Google Scholar]
- 39.Elias PM, Wakefield JS, Skin barrier function. In: Krutmann J, Humbert P, ed. Nutrition for healthy skin: strategies for clinical and cosmetic practice. New York City, NY: Springer Publishing; 2011, p. 35–48. [Google Scholar]
- 40.Mitragotri S, Anissimov YG, Bunge AL, Frasch HF, et al. Mathematical models of skin permeability: an overview. Inter J Pharm 2011;418:115–129. [DOI] [PubMed] [Google Scholar]
- 41.Maity N, Nema NK, Abedy MK, et al. Exploring tagetes erecta linn flower for the elastase, hyaluronidase and MMP-1 inhibitory activity. J Ethnopharmacol 2011;137:1300–1305. [DOI] [PubMed] [Google Scholar]
- 42.Fraternale D, Flamini G, Ascrizzi R.. In Vitro Anticollagenase and antielastase activities of essential oil of Helichrysum italicum subsp. italicum (Roth) G. Don. J Med Food 2019;22:1041–1046. [DOI] [PubMed] [Google Scholar]
- 43.Oguro T, Liu J, Klaassen CD, Yoshida T.. Inhibitory effect of oleanolic acid on 12-O-tetradecanoylphorbol-13-acetate-induced gene expression in mouse skin. Toxicol Sci 1998;45:88–93. [DOI] [PubMed] [Google Scholar]