

Abstract
The enigmatic eosinophil has emerged as an exciting component of the immune system, involved in a plethora of homeostatic and inflammatory responses. Substantial progress has been achieved through experimental systems manipulating eosinophils in vivo, initially in mice and more recently in humans. Researchers using eosinophil knockout mice have identified a contributory role for eosinophils in basal and inflammatory processes and protective immunity. Primarily fueled by the purported proinflammatory role of eosinophils in eosinophil-associated diseases, a series of anti-eosinophil therapeutics have emerged as a new class of drugs. These agents, which dramatically deplete eosinophils, provide a valuable opportunity to characterize the consequences of eosinophil knockout humans. Herein, we comparatively describe mouse and human eosinophil knockouts. We put forth the view that human eosinophils negatively contribute to a variety of diseases and, unlike mouse eosinophils, do not yet have an identified role in physiological health; thus, clarifying all roles of eosinophils remains an ongoing pursuit.
Keywords: eosinophil, eosinophil knockout, biologic agents, eosinophil-deficient mice, eosinophil-associated diseases, IL-5
1. INTRODUCTION
The eosinophil was first described by Paul Ehrlich in the late 1800s and has been an enigmatic cell that normally accounts for a small number of circulating leukocytes in most species (1–9). The eosinophil was identified on the basis of its intense staining with the acidic coal tar dyes (e.g., eosin). It is now appreciated that the vast majority of the cell’s protein content is derived from cationic basic charged constituents that are stored in granules, which avidly bind the dye eosin, accounting for the cell’s unique staining characteristics. These cationic proteins include major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil-derived neurotoxin (EDN), and eosinophil peroxidase (EPX). Interestingly, each of these proteins is cytotoxic to a variety of targets, including host tissue, and ECP and EPX are members of the pancreatic ribonuclease family. Evolutionarily, eosinophil-like cells have been maintained in vertebrates, including reptiles and fish, over millions of years, suggesting a beneficial role for this cell. A prevailing theory is that the eosinophil participates in protective immunity against parasites, especially helminths, as the cationic proteins in the granules get discharged onto parasites and have the capacity to kill these and other pathogens. Dysregulation of eosinophils is predicted to contribute to many eosinophil-associated diseases (EADs) (10, 11). However, definition and validation of this role of eosinophils have been challenged, as the evidence regarding their function and necessity in both mice and humans depleted of eosinophils is mixed. A variety of experimental systems, including constitutive and conditionally eosinophil-deficient mouse strains, have demonstrated that eosinophil-deficient mice have altered immune and physiologic responses in specific homeostatic processes and in a variety of disease models, including both EAD and non-EAD models. At the same time, data derived from eosinophil-depleted humans following anti-eosinophil biologic therapy have revealed eosinophils to be proinflammatory and destructive in EADs; evidence for a role in human physiological health is inconclusive. These findings bring into question the many unique roles of eosinophils as described in mice that are not evident in studies of eosinophil-depleted humans.
The concept for this article was formulated when the authors were convened as advisors to a drug company (AstraZeneca) with the objective of considering the broad topic of eosinophil immune dysfunction. During their deliberations, the authors realized the opportunity to synthesize emerging data concerning the role of eosinophils in health and disease, particularly the timely data concerning the impact of recently approved, eosinophil-depleting biological agents. Herein, without input from the drug company, we provide an overview of the consequences of manipulating eosinophil levels in mice and humans. We focus on considering the long-standing question of the role of human eosinophils in health and disease.
1.1. Development and Localization
Prior to circulation and recruitment to the tissue, eosinophils originate in the bone marrow from granulocyte/macrophage progenitor (GMP) precursors, followed by maturation of common eosinophil, mast cell, and basophil progenitors and subsequent eosinophil lineage–committed progenitor cells (EoPs). Eosinophil development occurs via decisive steps in cell fate driven by the action of primary lineage-determining transcription factors, most notably GATA-1, and contributions by GATA-2, ETS factor PU.1, the CCAAT-enhancer-binding protein (C/EBP) family members C/EBPα and C/EBPε, FOG1, IRF8, X-box binding protein 1 (XBP1), and members of the Ikaros zinc-finger (IKZF) family (12–24). Under homeostatic conditions, eosinophilopoiesis is regulated in part by a unique combinatorial program of these transcription factors, including the requisite expression of GATA-1, which occurs through the use of an eosinophil lineage–specific double palindromic enhancer in the GATA-1 gene itself. These transcription factors define the eosinophil lineage, including those encoding eosinophil granule cationic proteins, such as MBP, EPX, the Charcot–Leyden crystal protein (CLC)/galectin-10, the eotaxin receptor CCR3 (CD193), and IL-5 receptor α (IL-5Rα, also known as CD125) (5). Baseline eosinophilopoiesis is also regulated in part at the level of microRNAs and long noncoding RNAs (25, 26). EoPs express the IL-5 receptor, and IL-5 drives progenitor cell expansion and development into mature eosinophils. IL-5 also regulates eosinophil migration from the bone marrow into the circulation and the survival of tissue eosinophils. IL-5 is produced by cells of both the innate and adaptive immune systems, including mast cells, group 2 innate lymphoid cells (ILC2s), and activated T helper 2 (Th2) lymphocytes (27). In addition to IL-5, IL-3 and granulocyte-macrophage colony-stimulating factor (GM-CSF) have been shown, at least in vitro, to drive both murine and human EoPs to terminally differentiate and aid in survival. Recruitment of eosinophils into the tissue and their activation are also regulated by chemokines, especially the eotaxin family of eosinophil-selective chemoattractants C-C motif chemokine ligands (CCLs): CCL11 (eotaxin-1) and CCL24 (eotaxin-2) in both humans and mice and CCL26 (eotaxin-3) in humans only (28). These chemokines bind to the eosinophil-selective CCR3. Under homeostatic conditions, eosinophil recruitment is regulated by constitutive expression of CCL11 within the gastrointestinal tract (5) and elsewhere, including adipose tissue, thymus, and uterus (29). Similarly, under inflammatory conditions, an array of chemotactic proteins participate in eosinophil recruitment to the site of inflammation (30, 31). Notably, the eotaxin chemokines are markedly induced by IL-13, providing a synergistic mechanism by which Th2 cells and ILC2s, coproducing IL-5 and IL-13, regulate tissue eosinophilia.
1.2. Eosinophil Pleiotropy and Heterogeneity
Eosinophils have been classically viewed as terminally differentiated cells that respond to type 2 microenvironments generated by parasites or allergic eosinophilic inflammation and cells that uniformly release granule contents as an immediate and final effector function. However, this view is slowly changing as evidence emerges that these postmitotic cells undergo phenotypic changes in response to tissue microenvironmental cues, including altered morphology, enhanced responsiveness to cytokines (e.g., priming), prolonged survival for weeks, and various degrees of cellular activation (4, 32–35). Additionally, eosinophils are capable of expressing diverse sets of proteins (e.g., surface markers, cytokines), mRNA transcripts, and lipids in response to different stimuli; in addition they release specific granule proteins (Figure 1). In the lungs of mice (36–39) and humans (35, 40), differential expression of receptors and activation states have been identified. IL-33 is an innate proatopy cytokine (41) that is upregulated in asthma, induces a wide range of transcripts in eosinophils, and activates eosinophils such that they can promote type 2 inflammation in the lungs (42–44) (Figure 2).
Figure 1.
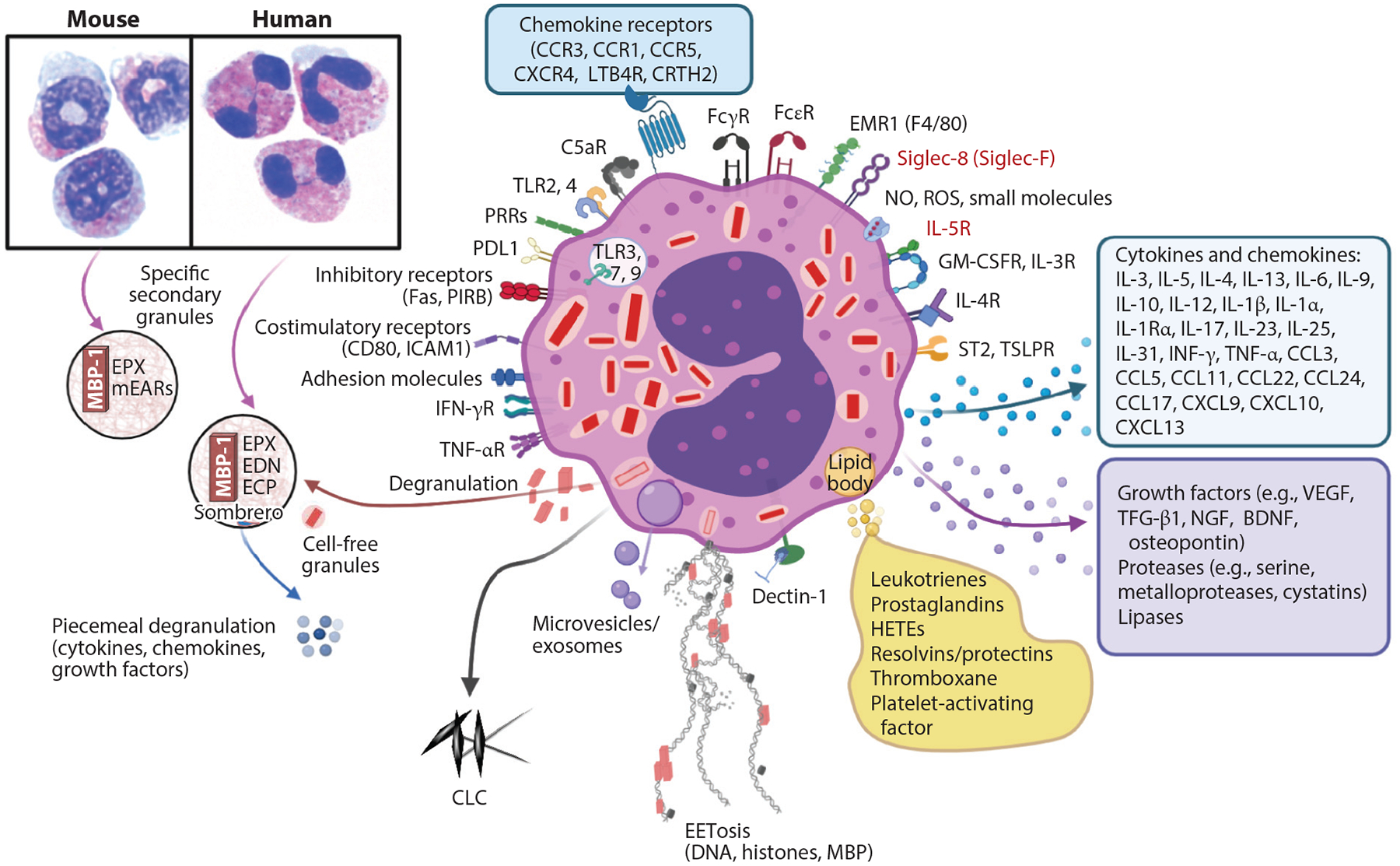
Eosinophil biology. Mouse and human peripheral blood eosinophils show similarities in eosin staining (pink) and differences in nuclei (blue), with mouse nuclei in a circular or figure-eight shape and human nuclei being bilobed, as seen by standard light microscopy. Secondary granules in both contain MBP-1, which creates a crystalline core visible by electron microscopy, and the matrix contains EPX. Human eosinophils have ECP and EDN, whereas mouse eosinophils have only divergent mEARs. Eosinophils undergo several forms of degranulation, including classical exocytosis with release of entire granule contents or piecemeal degranulation, which may occur through formation of sombrero vesicles or exosomes, resulting in the differential release of cytokines, chemokines, or growth factors. Finally, eosinophils may undergo cytolysis, which includes release of cell-free granules and may also involve EETosis whereby DNA, histones, and granule proteins are released, forming extracellular nets. Mediators from lipid bodies are shown, as well as the many cytokines, chemokines, and growth factors. Siglec-8, IL-5R (receptors in red), and IL-5 are targets for monoclonal antibodies to deplete eosinophils. This figure shows some key representative eosinophil molecules and not a complete list. Abbreviations: BDNF, brain-derived neurotrophic factor; CCL, C-C motif chemokine ligand; CCR, C-C motif chemokine receptor; CLC, Charcot–Leyden crystal protein; CRTH2, chemoattractant receptor-homologous molecule expressed on Th2 cells; CXCL, C-X-C motif chemokine ligand; CXCR, C-X-C motif chemokine receptor; ECP, eosinophil cationic protein; EDN, eosinophil-derived neurotoxin; EPX, eosinophil peroxidase; FcεR, Fc-epsilon receptor; FcγR, Fc-gamma receptor; GM-CSFR, granulocyte-macrophage colony-stimulating factor receptor; HETE, hydroxyeicosatetraenoic acid; ICAM1, intercellular adhesion molecule 1; LTB4R, leukotriene B4 receptor; MBP-1, major basic protein 1; mEAR, mouse eosinophil-associated ribonuclease; NGF, nerve growth factor; NO, nitric oxide; PIRB, paired immunoglobulin-like receptor B; PDLI, programmed death ligand 1; PRR, pathogen recognition receptor; ROS, reactive oxygen species; Siglec, sialic acid–binding immunoglobulin-like lectin; ST2, suppressor of tumor 2 [also known as IL-1RL1 (interleukin-1 receptor-like 1)]; TGF-β1, transforming growth factor beta 1; TLR, Toll-like receptor; TSLPR, thymic stromal lymphopoietin receptor; VEGF, vascular endothelial growth factor. Figure adapted from images created with BioRender.com.
Figure 2.
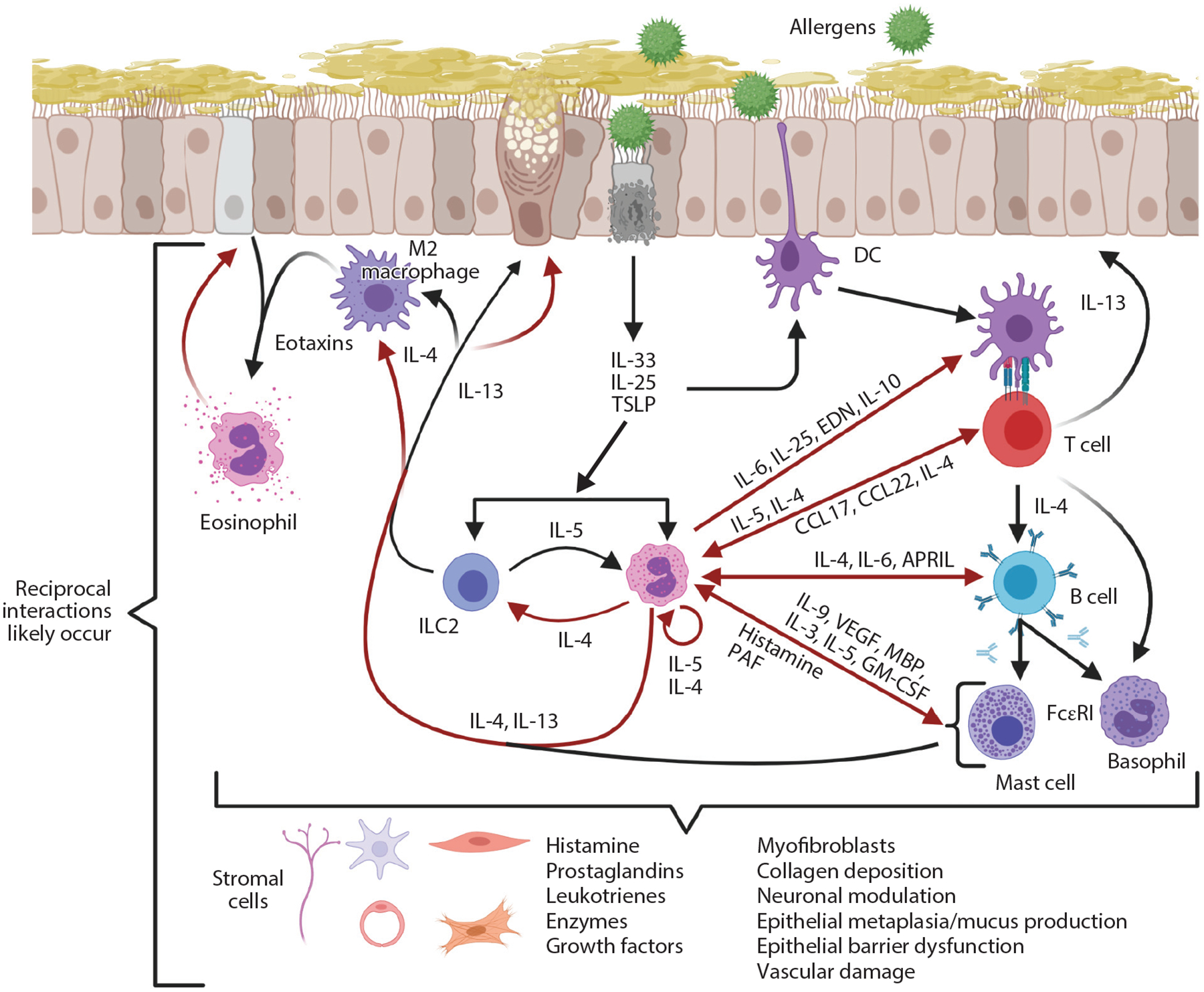
Type 2 immune responses in allergy. Allergens induce epithelial responses that lead to production of IL-33, IL-25, and TSLP, which are immediate cytokine signals released upon epithelial activation or damage. A wide variety of cells, including ILC2s, eosinophils, DCs, eosinophils, M2 macrophages, mast cells, basophils, and Th2 cells, respond to these cytokines. In principle, this can result in reciprocal interactions between these cells to release additional mediators. IL-13 enhances expression of eotaxins to promote eosinophil recruitment and leads to goblet cell metaplasia/mucus production. Red arrows represent products derived from eosinophils. Abbreviations: APRIL, a proliferation-inducing ligand; CCL, C-C motif chemokine ligand; DC, dendritic cell; EDN, eosinophil-derived neurotoxin; FcεRI, Fc-epsilon receptor I; GM-CSF, granulocyte-macrophage colony-stimulating factor; ILC2, group 2 innate lymphoid cell; MBP, major basic protein; PAF, platelet-activating factor; TSLP, thymic stromal lymphopoietin; VEGF, vascular endothelial growth factor. Figure adapted from images created with BioRender.com.
Unique activation states of eosinophils have been found in gastrointestinal disease as well. For example, esophageal eosinophils, elicited by epithelial IL-13 overexpression in vivo, express an increased number of genes compared with purified bone marrow eosinophils (45). The upregulated genes in esophageal eosinophils encode multiple cell surface receptors: cytokine and chemokine receptors, immunoglobulin-like receptors, and cell adhesion and migration molecules. Tissue eosinophils are dynamically regulated by a combination of activation and inhibitory receptors, underscoring their pleiotropic capacity (see the sidebar titled Technological Difficulties Unique to Eosinophils for Single-Cell Transcriptomics). For example, eosinophils incapable of inhibition due to loss of the paired immunoglobulin-like receptor B (PIRB) exhibit an activated phenotype and a modified transcriptomic signature that is associated with more severe experimental allergy models such as eosinophilic esophagitis (EoE) (45). Likewise, intestinal eosinophils are phenotypically distinguishable from autologous blood eosinophils and express antigen presentation markers not seen on blood eosinophils (46).
TECHNOLOGICAL DIFFICULTIES UNIQUE TO EOSINOPHILS FOR SINGLE-CELL TRANSCRIPTOMICS.
A commonly used technique called single-cell RNA sequencing (scRNA-seq) enables single-cell resolution of whole transcriptomics to characterize and identify individual cells from the overall heterogeneous population (257). New classification systems for immune cells have been developed from this technique (258), and specific disease states of immune cells have been identified (259). Successful scRNA-seq of tissue-derived eosinophils is a technical challenge limiting our ability to define eosinophil activities in situ in health and disease. Several factors, including the large number of intrinsic RNases in eosinophils as well as the easily triggered degranulation of human eosinophils, are proposed to contribute to this technological difficulty (11). Thus, published reports of scRNA-seq on tissue biopsies may underestimate the number or contribution of eosinophils. In light of these difficulties, new technologies with DNA that measure epigenetic methylation changes or chromosome accessibility may enable greater insight into cell-specific activities of eosinophils (260).
In addition to type 2 responses, eosinophils exposed to viral (47–51), bacterial, or fungal pathogens (52–54) and eosinophils involved in cancer or autoimmunity (reviewed in 9, 55, 56) undergo unique phenotypic changes associated with their ability to differentially regulate activation or suppression of the inflammatory process. Taken together, the emerging evidence indicates that eosinophils are pleiotropic cells, capable of dramatically and dynamically modifying their transcriptomic content in response to microenvironment and inflammatory signals.
2. ROLE OF EOSINOPHILS IN MICE
Although there are differences between mouse and human eosinophils, the similarities (reviewed in 2, 57, 58) provide an advantage that has guided research toward defining the importance or irrelevance of eosinophils with the use of novel strains of eosinophil knockout mice.
2.1. Generation of Eosinophil Knockout Mice
The process of manipulating eosinophils evolved from findings by several laboratories that IL-5 is a key mediator of eosinophil differentiation and hematopoiesis. Early in vitro studies demonstrated that IL-5 is more critical than GM-CSF or IL-3 for expansion and final differentiation and survival of eosinophils. Newly developed antibodies to IL-5 (e.g., TRFK5) were tested in parasite-infected mice and demonstrated that eosinophilia can be reduced, also suggesting that this cytokine is a clear target for inhibiting human eosinophilia in vivo (59, 60). Complete IL-5 (61) and IL-5Rα (62) knockout strains were generated in 1996 to test the role of IL-5 in greater detail. These mice had near wild-type levels of bone marrow eosinophils yet moderately reduced peripheral blood eosinophils and markedly impaired eosinophilia induction upon challenges in translational models, such as parasite infection or allergen exposure (60, 63, 64). The combination of off-target effects in IL-5/IL-5Rα–deficient mice (64–66) and the ability of eosinophils to survive in the absence of IL-5/IL-5Rα (67, 68) resulted in unresolved questions about the role of eosinophils.
In 2004, articles in Science described the first congenic eosinophil-deficient mice, ΔdblGATA-1 (69) and PHIL (70), using a mouse model of asthma. GATA-1 is an X-linked transcription factor whose cis-binding sites have been mutated to generate uniquely erythroid- and megakaryocyte-deficient mice (GATA-1 ΔneoΔHS/GATAlow) (71) or eosinophil-deficient mice (ΔdblGATA-1) (15, 72). PHIL mice express diphtheria toxin A (DTA) downstream of an EPX promoter, resulting in autonomous eosinophil death in the bone marrow. An additional congenic eosinophil-deficient strain is the MBP-1−/−EPX−/− double knockout mouse (73), which fails to produce eosinophils owing to unknown mechanisms related to impaired granulopoiesis. All three of these genetic strains are overall highly specific for eosinophil deficiency, yet they have intrinsic features that require consideration when results are interpreted.
Two knock-in strains, iPHIL (74) and eoCre (75), provide new means to bypass some of the issues of ΔdblGATA-1 and PHIL. iPHIL mice express the human diphtheria toxin receptor (DTR) downstream of the endogenous EPX promoter. Inducible depletion occurs upon administration of diphtheria toxin (DT), resulting in targeted cell death in the bone marrow. eoCre mice express Cre recombinase downstream of the endogenous EPX promoter, permitting removal of genes only in eosinophils by Cre/loxP mechanisms (57). Inducible depletion of eosinophils may be achieved by crossing eoCre mice with mice having genes surrounded by loxP sites that enable their expression. For example, eosinophil-specific expression of DTR (52) or human Siglec-8 (sialic acid–binding immunoglobulin-like lectin 8) (76) would permit inducible depletion by DT or antibody to Siglec-8, respectively. These new models provide the most versatility for defining eosinophil-specific effector functions in an otherwise normal animal. Table 1 summarizes beneficial and confounding features of mouse strains genetically engineered to modify eosinophils.
Table 1.
Eosinophil-deficient mouse strains
| Strain | Construct mechanism | Eosinophil levels | Advantages | Constraints/off target | References |
|---|---|---|---|---|---|
| IL-5−/− and IL-5Rα−/− | Knockout of IL-5 or IL-5Rα, which are important in eosinophil hematopoiesis, survival, and priming | BM: normal Blood: reduced Tissues: reduced |
|
|
60–68 |
| ΔdblGATA-1 | Mutation in the GATA-1a cis-binding site needed for EoP development. Not to be confused with GATA-1 ΔneoΔHS/GATA-1low | BM: deficient Blood: deficient Tissues: deficient |
|
|
15, 71, 72, 261–263 |
| MBP-1−/−/EPX−/− | Double knockout of eosinophil granule proteins MBP-1 and EPX results in death of EoPs | BM: deficient Blood: deficient Tissues: deficient |
|
|
73 |
| PHIL | Transgenic mouse expressing DTA downstream of an EPX promoter | BM: deficient Blood: deficient Tissues: deficient |
|
|
70 |
| iPHIL | Knock-in of EPX promoter driving DTR (injection of DT targets eosinophils) | BM: deficient Blood: deficient Tissues: deficient |
|
|
74 |
| eoCre | Knock-in of EPX promoter driving expression of Cre (eosinophil-specific Cre) Depletion occurs by crossing to ROSA-fl-stop-fl-DTA or ROSA-fl-stop-fl-hSiglec-8+αSiglec-8 treatment or Xbp1fl/fl |
BM: deficient Blood: deficient Tissues: deficient |
|
|
24, 52, 75, 76 |
Abbreviations: BM, bone marrow; Cre, Cre recombinase; DT, diphtheria toxin; DTA, diphtheria toxin A; DTR, diphtheria toxin receptor; eoCre, Epxtm1.1(cre)JLee; EoP, eosinophil progenitor; EPX, eosinophil peroxidase; iPHIL, Epxtm2.1(HBEGF)JLee; MBP-1, major basic protein 1; PHIL, Tg(EPO-DTA)#NAL.
Xbp1 and GATA-1 are transcription factors.
2.2. Homeostatic Functions for Eosinophils
Eosinophils have effector activities that suggest a necessary role for these cells in maintaining homeostasis or preventing disease (2–6, 8, 9). The significance of these findings to health is difficult to discern, as most eosinophil-deficient mice breed at normal rates, wean pups, live a normal life span, and have no overt changes in vitality in the unstressed and environmentally controlled life of a laboratory mouse (see also 77). Regardless, some key concepts of eosinophil functions in homeostasis and potential roles in disease stand out as areas warranting further study in consideration of their roles in human health.
2.2.1. Eosinophils, microbiome, mycobiome, and Th17 responses.
Eosinophils reside in the gastrointestinal tract, from the stomach to the rectum, of both mice and humans, with the largest numbers in the small intestine (78). Early studies with congenic eosinophil-deficient IL-5Rα−/−, PHIL, and ΔdblGATA-1 mice (65, 66, 79–81) found that they had smaller Peyer patches, reduced IgA+ B cells, and altered microbiome composition relative to wild-type littermates. Several mechanisms (Figure 3a) have been proposed for eosinophil modulation of IgA: (a) indirect activities of eosinophils on bacteria resulting in downstream T follicular helper (Tfh) cell–induced IgA production (81); (b) Toll-like receptor (TLR) activation of eosinophils to release a proliferation-inducing ligand (APRIL), IL-6, and transforming growth factor β1 (TGF-β1), which then induce IgA class switching and production by B cells (79, 82); and (c) eosinophil-derived, IL-1β–mediated increases in the numbers of group 3 innate lymphoid cells (ILC3s) (80), which express IL-17, a mediator of IgA production (83, 84). In contrast to these pathways promoting IgA and Th17 responses, intestinal eosinophils uniquely produce IL-1 receptor antagonist IL-1Rα (85), which inhibits IL-1β activity and suppresses Th17 responses. These collective findings indicate a dual function for eosinophils in gastrointestinal homeostasis that may be pro-Th17 or anti-Th17.
Figure 3.
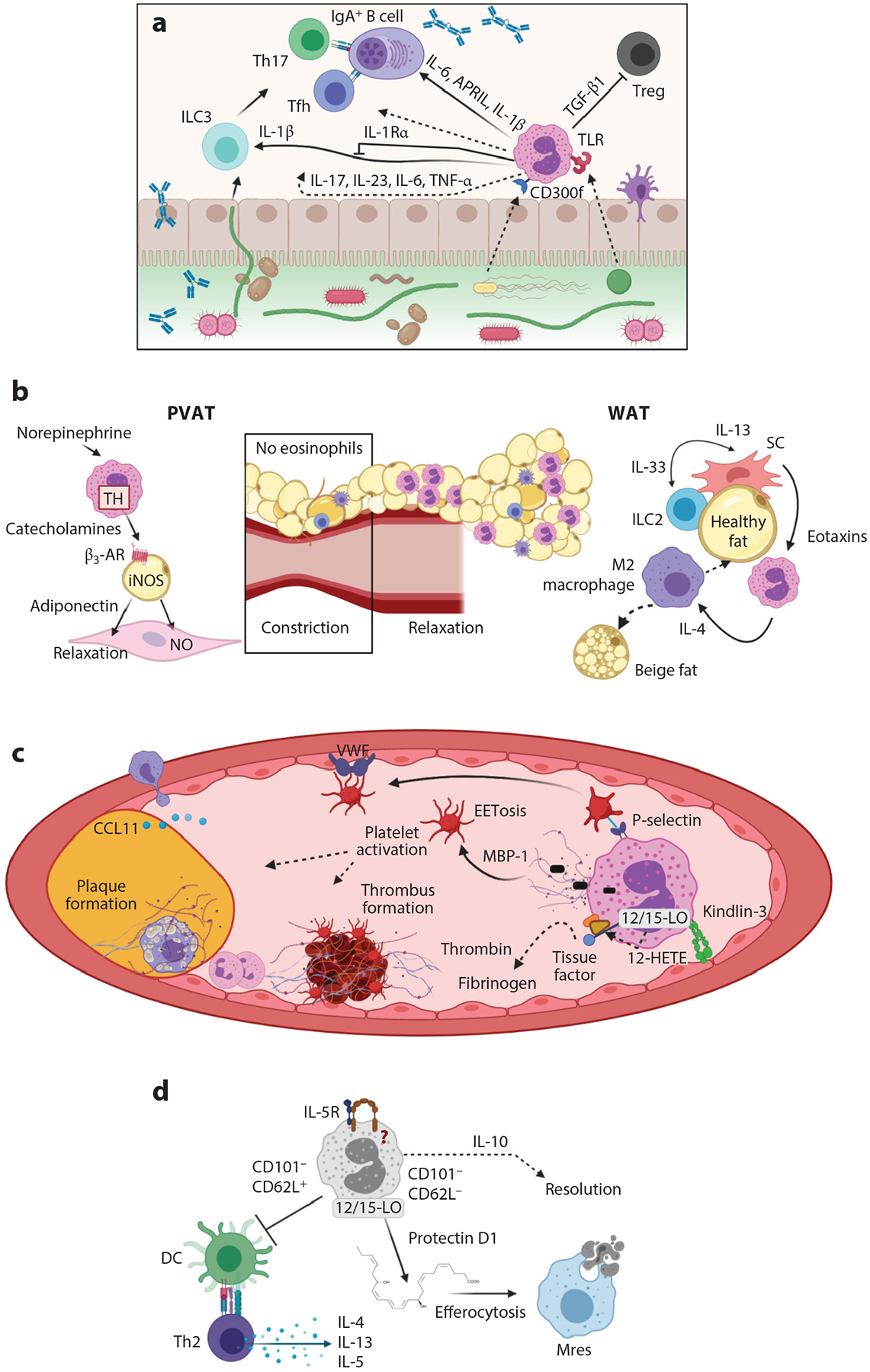
Eosinophil activities in homeostasis in mice. (a) Eosinophil activities with the microbiome, B cells, and Th17 responses in the gastrointestinal tract. Eosinophils express receptors such as TLRs and CD300f that recognize bacterial components to signal for immune responses. Eosinophils release the cytokines IL-6, APRIL, and IL-1β to influence B cell IgA production. This may happen through Tfh cells or Th17 cells, which express IL-17. Eosinophil release of IL-1β increases the activity of ILC3s; TGF-β1, which inhibits Tregs; and IL-17, IL-23, IL-6, and TNF-α, which may directly participate in the above responses. (b) Eosinophil functions in metabolic and vascular health. Eosinophils induce relaxation of blood vessels in PVAT through use of TH to release catecholamines that bind β3-AR, which stimulates iNOS in adipocytes to make NO that results in smooth muscle cell relaxation. In WAT, ILC2s produce IL-13 in response to the IL-33 produced by SCs. The IL-13 induces production of eotaxins that recruit eosinophils into fat. These eosinophils release IL-4 to polarize macrophages into alternatively activated M2 macrophages that contribute to transformation of white to beige fat cells, which are thermogenic cells that aid in metabolic health. (c) Eosinophils have multiple functions in vascular health. Eosinophils release a 12-hydroxyeicosatetraenoic acid (HETE) product that is generated from 12/15-LO and ultimately aids in tissue factor activation and development of thrombi through thrombin and fibrinogen activation. Eosinophils activate platelets through P-selectin promoting platelet binding to VWF in endothelial cells, which also aids in clot and plaque formation. Through integrin kindlin-3 binding of the endothelium, eosinophils are activated to undergo EETosis, releasing MBP-1 that further activates platelets and promotes atherosclerotic inflammation. (d) Eosinophils as lung-resident cells. Resident lung eosinophils are different from inflammatory recruited eosinophils due to their differential expression of CD101. CD101− cells that are CD62L+ suppress DC activation, which in turn inhibits Th2 cell release of cytokines IL-4, IL-13, and IL-5. CD101− eosinophils that are CD62L− are reported as important in resolution of inflammation in that they produce protectin D1 through 12/15-LO activity. Protectin D1 promotes efferocytosis, whereby Mres phagocytose dead neutrophils. Eosinophil-derived IL-10 is also important in resolution functions of eosinophils in the lung. Dashed arrows indicate either less-defined or indirect pathways of eosinophils in the roles presented. Abbreviations: APRIL, a proliferation-inducing ligand; β3-AR, beta 3 adrenergic receptor; CCL, C-C motif chemokine ligand; DC, dendritic cell; ILC2, group 2 innate lymphoid cell; iNOS, inducible nitric oxide synthase; MBP-1, major basic protein 1; Mres, resolution macrophage; NO, nitric oxide; PVAT, perivascular adipose tissue; SC, stromal vascular cell; Tfh, T follicular helper; TGF-β1, transforming growth factor beta 1; Th, T helper; TH, tyrosine hydroxylase; TLR, Toll-like receptor; TNF-α, tumor necrosis factor alpha; Treg, regulatory T cell; 12/15-LO, 12/15 lipoxygenase; VWF, Von Willebrand factor; WAT, white adipose tissue. Figure adapted from images created with BioRender.com.
The role of eosinophils as a modulator of Th17 pathways is not unique to the gastrointestinal tract. Eosinophils either suppress or promote Th1/Th17 responses depending on the antigen—ovalbumin (86) or Aspergillus fumigatus (50, 53, 54), respectively—in mouse allergic respiratory inflammation models. Eosinophils in A. fumigatus allergy models produce IL-17, IL-23, IL-27, and CXCL13. This finding brings into question the role of the mycobiome in eosinophil functions during immune responses as well (83, 87). Although humans and mice have distinct microbiomes (88), microbiome/mycobiome changes have also been considered contributors to asthma responses in humans (89, 90) and IgA levels (91), suggesting some commonality and a greater need to understand immune interactions between eosinophils and the gastrointestinal microbiome/mycobiome in health and disease.
2.2.2. Eosinophils and metabolism.
Eosinophils are found in adipose tissue in humans (92) and mice (93). Mouse models strongly indicate that type 2 responses in the fat are beneficial to metabolic health, leading to improved thermogenesis and glucose sensitivity and reduced weight gain (94) (Figure 3b). Deficiencies in eosinophils at homeostasis (i.e., normal diet) lead to reduced expression of favorable lipid metabolism genes in the gastrointestinal tract and fat (95) and reduced production of thermogenic fat (96–98). Several reports, though, show conflicting data on the role of eosinophils in adipose tissue and metabolic health, particularly in models of obesity (93, 95, 99, 100). Several reasons for these variations exist, such as the methods of eosinophil manipulation in these models (i.e., adoptive transfers or gene knockouts that affect other cells) and the contribution of the microbiome to gastrointestinal-adipose immune interactions (101). For example, modulation of microbiota by either calorie restriction (102) or ablation of commensal bacteria with antibiotics (103) induced recruitment of eosinophils to adipose tissue, concordant with increases in M2 macrophages and improved insulin responses. Finally, the contributions of eosinophils to metabolism are not exclusive to adipose tissue or the gastrointestinal tract, as the same type 2 environment that is proposed to favor metabolic health of adipose tissue increases the risk of fibrosis and eosinophil infiltration in the liver of patients with nonalcoholic fatty liver disease and in the esophagus of patients with EoE and in mouse models (104–106).
2.2.3. Eosinophils, vasculature, and nerves.
A recent set of studies have identified a role for eosinophils in both physiologic vascular tone and hemostasis, as well as potential negative effects by eosinophils in the settings of thrombus formation and vascular sclerosis (Figure 3c). Eosinophils are present in perivascular adipose tissue (PVAT) (107), which is found most predominantly around the aorta and other arteries of the heart (108). PVAT eosinophils induce release of adiponectin and nitric oxide, which are critical for vascular smooth muscle relaxation. Vascular constriction and relaxation and thrombus formation are needed to prevent excessive blood loss. Eosinophil-deficient mice have prolonged bleeding after an induced venous injury and fail to form a sufficient intravascular thrombus. In addition to platelet-eosinophil interactions, eosinophil-derived lipids produced by 12/15-lipoxygenase are necessary for promoting tissue factor activation and generating thrombin and fibrinogen coagulation in the blood vessel (109). Crossing eosinophil-deficient mice with an atherosclerosis model (ApoE−/−) demonstrated that eosinophils are recruited to sites of injured endothelium, are induced to release DNA, activate platelets through MBP-1 binding, and promote atherosclerosis (110).
Some of these same mechanisms may promote the vascular injury and vascular permeability that occur in some eosinophilic skin diseases. For example, eosinophil IgE binding induces clotting reactions in a model of bullous pemphigoid (111), and thrombus formation promoted by eosinophils is important in urticaria (112) and chronic rhinosinusitis (113). In mice, contact dermatitis results in increased eosinophil-dependent vascular permeability and ear swelling (114, 115). It is plausible that eosinophils perform similar activities that increase vascular permeability in inflammatory bowel disease (116), eosinophilic granulomatosis with polyangiitis (EGPA) (117, 118), and the blood-brain barrier upon injury (119).
Eosinophil-nerve interactions are important in physiological homeostasis of the vasculature system and multiple organ systems at homeostasis and in disease (120, 121). Eosinophils express numerous nerve-related proteins (122, 123); neuronal receptors (124); and IL-31 (125), a mediator of itch, suggesting reciprocal function for eosinophils and nerves. Pan et al. (126) recently demonstrated that eosinophil-derived IL-4 is directly responsible for nerve regeneration, suggesting that eosinophils may both promote and resolve neuronal injury and inflammation depending on the tissue and pathological context. For example, in skin disease models, eosinophil-deficient mice demonstrate attenuated dendrite growth as measured by substance P (115, 127). A recent study using 3D imaging demonstrated that at homeostasis, the synaptic vesicle ends of nerves directly contact eosinophils in the lamina propria of the ileal mucosa, and they are proposed to contribute to enteric nerve health (128).
2.2.4. Resident pulmonary eosinophils.
Eosinophils are present in the lungs of mice and humans at homeostasis. In mice, this was first shown by flow cytometry of naive lungs (36, 129) followed by intravital imaging identifying eosinophils as patrolling the lungs at homeostasis (32). Two recent studies suggest that identifying features of lung-resident eosinophils are that they are CD101− and that they are absent in congenic eosinophil-deficient mice (i.e., ΔdblGATA-1) (Figure 3d). Mesnil et al. (37) demonstrated by ex vivo and adoptive transfer techniques that these regulatory CD62L+ CD101− eosinophils inhibit type 2 responses to allergen through suppression of dendritic cell activation, thus prohibiting Th2 activation in response to allergens. The role of IL-5Rα is unknown, as the resident lung eosinophils are unaffected by IL-5 deficiency, and evidence for a human version of the regulatory eosinophil has not been obtained. In the second model, an acute lung injury lipopolysaccharide model, a population of CD62L− CD101− eosinophils was found to produce elevated levels of protectin D1 (39), a lipid previously shown to be made by eosinophils and to help promote macrophage uptake of neutrophils (130, 131). This would suggest eosinophils are important in resolution of inflammation, as shown by others (132, 133). Moreover, the contributions of these resident cells to immune responses that develop in the setting of infection; lung transplantation; and pulmonary diseases, such as chronic obstructive pulmonary disease (COPD), require further study.
2.3. Eosinophil Deficiency in Mouse Models of Eosinophilic Disease
2.3.1. Asthma.
Mouse models of asthma have evolved over the years, originally focusing on the primary role of eosinophils as proinflammatory cells contributing to airway damage and hyperreactivity and eventually supporting a consensus that their role spans a diverse set of functions ranging from proinflammatory to immune modulating. Early studies with ovalbumin models of asthma, in which degranulation was insignificant, pointed to eosinophils as immune remodeling and repair cells and not only destructive cells (69, 70, 134). Jacobsen et al. and several others demonstrated that eosinophils promote Th2 cell polarization, Th2 cell recruitment to the lung, type 2 cytokine and chemokine production, and M2 macrophage polarization in type 2 allergic asthma models (42, 43, 86, 135–137). More recent findings suggest that eosinophils also modulate ILC2 accumulation and activation in the airways in response to IL-33 or allergen (138). The importance of eosinophil immune activities was further demonstrated in mice with IL-5 and eotaxin-2 dual overexpression that developed significant degranulation, pulmonary remodeling, and lung dysfunction (139, 140). In particular, eosinophil-derived IL-13 was more critical to airway destruction and mucus production than release of either MBP or EPX from eosinophils in this model (141, 142). Despite these findings, eosinophil-independent inflammation can occur in asthma models (143). Moreover, the mechanisms of degranulation for mouse eosinophils as compared to human eosinophils are strikingly distinct and mice lack EDN, ECP, and CLC; altogether these findings lead one to be cautious in interpreting the significance (or insignificance) of degranulation in mouse models.
Eosinophils may have an underappreciated role in heterogeneous asthma endotypes and in exacerbating pulmonary infections. Eosinophils have the capacity to release IL-6 or type 17 mediators that occur in certain patients with exacerbation-prone asthma (144). Moreover, eosinophils have unique functions in viral infections (47–49, 51), where they may produce inducible nitric oxide synthase, take up viral particles, and activate CD8 T cells to kill viruses. In these nuanced disease processes, eosinophils may be contributory cells rather than primary mediators.
2.3.2. Gastrointestinal disease.
Inflammatory bowel disease has an eosinophil component (145), as do eosinophilic gastrointestinal diseases (EGIDs) (78). Discordant results are published concerning the role of eosinophils in colitis models (131, 146), and few studies indicate that eosinophils may have a role in models of Crohn’s disease (147, 148). Given the recent understanding of the role of the microbiome and mycobiome, it is possible that the mice in each study had unique commensal bacteria and fungi leading to worse or better health. Translational models for EGIDs are limited regarding the use of eosinophil knockout strains, yet a few studies suggest a role for eosinophils (45). A pathogenic effector role for eosinophils in gastric dysmotility associated with axonal necrosis has been described (149). Additionally, a role for eosinophils in eliciting allergen-mediated esophageal remodeling, including fibrosis, has been reported (150). Yet, IL-13–elicited EoE in mice is unabated in the absence of eosinophils (105). Conversely, in a humanized transgenic mouse model of eosinophilic gastroenteritis, antibodies targeting human Siglec-8-expressing eosinophils yielded depleted eosinophils and reduced levels of inflammatory mediators, suggesting a role for eosinophils in mediating disease pathologies (151).
2.4. Cancer
Understanding of the role of eosinophils in human tumor biology and immunology is actively evolving (2, 55). Eosinophils release mediators, such as granule proteins and enzymes; growth factors, such as TGF-β and vascular endothelial growth factor (VEGF); and metalloproteases that remodel the extracellular matrix to promote or inhibit tumor growth through a multitude of mechanisms. Eosinophils recruit to both necrotic and live regions of tumors, depending on the tumor, including the stromal and nonstromal portions of tumors in both humans and mice. Eosinophil-deficient mice do not develop tumors at homeostasis, but eosinophils have either a pro- or antitumorigenic role, depending upon the model. For example, tumor-infiltrating eosinophils degranulate and are essential for colon tumor rejection through an IFN-γ-linked antitumor effect that is independent of CD8 T cells (152). Conversely, in other cancer models, eosinophils promote IFN-γ-induced recruitment and activation of CD8 T cells, which mediate tumor killing (153). As an additional mechanism of antitumorigenic activity, eosinophils directly participate in tumor killing following activation by IL-33 (154). Emerging data suggest that eosinophils can indeed be predictive biomarkers and effector cells in immunotherapy, especially in response to immune checkpoint blockade therapy (155). The implications are significant, as tumor-associated tissue eosinophils are prevalent in a wide variety of human cancers and as eosinophils have the capacity to survive in tumors in the absence of IL-5. Their localization in tumors and their numbers as determined by histology correlate with effects that are beneficial (e.g., in colon cancer, breast cancer, melanoma), detrimental (e.g., in lung cancer, Hodgkin lymphoma), or of unclear prognostic significance (e.g., in brain cancer and pancreatic cancer). For example, in bladder cancer, eosinophils are found at increased numbers in patients whose disease responds better to therapy (156). The extensive presence of eosinophils in human cancer suggests that cancer could be considered a variant of organ EADs, wherein eosinophils are effector cells in the immune pathology.
2.5. Injury, Transplantation, and Autoimmunity
Emerging studies are demonstrating that eosinophils may uniquely benefit ischemia reperfusion injury repair (157) and allograft lung transplantations (158). Eosinophils are required to suppress CD8 T cell–mediated allograft rejection in mouse models of lung transplantation. Additionally, eosinophils may contribute to muscle injury repair in diseases such as muscular dystrophy by activating fibro-adipocyte progenitors for repair (159). Yet, eosinophils may equally participate in detrimental responses to autoimmune diseases (56, 119), indicating that the complexity of eosinophil functions is potentially wider and more diverse than previously appreciated. These diseases are very rare and tissue specific and thus may require special consideration for model building to deeply understand the role of eosinophils.
3. EOSINOPHIL DEPLETION IN HUMANS
3.1. Generation of Eosinophil-Depleting Monoclonal Antibodies
The observations in mice demonstrating a role for eosinophils in the airway hyperresponsiveness of asthma models led to the pursuit of monoclonal antibody therapies to deplete eosinophils in humans (Figure 4; Table 2). The first monoclonal antibody developed for clinical use is mepolizumab, which neutralizes IL-5. The preclinical trials in cynomolgus monkeys demonstrated safety in primates and identified dosages resulting in significant decreases in blood eosinophilia and eosinophil migration into the lung (160). In humans, mepolizumab induced maturational arrest of EoPs in the bone marrow (161) and reduced levels of blood and tissue eosinophils with a negative impact on their activation status (40, 162, 163). A second monoclonal antibody targeting neutralization of IL-5, reslizumab, was also developed. Mutagenesis experiments with recombinant IL-5 demonstrated that reslizumab neutralizes IL-5 by binding to amino acids 89–92 and prevents its binding to the IL-5 receptor (164). Reslizumab exhibits greater binding affinity for IL-5 than does mepolizumab (165). Finally, a third monoclonal antibody, benralizumab, is a humanized, nonfucosylated antibody that targets the IL-5Rα chain. The lack of fucosylation leads to enhanced antibody-dependent cellular cytotoxicity (ADCC), and by targeting the IL-5 receptor, benralizumab actively kills eosinophils and also basophils. Benralizumab binds to a site on the IL-5Rα chain in close proximity to the IL-5 binding site; however, its primary mechanism of depletion is inducing cell death via ADCC rather than affecting IL-5 binding to the receptor (166). Notably, a single intravenous administration of benralizumab results in depletion of peripheral blood eosinophils for up to 12 weeks, depending on the dose (167). Repeat subcutaneous dosing can produce significant reductions in tissue eosinophilia within 12 weeks (168). Interestingly, the initial clinical trials to deplete eosinophils in patients with asthma did not meet end points of reducing airway hyperresponsiveness or lung function improvement (163, 169). However, eventually all three anti-eosinophil antibodies were shown to dramatically improve various clinical, biological, and pathological features of a subgroup of patients with asthma and were approved by the United States Food and Drug Administration (FDA) between 2015 and 2017 for treatment of eosinophilic asthma. Shortly after, they were approved by the European Medicines Agency (EMA).
Figure 4.
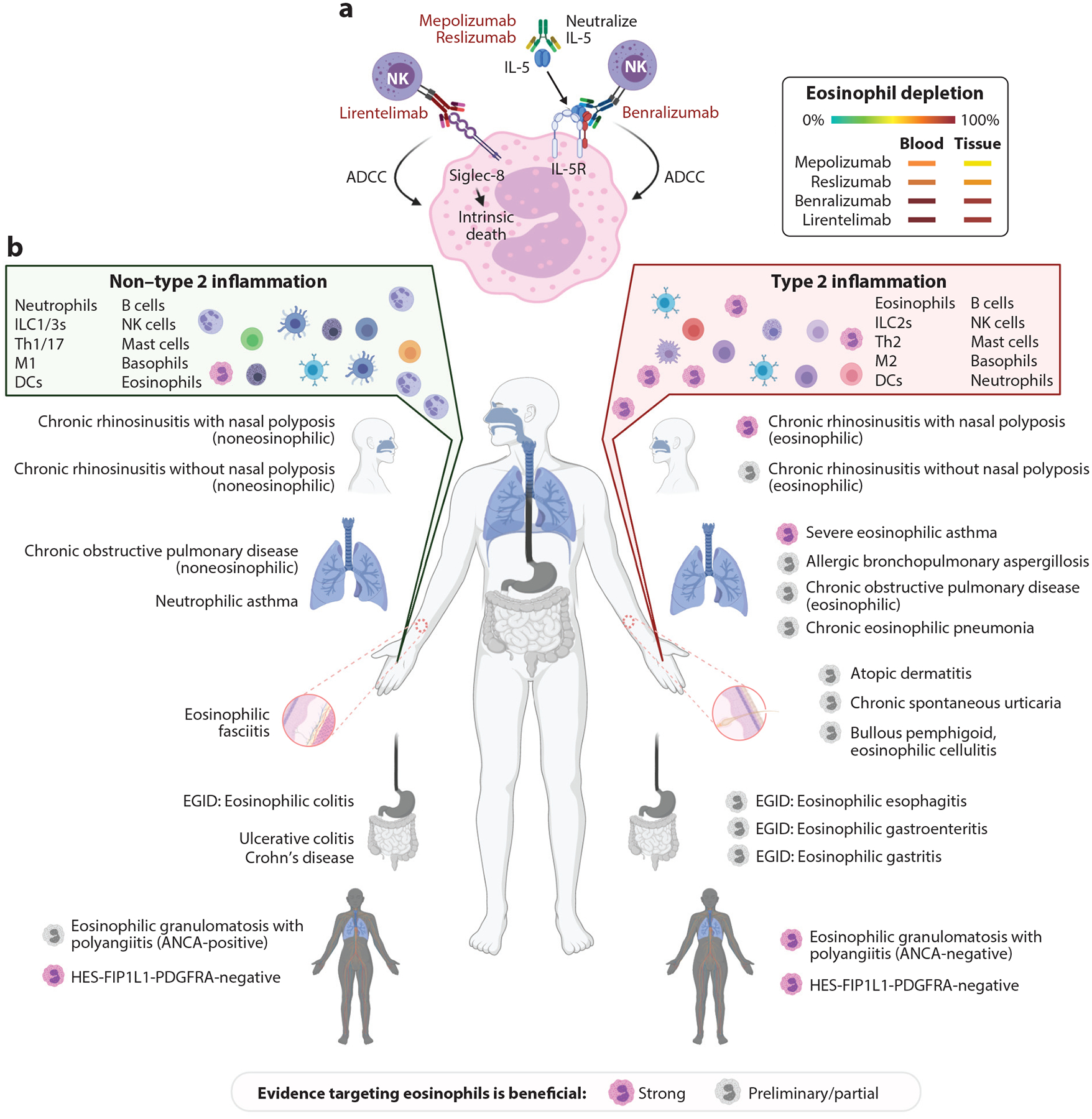
Monoclonal antibodies that target eosinophils and eosinophil-associated diseases. (a) Biologics that target eosinophils for depletion. Mepolizumab and reslizumab are monoclonal antibodies that neutralize IL-5, leading to inhibition of eosinophilopoiesis and impaired eosinophil activation and survival. Benralizumab is a monoclonal antibody that targets the IL-5Rα component. Benralizumab induces Fc-mediated killing by NK cells that mediate ADCC. Lirentelimab binds Siglec-8. This antibody induces intrinsic cell death and mediates ADCC. Eosinophil depletion that occurs in the blood or tissues is shown, with green representing no depletion and red representing complete depletion. (b) Eosinophil-associated diseases and their diversity. Eosinophils classically participate in type 2 diseases and also in non–type 2 diseases. Pathological responses may be eosinophil dependent or independent; in the latter case, other effector cells direct the disease phenotype. Pink eosinophils denote that substantial clinical studies demonstrate clear evidence of some benefit by antibodies targeting eosinophils. Gray eosinophils denote that preliminary clinical studies or a portion of the clinical study outcomes indicate that targeting eosinophils leads to improvement. Please note that studies indicating lack of response to anti-eosinophil therapy are not reflected in this figure. Please also note that HES is on both sides of the figure, as both type 2 and non–type 2 mechanisms are involved and are amenable to anti-eosinophil therapy. Abbreviations: ADCC, antibody-dependent cellular cytotoxicity; ANCA, antineutrophil cytoplasmic antibody; DC, dendritic cell; EGID, eosinophilic gastrointestinal disease; HES, hypereosinophilic syndrome; ILC2, group 2 innate lymphoid cell; M1 macrophage, classically activated macrophage; M2 macrophage, alternatively activated macrophage; NK, natural killer; Siglec-8, sialic acid–binding immunoglobulin-like lectin 8; Th1, T helper 1. Figure adapted from images created with BioRender.com.
Table 2.
Anti-eosinophil therapeutics
| Therapeutic parameters | Mepolizumab | Reslizumab | Benralizumab | Lirentelimab (AK002) |
|---|---|---|---|---|
| Target | IL-5 | IL-5 | IL-5Rα | Siglec-8 |
| Antibody (parent) | Humanized IgG1κ (murine 2B6) | Humanized IgG4κ (rat 39D10) | Humanized nonfucosylated IgG1κ | Humanized nonfucosylated IgG1κ |
| Indicated for severe eosinophilic asthma | Yes (100 mg SC, four doses weekly) | Yes (3 mg/kg IV, four doses weekly) | Yes (30 mg SC, three doses weekly, then every eight weeks) | No |
| Indicated for EGPA/HES | Yes | Noa | Noa | No |
| Other indications being pursued | HES, EoE, CRSwNP | CRSwNP | HES, EGID, CRSwNP | EGID |
| Pediatric indication | 6 years and above | No | 12 years and above | No |
| Eosinophil reduction (blood/tissue) | Yes (↓) | Yes (↓↓) | Near complete (↓↓↓) | Near complete (↓↓↓) |
| Effect on other cells | No | No | Reduction of basophils | Inhibition of mast cells |
Abbreviations: CRSwNP, chronic rhinosinusitis with nasal polyposis; EGID, eosinophilic gastrointestinal disease; EGPA, eosinophilic granulomatosis with polyangiitis; EoE, eosinophilic esophagitis; HES, hypereosinophilic syndrome; IV, intravenous; SC, subcutaneous; Siglec-8, sialic acid–binding immunoglobulin-like lectin 8.
Pending studies.
A new, currently unapproved monoclonal antibody, lirentelimab (AK002), has been developed on the basis of the discovery of Siglec-8—an inhibitory receptor selectively expressed on mature eosinophils and mast cells and with low expression on basophils (151, 170–172). AK002 is a first-in-class, humanized, nonfucosylated IgG1 anti–Siglec-8 monoclonal antibody that depletes eosinophils in blood and tissues via natural killer cell–mediated ADCC and apoptosis (151, 171). AK002 and other anti–Siglec-8 antibodies also inhibit mast cell activation, thereby reducing degranulation, secretion of inflammatory mediators, and recruitment of additional mast cells, eosinophils, and other immune cells to the tissue (151, 171). A recent double-blind phase 2 trial demonstrated efficacy for eosinophilic gastritis and eosinophilic duodenitis (173). This study demonstrated nearly complete ablation of eosinophils from the blood and gastrointestinal tissue and clinical improvements.
3.2. Positive Effects of Eosinophil Depletion on Disease States
Eosinophilic immune dysfunction is observed across a spectrum of conditions, and depending on the relative contribution of eosinophils to pathogenesis and disease manifestations, these disorders can be divided into eosinophil-dependent and -independent diseases. In the most extreme form, hypereosinophilia is considered the predominant cause of tissue damage, dysfunction, and disease manifestations; this is likely best exemplified by idiopathic hypereosinophilic syndrome. In more common conditions, blood and/or tissue hypereosinophilia accompanies a chronic inflammatory disease that also involves other cell types and disease mechanisms. Examples include severe eosinophilic asthma, chronic rhinosinusitis with nasal polyposis (CRSwNP), EoE, EGPAs, bullous pemphigoid, and atopic dermatitis. These disorders have complex pathogenic mechanisms, including but not limited to eosinophil-mediated organ damage and dysfunction. Eosinophil expansion can also occur in the setting of a number of secondary disease processes, such as the severe form of drug allergy DRESS (drug reaction with eosinophilia and systemic symptoms) and paraneoplastic hypereosinophilia associated with T cell lymphoma. Within these categories, a range of immune responses occur as well, from classical type 2 to non–type 2 responses, indicating different immune mechanisms of eosinophil effector functions likely occur in these diseases (Figure 4).
3.2.1. Severe eosinophilic asthma.
A reconsideration of the role of inflammation in airway disease has led to progress, with many targeted therapies now under development. A key change has been the identification of clinically relevant phenotypes and endotypes based on the underlying pathogenesis (174, 175). A new taxonomy of airway diseases based on biological mechanisms has been needed to identify the efficacy and optimize the use of biological therapies, and biomarker assessment has a central role (175, 176). The first controlled trial with mepolizumab showed that it dramatically reduced the blood and sputum eosinophil counts in patients with mild allergic asthma but did not improve the airway response to inhaled allergen, or airway responsiveness in general (169). A subsequent large randomized control trial looking at the safety and efficacy of mepolizumab in patients with moderate asthma showed again that blood and sputum eosinophil counts are significantly lowered by the drug; however, the clinical end points of lung function and quality of life did not significantly improve (177). This disappointing result—greatly reduced numbers of eosinophils not translating to improved symptoms—led to questions of whether eosinophilic inflammation has as important a role in asthma as initially thought. Two alternative explanations for the observed lack of clinical efficacy were that the drug was used in patients who did not have active eosinophilic inflammation and that the studies focused on the wrong outcomes.
Patients with active eosinophilic disease are at risk of exacerbation, and this outcome can be dissociated from symptoms and lung function. When focusing on patients with elevated sputum eosinophil percentages of >3% (normal is <2%), elevated blood eosinophil counts (>150 cells/μL of blood on inhaled glucocorticoid therapy), and/or severe asthma, a combination of benefits is observed in multiple clinical trials with the three recently FDA-approved anti-eosinophil therapeutics. These benefits include risk reduction of severe exacerbations, modest improvements in asthma-specific quality-of-life scores, oral corticosteroid (OCS) reduction, and modest improvements in FEV1 (178–183). Interestingly, the blood eosinophil count was predictive of response to anti–IL-5 therapy, suggesting that the treatment targets circulating eosinophils.
Nearly 30% of patients with COPD have an eosinophilia that may or may not be associated with asthma, resulting in a highly heterogeneous population. In the METREO trial (NCT02105948), mepolizumab was demonstrated to result in a mild reduction in exacerbations in COPD patients with blood eosinophilia (>300 cells/μL), although this was not reached in another mepolizumab trial [METREX (NCT02105961)] (184). Benralizumab treatment also failed to result in significant primary outcomes in the GALATHEA (NCT02138916) and TERRANOVA (NCT02155660) trials, unless patients were stratified specifically by exacerbation rates in years prior (3), blood eosinophilia >220 cells/μL, and triple inhaled therapy (185, 186). This suggests further studies on patients with stratified eosinophilic COPD are needed to define the potential of these eosinophil biologics.
3.2.2. Chronic rhinosinusitis with nasal polyps.
Chronic rhinosinusitis is divided into two major macroscopic phenotypes according to the presence (CRSwNP) or absence (CRSsNP) of noncancerous growths in the lining of the nose and surrounding sinuses (i.e., nasal polyps) (187). Most of the cases in Europe and North America are characterized by a chronic type 2 inflammatory response with tissue eosinophilia and significantly elevated levels of tissue expression of IL-5 and CLC (188, 189). CRSwNP in Asia was historically described as a predominantly neutrophilic disease (190); however, in the last 20 years, a shift toward an increased proportion of patients with tissue eosinophilia has been documented in several Asian countries (191–193). Pathophysiologically, eosinophils accumulate and display evidence of prolonged survival in sino-nasal mucosae (194) of patients with CRSwNP and release into the tissue several inflammatory mediators that are thought to be, at least partially, responsible for many of the pathological features and clinical consequences of the chronic inflammation (189, 195, 196). Moreover, eosinophils from nasal polyps are activated (197) and able to promote both innate and adaptive immune responses (198), fibrin formation (113), and tissue remodeling (199), therefore possibly directly contributing to CRSwNP pathogenesis.
Mepolizumab has been tested for efficacy and safety in patients with CRSwNP in two clinical trials, showing significant reduction in nasal polyp size, sino-nasal symptoms, and need for new sinus surgery (200, 201). A significant reduction in concentration of soluble IL-5 in nasal lavage fluid has been detected in patients treated with mepolizumab compared with those on placebo, but a significant reduction in ECP in nasal lavage fluid has not been observed. Blood eosinophils and serum-soluble IL-5 were reduced, suggesting that mepolizumab has a greater effect on peripheral eosinophilia compared to tissue cell infiltration in CRSwNP (200). A phase 3 clinical trial testing subcutaneous mepolizumab in patients with severe CRSwNP has recently been completed, but the results are not yet published (NCT03085797). Thus far, reslizumab has been tested only in a small, double-blind, placebo-controlled study with 24 patients investigating drug safety and pharmacokinetic properties. The study found a significant reduction in nasal polyp size in treated patients compared to those on placebo, particularly in those with the highest concentrations of IL-5 in nasal lavage (202). Benralizumab is currently under evaluation for the treatment of CRSwNP in several clinical trials, two of which have recently been completed, but whose results are not yet published (NCT03401229, NCT03450083, NCT04185012).
3.2.3. Eosinophilic skin disease.
Chronic dermatologic conditions with significant eosinophilic infiltrates include those occurring in the setting of atopic and allergic immune responses and certain autoimmune disorders and primary immunodeficiencies. The most common form of eosinophilic skin disease is atopic dermatitis, a form of atopic eczema, with a type 2 immune response component at early stages leading to skin damage and itch. In two small, randomized, double-blind clinical trials, treatment with mepolizumab for both shorter (750 mg intravenous, 2 weeks) and longer (100 mg subcutaneous, 16 weeks) durations successfully reduced blood eosinophilia but did not induce significant clinical improvement (i.e., with regard to physician global assessment, clinical scores, size of lesions, and pruritus) compared to placebo. A marginal potential benefit was suggested in terms of disease activity in the study with the higher dosing regimen, but exposure to treatment was too short to allow any meaningful conclusions in such a chronic and spontaneously fluctuating disease (203). The impact of treatment on tissue eosinophilia was not assessed, nor was the possibility that treatment could reduce occurrence of disease flares rather than baseline disease activity (NCT03055195) (204). The role of lymphocytes, mast cells, and other cells in the immune pathology may be more significant than that of eosinophils in pathogenesis of this complex disorder.
Chronic spontaneous urticaria is characterized by recurrent wheals, angioedema, and pruritis for greater than six weeks, with some patients experiencing remitting and relapsing disease over a life span. Although mast cells are considered a significant mediator of disease manifestations, benralizumab was shown to be efficacious in reducing the urticaria activity score [(UAS7) incorporating severity of wheals and pruritus] in patients with persistent disease activity despite antihistamine treatment in a small pilot study (205). This suggests a more prominent role for eosinophils in pathogenesis of this disease (NCT03183024). A phase 2 trial is underway with lirentelimab (NCT03436797), in addition to a phase 1 trial with mepolizumab (NCT03494881). Correlating the occasion of eosinopenia (50 cells/μL), which is characteristic of 10% of these patients (206), with the effectiveness of a specific biologic may provide additional insight into treatment stratification. An additional complex dermatological disease, bullous pemphigoid, is characterized by eosinophilic infiltration and type 2 immune responses that co-occur with autoimmune features to culminate in blister formation (207, 208). The one clinical study to date targeting eosinophils in bullous pemphigoid (NCT01705795) (209) has been completed with mepolizumab and failed to show changes in relapsing disease, suggesting a potential need for either patient stratification or treatment that targets additional cells (e.g., lirentelimab).
3.2.4. Eosinophilic gastrointestinal diseases.
Abundance of eosinophils is associated with an emerging class of diseases referred to as EGIDs, such as EoE, eosinophilic gastritis, eosinophilic gastroenteritis, and eosinophilic colitis. In these disorders, the eosinophil is thought to be one of the key effector cells and is therefore an obvious target for therapy.
Most data on the role of eosinophils and anti-eosinophil treatment come from studies of EoE, a chronic, food antigen–mediated disease characterized by esophageal dysfunction and intraepithelial eosinophil accumulation [≥15 eosinophils per high-power field (hpf)] (210). Its etiology involves a complex interplay of genetic and environmental factors (211). It is associated with strong heritability and is characterized by high sibling recurrence risk (212) and early-life environmental exposures (213). Genetic variants at 2p23 and 5q22 promote disease susceptibility, likely through the esophageal epithelial proteins calpain 14 (CAPN14) and thymic stromal lymphopoietin (TSLP), respectively (214–217). EoE molecular pathophysiology is underscored by esophageal epithelial dysfunction and CD4 T cell–associated type 2 immune responses associated with CAPN14 (215–217) and TSLP (214, 216, 218–220) induction. Reduction of eosinophils using dietary or medical treatment is accompanied by improvement of symptoms, reduction in overall histological disease severity, type 2 inflammatory pathway gene expression, and endoscopic improvement. However, most treatments do not selectively target the eosinophil but instead broadly affect the entire type 2 pathway.
Mepolizumab was first assessed in a small open-label series of adults with longstanding, symptomatic EoE. Four weeks after three infusions of intravenous mepolizumab, four of four patients demonstrated a clinical response (221). A large decrease in esophageal eosinophilia was observed, but peak eosinophil counts remained over the diagnostic threshold of 15 eosinophils/hpf. Two subsequent placebo-controlled trials with adults and children with EoE found a lack of clinical improvement with mepolizumab, despite reduction in eosinophils in most patients (222, 223).
Reslizumab was first evaluated in a randomized controlled trial with a large sample of pediatric patients with EoE. The proportion of patients with a reduced peak eosinophil count was significantly larger among those treated with reslizumab compared to those who received a placebo, but patients did not reach the threshold of clinical response (224). Six patients from one site continued to receive reslizumab in an open-label extension phase. Additionally, four patients were treated with reslizumab on the grounds of compassionate use. After nine years of treatment, reslizumab was still associated with a persistent reduced eosinophil count and was associated with a substantial improvement in EoE symptoms (224).
The results with monoclonal antibodies targeting IL-5 are thus mixed, with sometimes an incomplete inhibition of eosinophils or an insufficient effect on symptoms in an important subset of patients. A recent phase 2 trial with the Siglec-8 inhibitor lirentelimab and patients with eosinophilic gastritis and eosinophilic duodenitis, which have more systemic manifestations than does EoE (225), showed an almost complete abolition of eosinophils (>95% reduction) and a strong reduction in symptoms in patients treated with lirentelimab, which also blocks mast cells (151). The striking positive results from early clinical trials with anti–IL-13 and anti–IL-4R therapeutics are evidence that the clinical and histological features of EoE may be more causally related to type 2 inflammation than to eosinophils alone (226–228).
3.2.5. Hypereosinophilic syndromes.
Depending on the pathogenic mechanisms involved in eosinophil expansion, hypereosinophilic syndromes (HESs) are classified as primary or neoplastic (HESN, clonal eosinophil expansion), secondary or reactive (HESR, polyclonal eosinophil expansion in response to overexpression of eosinophilopoietic factors), or idiopathic (HESId, mechanisms of eosinophil expansion unknown) (10). HESId is commonly restricted to a single organ, such as in chronic eosinophilic pneumonia, gastroenteritis, and dermatitis, whereas the other forms of HES tend to have multi-organ involvement. The most common form of HESN is FIP1L1-PDGFRA+ chronic eosinophilic leukemia, and first-line treatment is undisputedly imatinib mesylate, a tyrosine kinase inhibitor that targets the product of this fusion gene (229). Lymphocytic variant HES (L-HES), driven by dysregulated (often clonal) T cells producing type 2 cytokines including IL-5, is the best-documented form of HESR (230). Patients with L-HES or HESId (i.e., the majority of patients with HES) often require maintenance OCS therapy and/or cytotoxic or immunomodulatory agents for disease control. The disease course is often debilitating because of recurrent or persistent disease activity and/or treatment-related morbidity.
Novel therapeutic options with less toxicity are essential to improve outcomes of patients with HES. Given the purported critical pathogenic role of eosinophils, this disorder is particularly appealing to test proof-of-concept of eosinophil-targeted therapy. Strategically, these patients with HES were important in the early phases of development of therapeutic monoclonal antibodies. After promising results of small open-label pilot studies evaluating efficacy of anti–IL-5 in subjects with HES (231, 232), a large-scale, phase 3, randomized, placebo-controlled clinical trial was conducted in the early 2000s (NCT00086658). Patients with steroid-dependent, FIP1L1-PDGFRA− HES received either 750 mg intravenous mepolizumab or placebo every 4 weeks for 32 weeks, and baseline OCS treatment was successfully tapered during the treatment period in patients receiving mepolizumab compared with those receiving a placebo (233). It took more than ten years to set up a second phase 3 clinical trial evaluating efficacy of mepolizumab in patients with FIP1L1-PDGFRA− HES (NCT02836496). In this trial, patients whose disease was uncontrolled (i.e., who experienced at least two flares during the 12 months prior to enrollment and whose eosinophil counts at inclusion were at least 1,000/μL) received either 300 mg subcutaneous mepolizumab or placebo every 4 weeks for 32 weeks. The proportion of patients who experienced disease flares and the flare rate during the treatment period were significantly lower in the active treatment arm, firmly establishing efficacy of anti–IL-5 treatment for this rare condition and leading to FDA approval (234).
Evidence that biologics targeting the IL-5 receptor may also benefit patients with HES was recently shown in a single-center, phase 2 pilot study evaluating safety and efficacy of 30 mg subcutaneous benralizumab every four weeks in FIP1L1-PDGFRA− disease (235), and a large-scale, multicenter, phase 3, randomized, placebo-controlled clinical trial is in preparation (NATRON, NCT04191304).
The combined favorable impacts of benralizumab treatment on blood eosinophilia, disease activity, and requirement for background OCS therapy are progress toward the long-held prospect of improved management of FIP1L1-PDGFRA− HES. However, observations during the above clinical trials as well as a retrospective analysis of patients enrolled in the mepolizumab compassionate use program indicate that treatment responses may vary depending on disease variants. Although the majority of patients fall in the category of HESId and respond remarkably well to therapy targeting IL-5 or its receptor, those with myeloproliferative/HESN (235, 236) or lymphocytic/HESR (235–237) are refractory or present with suboptimal clinical and/or hematological responses. Biomarker substudies conducted in parallel with these clinical trials should improve our understanding of the mechanisms involved in reduced efficacy of IL-5(R)–targeted therapy in such patients.
3.2.6. Eosinophilic granulomatosis with polyangiitis.
EGPA is a rare and heterogeneous eosinophilic condition characterized by severe eosinophilic asthma associated with blood and tissue hypereosinophilia, granulomatous eosinophilic inflammation, and vasculitis (238). EGPA is classified among the antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAVs), although the prevalence of ANCA (anti-myeloperoxidase, roughly 30–40%) is much lower in EGPA than in the other AAVs (239). The hypereosinophilic disease component is similar to HES, with eosinophilic infiltration of various tissues and organs, including most commonly (but not restricted to) the lungs with involvement of the paranasal sinuses. The vasculitic component may comprise both eosinophilic and non-eosinophilic invasion of small vessel walls, involving most commonly the skin, peripheral nerves, gastrointestinal tract, and kidneys. In clinical practice, biopsy-proven vasculitis is not mandatory for diagnosis of EGPA, and typical surrogates for vasculitic involvement include mononeuritis multiplex and palpable purpura. In many cases, however, diagnosis is made even in the absence of such evidence if patients have a combination of severe adult-onset asthma, chronic rhinosinusitis, and blood hypereosinophilia with various other manifestations of eosinophilic organ infiltration. In such cases, there is significant diagnostic overlap with HES and chronic eosinophilic pneumonia, and this is consistent with the absence of specific biomarkers distinguishing EGPA from the other eosinophilic conditions with lung involvement (240). Observational studies have shown that ANCA status partially accounts for disease heterogeneity, with ANCA-positive patients showing more frequent vasculitic disease complications such as peripheral neuropathy and glomerulonephritis, whereas those without ANCA are more likely to experience eosinophilic myocarditis (241). The importance of ANCA was recently corroborated by a large-scale genome-wide association study conducted with more than 650 patients with EGPA. Some of the observed genetic polymorphisms thought to be relevant in terms of pathogenesis differed according to ANCA status and, interestingly, overlapped with known type 2 immunity–associated loci (242). With HLA-DQ gene polymorphisms in ANCA-positive disease and barrier function gene polymorphisms in ANCA-negative disease, as well as a series of anomalies involving eosinophil/type 2 immunity across both groups, it is clear that EGPA as currently defined encompasses disorders with complex and heterogeneous pathogenic mechanisms. Furthermore, although eosinophils are prominent cells in EGPA, invading both tissues and vessel walls, there is evidence that other cells and mediators are involved in pathogenesis, including type 2 cells (ILC2s and Th2 cells) and Th1, Th17, and B cells (238).
Given the putative role of IL-5 in EGPA exacerbations (243) and the efficacy of IL-5R targeting in patients with severe eosinophilic asthma and HES, both of which share characteristics with EGPA, clinical trials have been designed to evaluate antibodies targeting the IL-5 pathway in patients with this disease. Clinical efficacy of anti–IL-5 antibodies in patients with EGPA has been demonstrated in individual case reports; pilot studies (244, 245); and a phase 3, randomized, placebo-controlled clinical trial comparing efficacy of 300 mg subcutaneous mepolizumab versus placebo every 4 weeks for 52 weeks in patients with treatment-refractory or relapsing disease (MIRRA, NCT02020889). The latter trial showed significant benefit with active treatment in terms of accrued time in remission, proportion of patients in remission, relapse rate, and OCS tapering (246), leading to the approval of mepolizumab for treatment of EGPA in the United States. Efficacy of 30 mg subcutaneous benralizumab every 4 weeks is now being compared to that of mepolizumab in patients with EGPA in the setting of an ongoing phase 3 randomized clinical trial (MANDARA, NCT04157348). An ambitious mechanistic substudy is being conducted in parallel, investigating mechanisms of action of these agents and cellular and molecular predictors of treatment responses, and may improve our understanding of the relative contribution of eosinophils to specific EGPA disease components and variants.
Indeed, data collected during the MIRRA trial have not satisfactorily addressed the concern that eosinophil-independent vasculitic complications may occur during IL-5–targeted therapy, especially if improvement in eosinophil-mediated disease manifestations results in tapering of background therapy. Although mepolizumab was reported to have a favorable effect on both asthma/sinonasal and vasculitic disease relapses, the nature of vasculitic complications that were controlled by treatment was not detailed. The proportion of ANCA-positive patients enrolled in this trial was only 10%, precluding subgroup analyses to explore differential efficacy of treatment for infiltrative versus vasculitic components of disease and according to ANCA status. Furthermore, disease relapses were reduced but not abolished in the active treatment arm, raising questions similar to those regarding eosinophilic asthma and the complex interplay between triggers, cells, and mediators that eventually results in a clinical exacerbation.
4. CONCLUSION
4.1. Health Status of Eosinophil Knockout Humans
Allied to an appreciation that the eosinophil may have a role in health has been a degree of apprehension that therapies targeting the eosinophil may inadvertently cause harm. In particular, these concerns have centered on the theoretical potential for impaired host defense against infection, impaired metabolism, and an increased risk of or poor outcome with malignancy. However, robust evidence of these possibilities in humans appears elusive (247).
Early reports of patients rendered eosinopenic due to immunodeficiency or immunoglobulin G–mediated eosinophil precursor destruction did not highlight any clinical sequelae attributable to the eosinophil reduction (248, 249). OCS used for decades in the setting of many severe respiratory, rheumatological, and other organ-specific conditions effectively depleted eosinophils (250) without any clear evidence of increased malignancy rates (251). While both mepolizumab and reslizumab reduce eosinophil numbers, the almost complete eosinophil depletion that occurs with benralizumab and lirentelimab presents the optimal opportunity to study the implications of eosinopenia in humans.
Over 8,000 patients have received benralizumab following enrollment in a clinical development program (247), and the combination of prospective phase 3 trial data in both asthma (SIROCCO and CALIMA trials and subsequent BORA extension trial) (182, 183, 252) and COPD (GALATHEA and TERRANOVA trials) (186) along with additional real-world (253, 254) and postmarketing data provides insights into the potential risks of eosinophil depletion. In the asthma studies, treatment with benralizumab was well-tolerated, with overall adverse event types and frequencies similar to those of placebo. The additional 56 weeks of benralizumab exposure as part of the BORA extension study likewise did not highlight any new safety concerns, providing additional reassurance. Evidence of malignancy was assessed by an independent safety event adjudication committee throughout the benralizumab phase 3 asthma program. The incidence rates of malignancy in the benralizumab treatment arms of the one-year placebo-controlled trials and 56-week extension period were 4/1,663 (0.2%) and 12/1,576 (0.8%), respectively (182, 183, 252). The incidence of malignancy for patients continuously exposed to benralizumab from the start of the controlled treatment period through the end of the extension period (up to two years) was 8/1,030 (0.8%), and there were no clear trends in organs or tissue types affected.
The two 56-week, placebo-controlled COPD trials of benralizumab, GALATHEA and TERRANOVA (186), included 2,792 patients, and the reported malignancy rates were 1.7% in the benralizumab arm and 2.1% in the placebo arm. Importantly, the subjects recruited to the COPD studies were at intrinsically higher risk of malignancy than those in the asthma studies, both being older (mean age 65 years as compared to 49 years) and having significant smoking histories. Indeed, a third of the subjects were active smokers at the time of recruitment, while fewer than 1% of the asthma subjects were current smokers. Yet, caution is needed when interpreting these figures, as observation times were comparatively short and statistical power for rare events is lacking. The extended COLUMBA (255) study of mepolizumab in severe asthma offers some additional reassurance in this regard, with no specific concerns relating to malignancy or infection identified following the treatment of 347 patients for an average of 3.5 years (4.5 years maximum), albeit eosinopenia was incomplete (i.e., absolute blood count did not fall to zero) in this study. Finally, postmarketing data (while naturally limited by a dependence on continuous reporting) include only 17 cases of spontaneous malignancies worldwide during more than 35,000 patient-years of exposure since launch (247).
The potential diminution of antihelminth defense with anti–IL-5 or anti–IL-5R monoclonal antibodies has also been a concern for clinicians, especially in regions where such infections are endemic. However, no helminth infections were reported for the phase 3 and extension studies. Importantly, CALIMA/SIROCCO included almost 500 subjects from South American countries, including Argentina (n = 269), Brazil (n = 36), Chile (n = 31), and Peru (n = 97), as well as subjects from other countries with a high incidence of helminth infections, including the Philippines (n = 61), Vietnam (n = 15), and South Africa (n = 26). In addition, evidence that eosinophil depletion might impair antiviral responses is lacking; there is a similar rate of herpes zoster infection (0.5%) in the placebo and benralizumab arms of the CALIMA/SIROCCO studies. No safety concern has yet been reported for patients with coronavirus disease 2019 (COVID-19) and eosinophilic disease, including those on anti-eosinophil therapy (49).
Monoclonal antibodies, such as mepolizumab and benralizumab, are transported across the placenta during the third trimester. The consequences of therapeutic eosinophil ablation with these agents for the developing human fetus are largely unknown. In a long-term safety trial of mepolizumab for HES, one patient gave birth to a healthy neonate (256). A pre- and postnatal study of benralizumab in cynomolgus monkeys found no evidence of fetal harm following exposures up to 310 times higher than the licensed human dosage (247). Postmarketing surveillance studies investigating pregnancy and infant outcomes for women exposed to mepolizumab and benralizumab are currently ongoing.
4.2. Mouse Versus Human Eosinophil Knockout
How does one align the seemingly contradictory findings discussed herein that eosinophils have a role in health but that their therapeutic depletion in humans does not appear to cause harm? One possibility is that the former is largely derived from murine studies and that the translational significance of these insights to humans remains questionable. Another is that there is sufficient redundancy within the immune system to counteract drug-induced eosinopenia.
Several caveats should be considered regarding the disparities between studies of eosinophil depletion in humans and the roles of eosinophils in health and disease in animal models. First and foremost, the animal models of IL-5 antibody treatment and use of IL-5−/− or IL-5Rα−/− mice faithfully predicted a role for eosinophils in certain models of EADs. Similar to the case of incomplete eosinophil deficiency in IL-5–deficient mice, biologics targeting IL-5 (mepolizumab or reslizumab) failed to achieve full depletion of eosinophilia in inflamed tissues (163). Therefore, some conclusions derived from mepolizumab or reslizumab studies with regard to the relative contribution of eosinophils to disease phenotype and activity may not apply to benralizumab or lirentelimab, as the former two may not represent an ideal model of eosinopenia in humans. This is particularly relevant given that eosinophils can survive in tissue without IL-5 and may leave behind cell-free granules that continue to inflame.
The goal of the human studies presented herein for these biologics has been to treat EADs and ensure safety in treatment and placebo cohorts. Here the data are quite similar to those for mice. Eosinophil-depleted mice, whether eosinophil depletion is congenic or induced, do not develop significant ill health simply due to the absence of eosinophils. For example, the findings with benralizumab strongly suggest eosinophil depletion is unlikely to trigger or enable cancers and that the role of eosinophils in immunosurveillance of cancer is not critical. This would be in agreement with studies in animal models, as eosinophil-deficient mice do not spontaneously develop cancer. Rather, for both mouse and human studies, one would be required to ask what the role of eosinophils is in disease settings such as bladder, pancreatic, and intestinal cancers, where eosinophils are a significant infiltrate, and specifically include assessment of the association between eosinophil counts and activation with patient outcome among primary study end points. Therefore, the existing studies are excellent evidence that eosinophil depletion in and of itself does not potentiate ill effect, yet the role of eosinophils in a variety of diseases, particularly non-EADs, requires additional consideration and longer-term studies to clarify nuances in diseases that may take decades to develop in predisposed individuals.
4.3. Eosinophil-Independent Eosinophilic Diseases
Despite the demonstrable benefit of eosinophil depletion in clinical trials, a substantial number of patients with the same diseases targeted in those trials do not respond to eosinophil depletion. These can be categorized as (a) cases where eosinophil depletion in tissue does not occur or is incomplete and (b) cases where eosinophils are depleted but pathologies and symptoms persist. Limited depletion of tissue eosinophilia may occur due to downregulation of IL-5Rα, IL-5–independent eosinophil development and survival, and/or impaired ADCC mechanisms (67). In the case of apparent eosinophil-independent disease mechanisms, the quantity of eosinophils in tissue may not be correlated with their contribution to the immune and inflammation responses. If, for example, eosinophils display predominantly immune regulatory or immune suppressive activities in a given disease, very few eosinophils may be required to mediate large effects. The lack of efficacy of eosinophil-targeted therapy in such instances would therefore not signify that eosinophils do not have a major (albeit regulatory) role in pathogenesis. Yet, when eosinophil depletion is complete, the role of eosinophils may be more nuanced or indeed irrelevant. Several scenarios may occur that include occult eosinophil activities, such as persistence of cell-free granules that mediate some level of effector functions in the absence of eosinophils, giving the impression of inflammation that is eosinophil independent. Conversely, eosinophils may indeed be irrelevant to disease progression and not primary promoters of the inflammatory process; rather, cells such as ILCs and T cells may predominate. This may help explain the mixed results of eosinophil-depleting therapies in asthma, atopic dermatitis, COPD, CRSwNP, and EGIDs. EGPA pathology spans both allergic, eosinophil-mediated mechanisms and vasculitic mechanisms, with eosinophil depletion not necessarily addressing the latter in cases nonresponsive to anti–IL-5 or anti–IL-5R therapies. Finally, the remodeling that generates significant symptoms in some diseases, such as EoE and severe chronic asthma, may be irreversible at some disease stages; thus, removing the effector cells of the disease is a belated effort. In such cases, physical treatment, such as bronchoplasty and esophageal balloon dilation in asthma and EoE, respectively, relieves symptoms. Furthermore, it is possible that future trials combining anti–IL-5 therapies with other biologics, such as dupilumab, will enhance improvement of symptoms and pathologies.
5. FINAL REMARKS
Significant gains have been made translating findings in mice to treatment of eosinophilic diseases in humans. IL-5– and IL-5R–targeted therapies are effective in a number of patients with EADs and have a good safety profile in humans. As in mice, the dependence of eosinophils on IL-5 is incomplete in humans, providing a partial explanation of the reduced ability of mepolizumab and reslizumab to produce true eosinophil knockout humans. Notably, benralizumab and lirentelimab, which bind IL-5Rα and Siglec-8, respectively, kill eosinophils, thereby generating a near–eosinophil knockout human. These biologics permit insights into the enigmatic role of the eosinophil in humans, particularly regarding whether humans require eosinophils for health and disease. Findings from these studies have proven that human eosinophils have a contributory disease-causing role in a number of common and rare diseases, such as severe asthma and hypereosinophilic diseases, respectively. These studies refute a key role for human eosinophils in contributing to physiological health, as eosinophil knockout humans develop no overt disease predispositions, although additional data are warranted. A caveat to the lack of adverse events in humans is that clinical trials and postmarketing surveillance studies may have selection bias for individuals with higher socioeconomic status (e.g., those with healthcare insurance) and thus may not be applicable to other populations that may suffer from different diseases and/or manifestations, such as parasitic infections. We predict that eosinophil knockout humans will also help stratify EADs into eosinophil-dependent and eosinophil-independent processes, and we anticipate that this will lead to reclassification of at least some diseases formerly considered to be primary eosinophilic diseases.
SUMMARY POINTS.
Mouse models have paved the way for discovery of a variety of biologics that deplete eosinophils in humans.
A new class of precision drugs, referred to as anti-eosinophil therapeutics, is available to treat a variety of eosinophil-associated diseases.
These new anti-eosinophil biologics result in something close to an eosinophil knockout human.
Animal models suggest that eosinophils participate in some homeostatic processes that are made evident when challenged by external stimuli, injury, or disease.
Monoclonal antibodies targeting human eosinophils reduce asthma symptoms and exacerbations, as well as manifestations of a number of other eosinophil-associated diseases, and their safety profile has no adverse events.
Despite the success of these monoclonal antibodies, human eosinophil depletion does not reduce pathologies in all eosinophil-associated diseases, underscoring heterogeneous and complex disease mechanisms.
Eosinophil knockout humans have no overt medical problems, suggesting that the primary role of eosinophils is promoting selective eosinophilic inflammatory diseases and that eosinophils do not have primary roles in maintenance of health.
It is unclear whether biologics can alter the outcomes of patients with non-eosinophil-associated diseases that have an extensive eosinophil presence, such as cancer.
ACKNOWLEDGMENTS
Shawna Hottinger, a medical writer employed by Cincinnati Children’s Hospital Medical Center, provided editorial assistance for this review. E.A.J. is supported by NIAID R01 AI145108, R21 AI132840, and R01 DK121330 and by the Mayo Foundation. E.A.J. would like to thank her late colleague Dr. James J. Lee, who created many of the strains mentioned in this manuscript and helped bring forth the idea that eosinophils have multiple roles in health and disease. A.J.B. is supported by the Vidi grant 91718300 from the Netherlands Organisation for Scientific Research NWO. F.R. is supported by the Belgian National Fund for Scientific Research (FNRS), grant numbers F 5/4/150/5 and FC 54372. M.E.R. is supported by NIH R01 AI045898, U19 AI070235, R01 AI057803, U54 AI117804, R01 AI124355, and R01 DK107502; the Campaign Urging Research for Eosinophilic Disease (CURED); and the Sunshine Charitable Foundation and its supporters and Denise and David Bunning.
DISCLOSURE STATEMENT
E.A.J. has received advisory board fees from AstraZeneca and speaker fees from GlaxoSmithKline (GSK). D.J.J. has received advisory board and speaker fees from AstraZeneca, GSK, Sanofi, Novartis, and Teva Pharmaceuticals. E.H. has received advisory board support from AstraZeneca, GSK, Novartis, Sanofi, Circassia, Stallergenes-Greer, and Nestlè Purina. S.K.M. has received consulting and speaker fees from AstraZeneca and GSK. A.J.B. has received research support from Nutricia, Bayer, Norgine, and SST and consulting/speaking fees from AstraZeneca, Alimentiv, Dr. Falk Pharma, Celgene, Arena Pharmaceuticals, Regeneron, Medtronic, and Laborie and has an equity interest in SST. In the last 5 years, I.D.P. has received speaker’s honoraria for speaking at sponsored meetings from AstraZeneca, Boehringer Ingelheim, Aerocrine, Almirall, Novartis, Teva, Chiesi, Sanofi/Regeneron, Menarini, and GSK and payments for organizing educational events from AstraZeneca, GSK, Sanofi/Regeneron, and Teva. He has received honoraria for attending advisory panels with Genentech, Sanofi/Regeneron, AstraZeneca, Boehringer Ingelheim, GSK, Novartis, Teva, Merck, Circassia, Chiesi, and Knopp and payments to support FDA approval meetings from GSK. He has received sponsorship to attend international scientific meetings from Boehringer Ingelheim, GSK, AstraZeneca, Teva, and Chiesi. He has received a grant from Chiesi to support a phase 2 clinical trial in Oxford, United Kingdom. He is co–patent holder of the rights to the Leicester Cough Questionnaire and has received payments for its use in clinical trials from Merck, Bayer, and Insmed. In 2014–2015, he was an expert witness for a patent dispute involving AstraZeneca and Teva. P.A. has received research support from the National Institutes of Health; has received research support and consultancy fees from and has been on advisory boards for AstraZeneca and GSK; has received consultancy fees from Ambrx; receives royalties from UpToDate; and has received honoraria from WebMD/Medscape, AHK, Prime CME, Rockpointe, MJH Life Sciences, and Vindico. F.R. is a consultant for GSK and AstraZeneca and receives royalties from UpToDate. M.E.R. is a consultant for Pulm One, Spoon Guru, ClostraBio, Serpin Pharm, Allakos, Celgene, AstraZeneca, Arena Pharmaceuticals, Ellodi Pharma, GSK, Sanofi/Regeneron, Guidepoint, and Suvretta Capital Management and has an equity interest in the first five listed and royalties from reslizumab (Teva Pharmaceuticals), PEESSv2 (Mapi Research Trust), and UpToDate; M.E.R. is an inventor on patents owned by Cincinnati Children’s Hospital Medical Center.
LITERATURE CITED
- 1.Acharya KR, Ackerman SJ. 2014. Eosinophil granule proteins: form and function. J. Biol. Chem 289:17406–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klion AD, Ackerman SJ, Bochner BS. 2020. Contributions of eosinophils to human health and disease. Annu. Rev. Pathol. Mech. Dis 15:179–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JJ, Jacobsen EA, McGarry MP, Schleimer RP, Lee NA. 2010. Eosinophils in health and disease: the LIAR hypothesis. Clin. Exp. Allergy 40:563–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weller PF, Spencer LA. 2017. Functions of tissue-resident eosinophils. Nat. Rev. Immunol 17:746–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothenberg ME, Hogan SP. 2006. The eosinophil. Annu. Rev. Immunol 24:147–74 [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg HF, Dyer KD, Foster PS. 2013. Eosinophils: changing perspectives in health and disease. Nat. Rev. Immunol 13:9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kay AB. 2015. The early history of the eosinophil. Clin. Exp. Allergy 45:575–82 [DOI] [PubMed] [Google Scholar]
- 8.O’Sullivan JA, Bochner BS. 2018. Eosinophils and eosinophil-associated diseases: an update. J. Allergy Clin. Immunol 141:505–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobsen EA, Helmers RA, Lee JJ, Lee NA. 2012. The expanding role(s) of eosinophils in health and disease. Blood 120:3882–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valent P, Klion AD, Horny HP, Roufosse F, Gotlib J, et al. 2012. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J. Allergy Clin. Immunol 130:607–12.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khoury P, Akuthota P, Ackerman SJ, Arron JR, Bochner BS, et al. 2018. Revisiting the NIH Taskforce on the Research needs of Eosinophil-Associated Diseases (RE-TREAD). J. Leukoc. Biol 104:69–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulessa H, Frampton J, Graf T. 1995. GATA-1 reprograms avian myelomonocytic cell lines into eosinophils, thromboblasts, and erythroblasts. Genes Dev 9:1250–62 [DOI] [PubMed] [Google Scholar]
- 13.Hirasawa R, Shimizu R, Takahashi S, Osawa M, Takayanagi S, et al. 2002. Essential and instructive roles of GATA factors in eosinophil development. J. Exp. Med 195:1379–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNagny K, Graf T. 2002. Making eosinophils through subtle shifts in transcription factor expression. J. Exp. Med 195:F43–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu C, Cantor AB, Yang H, Browne C, Wells RA, et al. 2002. Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J. Exp. Med 195:1387–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drissen R, Buza-Vidas N, Woll P, Thongjuea S, Gambardella A, et al. 2016. Distinct myeloid progenitor-differentiation pathways identified through single-cell RNA sequencing. Nat. Immunol 17:666–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Dijk TB, Caldenhoven E, Raaijmakers JA, Lammers JW, Koenderman L, et al. 1998. The role of transcription factor PU.1 in the activity of the intronic enhancer of the eosinophil-derived neurotoxin (RNS2) gene. Blood 91:2126–32 [PubMed] [Google Scholar]
- 18.Bouffi C, Kartashov AV, Schollaert KL, Chen X, Bacon WC, et al. 2015. Transcription factor repertoire of homeostatic eosinophilopoiesis. J. Immunol 195:2683–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bedi R, Du J, Sharma AK, Gomes I, Ackerman SJ. 2009. Human C/EBP-epsilon activator and repressor isoforms differentially reprogram myeloid lineage commitment and differentiation. Blood 113:317–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamanaka R, Barlow C, Lekstrom-Himes J, Castilla LH, Liu PP, et al. 1997. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. PNAS 94:13187–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lekstrom-Himes JA. 2001. The role of C/EBP(epsilon) in the terminal stages of granulocyte differentiation. Stem Cells 19:125–33 [DOI] [PubMed] [Google Scholar]
- 22.Gombart AF, Kwok SH, Anderson KL, Yamaguchi Y, Torbett BE, et al. 2003. Regulation of neutrophil and eosinophil secondary granule gene expression by transcription factors C/EBP epsilon and PU.1. Blood 101:3265–73 [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg HF, Gallin JI. 1993. Neutrophil-specific granule deficiency includes eosinophils. Blood 82:268–73 [PubMed] [Google Scholar]
- 24.Bettigole SE, Lis R, Adoro S, Lee AH, Spencer LA, et al. 2015. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat. Immunol 16:829–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu TX, Lim EJ, Besse JA, Itskovich S, Plassard AJ, et al. 2013. MiR-223 deficiency increases eosinophil progenitor proliferation. J. Immunol 190:1576–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu TX, Lim EJ, Itskovich S, Besse JA, Plassard AJ, et al. 2013. Targeted ablation of miR-21 decreases murine eosinophil progenitor cell growth. PLOS ONE 8:e59397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barlow JL, McKenzie AN. 2014. Type-2 innate lymphoid cells in human allergic disease. Curr. Opin. Allergy Clin. Immunol 14:397–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmermann N, Hershey GK, Foster PS, Rothenberg ME. 2003. Chemokines in asthma: cooperative interaction between chemokines and IL-13. J. Allergy Clin. Immunol 111:227–42 [DOI] [PubMed] [Google Scholar]
- 29.Matthews AN, Friend DS, Zimmermann N, Sarafi MN, Luster AD, et al. 1998. Eotaxin is required for the baseline level of tissue eosinophils. PNAS 95:6273–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabroe I, Hartnell A, Jopling LA, Bel S, Ponath PD, et al. 1999. Differential regulation of eosinophil chemokine signaling via CCR3 and non-CCR3 pathways. J. Immunol 162:2946–55 [PubMed] [Google Scholar]
- 31.Menzies-Gow A, Ying S, Sabroe I, Stubbs VL, Soler D, et al. 2002. Eotaxin (CCL11) and eotaxin-2 (CCL24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early- and late-phase allergic reactions following cutaneous injection in human atopic and nonatopic volunteers. J. Immunol 169:2712–18 [DOI] [PubMed] [Google Scholar]
- 32.Chojnacki A, Wojcik K, Petri B, Aulakh G, Jacobsen EA, et al. 2019. Intravital imaging allows real-time characterization of tissue resident eosinophils. Commun. Biol 2:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdala-Valencia H, Coden ME, Chiarella SE, Jacobsen EA, Bochner BS, et al. 2018. Shaping eosinophil identity in the tissue contexts of development, homeostasis, and disease. J. Leukoc. Biol 104:95–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Hulst G, Batugedara HM, Jorssen J, Louis R, Bureau F, et al. 2020. Eosinophil diversity in asthma. Biochem. Pharmacol 179:113963. [DOI] [PubMed] [Google Scholar]
- 35.Johansson MW. 2017. Eosinophil activation status in separate compartments and association with asthma. Front. Med 4:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdala Valencia H, Loffredo LF, Misharin AV, Berdnikovs S. 2016. Phenotypic plasticity and targeting of Siglec-Fhigh CD11clow eosinophils to the airway in a murine model of asthma. Allergy 71:267–71 [DOI] [PubMed] [Google Scholar]
- 37.Mesnil C, Raulier S, Paulissen G, Xiao X, Birrell MA, et al. 2016. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J. Clin. Investig 126:3279–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rothenberg ME. 2016. A hidden residential cell in the lung. J. Clin. Investig 126:3185–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu C, Weng QY, Zhou LR, Cao C, Li F, et al. 2020. Homeostatic and early recruited CD101− eosinophils suppress endotoxin-induced acute lung injury. Eur. Respir. J 56(5):1902354. [DOI] [PubMed] [Google Scholar]
- 40.Kelly EA, Esnault S, Liu LY, Evans MD, Johansson MW, et al. 2017. Mepolizumab attenuates airway eosinophil numbers, but not their functional phenotype, in asthma. Am. J. Respir. Crit. Care Med 196:1385–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drake LY, Kita H. 2017. IL-33: biological properties, functions, and roles in airway disease. Immunol. Rev 278:173–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacobsen EA, Doyle AD, Colbert DC, Zellner KR, Protheroe CA, et al. 2015. Differential activation of airway eosinophils induces IL-13-mediated allergic Th2 pulmonary responses in mice. Allergy 70:1148–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. 2010. IL-33 exacerbates eosinophil-mediated airway inflammation. J. Immunol 185:3472–80 [DOI] [PubMed] [Google Scholar]
- 44.Bouffi C, Rochman M, Zust CB, Stucke EM, Kartashov A, et al. 2013. IL-33 markedly activates murine eosinophils by an NF-κB-dependent mechanism differentially dependent upon an IL-4-driven autoinflammatory loop. J. Immunol 191:4317–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ben Baruch-Morgenstern N, Mingler MK, Stucke E, Besse JA, Wen T, et al. 2016. Paired Ig-like receptor B inhibits IL-13-driven eosinophil accumulation and activation in the esophagus. J. Immunol 197:707–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xenakis JJ, Howard ED, Smith KM, Olbrich CL, Huang Y, et al. 2018. Resident intestinal eosinophils constitutively express antigen presentation markers and include two phenotypically distinct subsets of eosinophils. Immunology 154:298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drake MG, Bivins-Smith ER, Proskocil BJ, Nie Z, Scott GD, et al. 2016. Human and mouse eosinophils have antiviral activity against parainfluenza virus. Am. J. Respir. Cell Mol. Biol 55:387–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.LeMessurier KS, Rooney R, Ghoneim HE, Liu B, Li K, et al. 2020. Influenza A virus directly modulates mouse eosinophil responses. J. Leukoc. Biol 108:151–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindsley AW, Schwartz JT, Rothenberg ME. 2020. Eosinophil responses during COVID-19 infections and coronavirus vaccination. J. Allergy Clin. Immunol 146:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Percopo CM, Dyer KD, Ochkur SI, Luo JL, Fischer ER, et al. 2014. Activated mouse eosinophils protect against lethal respiratory virus infection. Blood 123:743–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samarasinghe AE, Melo RC, Duan S, LeMessurier KS, Liedmann S, et al. 2017. Eosinophils promote antiviral immunity in mice infected with influenza A virus. J. Immunol 198:3214–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnold IC, Artola-Boran M, Tallon de Lara P, Kyburz A, Taube C, et al. 2018. Eosinophils suppress Th1 responses and restrict bacterially induced gastrointestinal inflammation. J. Exp. Med 215:2055–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guerra ES, Lee CK, Specht CA, Yadav B, Huang H, et al. 2017. Central role of IL-23 and IL-17 producing eosinophils as immunomodulatory effector cells in acute pulmonary aspergillosis and allergic asthma. PLOS Pathog 13:e1006175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malacco N, Rachid MA, Gurgel I, Moura TR, Sucupira PHF, et al. 2018. Eosinophil-associated innate IL-17 response promotes Aspergillus fumigatus lung pathology. Front. Cell Infect. Microbiol 8:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grisaru-Tal S, Itan M, Klion AD, Munitz A. 2020. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 20:594–607 [DOI] [PubMed] [Google Scholar]
- 56.Diny NL, Rose NR, Cihakova D. 2017. Eosinophils in autoimmune diseases. Front. Immunol 8:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JJ, Jacobsen EA, Ochkur SI, McGarry MP, Condjella RM, et al. 2012. Human versus mouse eosinophils: “That which we call an eosinophil, by any other name would stain as red.” J. Allergy Clin. Immunol 130:572–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jacobsen EA, Lee NA, Lee JJ. 2014. Re-defining the unique roles for eosinophils in allergic respiratory inflammation. Clin. Exp. Allergy 44:1119–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kung TT, Stelts DM, Zurcher JA, Adams GK 3rd, Egan RW, et al. 1995. Involvement of IL-5 in a murine model of allergic pulmonary inflammation: prophylactic and therapeutic effect of an anti-IL-5 antibody. Am. J. Respir. Cell Mol. Biol 13:360–65 [DOI] [PubMed] [Google Scholar]
- 60.Matthaei KI, Foster P, Young IG. 1997. The role of interleukin-5 (IL-5) in vivo: studies with IL-5 deficient mice. Mem. Inst. Oswaldo Cruz 92(Suppl. 2):63–68 [DOI] [PubMed] [Google Scholar]
- 61.Kopf M, Brombacher F, Hodgkin PD, Ramsay AJ, Milbourne EA, et al. 1996. IL-5-deficient mice have a developmental defect in CD5+ B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity 4:15–24 [DOI] [PubMed] [Google Scholar]
- 62.Yoshida T, Ikuta K, Sugaya H, Maki K, Takagi M, et al. 1996. Defective B-1 cell development and impaired immunity against Angiostrongylus cantonensis in IL-5Rα-deficient mice. Immunity 4:483–94 [DOI] [PubMed] [Google Scholar]
- 63.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. 1996. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J. Exp. Med 183:195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takatsu K 2011. Interleukin-5 and IL-5 receptor in health and diseases. Proc. Jpn. Acad. Ser. B 87:463–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bao S, Beagley KW, Murray AM, Caristo V, Matthaei KI, et al. 1998. Intestinal IgA plasma cells of the B1 lineage are IL-5 dependent. Immunology 94:181–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hiroi T, Yanagita M, Iijima H, Iwatani K, Yoshida T, et al. 1999. Deficiency of IL-5 receptor alpha-chain selectively influences the development of the common mucosal immune system independent IgA-producing B-1 cell in mucosa-associated tissues. J. Immunol 162:821–28 [PubMed] [Google Scholar]
- 67.Liu LY, Sedgwick JB, Bates ME, Vrtis RF, Gern JE, et al. 2002. Decreased expression of membrane IL-5 receptor α on human eosinophils: I. Loss of membrane IL-5 receptor α on airway eosinophils and increased soluble IL-5 receptor α in the airway after allergen challenge. J. Immunol 169:6452–58 [DOI] [PubMed] [Google Scholar]
- 68.Sanderson CJ. 1992. Interleukin-5, eosinophils, and disease. Blood 79:3101–9 [PubMed] [Google Scholar]
- 69.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, et al. 2004. A critical role for eosinophils in allergic airways remodeling. Science 305:1776–79 [DOI] [PubMed] [Google Scholar]
- 70.Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, et al. 2004. Defining a link with asthma in mice congenitally deficient in eosinophils. Science 305:1773–76 [DOI] [PubMed] [Google Scholar]
- 71.McDevitt MA, Shivdasani RA, Fujiwara Y, Yang H, Orkin SH. 1997. A “knockdown” mutation created by cis-element gene targeting reveals the dependence of erythroid cell maturation on the level of transcription factor GATA-1. PNAS 94:6781–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crispino JD, Horwitz MS. 2017. GATA factor mutations in hematologic disease. Blood 129:2103–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doyle AD, Jacobsen EA, Ochkur SI, McGarry MP, Shim KG, et al. 2013. Expression of the secondary granule proteins major basic protein 1 (MBP-1) and eosinophil peroxidase (EPX) is required for eosinophilopoiesis in mice. Blood 122:781–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jacobsen EA, Lesuer WE, Willetts L, Zellner KR, Mazzolini K, et al. 2014. Eosinophil activities modulate the immune/inflammatory character of allergic respiratory responses in mice. Allergy 69:315–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doyle AD, Jacobsen EA, Ochkur SI, Willetts L, Shim K, et al. 2013. Homologous recombination into the eosinophil peroxidase locus generates a strain of mice expressing Cre recombinase exclusively in eosinophils. J. Leukoc. Biol 94:17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Knuplez E, Krier-Burris R, Cao Y, Marsche G, O’Sullivan J, et al. 2020. Superior mouse eosinophil depletion in vivo targeting transgenic Siglec-8 instead of endogenous Siglec-F: mechanisms and pitfalls. J. Leukoc. Biol 108:43–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gleich GJ, Klion AD, Lee JJ, Weller PF. 2013. The consequences of not having eosinophils. Allergy 68:829–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jung Y, Rothenberg ME. 2014. Roles and regulation of gastrointestinal eosinophils in immunity and disease. J. Immunol 193:999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chu VT, Beller A, Rausch S, Strandmark J, Zanker M, et al. 2014. Eosinophils promote generation and maintenance of immunoglobulin-A-expressing plasma cells and contribute to gut immune homeostasis. Immunity 40:582–93 [DOI] [PubMed] [Google Scholar]
- 80.Jung Y, Wen T, Mingler MK, Caldwell JM, Wang YH, et al. 2015. IL-1β in eosinophil-mediated small intestinal homeostasis and IgA production. Mucosal Immunol 8:930–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Beller A, Kruglov A, Durek P, von Goetze V, Werner K, et al. 2020. Specific microbiota enhances intestinal IgA levels by inducing TGF-beta in T follicular helper cells of Peyer’s patches in mice. Eur. J. Immunol 50:783–94 [DOI] [PubMed] [Google Scholar]
- 82.Calcinotto A, Brevi A, Chesi M, Ferrarese R, Garcia Perez L, et al. 2018. Microbiota-driven interleukin-17-producing cells and eosinophils synergize to accelerate multiple myeloma progression. Nat. Commun 9:4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. 2010. Adaptive immune regulation in the gut: T cell–dependent and T cell–independent IgA synthesis. Annu. Rev. Immunol 28:243–73 [DOI] [PubMed] [Google Scholar]
- 84.Douzandeh-Mobarrez B, Kariminik A. 2019. Gut microbiota and IL-17A: physiological and pathological responses. Probiot. Antimicrob. Proteins 11:1–10 [DOI] [PubMed] [Google Scholar]
- 85.Sugawara R, Lee EJ, Jang MS, Jeun EJ, Hong CP, et al. 2016. Small intestinal eosinophils regulate Th17 cells by producing IL-1 receptor antagonist. J. Exp. Med 213:555–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jacobsen EA, Zellner KR, Colbert D, Lee NA, Lee JJ. 2011. Eosinophils regulate dendritic cells and Th2 pulmonary immune responses following allergen provocation. J. Immunol 187:6059–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Macpherson AJ, Yilmaz B, Limenitakis JP, Ganal-Vonarburg SC. 2018. IgA function in relation to the intestinal microbiota. Annu. Rev. Immunol 36:359–81 [DOI] [PubMed] [Google Scholar]
- 88.Hugenholtz F, de Vos WM. 2018. Mouse models for human intestinal microbiota research: a critical evaluation. Cell Mol. Life Sci 75:149–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barcik W, Boutin RCT, Sokolowska M, Finlay BB. 2020. The role of lung and gut microbiota in the pathology of asthma. Immunity 52:241–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Durack J, Christian LS, Nariya S, Gonzalez J, Bhakta NR, et al. 2020. Distinct associations of sputum and oral microbiota with atopic, immunologic, and clinical features in mild asthma. J. Allergy Clin. Immunol 146(5):1016–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grosserichter-Wagener C, Radjabzadeh D, van der Weide H, Smit KN, Kraaij R, et al. 2019. Differences in systemic IgA reactivity and circulating Th subsets in healthy volunteers with specific microbiota enterotypes. Front. Immunol 10:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hernandez JD, Tew BY, Li T, Gooden GC 2nd, Ghannam H, et al. 2020. A FACS-based approach to obtain viable eosinophils from human adipose tissue. Sci. Rep 10:13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, et al. 2011. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332:243–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chung KJ, Nati M, Chavakis T, Chatzigeorgiou A. 2018. Innate immune cells in the adipose tissue. Rev. Endocr. Metab. Disord 19:283–92 [DOI] [PubMed] [Google Scholar]
- 95.Lee EH, Itan M, Jang J, Gu HJ, Rozenberg P, et al. 2018. Eosinophils support adipocyte maturation and promote glucose tolerance in obesity. Sci. Rep 8:9894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Knights AJ, Vohralik EJ, Houweling PJ, Stout ES, Norton LJ, et al. 2020. Eosinophil function in adipose tissue is regulated by Kruppel-like factor 3 (KLF3). Nat. Commun 11:2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, et al. 2014. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 157:1292–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rozenberg P, Reichman H, Zab-Bar I, Itan M, Pasmanik-Chor M, et al. 2017. CD300f:IL-5 cross-talk inhibits adipose tissue eosinophil homing and subsequent IL-4 production. Sci. Rep 7:5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bolus WR, Gutierrez DA, Kennedy AJ, Anderson-Baucum EK, Hasty AH. 2015. CCR2 deficiency leads to increased eosinophils, alternative macrophage activation, and type 2 cytokine expression in adipose tissue. J. Leukoc. Biol 98:467–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, et al. 2013. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med 210:535–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tilg H, Zmora N, Adolph TE, Elinav E. 2020. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol 20:40–54 [DOI] [PubMed] [Google Scholar]
- 102.Fabbiano S, Suarez-Zamorano N, Chevalier C, Lazarevic V, Kieser S, et al. 2018. Functional gut microbiota remodeling contributes to the caloric restriction-induced metabolic improvements. Cell Metab 28:907–21.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Suarez-Zamorano N, Fabbiano S, Chevalier C, Stojanovic O, Colin DJ, et al. 2015. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat. Med 21:1497–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hart KM, Fabre T, Sciurba JC, Gieseck RL 3rd, Borthwick LA, et al. 2017. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-β. Sci. Transl. Med 9(396):eaal3694. [DOI] [PubMed] [Google Scholar]
- 105.Zuo L, Fulkerson PC, Finkelman FD, Mingler M, Fischetti CA, et al. 2010. IL-13 induces esophageal remodeling and gene expression by an eosinophil-independent, IL-13Rα2–inhibited pathway. J. Immunol 185:660–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.O’Shea KM, Aceves SS, Dellon ES, Gupta SK, Spergel JM, et al. 2018. Pathophysiology of eosinophilic esophagitis. Gastroenterology 154:333–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Withers SB, Forman R, Meza-Perez S, Sorobetea D, Sitnik K, et al. 2017. Eosinophils are key regulators of perivascular adipose tissue and vascular functionality. Sci. Rep 7:44571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Agabiti-Rosei C, Paini A, De Ciuceis C, Withers S, Greenstein A, et al. 2018. Modulation of vascular reactivity by perivascular adipose tissue (PVAT). Curr. Hypertens. Rep 20:44. [DOI] [PubMed] [Google Scholar]
- 109.Uderhardt S, Ackermann JA, Fillep T, Hammond VJ, Willeit J, et al. 2017. Enzymatic lipid oxidation by eosinophils propagates coagulation, hemostasis, and thrombotic disease. J. Exp. Med 214:2121–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marx C, Novotny J, Salbeck D, Zellner KR, Nicolai L, et al. 2019. Eosinophil-platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood 134:1859–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin L, Hwang BJ, Culton DA, Li N, Burette S, et al. 2018. Eosinophils mediate tissue injury in the autoimmune skin disease bullous pemphigoid. J. Investig. Dermatol 138:1032–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sakurai Y, Morioke S, Takeda T, Takahagi S, Hide M, et al. 2015. Increased thrombin generation potential in patients with chronic spontaneous urticaria. Allergol. Int 64:96–98 [DOI] [PubMed] [Google Scholar]
- 113.Shimizu S, Ogawa T, Takezawa K, Tojima I, Kouzaki H, et al. 2015. Tissue factor and tissue factor pathway inhibitor in nasal mucosa and nasal secretions of chronic rhinosinusitis with nasal polyp. Am. J. Rhinol. Allergy 29:235–42 [DOI] [PubMed] [Google Scholar]
- 114.Naidoo K, Jagot F, van den Elsen L, Pellefigues C, Jones A, et al. 2018. Eosinophils determine dermal thickening and water loss in an MC903 model of atopic dermatitis. J. Investig. Dermatol 138:2606–16 [DOI] [PubMed] [Google Scholar]
- 115.Lee JJ, Protheroe CA, Luo H, Ochkur SI, Scott GD, et al. 2015. Eosinophil-dependent skin innervation and itching following contact toxicant exposure in mice. J. Allergy Clin. Immunol 135:477–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lagrange J, Lacolley P, Wahl D, Peyrin-Biroulet L, Regnault V. 2020. Shedding light on hemostasis in patients with inflammatory bowel diseases. Clin. Gastroenterol. Hepatol In press [DOI] [PubMed] [Google Scholar]
- 117.Mastalerz L, Celinska-Lwenhoff M, Krawiec P, Batko B, Tlustochowicz W, et al. 2015. Unfavorably altered fibrin clot properties in patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome): association with thrombin generation and eosinophilia. PLOS ONE 10:e0142167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nishi R, Koike H, Ohyama K, Fukami Y, Ikeda S, et al. 2020. Differential clinicopathologic features of EGPA-associated neuropathy with and without ANCA. Neurology 94:e1726–37 [DOI] [PubMed] [Google Scholar]
- 119.Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, et al. 2002. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 125:1450–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Talbot S, Foster SL, Woolf CJ. 2016. Neuroimmunity: physiology and pathology. Annu. Rev. Immunol 34:421–47 [DOI] [PubMed] [Google Scholar]
- 121.Drake MG, Lebold KM, Roth-Carter QR, Pincus AB, Blum ED, et al. 2018. Eosinophil and airway nerve interactions in asthma. J. Leukoc. Biol 104:61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Guseva D, Rudrich U, Kotnik N, Gehring M, Patsinakidis N, et al. 2020. Neuronal branching of sensory neurons is associated with BDNF-positive eosinophils in atopic dermatitis. Clin. Exp. Allergy 50:577–84 [DOI] [PubMed] [Google Scholar]
- 123.Kobayashi H, Gleich GJ, Butterfield JH, Kita H. 2002. Human eosinophils produce neurotrophins and secrete nerve growth factor on immunologic stimuli. Blood 99:2214–20 [DOI] [PubMed] [Google Scholar]
- 124.Dileepan M, Ge XN, Bastan I, Greenberg YG, Liang Y, et al. 2020. Regulation of eosinophil recruitment and allergic airway inflammation by tropomyosin receptor kinase A. J. Immunol 204:682–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rudrich U, Gehring M, Papakonstantinou E, Illerhaus A, Engmann J, et al. 2018. Eosinophils are a major source of interleukin-31 in bullous pemphigoid. Acta Derm. Venereol 98:766–71 [DOI] [PubMed] [Google Scholar]
- 126.Pan D, Hunter DA, Schellhardt L, Fuchs A, Halevi AE, et al. 2020. T cells modulate IL-4 expression by eosinophil recruitment within decellularized scaffolds to repair nerve defects. Acta Biomater 112:149–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Foster EL, Simpson EL, Fredrikson LJ, Lee JJ, Lee NA, et al. 2011. Eosinophils increase neuron branching in human and murine skin and in vitro. PLOS ONE 6:e22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nakanishi S, Mantani Y, Haruta T, Yokoyama T, Hoshi N. 2020. Three-dimensional analysis of neural connectivity with cells in rat ileal mucosa by serial block-face scanning electron microscopy. J. Vet. Med. Sci 82:990–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Carlens J, Wahl B, Ballmaier M, Bulfone-Paus S, Forster R, et al. 2009. Common gamma-chain-dependent signals confer selective survival of eosinophils in the murine small intestine. J. Immunol 183:5600–7 [DOI] [PubMed] [Google Scholar]
- 130.Yamada T, Tani Y, Nakanishi H, Taguchi R, Arita M, et al. 2011. Eosinophils promote resolution of acute peritonitis by producing proresolving mediators in mice. FASEB J 25:561–68 [DOI] [PubMed] [Google Scholar]
- 131.Masterson JC, McNamee EN, Fillon SA, Hosford L, Harris R, et al. 2015. Eosinophil-mediated signalling attenuates inflammatory responses in experimental colitis. Gut 64:1236–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Takeda K, Shiraishi Y, Ashino S, Han J, Jia Y, et al. 2015. Eosinophils contribute to the resolution of lung-allergic responses following repeated allergen challenge. J. Allergy Clin. Immunol 135:451–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Isobe Y, Kato T, Arita M. 2012. Emerging roles of eosinophils and eosinophil-derived lipid mediators in the resolution of inflammation. Front. Immunol 3:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM, et al. 2001. Extensive eosinophil degranulation and peroxidase-mediated oxidation of airway proteins do not occur in a mouse ovalbumin-challenge model of pulmonary inflammation. J. Immunol 167:1672–82 [DOI] [PubMed] [Google Scholar]
- 135.Jacobsen EA, Ochkur SI, Pero RS, Taranova AG, Protheroe CA, et al. 2008. Allergic pulmonary inflammation in mice is dependent on eosinophil-induced recruitment of effector T cells. J. Exp. Med 205:699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Walsh ER, Sahu N, Kearley J, Benjamin E, Kang BH, et al. 2008. Strain-specific requirement for eosinophils in the recruitment of T cells to the lung during the development of allergic asthma. J. Exp. Med 205:1285–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fulkerson PC, Fischetti CA, Rothenberg ME. 2006. Eosinophils and CCR3 regulate interleukin-13 transgene-induced pulmonary remodeling. Am. J. Pathol 169:2117–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jacobsen EA, LeSuer WE, Nazaroff CD, Ochkur SI, Doyle AD, et al. 2019. Eosinophils induce recruitment and activation of ILC2s. J. Allergy Clin. Immunol 143(2 Suppl.):AB289 (Abstr.) [Google Scholar]
- 139.Ochkur SI, Jacobsen EA, Protheroe CA, Biechele TL, Pero RS, et al. 2007. Coexpression of IL-5 and eotaxin-2 in mice creates an eosinophil-dependent model of respiratory inflammation with characteristics of severe asthma. J. Immunol 178:7879–89 [DOI] [PubMed] [Google Scholar]
- 140.Mould AW, Ramsay AJ, Matthaei KI, Young IG, Rothenberg ME, et al. 2000. The effect of IL-5 and eotaxin expression in the lung on eosinophil trafficking and degranulation and the induction of bronchial hyperreactivity. J. Immunol 164:2142–50 [DOI] [PubMed] [Google Scholar]
- 141.Jacobsen EA, Ochkur SI, Doyle AD, LeSuer WE, Li W, et al. 2017. Lung pathologies in a chronic inflammation mouse model are independent of eosinophil degranulation. Am. J. Respir. Crit. Care Med 195:1321–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Doyle AD, Mukherjee M, LeSuer WE, Bittner TB, Pasha SM, et al. 2019. Eosinophil-derived IL-13 promotes emphysema. Eur. Respir. J 53:1801291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fattouh R, Al-Garawi A, Fattouh M, Arias K, Walker TD, et al. 2011. Eosinophils are dispensable for allergic remodeling and immunity in a model of house dust mite-induced airway disease. Am. J. Respir. Crit. Care Med 183:179–88 [DOI] [PubMed] [Google Scholar]
- 144.Peters MC, Mauger D, Ross KR, Phillips B, Gaston B, et al. 2020. Evidence for exacerbation-prone asthma and predictive biomarkers of exacerbation frequency. Am. J. Respir. Crit. Care Med 202(7):973–82. 10.1164/rccm.201909-1813OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Filippone RT, Sahakian L, Apostolopoulos V, Nurgali K. 2019. Eosinophils in inflammatory bowel disease. Inflamm. Bowel Dis 25:1140–51 [DOI] [PubMed] [Google Scholar]
- 146.Vieira AT, Fagundes CT, Alessandri AL, Castor MG, Guabiraba R, et al. 2009. Treatment with a novel chemokine-binding protein or eosinophil lineage-ablation protects mice from experimental colitis. Am. J. Pathol 175:2382–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Radnai B, Sturm EM, Stancic A, Jandl K, Labocha S, et al. 2016. Eosinophils contribute to intestinal inflammation via chemoattractant receptor-homologous molecule expressed on Th2 cells, CRTH2, in experimental Crohn’s disease. J. Crohn’s Colitis 10:1087–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.De Salvo C, Wang XM, Pastorelli L, Mattioli B, Omenetti S, et al. 2016. IL-33 drives eosinophil infiltration and pathogenic type 2 helper T-cell immune responses leading to chronic experimental ileitis. Am. J. Pathol 186:885–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hogan SP, Mishra A, Brandt EB, Royalty MP, Pope SM, et al. 2001. A pathological function for eotaxin and eosinophils in eosinophilic gastrointestinal inflammation. Nat. Immunol 2:353–60 [DOI] [PubMed] [Google Scholar]
- 150.Mishra A, Rothenberg ME. 2003. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 125:1419–27 [DOI] [PubMed] [Google Scholar]
- 151.Youngblood BA, Brock EC, Leung J, Falahati R, Bochner BS, et al. 2019. Siglec-8 antibody reduces eosinophils and mast cells in a transgenic mouse model of eosinophilic gastroenteritis. JCI Insight 4:e126219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Reichman H, Itan M, Rozenberg P, Yarmolovski T, Brazowski E, et al. 2019. Activated eosinophils exert antitumorigenic activities in colorectal cancer. Cancer Immunol. Res 7:388–400 [DOI] [PubMed] [Google Scholar]
- 153.Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, et al. 2015. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8+ T cells. Nat. Immunol 16:609–17 [DOI] [PubMed] [Google Scholar]
- 154.Hollande C, Boussier J, Ziai J, Nozawa T, Bondet V, et al. 2019. Inhibition of the dipeptidyl peptidase DPP4 (CD26) reveals IL-33-dependent eosinophil-mediated control of tumor growth. Nat. Immunol 20:257–64 [DOI] [PubMed] [Google Scholar]
- 155.Simon SCS, Hu X, Panten J, Grees M, Renders S, et al. 2020. Eosinophil accumulation predicts response to melanoma treatment with immune checkpoint inhibitors. Oncoimmunology 9:1727116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nunez-Nateras R, Castle EP, Protheroe CA, Stanton ML, Ocal TI, et al. 2014. Predicting response to bacillus Calmette-Guerin (BCG) in patients with carcinoma in situ of the bladder. Urol. Oncol 32:45.e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cameron GJM, Cautivo KM, Loering S, Jiang SH, Deshpande AV, et al. 2019. Group 2 innate lymphoid cells are redundant in experimental renal ischemia-reperfusion injury. Front. Immunol 10:826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Onyema OO, Guo Y, Mahgoub B, Wang Q, Manafi A, et al. 2019. Eosinophils downregulate lung alloimmunity by decreasing TCR signal transduction. JCI Insight 4:e128241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, et al. 2013. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153:376–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hart TK, Cook RM, Zia-Amirhosseini P, Minthorn E, Sellers TS, et al. 2001. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J. Allergy Clin. Immunol 108:250–57 [DOI] [PubMed] [Google Scholar]
- 161.Menzies-Gow A, Flood-Page P, Sehmi R, Burman J, Hamid Q, et al. 2003. Anti-IL-5 (mepolizumab) therapy induces bone marrow eosinophil maturational arrest and decreases eosinophil progenitors in the bronchial mucosa of atopic asthmatics. J. Allergy Clin. Immunol 111:714–19 [DOI] [PubMed] [Google Scholar]
- 162.Stein ML, Villanueva JM, Buckmeier BK, Yamada Y, Filipovich AH, et al. 2008. Anti-IL-5 (mepolizumab) therapy reduces eosinophil activation ex vivo and increases IL-5 and IL-5 receptor levels. J. Allergy Clin. Immunol 121:1473–83.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. 2003. Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am. J. Respir. Crit. Care Med 167:199–204 [DOI] [PubMed] [Google Scholar]
- 164.Zhang J, Kuvelkar R, Murgolo NJ, Taremi SS, Chou CC, et al. 1999. Mapping and characterization of the epitope(s) of Sch 55700, a humanized mAb, that inhibits human IL-5. Int. Immunol 11:1935–44 [DOI] [PubMed] [Google Scholar]
- 165.Liddament M, Husten J, Estephan T, Laine D, Mabon D, et al. 2019. Higher binding affinity and in vitro potency of reslizumab for interleukin-5 compared with mepolizumab. Allergy Asthma Immunol. Res 11:291–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kolbeck R, Kozhich A, Koike M, Peng L, Andersson CK, et al. 2010. MEDI-563, a humanized anti-IL-5 receptor alpha mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J. Allergy Clin. Immunol 125:1344–53.e2 [DOI] [PubMed] [Google Scholar]
- 167.Busse WW, Katial R, Gossage D, Sari S, Wang B, et al. 2010. Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti-IL-5 receptor alpha antibody, in a phase I study of subjects with mild asthma. J. Allergy Clin. Immunol 125:1237–44.e2 [DOI] [PubMed] [Google Scholar]
- 168.Laviolette M, Gossage DL, Gauvreau G, Leigh R, Olivenstein R, et al. 2013. Effects of benralizumab on airway eosinophils in asthmatic patients with sputum eosinophilia. J. Allergy Clin. Immunol 132:1086–96.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Leckie MJ, ten Brinke A, Khan J, Diamant Z, O’Connor BJ, et al. 2000. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 356:2144–48 [DOI] [PubMed] [Google Scholar]
- 170.Kiwamoto T, Kawasaki N, Paulson JC, Bochner BS. 2012. Siglec-8 as a drugable target to treat eosinophil and mast cell-associated conditions. Pharmacol. Ther 135:327–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Youngblood BA, Brock EC, Leung J, Falahati R, Bryce PJ, et al. 2019. AK002, a humanized sialic acid-binding immunoglobulin-like lectin-8 antibody that induces antibody-dependent cell-mediated cytotoxicity against human eosinophils and inhibits mast cell-mediated anaphylaxis in mice. Int. Arch. Allergy Immunol 180:91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kikly KK, Bochner BS, Freeman SD, Tan KB, Gallagher KT, et al. 2000. Identification of SAF-2, a novel siglec expressed on eosinophils, mast cells, and basophils. J. Allergy Clin. Immunol 105:1093–100 [DOI] [PubMed] [Google Scholar]
- 173.Dellon ES, Peterson KA, Murray JA, Falk GW, Gonsalves N, et al. 2020. Anti–Siglec-8 antibody for eosinophilic gastritis and duodenitis. N. Engl. J. Med 383:1624–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, et al. 2015. Asthma. Nat. Rev. Dis. Primers 1:15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yancey SW, Keene ON, Albers FC, Ortega H, Bates S, et al. 2017. Biomarkers for severe eosinophilic asthma. J. Allergy Clin. Immunol 140:1509–18 [DOI] [PubMed] [Google Scholar]
- 176.Ray A, Camiolo M, Fitzpatrick A, Gauthier M, Wenzel SE. 2020. Are we meeting the promise of endotypes and precision medicine in asthma? Physiol. Rev 100:983–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Flood-Page P, Swenson C, Faiferman I, Matthews J, Williams M, et al. 2007. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am. J. Respir. Crit. Care Med 176:1062–71 [DOI] [PubMed] [Google Scholar]
- 178.Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, et al. 2009. Mepolizumab and exacerbations of refractory eosinophilic asthma. N. Engl. J. Med 360:973–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, et al. 2009. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N. Engl. J. Med 360:985–93 [DOI] [PubMed] [Google Scholar]
- 180.Castro M, Mathur S, Hargreave F, Boulet LP, Xie F, et al. 2011. Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am. J. Respir. Crit. Care Med 184:1125–32 [DOI] [PubMed] [Google Scholar]
- 181.Castro M, Zangrilli J, Wechsler ME, Bateman ED, Brusselle GG, et al. 2015. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir. Med 3:355–66 [DOI] [PubMed] [Google Scholar]
- 182.FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, et al. 2016. Benralizumab, an anti-interleukin-5 receptor alpha monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 388:2128–41 [DOI] [PubMed] [Google Scholar]
- 183.Bleecker ER, FitzGerald JM, Chanez P, Papi A, Weinstein SF, et al. 2016. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting β2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet 388:2115–27 [DOI] [PubMed] [Google Scholar]
- 184.Pavord ID, Chanez P, Criner GJ, Kerstjens HAM, Korn S, et al. 2017. Mepolizumab for eosinophilic chronic obstructive pulmonary disease. N. Engl. J. Med 377(17):1613–29 [DOI] [PubMed] [Google Scholar]
- 185.Criner GJ, Celli BR, Singh D, Agusti A, Papi A, et al. 2020. Predicting response to benralizumab in chronic obstructive pulmonary disease: analyses of GALATHEA and TERRANOVA studies. Lancet Respir. Med 8(2):158–70 [DOI] [PubMed] [Google Scholar]
- 186.Criner GJ, Celli BR, Brightling CE, Agusti A, Papi A, et al. 2019. Benralizumab for the prevention of COPD exacerbations. N. Engl. J. Med 381:1023–34 [DOI] [PubMed] [Google Scholar]
- 187.Fokkens WJ, Lund VJ, Hopkins C, Hellings PW, Kern R, et al. 2020. European Position Paper on Rhinosinusitis and Nasal Polyps 2020. Rhinology 58:1–464 [DOI] [PubMed] [Google Scholar]
- 188.Tan BK, Klingler AI, Poposki JA, Stevens WW, Peters AT, et al. 2017. Heterogeneous inflammatory patterns in chronic rhinosinusitis without nasal polyps in Chicago, Illinois. J. Allergy Clin. Immunol 139:699–703.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bochner BS, Stevens WW. 2021. Biology and function of eosinophils in chronic rhinosinusitis with or without nasal polyps. Allergy Asthma Immunol. Res 13(1):8–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang Y, Gevaert E, Lou H, Wang X, Zhang L, et al. 2017. Chronic rhinosinusitis in Asia. J. Allergy Clin. Immunol 140:1230–39 [DOI] [PubMed] [Google Scholar]
- 191.Kim SJ, Lee KH, Kim SW, Cho JS, Park YK, et al. 2013. Changes in histological features of nasal polyps in a Korean population over a 17-year period. Otolaryngol. Head Neck Surg 149:431–37 [DOI] [PubMed] [Google Scholar]
- 192.Wang W, Gao Y, Zhu Z, Zha Y, Wang X, et al. 2019. Changes in the clinical and histological characteristics of Chinese chronic rhinosinusitis with nasal polyps over 11 years. Int. Forum. Allergy Rhinol 9:149–57 [DOI] [PubMed] [Google Scholar]
- 193.Katotomichelakis M, Tantilipikorn P, Holtappels G, De Ruyck N, Feng L, et al. 2013. Inflammatory patterns in upper airway disease in the same geographical area may change over time. Am. J. Rhinol. Allergy 27:354–60 [DOI] [PubMed] [Google Scholar]
- 194.Simon HU, Yousefi S, Schranz C, Schapowal A, Bachert C, et al. 1997. Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J. Immunol 158:3902–8 [PubMed] [Google Scholar]
- 195.Persson EK, Verstraete K, Heyndrickx I, Gevaert E, Aegerter H, et al. 2019. Protein crystallization promotes type 2 immunity and is reversible by antibody treatment. Science 364(6442):eaaw4295. [DOI] [PubMed] [Google Scholar]
- 196.Perez-Novo CA, Watelet JB, Claeys C, Van Cauwenberge P, Bachert C. 2005. Prostaglandin, leukotriene, and lipoxin balance in chronic rhinosinusitis with and without nasal polyposis. J. Allergy Clin. Immunol 115:1189–96 [DOI] [PubMed] [Google Scholar]
- 197.Yun Y, Kanda A, Kobayashi Y, Van Bui D, Suzuki K, et al. 2020. Increased CD69 expression on activated eosinophils in eosinophilic chronic rhinosinusitis correlates with clinical findings. Allergol. Int 69:232–38 [DOI] [PubMed] [Google Scholar]
- 198.Gevaert E, Delemarre T, De Volder J, Zhang N, Holtappels G, et al. 2020. Charcot-Leyden crystals promote neutrophilic inflammation in patients with nasal polyposis. J. Allergy Clin. Immunol 145:427–30.e4 [DOI] [PubMed] [Google Scholar]
- 199.Watelet JB, Bachert C, Claeys C, Van Cauwenberge P. 2004. Matrix metalloproteinases MMP-7, MMP-9 and their tissue inhibitor TIMP-1: expression in chronic sinusitis versus nasal polyposis. Allergy 59:54–60 [DOI] [PubMed] [Google Scholar]
- 200.Gevaert P, Van Bruaene N, Cattaert T, Van Steen K, Van Zele T, et al. 2011. Mepolizumab, a humanized anti-IL-5 mAb, as a treatment option for severe nasal polyposis. J. Allergy Clin. Immunol 128:989–95. e1–8 [DOI] [PubMed] [Google Scholar]
- 201.Bachert C, Sousa AR, Lund VJ, Scadding GK, Gevaert P, et al. 2017. Reduced need for surgery in severe nasal polyposis with mepolizumab: randomized trial. J. Allergy Clin. Immunol 140:1024–31.e14 [DOI] [PubMed] [Google Scholar]
- 202.Gevaert P, Lang-Loidolt D, Lackner A, Stammberger H, Staudinger H, et al. 2006. Nasal IL-5 levels determine the response to anti-IL-5 treatment in patients with nasal polyps. J. Allergy Clin. Immunol 118:1133–41 [DOI] [PubMed] [Google Scholar]
- 203.Oldhoff JM, Darsow U, Werfel T, Katzer K, Wulf A, et al. 2005. Anti-IL-5 recombinant humanized monoclonal antibody (mepolizumab) for the treatment of atopic dermatitis. Allergy 60(5):693–96 [DOI] [PubMed] [Google Scholar]
- 204.Kang EG, Narayana PK, Pouliquen IJ, Lopez MC, Ferreira-Cornwell MC, et al. 2020. Efficacy and safety of mepolizumab administered subcutaneously for moderate to severe atopic dermatitis. Allergy 75(4):950–53 [DOI] [PubMed] [Google Scholar]
- 205.Bernstein JA, Singh U, Rao MB, Berendts K, Zhang X, et al. 2020. Benralizumab for chronic spontaneous urticaria. N. Engl. J. Med 383(14):1389–91 [DOI] [PubMed] [Google Scholar]
- 206.Kolkhir P, Church MK, Altrichter S, Skov PS, Hawro T, et al. 2020. Eosinopenia, in chronic spontaneous urticaria, is associated with high disease activity, autoimmunity, and poor response to treatment. J. Allergy Clin. Immunol. Pract 8(1):318–25 [DOI] [PubMed] [Google Scholar]
- 207.Amber KT, Valdebran M, Kridin K, Grando SA. 2018. The role of eosinophils in bullous pemphigoid: a developing model of eosinophil pathogenicity in mucocutaneous disease. Front. Med 5:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, et al. 2017. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy 72(7):1105–13 [DOI] [PubMed] [Google Scholar]
- 209.Simon D, Yousefi S, Cazzaniga S, Bürgler C, Radonjic S, et al. 2020. Mepolizumab failed to affect bullous pemphigoid: a randomized, placebo-controlled, double-blind phase 2 pilot study. Allergy 75(3):669–72 [DOI] [PubMed] [Google Scholar]
- 210.Dellon ES, Liacouras CA, Molina-Infante J, Furuta GT, Spergel JM, et al. 2018. Updated international consensus diagnostic criteria for eosinophilic esophagitis: proceedings of the AGREE conference. Gastroenterology 155:1022–33.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lyles J, Rothenberg M. 2019. Role of genetics, environment, and their interactions in the pathogenesis of eosinophilic esophagitis. Curr. Opin. Immunol 60:46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Blanchard C, Wang N, Rothenberg ME. 2006. Eosinophilic esophagitis: pathogenesis, genetics, and therapy. J. Allergy Clin. Immunol 118:1054–59 [DOI] [PubMed] [Google Scholar]
- 213.Alexander ES, Martin LJ, Collins MH, Kottyan LC, Sucharew H, et al. 2014. Twin and family studies reveal strong environmental and weaker genetic cues explaining heritability of eosinophilic esophagitis. J. Allergy Clin. Immunol 134:1084–92.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rothenberg ME, Spergel JM, Sherrill JD, Annaiah K, Martin LJ, et al. 2010. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat. Genet 42:289–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kottyan LC, Davis BP, Sherrill JD, Liu K, Rochman M, et al. 2014. Genome-wide association analysis of eosinophilic esophagitis provides insight into the tissue specificity of this allergic disease. Nat. Genet 46:895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sleiman PM, Wang ML, Cianferoni A, Aceves S, Gonsalves N, et al. 2014. GWAS identifies four novel eosinophilic esophagitis loci. Nat. Commun 5:5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Litosh VA, Rochman M, Rymer JK, Porollo A, Kottyan LC, et al. 2017. Calpain-14 and its association with eosinophilic esophagitis. J. Allergy Clin. Immunol 139:1762–71.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Allakhverdi Z, Comeau MR, Jessup HK, Yoon BR, Brewer A, et al. 2007. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med 204:253–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wong CK, Hu S, Cheung PF, Lam CW. 2010. Thymic stromal lymphopoietin induces chemotactic and prosurvival effects in eosinophils: implications in allergic inflammation. Am. J. Respir. Cell Mol. Biol 43:305–15 [DOI] [PubMed] [Google Scholar]
- 220.Noti M, Wojno ED, Kim BS, Siracusa MC, Giacomin PR, et al. 2013. Thymic stromal lymphopoietin-elicited basophil responses promote eosinophilic esophagitis. Nat. Med 19:1005–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Stein ML, Collins MH, Villanueva JM, Kushner JP, Putnam PE, et al. 2006. Anti-IL-5 (mepolizumab) therapy for eosinophilic esophagitis. J. Allergy Clin. Immunol 118:1312–19 [DOI] [PubMed] [Google Scholar]
- 222.Straumann A, Conus S, Grzonka P, Kita H, Kephart G, et al. 2010. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo-controlled, double-blind trial. Gut 59:21–30 [DOI] [PubMed] [Google Scholar]
- 223.Assa’ad AH, Gupta SK, Collins MH, Thomson M, Heath AT, et al. 2011. An antibody against IL-5 reduces numbers of esophageal intraepithelial eosinophils in children with eosinophilic esophagitis. Gastroenterology 141:1593–604 [DOI] [PubMed] [Google Scholar]
- 224.Spergel JM, Rothenberg ME, Collins MH, Furuta GT, Markowitz JE, et al. 2012. Reslizumab in children and adolescents with eosinophilic esophagitis: results of a double-blind, randomized, placebo-controlled trial. J. Allergy Clin. Immunol 129:456–63.e3 [DOI] [PubMed] [Google Scholar]
- 225.Shoda T, Wen T, Caldwell JM, Collins MH, Besse JA, et al. 2020. Molecular, endoscopic, histologic, and circulating biomarker-based diagnosis of eosinophilic gastritis: multi-site study. J. Allergy Clin. Immunol 145:255–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hirano I, Dellon ES, Hamilton JD, Collins MH, Peterson K, et al. 2020. Efficacy of dupilumab in a phase 2 randomized trial of adults with active eosinophilic esophagitis. Gastroenterology 158:111–22.e10 [DOI] [PubMed] [Google Scholar]
- 227.Hirano I, Collins MH, Assouline-Dayan Y, Evans L, Gupta S, et al. 2019. RPC4046, a monoclonal antibody against IL13, reduces histologic and endoscopic activity in patients with eosinophilic esophagitis. Gastroenterology 156:592–603.e10 [DOI] [PubMed] [Google Scholar]
- 228.Rothenberg ME, Wen T, Greenberg A, Alpan O, Enav B, et al. 2015. Intravenous anti-IL-13 mAb QAX576 for the treatment of eosinophilic esophagitis. J. Allergy Clin. Immunol 135:500–7 [DOI] [PubMed] [Google Scholar]
- 229.Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, et al. 2003. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N. Engl. J. Med 348:1201–14 [DOI] [PubMed] [Google Scholar]
- 230.Roufosse F, Cogan E, Goldman M. 2007. Lymphocytic variant hypereosinophilic syndromes. Immunol. Allergy Clin. N. Am 27:389–413 [DOI] [PubMed] [Google Scholar]
- 231.Garrett JK, Jameson SC, Thomson B, Collins MH, Wagoner LE, et al. 2004. Anti-interleukin-5 (mepolizumab) therapy for hypereosinophilic syndromes. J. Allergy Clin. Immunol 113:115–19 [DOI] [PubMed] [Google Scholar]
- 232.Plotz SG, Simon HU, Darsow U, Simon D, Vassina E, et al. 2003. Use of an anti-interleukin-5 antibody in the hypereosinophilic syndrome with eosinophilic dermatitis. N. Engl. J. Med 349:2334–39 [DOI] [PubMed] [Google Scholar]
- 233.Rothenberg ME, Klion AD, Roufosse FE, Kahn JE, Weller PF, et al. 2008. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N. Engl. J. Med 358:1215–28 [DOI] [PubMed] [Google Scholar]
- 234.Roufosse F, Kahn J-E, Rothenberg ME, Wardlaw AJ, Klion AD, et al. 2020. Efficacy and safety of mepolizumab in hypereosinophilic syndrome: a phase III, randomized, placebo-controlled trial. J. Allergy Clin. Immunol 146(6):1397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kuang FL, Legrand F, Makiya M, Ware J, Wetzler L, et al. 2019. Benralizumab for PDGFRA-negative hypereosinophilic syndrome. N. Engl. J. Med 380:1336–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kuang FL, Fay MP, Ware J, Wetzler L, Holland-Thomas N, et al. 2018. Long-term clinical outcomes of high-dose mepolizumab treatment for hypereosinophilic syndrome. J. Allergy Clin. Immunol. Pract 6:1518–27.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Roufosse F, de Lavareille A, Schandene L, Cogan E, Georgelas A, et al. 2010. Mepolizumab as a corticosteroid-sparing agent in lymphocytic variant hypereosinophilic syndrome. J. Allergy Clin. Immunol 126:828–35.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Khoury P, Grayson PC, Klion AD. 2014. Eosinophils in vasculitis: characteristics and roles in pathogenesis. Nat. Rev. Rheumatol 10:474–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, et al. 2013. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 65:1–11 [DOI] [PubMed] [Google Scholar]
- 240.Khoury P, Zagallo P, Talar-Williams C, Santos CS, Dinerman E, et al. 2012. Serum biomarkers are similar in Churg-Strauss syndrome and hypereosinophilic syndrome. Allergy 67:1149–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sable-Fourtassou R, Cohen P, Mahr A, Pagnoux C, Mouthon L, et al. 2005. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann. Intern. Med 143:632–38 [DOI] [PubMed] [Google Scholar]
- 242.Lyons PA, Peters JE, Alberici F, Liley J, Coulson RMR, et al. 2019. Genome-wide association study of eosinophilic granulomatosis with polyangiitis reveals genomic loci stratified by ANCA status. Nat. Commun 10:5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jakiela B, Szczeklik W, Plutecka H, Sokolowska B, Mastalerz L, et al. 2012. Increased production of IL-5 and dominant Th2-type response in airways of Churg-Strauss syndrome patients. Rheumatology 51:1887–93 [DOI] [PubMed] [Google Scholar]
- 244.Kim S, Marigowda G, Oren E, Israel E, Wechsler ME. 2010. Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. J. Allergy Clin. Immunol 125:1336–43 [DOI] [PubMed] [Google Scholar]
- 245.Moosig F, Gross WL, Herrmann K, Bremer JP, Hellmich B. 2011. Targeting interleukin-5 in refractory and relapsing Churg-Strauss syndrome. Ann. Intern. Med 155:341–43 [DOI] [PubMed] [Google Scholar]
- 246.Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, et al. 2017. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N. Engl. J. Med 376:1921–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Jackson DJ, Korn S, Mathur SK, Barker P, Meka VG, et al. 2020. Safety of eosinophil-depleting therapy for severe, eosinophilic asthma: focus on benralizumab. Drug Saf 43:409–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Nakahata T, Spicer SS, Leary AG, Ogawa M, Franklin W, et al. 1984. Circulating eosinophil colony-forming cells in pure eosinophil aplasia. Ann. Intern. Med 101:321–24 [DOI] [PubMed] [Google Scholar]
- 249.Krantz SB, Kao V. 1967. Studies on red cell aplasia: I. Demonstration of a plasma inhibitor to heme synthesis and an antibody to erythroblast nuclei. PNAS 58:493–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Prazma CM, Bel EH, Price RG, Bradford ES, Albers FC, et al. 2019. Oral corticosteroid dose changes and impact on peripheral blood eosinophil counts in patients with severe eosinophilic asthma: a post hoc analysis. Respir. Res 20:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Al Efraij K, Johnson KM, Wiebe D, Sadatsafavi M, FitzGerald JM. 2019. A systematic review of the adverse events and economic impact associated with oral corticosteroids in asthma. J. Asthma 56:1334–46 [DOI] [PubMed] [Google Scholar]
- 252.Busse WW, Bleecker ER, FitzGerald JM, Ferguson GT, Barker P, et al. 2019. Long-term safety and efficacy of benralizumab in patients with severe, uncontrolled asthma: 1-year results from the BORA phase 3 extension trial. Lancet Respir. Med 7:46–59 [DOI] [PubMed] [Google Scholar]
- 253.Kavanagh JE, d’Ancona G, Elstad M, Green L, Fernandes M, et al. 2020. Real-world effectiveness and the characteristics of a “super-responder” to mepolizumab in severe eosinophilic asthma. Chest 158:491–500 [DOI] [PubMed] [Google Scholar]
- 254.Kavanagh JE, Hearn AP, Dhariwal J, d’Ancona G, Douiri A, et al. 2021. Real world effectiveness of benralizumab in severe eosinophilic asthma. Chest 159:496–506 [DOI] [PubMed] [Google Scholar]
- 255.Khatri S, Moore W, Gibson PG, Leigh R, Bourdin A, et al. 2019. Assessment of the long-term safety of mepolizumab and durability of clinical response in patients with severe eosinophilic asthma. J. Allergy Clin. Immunol 143:1742–51.e7 [DOI] [PubMed] [Google Scholar]
- 256.Roufosse FE, Kahn JE, Gleich GJ, Schwartz LB, Singh AD, et al. 2013. Long-term safety of mepolizumab for the treatment of hypereosinophilic syndromes. J. Allergy Clin. Immunol 131:461–67.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Efremova M, Vento-Tormo R, Park JE, Teichmann SA, James KR. 2020. Immunology in the era of single-cell technologies. Annu. Rev. Immunol 38:727–57 [DOI] [PubMed] [Google Scholar]
- 258.Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL. 2019. The myeloid cell compartment—cell by cell. Annu. Rev. Immunol 37:269–93 [DOI] [PubMed] [Google Scholar]
- 259.Wen T, Rothenberg ME. 2019. Cell-by-cell deciphering of T cells in allergic inflammation. J. Allergy Clin. Immunol 144:1143–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Philpott M, Cribbs AP, Brown T Jr., Brown T Sr., Oppermann U. 2020. Advances and challenges in epigenomic single-cell sequencing applications. Curr. Opin. Chem. Biol 57:17–26 [DOI] [PubMed] [Google Scholar]
- 261.Nei Y, Obata-Ninomiya K, Tsutsui H, Ishiwata K, Miyasaka M, et al. 2013. GATA-1 regulates the generation and function of basophils. PNAS 110:18620–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Sharma S, Tomar S, Dharne M, Ganesan V, Smith A, et al. 2019. Deletion of dblGata motif leads to increased predisposition and severity of IgE-mediated food-induced anaphylaxis response. PLOS ONE 14:e0219375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Takemoto CM, Brandal S, Jegga AG, Lee YN, Shahlaee A, et al. 2010. PU.1 positively regulates GATA-1 expression in mast cells. J. Immunol 184:4349–61 [DOI] [PubMed] [Google Scholar]