

ABSTRACT
Inhibitors of programmed cell death protein 1 (PD-1) and its ligand (PD-L1) have dramatically changed the treatment landscape for patients with cancer. Clinical activity of anti-PD-(L)1 antibodies has resulted in increased median overall survival and durable responses in patients across selected tumor types. To date, 6 PD-1 and PD-L1, here collectively referred to as PD-(L)1, pathway inhibitors are approved by the US Food and Drug Administration for clinical use. The availability of multiple anti-PD-(L)1 antibodies provides treatment and dosing regimen choice for patients with cancer. Here, we describe the nonclinical characterization of dostarlimab (TSR-042), a humanized anti-PD-1 antibody, which binds with high affinity to human PD-1 and effectively inhibits its interaction with its ligands, PD-L1 and PD-L2. Dostarlimab enhanced effector T-cell functions, including cytokine production, in vitro. Since dostarlimab does not bind mouse PD-1, its single-agent antitumor activity was evaluated using humanized mouse models. In this model system, dostarlimab demonstrated antitumor activity as assessed by tumor growth inhibition, which was associated with increased infiltration of immune cells. Single-dose and 4-week repeat-dose toxicology studies in cynomolgus monkeys indicated that dostarlimab was well tolerated. In a clinical setting, based on data from the GARNET trial, dostarlimab (Jemperli) was approved for the treatment of adult patients with mismatch repair–deficient recurrent or advanced endometrial cancer that had progressed on or following prior treatment with a platinum-containing regimen. Taken together, these data demonstrate that dostarlimab is a potent anti-PD-1 receptor antagonist, with properties that support its continued clinical investigation in patients with cancer.
KEYWORDS: Anti-PD-1 antibody, cancer, characterization, dostarlimab, TSR-042, solid tumors, immune checkpoint
Introduction
Immunotherapy has drastically changed the therapeutic landscape for patients with cancer. Immunotherapies targeting checkpoint proteins such as programmed cell death protein-1 (PD-1) and cytotoxic T-lymphocyte–associated protein-4 (CTLA-4) have resulted in increased median overall survival and durable responses in patients across multiple tumor types.1
PD-1 is an inhibitory immune checkpoint receptor expressed on activated T cells. Through interactions with its ligands, programmed cell death ligands 1 and 2 (PD-L1 and PD-L2), PD-1 suppresses activated effector T-cell functions, including proliferation, cytokine production, and cytotoxic activity. Upregulation of PD-L1 by tumor cells is one of the mechanisms by which tumor cells can evade the immune system and interfere with cancer-specific immune responses.2,3 Preclinical and clinical studies have demonstrated that therapies that bind to either the PD-1 receptor or ligand, and effectively block the receptor–ligand interaction, can activate the antitumor immune response and increase patient survival across a variety of cancers.4 To date, there are 6 PD-1 and PD-L1, collectively referred to here as PD-(L)1, inhibitors approved by the US Food and Drug Administration for clinical use.5–10 This competitive landscape of anti-PD-1 antibodies provides patients with cancer with possibilities for different dosing regimens, tolerability profiles, disease-specific treatments, and pricing options. However, not all patients respond to a single treatment, indicating a need for more optimized therapeutic agents.
Dostarlimab (TSR-042, JEMPERLI) is an approved, humanized, monoclonal antibody (mAb) of the immunoglobulin G (IgG) 4κ isotype designed to bind to PD-1 and block interaction with its ligands, PD-L1 and PD-L2. Dostarlimab was derived from a mouse mAb that was humanized by grafting the heavy- and light-chain complementarity-determining regions onto the germline variable region frameworks of their nearest human species orthologs, followed by affinity maturation via mammalian cell display and somatic hypermutation, using the AnaptysBio SHM-XEL system.11,12 Owing to the lack of cross-reactivity of dostarlimab to mouse PD-1, its single-agent antitumor activity was evaluated using humanized mouse models. In this model system, dostarlimab demonstrated antitumor activity as assessed by tumor growth inhibition, which was associated with increased infiltration of immune cells. These data demonstrate that dostarlimab is a potent anti-PD-1 receptor antagonist, with properties that support its continued clinical investigation in patients with cancer.
Dostarlimab is currently being evaluated in several solid tumor types in the Phase 1 GARNET trial (NCT02715284), a dose escalation and safety/efficacy cohort expansion study.13,14 Dostarlimab has been approved in patients with deficient mismatch repair endometrial cancer after demonstrating clinically meaningful antitumor activity, with an objective response rate of 42.3%, a disease control rate of 57.7%, and a safety profile consistent with that of approved anti-PD-1 drugs.15,16
Results
Binding of dostarlimab to human and cynomolgus monkey PD-1
Binding profiles of dostarlimab to human and monkey PD-1 proteins were assessed by 2 orthogonal methods: surface plasmon resonance (SPR) and flow cytometry.
Biochemical-binding characteristics of dostarlimab to PD-1
SPR was used to assess the biochemical-binding characteristics of dostarlimab to the extracellular domain of purified human and cynomolgus monkey PD-1 protein. The binding affinities of dostarlimab to human and cynomolgus monkey PD-1 were 0.3 and 0.5 nM, respectively, suggesting strong binding affinity to PD-1 proteins. For human PD-1, dostarlimab displayed an association rate of 5.7 × 105 (M–1s–1) and dissociation rate of 1.7 × 10–4 (s–1), suggesting rapid association with the target and slower dissociation from target (Table 1). Binding kinetics to human and cynomolgus monkey PD-1 were similar, with less than a 2-fold difference in observed affinity constant (KD) values. High binding affinity to cynomolgus PD-1 also supports using the monkey as a relevant species for preclinical toxicologic evaluation.
Table 1.
Biochemical and cell-based binding of dostarlimab to PD-1
| Kinetic parameters (SPR) |
PD-1–expressing CHO cellsa | |||
|---|---|---|---|---|
| Kassoc (M–1s–1) | Kdissoc (s–1) | KD (nM) | EC50 (nM) | |
| Human PD-1 | 5.7 × 105 | 1.7 × 10–4 | 0.30 | 2.0 |
| Cynomolgus PD-1 | 4.3 × 105 | 2.3 × 10–4 | 0.53 | 3.4 |
aDostarlimab binding was not detected on nontransfected CHO cells.
CHO: Chinese hamster ovary; EC50: half-maximal effective concentration; Kassoc: association rate constant; KD: dissociation constant; Kdissoc: dissociation rate constant; SPR: surface plasmon resonance.
Cell surface binding of dostarlimab to PD-1
Dostarlimab binds to recombinant human and cynomolgus monkey cell surface PD-1 expressed on CHO-K1 cells with calculated half-maximal effective concentration (EC50) values of 2.0 and 3.4 nM, respectively. These data are consistent with the amino acid sequence identity between the extracellular domain of human and cynomolgus monkey PD-1 (97%). We also measured dostarlimab binding to endogenous PD-1 on human and cynomolgus monkey peripheral blood mononuclear cells (PBMCs). On gated CD3+ T cells, dose-dependent receptor occupancy was observed in 3 donors, which saturated at dostarlimab concentrations above 14 nM (2000 ng/mL; Figure 1).
Figure 1.
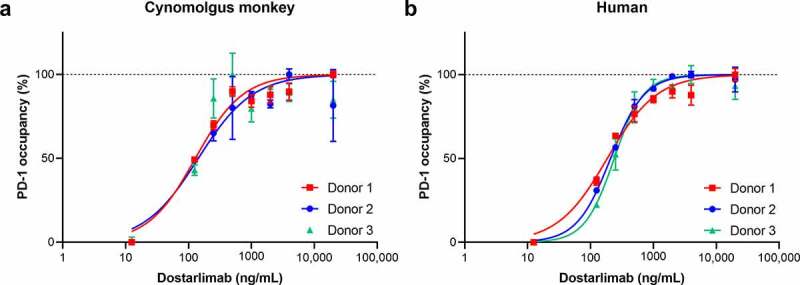
Dostarlimab binding to native PD-1 receptor expressed on human and cynomolgus monkey CD3+ T cells. Dostarlimab binds with high affinity to native human and cynomolgus monkey PD-1 expressed on the surface of CD3+ T cells. Occupancy of dostarlimab on cynomolgus monkey and human CD3+ T cells was assessed by preincubating PBMCs from either (a) cynomolgus monkey or (b) human donors with dostarlimab at varying concentrations and then incubating with either a saturating concentration (20 μg/mL) of dostarlimab or IgG4 isotype control before detection with either FITC-anti-CD3 or PE-anti-IgG4 antibodies and analysis by flow cytometry. The number of CD3+/IgG4+ cells was quantified, and percentage receptor occupancy was calculated as described in Materials and Methods. FITC: fluorescein isothiocyanate; IgG: immunoglobulin G; PBMC: peripheral blood mononuclear cells; PD-1: programmed cell death protein 1; PE: phycoerythrin
Effect of dostarlimab on interaction of PD-1 with its ligands PD-L1 and PD-L2
Compared with an IgG4 isotype control, dostarlimab efficiently blocked PD-1/PD-L1 and PD-1/PD-L2 binding, with calculated half-maximal inhibitory concentration (IC50) values of 1.8 and 1.5 nM, respectively (Figure 2a). In a cell-based functional reporter assay, dostarlimab potently inhibited the interaction between PD-1 and PD-L1 with an EC50 of approximately 2 nM (Figure 2b). This result was consistent with data generated using soluble PD-L1 (Figure 2a).
Figure 2.
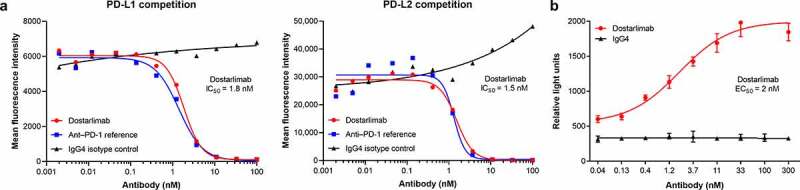
Dostarlimab disrupts binding of PD-1 to its ligands. Dostarlimab potently blocks the interaction of PD-1 with its ligands PD-L1 and PD-L2 and inhibits PD-1/PD-L1-driven signaling. (a) Dostarlimab inhibits the binding of labeled PD-L1-mFc-DyL650 and PD-L2-mFc-DyL650 to CHO-K1 cells stably expressing recombinant human PD-1. (b) Dostarlimab relieves PD-1/PD-L1-mediated repression of an NFAT-luciferase reporter downstream of TCR signaling. For a detailed description of the assay, see Materials and Methods. CHO: Chinese hamster ovary; EC50: half-maximal effective concentration; IC50: half-maximal inhibitory concentration; NFAT: nuclear factor of activated T cells response element; PD-1: programmed cell death protein 1; PD-L1(2): programmed cell death ligand 1(2); RLU: relative light units; TCR: T-cell receptor
Fc domain-mediated functions of dostarlimab
Immunoglobulins bound to cell surface receptors, such as PD-1, can attract natural killer cells, macrophages, and monocytes. Binding of these cells via fragment crystallizable gamma (Fcγ) receptors (particularly FcγRI and FcγRIIIA) to the Fc portion of immunoglobulins can initiate antibody-dependent cellular cytotoxicity (ADCC).17 However, complement-dependent cytotoxicity (CDC) is initiated by binding of complement component 1q (C1q) to the Fc portion of immunoglobulins. Different IgG isotypes bind to Fc receptors and complement proteins with different affinities, thereby eliciting different levels of effector function. IgG4 isotypes have very low effector function and hence are a valuable backbone for therapeutic antibodies that require minimal Fc function.18 Therefore, IgG4 isotype was selected for dostarlimab to ensure that it did not mediate clearance of tumor-reactive T cells.
To evaluate Fc-mediated functions of dostarlimab, binding to multiple Fc receptors and C1q protein was assessed. In comparison to a human IgG1 control antibody (rituximab [MabThera]; Roche) and consistent with expectations of a human IgG4 antibody, C1q showed no binding to dostarlimab (Supplementary Figure S1). We measured the binding properties of dostarlimab to Fcγ receptors using enzyme-linked immunosorbent assays (ELISA) and SPR methodologies. Whereas Humira (adalimumab) and MabThera, which are both IgG1 antibodies, effectively bound FcγRIIIA and FcγRI receptors, respectively, dostarlimab binding affinity estimates against FcγRIIIA were minimal and slightly reduced against the FcγRI receptor, as expected for an IgG4 antibody (Supplementary Table S1 and Supplementary Figure S2).
Effect of dostarlimab on T-cell activation in vitro
The effects of dostarlimab on T-cell function were assessed by three orthogonal methods: (1) mixed lymphocyte reaction (MLR); (2) in vitro polyclonal T-cell activation assay using staphylococcal enterotoxin B (SEB); and (3) an antigen-dependent T-cell activation system using a combination of influenza/purified protein derivative/tetanus toxoid (flu/PPD/TT) antigens.
The functional antagonist activity of dostarlimab in augmenting in vitro responses of primary human CD4+ T cells was tested in a human CD4+ T-cell MLR assay. Antagonism of PD-1 in this assay has been shown to result in increased T-cell activation, as measured by increased interleukin (IL)-2 production.19 Antagonism of PD-1 by dostarlimab increased IL-2 production with an EC50 of approximately 1 nM (n = 4; range, 0.13–2 nM). An illustrative example is shown in Figure 3a.
Figure 3.
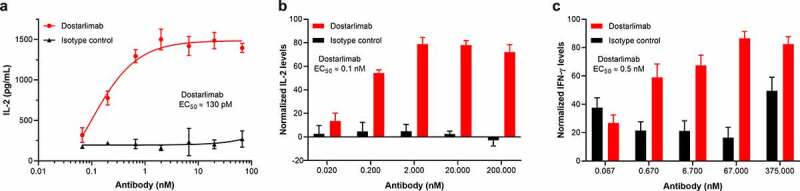
Functional activity of dostarlimab in primary T-cell–based assays. The functional activity of dostarlimab was evaluated in several in vitro assays, including (a) allogeneic MLR, (b) SEB activation, and (c) flu/PPD/TT activation assays. In the MLR assay, dendritic cells and allogeneic CD4+ T cells were incubated in the presence of dostarlimab or isotype control at the indicated concentrations for 48 hours, and activation of T cells was determined by quantifying the level of IL-2 secretion. Representative data from 4 donor combinations is shown in (a). PBMCs from 5 and 3 donors, respectively, were incubated as described in Materials and Methods with either SEB or flu/PPD/TT in the presence of dostarlimab or isotype control at the indicated concentrations for 72 hours and 5 days, respectively. Activation of T cells was determined by measuring the level of IL-2 or IFN-γ secretion. Results shown are the normalized mean ± SEM values across all donors. IFN: interferon; IL: interleukin; MLR: mixed lymphocyte reaction; PBMC: peripheral blood mononuclear cell; PPD: purified protein derivative from Mycobacterium tuberculosis tuberculin; SEB: staphylococcal enterotoxin B; SEM: standard error of the mean; TT: tetanus toxoid antigen
The immunostimulatory capacity of dostarlimab was tested in a functional T-cell activation assay. The addition of dostarlimab to SEB-stimulated PBMCs led to increased IL-2 production (Figure 3b), with an EC50 of approximately 0.1 nM, which supports induction of increased T-cell activation by dostarlimab.
The ability of dostarlimab to enhance antigen-specific (flu/PPD/TT) T-cell activation was also evaluated. In this assay, consistent with its ability to functionally antagonize PD-1, dostarlimab treatment enhanced interferon (IFN)-γ release with an EC50 of approximately 0.5 nM (Figure 3c). Taken together, all these functional assay systems support that dostarlimab can enhance T-cell function in vitro.
Dostarlimab lacks T-cell agonist activity
To determine whether dostarlimab could stimulate T cells in the absence of T-cell receptor (TCR) activation, we tested dostarlimab in a cytokine release assay.20 In this assay, stimulation of healthy donor PBMCs with anti-CD3– and anti-CD28-coated beads for 48 hours induced a dose-dependent increase in IFN-γ and IL-2 production. Dostarlimab incubated with PBMCs at doses of 1 to 100 μg/mL did not induce significant production of either cytokine. Multiplexed analysis of culture supernatants from dostarlimab-treated PBMCs confirmed no significant production of IFN-γ, tumor necrosis factor alpha (TNF-α), IL-2, IL-4, IL-6, or IL-10 (Supplementary Table S2). These data support lack of T-cell agonist activity of dostarlimab.
Antitumor activity and pharmacodynamics of dostarlimab monotherapy in humanized mouse models
As dostarlimab does not cross-react with mouse PD-1, the ability of dostarlimab to exhibit antitumor activity in vivo was assessed in humanized mouse models representative of human non–small cell lung cancer and breast cancer.21,22 Briefly, highly immunodeficient NOG-EXL mice that express human granulocyte-macrophage colony-stimulating factor (GM-CSF) and human IL-3 (Taconic Bioscience, NY) were humanized by engraftment of CD34+ human hematopoietic stem cells from cord blood, resulting in functional human immune cells in the host mice.23 Compared to treatment with the IgG4 isotype control, dostarlimab monotherapy resulted in greater inhibition of tumor growth in an A549 lung cancer model (tumor growth inhibition [TGI] of 62% at termination; Figure 4b). Pharmacodynamic changes in tumor-associated CD45+ immune cells after treatment with dostarlimab were assessed at the end of the study. The antitumor activity of dostarlimab was associated with a reduction in tumor-associated regulatory T cells and a trend toward increased tumor-infiltrating CD8+ T cells (Figure 4c). Relative to isotype control, dostarlimab monotherapy also inhibited tumor growth of MDA-MB-436 breast cancer model (TGI of 53%; Figure 5b).
Figure 4.
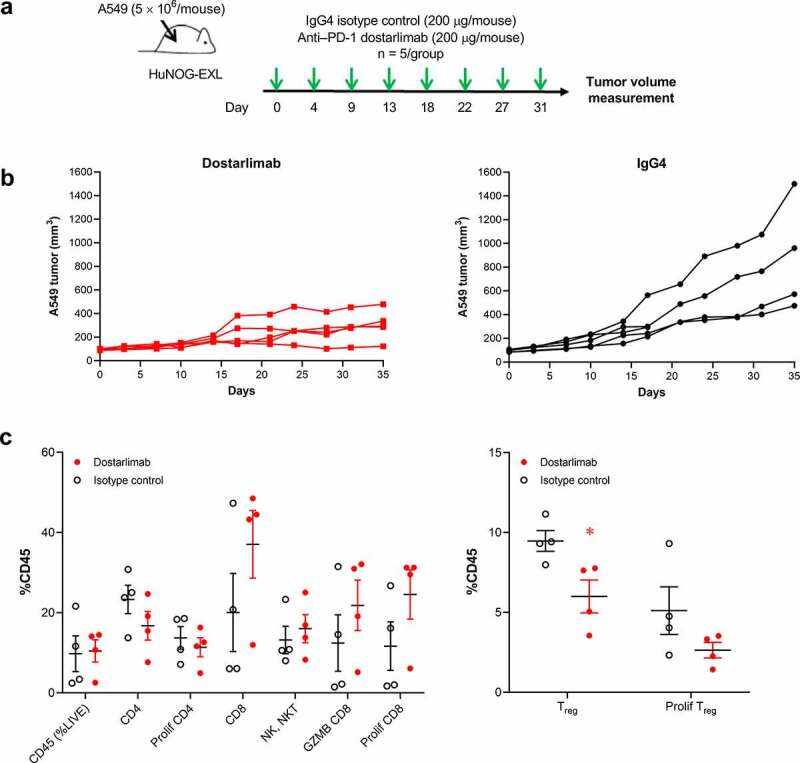
Antitumor and pharmacodynamic activity of dostarlimab monotherapy in a humanized mouse model of non–small cell lung cancer. (a) Study schema: dostarlimab or IgG4 isotype control (200 µg/mouse, twice weekly) was dosed to established A549 tumors (80–120 mm3) grown as xenografts in humanized NOG-EXL mice (Taconic), and (b) antitumor activity and (c) pharmacodynamic effects in the tumor. Human CD4+, CD8+, NK/NKT, and activated CD8+ (GZMB, CD8+) cells in the tumors were quantified by flow cytometry as percentage of human CD45+ cells. *p < .05 (unpaired t test). PD-1: programmed cell death protein 1; CD-8: cluster of differentiation-8; GZMB: Granzyme B; IgG: immunoglobulin G; NK: natural killer; Treg: regulatory T cells
Figure 5.
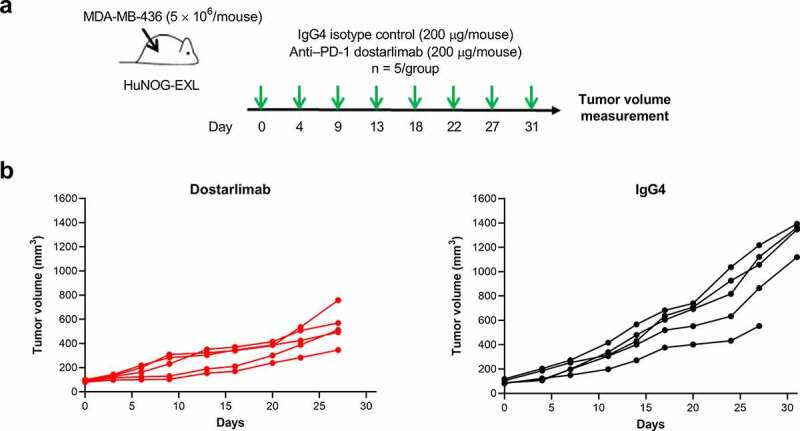
Antitumor activity of dostarlimab monotherapy in a humanized mouse model of breast cancer. (a) Study schema; dostarlimab or IgG4 isotype control (200 μg/mouse, twice weekly) was dosed to already established MDA-MB-436 tumors (80–120 mm3) grown as xenografts in humanized NOG-EXL mice (Taconic), and (b) antitumor activity. PD-1: programmed cell death protein 1; IgG: immunoglobulin G
Pharmacokinetics and toxicity of dostarlimab in cynomolgus monkeys
The pharmacokinetics and toxicology of dostarlimab were assessed in cynomolgus monkeys. In a single-dose pharmacokinetics study, dostarlimab (10, 30, and 100 mg/kg) was administered to monkeys via intravenous (IV) infusion for 15 minutes. The concentration–time profiles of dostarlimab were characterized by an initial brief distribution phase, followed by a linear beta elimination phase and a terminal antidrug antibody-mediated elimination phase (Figure 6). Pharmacokinetic parameters are summarized in Table 2. The observed time to maximum concentration (Tmax) was 0.5–1.0 hour post-dose, and mean volume of distribution at steady state (Vss) was 46.5, 48.5, and 43.9 mL/kg after the administration of 10, 30, and 100 mg/kg, respectively. Mean IV clearances (CLs) were 0.215, 0.186, and 0.258 mL/h/kg for the 10-, 30-, and 100-mg/kg dose groups, respectively. Individual terminal half-lives ranged from 14.9 to 283 hours. Overall, as the dosage increased from 10 to 100 mg/kg, exposure to dostarlimab (maximum concentration [Cmax] and area under the concentration–time curve [AUC] from 0 to 1320 hours [AUC0–1320h]) increased in an approximately dose-proportional manner, without the observation of target-mediated disposition.24 Based on the CL and Vss, dostarlimab is a typical mAb.25 No sex-specific differences were observed at any of the three doses evaluated.
Figure 6.
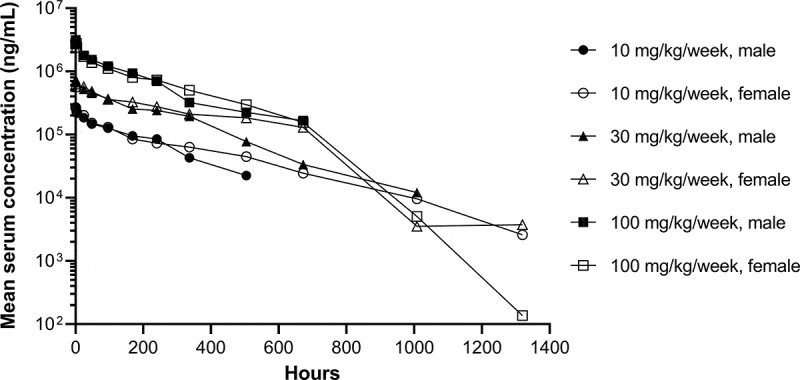
Concentration–time profiles of single-dose dostarlimab. Mean serum concentration–time profiles of dostarlimab after single intravenous infusion of dostarlimab to male and female cynomolgus monkeys. Concentration–time profiles were plotted through the first post-dose time point that was below the limit of quantification, if applicable. The lower limit of quantification was 31.0 ng/mL
Table 2.
Mean pharmacokinetic parameters for treatment groups of dostarlimab after single-dose intravenous infusion to monkeys
| Dose (mg/kg) | Sex | Cmax (μg/mL) | Tmax (h) | t1/2 (h) | AUC0–1320 (h × μg/mL) | Vss (mL/kg) | MRT (h) | CL (mL/h/kg) |
| 10 | Male | 265 ± 9.87 | 0.5 (0.5–1.0) | 31.0 (14.9–97.0) | 45,700 ± 21,800 | 39.9 ± 8.17 | 193 ± 129 | 0.248 ± 0.0933 |
| Female | 274 ± 6.81 | 0.5 (0.5–1.0) | 168.6 (72.1–282.6) | 57,400 ± 14,400 | 53.1 ± 5.28 | 314 ± 110 | 0.181 ± 0.0547 | |
| Combined | 269 ± 9.04 | 0.5 (0.5–1.0) | 84.5 (14.9–282.6) | 51,600 ± 17,700 | 46.5 ± 9.49 | 254 ± 126 | 0.215 ± 0.0778 | |
| 30 | Male | 758 ± 77.0 | 0.5 (0.5–1.0) | 58.5 (26.8–243.2) | 145,000 ± 34,100 | 48.5 ± 7.90 | 242 ± 99.0 | 0.213 ± 0.0465 |
| Female | 693 ± 44.7 | 0.5 (0.5–0.5) | 123.0 (86.7–217.2) | 189,000 ± 12,400 | 48.6 ± 6.81 | 307 ± 30.5 | 0.158 ± 0.00896 | |
| Combined | 725 ± 66.6 | 0.5 (0.5–1.0) | 104.9 (26.8–243.2) | 167,000 ± 33,200 | 48.5 ± 6.61 | 275 ± 74.5 | 0.186 ± 0.0424 | |
| 100 | Male | 3400 ± 80.0 | 1.0 (0.5–4.0) | 56.7 (31.1–138.7) | 426,000 ± 224,000 | 41.0 ± 39.4 | 282 ± 170 | 0.274 ± 0.114 |
| Female | 2930 ± 135 | 1.0 (0.5–1.0) | 60.7 (19.9–66.2) | 437,000 ± 126,000 | 47.5 ± 48.4 | 271 ± 213 | 0.242 ± 0.0685 | |
| Combined | 3170 ± 274 | 1.0 (0.5–4.0) | 58.7 (19.9–138.7) | 432,000 ± 163,000 | 43.9 ± 7.22 | 192 ± 79.9 | 0.258 ± 0.0863 |
Data are presented as mean ± standard deviation for Cmax, AUC0–1320, Vss, MRT, and CL values, and median (range) for Tmax and t1/2 values. AUC0–1320: area under the concentration–time curve from time 0 to 1320 hours; CL: clearance; Cmax: maximum concentration; MRT: mean residence time; t1/2: half-life; Tmax: time to Cmax; Vss: volume of distribution at steady state.
In a 1-month repeat-dose toxicology study, dostarlimab was well tolerated when administered once weekly to male and female cynomolgus monkeys via IV infusion at doses of 10, 30, or 100 mg/kg. No unexpected deaths, no drug-related clinical signs, and no effect on body weight were observed during the study. The exposure (Cmax and AUC from 0 to 168 hours [AUC0–168h]) increased in an approximate proportion to the dose on days 1 and 22. There were no sex-specific differences in exposure between males and females (Table 3). The Tmax ranged from 0.5 to 8 hours. Drug accumulation was observed with the once-weekly dosing schedule (Table 3). The accumulation ratio was 2.9 (10 mg/kg), 2.4 (30 mg/kg), and 2.1 (100 mg/kg) when AUC0–168h was compared between day 22 (the fourth dose) and day 1 (the first dose).
Table 3.
Mean pharmacokinetic parameters for treatment groups of dostarlimab after intravenous infusion to monkeys at day 1 and day 22 in 1-month repeat-dose toxicology study
| Dose (mg/kg) | Study day | Sex | Cmax (μg/mL) | Tmax (h) | t1/2 (h) | AUC0–168 (h*μg/mL) |
|---|---|---|---|---|---|---|
| 10 | 1 | Male | 257 ± 77.8 | 0.5 (0.5–4.0) | 260.1 (140.7–597.2) | 17,700 ± 3870 |
| Female | 273 ± 27.2 | 0.5 (0.5–4.0) | 211.9 (133.9–346.6) | 19,800 ± 2620 | ||
| Combined | 265 ± 56.1 | 0.5 (0.5–4.0) | 211.9 (133.9–597.2) | 18,800 ± 3340 | ||
| 22 | Male | 451 ± 33.5 | 0.5 (0.5–0.5) | 249.3 (191.3–424.3) | 54,600 ± 5110 | |
| Female | 488 ± 49.3 | 0.5 (0.5–4.0) | 187.3 (175.3–542.3) | 54,300 ± 8540 | ||
| Combined | 469 ± 44.5 | 0.5 (0.5–4.0) | 217.8 (175.3–542.3) | 54,400 ± 6710 | ||
| 30 | 1 | Male | 691 ± 76.6 | 0.5 (0.5–0.5) | 184.5 (109.4–294.1) | 54,300 ± 5730 |
| Female | 873 ± 77.6 | 4.0 (0.5–8.0) | 137.7 (107.8–224.0) | 68,000 ± 9350 | ||
| Combined | 782 ± 120 | 0.5 (0.5–8.0) | 174.9 (107.8–294.1) | 61,200 ± 10,300 | ||
| 22 | Male | 1260 ± 251 | 4.0 (0.5–8.0) | 443.6 (111.8–810.2) | 147,000 ± 57,700 | |
| Female | 1380 ± 197 | 4.0 (0.5–8.0) | 222.0 (192.9–438.6) | 147,000 ± 22,100 | ||
| Combined | 1320 ± 224 | 4.0 (0.5–8.0) | 318.2 (111.8–810.2) | 147,000 ± 41,700 | ||
| 100 | 1 | Male | 2380 ± 475 | 0.5 (0.5–0.5) | 227.5 (165.5–305.1) | 193,000 ± 14,800 |
| Female | 2850 ± 718 | 0.5 (0.5–8.0) | 144.1 (110.3–191.0) | 201,000 ± 16,800 | ||
| Combined | 2620 ± 630 | 0.5 (0.5–8.0) | 172.3 (110.3–305.1) | 197,000 ± 15,600 | ||
| 22 | Male | 4080 ± 313 | 4.0 (4.0–8.0) | 286.6 (161.2–468.6) | 473,000 ± 69,000 | |
| Female | 4060 ± 1470 | 0.5 (0.5–8.0) | 175.3 (57.1–266.0) | 365,000 ± 191,000 | ||
| Combined | 4070 ± 1010 | 4.0 (0.5–8.0) | 212.8 (57.1–468.6) | 419,000 ± 148,000 |
Data are presented as mean ± standard deviation for Cmax and AUC0–168 values, and median (range) for Tmax and t1/2 values. AUC0–168: area under the concentration–time curve from time 0 to 168 hours; Cmax: maximum concentration; t1/2: half-life; Tmax: time to Cmax.
Discussion
Preclinical characterization of dostarlimab demonstrated a favorable anti-PD-1 antibody profile, including effectively binding to PD-1 and successfully antagonizing the interaction with PD-L1 and PD-L2. Dostarlimab binds with high affinity to the human PD-1 receptor, with a binding affinity (KD) of 300 pM. This binding profile is similar to preclinical data reported for the approved PD-1 therapies nivolumab, pembrolizumab, and cemiplimab.26–29 IgG4 isotype was selected for dostarlimab to produce the most reliable and efficacious therapeutic modality. All three approved anti-PD-1 antibodies (pembrolizumab, nivolumab, and cemiplimab) are IgG4 modalities; anti-PD-L1 antibodies (atezolizumab, avelumab, and durvalumab) are IgG1 isotypes. In a therapeutic setting where immune enhancement is the outcome, chronic administration of IgG1 Fc with effectorless mutations could introduce additional immunogenic sites. IgG2 antibodies are more challenging to manufacture than IgG1 or IgG4.30
Dostarlimab also demonstrated effective enhancement of T-cell activation in multiple in vitro functional assay systems using primary human T cells. In addition, while dostarlimab enhanced T-cell activation in antigen-dependent systems, it did not have any direct nonspecific effects on T-cell responses as demonstrated by the lack of cytokine release upon exposure to antibody alone. Dostarlimab displayed effective antitumor activity in humanized mouse tumor models and showed a consistent pharmacokinetic and pharmacodynamic profile, with low potential for off-target effects.
Although increasing the activation state of the immune system is an effective antitumor strategy, anti-PD-(L)1 therapies are associated with a number of adverse events, including pneumonitis, colitis, hypothyroidism, and infusion-site reactions.31 These events are likely related to the binding to the target, and associated pharmacodynamic effects, such as immune-related adverse events of the anti-PD-(L)1 therapies.32 The other critical aspect in predicting the safety profile of a new anti-PD-(L)1 therapy is cytotoxicity due to the antibody’s binding to complement or FCγ receptors. Consistent with its IgG4 framework, dostarlimab had little to no binding to complement protein C1q or Fcγ, receptors that elicit CDC or ADCC, respectively,19 and hence is unlikely to result in the depletion of antitumor effector T cells.
In the single-dose pharmacokinetics study, large variations in t1/2 were observed (Table 2). Males had shorter t1/2 than females in both 10-mg/kg and 30-mg/kg groups, with similar results for 100 mg/kg. The large variation may be due to the effects of antidrug antibodies (ADAs). At days 29, 42, and 56, a higher percentage of male cynomolgus monkeys had confirmed ADAs than female cynomolgus monkeys in both 10-mg/kg and 30-mg/kg groups. For the 100-mg/kg group, both male and female cynomolgus monkeys had the same numbers of ADA-positive animals for the whole pharmacokinetics/ADA sampling process.
The cynomolgus monkey was selected as the relevant species to evaluate dostarlimab toxicity. The selection was based on DNA homology of PD-1 extracellular domains of different species, cross-species PBMC binding by dostarlimab, and PD-1 binding affinity and receptor occupancy of dostarlimab. Furthermore, the results obtained from the human and cynomolgus monkey tissue cross-reactivity studies indicated that dostarlimab stained similar cell types (including lymphocytes and monocytes) in human and cynomolgus monkey tissues. Since dostarlimab is not active in rats or mice and has low binding to dog PBMCs, the cynomolgus monkey serves as the only species for the toxicology program. This selection is in accordance with International Conference on Harmonization S6(R1) Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals, which states that although safety evaluation programs should normally include two relevant species, in certain justified cases one relevant species may suffice (e.g., when only one relevant species can be identified or when the biological activity of the biopharmaceutical is well understood). Pharmacokinetic and toxicologic evaluation of dostarlimab in single- and 4-week repeat-dose toxicology studies in cynomolgus monkeys indicated that dostarlimab was well tolerated with a dose proportional and typical mAb pharmacokinetic profile.25 These studies demonstrate a favorable risk:benefit ratio for dostarlimab and support clinical studies. Taken together, these data demonstrate that dostarlimab is a potent anti-PD-1 receptor antagonist, with properties that support its clinical investigation in patients with cancers.
The preclinical data presented here support dostarlimab’s first-in-human dose selection and demonstrate that there is a sufficient safety margin for the drug to be further evaluated in the human dose-finding parts 1 and 2A of the Phase 1 GARNET trial.33 The 1-mg/kg starting dose in the GARNET trial represents a 100-fold dose multiple (i.e., 100 mg/kg/1 mg/kg) compared to the highest dose level tested in the monkeys. Moreover, the patients can get some clinical benefit at the proposed starting dose of 1 mg/kg since it is within the efficacious dose range of the approved anti-PD-1 agent, pembrolizumab.34 After the part 1 dose escalation and part 2A safety and pharmacokinetics confirmation, a patient central therapeutic dose – four doses of dostarlimab administered every 3 weeks at 500 mg followed by every 6 weeks at 1000 mg – was established. The GARNET trial is the first trial to test a 6-week anti-PD-1 dosing regimen in patients. Based on data from the GARNET trial, dostarlimab was approved for the treatment of adult patients with mismatch repair–deficient recurrent or advanced endometrial cancer that have progression on or following prior treatment with platinum-containing regimen.16
In ongoing clinical studies, dostarlimab has demonstrated robust and durable responses and a manageable safety profile with adverse events characteristic of other anti-PD-1 therapies.13,14 Dostarlimab shows promise as an anti–PD-1 therapy, and clinical trials are ongoing in which dostarlimab is being evaluated for use as monotherapy and combination therapy across multiple tumor types, including RUBY (NCT03981796), FIRST (NCT03602859), IOLite (NCT03307785), and MOONSTONE (NCT03955471).13,14
Materials and methods
Preparation of dostarlimab
Dostarlimab, a humanized mouse mAb to human PD-1, was generated from human (gamma)4 (S228P)/(kappa) constant regions (designated IgG4 [S228P]) and optimized variable regions, using AnaptysBio’s SHM-XEL platform.35 To support non-GLP preclinical studies, a research lot of dostarlimab was produced by stable transfection of CHO-S cells with a single, linearized vector encoding the antibody heavy and light chains, using Lipofectamine LTX (Invitrogen #15338500) according to the manufacturer’s instructions. The pool of transfected cells was subcloned, and a stable clone was selected for scale-up production based on mAb expression levels and growth characteristics. The CHO-S subclone was seeded into a 20-L WAVE Bioreactor and monitored daily for cell density and human IgG in the culture medium. Antibody in culture supernatant was purified on a MabSelect protein A affinity medium column, a high-flow agarose matrix designed for capturing antibodies in large sample volumes (GE Healthcare Life Sciences; 250-mL column). Glycine (100 mM, pH 3.0)-eluted fractions were neutralized, pooled, and buffer exchanged by tangential flow filtration (Millipore) into PBS, pH 7.4. Antibody concentrations were determined by absorption at 280 nm.
Binding assays
PD-1-binding assays
The ability of dostarlimab to bind to human and cynomolgus monkey PD-1 was assessed by flow cytometry and SPR. SPR studies were carried out using a Biacore T200, and kinetic constants were determined using the corresponding evaluation software. Experimental parameters were chosen to ensure that saturation would be reached at the highest antigen concentrations and maximum analyte-binding capacity (Rmax) values would be kept under 100 response units. Anti-human IgG (Fc-specific, ≈10,000 response units; GE Healthcare Life Sciences) was immobilized on a Biacore CM5 chip using N-(3-dimethylaminopropyl)-N´-ethylcarbodiimide hydrochloride)–activated amine coupling chemistry. Dostarlimab (0.5 μg/mL, 60-second capture time) was then captured onto this surface at 25°C. Soluble monomeric human or cynomolgus monkey PD-1 in a 2-fold serial dilution from 25 to 0.78 nM with a duplicate at 6.25 nM was flowed over captured antibody for 240 seconds at a rate of 30 μL/min, and dissociation was monitored for 600 seconds. Capture and analyte binding were performed in HBS-EP+ buffer (10 mM N-[2-hydroxyethyl]piperazine-N´[2-ethansulfonic acid] [HEPES], pH 7.6, 150 mM sodium chloride [NaCl], 3 mM ethylenediaminetetraacetic acid [EDTA], 0.05% surfactant P20; Teknova). Chips were regenerated between each run using 3 M magnesium chloride (30-second contact time followed by 60-second wash at 30 μL/min). The resulting sensorgrams were fitted globally using a 1:1 binding model to calculate on and off rates (kassoc and kdissoc, respectively) and dissociation constants as a measure of overall affinity (KD).
For the flow cytometry–based binding studies, dostarlimab (0.1–100 nM, 3-fold dilutions) was added to Chinese hamster ovary (CHO)-K1 cells (1 × 105) stably expressing either full-length human or cynomolgus monkey PD-1 and incubated on ice for 30 minutes. Cells were washed twice in phosphate-buffered saline (PBS) with 1% bovine serum albumin (BSA) and incubated for 30 minutes on ice with phycoerythrin (PE)-conjugated goat anti-human IgG Fc (1:100; Southern Biotech, Cat#2014-09) or PE-conjugated mouse anti-human IgG4 Fc (1:500; Southern Biotech, Cat#9200-09) to detect antibody binding. Cells were washed and resuspended in the presence of propidium iodide to exclude dead cells or were fixed in PBS with 1% paraformaldehyde and analyzed for fluorescence on a BD FACSArray (BD Biosciences). Data were analyzed and graphed for median fluorescence intensity, and curves were fit for EC50 calculation in GraphPad Prism (GraphPad Software, Inc.) using a nonlinear (sigmoidal) regression analysis.
To determine the binding of dostarlimab to native PD-1, cryopreserved PBMCs from healthy human donors or cynomolgus monkeys (n = 3) were thawed and rested overnight at 37°C/5% carbon dioxide (CO2). Then, 4 × 105 cells in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% BSA were treated with human A/B serum (Innovative Research) to block Fc receptors and incubated with dostarlimab (20 μg/mL–12.5 ng/mL) for 30 minutes at 4°C followed by washing 3 times with MACS buffer (Miltenyi). The samples were then split evenly, and 1 set of cells was incubated with a saturating concentration of dostarlimab (20 μg/mL) and another with IgG4 control antibody (20 μg/mL; Eureka Therapeutics) for 30 minutes at 4°C. Cells were then washed 4 times with MACS buffer (Miltenyi), stained with fluorescein isothiocyanate (FITC)-anti-CD3 and PE-anti-IgG4 antibodies (both from SouthernBiotech, Cat#9515-02 and Cat#9200-09, respectively), and analyzed by flow cytometry. The number of CD3+/IgG4+ cells was determined for each condition, and the percentage of dostarlimab occupancy was determined by dividing the number of CD3+/IgG4+ cells at a given concentration of antibody incubation by the number of CD3+/IgG4+ cells at a saturating concentration of dostarlimab incubation.
PD-1/PD-L1 and PD-1/PD-L2 receptor-ligand competition assays
Human PD-L1 mouse IgG1 Fcγ-fusion protein and human PD-L2 mouse IgG1 Fcγ-fusion protein were expressed, purified, and labeled with DyLight 650 (Thermo Fisher). A dose-response analysis of PD-L1 and PD-L2 binding to PD-1–expressing CHO-K1 cells was carried out, and it was determined that binding of PD-L1-Fc and PD-L2-Fc fusion proteins to cell surface PD-1 did not saturate, despite the high density of expression of PD-1 on CHO-K1 cells. To produce a sensitive competition assay with a reproducible signal within a larger dynamic range, an approximate EC30 value for both reagents was calculated and selected for the standard assay conditions. To quantify blocking of ligand binding to PD-1 in CHO cells, dostarlimab was premixed in 3-fold dilutions from 100 to 0.0017 nM with the EC30 concentration of PD-L1-mFc-DyL650 (10 nM) or PD-L2-mFc-DyL650 (5 nM). The mixture was added to human PD-1 CHO-K1 cells (3 × 105) and incubated for 30 minutes at 4°C. Cells were washed once, resuspended in PBS with 1% BSA in the presence of propidium iodide, and DyL650-PD-L1 or DyL650-PD-L2 binding was analyzed on a BD FACSArray (BD Bioscience), excluding dead cells. Data were analyzed for DyL650-PD-L1 or DyL650-PD-L2 median fluorescence intensity. Curves were fitted for IC50 calculation using a nonlinear regression analysis in GraphPad Prism. A cell-based PD-1/PD-L1 functional reporter assay was also used. In this system (PD-1/PD-L1 Blockade Bioassay; Promega, Cat#J1250), CHO cells were engineered to express recombinant human PD-L1 and a proprietary cell surface protein designed to activate TCRs in an antigen-independent manner. Jurkat cells were engineered to express PD-1 and a luciferase reporter driven by a nuclear factor of activated T cells response element (NFAT-RE). When the 2 cell types are co-cultured, TCR signaling-driven NFAT-RE–mediated luminescence is inhibited by the PD-1/PD-L1 interaction. To test the PD-1/PD-L1 blocking activity of dostarlimab, PD-L1–expressing CHO cells were plated and incubated at 37°C/5% CO2 for 16–20 hours. Dostarlimab or isotype control antibody (3-fold dilutions from 300 nM) and PD-1–expressing Jurkat reporter cells were added, and plates incubated for 6 hours at 37°C/5% CO2. Bio-Glo reagent (Promega) was added, and luminescence was read on a luminometer plate reader. Curves were fitted for EC50 calculation using a nonlinear regression analysis in GraphPad Prism.
CDC/ADCC-binding assays
To assess the potential for complement fixation by dostarlimab, an ELISA-based C1q binding assay was employed. Dostarlimab or a positive control (MabThera) was coated onto a 96-well polystyrene plate at 8 concentrations: 10, 7.7, 5.9, 4.6, 3.5, 2.7, 2.1, and 1.6 µg/mL. The plate was incubated overnight (2–8°C) and then aspirated. The plate was blocked for 1 hour at room temperature with 1X PBS supplemented with 0.3% BSA and 0.05% Tween 20 before being washed with 10 mM phosphate buffer at pH 7.4 supplemented with 140 mM NaCl, 2.7 mM potassium chloride, and 0.05% Tween 20. Recombinant human C1q (10 µg/mL; Quidel) was added to each well and incubated for 2 hours at room temperature. After incubation, the plate was washed and a sheep anti-human C1q/horseradish peroxidase conjugate (0.5 µg/mL; Abcam, Cat#ab46191) added. The plate was incubated for 1 hour at room temperature, washed, and SuperSignal ELISA Femto Substrate (1:1 mixture of luminol/enhancer and stable peroxide solution; Thermo Fisher Scientific) added to all wells. The plate was read within 1–5 minutes on a luminometer (MS SpectraMax), measuring total light output in relative light units (RLU). Four parameter nonlinear regression analyses were performed on each antibody titration, and EC50 values were calculated.
Binding of dostarlimab to FcγRI (Cat#1257-FC-050), FcγRIIa (Cat#9595-CD-050), FcγRIIb (Cat#1875-CD-050), FcγRIIIa (Cat#4325-FC-050), and FcγRIIIb (Cat#1597-FC-050; all from R&D Systems) was assessed by SPR using Biacore T200 and Biacore 4000 instruments in which the appropriate Fcγ receptors were immobilized to a Biacore CM5 sensor chip via amine coupling. MabThera, or Humira (adalimumab; Midwinter Solutions Ltd), an IgG1 antibody known to bind all Fcγ receptors, was used as the reference control in the assays to evaluate assay performance. Dostarlimab (0.041–10 μM) was flowed over the captured analyte for 60–300 seconds at a rate of 30 μL/min, and dissociation monitored for 60–600 seconds. Capture and analyte binding were performed in HBS-EP+ buffer (10 mM HEPES, pH 7.6, 150 mM NaCl, 3 mM EDTA, 0.05% surfactant P-20; Teknova). Chips were regenerated between each run using 1 or 2.5 mM sodium hydroxide (30-second contact time followed by a 60-second wash at 30 μL/min). The resulting sensorgrams were fitted globally using a 1:1 binding model to calculate on and off rates (kassoc and kdissoc, respectively) and dissociation constants as a measure of overall affinity (KD).
In vitro T-cell assays
Mixed lymphocyte reaction
CD4+ T cells were isolated from the PBMCs of a healthy human donor isolated from a Leukopak obtained from the San Diego Blood Bank (CD4+ T Cell Isolation Kit, human; Miltenyi Biotec), and dendritic cells were derived from a different donor, via isolation of monocytes, followed by 7 days of culture with GM-CSF and IL-4, a widely used protocol for generation of monocyte-derived dendritic cells.36 Dendritic cells (1 × 104/well) and allogeneic CD4+ T cells (1 × 105/well) were incubated in the presence of various concentrations of dostarlimab or isotype control antibody for 48 hours, and T-cell activation was quantified by the level of IL-2 secreted in culture supernatants as determined by ELISA (DuoSet, R&D Systems).
SEB stimulation
PBMCs from healthy human donors (n = 5), sourced from multiple commercial suppliers (Lonza; Stem Cell Technologies), were plated at 100,000 cells/well in 96-well flat-bottomed plates and stimulated with 100 ng/mL SEB for 3 days in the presence of dostarlimab or isotype control antibody at 37°C and 5% CO2, followed by measurement of IL-2 in cell culture supernatants using a cytometric bead array (BD Biosciences). PD-1 expression in individual donors was verified by flow cytometry. PD-1 expression was less than 5% in unstimulated CD3+ T cells and increased to >30% upon SEB stimulation (data not shown). To account for variability in levels of IL-2 production among donors, data were background subtracted and normalized to the maximum response achieved in each donor.
Flu/TT/PPD
PBMCs from three individual donors were stimulated with whole-protein preparations of influenza antigen (seasonal split virion influenza vaccine, 2016, purchased from Fluarix Tetra), tetanus toxoid antigen (purchased from NIBSC, UK, code 02/232), and purified protein derivative of Mycobacterium tuberculosis tuberculin (purchased from NIBSC, UK, code PPDT) at 0.3 μg/mL, 5 Lf/mL, and 2 μg/mL, respectively, in the presence of the anti-PD-1 antibody dostarlimab or control isotype antibody IgG4. Dostarlimab or the control IgG4 was tested over a 4-point dose–response curve, with a range of 10,000–10 ng/mL (10-fold dilution) and at 56,250 ng/mL. After 5 days in culture, IFN-γ production was determined by Luminex technology. Each condition was plated in duplicate for the collection of supernatants for cytokine analysis. To account for variability in levels of IFN-γ production among donors, data were background subtracted using a “cells only” control for the baseline and normalized to the maximum response achieved in each donor.
In vivo humanized mouse models of cancer
Twelve-week-old humanized NOG-EXL mice (highly immunodeficient NOG mice) expressing human GM-CSF and human IL-3, engrafted with CD34+ human cord blood stem cells (Taconic), were acclimated as per the Institutional Animal Care and Use Committee guidelines before implantation with either A549 (lung cancer) or MDA-MB-436 (breast cancer) cell lines. Cell lines were obtained from the American Type Culture Collection and grown in monolayer culture in RPMI 1640 medium supplemented with 10% fetal bovine serum, harvested by trypsinization, and 5 × 106 cells were implanted subcutaneously in the right flank of each mouse. Mice were randomized at tumor volumes between 80 and 120 mm3 into groups of 5 mice each. Each group was administered either human IgG4 isotype control or dostarlimab intraperitoneally at a dose of 200 μg/mouse twice weekly throughout the study duration (Figures 4a and 5a). Tumor and body weight measurements were collected twice weekly, and tumor volumes were calculated using the equation (L× W2)/2, where L and W refer to the length and width dimensions, respectively. The general health of mice was monitored daily, and all experiments were conducted in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care and the Institutional Animal Care and Use Committee guidelines. The mice were euthanized a day after the last dose, and tumors were collected in ice-cold RPMI 1640 medium and processed immediately for flow cytometry.
Tumor processing and flow cytometry
Tumors were disaggregated using the GentleMACS Mouse Tumor Dissociation kit (Miltenyi). Spleens were dissociated through a 70-µm nylon cell strainer and resuspended in red blood cell lysis buffer (Sigma) for 1 minute. All cells were then stained with a viability dye (Thermo Fisher Scientific) and blocked with Fc block (eBioscience) before staining with fluorescence-conjugated antibodies in flow cytometry staining buffer. The cells were stained for T-cell markers, including CD45, CD3, CD4, CD8, FOXP3, CCR7, Ki67, and CD45RA (eBioscience). Intracellular staining was performed using the FoxP3/Transcription Factor Staining Buffer Set (eBioscience) for Granzyme B to identify activated CD8+ cytotoxic T cells and FoxP3 to identify Treg cells, and cells were fixed. Myeloid markers included CD68, HLA-DR, CD33, CD209, and CD56. Counting beads (123Count eBeads; eBioscience) were added to the samples before acquisition on an LSRII (BD Biosciences), and data analysis was performed using FlowJo (TreeStar).
Pharmacokinetics, toxicity, and immunogenicity assessment of dostarlimab in cynomolgus monkeys
In a single-dose pharmacokinetic study, cynomolgus monkeys (Macaca fascicularis) received dostarlimab at 10, 30, or 100 mg/kg (3/sex/group) by IV administration (15-minute infusion). Dostarlimab for the single-dose pharmacokinetics study was provided by Anaptys Bio, Inc., with a purity of 97%. It was formulated in a PBS (pH = 7.4) with a concentration of 8.5 mg/mL. Blood samples were collected pre-dose and at various times for 55 days after infusion. Dostarlimab concentrations in serum were measured by ELISA. Dostarlimab, captured on the ELISA plate coated with the extracellular domain of human PD-1, was detected with a biotinylated mouse anti-human IgG4-specific mAb (Abcam, Cat#GR202921-1), which in turn was detected with streptavidin conjugated to horseradish peroxidase. Antidrug antibodies were measured using a qualified ELISA with spectrophotometry, in which a goat anti-human IgG, F(ab´)2-fragment-specific polyclonal antibody (Jackson ImmunoResearch Laboratories, Cat#116287) spiked into pooled normal cynomolgus monkey serum served as a positive control, and biotinylated dostarlimab was used for detection.
In a 4-week toxicology study, cynomolgus monkeys (6/sex/group) received 4 weekly IV infusions of 0 (vehicle), 10, 30, or 100 mg/kg dostarlimab over 15 minutes, administered at a constant volume of 4 mL/kg, followed by a 4-week recovery period. Dostarlimab for the 28-day toxicology study was provided by TESARO, Inc., with a purity of 99.3%. It was formulated in a citrate buffer solution (pH = 6.0 ± 0.5) with a concentration of 20.8 mg/mL. Toxicity was assessed based on mortality, clinical observations, body weight, body weight changes, food evaluation, ophthalmologic examinations, safety pharmacology parameters (neurologic examinations, electrocardiography, blood pressure, and respiration), clinical pathology, gross pathology, organ weight, and histopathology.
Ethical statement
Humanized NOG-EXL mice (highly immunodeficient NOG mice) expressing human GM-CSF and human IL-3, engrafted with CD34 human stem cells were procured from Taconic Biosciences (Albany, NY). Cynomolgus monkeys (Macaca fascicularis) were obtained from Hainan Jingang Biotech Co., Ltd., (Hainan, China). All animal studies were conducted in contract research facilities fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All procedures were in accordance with the Guide for the Care and Use of Laboratory Animals and Animal Welfare and approved by the local Institutional Animal Care and Use Committee (IACUC). The 4-week toxicology study in monkeys was conducted in compliance with the Organization for Economic Co-Operation and Development (OECD): Principles of Good Laboratory Practice, ENV/MC/CHEM (98) 17 (revised in 1997, issued January 1998).
Supplementary Material
Acknowledgments
The authors would like to thank Jonathan Travers, PhD, and Ashley Milton, PhD, for the critical review of the manuscript, and Tina Talreja, MBA, and Haley Laken, PhD, for their support of the program. The authors also thank Joseph Sheffer, MA, for scale-up antibody expression in CHO cells and Patricia McNeeley, BS, for primary cell assays. The authors also thank the staff of WuXi AppTec (Suzhou) Co, Ltd., for the excellent work they have accomplished for PK/TK/Immunogenicity. Writing and editorial support, coordinated by Hasan Jamal, MSc, of GlaxoSmithKline (Waltham, MA), was provided by Nicole Renner, PhD, Eric Scocchera, PhD, and Anne M. Cooper, MA, of Ashfield Healthcare Communications (Middletown, CT).
Funding Statement
This work was supported by GlaxoSmithKline. Trademarks are owned by or licensed to the GSK group of companies.
Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; AUC, area under the concentration–time curve; BSA, bovine serum albumin; C1q, complement component 1q; CD, cluster of differentiation; CDC, complement-dependent cytotoxicity; CHO, Chinese hamster ovary; CL, clearance; Cmax, maximum concentration; CTLA-4, cytotoxic T-lymphocyte–associated protein-4; EC50, half-maximal effective concentration; EDTA, ethylenediaminetetraacetic acid; ELISA, enzyme-linked immunosorbent assay; Fcγ, fragment crystallizable gamma; FITC, fluorescein isothiocyanate; flu, influenza antigen; GM-CSF, granulocyte-macrophage colony-stimulating factor; HEPES, N-[2-hydroxyethyl]piperazine-N´[2-ethansulfonic acid]; IC50, half-maximal inhibitory concentration; IFN, interferon; IgG, immunoglobulin G; IL, interleukin; IV, intravenous; Kassoc, association rate constant; KD, affinity constant; Kdissoc, dissociation rate constant; mAb, monoclonal antibody; MLR, mixed lymphocyte reaction; MRT, mean residence time; NaCl, sodium chloride; NFAT-RE, nuclear factor of activated T cells response element; PBMC, peripheral blood mononuclear cell; PBS, phosphate-buffered saline; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; PD-L2, programmed cell death ligand 2; PE, phycoerythrin; PPD, purified protein derivative from Mycobacterium tuberculosis tuberculin; RLU, relative light units; Rmax, maximum analyte-binding capacity; RPMI, Roswell Park Memorial Institute; SEB, staphylococcal enterotoxin B; SEM, standard error of the mean; SHM, somatic hypermutation; SPR, surface plasmon resonance; TCR, T-cell receptor; TGI, tumor growth inhibition; Tmax, observed time to maximum concentration; Treg, regulatory T cells; TT, tetanus toxoid antigen; t1/2, half-life; Vss, mean volume of distribution at steady state.
Disclosure statement
All authors are present or former employees of GlaxoSmithKline or AnaptysBio, Inc.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Ghahremanloo A, Soltani A, Modaresi SMS, Hashemy SI.. Recent advances in the clinical development of immune checkpoint blockade therapy. Cell Oncol (Dordr). 2019;42(5):609–12. doi: 10.1007/s13402-019-00456-w. [DOI] [PubMed] [Google Scholar]
- 2.Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114(8):1545–52. doi: 10.1182/blood-2009-03-206672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125(9):3384–91. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centanni M, Moes D, Troconiz IF, Ciccolini J, van Hasselt JGC. Clinical pharmacokinetics and pharmacodynamics of immune checkpoint inhibitors. Clin Pharmacokinet. 2019;58(7):835–57. doi: 10.1007/s40262-019-00748-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.US Food and Drug Administration . Opdivo (nivolumab) label. [accessed 2020 Sept 5]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125554s083lbl.pdf.
- 6.US Food and Drug Administration . Keytruda (pembrolizumab) label. [accessed 2020 Sept 5]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125514s084lbl.pdf.
- 7.US Food and Drug Administration . Tecentriq (atezolizumab) label. [accessed 2020 Sept 5]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761034s028lbl.pdf.
- 8.US Food and Drug Administration . Bavencio (avelumab) label. [accessed 2020 Sept 5]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761049s009lbl.pdf.
- 9.US Food and Drug Administration . Imfinzi (durvalumab) label. [accessed 2020 Sept 5]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761069s020lbl.pdf.
- 10.US Food and Drug Administration . Libtayo (cemiplimab) label. [accessed 2020 Sept 5]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761097s005lbl.pdf.
- 11.Bowers PM, Horlick RA, Neben TY, Toobian RM, Tomlinson GL, Dalton JL, Jones HA, Chen A, Altobell L, Zhang X, et al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc Natl Acad Sci U S A. 2011;108(51):20455–60. doi: 10.1073/pnas.1114010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horlick RA, Macomber JL, Bowers PM, Neben TY, Tomlinson GL, Krapf IP, Dalton JL, Verdina P, King DJ. Simultaneous surface display and secretion of proteins from mammalian cells facilitate efficient in vitro selection and maturation of antibodies. J Biol Chem. 2013;288(27):19861–69. doi: 10.1074/jbc.M113.452482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oaknin A. Preliminary safety, efficacy, and pharmacokinetic/pharmacodynamic characterization from GARNET, a phase 1/2 clinical trial of the anti-PD-1 monoclonal antibody, dostarlimab, in patients with recurrent or advanced MSI-H and MSS endometrial cancer (EC). Paper presented at: SGO Annual Meeting on Women’s Cancer; 2019 Mar 16–19; Honolulu, HI. [Google Scholar]
- 14.Perez DSJ, Pikiel J, Barretina-Ginesta MP, Trigo J, Guo W, Lu S, Jenkins D, Jen KY, Danaee H, Dunlap S, et al. GARNET: preliminary safety, efficacy, pharmacokinetic, and biomarker characterization from a phase 1 clinical trial of TSR-042 (anti-PD-1 monoclonal antibody) in patients with recurrent/advanced NSCLC. Paper presented at: SITC 2018 Annual Meeting; 2018 Nov 7–11; Washington, DC. [Google Scholar]
- 15.Oaknin A, Tinker AV, Gilbert L, Samouëlian V, Mathews C, Brown J, Barretina-Ginesta MP, Moreno C, Gravina A, Abdeddaim C, et al. Clinical activity and safety of the anti-programmed death 1 monoclonal antibody dostarlimab for patients with recurrent or advanced mismatch repair-deficient endometrial cancer: a nonrandomized phase 1 clinical trial. JAMA Oncol. 2020;6:1766. doi: 10.1001/jamaoncol.2020.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.US Food and Drug Administration . Jemperli (dostarlimab) label. [accessed 2020June 10]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761174s000lbl.pdf.
- 17.Chenoweth AM, Wines BD, Anania JC, Mark Hogarth P. Harnessing the immune system via FcgammaR function in immune therapy: a pathway to next-gen mAbs. Immunol Cell Biol. 2020;98(4):287–304. doi: 10.1111/imcb.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crescioli S, Correa I, Karagiannis P, Davies AM, Sutton BJ, Nestle FO, Karagiannis SN. IgG4 characteristics and functions in cancer immunity. Curr Allergy Asthma Rep. 2016;16(1):7. doi: 10.1007/s11882-015-0580-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daëron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113(16):3716–25. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 20.Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, Mistry Y, Dilger P, Liefooghe E, Cludts I, et al. “Cytokine storm” in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol. 2007;179(5):3325–31. doi: 10.4049/jimmunol.179.5.3325. [DOI] [PubMed] [Google Scholar]
- 21.De La Rochere P, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized mice for the study of immuno-oncology. Trends Immunol. 2018;39(9):748–63. doi: 10.1016/j.it.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Jiang D, England C, Ehlerding E, Graves S, Hernandez R, Cai W. Targeting programmed cell death 1 receptor (PD-1) expression in lung cancer using a humanized mouse model. J Nucl Med. 2017;58:179–86. [Google Scholar]
- 23.Ito R, Takahashi T, Katano I, Kawai K, Kamisako T, Ogura T, Ida-Tanaka M, Suemizu H, Nunomura S, Ra C, et al. Establishment of a human allergy model using human IL-3/GM-CSF-transgenic NOG mice. J Immunol. 2013;191(6):2890–99. doi: 10.4049/jimmunol.1203543. [DOI] [PubMed] [Google Scholar]
- 24.Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–59. doi: 10.2165/11535960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 25.Betts A, Keunecke A, van Steeg TJ, van der Graaf PH, Avery LB, Jones H, Berkhout J. Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: a comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. MAbs. 2018;10(5):751–64. doi: 10.1080/19420862.2018.1462429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fessas P, Lee H, Ikemizu S, Janowitz T. A molecular and preclinical comparison of the PD-1-targeted T-cell checkpoint inhibitors nivolumab and pembrolizumab. Semin Oncol. 2017;44(2):136–40. doi: 10.1053/j.seminoncol.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q, Qi J, Wong G, Zhu X, Liu WJ, et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat Commun. 2017;8:14369. doi: 10.1038/ncomms14369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longoria TC, Tewari KS. Evaluation of the pharmacokinetics and metabolism of pembrolizumab in the treatment of melanoma. Expert Opin Drug Metab Toxicol. 2016;12(10):1247–53. doi: 10.1080/17425255.2016.1216976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burova E, Hermann A, Waite J, Potocky T, Lai V, Hong S, Liu M, Allbritton O, Woodruff A, Wu Q, et al. Characterization of the anti-PD-1 antibody REGN2810 and its antitumor activity in human PD-1 knock-in mice. Mol Cancer Ther. 2017;16(5):861–70. doi: 10.1158/1535-7163.MCT-16-0665. [DOI] [PubMed] [Google Scholar]
- 30.Muhammed Y. The best IgG subclass for the development of therapeutic monoclonal antibody drugs and their commercial production: a review. Immunome Res. 2020;16:173. [Google Scholar]
- 31.Luo W, Wang Z, Tian P, Li W. Safety and tolerability of PD-1/PD-L1 inhibitors in the treatment of non-small cell lung cancer: a meta-analysis of randomized controlled trials. J Cancer Res Clin Oncol. 2018;144(10):1851–59. doi: 10.1007/s00432-018-2707-4. [DOI] [PubMed] [Google Scholar]
- 32.Weinmann SC, Pisetsky DS. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology (Oxford). 2019;58(suppl7):vii59–vii67. doi: 10.1093/rheumatology/kez308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sachdev JC, Patnaik A, Waypa J, Pelusi J, Beeram M, Im E, Jenkins D, McEachern K, Lu S, Guo W, et al. Safety, pharmacodynamic, and pharmacokinetic profile of TSR-042, an anti–PD–1 monoclonal antibody, in patients (pts) with advanced solid tumors. Ann Oncol. 2017;28(suppl 5):v403–v427. doi: 10.1093/annonc/mdx376.050. [DOI] [Google Scholar]
- 34.US Food and Drug Administration . Keytruda (pembrolizumab): clinical pharmacology BLA review. [accessed 2020Feb 12]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125514Orig1s000ClinPharmR.pdf.
- 35.Bowers PM, Horlick RA, Kehry MR, Neben TY, Tomlinson GL, Altobell L, Zhang X, Macomber JL, Krapf IP, Wu BF, et al. Mammalian cell display for the discovery and optimization of antibody therapeutics. Methods. 2014;65(1):44–56. doi: 10.1016/j.ymeth.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 36.Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology. 8th. Philadelphia (PA): Elsevier Saunders; 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.